 Alejandro Ciocci Pardo*
Alejandro Ciocci Pardo* Leandro A. Díaz ZegarraLuisa F. González ArbeláezErnesto A. AielloSusana M. Mosca
Leandro A. Díaz ZegarraLuisa F. González ArbeláezErnesto A. AielloSusana M. Mosca- Centro de Investigaciones Cardiovasculares “Dr Horacio E Cingolani”, CCT-CONICET, Facultad de Ciencias Médicas, Universidad Nacional de La Plata. La Plata, Buenos Aires, Argentina
The increase of intracellular Ca2+ concentration, produced principally by its influx through the L-type Ca2+ channels, is one of the major contributors to the ischemia-reperfusion injury. The inhibition of those channels in different experimental models was effective to ameliorate the post-ischemic damage. However, at a clinical level, the results were contradictory. Recent results of our group obtained in an ¨ex vivo¨ heart model demonstrated that a chemical derived from acetazolamide, the N-methylacetazolamide (NMA) protected the heart against ischemia-reperfusion injury, diminishing the infarct size and improving the post-ischemic recovery of myocardial function and mitochondrial dynamic. A significant inhibitory action on L-type Ca2+ channels was also detected after NMA treatment, suggesting this action as responsible for the beneficial effects on myocardium exerted by this compound. Although these results were promising, the effectiveness of NMA in the treatment of ischemic heart disease in humans as well as the advantages or disadvantages in comparison to the classic calcium antagonists needs to be investigated.
Introduction
Ischemic heart disease (IHD) is one of the most frequent causes of heart failure and remains the leading reason of mortality worldwide (Bauersachs et al., 2019). IHD is normally attributed to coronary artery disease leading to a diminution of blood supply, myocyte death and contractile impairment (Severino et al., 2020). Although reperfusion appears as the best strategy to attenuate the ischemic damage, it produces an additional damage called reperfusion injury. Several interventions and treatments have been developed to attenuate the ischemia-reperfusion injury. In this sense, it was previously described that ischemic pre-and postconditioning confers cardioprotection through the activation of several protective mechanisms by the application of short periods of ischemia-reperfusion, before or after the main insult, respectively (Heusch, 2015; Donato et al., 2017; Wu et al., 2021). Although numerous factors have been implicated in the ischemia-reperfusion injury, Ca2+ overload appears to play a crucial role (Dibb et al., 2007; Murphy and Steenbergen, 2008; Kalogeris et al., 2016; Pittas et al., 2018).
Ca2+ overload: role of l-type Ca2+ channel
In cardiac muscle, membrane depolarization produces the influx of extracellular Ca2+ through the cardiac α1 subunit of voltage-gated L-type Ca2+ channel (CaC) which activates ryanodine receptor 2 (RyR2). Ca2+ release triggers shortening of the contractile unit, the sarcomere, resulting in the force generation. Muscle relaxation occurs when Ca2+ is removed from the contractile unit through the combined action of Ca2+ pumps and Na+/Ca2+ exchangers (Barry and Bridge, 1993; Dibb et al., 2007).
The purified CaC contains five subunits, the principal or pore-forming subunit, α1 and different auxiliary subunits, α2, β, δ, and γ. The auxiliary subunits are non-covalently linked to the α1 subunit, modulating the biophysical properties and trafficking of the α1 subunit onto the membrane (Catterall, 1995; Striessnig et al., 2015). The α1 subunit corresponds to the pore-forming segment of CaC that allows the passage of Ca2+ ions and is composed of approximately 2000 amino acids. The other components serve as auxiliary subunits modifying the channel function.
Ca2+ homeostasis is particularly important for myocardial cell structure and function. During ischemia-reperfusion this process is altered and the intracellular Ca2+ concentration increases generating the previously mentioned Ca2+ overload (Smith and Eisner, 2019; Wang et al., 2020). Therefore, the prevention and or the treatment of Ca2+ overload can protect cardiac myocytes against ischemia-reperfusion injury (Han et al., 2019; Wang et al., 2020). One useful approach to prevent or treat Ca2+ overload is the sarcolemmal Ca2+ channels blockers, demonstrating that CaC is an important target to protect the heart against ischemia-reperfusion injury (Talukder et al., 2009). These drugs such as dihydropyridines (DHPs) or phenylalkylamines, bind to a region close to the pore (α1-subunit) decreasing the Ca2+ entry to the cell.
Previous studies from our and other laboratories demonstrated in ¨ex vivo¨ experimental models the beneficial effects of Ca2+ channels blockers on the hearts during reperfusion (Moreyra et al., 1994; Chiappe de Cingolani et al., 1996; Simonovic and Jeremic, 2017) showing a reduction of infarct size and the postischemic contractile dysfunction. At a clinical level a reduction in myocardial oxygen consumption due to negative inotropic and chronotropic effects of CaC blockers were referred as anti-ischemic effects of these compounds (Gross et al., 1999). However, the use of some of them in patients did not modify the survival and post-infarct cellular injury (Elliott and Ram, 2011; Sueta et al., 2017). In other words, the clinical use of traditional CaC blockers in myocardial infarct is still in dispute because of their marked hemodynamic effects. Therefore, the exploration of effective therapeutic strategies against these cardiovascular disorders is still an essential research direction.
N-methylacetazolamide (NMA)
N-methylacetazolamide (NMA) belongs to a group of chemicals derived from the carbonic anhydrase (CA) inhibitor acetazolamide. In this case, a methyl group substituted one H+ in the sulfonamide moiety of acetazolamide. This substitution results in an approximately 200-fold decrease in binding affinity to CA maintaining the physical–chemical properties of acetazolamide (Maren, 1956; Teppema1 and Swenson, 2015).
The lack of inhibitory action of NMA on CA was revealed in our experiments on isolated papillary muscles subjected to an acid load, which showed that this drug did not contribute to the H+ efflux or the intracellular pH recovery (Ciocci Pardo et al., 2022).
Cardiac effects
We recently demonstrated the beneficial effects of NMA on the alterations subsequent to ischemia and reperfusion in the isolated rat heart (Ciocci Pardo et al., 2022). Our experiments showed that the treatment of hearts during the first 10 min of reperfusion with NMA 5 μM was able to decrease the infarct size (measured by TTC staining technique and expressed as percentage of risk area) produced by 30 min of global ischemia and 60 min of reperfusion. Thus, while in untreated hearts an infarction of approximately 35% was detected, in those treated with NMA this value was lower, of approximately 20%. NMA also significantly improved the postischemic myocardial function. An increase of systolic function (assessed by left ventricular developed pressure, LVDP) and a decrease diastolic stiffness (assessed by left ventricular end diastolic, LVEDP) were some beneficial effects observed in NMA treated hearts. That is, acute treatment with NMA was efficient to decrease the cell death and myocardial dysfunction following to ischemia-reperfusion.
Molecular mechanisms
It was previously documented that the increase in intracellular Ca2+ concentration induced by hypoxia -measured by the use of Ca2+sensitive dye fura2-was markedly reduced in pulmonary arterial smooth muscle cells treated with NMA (Shimoda et al., 2007). These authors concluded that this action of NMA was independent of CA inhibition. In isolated myocytes we observed a similar action detecting a decrease of CaC current, measured by patch clamp technique in whole cell configuration, after NMA treatment (Figure 1). The inhibition of CaC current is rapidly turned on, suggesting the direct binding of NMA to the channels. On the other hand, voltage-dependence of activation and inactivation were not affected by NMA. Thus, a direct effect of the compound on the pore without affecting the biophysical properties of the channel is most likely (Ciocci Pardo et al., 2022).
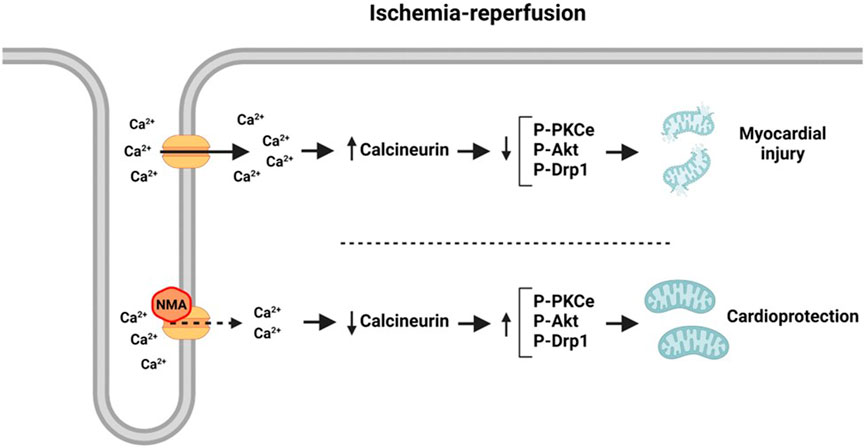
FIGURE 1. Schematic representation of L-type Ca2+ channel structure and its typical current in untreated and NMA treated hearts.
Taking into account these data, our first conclusion was that a diminution of Ca2+ overload, probably mediated by a direct binding of NMA to CaC would be a possible mechanism involved in the beneficial effects achieved by NMA on ischemic myocardium. The increase in cytosolic Ca2+ during ischemia has been associated with an enhancement of sarcoplasmic reticulum (SR) Ca2+ load, which is released at the onset of reperfusion and produces an abrupt rise in cytosolic Ca2+ and the consequent decrease in SR Ca2+ content and Ca2+ transient (Federico et al., 2020). Although the action of NMA on SR was not examined we can speculate that the diminution of Ca2+ entry through a direct action of NMA on CaC could attenuate the SR Ca2+ release, thus decreasing the intracellular Ca2+ concentration, crucial factor of myocardial damage.
The preservation of mitochondrial function is the main mechanism to protect the heart against ischemia-reperfusion injury (Honda et al., 2005; Anzell et al., 2018). Several protein kinases activated immediately prior to or at the time of reperfusion have been implicated in the pathways leading to cardioprotection (Altamirano et al., 2015). Thus, phosphatidylinositol-3-kinase (PI3K/Akt) and PKCε are contributing to myocyte defense against Ca2+ overload (Mochly-Rosen et al., 2012; Duan et al., 2015). On the other hand, it was previously demonstrated that calcineurin activation contributes to myocardial postischemic damage probably associated to an increase of Ca2+ intracellular concentration via CaC (Lakshmikuttyamma et al., 2003; Tandan et al., 2009).
Cytosolic Ca2+ overload is a key stimulus to open the mitochondrial permeability transition pore (mPTP) and the activation of mitochondrial fragmentation -assessed by dephosphorylation at Ser637Drp1 (a dynamin-related protein-1)- both events occurring during ischemia-reperfusion (Youle and van der Bliek, 2012; Hernandez-Resendiz et al., 2020; Ramachandra et al., 2020). The activation of pathways initiated by the kinases mentioned above and the inactivation of calcineurin have been involved in the prevention or attenuation of those mitochondrial detrimental actions (Baines et al., 2003; Cereghetti et al., 2008; Miyamoto et al., 2008; Maneechote et al., 2017; Wang et al., 2017). Our data demonstrated that the cardioprotection afforded by NMA involve pathways activated by Akt and PKCε and calcineurin inactivation having the mitochondria as a crucial end point. At the level of this organelle, our results also show an increase of Drp1 phosphorylation suggesting that an attenuation of mitochondrial fission could be a possible mechanism involved in the diminution of post-ischemic damage NMA-mediated (Figure 2).
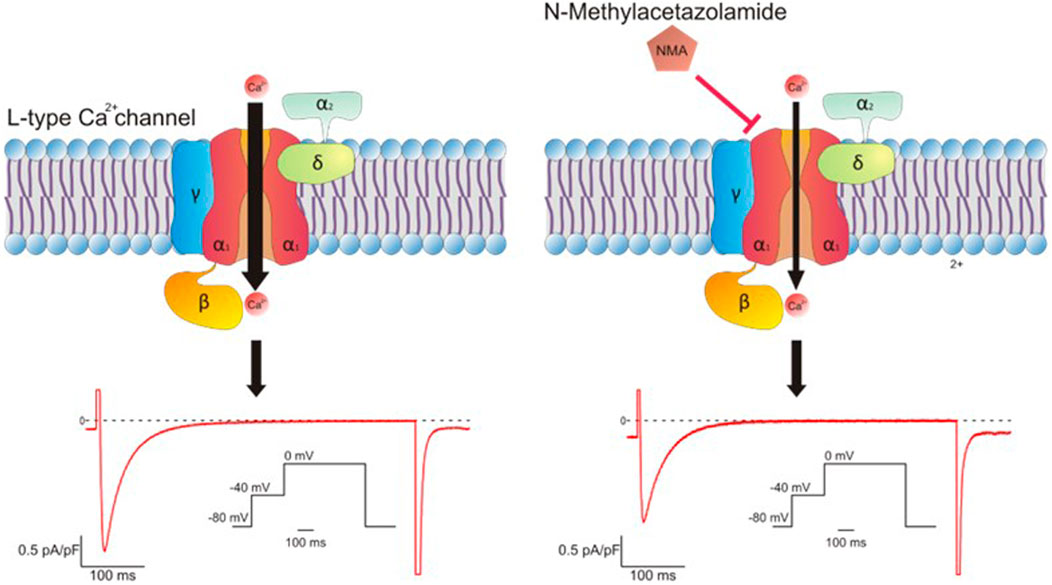
FIGURE 2. Possible pathway of the cardioprotection NMA-mediated.
Conclusion
Taking into account previous data (Ciocci Pardo et al., 2022), we consider that NMA, through the blockade of L-type Ca2+ channel in a similar manner to the classic calcium antagonists, represents an attractive alternative to ameliorate the postischemic impairment. According to our experience the main difference between those drugs, such as dihydropyridine (DHP) compounds, and NMA was the administration time; while the classic calcium blockers were given prior to ischemia, NMA was administered at the beginning of reperfusion. This fact is very important, making ¨a priori¨ the NMA a superior tool for the treatment of ischemic heart disease in patients. Although this fact is very important, clinical trials will be mandatory to demonstrate the effectiveness of NMA in humans and its advantages or disadvantages in comparison to other calcium antagonists.
Ethics statement
The experiments referenced in this review were performed in accordance with the Guide for Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 2011) and approved by the Ethics committee of the Faculty of Medicine, La Silver, Argentina (CICUAL # P01-05-15).
Author contributions
SM and EA Conceptualization and funding acquisition, AP, LD, and LG Investigation and prepared figures; SM approved final version of manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Grant M-203 from the Universidad Nacional de La Plata of Argentina to SM and grant PICT 00958 from FONCyT (Fondo para la Investigación Científica y Tecnológica) of Argentina to AP.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Altamirano, F., Wang, Z. V., and Hill, J. A. (2015). Cardioprotection in ischaemia-reperfusion injury: novel mechanisms and clinical translation. J. Physiol. 593, 3773–3788. doi:10.1113/JP270953
Anzell, A. R., Maizy, R., Przyklenk, K., and Sanderson, T. H. (2018). Mitochondrial quality control and disease: insights into ischemia-reperfusion injury. Mol. Neurobiol. 55, 2547–2564. doi:10.1007/s12035-017-0503-9
Baines, C. P., Song, C. X., Zheng, Y. T., Wang, G. W., Zhang, J., Wang, O. L., et al. (2003). Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ. Res. 92, 873–880. doi:10.1161/01.RES.0000069215.36389.8D
Barry, W. H., and Bridge, J. H. (1993). Intracellular calcium homeostasis in cardiac myocytes. Circulation 87, 1806–1815. doi:10.1161/01.cir.87.6.1806
Bauersachs, R., Zeymer, U., Brière, J. B., Marre, C., Bowrin, K., and Huelsebeck, M. (2019). Burden of coronary artery disease and peripheral artery disease: a literature review. Cardiovasc Ther. 2019, 8295054. doi:10.1155/2019/8295054
Catterall, W. A. (1995). Structure and function of voltage-gated ion channels. Annu. Rev. Biochem. 64, 493–531. doi:10.1146/annurev.bi.64.070195.002425
Cereghetti, G. M., Stangherlin, A., de Brito, O. M., Chang, C. R., Blackstone, C., Bernardi, P., et al. (2008). Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U. S. A. 105, 15803–15808. doi:10.1073/pnas.0808249105
Chiappe de Cingolani, G. E., Mosca, S. M., Moreyra, A. E., and Cingolani, H. E. (1996). Chronic nifedipine treatment diminishes cardiac inotropic response to nifedifine: functional upregulation of dihydropyridine receptors. J. Cardiovasc Pharmacol. 27, 240–246. doi:10.1097/00005344-199602000-00010
Ciocci Pardo, A., Díaz Zegarra, L. A., González Arbeláez, L. F., Ibáñez, A. M., Díaz, R. G., Aiello, E. A., et al. (2022). Cardioprotective effects of N-methylacetazolamide mediated by inhibition of L-type Ca2+ channel current. BBA-General Subj. 1866, 130098. doi:10.1016/j.bbagen.2022.130098
Dibb, K. M., Graham, H. K., Venetucci, L. A., Eisner, D. A., and Trafford, A. W. (2007). Analysis of cellular calcium fluxes in cardiac muscle to understand calcium homeostasis in the heart. Cell Calcium 42, 503–512. doi:10.1016/j.ceca.2007.04.002
Donato, M., Evelson, P., and Gelpi, R. J. (2017). Protecting the heart from ischemia/reperfusion injury: an update on remote ischemic preconditioning and postconditioning. Curr. Opin. Cardiol. 32, 784–790. doi:10.1097/HCO.0000000000000447
Duan, Q., Madan, N. D., Wu, J., Kalisz, J., Doshi, K. Y., Haldar, S. M., et al. (2015). Role of phosphoinositide 3-kinase IA (PI3K-IA) activation in cardioprotection induced by ouabain preconditioning. J. Mol. Cell Cardiol. 80, 114–125. doi:10.1016/j.yjmcc.2014.12.021
Elliott, W. J., and Ram, C. V. S. (2011). Calcium channel blockers. J. Clin. Hypertens. 13, 687–689. doi:10.1111/j.1751-7176.2011.00513.x
Federico, M., Valverde, C. A., Mattiazzi, A., and Palomeque, J. (2020). Unbalance between sarcoplasmic reticulum Ca2+ uptake and release: a first step toward Ca2+ triggered arrhythmias and cardiac damage. Front. Physiol. 10, 1630. doi:10.3389/fphys.2019.01630
Gross, G. J., Kersten, J. R., and Warltier, D. C. (1999). Mechanisms of postischemic contractile dysfunction. Ann. Thorac. Surg. 68, 1898–1904. doi:10.1016/s0003-4975(99)01035-8
Han, X., Li, M., Zhao, Z., Zhang, Y., Zhang, J., Zhang, X., et al. (2019). Mechanisms underlying the cardio-protection of total ginsenosides against myocardial ischemia in rats in vivo and in vitro: possible involvement of L-type Ca2+ channels, contractility and Ca2+ homeostasis. J. Pharmacol. Sci. 139, 240–248. doi:10.1016/j.jphs.2019.02.001
Hernandez-Resendiz, S., Prunier, F., Girao, H., Dorn, G., and Hausenloy, D. J.EU-CARDIOPROTECTION COST Action CA16225 (2020). Targeting mitochondrial fusion and fission proteins for cardioprotection. J. Cell Mol.Med 24, 6571–6585. doi:10.1111/jcmm.15384
Heusch, G. (2015). Molecular basis of cardioprotection: signal transduction in ischemic pre-post-and remote conditioning. Circ. Res. 116, 674–699. doi:10.1161/CIRCRESAHA.116.305348
Honda, H. M., Korge, P., and Weiss, J. N. (2005). Mitochondria and ischemia/reperfusion injury. Ann. N. Y. Acad. Sci. 1047, 248–258. doi:10.1196/annals.1341.022
Kalogeris, T., Baines, C. P., Krenz, M., and Korthuis, R. J. (2016). Ischemia/reperfusion. Compr. Physiol. 7 (1), 113–170. doi:10.1002/cphy.c160006
Lakshmikuttyamma, A., Selvakumar, P., Kakkar, R., Kanthan, R., Wang, R., and Sharma, R. K. (2003). Activation of calcineurin expression in ischemia-reperfused rat heart and in human ischemic myocardium. J. Cell Biochem. 90, 987–997. doi:10.1002/jcb.10722
Maneechote, C., Palee, S., Chattipakorn, S. C., and Chattipakorn, N. (2017). Roles of mitochondrial dynamics modulators in cardiac ischaemia/reperfusion injury. J. Cell Mol. Med. 21, 2643–2653. doi:10.1111/jcmm.13330
Maren, T. H. (1956). Carbonic anhydrase inhibition. V. N5-substituted 2-acetylamino-1,3,4-thiadiazole-5-sulfonamides: metabolic conversion and use as control substances. J. Pharmacol. Exp. Ther. 117, 385–401.
Miyamoto, S., Murphy, A. N., and Brown, J. H. (2008). Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 15, 521–529. doi:10.1038/sj.cdd.4402285
Mochly-Rosen, D., Das, K., and Grimes, K. V. (2012). Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 11, 937–957. doi:10.1038/nrd3871
Moreyra, A. E., Gelpi, R. J., Mosca, S. M., and Cingolani, H. E. (1994). Chronic administration of nicardipine attenuates myocardial stunning in isolated rabbit hearts. J. Mol. Cell Cardiol. 26, 979–984. doi:10.1006/jmcc.1994.1118
Murphy, E., and Steenbergen, C. (2008). Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 88, 581–609. doi:10.1152/physrev.00024.2007
Pittas, K., Vrachatis, D. A., Angelidis, C., Tsoucala, S., Giannopoulos, G., and Deftereos, S. (2018). The role of calcium handling mechanisms in reperfusion injury. Curr. Pharm. Des. 24, 4077–4089. doi:10.2174/1381612825666181120155953
Ramachandra, C. J. A., Hernandez-Resendiz, S., Crespo-Avilan, G. E., Lin, Y-H., and Hausenloy, D. J. (2020). Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 57, 102884. doi:10.1016/j.ebiom.2020.102884
Severino, P., D'Amato, A., Pucci, M., Infusino, F., Birtolo, L. I., Mariani, M. V., et al. (2020). Ischemic heart disease and heart failure: role of coronary ion channels. Int. J. Mol. Sci. 21, 3167. doi:10.3390/ijms21093167
Shimoda, L. A., Luke, T., Sylvester, J. T., Shih, H. W., Jain, A., and Swenson, E. R. (2007). Inhibition of hypoxia-induced calcium responses in pulmonary arterial smooth muscle by acetazolamide is independent of carbonic anhydrase inhibition. Am. J. Physiol. Lung Cell Mol. Physiol. 292, 1002–1012. doi:10.1152/ajplung.00161.2006
Simonovic, N., and Jeremic, J. (2017). Role of calcium channel blockers in myocardial preconditioning. Ser. J. Exp. Clin. Res. 18, 281–287. doi:10.1515/sjecr-2016-0073
Smith, G. L., and Eisner, D. A. (2019). Calcium buffering in the heart in health and disease. Circulation 139, 2358–2371. doi:10.1161/CIRCULATIONAHA.118.039329
Striessnig, J., Ortner, N. J., and Alexandra Pinggera, A. (2015). Pharmacology of L-type calcium channels: novel drugs for old targets? Curr. Mol. Pharmacol. 8, 110–122. doi:10.2174/1874467208666150507105845
Sueta, D., Tabata, N., and Hokimoto, S. (2017). Clinical roles of calcium channel blockers in ischemic heart diseases. Hypertens. Res. 40, 423–428. doi:10.1038/hr.2016.183
Talukder, M. A. H., Zweier, J. L., and Periasamy, M., (2009). Targeting calcium transport in ischaemic heart disease. Circ. Res. 84, 345–352. doi:10.1093/cvr/cvp264
Tandan, S., Wang, Y., Wang, T. T., Jiang, N., Hall, D. D., Hell, J. W., et al. (2009). Physical and functional interaction between calcineurin and the cardiac L-type Ca2+ channel. Circ. Res. 105, 51–60. doi:10.1161/CIRCRESAHA.109.199828
Teppema1, L. J., and Swenson, E. R. (2015). The noncarbonic anhydrase inhibiting acetazolamide analog N-methylacetazolamide reduces the hypercapnic, but not hypoxic, ventilatory response. Physiol. Rep. 3, e12484. doi:10.14814/phy2.12484
Wang, R., Wang, M., He, S., Sun, G., and Sun, X. (2020). Targeting calcium homeostasis in myocardial ischemia/reperfusion injury: an overview of regulatory mechanisms and therapeutic reagents. Front. Pharmacol. 11, 872. doi:10.3389/fphar.2020.00872
Wang, S., Zhang, F., Zhao, G., Cheng, Y., Wu, T., Wu, B., et al. (2017). Mitochondrial PKC-ε deficiency promotes I/R-mediated myocardial injury via GSK3β-dependent mitochondrial permeability transition pore opening. J. Cell Mol. Med. 21, 2009–2021. doi:10.1111/jcmm.13121
Wu, Y., Liu, H., and Wang, X. (2021). Cardioprotection of pharmacological postconditioning on myocardial ischemia/reperfusion injury. Life Sci. 264, 118628. doi:10.1016/j.lfs.2020.118628
Keywords: N-methylacetazolamide, myocardial ischaemia, reperfusion injury, calcium, L-type Ca2+ channel (CaL), cardioprotection
Citation: Pardo AC, Díaz Zegarra LA, González Arbeláez LF, Aiello EA and Mosca SM (2023) Is N-methylacetazolamide a possible new therapy against ischemia-reperfusion injury?. Front. Pharmacol. 14:1223132. doi: 10.3389/fphar.2023.1223132
Received: 20 May 2023; Accepted: 24 July 2023;
Published: 10 August 2023.
Edited by:
Claudia Capurro, National Scientific and Technical Research Council (CONICET), ArgentinaReviewed by:
Maria Teresa Politi, University of Buenos Aires, ArgentinaLi Chu, Hebei Medical University, China
Copyright © 2023 Pardo, Díaz Zegarra, González Arbeláez, Aiello and Mosca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alejandro Ciocci Pardo, YWxlamFuZHJvLmNpb2NjaXBhcmRvQHVuaXBkLml0