 Zihan Liu1,2†
Zihan Liu1,2† Zhanjun Dong
Zhanjun Dong- 1Graduate School, Hebei Medical University, Shijiazhuang, China
- 2Department of Pharmacy, Hebei General Hospital, Hebei Key Laboratory of Clinical Pharmacy, Shijiazhuang, China
Background: Apixaban (API) and rivaroxaban (RIVA) are orally available inhibitors of coagulation factor Xa and are commonly used to treat cancer-related venous thrombosis. Ribociclib (RIBO), a first-line treatment for hormone receptor-positive/human epidermal growth factor receptor 2 negative (HR+/HER2−) advanced breast cancer, is an inhibitor of CYP3A4, P-gp, and BCRP. Given the potential for these drugs to be co-administered in clinical settings, there is limited information regarding the pharmacokinetic drug-drug interactions (DDIs) between ribociclib and these anticoagulants. This study aimed to evaluate the extent of DDIs between ribociclib and rivaroxaban or apixaban in rats and to explore the optimization of drug dosing strategies.
Methods: Male Sprague-Dawley rats were divided into 9 groups (n = 6), receiving ribociclib, apixaban, rivaroxaban, ribociclib with rivaroxaban, ribociclib with apixaban, and combinations with reduced doses and time intervals. Blood concentrations were measured using ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). Pharmacokinetic parameters such as AUC, Cmax, CLz/F, and Vz/F.
Results: Ribociclib significantly increased exposure to both rivaroxaban and apixaban, with a greater impact on rivaroxaban. Specifically, ribociclib increased the AUC0-t, AUC0-
Discussion: This research highlights the need for careful consideration of dosing regimens to minimize toxicity risk and optimize the safety of clinical co-administration of ribociclib with rivaroxaban.
1 Introduction
Venous thromboembolism (VTE) is the most common complication of cancer, with approximately 15% of patients with cancer experiencing at least one cancer-associated thrombosis (CAT). Conventional cancer treatments often exacerbate the pre-CAT state (Amin et al., 2024; Dickson et al., 2022; Chen, 2024). While CAT is relatively rare in breast cancer compared to other solid cancers, breast cancer remains the most common cancer among women worldwide, and the number of breast cancer-associated thrombosis (BrCAT) is significantly high (Bray et al., 2024). The treatment of CAT is associated with a higher incidence of thrombotic recurrence and/or major bleeding compared to non-cancer VTE (Fernandes et al., 2019; Riaz et al., 2023). According to clinical guidelines, low molecular heparin (LMWH) has historically been the preferred regimen for the treatment of CAT (Kahn et al., 2012; Key et al., 2020; Mandala et al., 2011). However, the necessity and high cost of daily subcutaneous injections have reduced patient compliance (Wittkowsky, 2006). Recent randomized clinical trials have demonstrated that oral direct anticoagulants (DOACs) are as effective as LMWH in the acute management of CAT, with a lower rate of VTE recurrence (Agnelli et al., 2020; Schrag et al., 2023; Marcucci et al., 2022; Amin et al., 2024). DOACs also offer practical advantages, such as not requiring frequent monitoring of the International Normalized Ratio (INR), which enhances ease of use and cost-effectiveness for patients. As a result, international clinical practice guidelines have increasingly supported the use of DOACs as an alternative to LMWH monotherapy for both the initial and long-term treatment of CAT. Despite their benefits, the adoption of DOACs necessitates careful consideration due to certain limitations. Notably, DOACs are associated with a higher risk of bleeding and potential drug-drug interactions (DDIs) with some anticancer therapies (Frere et al., 2022; Lyman et al., 2021).
Apixaban is a small-molecule, selective factor Xa inhibitor that inhibits both free and clot-bound factor Xa and has been approved for the clinical treatment of several thromboembolic diseases, including the prevention of VTE (Jiang et al., 2009). Apixaban is primarily metabolised by the cytochrome P450 (CYP) enzyme CYP3A4/5, with lesser involvement of CYP1A2, CYP2C8, CYP2C9, CYP2C19, and CYP2J2 (Byon et al., 2019). Additionally, it is a substrate for P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) (Zhang et al., 2013). Rivaroxaban is another direct factor Xa inhibitor, with a higher selectivity for this coagulation factor. It is rapidly absorbed, reaching maximum plasma concentration (Cmax) within 2–4 h after administration (Kvasnicka et al., 2017). Its pharmacokinetics and pharmacodynamics are proportional to the dose. Rivaroxaban is metabolized by CYP3A4/5 and CYP2J2, with CYP3A4/5 accounting for approximately 18% of the total elimination and CYP2J2 for approximately 14% (Mueck et al., 2013). Additionally, non-CYP-dependent pathways also contribute to the elimination of rivaroxaban. Combinations therapies can lead to DDIs that affect the exposure or pharmacological activity of DOACs. Both potent inhibitors and inducers of P-gp or CYP3A4 are known to significantly influence DOAC pharmacokinetics and efficacy (Bellesoeur et al., 2018).
The efficacy of cyclin-dependent kinase (CDK) 4/6 inhibitors in treating breast cancer is well established, making them the first-line therapy for hormone receptor-positive/human epidermal growth factor receptor 2 negative (HR+/HER2−) breast cancer (Pavlovic et al., 2023; Mayer et al., 2021; Lee et al., 2023). Among these, ribociclib is a novel CDK4/6 inhibitor that has shown efficacy in the treatment of advanced breast cancer and was approved by the Food and Drug Administration (FDA) in 2017. The recommended starting dose of ribociclib is 600 mg once daily (Braal et al., 2021). Ribociclib is mainly metabolized by CYP3A4 and is a substrate for P-gp. At the dose of 600 mg, ribociclib is a potent inhibitor of CYP3A4, and significantly inhibits P-gp and BCRP (Sorf et al., 2018; Kulkarni and Singh, 2024).
Co-administration of ribociclib with midazolam, a sensitive CYP3A4 substrate, has been shown to increase midazolam exposure (Samant et al., 2020). Consequently, we postulate that the co-administration ribociclib with DOACs may alter the exposure or pharmacological activity of DOACs. However, there is currently a paucity of data on the DDIs between ribociclib and DOACs, such as rivaroxaban and apixaban. Such DDIs could increase the risk of haemorrhage recurrence or recurrent VTE (Sabatino et al., 2020). Furthermore, DDIs may decrease the efficacy and safety of anticancer treatments or other medications employed for managing comorbidities. At present, clinical evidence on ribociclib-DOAC interactions is currently limited, particularly in patients with cancer. It is therefore imperative that these interactions are comprehensively studied to ensure the safe co-administration of these drugs and inform dosage adjustments (Tsoukalas et al., 2022).
The objective of the present study was to examine the potential DDIs between ribociclib and rivaroxaban or apixaban. To this end, the study comprised several key steps aimed at evaluating the pharmacokinetic interactions between ribociclib, apixaban and rivaroxaban Rats were assigned to different treatment groups and administered the three drugs either as monotherapy or in combination with ribocicliband apixaban or rivaroxaban. Ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) was employed to detect the blood drug concentrations at different time points. Pharmacokinetic parameters were then calculated and observed drug interactions were subjected to statistical analysis. All materials, experimental procedures, and validation methods were rigorously designed and executed to ensure the accuracy, reliability, and reproducibility of the data obtained.
2 Materials and methods
2.1 Study design and treatments
2.1.1 Animals
Adult specific pathogen-free (SPF)-grade male Sprague-Dawley (SD) rats weighing 230 ± 30 g were provided by Beijing Huafukang Biotechnology Co., LTD., (Beijing, China; license number SCXK (Jing) 2019-0008). The study adhered to the ARRIVE guidelines (Percie et al., 2020), and all mouse procedures were conducted under humane process. The study was approved and supervised by the Animal Ethics Committee of Hebei General Hospital (Shijiazhuang, China) (No.2024DW-072).
Before the experiment, the rats were acclimated to standard laboratory conditions, including a 12-h light/dark cycle, a temperature of 23 ± 2°C, and a relative humidity of 50% ± 10%. They were provided with adequate food and water during a one-week adaptive feeding period. Food was withheld for 12 h before drug administration to ensure standardized conditions for the experiment.
2.1.2 Pharmacokinetic study in rats
Experimental animals were randomly divided into nine groups (n = 6, Table 1). Ribociclib was suspended in methylcellulose (MC), rivaroxaban was prepared in hydroxypropyl methylcellulose (HPMC), and apixaban was suspended in 5% DMSO in water. To achieve steady-state blood concentrations, a drug typically requires five to seven half-lives. Based on this principle, ribociclib was administered orally for eight consecutive days, while rivaroxaban and apixaban were administered orally for five consecutive days to ensure steady-state concentrations were reached before further analysis.
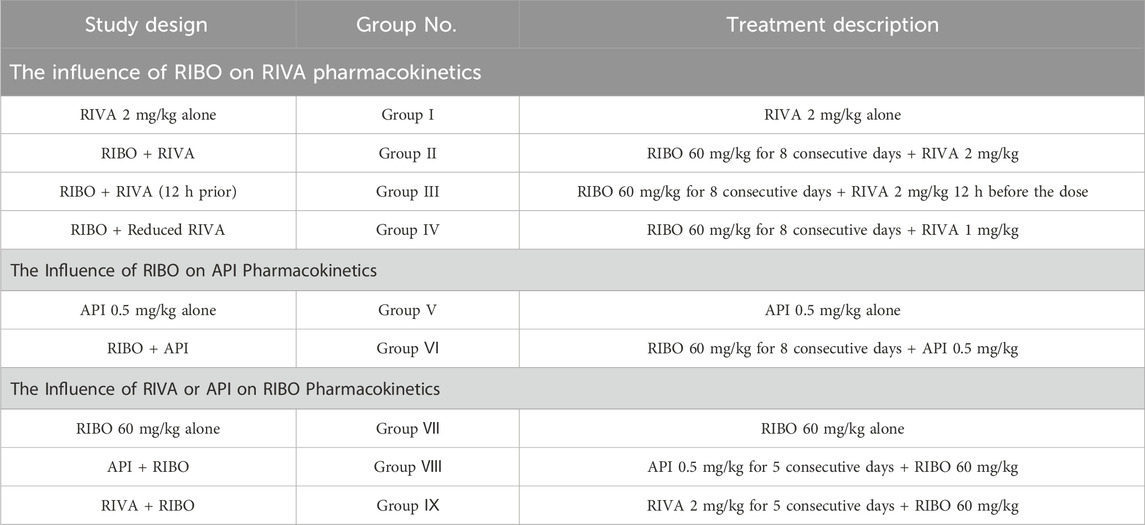
Table 1. Study design.
2.1.3 Blood sample
Approximately 0.1 mL of blood was collected into heparinized tubes via the orbitalat specified time points before and after the final drug administration. For apixaban, blood samples were collected at 0, 0.083, 0.167, 0.25, 0.333, 0.5, 0.75, 1, 3, 5, 7, 10, and 12 h. For rivaroxaban, sampling was conducted at 0, 0.167, 0.333, 0.5, 0.75, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, and 24 h. For ribociclib, samples were taken at 0, 0.5, 1, 2, 4, 5, 6, 8, 10, 12, 24, and 48 h. Blood samples were centrifuged at 3,500 rpm for 10 min, and the supernatant was collected and stored at −80°C until processing for UPLC-MS/MS.
2.2 UPLC-MS/MS assay
2.2.1 Chemicals and reagents
Ribociclib (purity>98%, Lot EMC104) was supplied by Bido Pharmaceutical Co., Ltd. Ribociclib-d6 (purity>99%, Lot IR-67675) and apixaban (purity ≥98%, Lot C15069980) were purchased from Shanghai Macklin Bio-Technology Co. Ltd., (Shanghai, China). Rivaroxaban (purity ≥99%, Lot H25J9Z64216) was provided by Shanghai yuan ye Bio-Technology Co. Ltd., (Shanghai, China). Rivaroxaban-d4 (purity >98%, Lot 21,702) was obtained from B1203 Life Science Park, SCT Creative Factory. (Shenzhen, China). Dimethyl sulfoxide (DMSO) was acquired from Beijing Solarbio Science Technology Co. Ltd., (Beijing, China). Acetonitrile, formic acid, ammonium acetate and methyl tert-butyl ether were of high-performance liquid chromatography (HPLC) grade and supplied by Fisher Scientific (Pittsburgh, PA, United States). Ultrapure water was obtained from Wahaha Group Co., Ltd., (Hangzhou, China).
2.2.2 Instrumentation and chromatographic conditions
Apixaban and rivaroxaban concentrations in plasma samples were determined using our previously established UPLC-MS/MS method (Wang et al., 2023). Ribociclib concentrations were quantified using an LC-30A UPLC system (Shimadzu, Kyoto, Japan) coupled with a Sciex Triple Quad 5500 tandem quadrupole mass spectrometer (AB Sciex, Framingham, MA, United States) equipped with an electrospray ionization interface. Chromatographic separation was performed using a Welch Boltimate column (2.1 mm × 100 mm, 2.7 μm) maintained at 40°C. The mobile phase was composed of water containing 0.1% formic acid (phase A) and acetonitrile (phase B). The flow rate was held constant at 0.3 mL/min. The gradient elution protocol was as follows: 0–2.5 min, 60% B; 2.5–3.5 min, 60%–90% B; 3.5–5.5 min, 90% B; 5.5–5.6 min, 90%–60% B; 5.6–6.6 min, 60% B. The injection volume was 1 μL.
The mass spectrometer operated in positive ion mode and multiple reaction monitoring (MRM) mode for detection and quantification. The monitored transitions of precursors to product ions were as follows: 441.3→332.1 for ribociclib-d6 and 435.3.→322.1 for ribociclib (Figure 1). The mass spectrometer conditions, including delustering potential (DP) and collision energy (CE) of the compounds, are shown in Table 2. Other parameters of the mass spectrometer were as follows: ion source gas 160.0 psi; ion source gas 2,50.0 psi; curtain gas, 25.0 psi. The source temperature was 500°C and the ion spray voltage was 5,500 V.
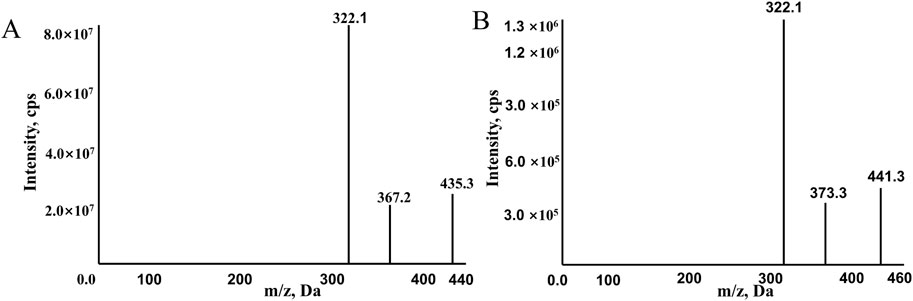
Figure 1. The mass spectra of RIBO (A) and RIBO-d6 (B).
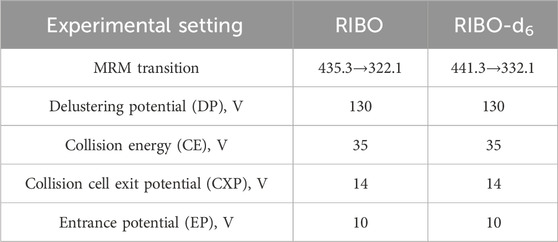
Table 2. The experimental setting for the tandem mass-spectrometer for the analytes and internal standards.
2.2.3 Preparation of calibration standards and quality control (QC) samples
Stock solutions of 1-mg/mL ribociclib and ribociclib-d6 (IS) were prepared separately in DMSO. Working solutions were prepared by diluting the standard stock solutions with 50% (v/v) acetonitrile in water. Calibration standards were prepared by spiking 5 μL of the working solution into 45 μL of blank rat plasma. The stock solutions were further diluted with 50% (v/v) acetonitrile in water to create working solutions of varying concentrations. The final concentrations for calibration curves were as follows: 10, 50, 100, 200, 400, 800, 2,000, and 4,000 ng/mL of ribociclib. Low-, medium-, and high-concentration QC samples were processed independently and contained 20, 500, and 3,000 ng/mL.
2.2.4 Method validation
The method was validated per the guidelines of the Food and Drug Administration (US-FDA, 2022) and Chinese Pharmacopoeia (2020). Selectivity was determined by analyzing blank plasma samples from six different batches of rats and plasma samples from six different batches of rats after administration of ribociclib. There should be no interference, with retention times such that the blank plasma peak area is less than 5% of the internal standard (IS) and less than 20% of the lower limit of quantification (LLOQ). The calibration curves were evaluated using 10–4,000 ng/mL of ribociclib. Linearity was assessed by plotting the peak area ratios of the analyte to the IS against nominal concentrations, using weighted (1/x2) least-squares linear regression. Accuracy and precision were determined for all concentrations derived from the calibration curves, ensuring that deviations were within 15% of the nominal concentration, except for the LLOQ, where the deviation should not exceed 20%. Precision and accuracy were assessed by analyzing plasma samples at low, medium, high, and LLOQ concentration levels over three consecutive days. The final precision and accuracy were determined by calculating the relative standard deviation (RSD) and relative error (RE) of the six samples. RSD and RE for QC concentrations were required to be within ±15%, and for the LLOQ, within ±20%. Matrix effects were assessed by comparing analyte peak areas in blank plasma samples of low, medium, and high concentration QC samples (n = 6) with analyte peak areas in the corresponding pure solutions. Extraction recovery was assessed by comparing the peak areas of analytes in extracted plasma samples from QC samples at three concentrations (n = 6) with the peak areas of analytes in blank plasma extracts at the same concentrations. Analyte stability was assessed at low, medium, and high QC concentrations under four different storage and handling conditions: room temperature (25°C) for 4 h, autosampler temperature (15°C) for 6 h, −80°C for 30 days, and three freeze-thaw cycles of plasma samples (−80°C–25°C). The RSD of all QC samples should be less than 15%, and the RE should be between 85% and 115% of the labeled concentration.
2.3 Quantitative real-time PCR (qRT-PCR) analysis
qRT-PCR analysis was used to determine the mRNA levels of Cyp3a1(CYP3A4) in the liver and Abcb1a (P-gp), Cyp3a1(CYP3A4), Abcg2(BCRP) in the intestines. Total RNA was extracted from frozen liver and intestine samples using the TRNzol Universal reagent, following the manufacturer’s instructions. The Bio Tek Epoch (Bio Tek Instruments, Inc., Winooski, VT, United States) was used to quantify the purity and concentration of the total RNA, based on the ratio of the absorbance at 260 and 280 nm. The RNA samples, each containing 1 µg of material, were converted to complementary DNA (cDNA) using the FastKing RT Kit. The real-time PCR assays were performed using a two-step amplification protocol on the SLAN-96S Real-Time PCR system (Shanghai Hongshi Medical Technology Co., Ltd., Shanghai, China). NADPH was employed as an internal control, and the PCR cycling criteria were as follows: 95°C for 15 min, followed by 40 cycles of 95°C for 10 s and 60°C for 32 s. The sequences of the primers are presented in Table 3.
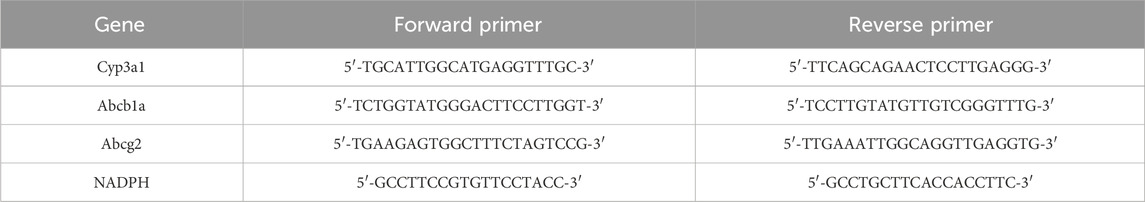
Table 3. Primer sequences used in quantitative RT-PCR.
2.4 Pharmacokinetic analysis
2.4.1 Plasma sample preparation
A 50 µL plasma sample was mixed with 5 µL of the IS working solution, followed by the addition of 150 µL of acetonitrile. The mixture was vortexed for 2 min and then centrifuged at 12,000 rpm for 10 min. Next, 70 µL of the supernatant was collected and mixed with 70 µL of 50% (v/v) of acetonitrile in water. The mixture was vortexed for 1 min, and then 1 µL was injected into the UPLC-MS/MS system for analysis.
2.4.2 Statistical analysis
Pharmacokinetic parameters were calculated based on a non-compartmental model using DAS 2.1.1 Software (Mathematical Pharmacology Professional Committee of China, Shanghai, China). The area under the concentration–time curve (AUC), maximum plasma concentration (Cmax), time to maximum plasma concentration (Tmax), the time required to eliminate half of the plasma drug concentration (t1/2), clearance of drug plasma volume per unit time (CLz/F), apparent volume distribution (Vz/F), mean residence time (MRT) and the absorption rate constant (Ka) are all expressed as mean ± standard deviation (SD). SPSS 25.0 statistical software (SPSS Inc., Chicago, IL, United States) was applied to statistically analyze the mainpharmacokinetic parameters. Shapiro-Wilk test was used to assess whether the data were normally distributed, t test was used for parameters conforming to the normal distribution, and Mann-Whitney U test was used for parameters not conforming to the normal distribution. Statistical comparisons were conducted using a analysis of variance, t-test or nonparametric rank-sum test depending on the data type. A P-value <0.05 was deemed statistically significant.
3 Results
3.1 Method development and optimization
A highly sensitive and reproducible UPLC-MS/MS method was developed to evaluate the pharmacokinetic interactions of ribociclib in rats. The chromatographic conditions were optimized to obtain good peak symmetry, high detection sensitivity and a short retention time. Given that acetonitrile exhibits superior elution ability relative to methanol, it was selected as the organic phase, and formic acid was introduced to augment the chromatographic signal and peak spectrum of ribociclib. A gradient elution method was employed, starting with 40% (v/v) acetonitrile at a flow rate of 0.3 mL/min. This approach yielded high detection sensitivity and a short retention time. Ribociclib-d6, a deuterated analogue of ribociclib with similar physicochemical properties and identical mass spectral characteristics was selected as the IS to enhance the accuracy and precision of the analysis. Figure 2 shows typical chromatograms of ribociclib and ribociclib-d6 in different plasma samples. No significant interference from endogenous substances was detected. Calibration curves were constructed using linear regression analysis across a concentration ranges of 10–4,000 ng/mL for ribociclib. A typical calibration curves was described by the equation Y = 0.00332 X + 0.00874 (r = 0.9987) for ribociclib. Intra- and inter-day precision values were below more than 6.3%, and the accuracies ranged from −7.6% to 10.7% for all investigated analyte concentrations in rat plasma. Matrix effects for ribociclib ranged from 90.1% to 106.5%, indicating no significant matrix effect existed in rat plasma. Recovery rates, normalized by IS peak area, ranged from 97.7% to 108.1%, and the RSD was less than 5.63% for the analyte. Stability tests for QC samples, conducted under various conditions—including room temperature, autosampler storage at 15°C, −80°C, and after three freeze-thaw cycles—demonstrated excellent stability. Relative error (RE) values were below 5.8%, and RSD values were less than 6.7%. These results indicate that the developed method is repeatable and reproducible.
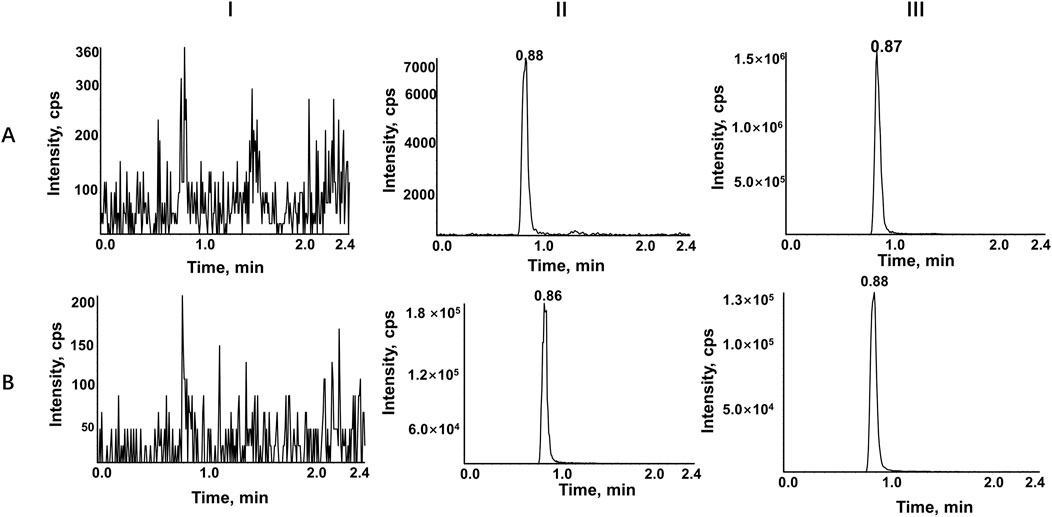
Figure 2. Representative chromatograms of RIBO (A), and RIBO-d6 (B). Ⅰ, a blank rat plasma sample; Ⅱ, a blank rat plasma sample spiked with the working solution at LLOQ level and IS; Ⅲ, a rat plasma sample after oral administration of 60 mg/kg RIBO.
3.2 Pharmacokinetic study
3.2.1 Effect of ribociclib on the pharmacokinetics of rivaroxaban
The mean plasma concentration-time profiles of rivaroxaban alone and in combination with multiple doses and timed administration of ribociclib are shown in Figure 3. The main pharmacokinetic parameters of rivaroxaban are summarized in Table 4. After multiple doses of ribociclib, the AUC0-t, AUC0-
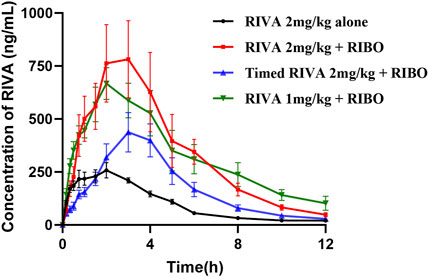
Figure 3. Mean plasma concentration-time profiles of RIVA alone and in combination with multiple doses and timed administration of RIBO.
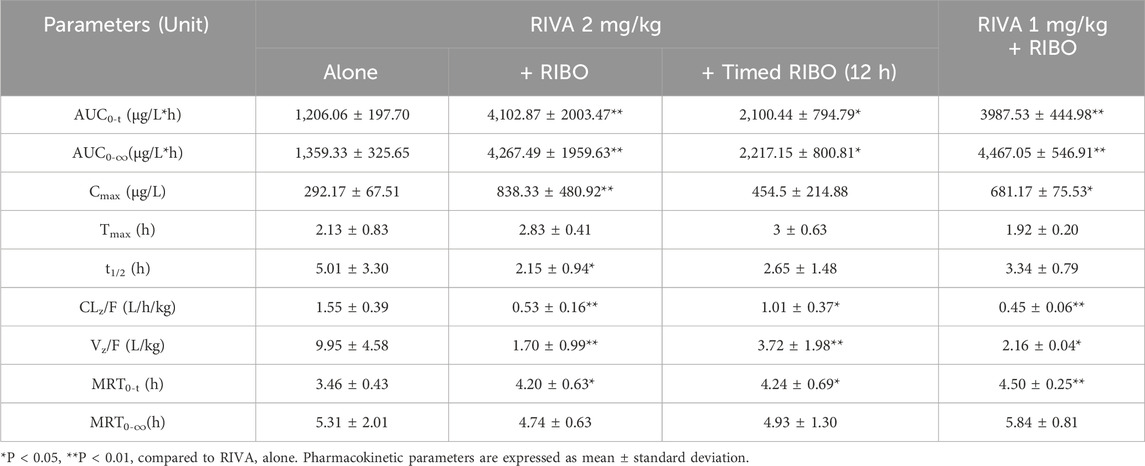
Table 4. Pharmacokinetic parameters of RIVA in rats when administered alone and following simultaneous and timed administration of RIBO.
3.2.2 Effect of ribociclib on the pharmacokinetics of apixaban
Figure 4 presents the mean plasma concentration-time curves of apixaban (0.5 mg/kg) administered alone or concomitantly with ribociclib, while Table 5 summarizes the main pharmacokinetic parameters of apixaban. After multiple administrations of ribociclib, the AUC0-
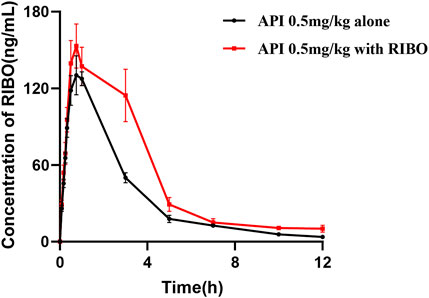
Figure 4. The mean plasma concentration-time graphs of API after oral administration alone and following multiple doses of RIBO.
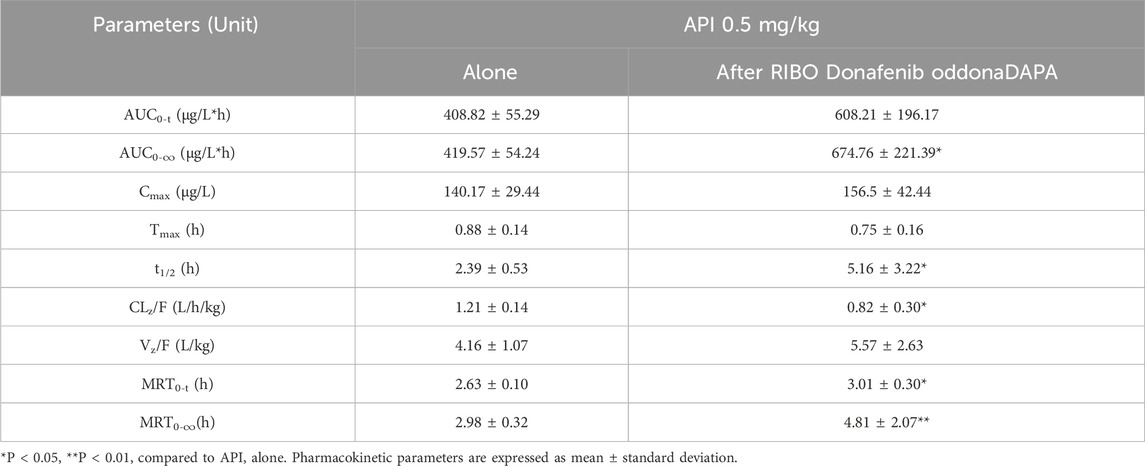
Table 5. Pharmacokinetic parameters of API in rats after oral administration alone and following multiple doses of RIBO.
3.2.3 Effect of rivaroxaban or apixaban on the pharmacokinetics of ribociclib
Figure 5 shows the plasma concentration-time curves of ribociclib after administration of ribociclib alone and in combination with multiple doses of rivaroxaban or apixaban, and the pharmacokinetic parameters are shown in Table 6. There were no significant changes in the pharmacokinetic parameters of ribociclib after co-administration with rivaroxaban or apixaban.
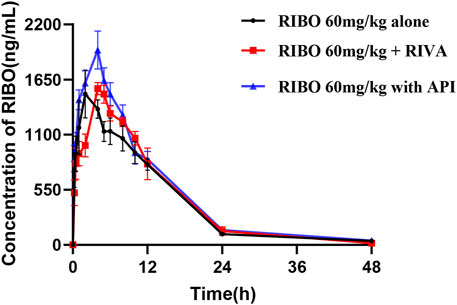
Figure 5. Mean plasma concentration-time profiles of RIBO after oral administration alone and following multiple doses of RIVA or API.
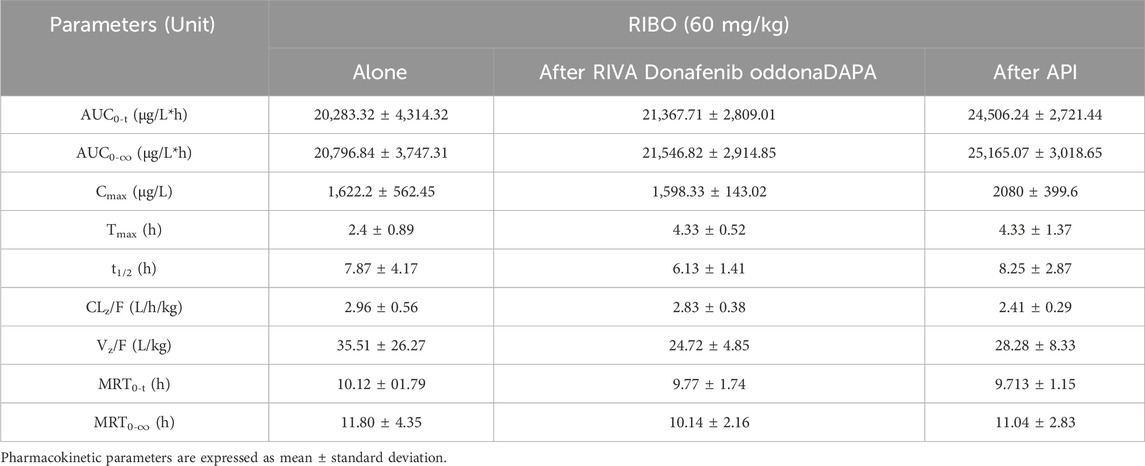
Table 6. Pharmacokinetic parameters of RIBO in rats after oral administration alone and following multiple doses of RIVA or API.
3.3 Messenger RNA (mRNA) expression in rat liver and intestines
To investigate the possible mechanism underlying the pharmacokinetic interactions between ribociclib and rivaroxaban or apixaban involving transporters and metabolic enzymes, we assessed the mRNA expression of Abcb1a, Abcg2, and Cyp3a1 in the liver and intestines of rats. Figure 6 illustrates that continuous administration of ribociclib for 8 days significantly suppressed the mRNA expression of intestinal Cyp3a1 and Abcg2, with inhibition rates of 82.6% (P < 0.001) and 45.0% (P = 0.001), respectively. However, no significant changes were observed in the mRNA expression of Cyp3a1 in the liver or Abcb1a in the intestine.
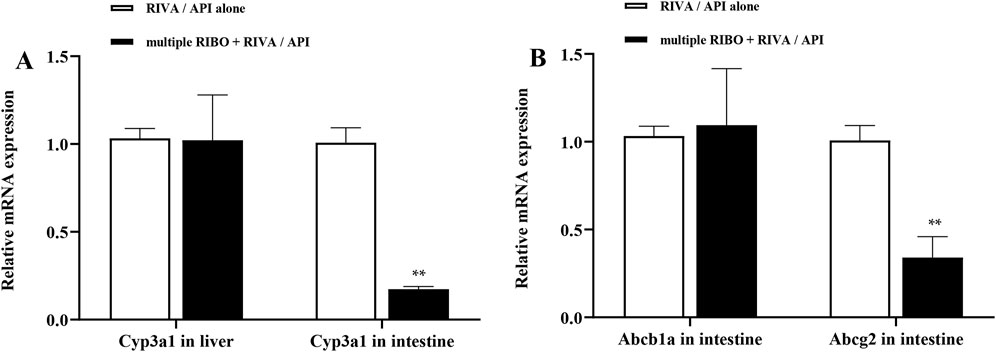
Figure 6. Relative mRNA expression in liver and intestine. (A) Effect of multiple-doses RIBO administration on mRNA expression of Cyp3a1 in liver and intestine; (B) Effect of multiple-doses RIBO administration on mRNA expression of Abcb1a and Abcg2 in intestine. **P < 0.01.
4 Discussion
This study demonstrated a significant pharmacokinetic interaction between ribociclib and the oral anticoagulants rivaroxaban, while showing less effect on apixaban. The difference in results may result from the different pharmacokinetics and pharmacodynamics associated with rivaroxaban and apixaban The substantial increase in drug exposure, particularly with rivaroxaban, raises concerns about an elevated risk of bleeding in patients with cancer. This study is one of the first to examine the interaction between ribociclib and DOACs to inform the use of anticoagulants in patients with BrCAT.
Multiple doses of ribociclib combined with rivaroxaban (2 mg/kg) resulted in a significant increase in exposure to rivaroxaban, with its AUC increasing nearly 2.4-fold and its Cmax increasing 1.9-fold compared to the control group. In addition, the CLZ/F of rivaroxaban decreased with the use of ribociclib. Multiple doses of ribociclib combined with apixaban (0.5 mg/kg) resulted in a 60.82% increase in the AUC of apixaban. This finding may be due to the inhibitory effect of ribociclib on metabolic enzyme,and transporters.
CYP enzymes, particularly CYP3A4, are central to the metabolism of numerous endogenous and exogenous substances. CYP3A4, expressed predominantly in the liver and the small intestine, plays a vital role in first-pass metabolism, impacting the bioavailability of many drugs. The uptake and elimination of DOACs like rivaroxaban and apixaban depend on P-gp and BCRP efflux transporter systems. Both CYP3A4 and these transporters are highly susceptible to induction or inhibition by various compounds, frequently leading to DDIs. Evidence from rat experiments with almonertinib, another inhibitor of CYP3A4, P-gp, and BCRP, showed over three fold increases in the AUC and Cmax of rivaroxaban and apixaban (Wang et al., 2023). Similarly, drugs that strongly induce or inhibit CYP3A4 and P-gp, such as phenytoin, rifampicin, carbamazepine, and ketoconazole, have been reported to significantly interact with apixaban and rivaroxaban (Byon et al., 2019; Cohen et al., 2021; Ngo et al., 2020). Studies have demonstrated that ribociclib acts as an inhibitor of CYP3A4, P-gp, and BCRP,as well as a substrate of P-gp (Sorf et al., 2018; Samant et al., 2020). However, in our study, we only observed ribociclib to inhibit CYP3A4 and BCRP mRNA expression in rat small intestine, with no effect on P-gp in rat small intestine and CYP3A4 in rat liver. We speculate that the reasons for the different expression of CYP3A4 mRNA in the intestine and liver and the different expression of P-gp mRNA and BCRP mRNA in the intestine are as follows. In vitro studies demonstrated that ribociclib (molecular weight 434) exhibits a half-maximal inhibitory concentration (IC50) of 12.8 µM against CYP3A4 (Sorf et al., 2018). In our investigation, the Cmax of ribociclib in rats administered 60 mg/kg reached approximately 1,500 ng/mL, which is significantly lower than its IC50 for CYP3A4 inhibition. This may explain the lack of apparent effect of ribociclib on liver CYP3A4 activity. In contrast, after oral administration of ribociclib, the drug concentration in the intestine can reach levels that inhibit the IC50 values of CYP3A4, P-gp, and BCRP (Sorf et al., 2018). However, in our study, no observable effect of ribociclib on P-gp mRNA expression in rat intestine was detected. This discrepancy may be attributed to ribociclib’s role as a substrate of P-gp, whereby its interaction with P-gp likely involves competition for substrate-binding sites rather than direct suppression of P-gp mRNA. Therefore, ribociclib may compete with rivaroxaban for P-gp binding, thereby reducing the efflux of rivaroxaban. These effects reduces the rate of rivaroxaban metabolism and excretion, resulting in a decrease in CLZ/F of rivaroxaban. Consequently, we speculate that the increased exposure to rivaroxaban and apixaban with ribociclib is primarily due to the inhibition of metabolic and efflux transport mechanisms.
The results of the rat experiments showed that the effect of ribociclib on exposure to rivaroxaban was significantly greater than that of apixaban. We offer several possible explanations for this observation. First, the differential roles of CYP3A4 and efflux transporters in the pharmacokinetics of these drugs may explain the variation. While fluconazole and voriconazole, moderate to strong CYP3A inhibitors, show minimal effects on apixaban pharmacokinetics, they exhibit a more pronounced impact on rivaroxaban (Rohr et al., 2022). This suggests that apixaban pharmacokinetic interaction is not dependent on the singular inhibition of CYP3A4. Furthermore, the renal clearance of rivaroxaban is highly dependent on active renal secretion via P-gp and BCRP, whereas apixaban exhibits a lower dependence on these transporters. Thus, ribociclib’s potential inhibitory effect on renal transporters may result in a greater impact on rivaroxaban exposure (Hindley et al., 2023; Bratsos, 2019). In addition, plasma protein binding was approximately 87% with apixaban and higher (approximately 95%) with rivaroxaban, suggesting that ribociclib-induced displacement of rivaroxaban from plasma proteins may lead to a greater interaction likelihood (Hindley et al., 2023). Notably, evidence indicates that inhibition of a single efflux transporter does not effectively suppress the transport of apixaban in Caco-2 cells expressing multiple transporters. However, simultaneous inhibition of P-gp and BCRP causes a more substantial reduction in apixaban efflux (Zhang et al., 2013). Intestinal efflux transport has been demonstrated to affect the absorption of rivaroxaban (Kou et al., 2021). This is also consistent with the PCR results that ribociclib inhibited intestinal BCRP expression but not intestinal P-gp. This selective inhibition may be the main reason the magnitude of the effect of ribociclib was smaller in the apixaban group than in the rivaroxaban group.
Based on these pharmacokinetic results, we investigated the effect of multiple doses of ribociclib on low-dose rivaroxaban (1 mg/kg) blood levels. The results showed that the AUC and Cmax of rivaroxaban increased by approximately 2.3-fold and 1.3-fold, respectively. This indicates that co-administration of low-dose rivaroxaban with potent inhibitors of CYP3A4, and BCRP does not achieve the intended reduction in exposure. Notably, the AUC of rivaroxaban at 1 mg/kg was not significantly different from that at 2 mg/kg, likely due to limited absorption at the higher dose. Rivaroxaban doses exceeding 20 mg exhibit low bioavailability, suggesting that dose and exposure are not proportional at higher levels. The increased bioavailability of the lower dose, combined with ribociclib’s inhibitory effects, led to similar AUC values between the 1 mg/kg and 2 mg/kg dose groups. Therefore, when combining ribociclib and rivaroxaban in clinical practice, it is necessary to actively monitor the blood concentration of rivaroxaban. Ribociclib and rivaroxaban can be administered at appropriate intervals to avoid toxic side effects. In addition, apixaban has a better safety profile when administered concomitantly with ribociclib than rivaroxaban and can be used as the preferred dosing regimen. These findings can guide the clinical use of rivaroxaban and apixaban in combination with ribociclib.
Multiple doses of rivaroxaban or apixaban did not alter the pharmacokinetic parameters of ribociclib. Previous studies indicate that ritonavir, a strong CYP3A inhibitor, increases ribociclib AUC by 3.2-fold, whereas rifampicin, a strong CYP3A inducer, reduces ribociclib AUC by 89% (Samant et al., 2020). In contrast, rivaroxaban and apixaban were not inhibited or induced by metabolism and transport only through CYP3A4, P-gp and BCRP. Even though there was a slight increase in Cmax in the apixaban group, probably due to competition for the same transporters and metabolizing enzymes, it was not statistically significant.
Moreover, it is essential to consider the limitations of this study. First, the observed pharmacokinetic interactions may differ between rats and humans due to species-specific differences in metabolism and transporter activity. Second, we did not use a rat model of breast cancer, and there may be variations in the metabolism and transport processes between healthy rats and those with breast cancer, potentially influencing the study outcomes. Third, the underlying mechanisms of this study are require further validation through more detailed and in-depth investigations.
5 Conclusion
In this study, we developed and validated a sensitive, rapid, reliable, and accurate UPLC-MS/MS method for the quantification of ribociclib in rat plasma. The method was successfully applied to the pharmacokinetic interaction study in this experiment. The experimental results showed that multiple doses of ribociclib led to increased exposure to rivaroxaban, potentially increasing the risk of hemorrhage. Furthermore, the study suggests that there may be no clinically significant drug interactions between apixaban and ribociclib. Importantly, this study provides insights that may assist in optimizing dosing regimens for patients with breast cancer-associated thrombosis, helping to adjust doses and minimize toxic side effects. Given that this experiment was conducted in rats, additional clinical validation is necessary to confirm these findings in human populations.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was approved by Animal Ethics Committee of Hebei General Hospital (Shijiazhuang, China). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
ZL: Conceptualization, Data curation, Investigation, Methodology, Project administration, Software, Writing–original draft. WD: Conceptualization, Data curation, Investigation, Methodology, Project administration, Software, Writing–original draft. QW: Conceptualization, Data curation, Investigation, Methodology, Project administration, Software, Writing–review and editing. ZW: Investigation, Methodology, Writing–review and editing. JA: Investigation, Methodology, Writing–review and editing. YM: Investigation, Methodology, Writing–review and editing. ZD: Conceptualization, Writing–review and editing. YL: Conceptualization, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We acknowledge the support of the Hebei General Hospital Clinical Research Center, Shijiazhuang, Hebei, China.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agnelli, G., Becattini, C., Meyer, G., Munoz, A., Huisman, M. V., Connors, J. M., et al. (2020). Apixaban for the treatment of venous thromboembolism associated with cancer. N. Engl. J. Med. 382 (17), 1599–1607. doi:10.1056/NEJMoa1915103
Amin, A., Naeem, M. O., Amin, L., Khaliq, S. U., Ahmad, A., Vohra, R. R., et al. (2024). Apixaban versus low molecular weight heparin in patients with cancer-associated venous thromboembolism: a systematic review and meta-analysis. Ann. Med. Surg. 86 (8), 4675–4683. doi:10.1097/MS9.0000000000002147
Bellesoeur, A., Thomas-Schoemann, A., Allard, M., Smadja, D., Vidal, M., Alexandre, J., et al. (2018). Pharmacokinetic variability of anticoagulants in patients with cancer-associated thrombosis: clinical consequences. Crit. Rev. Oncol. Hematol. 129, 102–112. doi:10.1016/j.critrevonc.2018.06.015
Braal, C. L., Jongbloed, E. M., Wilting, S. M., Mathijssen, R., Koolen, S., and Jager, A. (2021). Inhibiting CDK4/6 in breast cancer with palbociclib, ribociclib, and abemaciclib: similarities and differences. Drugs 81 (3), 317–331. doi:10.1007/s40265-020-01461-2
Bratsos, S. (2019). Pharmacokinetic properties of rivaroxaban in healthy human subjects. Cureus 11, e5484. doi:10.7759/cureus.5484
Bray, F., Laversanne, M., Sung, H., Ferlay, J., Siegel, R. L., Soerjomataram, I., et al. (2024). Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca. Cancer. J. Clin. 74 (3), 229–263. doi:10.3322/caac.21834
Byon, W., Garonzik, S., Boyd, R. A., and Frost, C. E. (2019). Apixaban: a clinical pharmacokinetic and pharmacodynamic review. Clin. Pharmacokinet. 58, 1265–1279. doi:10.1007/s40262-019-00775-z
Chen, Y. (2024). Letter Re: treating cancer-associated venous thromboembolism: a practical approach. Eur. J. Cancer. 210, 114304. doi:10.1016/j.ejca.2024.114304
Cohen, A., Sah, J., Lee, T., Rosenblatt, L., Hlavacek, P., Emir, B., et al. (2021). Effectiveness and safety of apixaban vs. Warfarin in venous thromboembolism patients with obesity and morbid obesity. J. Clin. Med. 10 (2), 200. doi:10.3390/jcm10020200
Dickson, K., Koom-Dadzie, K., Brito-Dellan, N., and Escalante, C. (2022). Risks, diagnosis, and management of recurrent cancer-associated thrombosis (CAT): a narrative review. Support. Care Cancer. 30 (10), 8539–8545. doi:10.1007/s00520-022-07160-w
Fernandes, C. J., Morinaga, L. T. K., Alves, J. J. L., Castro, M. A., Calderaro, D., Jardim, C. V. P., et al. (2019). Cancer-associated thrombosis: the when, how and why. Eur. Respir. Rev. 28 (151), 180119. doi:10.1183/16000617.0119-2018
Frere, C., Farge, D., Douketis, J., and Connors, J. M. (2022). The 2022 ITAC evidence-based clinical practice guidelines: new update from the International Initiative on Thrombosis and Cancer to improve the care in patients with cancer-associated thrombosis. J. Med. Vasc. 47 (3), 113–115. doi:10.1016/j.jdmv.2022.08.002
Hindley, B., Lip, G. Y. H., Mccloskey, A. P., and Penson, P. E. (2023). Pharmacokinetics and pharmacodynamics of direct oral anticoagulants. Expert Opin. Drug Metab. Toxicol. 19 (12), 911–923. doi:10.1080/17425255.2023.2287472
Jiang, X., Crain, E. J., Luettgen, J. M., Schumacher, W. A., and Wong, P. C. (2009). Apixaban, an oral direct factor Xa inhibitor, inhibits human clot-bound factor Xa activity in vitro. Thromb. Haemost. 101 (4), 780–782. doi:10.1160/th08-07-0486
Kahn, S. R., Lim, W., Dunn, A. S., Cushman, M., Dentali, F., Akl, E. A., et al. (2012). Prevention of VTE in nonsurgical patients: antithrombotic therapy and prevention of thrombosis, 9th ed: American college of chest physicians evidence-based clinical practice guidelines. Chest 141 (2 Suppl. l), e195S–e226S. doi:10.1378/chest.11-2296
Key, N. S., Khorana, A. A., Kuderer, N. M., Bohlke, K., Lee, A., Arcelus, J. I., et al. (2020). Venous thromboembolism prophylaxis and treatment in patients with cancer: ASCO clinical practice guideline update. J. Clin. Oncol. 38 (5), 496–520. doi:10.1200/JCO.19.01461
Kou, W., Sodhi, J. K., Wu, X. A., and Benet, L. Z. (2021). Investigating intestinal transporter involvement in rivaroxaban disposition through examination of changes in absorption. Pharm. Res. 38 (5), 795–801. doi:10.1007/s11095-021-03039-3
Kulkarni, A., and Singh, J. (2024). Predicting drug-drug interactions in breast cancer patients treated with CDK4/6 inhibitors and forward planning. Expert Opin. Drug Metab. Toxicol. 20 (4), 225–233. doi:10.1080/17425255.2024.2341810
Kvasnicka, T., Malikova, I., Zenahlikova, Z., Kettnerova, K., Brzezkova, R., Zima, T., et al. (2017). Rivaroxaban - metabolism, pharmacologic properties and drug interactions. Curr. Drug Metab. 18 (7), 636–642. doi:10.2174/1389200218666170518165443
Lee, P., Kistler, K. D., Douyon, L., Volodarsky, R., Young, A., Karve, S., et al. (2023). Systematic literature review of real-world effectiveness results data for first-line ibrutinib in chronic lymphocytic leukemia and small lymphocytic lymphoma. Drugs-Real World Outcomes 10 (1), 11–22. doi:10.1007/s40801-022-00332-4
Lyman, G. H., Carrier, M., Ay, C., Di Nisio, M., Hicks, L. K., Khorana, A. A., et al. (2021). American Society of Hematology 2021 guidelines for management of venous thromboembolism: prevention and treatment in patients with cancer. Blood Adv. 5 (4), 927–974. doi:10.1182/bloodadvances.2020003442
Mandala, M., Falanga, A., and Roila, F.ESMO Guidelines Working Group (2011). Management of venous thromboembolism (VTE) in cancer patients: ESMO Clinical Practice Guidelines. Ann. Oncol. 22 (Suppl. 6), vi85–92. doi:10.1093/annonc/mdr392
Marcucci, M., Etxeandia-Ikobaltzeta, I., Yang, S., Germini, F., Gupta, S., Agarwal, A., et al. (2022). Benefits and harms of direct oral anticoagulation and low molecular weight heparin for thromboprophylaxis in patients undergoing non-cardiac surgery: systematic review and network meta-analysis of randomised trials. BMJ 376, e066785. doi:10.1136/bmj-2021-066785
Mayer, E. L., Dueck, A. C., Martin, M., Rubovszky, G., Burstein, H. J., Bellet-Ezquerra, M., et al. (2021). Palbociclib with adjuvant endocrine therapy in early breast cancer (PALLAS): interim analysis of a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 22 (2), 212–222. doi:10.1016/S1470-2045(20)30642-2
Mueck, W., Kubitza, D., and Becka, M. (2013). Co-administration of rivaroxaban with drugs that share its elimination pathways: pharmacokinetic effects in healthy subjects. Br. J. Clin. Pharmacol. 76 (3), 455–466. doi:10.1111/bcp.12075
Ngo, L. T., Yang, S. Y., Tran, Q. T., Kim, S. K., Yun, H. Y., and Chae, J. W. (2020). Effects of carbamazepine and phenytoin on pharmacokinetics and pharmacodynamics of rivaroxaban. Pharmaceutics 12 (11), 1040. doi:10.3390/pharmaceutics12111040
Pavlovic, D., Niciforovic, D., Papic, D., Milojevic, K., and Markovic, M. (2023). CDK4/6 inhibitors: basics, pros, and major cons in breast cancer treatment with specific regard to cardiotoxicity - a narrative review. Ther. Adv. Med. Oncol. 15, 17588359231205848. doi:10.1177/17588359231205848
Percie, D. S. N., Hurst, V., Ahluwalia, A., Alam, S., Avey, M. T., Baker, M., et al. (2020). The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. BMJ Open Sci. 4 (1), e100115. doi:10.1136/bmjos-2020-100115
Riaz, I. B., Fuentes, H., Deng, Y., Naqvi, S., Yao, X., Sangaralingham, L. R., et al. (2023). Comparative effectiveness of anticoagulants in patients with cancer-associated thrombosis. JAMA Netw. Open 6 (7), e2325283. doi:10.1001/jamanetworkopen.2023.25283
Rohr, B. S., Foerster, K. I., Blank, A., Burhenne, J., Mahmoudi, M., Haefeli, W. E., et al. (2022). Perpetrator characteristics of azole antifungal drugs on three oral factor Xa inhibitors administered as a microdosed cocktail. Clin. Pharmacokinet. 61 (1), 97–109. doi:10.1007/s40262-021-01051-9
Sabatino, J., De Rosa, S., Polimeni, A., Sorrentino, S., and Indolfi, C. (2020). Direct oral anticoagulants in patients with active cancer: a systematic review and meta-analysis. JACC-Cardiooncol. 2 (3), 428–440. doi:10.1016/j.jaccao.2020.06.001
Samant, T. S., Huth, F., Umehara, K., Schiller, H., Dhuria, S. V., Elmeliegy, M., et al. (2020). Ribociclib drug-drug interactions: clinical evaluations and physiologically-based pharmacokinetic modeling to guide drug labeling. Clin. Pharmacol. Ther. 108 (3), 575–585. doi:10.1002/cpt.1950
Schrag, D., Uno, H., Rosovsky, R., Rutherford, C., Sanfilippo, K., Villano, J. L., et al. (2023). Direct oral anticoagulants vs low-molecular-weight heparin and recurrent VTE in patients with cancer: a randomized clinical trial. JAMA 329 (22), 1924–1933. doi:10.1001/jama.2023.7843
Sorf, A., Hofman, J., Kucera, R., Staud, F., and Ceckova, M. (2018). Ribociclib shows potential for pharmacokinetic drug-drug interactions being a substrate of ABCB1 and potent inhibitor of ABCB1, ABCG2 and CYP450 isoforms in vitro. Biochem. Pharmacol. 154, 10–17. doi:10.1016/j.bcp.2018.04.013
Tsoukalas, N., Brito-Dellan, N., Font, C., Butler, T., Rojas-Hernandez, C. M., Butler, T., et al. (2022). Complexity and clinical significance of drug-drug interactions (DDIs) in oncology: challenging issues in the care of patients regarding cancer-associated thrombosis (CAT). Support. Care Cancer. 30 (10), 8559–8573. doi:10.1007/s00520-022-07235-8
Wang, Z., Li, Y., He, X., Fu, Y., Li, Y., Zhou, X., et al. (2023). In vivo evaluation of the pharmacokinetic interactions between almonertinib and rivaroxaban, almonertinib and apixaban. Front. Pharmacol. 14, 1263975. doi:10.3389/fphar.2023.1263975
Wittkowsky, A. K. (2006). Barriers to the long-term use of low-molecular weight heparins for treatment of cancer-associated thrombosis. J. Thromb. Haemost. 4 (9), 2090–2091. doi:10.1111/j.1538-7836.2006.02073.x
Keywords: cancer-associated venous thromboembolism, drug-drug interaction, ribociclib, apixaban, rivaroxaban
Citation: Liu Z, Du W, Wang Q, Wang Z, An J, Ma Y, Dong Z and Li Y (2025) In Vivo pharmacokinetic interactions of ribociclib with rivaroxaban and apixaban in rats: implications for increased drug exposure and dose adjustments. Front. Pharmacol. 16:1530806. doi: 10.3389/fphar.2025.1530806
Received: 19 November 2024; Accepted: 10 March 2025;
Published: 31 March 2025.
Edited by:
Yurong Lai, Gilead, United StatesReviewed by:
Mamunur Rashid, University of Nebraska Medical Center, United StatesMyeongJin Yi, National Institute of Environmental Health Sciences (NIH), United States
Copyright © 2025 Liu, Du, Wang, Wang, An, Ma, Dong and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhanjun Dong, ZHpqaGJnaEAxMjYuY29t; Ying Li, bHl5YW9kYUAxMjYuY29t
†These authors have contributed equally to this work