 Minlong Wei1†Jinyun Lin1†Yi Zeng2Xiaojuan Wang2Jialu Wen1Jing Wang3
Minlong Wei1†Jinyun Lin1†Yi Zeng2Xiaojuan Wang2Jialu Wen1Jing Wang3 Wei Zou4Kang Tu1
Wei Zou4Kang Tu1 Menghua Liu1*
Menghua Liu1* Juan Li5*
Juan Li5*- 1Key Laboratory of Drug Metabolism Research and Evaluation of the State Drug Administration, Guangdong Provincial Key Laboratory of New Drug Screening, School of Pharmaceutical Sciences, Southern Medical University, Guangzhou, China
- 2Southern Medical University Hospital of Integrated Traditional Chinese and Western Medicine, Southern Medicine University, Guangzhou, China
- 3School of Traditional Chinese Medicine, Southern Medical University, Guangzhou, China
- 4Changsha Research and Development Center on Obstetric and Gynecologic Traditional Chinese Medicine Preparation, Hunan Provincial Maternal and Child Healthcare Hospital, Changsha, Hunan, China
- 5School of Nursing, Southern Medical University, Guangzhou, Guangdong, China
Chronic kidney disease (CKD) involves intricate pathological mechanisms that currently lack definitive therapeutic interventions to halt disease progression. Increasing evidence suggests that enzymatic post-translational modifications (ePTMs) of proteins play an important role in CKD. As a dynamic and reversible type of PTM, ePTMs offer advantages such as enzyme-specific catalysis, high reversibility, and precise regulation. Various forms of ePTMs have been reported in CKD, including methylation, acetylation, ubiquitination, enzymatic glycosylation, lactylation, palmitoylation, crotonylation, SUMOylation, and prenylation. Given the critical roles of these ePTMs in CKD, this review summarizes their molecular mechanisms in disease progression, explores their potential as diagnostic markers and therapeutic targets, and highlights advances in small-molecule drugs targeting ePTMs. It is important to note that most ePTMs remain in the early stages of research, with evidence of cross-regulation and synergistic effects among different modifications. Further investigation will require more basic studies and clinical trials. This review aims to help bridge the gap between basic research and clinical application of ePTMs in CKD, and to support the development of more effective treatment strategies.
1 Introduction
Chronic kidney disease (CKD), characterized by progressive loss of renal function, has become a global public health problem that affects approximately 10% of the world’s population (Miguel et al., 2025; Romagnani et al., 2025). With the disease progressing, patients with CKD usually face the risk of multiple adverse outcomes, including cognitive impairment, cardiovascular events, end-stage kidney disease (ESKD), and even death (KDIGO, 2024). Diabetes, hypertension, autoimmune diseases and genetic susceptibility are all major causes CKD. Its pathological process involves complex signaling pathways, such as inflammatory responses, oxidative stress, apoptosis and fibrosis (Romagnani et al., 2017; Burnier and Damianaki, 2023). Although various clinical interventions are available, effectively halting CKD progression remains challenging to date (Romagnani et al., 2025). There is an urgent need to delve into its molecular mechanism to find new therapeutic targets.
Post-translational modification (PTM) of proteins is a chemical modification process after the completion of protein translation, which can regulate the activity, stability, localization and intermolecular interactions of proteins (Wang et al., 2022). A growing number of research findings suggest PTMs plays an important role in the pathophysiological process of CKD (Noels et al., 2024), especially in diabetic nephropathy (DN), where modifications such as protein deubiquitinating modification affect the process of podocyte inflammation and injury (Zhao et al., 2024b). PTMs have been reported to be classified into two types: non-enzymatic PTMs and enzymatic PTMs (ePTMs) (Jennings et al., 2022). Non-enzymatic PTM is usually triggered by the direct reaction of proteins with active metabolites (Tang and Kalim, 2022). In CKD, oxidative stress and metabolic disorders accelerate a significant increase in non-enzymatic PTMs, intensifying the inflammatory response and fibrosis in renal tissue (Noels et al., 2024). Taking the advanced glycation end products as an example, they can activate downstream signaling pathways such as nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) by binding to the cell surface receptor, subsequently leading to the release of pro-inflammatory cytokines (Stinghen et al., 2016; Wang and Zhang, 2024).
EPTMs rely on the catalysis of specific enzymes and are characterized by strong reversibility and precise regulation (Li Z. et al., 2025). Here, nine key ePTMs are highlighted, including methylation, acetylation, ubiquitination, glycosylation, lactylation, palmitoylation, crotonylation, small ubiquitin-like modifier (SUMO)-mediated modification, and prenylation. They have received increasing attention in CKD, participating in core pathological processes such as fibrosis and inflammation, and showing potential as therapeutic targets (Laget et al., 2022). However, their mechanisms of action have not yet been systematically integrated. Notably, phosphorylation has been extensively studied in chronic kidney disease with many established databases (e.g., PhosphoSitePlus, and PhosphoGRID), so it is not listed separately here to avoid redundancy, but will be mentioned when it crosstalks with other ePTMs (Li K. et al., 2024; Šakić et al., 2024; Li J. et al., 2024). Accumulating evidence indicates these ePTMs are not merely passive markers of CKD progression (e.g., Gd-IgA1), but actively participate in disease etiology by regulating core pathological processes (Vaz de Castro et al., 2024; Li J. et al., 2024). For example, histone deacetylases (HDACs) regulate histone acetylation and deacetylation, and their inhibitors (such as vorinostat and romidepsin) have been approved by the U.S. Food and Drug Administration (FDA) for treating lymphoma. (Mabe et al., 2024; Roza et al., 2023). Recent studies have also explored inhibitors or activators targeting protein kinases, methyltransferases, and ubiquitin ligases to modulate ePTMs in disease treatment (Mabe et al., 2024). The development of such drugs requires a thorough understanding of these enzymes’ structure, function and roles in CKD.
Given the unique advantages of ePTMs in disease regulation, this review focuses on three key aspects: (1) the molecular mechanisms of ePTMs and their regulatory enzymes (e.g., acetyltransferases and deacetylases); (2) the roles of ePTMs in the development and progression of CKD, including tubulointerstitial fibrosis, inflammation, and metabolic disturbances; (3) the potential of ePTMs as diagnostic markers and therapeutic targets for CKD, with particular attention to current drug development and future directions in precision medicine. This review aims to bridge the gap between basic research and clinical application, laying the groundwork for the development of more effective therapies for CKD.
2 Methylation
2.1 Enzymatic mechanisms of methylation
Protein methylation is a major form of ePTMs involved in the pathogenesis of CKD (Noels et al., 2024). It entails the enzymatic transfer of a methyl group (–CH3) to specific amino acids, mainly lysine and arginine, in forms such as monomethylation (me1), dimethylation (me2), and trimethylation (me3). Lysine may undergo me1, me2, or me3, while arginine can be modified by me1, symmetric dimethylation (me2s), or asymmetric dimethylation (me2a) (Paik et al., 2007; Jin et al., 2023b). These modifications regulate protein function, interactions, and crosstalk with other PTMs, thus influencing various physiological and pathological processes (Jin et al., 2023b; Tan et al., 2024).
Protein lysine methyltransferases (PKMTs) and protein arginine methyltransferases (PRMTs) are the main enzymes responsible for methylation (Luo, 2018; Wu et al., 2021). Using S-adenosylmethionine (SAM) as a methyl donor, they catalyze methylation via a bimolecular nucleophilic substitution reaction, generating methylated proteins and S-adenosylhomocysteine (SAH). Over 50 PKMTs have been identified, many of which target histone and non-histone proteins (Schnee et al., 2024). The largest subgroup, Su(var)3-9, Enhancer-of-zeste, Trithorax (SET) domain, containing PKMTs, share a conserved domain that binds both substrate and SAM (Schnee et al., 2024). For instance, (Su(var)3-9, enhancer of zeste, Trithorax) domain-containing protein 7 (SETD7) and enhancer of zeste homolog 2 (EZH2) catalyze methylation at histone H3 lysine 4 (H3K4) and lysine 27 (H3K27), respectively (Sun et al., 2010; Su et al., 2022). PRMT1-9 are the known arginine methyltransferases, with PRMT1 generating asymmetric dimethylarginine, a contributor to endothelial dysfunction in CKD (Figure 1).
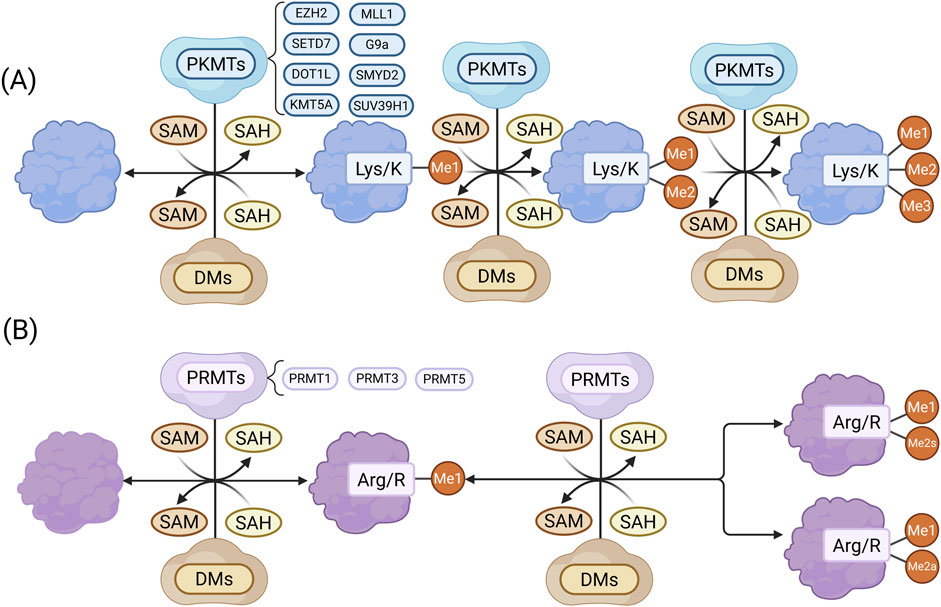
Figure 1. (A) Lysine methylation: PKMTs catalyze the transfer of a methyl group from SAM to lysine residues, generating me1 and SAH. The process may continue to form me2 and me3 proteins. (B) Arginine methylation: PRMTs transfer a methyl group from SAM to arginine residues, producing me1 proteins and SAH, followed by either me2s or me2a. Methylated proteins can be reversed by demethylases (DEs). (Created in https://BioRender.com).
Histone methylation regulates gene expression by altering chromatin structure. H3K4me3, mediated by SETD7, promotes transcription of pro-fibrotic genes like collagen1alpha (Col1α1) and connective tissue growth factor (CTGF) (Sun et al., 2010), whereas H3K27me3, catalyzed by EZH2, suppresses anti-fibrotic gene expression (Wu et al., 2024). Methylation of non-histone proteins, such as transcription 3 (STAT3) and nuclear factor-kappa B (NF-κB) p65, also affects signaling and cell cycle regulation (Li L. X. et al., 2017; Cui et al., 2022). Overall, protein methylation contributes to CKD by modulating gene expression and cellular function, offering potential therapeutic targets. Further studies are needed to clarify its roles and clinical applications in CKD.
2.2 Effect of methylation in the pathological progression of CKD
In CKD, histone methylation modulates gene transcription and contributes to disease progression (Table 1). The histone methyltransferase EZH2 catalyzes H3K27 trimethylation. Elevated levels of the long non-coding RNA (lncRNA) antisense non-coding RNA in the INK4 locus (ANRIL) in CKD recruit EZH2 to the brain-derived neurotrophic factor (BDNF) promoter, reducing BDNF expression and leading to endothelial dysfunction and mitochondrial imbalance (Su et al., 2022). In diabetic nephropathy (DN), lncRNA Dlx6os1 enhances EZH2-mediated H3K27me3 at the SRY-box transcription factor 6 (SOX6) promoter, promoting mesangial cell proliferation, fibrosis, and inflammation (Chen Y. X. et al., 2022). In the ischemia/reperfusion (I/R) and folic acid (FA)-induced acute kidney injury to chronic kidney disease (AKI-to-CKD) transition models, EZH2 can induce trimethylation of histone H3 and represses phosphatase and tensin homolog (PTEN) expression, subsequently activating epidermal growth factor receptor (EGFR)/ERK1/2/STAT3 signaling to drive epithelial–mesenchymal transition (EMT) and G2/M arrest, thereby mediating the progression from acute kidney injury to chronic kidney disease (Zhou et al., 2023). So, inhibiting EZH2 preserves mothers against decapentaplegic homolog (Smad7) and PTEN levels, blocking transforming growth factor beta (TGF-β)/Smad3, EGFR, and platelet-derived growth factor receptor beta (PDGFRβ) signaling and thereby alleviating fibrosis (Zhou et al., 2016). However, It has also been shown that long noncoding (lnc)RNA AFAP1-AS1 can interact with EZH2 to upregulate the level of H3K27me3 in the NOD-like receptor protein 3 (NLRP3) promoter region in M2 macrophage-derived exosomes, which inhibits the protein levels of NLRP3, cleaved caspase-1, gasdermin D (GSDMD)-N, as well as the levels of interleukin (IL)-18, IL-1β, and lactate dehydrogenase (LDH), and ultimately suppresses podocyte pyroptosis (Zhan et al., 2025).
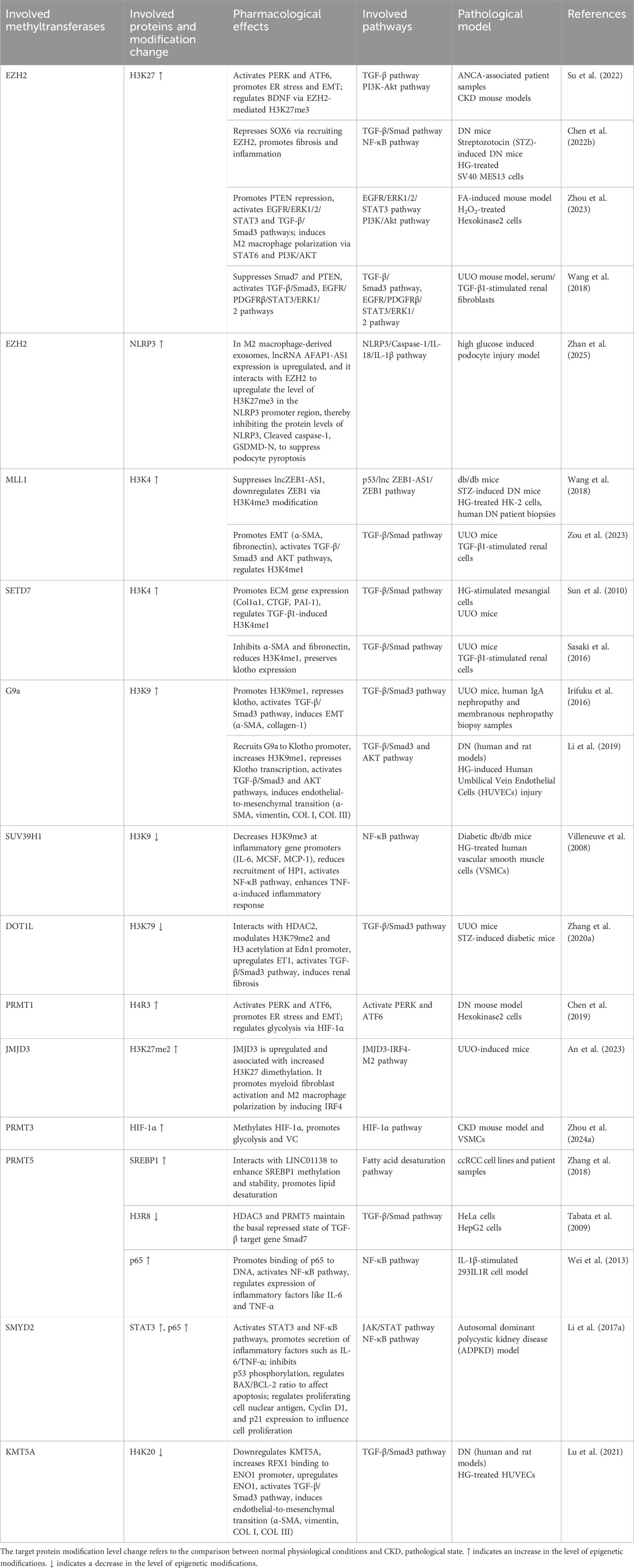
Table 1. Regulatory networks and pathological effects of PKMTs and PRMTs in CKD.
Mixed lineage leukemia 1 (MLL1), another histone methyltransferase, interacts with zinc finger E-box binding homeobox 1 antisense 1 (ZEB1-AS1) in DN to enhance H3K4me3 at the ZEB1 promoter, exerting anti-fibrotic effects (Wang et al., 2018). In unilateral ureteral obstruction (UUO) model, MLL1 and its cofactor menin increase H3K4me1 and activate transforming growth factor-beta1 (TGF-β1) signaling, inducing EMT-related transcription factors and fibrotic markers (Zou et al., 2023). High glucose (HG) also enhances H3K4 methylation at extracellular matrix (ECM) gene promoters via TGF-β1 signaling, promoting ECM production (Sun et al., 2010). TGF-β1 upregulates SET7/9 through Smad3, increasing H3K4me1 and activating pro-fibrotic genes (Sasaki et al., 2016). It also induces G9a, which raises H3K9me1 and suppresses the anti-fibrotic gene Klotho (Irifuku et al., 2016). LncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) recruits G9a to the Klotho promoter, further reducing its expression and contributing to HG-induced endothelial injury (Li et al., 2019). In DN, reduced of the suppressor of variegation 3-9 homolog 1 (SUV39H1) expression and lower H3K9me3 levels correlate with elevated inflammatory gene expression (Villeneuve et al., 2008). Disruptor of Telomeric Silencing 1-Like (DOT1L), which catalyzes H3K79me2, represses Endothelin 1 (EDN1) transcription; its deficiency upregulates EDN1 and promotes fibrosis (Zhang L. et al., 2020). Jumonji domain-containing protein-3 (JMJD3), a histone H3K27 demethylase, is significantly increased with elevated H3K27 dimethylation, promotes myeloid fibroblast activation and M2 macrophage polarization via the JMJD3-IRF4 axis in UUO mice, and this can be reversed by the JMJD3 inhibitor GSK-J4 (An et al., 2023).
Non-histone methylation also contributes to CKD pathogenesis. In DN, high glucose upregulates PRMT1, which mediates H4R3me2a modification at the activating transcription factor 6 (ATF6) promoter, activating the protein kinase R-like endoplasmic reticulum kinase (PERK) and ATF6 pathways and triggering the endoplasmic reticulum (ER) stress and apoptosis in tubular cells (Chen et al., 2019). PRMT1 also deposits H4R3me2 at the ATF6 promoter, inducing EMT and fibrosis (Chen et al., 2019). In CKD, PRMT3 stabilizes the transcription factor hypoxia-inducible factor-1alpha (HIF-1α) via methylation, enhancing glycolysis and promoting vascular smooth muscle cell (VSMC) osteogenic transformation and vascular calcification (VC) (Zhou G. et al., 2024). PRMT5 interacts with long intergenic non-coding RNA located on 1q21.2 (LINC01138) in renal carcinoma to induce Sterol regulatory element-binding protein 1 (SREBP1) me2s, supporting lipid synthesis and tumor proliferation (Zhang et al., 2018). It also mediates basal repression of the Samd7 promoter and enhances NF-κB signaling under IL-1β stimulation by catalyzing R30 dimethylation of p65 (Tabata et al., 2009; Wei et al., 2013).
SET and MYND domain-containing protein 2 (SMYD2) methylates p53 at K370, suppressing its transcriptional activity and promoting cyst epithelial cell survival (Li L. X. et al., 2017). It also modifies STAT3 and p65, activating genes linked to inflammation and proliferation (Li L. X. et al., 2017). In DN, HG reduces lysine methyltransferase 5A (KMT5A) expression, lowering H4K20me1 and lifting transcriptional repression of Enolase 1 (ENO1), which contributes to endothelial–mesenchymal transition and fibrosis (Lu et al., 2021). Collectively, histone and non-histone methylation regulate gene expression, signaling pathways, and phenotypic transitions, playing central roles in CKD development.
2.3 The therapeutic potential of methylation in CKD
Given the role of methylation in CKD progression, methyltransferase inhibitors show therapeutic potential by modulating histone and non-histone methylation (Table 2). The EZH2 inhibitor 3-deazaneplanocin A (3-DZNeP) attenuates fibrosis in UUO model by suppressing TGF-β/Smad3 and EGFR/PDGFR signaling, thereby reducing fibroblast activation and ECM deposition (Zhou et al., 2016). SET7/9 inhibitors such as sinefungin and the selective inhibitor PFI-2 reduce H3K4me1 levels and inhibit fibrosis. Sinefungin blocks TGF-β1-induced fibroblast activation and ECM production (e.g., α-SMA, collagen I) (Sasaki et al., 2016), while PFI-2 suppresses Th2 cytokines (IL-4, IL-13), inhibits M2-to-myofibroblast transition, and reduces nuclear NF-κB p65 translocation (Liu et al., 2021). The DOT1L inhibitor EPZ5676 decreases H3K79me2, stabilizes PTEN and Smad7, and suppresses EMT and fibroblast activation (Liu L. et al., 2019). PRMT1 inhibitor AMI-1 and G9a inhibitor BIX01294 lower H4R3me2a and H3K9me1 levels, respectively, thereby inhibiting Smad3 phosphorylation and TGF-β1-induced fibrosis (Zhu Y. et al., 2020; Irifuku et al., 2016). MLL1 complex inhibitors MI-503 and MM102 also reduce renal injury (Zhang et al., 2022; Zou et al., 2023). Additionally, the demethylase inhibitor GSK-J4 prevents H3K27 demethylation by targeting KDM6A, downregulating DKK1 and TGF-β1, and alleviating fibrosis in DN (Hung et al., 2022). Furthermore, methyltransferase inhibitors have also been shown to alleviate CKD-related complications. For example, PRMT3 inhibitor SGC707 reduces arginine methylation of HIF-1α and attenuates VC (Zhou G. et al., 2024). The SMYD2 inhibitor AZ505 lowers methylation of STAT3, NF-κB p65, and H3K36me3, thereby reducing tubular cell apoptosis and inflammation (Li L. X. et al., 2017; Cui et al., 2022). In general, these compounds targeting methyltransferases regulate histone and non-histone methylation through various signaling pathways such as TGF-β/Smad3 and EGFR/ERK1/2/STAT3, and exert ameliorative effects in pathological conditions of CKD such as renal fibrosis. This indicates that methylation is a driver of the progression of CKD and its complications, and regulating its related enzymes could be a potential research direction for CKD treatment. It should also be noted that most compounds that regulate methylation are still in preclinical stages such as laboratory or animal studies, and there is a long development process ahead before they can be practically applied in clinic.
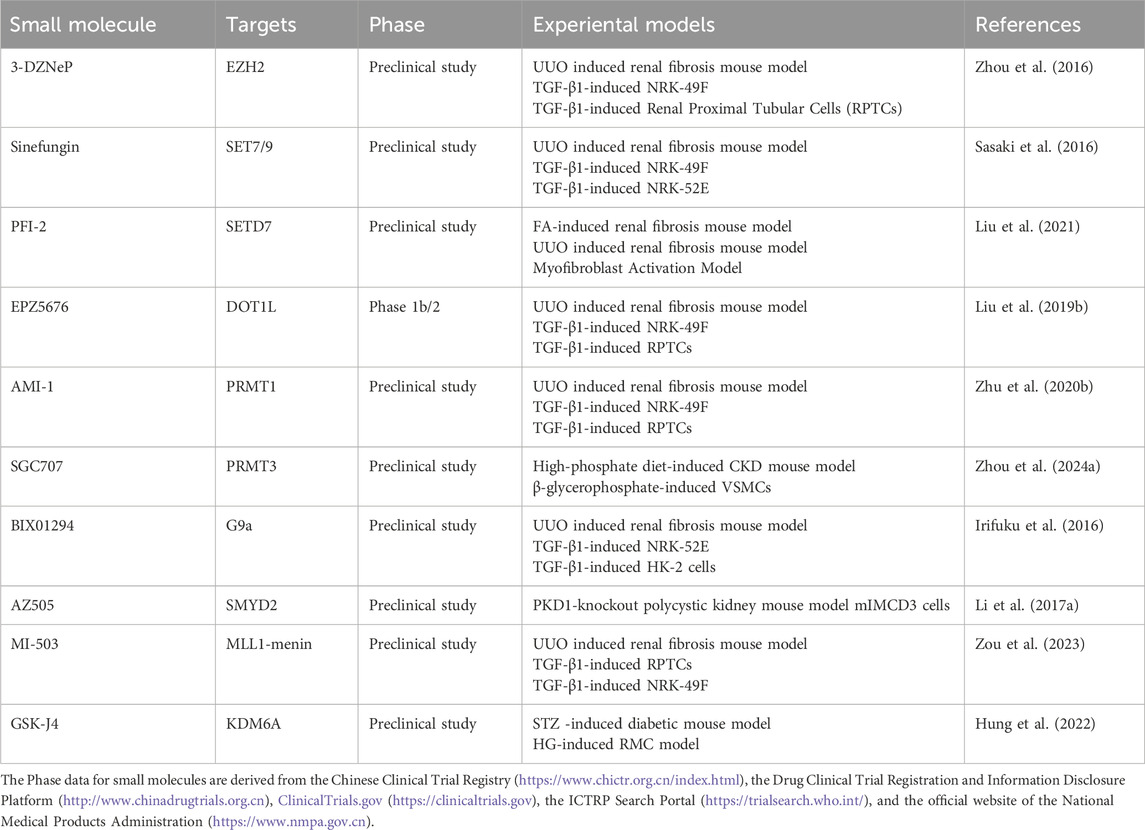
Table 2. Small molecules targeting methylation modifications in CKD and their target proteins.
3 Acetylation
3.1 Acetylation modification and its key enzymes involved
Acetylation is a reversible modification in which an acetyl group is transferred from acetyl-coenzyme A (CoA) to the N-terminus or ε-amino group of lysine residues. In CKD, widespread protein acetylation has been detected in renal tubular epithelial cells and contributes to disease progression (Pan et al., 2024; Tan et al., 2024). A proteomic study using immunoaffinity enrichment and liquid chromatography-mass spectrometry identified 2,012 acetylated proteins and 4,311 acetylation sites in mouse tubular epithelial cells under high glucose conditions (Wan et al., 2021), with most located in the mitochondria, nucleus, and cytoplasm.
Histones are among the most extensively acetylated proteins. Their acetylation is regulated by histone acetyltransferases (HATs), which add acetyl groups, and HDACs, which remove them (Figure 2). HATs, or “writers,” catalyze lysine acetylation, neutralizing histone charge, loosening chromatin, and promoting transcription by enhancing DNA accessibility (Singh et al., 2024). HATs are classified into three main families: p300/CREB-binding protein (CBP), MYST (e.g., Esa1, Sas2, Tip60, MOF, MOZ, MOR), and GCN5-related N-acetyltransferase (GNAT) (e.g., GCN5, PCAF, Elp3, Hpa2, Hat1) (White et al., 2024). HDACs, or “erasers,” remove acetyl groups, leading to chromatin condensation and transcriptional repression (Seto and Yoshida, 2014). Eighteen HDACs have been identified and are grouped into four classes based on structure and localization: Class I (HDAC1, 2, 3, 8), Class IIa (HDAC4, 5, 7, 9), Class IIb (HDAC6, 10), Class III (sirtuins, SIRT1–7), and Class IV (HDAC11) (Singh et al., 2024).
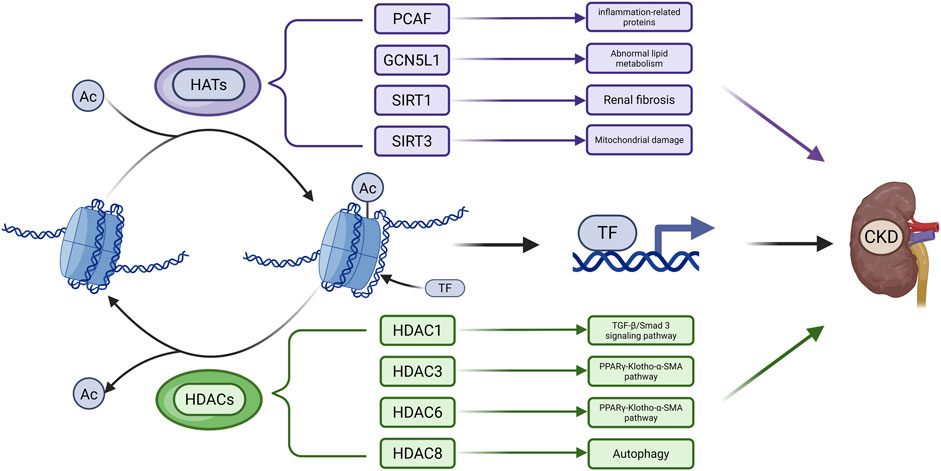
Figure 2. Main Mechanisms of Acetylation in CKD. Acetyl groups (Ac) are transferred to histone amino acid residues by HATs, leading to a more relaxed chromatin structure. This facilitates the binding of transcription factors (TFs) to DNA and thereby regulates gene expression. Acetylated proteins can be deacetylated by HDACs. The dysregulation of acetylation and deacetylation processes contributes to the accelerated progression of CKD. (Created in https://BioRender.com).
3.2 Effect of acetylation in the pathological progression of CKD
In CKD, an imbalance between HATs and HDACs disrupts gene regulation and accelerates disease progression (Table 3). While acetylation levels remain low in healthy kidneys, they are markedly increased in fibrotic renal tissue (Zhang et al., 2021b). Among HATs, acetyltransferase p300/CBP-associated factor (PCAF) and General control of amino acid synthesis 5 like 1 (GCN5L1) are implicated in CKD. PCAF is upregulated in the kidneys of db/db and lipopolysaccharide-treated mice, where it increases histone acetylation (e.g., H3K18ac) and activates inflammatory gene promoters such as Intercellular adhesion molecule-1 (ICAM-1) (Huang J. et al., 2015). GCN5L1 is highly expressed in kidneys of high-fat diet (HFD)-fed mice and regulates acetylation of fatty acid oxidation enzymes, including long-chain acyl coenzyme A dehydrogenase (LCAD) and beta-hydroxyacyl-CoA dehydrogenase (β-HAD), thereby modulating their activity (Lv et al., 2019).
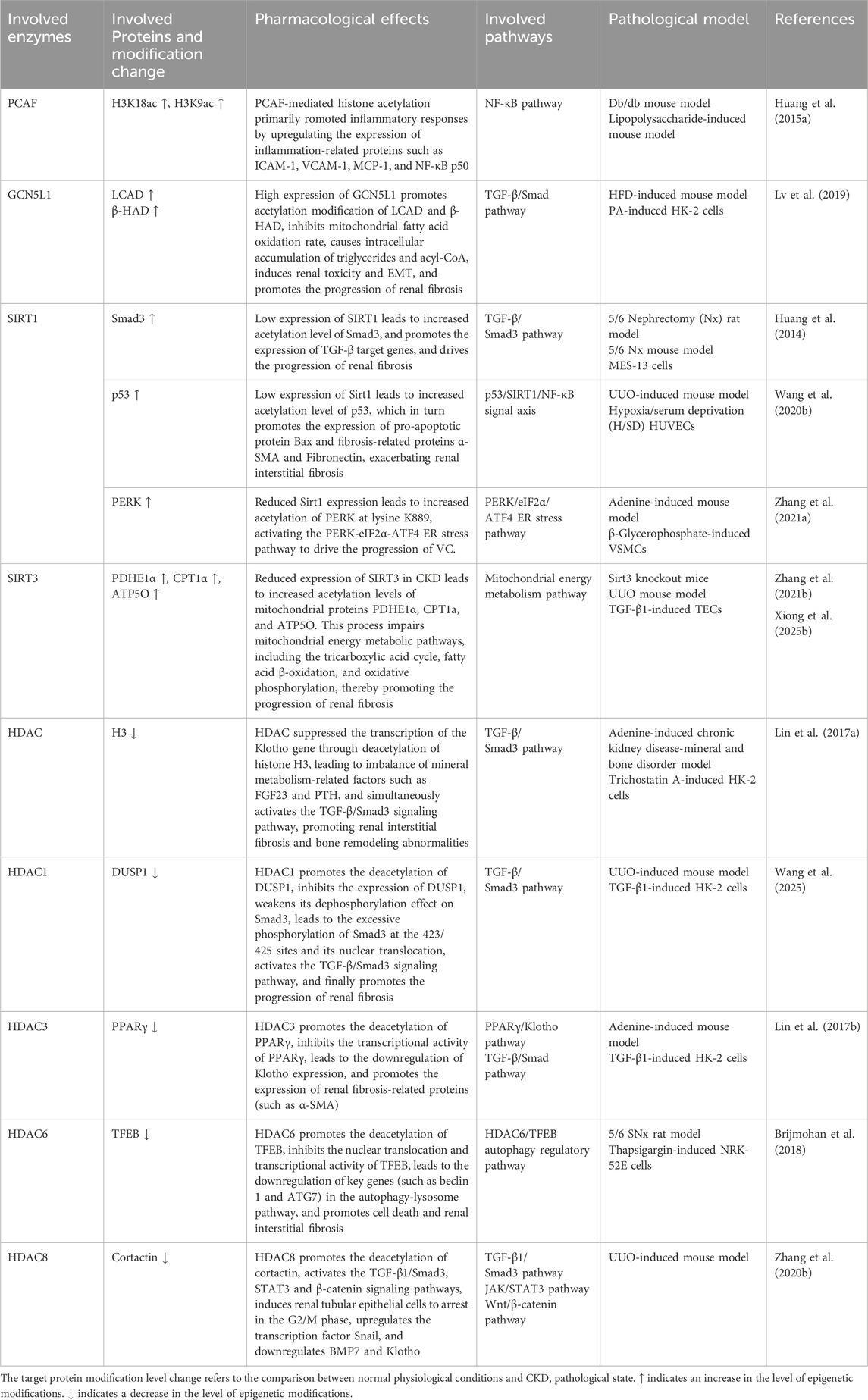
Table 3. Regulatory networks and pathological effects of HATs and HDACs in CKD.
HDACs such as SIRT1, SIRT3, HDAC1, HDAC3, HDAC6, and HDAC8 contribute to CKD pathogenesis by modulating histone and non-histone acetylation. SIRT1 knockout in the ischemia-reperfusion injury (IRI) model exacerbates renal injury and fibrosis by inhibiting H3K27 acetylation at the ATP citrate lyase (ACLY) promoter to impair fatty acid oxidation (FAO), binding to and deacetylating Smad3 to enhance its transcriptional activity, and deacetylating p53 at K382 and K320 to suppress apoptosis (Huang et al., 2014; Wang Y. et al., 2020). SIRT1 overexpression improves renal function, while acetylation at its K889 site regulates PERK activity, reduces ER stress, and mitigates VC (Zhang et al., 2021a). SIRT3 prevents hyperacetylation of pyruvate dehydrogenase (PDH), E1α, carnitine palmitoyl-transferase 1 A (CPT1A), and ATP synthase subunit O (ATP5O) in UUO and Sirt3-deficient mice (Zhang et al., 2021b). Acetylation at pyruvate dehydrogenase 1alpha (PDHE1α) K385 is essential for PDH function under pro-fibrotic stress (Zhang et al., 2021b). Several HDACs suppress protective gene expression. In adenine-fed mice, HDACs reduce Klotho expression by removing H3K9 acetylation. In UUO model, HDACs inhibit dual specificity phosphatase 1 (DUSP1), which is associated with renal dysfunction and fibrosis via Smad3 activation (Lin et al., 2017a; Wang et al., 2025). HDAC3 inhibition enhances Klotho expression by promoting peroxisome proliferator-activated receptor gamma (PPARγ) acetylation and DNA binding (Lin et al., 2017b), while HDAC6 inhibition promotes TFEB acetylation and nuclear translocation, activating autophagy (Brijmohan et al., 2018). TF3, which co-localizes with H3K27Ac, may preserve acetylation by recruiting CBP/p300 (Yang et al., 2025). HDAC1 also regulates DUSP1, and its loss contributes to fibrosis through Smad3 signaling (Wang et al., 2025). HDAC8 is upregulated in UUO and deacetylates cortactin, thereby activating TGF-β1/Smad3, STAT3, and β-catenin pathways while suppressing bone morphogenetic protein 7 (BMP7) and Klotho to promote interstitial fibrosis (Zhang Y. et al., 2020). In conclusion, HATs and HDACs have diverse and complex roles in CKD progression. Their targeted regulation may offer promising strategies for therapeutic intervention.
3.3 The therapeutic potential of acetylation in CKD
Several pharmacological agents targeting acetylation have demonstrated therapeutic potential in CKD models (Table 4). Trichostatin A, an HDAC1/2/3 inhibitor, alleviates fibrosis and mineral metabolism disorders in UUO and adenine-induced models by increasing histone and PPARγ acetylation, suppressing colony-stimulating factor-1 (CSF-1), and upregulating Klotho (Marumo et al., 2010; Azechi et al., 2013; Lin et al., 2017a; Lin et al., 2017b). Chidamide inhibits HDAC1, enhances histone acetylation at the DUSP1 promoter, and suppresses Smad3 signaling (Wang et al., 2025). Sulforaphane, via HDAC2 inhibition, upregulates BMP-7 expression through H3K9/14 acetylation and mitigates diabetic nephrofibrosis via the BMP7/Smad pathway (Kong et al., 2021). The HDAC3 inhibitor RGFP966 promotes PPARγ acetylation, increases Klotho, and inhibits TGF-β/Smad signaling (Chen et al., 2021; Lin et al., 2017b). HDAC4 inhibitors such as MC1568 and tacedinaline elevate histone H3 acetylation, suppress TGF-β1/Smad3 signaling, and restore Klotho expression (Shen et al., 2022; Xiong et al., 2019). Piceatannol downregulates HDAC4/5, inhibits the p38-MAPK pathway, and reduces ECM deposition (Choi et al., 2016). Selective HDAC6 inhibitors (ACY-1215, CKD-506, tubastatin A, and tubacin) enhance α-tubulin acetylation and inhibit NF-κB and TGF-β1/Smad3 pathways (Chen X. et al., 2020; Choi et al., 2018; Choi et al., 2015; Cebotaru et al., 2016). PCI34051, an HDAC8 inhibitor, restores cortactin acetylation and blocks TGF-β1/Smad3 signaling (Zhang Y. et al., 2020). Quisinostat, targeting HDAC11, restores Kruppel-like factor 15 (KLF15) activity and inhibits EMT (Mao et al., 2020).
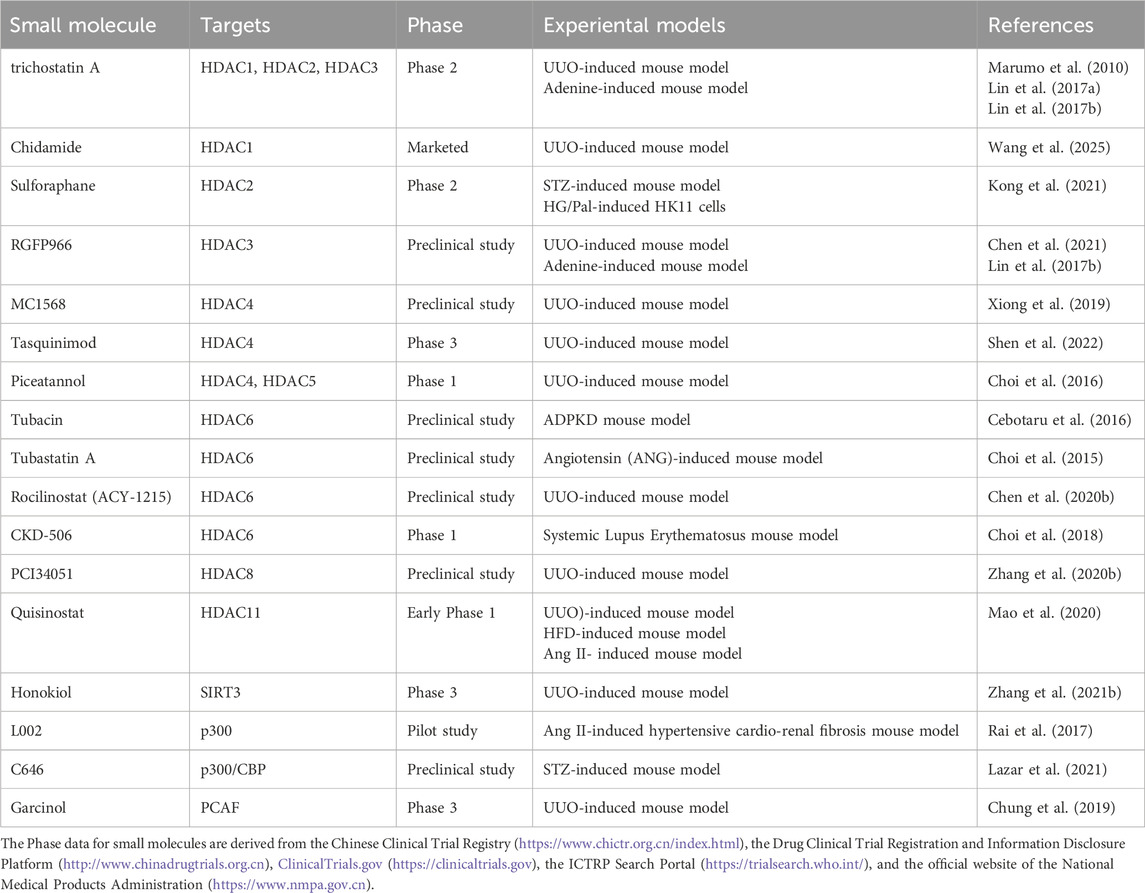
Table 4. Small molecules targeting acetylation in CKD and their target proteins.
Agents targeting HATs have also shown efficacy. Honokiol activates SIRT3, reduces mitochondrial protein acetylation, and improves energy metabolism (Zhang et al., 2021b). L002 and C646 competitively inhibit p300/CBP, reduce acetylation at H3K9, H4, and H3K27, and suppress the TGF-β/Smad pathway and oxidative stress (Rai et al., 2017; Lazar et al., 2021). Gambogic acid inhibits PCAF, reduces H3K9 acetylation, and suppresses NF-κB–mediated inflammation (Chung et al., 2019). It is encouraging that some ePTM-targeted compounds have entered the clinical stage. For instance, the HDAC inhibitor Chidamide, which is approved for peripheral T-cell lymphoma, has been marketed (Shi et al., 2017). Trichostatin A (Phase 3, for metastatic castration-resistant prostate cancer) and ACY-1215 (Phase 1/2, for lymphoma) are among those in clinical research stages (Sternberg et al., 2016; Amengual et al., 2021). However, that the approved indications for these drugs are mostly tumors or autoimmune diseases, and clinical trials specifically targeting CKD remain very limited. It should be clarified that acetylation imbalance is recognized as a driver of CKD progression, and targeted inhibition of relevant enzymes has demonstrated efficacy in UUO, DN, and adenine-induced pathological models. Importantly, some of these drugs, such as chidamide, undergo renal excretion, which underscores the necessity of further addressing safety concerns prior to their clinical translation for CKD.
4 Ubiquitination
4.1 Ubiquitination modification and its key enzymes involved
Protein ubiquitination is a reversible process regulated by ubiquitinating enzymes (E1, E2, E3) and deubiquitinating enzymes (DUBs). First identified in 1975 (Nakamura, 2018), this modification is essential for maintaining renal cell function and homeostasis (Meyer-Schwesinger, 2019; You et al., 2024) E1 enzymes activate ubiquitin (Ub) in an ATP-dependent manner, forming a thioester bond. Ub is then transferred to E2 conjugating enzymes, and finally attached to target proteins by E3 ligases, which confer substrate specificity (Liao et al., 2024; Gan et al., 2025) (Figure 3). DUBs reverse this process by hydrolyzing Ub from proteins. Ubiquitination plays a central role in protein degradation via the ubiquitin–proteasome system, where Ub-tagged proteins are recognized and degraded by the proteasome (Meyer-Schwesinger, 2019). In human cells, over 50 E2 enzymes and approximately 600 E3 ligases have been identified (Wang M. et al., 2020).
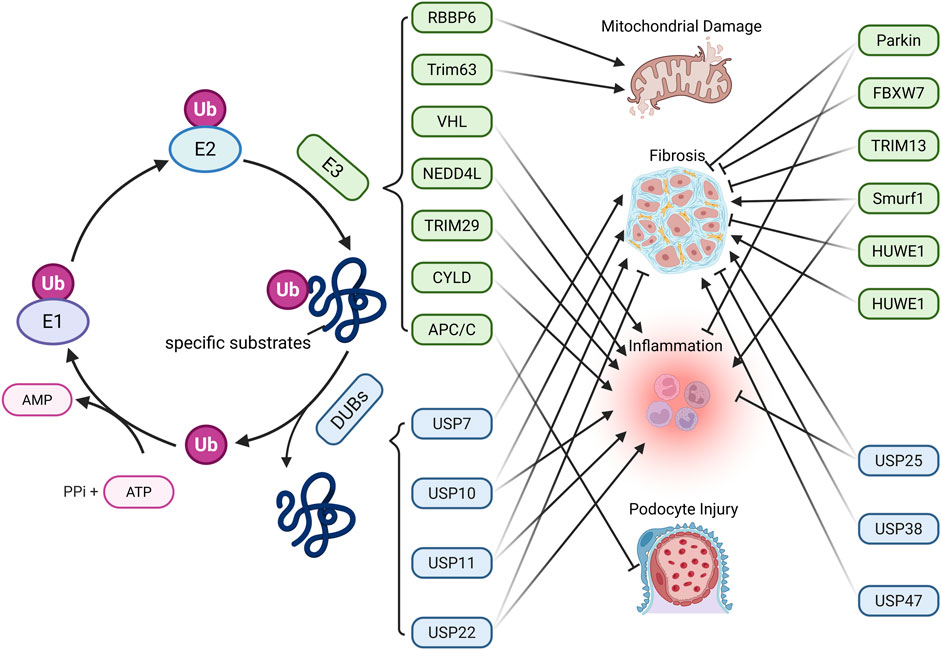
Figure 3. Main Mechanisms of ubiquitination-deubiquitination in CKD. Protein ubiquitination is mediated by ubiquitin-activating enzymes (E1), conjugating enzymes (E2), and ligases (E3). E1 activates Ub by forming a high-energy thioester bond with Ub in an ATP-dependent manner. The activated Ub is then transferred to E2 via a new thioester bond. Finally, E3 ligases facilitate the transfer of Ub to specific substrate proteins. Ubiquitinated proteins can be deubiquitinated by deubiquitinating enzymes (DUBs), enabling Ub recycling. The dynamic balance between protein ubiquitination and deubiquitination affects CKD progression through various pathways. (Created in https://BioRender.com).
4.2 Effect of ubiquitination in the pathological progression of CKD
Ubiquitination regulates renal fibrosis, inflammation, and sodium homeostasis in CKD by modulating key signaling pathways and cellular functions (Table 5). Among ubiquitination-related enzymes, E3 ligases are the most extensively studied. In DN, E3 ligases such as retinoblastoma binding protein 6 (RBBP6) (Hu et al., 2024), Von Hippel-Lindau (VHL) (Wang et al., 2019), tripartite motif containing (TRIM) 63 (Chen et al., 2023), NEDD4-like E3 ubiquitin protein ligase (NEDD4L) (Zhang et al., 2024), TRIM29 (Xu et al., 2023), SMAD-specific E3 ubiquitin protein ligase (SMURF) 1 (Lin et al., 2023), anaphase-promoting complex/cyclosome (APC/C) (Su et al., 2015), TRIM13 (Li et al., 2020), parkinson juvenile disease protein 2 (Parkin) (Chen K. et al., 2020), and F-Box and WD Repeat Domain Containing 7 (FBXW7) (Li et al., 2021) mediate the ubiquitination of targets including estrogen-related receptor alpha (ERRα), glucose-6-phosphate dehydrogenase (G6PD), PPARα, IkappaB kinase (IKK), IκBα, Takeda G protein-coupled receptor 5 (TGR5), nuclear factor erythroid 2-related factor 2 (Nrf2), C/EBP homologous protein (CHOP), cyclin B1, S-phase kinase-associated protein 2 (Skp2), Kelch-like ECH associated protein 1 (Keap1), GATA-binding protein 4 (GATA4), and SREBP-1 through distinct domains. These ubiquitination events contribute to mitochondrial dysfunction, oxidative stress, inflammation, and fibrosis.
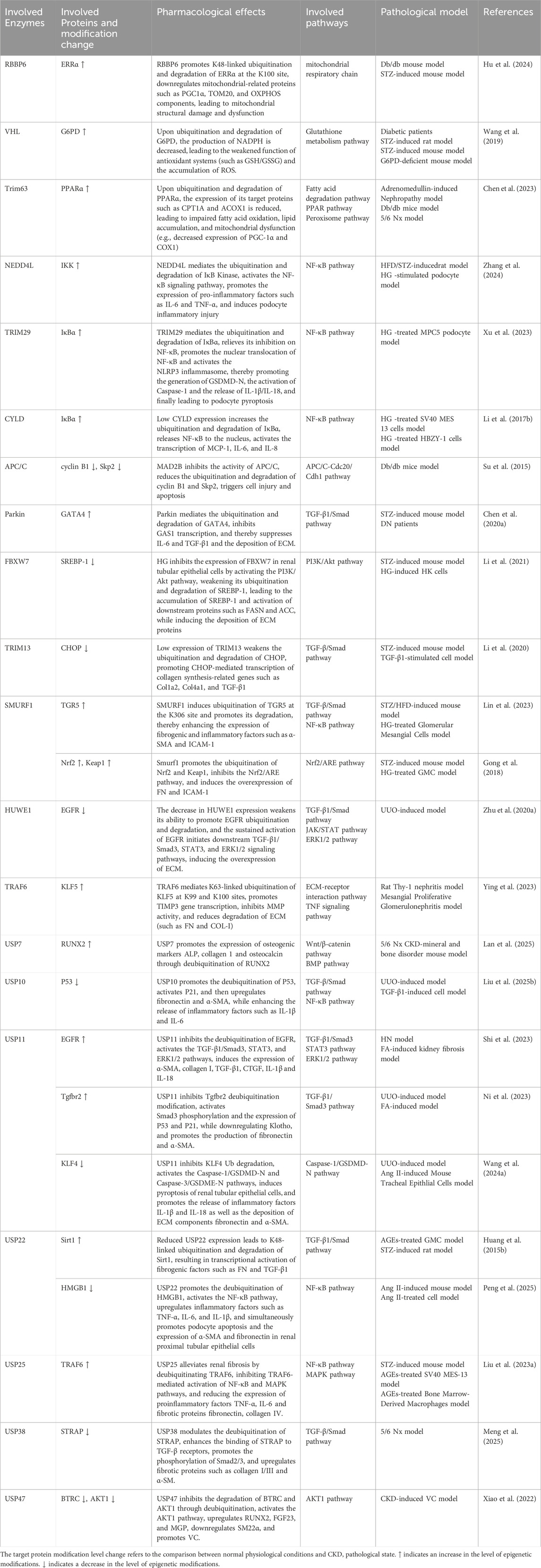
Table 5. Regulatory networks and pathological effects of ubiquitination in CKD.
In CKD, DUBs such as cylindromatosis (CYLD), ubiquitin-specific protease (USP) 11, and USP22 are downregulated. CYLD and USP22 deubiquitinate IκBα, EGFR, and Sirt1, leading to NF-κB activation and partial EMT, thereby promoting fibrosis (Li Y. et al., 2017; Huang K. P. et al., 2015). In contrast, USP25 is upregulated as a compensatory mechanism and inhibits NF-κB and MAPK pathways by deubiquitinating TNF receptor associated factor 6 (TRAF6), delaying disease progression (Liu B. et al., 2023). In the UUO model, USP11 promotes renal fibrosis and tubular cell senescence by stabilizing TGFbeta type II receptor (Tgfbr2) and Kruppel-like factor 4 (KLF4) via deubiquitination, through Smad3/p53 signaling (Ni et al., 2023). It also contributes to pyroptosis through caspase activation (Wang X. et al., 2024). USP10 stabilizes p53, inducing tubular senescence and ECM accumulation (Liu S. et al., 2025). Conversely, Hect, uba, and wwe domain containing 1 (HUWE1) enhances EGFR degradation through ubiquitination, exerting anti-fibrotic effects (Zhu Q. et al., 2020). In hyperuricemic nephropathy (HN), USP11 and USP22 deubiquitinate EGFR and high mobility group box 1 (HMGB1), respectively. This modification activates inflammatory signaling and the TGF-β1/Smad3 pathway, thereby promoting renal fibrosis (Shi et al., 2023; Peng et al., 2025). In mesangial proliferative glomerulonephritis and the rat Thy-1 nephritis model, TRAF6 facilitates ECM accumulation by mediating K63-linked ubiquitination of KLF5 (Ying et al., 2023). In CKD-associated vascular calcification, USP47 stabilizes BTRC and AKT1 through deubiquitination, promoting osteogenic transdifferentiation of vascular smooth muscle cells (Xiao et al., 2022). In CKD-related atrial fibrillation, USP38 targets serine-threonine kinase receptor associated protein (STRAP) for deubiquitination, thereby activating the TGF-β/Smad pathway and contributing to atrial fibrosis (Meng et al., 2025). Conversely, in CKD-related mineral and bone disorders, USP7 deubiquitinates runt-related transcription factor 2 (RUNX2) and helps improve abnormal bone metabolism (Lan et al., 2025). Angiotensin II type 1 receptor (AT1R) is a key effector in the local renal renin-angiotensin system (RAS) (Zhu et al., 2019). In the hypertensive kidney injury model, tissue transglutaminase (TG2) modifies AT1R through isopeptide bonds, causing its accumulation and heightened sensitivity to angiotensin II, thus contributing to the model’s pathological processes, whereas the TG2 inhibitor ERW1041E can ease related symptoms (Liu C. et al., 2019). The dopamine 5 receptor (D5R) promotes degradation of glycosylated AT1R via the ubiquitin-proteasome pathway. D5R deficiency leads to increased AT1R expression and higher blood pressure, and the AT1R antagonist losartan can reverse this hypertension (Li et al., 2008). These findings underscore the central role of ubiquitination and deubiquitination in CKD progression and highlight potential therapeutic targets.
4.3 The therapeutic potential of ubiquitination in CKD
The development of E1 and E2 inhibitors remains limited due to their broad impact on numerous proteins and cellular networks throughout the body. Although small-molecule drugs targeting E3 ligases or DUBs have demonstrated therapeutic potential in both preclinical and clinical studies (Table 6), only a few have progressed to clinical trials, largely due to the complexity of target structures and the presence of multiple active sites.
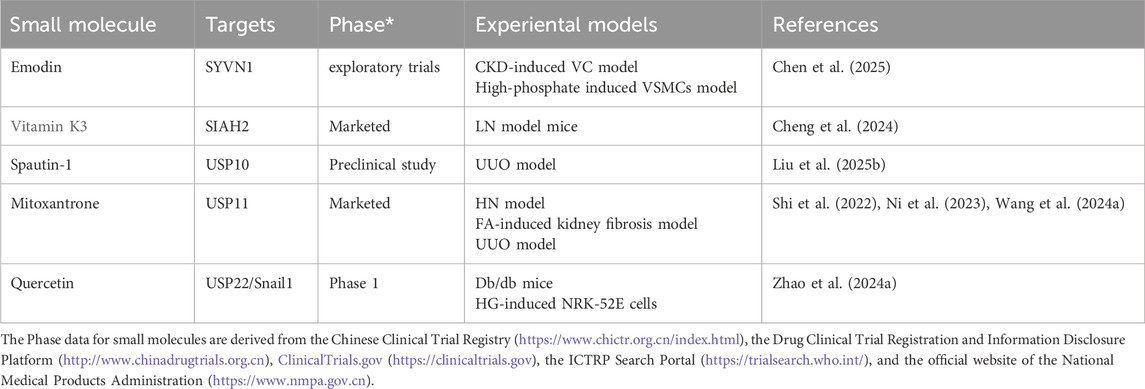
Table 6. Small molecules targeting ubiquitination in CKD.
Several DUB inhibitors have demonstrated renoprotective effects. Mitoxantrone, a USP11 inhibitor, prevents the deubiquitination of EGFR, Tgfbr2, and KLF4, thereby suppressing TGF-β1/Smad3, p53, and caspase-3/GSDME pathways. This reduces EMT, pyroptosis, and renal fibrosis (Shi et al., 2022; Ni et al., 2023; Wang X. et al., 2024). Quercetin inhibits USP22, promotes Snail1 degradation, and alleviates EMT and ECM accumulation in DN (Zhao X. et al., 2024). Spautin-1 enhances p53 ubiquitination by inhibiting USP10, suppressing pro-fibrotic and inflammatory gene expression in tubular epithelial cells (Liu S. et al., 2025). Targeting E3 ligases has also shown potential. Vitamin K3 inhibits seven in Absentia Homolog 2 (SIAH2), restores large tumor suppressor kinase 2 (LATS2) expression, and reduces Yes-associated protein (YAP) activity, thereby attenuating renal fibrosis in lupus nephritis (LN) (Cheng et al., 2024). Emodin enhances the interaction between the estrogen receptor and the E3 ligase SYVN1, promoting ERα ubiquitination and degradation, thereby alleviating vascular calcification in CKD (Chen et al., 2025). In addition, some ubiquitin-proteasome system inhibitors such as bortezomib and carfilzomib block the 26S proteasome, suppressing NF-κB and TGF-β/Smad signaling and reducing ECM accumulation, thus mitigating renal fibrosis (Sawa-Aihara et al., 2023; Zeniya et al., 2017). Although therapeutic strategies targeting ubiquitination still face challenges, the renoprotective effects of related inhibitors in pathological states such as DN, HN, and LN models, indicate that abnormal ubiquitination is a driver of CKD progression under these pathological conditions. Currently, Vitamin K3 is approved for the treatment of bleeding caused by vitamin K deficiency, and Mitoxantrone is approved for the treatment of various tumors (Dezee et al., 2006; Deng et al., 2021). The elimination of both in the body involves the kidneys, and if used for the treatment of chronic kidney disease, potential safety risks may exist based on their pharmacokinetics (PK) (Ehninger et al., 1990; Hassan, 2013).
5 Glycosylation
5.1 Glycosylation modification and its key enzymes involved
Glycosylation plays diverse and complex roles in CKD pathogenesis. Intracellular glycosylation mainly includes four types: N-glycosylation (linked to asparagine), O-glycosylation (linked to serine or threonine), C-glycosylation (linked to tryptophan), and glycosylphosphatidylinositol (GPI)-anchored glycosylation, with N- and O-glycosylation being the most common forms (He M. et al., 2024).
Two key enzyme classes regulate glycosylation in CKD: glycosyltransferases (GTs) and glycosidases (GHs) (Figure 4). GTs catalyze the transfer of sugar moieties from activated donors (e.g., UDP-Gal, GDP-Man) to acceptors such as proteins, lipids, and nucleic acids (Lairson et al., 2008). For example, α-1,6-fucosyltransferase mediates N-glycosylation of TGF-β receptor II in UUO models (Shen et al., 2013). Structurally, GTs are classified into GT-A, GT-B, and GT-C families (Lairson et al., 2008). GHs, or glycoside hydrolases, cleave glycosidic bonds by hydrolyzing the linkage between sugar residues or between sugars and aglycones (Cantarel et al., 2009). The GH family is large, comprising 191 subfamilies (GH1–GH191) and nearly two million annotated modules according to the CAZy database (https://www.cazy.org/Glycoside-Hydrolases.html). Alterations in GH activity are closely associated with disease progression. For example, in IgA nephropathy, elevated urinary levels of N-acetyl-β-D-glucosaminidase, released from injured proximal tubular epithelial cells, serve as a biomarker for tubulointerstitial involvement and renal function decline (Liu X. et al., 2023).
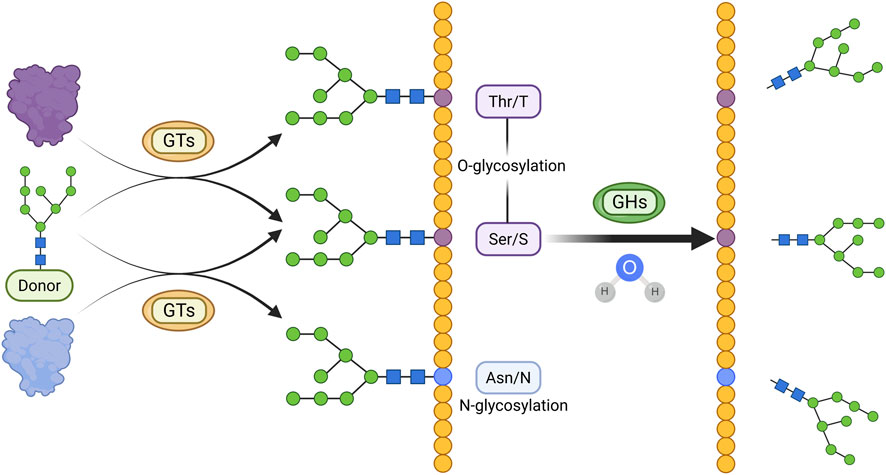
Figure 4. Main Mechanisms of glycosylation in CKD. Glycosyl groups are transferred from activated sugar donors to threonine/serine or asparagine residues by GTs, resulting in O-glycosylation or N-glycosylation, respectively. Glycosylated proteins can undergo hydrolysis of glycosidic bonds catalyzed by GHs. (Created in https://BioRender.com).
5.2 Effect of glycosylation in the pathological progression of CKD
Studies have revealed widespread alterations in the glycosylation profiles of plasma, urine, and kidney tissues in CKD, suggesting potential diagnostic utility. A clinical study identified 62 glycoproteins and 172 N-glycopeptides altered in individuals prior to clinical CKD onset (Santiago-Hernandez et al., 2024). Glycosylation of insulin-like growth factor Ⅱ correlates with age and eGFR, offering potential as a marker for disease progression and cardiorenal risk (Lohia et al., 2023). Specific lipid glycosylation patterns, such as decreased plasma lactosylceramide and elevated C18:1-hexosylceramide, are associated with macroalbuminuria and CKD progression, respectively (Lopes-Virella et al., 2024). A cross-sectional study identified three N-glycans (GP12, GP16, and GP22) as being associated with renal function (Adua et al., 2018). In patients with LN, abnormal cellular mannosylation has emerged as a potential biomarker capable of predicting CKD development (Alves et al., 2021). In IgA nephropathy (IgAN), reduced α-2,6-sialylation and increased galactose-deficient IgA1 (Gd-IgA1) are observed, though the prognostic value of Gd-IgA1 remains inconclusive (Ding et al., 2007; Vaz de Castro et al., 2024). Large-cohort analyses indicate that IgG glycosylation features, including altered galactosylation, sialylation, and bisecting GlcNAc, are associated with kidney function and improve disease prediction when combined with clinical data (Barrios et al., 2016; Zhao et al., 2024c). In animal models, both N- and O-glycan profiles of the renal brush-border membrane are altered in CKD and diabetes, with increased fucosylation and sialylation (Yu et al., 2021a; Yu et al., 2021b). These findings suggest that glycosylation patterns vary by disease subtype and may aid early detection and risk stratification.
Beyond biomarkers, glycosylation actively contributes to CKD pathogenesis, particularly in tubulointerstitial injury and fibrosis (Table 7). TGF-β1 enhances N-glycosylation of lysyl oxidase-like 2 (LOXL2) via N-acetylglucosaminyltransferase V (MGAT5), promoting fibrosis (Kamiya et al., 2023). Aberrant IgG glycosylation reduces Fcγ receptor binding and alters complement activation, contributing to inflammation in IgAN and membranous nephropathy (Barrios et al., 2016; Zhao et al., 2024c; Zhang et al., 2021c). In autosomal dominant polycystic kidney disease, mislocalization of ALG5 disrupts N-glycosylation and GPI anchoring of uromodulin (UMOD), leading to ER retention (Elhassan et al., 2024). In ESKD, peritoneal dialysis modifies transferrin (Tf) glycosylation, potentially impairing iron metabolism (Miljuš et al., 2024). Polypeptide N-acetylgalactosaminyltransferase 11 (Galnt11) deficiency reduces O-glycosylation of megalin, impairing proximal tubular reabsorption and causing proteinuria (Tian et al., 2019). Similarly, the hypertensive state can induce O-GlcNAcylation of megalin, reduce its surface expression, and inhibit the endocytosis and reabsorption of albumin and other proteins, ultimately leading to the development of proteinuria (Silva-Aguiar et al., 2018). In C57BL/6J mice with heminephrectomy, indoxyl sulfate promotes fibroblast growth factor 23 (FGF23) glycosylation and cardiac hypertrophy via the AhR–FGF23–FGFR4 axis (Kishimoto et al., 2023; Ho and Bergwitz, 2021). Under high-phosphate conditions, N- and O-glycosylation of cytochrome p450 27B1 (CYP27B1) enhances RUNX2 expression, while O-Linked N-acetylglucosamine transferase (OGT)-mediated YAP glycosylation inhibits autophagy, accelerating disease progression under hyperphosphatemia with CKD (Yimamu et al., 2022; Xu et al., 2020). In the renal proximal tubule epithelial cells of spontaneously hypertensive rats (SHR), high concentrations of H2O2 promote the accumulation of glycosylated AT1R in lipid rafts, thereby enhancing its sensitivity to Ang II, however, NADPH-oxidase inhibitor apocynin can reverse this phenomenon (Pedrosa et al., 2008). Given that hypertension is often accompanied by renal injury, it is speculated that such glycosylation modification may be potentially associated with renal injury, but the specific mechanism remains to be further confirmed by studies.
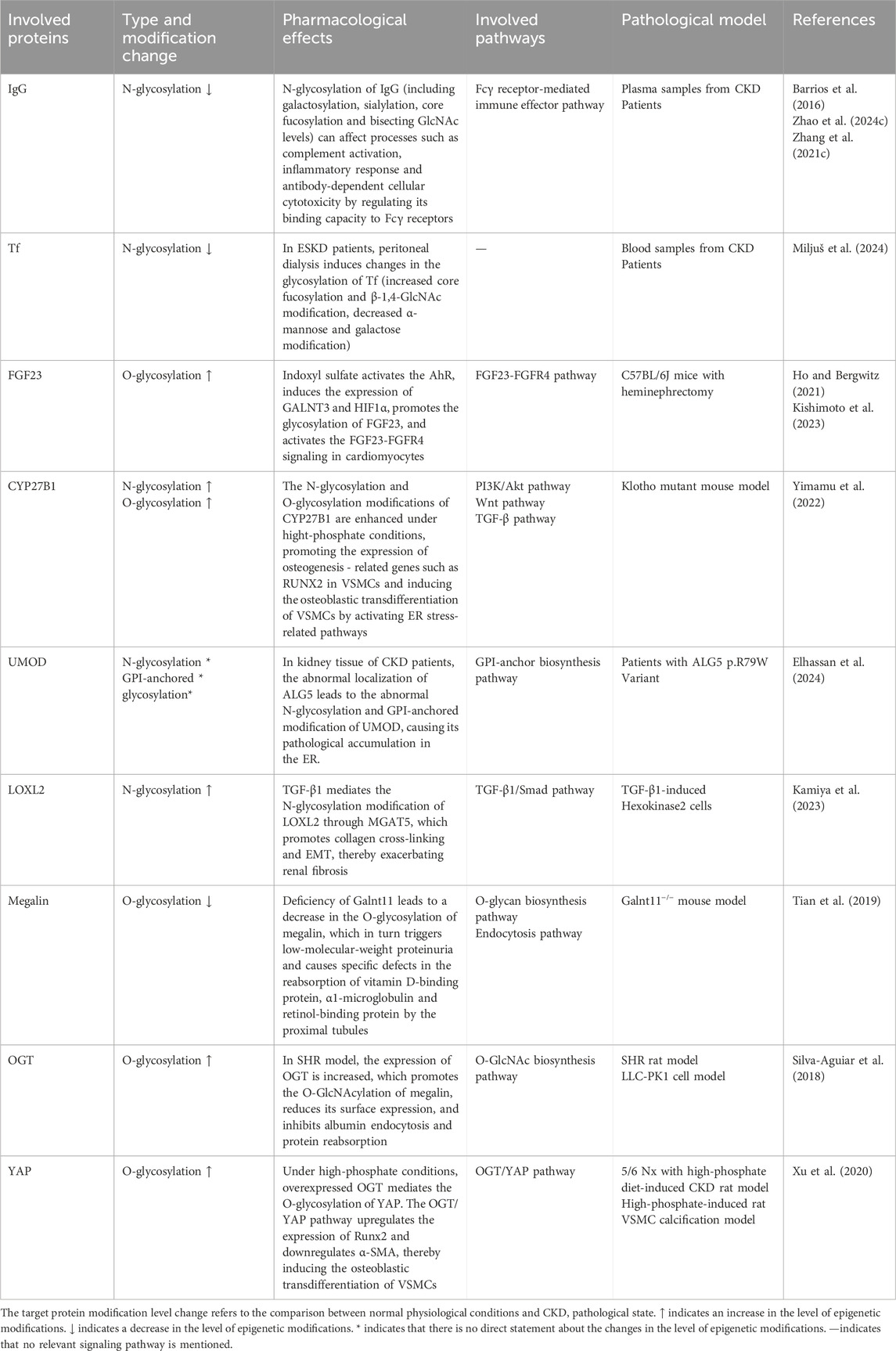
Table 7. Regulatory networks and pathological effects of glycosylation in CKD.
5.3 The therapeutic potential of glycosylation in CKD
Given the regulatory role of glycosylation in CKD, targeting this process has gained attention as a potential therapeutic strategy. Several compounds have shown renoprotective effects in preclinical or clinical studies. In HK-2 cells, the MGAT5 inhibitor glucosamine hydrochloride reduces LOXL2 secretion by inhibiting its N-glycosylation, thereby attenuating TGF-β1-induced fibrosis (Kamiya et al., 2023). In spontaneously hypertensive rats (SHR), 6-diazo-5-oxo-L-norleucine inhibits lutamine-fructose-6-phosphate amidotransferase activity, decreases O-GlcNAcylation in the renal cortex, restores megalin localization, and reduces proteinuria (Silva-Aguiar et al., 2018). Tunicamycin, an N-glycosylation inhibitor, blocks tissue factor glycosylation and improves coagulation abnormalities in CKD (Humphries et al., 2021). In IgA nephropathy, prednisone reduces aberrant O-glycosylation of IgA1, possibly by modulating enzymes such as C1GalT1 and ST6GalNAc-II, though the mechanism remains unclear (Kosztyu et al., 2018). These indicate that abnormal glycosylation is involved in the progression of CKD under these pathological states, and interventions targeting glycosyltransferases, glycosidases, etc., are potential research mechanisms for the treatment of CKD.
However, the specific mechanisms by which glycosylation contributes to CKD pathophysiology have not yet been fully elucidated. Metabolic disorders commonly associated with CKD, such as diabetes, can exacerbate protein dysfunction through glycation under high-glucose conditions (Hoffmann et al., 2016), further complicating disease progression. Therefore, the development of therapeutic agents targeting glycosylation requires a comprehensive understanding of the specific roles of glycosylation in CKD. This includes distinguishing between protective and deleterious glycosylation events and accurately identifying therapeutic targets.
6 Lactylation
6.1 Enzymatic mechanisms of lactylation
Lactylation is a lactate-derived PTM involving the enzymatic transfer of a lactyl group to lysine residues, forming ester bonds and resulting in lactylated proteins (Li S. et al., 2025). This modification influences gene expression, signal transduction, and cellular metabolism by altering protein charge, conformation, and interaction patterns (Hou et al., 2025). Lactylation is dynamic and reversible, regulated by specific lactyltransferases (writers) and delactylases (erasers) (Fan et al., 2023).
The donor molecule for lactylation is lactyl-CoA, synthesized by acyl-CoA synthetase, which catalyzes the conjugation of lactate with CoA (Li H. et al., 2024). Lactate, mainly produced via glycolysis, enters cells through monocarboxylate transporters (MCT1–MCT5), supporting intracellular lactyl-CoA production and linking cellular metabolism to epigenetic regulation (Wang Y. et al., 2024; Chen L. et al., 2022). Lactyltransferases catalyzing this modification include P300, CBP, lysine acetyltransferase (KAT) 5, KAT8, alanyl-tRNA synthetase (AARS) 1, AARS2, and GCN5, while delactylases such as HDAC1, HDAC2, HDAC3, HDAC8, and sirtuins (SIRT1, SIRT2, SIRT3) remove lactyl groups (Jin et al., 2023a; Iozzo et al., 2025; Zong et al., 2025). These enzymes modulate transcriptional programs and cellular functions by regulating the lactylation status of target proteins (Figure 5).
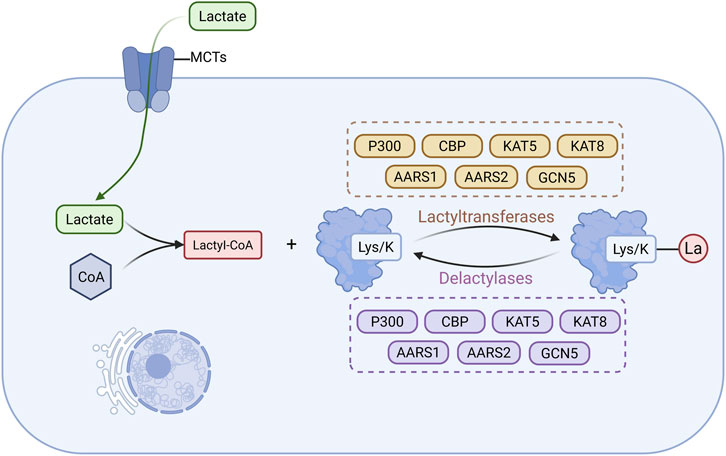
Figure 5. Main Mechanisms of lactylation in CKD. Lactate produced via glycolysis enters cells through monocarboxylate transporters to support the synthesis of lactyl-CoA, which subsequently serves as the donor of lactyl groups (La) that are enzymatically added to lysine residues on proteins by lactyltransferases. Lactylated proteins can also undergo delactylation through the action of delactylases. (Created in https://BioRender.com).
6.2 Effect of lactylation in the pathological progression of CKD
Research on protein lactylation in CKD is limited, with current studies mainly focusing on the impact of elevated lactate levels on fibrosis and inflammation (Li X. et al., 2025). Glycolytic enzymes contribute to CKD progression by promoting lactate accumulation and lactylation (Wang Y. et al., 2024). In FA nephropathy, phosphofructokinase-2/fructose-2,6-bisphosphatase 3 (PFKFB3) enhances glycolysis, increases lactate production, and induces H4K12 and H4K5 lactylation. This activates the NF-κB pathway and upregulates pro-inflammatory genes such as IκB, Rela, and Relb (Wang Y. et al., 2024). In the UUO model, pyruvate kinase M2 (PKM2) promotes H3K18 lactylation, enhances TGF-β1 transcription, and activates Smad3 signaling, facilitating macrophage-to-myofibroblast transition (Xiang et al., 2024). Meanwhile, the gut microbiota metabolite trimethylamine N-Oxide (TMAO) can alter the pyruvate metabolism of renal cells, leading to increased lactic acid accumulation, and then through the lactylation modification of histone H4, promote macrophage M2 polarization by directly binding to the promoters of IL-10 and TGF-β, resulting renal fibrosis (Tang et al., 2025). Whereas knockout of lactate transferase can reduce M2 macrophage infiltration and renal fibrosis (Tang et al., 2025). In diabetes, lactate induces K182 lactylation of ACSF2, reducing its activity and increasing mitochondrial reactive oxygen species (ROS) (Chen J. et al., 2024). In ischemic-reperfusion injury (IRI), citrate synthase lactylation impairs its function, activates the NLRP3 inflammasome, and accelerates AKI to CKD progression (Chen, 2022). These findings suggest that lactylation contributes to CKD by promoting inflammation, fibrosis, and mitochondrial dysfunction. Targeting glycolytic enzymes or interfering with lactylation may offer therapeutic potential.
6.3 The therapeutic potential of lactylation in CKD
In CKD models, several compounds have shown therapeutic potential by modulating lactate transport, glycolytic activity, or enzymes involved in protein lactylation (Nishima and Tanaka, 2024). Glycolysis inhibitors, such as oxamate (LDH inhibitor) (Ye et al., 2016), 3PO (Clem et al., 2008) and PFK15 (Clem et al., 2013) (PFKFB3 inhibitors), and shikonin (PKM2 inhibitor) (Xiang et al., 2025), suppress lactate production, leading to reduced histone lactylation (e.g., H3K18la and H4K12la), downregulates the NF-κB and TGF-β1/Smad3 signaling pathways and inhibits macrophage-to-myofibroblast transition (Wang Y. et al., 2024; Nishima and Tanaka, 2024). Inhibition of glucose transporter 1 (GLUT1) with BAY-876 lowers intracellular lactate levels and decreases H4K12la in renal tubular epithelial cells (Qiao et al., 2024). Additionally, HDAC inhibitors such as trichostatin A (TSA) and RGFP966 enhance delactylation and suppress NF-κB-mediated gene expression by reducing H4K12la levels (Cianciolo Cosentino et al., 2013; Chen et al., 2021). This indicates that lactylation modifications play a driving role in the occurrence and development of CKD and can serve as a new strategy for the treatment of chronic kidney disease.
7 Palmitoylation
Palmitoylation is a reversible post-translational modification involving the covalent attachment of a 16-carbon palmitate to cysteine residues via thioester bonds. Although relatively underexplored in CKD, this modification increases protein hydrophobicity and affects localization, stability, and protein–protein interactions (Fraser et al., 2020; Fransisco et al., 2024). Palmitoylation is dynamically regulated by palmitoyltransferases (PATs) and depalmitoylases (Pei and Piao, 2024). Most PATs belong to the DHHC (Asp-His-His-Cys) family, with 23 DHHC enzymes identified in human cells, each showing distinct localization and substrate specificity (Tabaczar et al., 2017; De and Sadhukhan, 2018). T These enzymes transfer palmitate from palmitoyl-CoA to target proteins (Ko and Dixon, 2018). Depalmitoylases, such as acyl protein thioesterase (APT) 1, APT2, and α/β-hydrolase domain proteins (e.g., alpha/beta hydrolase fold domain (ABHD) 10, ABHD17A/B/C), remove palmitate by cleaving the thioester bond (Zhang and Hang, 2017; Cao et al., 2019) (Figure 6).
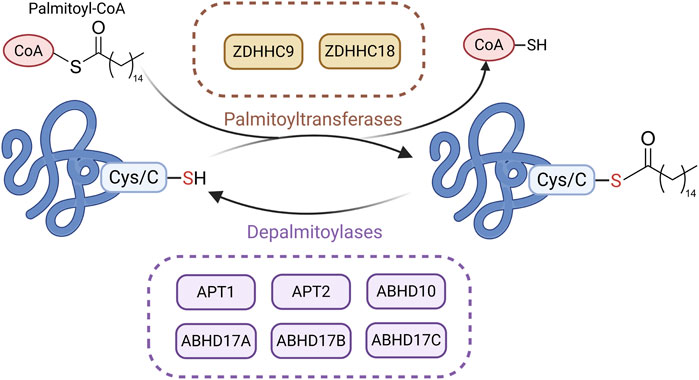
Figure 6. Main Mechanisms of palmitoylation in CKD. Under the action of palmitoyltransferases, palmitate is transferred from palmitoyl-CoA to the cysteine residues of substrate proteins via thioester bonds. Palmitoylated proteins can undergo depalmitoylation through the hydrolytic removal of palmitate by depalmitoylases. (Created in https://BioRender.com).
Recent studies suggest that palmitoylation plays a regulatory role in CKD progression. In UUO and IRI models, APT1 enhances β-catenin accumulation through depalmitoylation, thereby activating Wnt/β-catenin signaling and promoting fibrosis. This process can be blocked by the APT1 inhibitor ML348 (Gu M. et al., 2023). In contrast, DHHC9 promotes β-catenin palmitoylation, suppressing wingless-type MMTV integration site family (Wnt) signaling and reducing fibrosis (Gu M. et al., 2023). In UUO and FA-induced models, zinc finger DHHC-type containing (ZDHHC) 18 is upregulated and mediates Harvey rat sarcoma viral oncogene homolog (HRAS) palmitoylation, activating the MEK/ERK pathway and promoting EMT and fibrosis. Inhibiting ZDHHC18 or mutating HRAS palmitoylation sites alleviates fibrosis (Lu et al., 2025). Although research on palmitoylation in CKD is still in its early stages, these findings indicate that abnormal palmitoylation drives CKD progression in models such as UUO, IRI, and FA, thereby highlighting palmitoylation as a potential therapeutic target in CKD.
8 Crotonylation
Crotonylation is a histone lysine acylation modification first identified in 2011. It regulates gene expression by transferring a crotonoyl group (–CO–CH = CH–CH3) to lysine residues on histones, such as H3K9 and H3K18 (Tan et al., 2011). This process is dynamically controlled by a “writer–eraser–reader” system. Crotonyl-CoA (Cr-CoA) serves as the donor molecule, and histone crotonyltransferases, including p300/CBP and metal-organic framework (MOF), catalyze the modification. De-crotonylases such as HDAC1/2/3 and SIRT1/2/3 remove it, while reader proteins with the Yaf9, ENL, AF9, Taf14, Sas5 (YEATS) domains (e.g., ALL1-fused gene from chromosome 9 (AF9), YEATS2) or double plant homeodomain (PHD) fingers (e.g., monocytic leukemia zinc finger protein (MOZ), double PHD fingers 2 (DPF2) recognize crotonylated lysines and mediate transcriptional activation (Xie et al., 2024) (Figure 7).
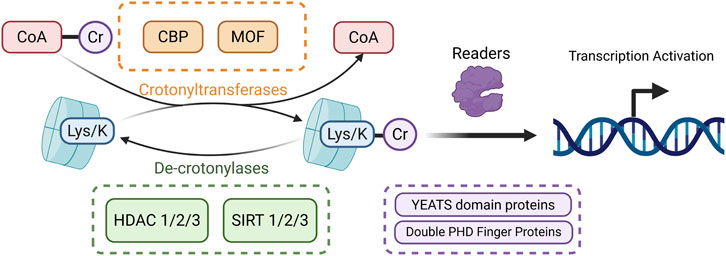
Figure 7. Mechanism of histone crotonylation. The crotonyl group is transferred from Cr-CoA to lysine residues on histones by crotonyltransferases, leading to histone crotonylation. The resulting crotonylated proteins are recognized by specific reader proteins (shown in purple), which facilitate transcriptional activation. Crotonylated proteins can be reverted through the removal of crotonyl groups by de-crotonylases, completing the dynamic regulation of this modification. (Created in https://BioRender.com).
Emerging evidence suggests that crotonylation contributes to CKD progression via multiple pathways. In patients with chronic kidney failure, 772 crotonylation sites are upregulated in peripheral blood mononuclear cells, with associated proteins enriched in pathways related to platelet granules and cell adhesion, implicating crotonylation in renal fibrosis (Huang et al., 2021). In a renal transplant ischemia–reperfusion model, neuropilin-1 (NRP1) is overexpressed and promotes energy metabolism dysfunction by reducing cytochrome Cox4i1 crotonylation via NF-κB activation. This aggravates fibrosis through the TGF-β/Smad3/PDGFβ pathway (Li Y. et al., 2024). In UUO and FA–induced models, acyl-CoA synthetase short-chain family member 2 (ACSS2, a Cr-CoA synthase) increases Cr-CoA production, enhances H3K9 crotonylation, and activates IL-1β transcription, leading to macrophage activation and tubular senescence. These effects can be reversed by the ACSS2 inhibitor VY-3-249, which attenuates fibrosis (Li L. et al., 2024). Conversely, in DN models, oral sodium crotonate increases H3K18 crotonylation via ACSS2, suppresses proinflammatory cytokines (IL-1β, IL-6), downregulates fibrotic markers (TGF-β1, α-SMA), and improves renal function (He Y. et al., 2024). Despite these findings, the overlap between crotonylation and other lysine modifications, such as acetylation, poses challenges for developing selective therapies. Moreover, the involvement of crotonylation in multiple interconnected pathways complicates the design of precise and safe interventions.
9 SUMOylation
SUMOylation, similar to ubiquitination, is a post-translational modification in which the SUMO (Small Ubiquitin-like Modifier) protein is covalently attached to lysine residues of target proteins through a cascade involving E1, E2, and E3 enzymes (Sheng et al., 2021). DeSUMOylation and precursor processing are catalyzed by SUMO-specific protease (SENP, Figure 8) (Zhou H. et al., 2024). Five SUMO isoforms (SUMO1, SUMO2, SUMO3, SUMO4, and SUMO5) have been identified in mammalian cells, with most SUMOylated proteins located in the nucleus (Sheng et al., 2021; Sheng et al., 2019). In humans, the SENP family includes SENP1, SENP2, SENP3, SENP5, SENP6, and SENP7(Wu and Huang, 2023).
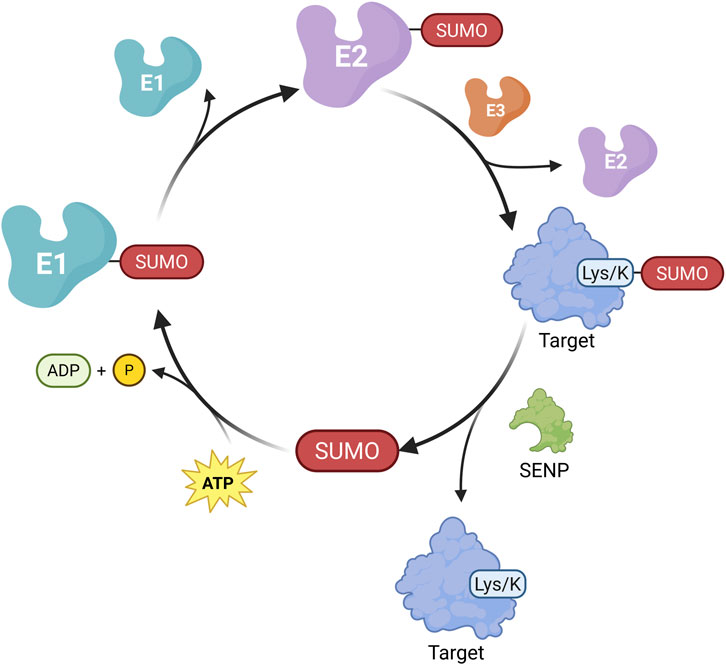
Figure 8. Mechanism of Protein SUMOylation. Similar to ubiquitination, the SUMOylation process is initiated by the activation of SUMO by the E1-activating enzyme in an ATP-dependent manner, forming a thioester bond between SUMO and E1. The activated SUMO is then transferred to the E2-conjugating enzyme. Subsequently, under the catalysis of an E3 ligase, SUMO is covalently attached to the lysine residue of the target protein, thereby completing the SUMOylation modification. SUMO-conjugated proteins can undergo deSUMOylation through the action of SENP, resulting in the removal of SUMO moieties. (Created in https://BioRender.com).
In CKD, SUMOylation is dysregulated and contributes to disease progression. In renal fibrosis models, such as UUO and unilateral ischemia-reperfusion injury, ATF4 binds to heat shock protein family A (Hsp70) member 5 (HSPA5) and promotes its SUMOylation, which exacerbates ferroptosis via the HSPA5 signaling pathway (Huang et al., 2025). β-catenin represses SUMO3 transcription, reducing SUMO3-mediated modification of live kinase B1 (LKB1), thereby impairing AMP-activated protein kinase (AMPK) activation and fatty acid oxidation (Chen S. et al., 2024). The E2 enzyme ubiquitin-conjugating enzyme 9 (UBC9) catalyzes the SUMOylation of the nuclear receptor nuclear receptor subfamily 5 group A member 2 (NR5A2), enhancing its binding to the calreticulin gene promoter, promoting its transcription, and upregulating the expression of fibrosis-related genes such as collagen type 1 alpha 1 (Col1α1) and TGFβ1 (Politis and Charonis, 2022; Arvaniti et al., 2016). In DN models, high glucose induces SUMOylation of IκBα, HIF-1α, Smad4, and STAT1, activating NF-κB and TGF-β signaling pathways and promoting inflammation and fibrosis (Huang et al., 2013; Wusiman et al., 2025; Gu C. et al., 2023; Zhou et al., 2014). High glucose also triggers the de-SUMOylation of RBMX, leading to mitochondrial dysfunction and contributing to tubulointerstitial fibrosis (Yang et al., 2024).
Targeting SUMOylation has shown potential in reducing renal fibrosis. For example, the UBC9 inhibitor 2-D08 specifically blocks NR5A2 SUMOylation, thereby attenuating fibrosis in the UUO model (Arvaniti et al., 2016). Some natural compounds, including Ginkgolic acid and Astragaloside IV, appear to influence SUMOylation, though their effects in CKD models remain unconfirmed (Wang B. S. et al., 2021; Yu et al., 2022).
10 Prenylation
Recent studies have shown that protein prenylation is closely linked to CKD progression. This post-translational modification involves the covalent attachment of isoprenyl groups (farnesyl or geranylgeranyl) to the C-terminal CAAX (C represents cysteine, A refers to aliphatic amino acids, and X typically indicates methionine or serine) motif of target proteins (Mohamed et al., 2024; Khwaja et al., 2006) (Figure 9). Prenylation facilitates membrane anchoring and activates downstream signaling pathways (Khwaja et al., 2006). The process is mainly catalyzed by four types of prenyltransferases: farnesyltransferase (FTase), geranylgeranyltransferase (GGTase)-type I, GGTase type-II, and GGTase-III (Marchwicka et al., 2022) Their activity depends on intermediates produced by the mevalonate pathway, where HMG-CoA reductase acts as the rate-limiting enzyme (Khwaja et al., 2006; Buemi et al., 2002).
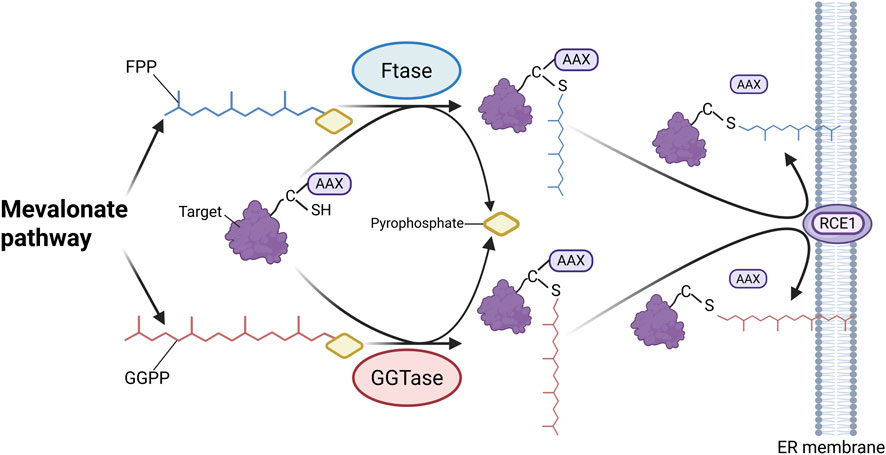
Figure 9. Mechanism of protein prenylation. Isoprenoid carriers, including FPP and GGPP, are synthesized via the mevalonate pathway. FTase and GGTase specifically recognize the CAAX (C represents cysteine) motif at the C-terminus of target proteins and catalyze the covalent attachment of farnesyl or geranylgeranyl groups to cysteine residues, anchoring the modified proteins to the endoplasmic reticulum membrane. Subsequently, the -AAX residues are cleaved by RAS-converting CAAX endopeptidase 1 (RCE1), completing the prenylation process. (Created in https://BioRender.com).
In fibrotic animal and cellular models, aberrant prenylation of proteins such as H-Ras, Ki-Ras, and RhoA has been shown to activate the NF-κB, TGF-β1/Smad, and MAPK signaling pathways, thereby promoting renal tubular EMT, fibroblast activation, and ECM deposition (Rodríguez-Peña et al., 2014; Sharpe et al., 1999; Khwaja et al., 2006). Small-molecule inhibitors targeting this process have shown therapeutic potential. Statins (e.g., lovastatin and atorvastatin) inhibit HMG-CoA reductase, reducing the synthesis of farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), and thereby suppressing Ras and RhoA prenylation. This attenuates mesangial cell proliferation, inflammation, and ECM accumulation (Essig et al., 1998; Buemi et al., 2002; Rodríguez-Peña et al., 2014). The FTase inhibitor L-744,832 directly blocks Ras farnesylation and reduces fibronectin levels (Rodríguez-Peña et al., 2014). GGTI-298, a GGTase inhibitor, disrupts RhoA-mediated Erk1/2 and Akt activation, thus limiting fibrosis (Khwaja et al., 2006). In the 5/6 Nx model, chaetomellic acid A selectively inhibits H-Ras farnesylation and alleviates glomerulosclerosis and arteriolosclerosis via the TGF-β1 pathway (Nogueira et al., 2017). Collectively, these findings support the potential of targeting dysregulated prenylation as an antifibrotic strategy in CKD.
11 Other ePTMs
In addition to the aforementioned ePTMs, other modifications, such as myristoylation, succinylation, and sulfenylation, also contribute to CKD progression by modulating protein function and interactions. Myristoylation involves the covalent attachment of myristic acid to the N-terminal glycine residue of a protein, a reaction catalyzed by N-myristoyltransferase (Wang B. et al., 2021). In both CKD patients and animal models, myristoylated alanine-rich C-kinase substrate shows altered expression and has been proposed as a potential biomarker for chronic renal injury, although its mechanistic role remains unclear (Kim et al., 2021).
Succinylation refers to the transfer of a succinyl group (–CO–CH2–CH2–CO–) to lysine residues, mediated by succinyltransferases such as SUCL2 and GCN5, while desuccinylation is regulated by enzymes like SIRT5 (Zhang et al., 2011). In mouse models of IRI, global lysine succinylation is elevated, whereas SIRT5 deficiency improves renal function, suggesting a role for succinylation in the transition from AKI to CKD (Chiba et al., 2019).
Sulfenylation is a PTM where cysteine thiols (-SH) are oxidized to sulfenic acids (-SOH), regulated by redox environments and related enzymes (Mu et al., 2024). Direct research on its association with CKD remains limited. In human kidney microsomes oxidative stress triggers sulfenylation of CYP2C8, CYP2D6, CYP3A4, and CYP4A11 (Albertolle et al., 2018; Albertolle et al., 2017). This may reduce their activity, impairing renal metabolism of endogenous substances and detoxification of exogenous drugs, thereby exacerbating kidney injury and contributing to the oxidative stress-metabolic disorder cycle in CKD. Under high glucose or diabetic conditions, sulfenylation at Cys358 of PKM2 inhibits its tetramer formation and activity which leads to accumulation of toxic glucose metabolites (e.g., methylglyoxal, sorbitol), mitochondrial dysfunction, and podocyte apoptosis, worsening renal pathology (Qi et al., 2017). Notably, the PKM2 activator TEPP-46 can reduce this modification, reversing damage (Qi et al., 2017). Thus, sulfenylation may participate in CKD progression by affecting renal metabolic enzymes and key signaling molecules, making its regulation a potential therapeutic target for CKD.
12 Crosstalk between different ePTMs
In CKD, ePTMs dynamically regulate protein function, signaling pathways, and gene expression, thereby contributing to renal fibrosis, inflammation, and metabolic dysregulation. Complex crosstalk occurs among distinct ePTMs, exhibiting model-specific regulatory patterns under different pathological contexts. Accordingly, elucidating the mechanisms of ePTM crosstalk across diverse CKD models is essential for a comprehensive understanding of disease progression and for the development of novel therapeutic strategies.
In DN, the interplay between methylation and other ePTMs appears to be particularly prominent. One mechanism involves the activities of methyltransferases are frequently regulated by ubiquitination and acetylation. For example, ubiquitination of histones H2AK119 and H2BK120 can increase the expression of the methyltransferases SET7/9 and SUV39H1, thereby enhancing H3K4 and H3K9 methylation, respectively, which promotes the transcription of fibrosis-related genes (Goru et al., 2016; Pan et al., 2024). In addition, the activity of the histone H3K4 methyltransferase Set1 is regulated by N-terminal acetylation mediated by N-acetyltransferases, which maintains H3K4 methylation, thereby activating the transcription of profibrotic genes and contributing to podocyte injury in DN (Woo et al., 2024; Sasaki et al., 2016; Zhang et al., 2023). Another mechanism arises from the competition between methylation and acetylation for the same target to influence disease progression. DOT1L, the only known H3K79 methyltransferase, competes with HDAC2 at the endothelin-1 promoter to regulate the balance between methylation and acetylation, thereby affecting fibrosis in DN (Zhang L. et al., 2020). Moreover, a novel ubiquitin-like modification, neddylation, has been shown to stabilize RhoA by preventing its ubiquitination, subsequently activating the ERK1/2 pathway and driving fibrosis (Li X. Q. et al., 2025).
In HN, reports on ePTM crosstalk are relatively limited and mainly involve phosphorylation, ubiquitination, and acetylation. For example, the deubiquitinase OTUD6A removes ubiquitin from STAT3, thereby enhancing its phosphorylation, nuclear translocation, and promotion of Ang II–induced fibrosis (Sun et al., 2024). In parallel, HDAC6 deacetylates Smad2/3, enhancing their phosphorylation and binding activity at profibrotic gene promoters, thus activating the TGF-β/Smad pathway (Choi et al., 2015). In HN, AT1R is a central regulatory molecule that mediates Ang II signaling to modulate renal hemodynamics, sodium reabsorption, and fibrosis, thereby influencing CKD progression (Wehbi et al., 2001). Its function is precisely regulated by glycosylation, ubiquitination, and phosphorylation. The phosphorylation state affects the efficiency and sites of glycosylation, while glycosylation may alter the receptor conformation to influence the exposure of phosphorylation sites, subsequently regulating the membrane localization and functional activity of AT1R, which affects disease progression under pathological conditions such as hypertension (Al-Qattan et al., 2006; Pedrosa et al., 2008). Additionally, phosphorylation can recruit E3 ubiquitin ligases to promote ubiquitination and degradation, while glycosylation may maintain receptor stability and affect the ubiquitination process (Liu C. et al., 2019; Li et al., 2008). Moreover, these three modifications precisely regulate the signal transduction specificity of AT1R through dynamic interactions. For instance, the phosphorylation “barcode” determines the downstream β-arrestin-mediated signaling pathway, glycosylation ensures the correct folding and localization of the receptor, and ubiquitination participates in receptor desensitization and degradation balance, collectively influencing its function under related pathological conditions (Gareri et al., 2024; Liu C. et al., 2019; Li et al., 2008).
In autoimmune kidney diseases, studies on ePTM crosstalk have so far been largely confined to LN. In LN, ePTM interactions play a critical role in regulating mesangial cell hyperproliferation and inflammatory responses. Recent study demonstrates that lactylation of PBX1 enhances its interaction with the E3 ubiquitin ligase TRIM21, thereby promoting PBX1 ubiquitination and degradation, which subsequently reduces the transcription of the cell cycle inhibitor P27 and drives abnormal mesangial cell proliferation (Liu E. et al., 2025). In addition, crotonylation competes with acetylation at the same lysine residues, thereby influencing inflammatory and fibrotic processes associated with autoimmune kidney disease (Zeng et al., 2023).
Renal fibrosis is a common pathological process across all types of CKD, and the UUO model, as a classic fibrosis model, has been widely used to investigate ePTM crosstalk in CKD. In the UUO model, multiple ePTMs orchestrate the fine-tuned regulation of fibrosis-related signaling pathways, particularly at the level of transcription factors and histone modifications. For instance, PRMT1 catalyzes the methylation of bromodomain protein 4 and enhances its phosphorylation, which in turn increases the acetylation of Snail, thereby promoting Snail-mediated EMT and fibrosis (Xiong C. et al., 2025). EZH2 aggravates fibrosis progression by downregulating PTEN and increasing STAT3 and ERK1/2 phosphorylation (Zhou et al., 2016). In contrast, JMJD3-mediated demethylation of H3K27me3 inhibits AKT and ERK1/2 phosphorylation, thereby suppressing fibrosis progression (Yu C. et al., 2021). At the signaling protein level, O-GlcNAcylation of serine/threonine kinase (RAF1) prevents its ubiquitination, stabilizing RAF1 and activating the Ras/RAF1/ERK pathway to drive fibrosis (Feng et al., 2020). Conversely, SIRT2 deacetylates SMAD2/3 and promotes their ubiquitination and degradation, exerting an antifibrotic effect (Yang et al., 2023). In contrast, HDACs promote fibrosis by DUSP1, which enhances Smad3 phosphorylation and activates the TGF-β/Smad pathway (Wang et al., 2025). Similarly, FAT10 overexpression increases checkpoint kinase 1 (CHK1) levels by reducing USP7 ubiquitination, thereby amplifying TGF-β signaling and promoting fibrosis (Shao et al., 2022). Palmitoylation also plays a critical role in the regulation of the TGF-β/Smad pathway. ZDHHC18-mediated palmitoylation of HRAS promotes downstream MEK/ERK phosphorylation and aggravates fibrosis (Lu et al., 2025), while downregulation of DHHC9 reduces palmitoylation and ubiquitination of β-catenin, leading to its accumulation and Smad2/3 activation, further exacerbating fibrosis (Gu M. et al., 2023). Moreover, macrophage polarization represents a decisive factor in UUO-induced fibrosis (Wehbi et al., 2001). Among macrophages, M1 macrophages secrete pro-inflammatory factors in the early stage, exacerbating renal injury, while M2 macrophages can promote tissue regeneration by secreting related factors, but their persistent infiltration can aggravate renal fibrosis (Gao et al., 2025; Tan et al., 2025). In this process, various epigenetic modifications, such as histone lactylation, acetylation, and methylation, regulate the expression of genes related to macrophage polarization, thereby influencing chronic kidney disease (Gao et al., 2025; Tang et al., 2025; An et al., 2023; Zhan et al., 2025). Meanwhile, there exist cross-regulations between metabolism and epigenetics (e.g., TMAO can induce lactate secretion and promote histone lactylation) and intercellular epigenetic signal transmission (e.g., regulation by exosomal non-coding RNAs). These regulations collectively participate in the pathological process of chronic kidney disease by modulating macrophage polarization (Tan et al., 2025; Tang et al., 2025; Zhan et al., 2025). Drugs targeting epigenetic regulation, such as SIRT6 agonists, JMJD3 inhibitors, and TMAO inhibitors, can regulate the balance of macrophage polarization, providing potential strategies for the treatment of CKD (Gao et al., 2025; Tang et al., 2025; An et al., 2023). In summary, ePTMs in the UUO model drive fibrosis not only through regulation of transcription factors and signaling pathways but also by shaping macrophage polarization, together forming a complex pathological network of renal fibrosis.
In summary, ePTM crosstalk in CKD forms a highly complex regulatory network, influencing disease progression through the modulation of protein expression and signal transduction. Elucidating these ePTM interactions not only deepens our understanding of the molecular drivers underlying CKD but also provides a theoretical foundation for the development of multi-target combination therapies.
13 Conclusion and future perspectives
EPTMs play a crucial role in the pathogenesis of CKD by regulating key pathological processes such as renal fibrosis, inflammation, metabolic dysregulation, and cellular stress responses (Laget et al., 2022). In this review, we summarize the mechanisms and clinical significance of major ePTMs, including methylation, acetylation, ubiquitination, enzymatic glycosylation, lactylation, palmitoylation, crotonylation, SUMOylation, and prenylation, in CKD. Despite increasing attention, most ePTMs in CKD remain in the early stages of investigation, and their complex regulatory networks and cell type-specific functions require further elucidation. The crosstalk between different ePTMs represents a critical yet underexplored aspect of CKD pathogenesis. This intricate interplay complicates therapeutic strategies, as targeting a single modification may inadvertently interfere with interconnected pathways and lead to unintended effects (Fontecha-Barriuso et al., 2018).
Emerging evidence highlights the therapeutic potential of targeting ePTMs in the treatment of CKD. Small-molecule inhibitors of methyltransferases (e.g., 3-DZNeP targeting EZH2 (Zhou et al., 2016)), HDACs (e.g., ACY-1215 targeting HDAC6 (Chen X. et al., 2020)), and ubiquitin-related enzymes (e.g., mitoxantrone targeting USP11 (Shi et al., 2023)) have shown encouraging effects in preclinical models by mitigating fibrosis, inflammation, and organ injury. In addition, agents targeting glycosylation (e.g., glucosamine hydrochloride (Kamiya et al., 2023)) and lactylation (e.g., shikonin (Xiang et al., 2025)) provide novel avenues for modulating CKD-associated pathways.
Despite these advances, several challenges hinder the clinical translation of ePTM-targeted therapies. First, the crosstalk among different ePTMs, including the overlapping enzymatic machinery of crotonylation and acetylation (Xie et al., 2024)) complicates selective targeting and increases the risk of off-target effects. Second, the regulatory networks of ePTMs are complex, with individual enzymes often acting on multiple substrates and pathways. For example, PRMT5 modulates both SREBP1 and NF-κB signaling (Zhang et al., 2018; Wei et al., 2013), highlighting the need for a more comprehensive understanding of context-specific functions. Third, most current findings are derived from animal models, and the safety and efficacy of ePTM-based interventions in humans remain to be rigorously validated. Notably, CKD patients with impaired renal function and metabolic disorders may face unique PK/pharmacodynamic (PK/PD) challenges, such as drug accumulation or altered target binding in fibrotic microenvironments, which further limit translational progress (Franchi et al., 2022). A notable example is the discontinuation of the JNK inhibitor CC-930 due to hepatotoxicity in clinical trials (Qian et al., 2023). Future research should aim to identify tissue- and cell-type-specific patterns of ePTMs to facilitate the development of precision medicine approaches. It is also essential to design highly selective inhibitors using advanced molecular technologies and to investigate combination therapies that target multiple ePTMs simultaneously, thereby enhancing therapeutic efficacy while minimizing adverse effects.
In conclusion, ePTMs represent a promising frontier in CKD research, bridging the gap between molecular mechanisms and clinical application. Among the discussed ePTMs, methylation, acetylation, and ubiquitination are relatively well-studied with more preclinical evidence supporting their therapeutic potential. With continued efforts to elucidate their biological roles and refine targeting strategies, ePTM-based therapies may offer substantial benefits for improving outcomes in patients with CKD and addressing current therapeutic limitations.
Author contributions
MW: Writing – original draft. JiL: Writing – original draft. YZ: Investigation, Writing – review and editing. XW: Investigation, Writing – review and editing. JWe: Visualization, Writing – review and editing. JWa: Visualization, Writing – review and editing. WZ: Writing – review and editing. KT: Writing – review and editing. ML: Conceptualization, Supervision, Writing – review and editing. JuL: Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the Natural Science Foundation of Guangdong Province (No. 2023A1515011814) and the National Natural Science Foundation of China (No. 82374138).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adua, E., Anto, E. O., Roberts, P., Kantanka, O. S., Aboagye, E., and Wang, W. (2018). The potential of N-glycosylation profiles as biomarkers for monitoring the progression of Type II diabetes mellitus towards diabetic kidney disease. J. Diabetes Metab. Disord. 17, 233–246. doi:10.1007/s40200-018-0365-3
Al-Qattan, K. K., Al-Akhawand, S. J., and Mansour, M. H. (2006). Immunohistochemical localization of distinct angiotensin II AT1 receptor isoforms in the kidneys of the Sprague-Dawley rat and the desert rodent Meriones crassus. Anat. Histol. Embryol. 35, 130–138. doi:10.1111/j.1439-0264.2005.00649.x
Albertolle, M. E., Kim, D., Nagy, L. D., Yun, C. H., Pozzi, A., Savas, Ü., et al. (2017). Heme-thiolate sulfenylation of human cytochrome P450 4A11 functions as a redox switch for catalytic inhibition. J. Biol. Chem. 292, 11230–11242. doi:10.1074/jbc.M117.792200
Albertolle, M. E., Phan, T. T. N., Pozzi, A., and Guengerich, F. P. (2018). Sulfenylation of human liver and kidney microsomal cytochromes P450 and other drug-metabolizing enzymes as a response to redox alteration. Mol. Cell Proteomics 17, 889–900. doi:10.1074/mcp.RA117.000382
Alves, I., Santos-Pereira, B., Dalebout, H., Santos, S., Vicente, M. M., Campar, A., et al. (2021). Protein mannosylation as a diagnostic and prognostic biomarker of Lupus nephritis: an unusual glycan neoepitope in systemic Lupus erythematosus. Arthritis Rheumatol. 73, 2069–2077. doi:10.1002/art.41768
Amengual, J. E., Lue, J. K., Ma, H., Lichtenstein, R., Shah, B., Cremers, S., et al. (2021). First-in-Class selective HDAC6 inhibitor (ACY-1215) has a highly favorable safety profile in patients with relapsed and refractory lymphoma. Oncologist 26, 184–e366. doi:10.1002/onco.13673
An, C., Jiao, B., Du, H., Tran, M., Song, B., Wang, P., et al. (2023). Jumonji domain-containing protein-3 (JMJD3) promotes myeloid fibroblast activation and macrophage polarization in kidney fibrosis. Br. J. Pharmacol. 180, 2250–2265. doi:10.1111/bph.16096
Arvaniti, E., Vakrakou, A., Kaltezioti, V., Stergiopoulos, A., Prakoura, N., Politis, P. K., et al. (2016). Nuclear receptor NR5A2 is involved in the calreticulin gene regulation during renal fibrosis. Biochim. Biophys. Acta 1862, 1774–1785. doi:10.1016/j.bbadis.2016.06.013
Azechi, T., Kanehira, D., Kobayashi, T., Sudo, R., Nishimura, A., Sato, F., et al. (2013). Trichostatin A, an HDAC class I/II inhibitor, promotes Pi-induced vascular calcification via up-regulation of the expression of alkaline phosphatase. J. Atheroscler. Thromb. 20, 538–547. doi:10.5551/jat.15826
Barrios, C., Zierer, J., Gudelj, I., Štambuk, J., Ugrina, I., Rodríguez, E., et al. (2016). Glycosylation profile of IgG in moderate kidney dysfunction. J. Am. Soc. Nephrol. 27, 933–941. doi:10.1681/asn.2015010109
Brijmohan, A. S., Batchu, S. N., Majumder, S., Alghamdi, T. A., Thieme, K., McGaugh, S., et al. (2018). HDAC6 inhibition promotes transcription factor EB activation and is protective in experimental kidney disease. Front. Pharmacol. 9, 34. doi:10.3389/fphar.2018.00034
Buemi, M., Senatore, M., Corica, F., Aloisi, C., Romeo, A., Cavallaro, E., et al. (2002). Statins and progressive renal disease. Med. Res. Rev. 22, 76–84. doi:10.1002/med.10000
Burnier, M., and Damianaki, A. (2023). Hypertension as cardiovascular risk factor in chronic kidney disease. Circ. Res. 132, 1050–1063. doi:10.1161/circresaha.122.321762
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 37, D233–D238. doi:10.1093/nar/gkn663
Cao, Y., Qiu, T., Kathayat, R. S., Azizi, S. A., Thorne, A. K., Ahn, D., et al. (2019). ABHD10 is an S-depalmitoylase affecting redox homeostasis through peroxiredoxin-5. Nat. Chem. Biol. 15, 1232–1240. doi:10.1038/s41589-019-0399-y
Cebotaru, L., Liu, Q., Yanda, M. K., Boinot, C., Outeda, P., Huso, D. L., et al. (2016). Inhibition of histone deacetylase 6 activity reduces cyst growth in polycystic kidney disease. Kidney Int. 90, 90–99. doi:10.1016/j.kint.2016.01.026
Chen, K. (2022). Protein lactylation promotes AKI-CKD transition by activating NLRP3 inflammasome. J. Am. Soc. Nephrol. 33, 359. doi:10.1681/asn.20223311s1359b
Chen, Y. Y., Peng, X. F., Liu, G. Y., Liu, J. S., Sun, L., Liu, H., et al. (2019). Protein arginine methyltranferase-1 induces ER stress and epithelial-mesenchymal transition in renal tubular epithelial cells and contributes to diabetic nephropathy. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 2563–2575. doi:10.1016/j.bbadis.2019.06.001
Chen, K., Chen, J., Wang, L., Yang, J., Xiao, F., Wang, X., et al. (2020a). Parkin ubiquitinates GATA4 and attenuates the GATA4/GAS1 signaling and detrimental effects on diabetic nephropathy. Faseb J. 34, 8858–8875. doi:10.1096/fj.202000053R
Chen, X., Yu, C., Hou, X., Li, J., Li, T., Qiu, A., et al. (2020b). Histone deacetylase 6 inhibition mitigates renal fibrosis by suppressing TGF-β and EGFR signaling pathways in obstructive nephropathy. Am. J. Physiol. Ren. Physiol. 319, F1003–f1014. doi:10.1152/ajprenal.00261.2020
Chen, F., Gao, Q., Wei, A., Chen, X., Shi, Y., Wang, H., et al. (2021). Histone deacetylase 3 aberration inhibits Klotho transcription and promotes renal fibrosis. Cell Death Differ. 28, 1001–1012. doi:10.1038/s41418-020-00631-9
Chen, L., Huang, L., Gu, Y., Cang, W., Sun, P., and Xiang, Y. (2022a). Lactate-Lactylation hands between metabolic reprogramming and immunosuppression. Int. J. Mol. Sci. 23, 11943. doi:10.3390/ijms231911943
Chen, Y. X., Zhu, S. Y., Huang, C., Xu, C. Y., Fang, X. D., and Tu, W. P. (2022b). LncRNA Dlx6os1 accelerates diabetic nephropathy progression by epigenetically repressing SOX6 via recruiting EZH2. Kidney Blood Press Res. 47, 177–184. doi:10.1159/000520490
Chen, Q., Xie, C., Tang, K., Luo, M., Zhang, Z., Jin, Y., et al. (2023). The E3 ligase Trim63 promotes podocyte injury and proteinuria by targeting PPARα to inhibit fatty acid oxidation. Free Radic. Biol. Med. 209, 40–54. doi:10.1016/j.freeradbiomed.2023.09.039
Chen, J., Feng, Q., Qiao, Y., Pan, S., Liang, L., Liu, Y., et al. (2024a). ACSF2 and lysine lactylation contribute to renal tubule injury in diabetes. Diabetologia 67, 1429–1443. doi:10.1007/s00125-024-06156-x
Chen, S., Li, J., Liang, Y., Zhang, M., Qiu, Z., Liu, S., et al. (2024b). β-catenin-inhibited Sumoylation modification of LKB1 and fatty acid metabolism is critical in renal fibrosis. Cell Death Dis. 15, 769. doi:10.1038/s41419-024-07154-y
Chen, Y., Han, X., Hao, Y., Wang, X., Wu, F., Tan, S., et al. (2025). Emodin-induced ERα degradation via SYVN1 alleviates vascular calcification by preventing HIF-1α deacetylation in chronic kidney disease. Phytomedicine 145, 156915. doi:10.1016/j.phymed.2025.156915
Cheng, C., Yang, H., Yang, C., Xie, J., Wang, J., Cheng, L., et al. (2024). LATS2 degradation promoted fibrosis damage and rescued by vitamin K3 in lupus nephritis. Arthritis Res. Ther. 26, 64. doi:10.1186/s13075-024-03292-y
Chiba, T., Peasley, K. D., Cargill, K. R., Maringer, K. V., Bharathi, S. S., Mukherjee, E., et al. (2019). Sirtuin 5 regulates proximal tubule fatty acid oxidation to protect against AKI. J. Am. Soc. Nephrol. 30, 2384–2398. doi:10.1681/asn.2019020163
Choi, S. Y., Ryu, Y., Kee, H. J., Cho, S. N., Kim, G. R., Cho, J. Y., et al. (2015). Tubastatin A suppresses renal fibrosis via regulation of epigenetic histone modification and Smad3-dependent fibrotic genes. Vasc. Pharmacol. 72, 130–140. doi:10.1016/j.vph.2015.04.006
Choi, S. Y., Piao, Z. H., Jin, L., Kim, J. H., Kim, G. R., Ryu, Y., et al. (2016). Piceatannol Attenuates renal Fibrosis induced by unilateral ureteral obstruction via downregulation of histone deacetylase 4/5 or p38-MAPK signaling. PLoS One 11, e0167340. doi:10.1371/journal.pone.0167340
Choi, E. W., Song, J. W., Ha, N., Choi, Y. I., and Kim, S. (2018). CKD-506, a novel HDAC6-selective inhibitor, improves renal outcomes and survival in a mouse model of systemic lupus erythematosus. Sci. Rep. 8, 17297. doi:10.1038/s41598-018-35602-1
Chung, S., Kim, S., Son, M., Kim, M., Koh, E. S., Shin, S. J., et al. (2019). Inhibition of p300/CBP-Associated factor attenuates renal tubulointerstitial fibrosis through modulation of NF-kB and Nrf2. Int. J. Mol. Sci. 20, 1554. doi:10.3390/ijms20071554
Cianciolo Cosentino, C., Skrypnyk, N. I., Brilli, L. L., Chiba, T., Novitskaya, T., Woods, C., et al. (2013). Histone deacetylase inhibitor enhances recovery after AKI. J. Am. Soc. Nephrol. 24, 943–953. doi:10.1681/asn.2012111055
Clem, B., Telang, S., Clem, A., Yalcin, A., Meier, J., Simmons, A., et al. (2008). Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 7, 110–120. doi:10.1158/1535-7163.Mct-07-0482
Clem, B. F., O'Neal, J., Tapolsky, G., Clem, A. L., Imbert-Fernandez, Y., Kerr, D. A., et al. (2013). Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol. Cancer Ther. 12, 1461–1470. doi:10.1158/1535-7163.Mct-13-0097
Cui, B., Hou, X., Liu, M., Li, Q., Yu, C., Zhang, S., et al. (2022). Pharmacological inhibition of SMYD2 protects against cisplatin-induced acute kidney injury in mice. Front. Pharmacol. 13, 829630. doi:10.3389/fphar.2022.829630
De, I., and Sadhukhan, S. (2018). Emerging roles of DHHC-mediated protein S-palmitoylation in physiological and pathophysiological context. Eur. J. Cell Biol. 97, 319–338. doi:10.1016/j.ejcb.2018.03.005
Deng, L., Zhang, C., Ying, S., Cai, B., and Zhou, F. (2021). Effect of dose ratio on mitoxantrone and daunorubicin in Acute Myeloid leukemia: a systematic review and meta-analysis of randomized controlled trials. Clin. Lymphoma Myeloma Leuk. 21, e10–e20. doi:10.1016/j.clml.2020.08.001
Dezee, K. J., Shimeall, W. T., Douglas, K. M., Shumway, N. M., and O'Malley, P. G. (2006). Treatment of excessive anticoagulation with phytonadione (vitamin K): a meta-analysis. Arch. Intern Med. 166, 391–397. doi:10.1001/.391
Ding, J. X., Xu, L. X., Lv, J. C., Zhao, M. H., Zhang, H., and Wang, H. Y. (2007). Aberrant sialylation of serum IgA1 was associated with prognosis of patients with IgA nephropathy. Clin. Immunol. 125, 268–274. doi:10.1016/j.clim.2007.08.009
Ehninger, G., Schuler, U., Proksch, B., Zeller, K. P., and Blanz, J. (1990). Pharmacokinetics and metabolism of mitoxantrone. A review. Clin. Pharmacokinet. 18, 365–380. doi:10.2165/00003088-199018050-00003
Elhassan, E. A. E., Kmochová, T., Benson, K. A., Fennelly, N. K., Barešová, V., Kidd, K., et al. (2024). A novel monoallelic ALG5 variant causing late-onset ADPKD and tubulointerstitial fibrosis. Kidney Int. Rep. 9, 2209–2226. doi:10.1016/j.ekir.2024.04.031
Essig, M., Vrtovsnik, F., Nguyen, G., Sraer, J. D., and Friedlander, G. (1998). Lovastatin modulates in vivo and in vitro the plasminogen activator/plasmin system of rat proximal tubular cells: role of geranylgeranylation and rho proteins. J. Am. Soc. Nephrol. 9, 1377–1388. doi:10.1681/asn.V981377
Fan, H., Yang, F., Xiao, Z., Luo, H., Chen, H., Chen, Z., et al. (2023). Lactylation: novel epigenetic regulatory and therapeutic opportunities. Am. J. Physiol. Endocrinol. Metab. 324, E330–e338. doi:10.1152/ajpendo.00159.2022
Feng, D., Sheng-Dong, L., Tong, W., and Zhen-Xian, D. (2020). O-GlcNAcylation of RAF1 increases its stabilization and induces the renal fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165556. doi:10.1016/j.bbadis.2019.165556
Fontecha-Barriuso, M., Martin-Sanchez, D., Ruiz-Andres, O., Poveda, J., Sanchez-Niño, M. D., Valiño-Rivas, L., et al. (2018). Targeting epigenetic DNA and histone modifications to treat kidney disease. Nephrol. Dial. Transpl. 33, 1875–1886. doi:10.1093/ndt/gfy009
Franchi, F., Rollini, F., Been, L., Maaliki, N., Abou Jaoude, P., Rivas, A., et al. (2022). Impact of chronic kidney disease on the pharmacodynamic and pharmacokinetic effects of ticagrelor in patients with diabetes mellitus and coronary artery disease. Eur. Heart J. Cardiovasc Pharmacother. 8, 452–461. doi:10.1093/ehjcvp/pvab042
Fransisco, S. M., Abrami, L., Linder, M. E., Bamji, S. X., Dickinson, B. C., and van der Goot, F. G. (2024). Mechanisms and functions of protein S-acylation. Nat. Rev. Mol. Cell Biol. 25, 488–509. doi:10.1038/s41580-024-00700-8
Fraser, N. J., Howie, J., Wypijewski, K. J., and Fuller, W. (2020). Therapeutic targeting of protein S-acylation for the treatment of disease. Biochem. Soc. Trans. 48, 281–290. doi:10.1042/bst20190707
Gan, L., Luo, P., Li, W., Chen, Q., Cheng, L., Zhong, H., et al. (2025). The role of ubiquitination and deubiquitination in urological tumours. Front. Pharmacol. 16, 1532878. doi:10.3389/fphar.2025.1532878
Gao, X., Liu, X., Han, Z., Liao, H., and Li, R. (2025). Friend or foe? The role of SIRT6 on macrophage polarized to M2 subtype in acute kidney injury to chronic kidney disease. Ren. Fail 47, 2482121. doi:10.1080/0886022x.2025.2482121
Gareri, C., Pfeiffer, C. T., Jiang, X., Paulo, J. A., Gygi, S. P., Pham, U., et al. (2024). Phosphorylation patterns in the AT1R C-terminal tail specify distinct downstream signaling pathways. Sci. Signal 17, eadk5736. doi:10.1126/scisignal.adk5736
Gong, W., Chen, Z., Zou, Y., Zhang, L., Huang, J., Liu, P., et al. (2018). CKIP-1 affects the polyubiquitination of Nrf2 and Keap1 via mediating Smurf1 to resist HG-induced renal fibrosis in GMCs and diabetic mice kidneys. Free Radic. Biol. Med. 115, 338–350. doi:10.1016/j.freeradbiomed.2017.12.013
Goru, S. K., Kadakol, A., Pandey, A., Malek, V., Sharma, N., and Gaikwad, A. B. (2016). Histone H2AK119 and H2BK120 mono-ubiquitination modulate SET7/9 and SUV39H1 in type 1 diabetes-induced renal fibrosis. Biochem. J. 473, 3937–3949. doi:10.1042/bcj20160595
Gu, C., Gao, F., Zhang, S., Kang, L., Zhang, W., Feng, X., et al. (2023a). Role of SUMOylation of STAT1 in tubular epithelial-mesenchymal transition induced by high glucose. Mol. Med. Rep. 27, 42. doi:10.3892/mmr.2023.12929
Gu, M., Jiang, H., Tan, M., Yu, L., Xu, N., Li, Y., et al. (2023b). Palmitoyltransferase DHHC9 and acyl protein thioesterase APT1 modulate renal fibrosis through regulating β-catenin palmitoylation. Nat. Commun. 14, 6682. doi:10.1038/s41467-023-42476-z
Hassan, G. S. (2013). Menadione. Profiles Drug Subst. Excip. Relat. Methodol. 38, 227–313. doi:10.1016/b978-0-12-407691-4.00006-x
He, M., Zhou, X., and Wang, X. (2024a). Glycosylation: mechanisms, biological functions and clinical implications. Signal Transduct. Target Ther. 9, 194. doi:10.1038/s41392-024-01886-1
He, Y., Xie, Y., Zhou, T., Li, D., Cheng, X., Yang, P., et al. (2024b). Sodium crotonate alleviates diabetic kidney disease partially via the histone crotonylation pathway. Inflammation 48, 254–275. doi:10.1007/s10753-024-02047-w
Ho, B. B., and Bergwitz, C. (2021). FGF23 signalling and physiology. J. Mol. Endocrinol. 66, R23–r32. doi:10.1530/jme-20-0178
Hoffmann, B., Widlansky, M., and Greene, A. (2016). Hyperglycemia-induced glycosylation: a driving force for vascular dysfunction in diabetes? FASEB J. 30. doi:10.1096/fasebj.30.1_supplement.1282.13
Hou, Y., Liu, D., Guo, Z., Wei, C., Cao, F., Xu, Y., et al. (2025). Lactate and lactylation in AKI-to-CKD: epigenetic regulation and therapeutic opportunities. Cell Prolif. 58, e70034. doi:10.1111/cpr.70034
Hu, H., Hu, J., Chen, Z., Yang, K., Zhu, Z., Hao, Y., et al. (2024). RBBP6-Mediated ERRα degradation contributes to mitochondrial injury in renal tubular cells in diabetic kidney disease. Adv. Sci. (Weinh) 11, e2405153. doi:10.1002/advs.202405153
Huang, W., Xu, L., Zhou, X., Gao, C., Yang, M., Chen, G., et al. (2013). High glucose induces activation of NF-κB inflammatory signaling through IκBα sumoylation in rat mesangial cells. Biochem. Biophys. Res. Commun. 438, 568–574. doi:10.1016/j.bbrc.2013.07.065
Huang, X. Z., Wen, D., Zhang, M., Xie, Q., Ma, L., Guan, Y., et al. (2014). Sirt1 activation ameliorates renal fibrosis by inhibiting the TGF-β/Smad3 pathway. J. Cell Biochem. 115, 996–1005. doi:10.1002/jcb.24748
Huang, J., Wan, D., Li, J., Chen, H., Huang, K., and Zheng, L. (2015a). Histone acetyltransferase PCAF regulates inflammatory molecules in the development of renal injury. Epigenetics 10, 62–72. doi:10.4161/15592294.2014.990780
Huang, K. P., Chen, C., Hao, J., Huang, J. Y., Liu, P. Q., and Huang, H. Q. (2015b). AGEs-RAGE system down-regulates Sirt1 through the ubiquitin-proteasome pathway to promote FN and TGF-β1 expression in male rat glomerular mesangial cells. Endocrinology 156, 268–279. doi:10.1210/en.2014-1381
Huang, J., Tang, D., Zheng, F., Xu, H., and Dai, Y. (2021). Comprehensive analysis of lysine crotonylation modification in patients with chronic renal failure. BMC Nephrol. 22, 310. doi:10.1186/s12882-021-02445-4
Huang, Z., Zhou, L., Liu, B., Li, X., and Sang, Y. (2025). Endoplasmic reticulum stress aggravates ferroptosis via PERK/ATF4/HSPA5 pathway in UUO-induced renal fibrosis. Front. Pharmacol. 16, 1545972. doi:10.3389/fphar.2025.1545972
Humphries, T. L. R., Shen, K., Iyer, A., Johnson, D. W., Gobe, G. C., Nikolic-Paterson, D., et al. (2021). PAR2-Induced tissue factor synthesis by primary cultures of human kidney tubular epithelial cells is modified by glucose availability. Int. J. Mol. Sci. 22, 7532. doi:10.3390/ijms22147532
Hung, P. H., Hsu, Y. C., Chen, T. H., Ho, C., and Lin, C. L. (2022). The histone Demethylase inhibitor GSK-J4 is a therapeutic target for the kidney fibrosis of diabetic kidney disease via DKK1 modulation. Int. J. Mol. Sci. 23, 9407. doi:10.3390/ijms23169407
Iozzo, M., Pardella, E., Giannoni, E., and Chiarugi, P. (2025). The role of protein lactylation: a kaleidoscopic post-translational modification in cancer. Mol. Cell 85, 1263–1279. doi:10.1016/j.molcel.2025.02.011
Irifuku, T., Doi, S., Sasaki, K., Doi, T., Nakashima, A., Ueno, T., et al. (2016). Inhibition of H3K9 histone methyltransferase G9a attenuates renal fibrosis and retains klotho expression. Kidney Int. 89, 147–157. doi:10.1038/ki.2015.291
Jennings, E. Q., Fritz, K. S., and Galligan, J. J. (2022). Biochemical genesis of enzymatic and non-enzymatic post-translational modifications. Mol. Asp. Med. 86, 101053. doi:10.1016/j.mam.2021.101053
Jin, J., Bai, L., Wang, D., Ding, W., Cao, Z., Yan, P., et al. (2023a). SIRT3-dependent delactylation of cyclin E2 prevents hepatocellular carcinoma growth. EMBO Rep. 24, e56052. doi:10.15252/embr.202256052
Jin, J., Liu, X. M., Shao, W., and Meng, X. M. (2023b). Nucleic acid and protein methylation modification in renal diseases. Acta Pharmacol. Sin. 45, 661–673. doi:10.1038/s41401-023-01203-6
Kamiya, T., Kadowaki, M., Atobe, T., Kunieda, K., Morimoto, K., and Hara, H. (2023). Inhibition of N-glycosylation by glucosamine hydrochloride inhibits TGF-β1-induced LOXL2 secretion. J. Cell Biochem. 124, 797–807. doi:10.1002/jcb.30404
KDIGO (2024). KDIGO 2024 Clinical Practice Guideline for the evaluation and management of chronic kidney disease. Kidney Int. 105, S117–s314. doi:10.1016/j.kint.2023.10.018
Khwaja, A., Sharpe, C. C., Noor, M., and Hendry, B. M. (2006). The role of geranylgeranylated proteins in human mesangial cell proliferation. Kidney Int. 70, 1296–1304. doi:10.1038/sj.ki.5001713
Kim, J. E., Han, D., Jeong, J. S., Moon, J. J., Moon, H. K., Lee, S., et al. (2021). Multisample mass spectrometry-based approach for discovering injury markers in chronic kidney disease. Mol. Cell Proteomics 20, 100037. doi:10.1074/mcp.RA120.002159
Kishimoto, H., Nakano, T., Torisu, K., Tokumoto, M., Uchida, Y., Yamada, S., et al. (2023). Indoxyl sulfate induces left ventricular hypertrophy via the AhR-FGF23-FGFR4 signaling pathway. Front. Cardiovasc Med. 10, 990422. doi:10.3389/fcvm.2023.990422
Ko, P. J., and Dixon, S. J. (2018). Protein palmitoylation and cancer. EMBO Rep. 19, e46666. doi:10.15252/embr.201846666
Kong, L., Wang, H., Li, C., Cheng, H., Cui, Y., Liu, L., et al. (2021). Sulforaphane ameliorates diabetes-induced renal fibrosis through epigenetic Up-Regulation of BMP-7. Diabetes Metab. J. 45, 909–920. doi:10.4093/dmj.2020.0168
Kosztyu, P., Hill, M., Jemelkova, J., Czernekova, L., Kafkova, L. R., Hruby, M., et al. (2018). Glucocorticoids reduce aberrant O-Glycosylation of IgA1 in IgA nephropathy patients. Kidney Blood Press Res. 43, 350–359. doi:10.1159/000487903
Laget, J., Duranton, F., Argilés, À., and Gayrard, N. (2022). Renal insufficiency and chronic kidney disease - promotor or consequence of pathological post-translational modifications. Mol. Asp. Med. 86, 101082. doi:10.1016/j.mam.2022.101082
Lairson, L. L., Henrissat, B., Davies, G. J., and Withers, S. G. (2008). Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem. 77, 521–555. doi:10.1146/annurev.biochem.76.061005.092322
Lan, H., Yu, X. J., Ling, G., Zeng, Y., Yang, Y., He, M., et al. (2025). Targeting casein kinase 2 and ubiquitin-specific protease 7 to modulate RUNX2-mediated osteogenesis in chronic kidney disease. Mol. Med. 31, 214. doi:10.1186/s10020-025-01222-5
Lazar, A. G., Vlad, M. L., Manea, A., Simionescu, M., and Manea, S. A. (2021). Activated histone acetyltransferase p300/CBP-Related signalling pathways mediate Up-Regulation of NADPH oxidase, inflammation, and fibrosis in diabetic kidney. Antioxidants (Basel) 10, 1356. doi:10.3390/antiox10091356
Li, H., Armando, I., Yu, P., Escano, C., Mueller, S. C., Asico, L., et al. (2008). Dopamine 5 receptor mediates Ang II type 1 receptor degradation via a ubiquitin-proteasome pathway in mice and human cells. J. Clin. Invest 118, 2180–2189. doi:10.1172/jci33637
Li, L. X., Fan, L. X., Zhou, J. X., Grantham, J. J., Calvet, J. P., Sage, J., et al. (2017a). Lysine methyltransferase SMYD2 promotes cyst growth in autosomal dominant polycystic kidney disease. J. Clin. Invest 127, 2751–2764. doi:10.1172/jci90921
Li, Y., Huang, W., Xu, Y., Zhou, L., Liang, Y., Gao, C., et al. (2017b). CYLD deubiquitinase negatively regulates high glucose-induced NF-κB inflammatory signaling in mesangial cells. Biomed. Res. Int. 2017, 3982906. doi:10.1155/2017/3982906
Li, Y., Ren, D., and Xu, G. (2019). Long noncoding RNA MALAT1 mediates high glucose-induced glomerular endothelial cell injury by epigenetically inhibiting klotho via methyltransferase G9a. IUBMB Life 71, 873–881. doi:10.1002/iub.2009
Li, Y., Ren, D., Shen, Y., Zheng, X., and Xu, G. (2020). Altered DNA methylation of TRIM13 in diabetic nephropathy suppresses mesangial collagen synthesis by promoting ubiquitination of CHOP. EBioMedicine 51, 102582. doi:10.1016/j.ebiom.2019.11.043
Li, L., Yang, J., Li, F., Gao, F., Zhu, L., and Hao, J. (2021). FBXW7 mediates high glucose-induced SREBP-1 expression in renal tubular cells of diabetic nephropathy under PI3K/Akt pathway regulation. Mol. Med. Rep. 23, 233. doi:10.3892/mmr.2021.11872
Li, H., Sun, L., Gao, P., and Hu, H. (2024a). Lactylation in cancer: current understanding and challenges. Cancer Cell 42, 1803–1807. doi:10.1016/j.ccell.2024.09.006
Li, J., Zou, Y., Kantapan, J., Su, H., Wang, L., and Dechsupa, N. (2024b). TGF‑β/Smad signaling in chronic kidney disease: exploring post‑translational regulatory perspectives (Review). Mol. Med. Rep. 30, 143. doi:10.3892/mmr.2024.13267
Li, K., Xia, X., and Tong, Y. (2024c). Multiple roles of mitochondrial autophagy receptor FUNDC1 in mitochondrial events and kidney disease. Front. Cell Dev. Biol. 12, 1453365. doi:10.3389/fcell.2024.1453365
Li, L., Xiang, T., Guo, J., Guo, F., Wu, Y., Feng, H., et al. (2024d). Inhibition of ACSS2-mediated histone crotonylation alleviates kidney fibrosis via IL-1β-dependent macrophage activation and tubular cell senescence. Nat. Commun. 15, 3200. doi:10.1038/s41467-024-47315-3
Li, Y., Wang, Z., Xu, H., Hong, Y., Shi, M., Hu, B., et al. (2024e). Targeting the transmembrane cytokine co-receptor neuropilin-1 in distal tubules improves renal injury and fibrosis. Nat. Commun. 15, 5731. doi:10.1038/s41467-024-50121-6
Li, S., Dong, L., and Wang, K. (2025a). Current and future perspectives of lysine lactylation in cancer. Trends Cell Biol. 35, 190–193. doi:10.1016/j.tcb.2024.12.015
Li, X., Hu, L., Hu, Q., and Jin, H. (2025b). Lactate and lactylation in the kidneys: current advances and prospects (Review). Int. J. Mol. Med. 56, 121. doi:10.3892/ijmm.2025.5562
Li, X. Q., Jin, B., Liu, S. X., Zhu, Y., Li, N., Zhang, Q. Y., et al. (2025c). Neddylation of RhoA impairs its protein degradation and promotes renal interstitial fibrosis progression in diabetic nephropathy. Acta Pharmacol. Sin. 46, 1692–1705. doi:10.1038/s41401-024-01460-z
Li, Z., Zhu, T., Wu, Y., Yu, Y., Zang, Y., Yu, L., et al. (2025d). Functions and mechanisms of non-histone post-translational modifications in cancer progression. Cell Death Discov. 11, 125. doi:10.1038/s41420-025-02410-2
Liao, Y., Zhang, W., Liu, Y., Zhu, C., and Zou, Z. (2024). The role of ubiquitination in health and disease. MedComm 5 (2020), e736. doi:10.1002/mco2.736
Lin, W., Li, Y., Chen, F., Yin, S., Liu, Z., and Cao, W. (2017a). Klotho preservation via histone deacetylase inhibition attenuates chronic kidney disease-associated bone injury in mice. Sci. Rep. 7, 46195. doi:10.1038/srep46195
Lin, W., Zhang, Q., Liu, L., Yin, S., Liu, Z., and Cao, W. (2017b). Klotho restoration via acetylation of Peroxisome Proliferation-Activated Receptor γ reduces the progression of chronic kidney disease. Kidney Int. 92, 669–679. doi:10.1016/j.kint.2017.02.023
Lin, Z., Li, S., Xiao, H., Xu, Z., Li, C., Zeng, J., et al. (2023). The degradation of TGR5 mediated by Smurf1 contributes to diabetic nephropathy. Cell Rep. 42, 112851. doi:10.1016/j.celrep.2023.112851
Liu, C., Luo, R., Wang, W., Peng, Z., Johnson, G. V. W., Kellems, R. E., et al. (2019a). Tissue transglutaminase-mediated AT1 receptor sensitization underlies pro-inflammatory cytokine LIGHT-Induced hypertension. Am. J. Hypertens. 32, 476–485. doi:10.1093/ajh/hpz018
Liu, L., Zou, J., Guan, Y., Zhang, Y., Zhang, W., Zhou, X., et al. (2019b). Blocking the histone lysine 79 methyltransferase DOT1L alleviates renal fibrosis through inhibition of renal fibroblast activation and epithelial-mesenchymal transition. Faseb J. 33, 11941–11958. doi:10.1096/fj.201801861R
Liu, B., Nie, J., Liang, H., Liang, Z., Huang, J., Yu, W., et al. (2021). Pharmacological inhibition of SETD7 by PFI-2 attenuates renal fibrosis following folic acid and obstruction injury. Eur. J. Pharmacol. 901, 174097. doi:10.1016/j.ejphar.2021.174097
Liu, B., Miao, X., Shen, J., Lou, L., Chen, K., Mei, F., et al. (2023a). USP25 ameliorates diabetic nephropathy by inhibiting TRAF6-mediated inflammatory responses. Int. Immunopharmacol. 124, 110877. doi:10.1016/j.intimp.2023.110877
Liu, X., Gong, S., Ning, Y., Li, Y., Zhou, H., He, L., et al. (2023b). Urinary N-Acetyl-Beta-D-Glucosaminidase levels predict immunoglobulin a nephropathy remission status. BMC Nephrol. 24, 208. doi:10.1186/s12882-023-03262-7
Liu, E., Weng, C., Yan, C., Zhu, X., Li, X., Liu, M., et al. (2025a). Lactate programs PBX1 lactylation and mesangial proliferation in lupus nephritis. JCI Insight 10, e190838. doi:10.1172/jci.insight.190838
Liu, S., Yang, Y., Li, Q., Yu, L., Zong, Z., Zang, R., et al. (2025b). Ubiquitin-specific peptidase 10 promotes renal interstitial fibrosis progression through deubiquitinating and stabilizing P53 protein. Biochim. Biophys. Acta Mol. Basis Dis. 1871, 167660. doi:10.1016/j.bbadis.2025.167660
Lohia, S., Latosinska, A., Zoidakis, J., Makridakis, M., Mischak, H., Glorieux, G., et al. (2023). Glycosylation analysis of urinary peptidome highlights IGF2 glycopeptides in Association with CKD. Int. J. Mol. Sci. 24, 5402. doi:10.3390/ijms24065402
Lopes-Virella, M. F., Baker, N. L., Hunt, K. J., Hammad, S. M., Arthur, J., Virella, G., et al. (2024). Glycosylated sphingolipids and progression to kidney dysfunction in type 1 diabetes. J. Clin. Lipidol. 13, 481–491.e1. doi:10.1016/j.jacl.2019.03.005
Lu, L., Li, X., Zhong, Z., Zhou, W., Zhou, D., Zhu, M., et al. (2021). KMT5A downregulation participated in high Glucose-mediated EndMT via Upregulation of ENO1 Expression in Diabetic Nephropathy. Int. J. Biol. Sci. 17, 4093–4107. doi:10.7150/ijbs.62867
Lu, D., Aji, G., Li, G., Li, Y., Fang, W., Zhang, S., et al. (2025). ZDHHC18 promotes renal fibrosis development by regulating HRAS palmitoylation. J. Clin. Invest 135, e180242. doi:10.1172/jci180242
Luo, M. (2018). Chemical and biochemical perspectives of protein lysine methylation. Chem. Rev. 118, 6656–6705. doi:10.1021/acs.chemrev.8b00008
Lv, T., Hu, Y., Ma, Y., Zhen, J., Xin, W., and Wan, Q. (2019). GCN5L1 controls renal lipotoxicity through regulating acetylation of fatty acid oxidation enzymes. J. Physiol. Biochem. 75, 597–606. doi:10.1007/s13105-019-00711-6
Mabe, N. W., Perry, J. A., Malone, C. F., and Stegmaier, K. (2024). Pharmacological targeting of the cancer epigenome. Nat. Cancer 5, 844–865. doi:10.1038/s43018-024-00777-2
Mao, L., Liu, L., Zhang, T., Qin, H., Wu, X., and Xu, Y. (2020). Histone deacetylase 11 contributes to renal fibrosis by repressing KLF15 transcription. Front. Cell Dev. Biol. 8, 235. doi:10.3389/fcell.2020.00235
Marchwicka, A., Kamińska, D., Monirialamdari, M., Błażewska, K. M., and Gendaszewska-Darmach, E. (2022). Protein prenyltransferases and their inhibitors: structural and functional characterization. Int. J. Mol. Sci. 23, 5424. doi:10.3390/ijms23105424
Marumo, T., Hishikawa, K., Yoshikawa, M., Hirahashi, J., Kawachi, S., and Fujita, T. (2010). Histone deacetylase modulates the proinflammatory and -fibrotic changes in tubulointerstitial injury. Am. J. Physiol. Ren. Physiol. 298, F133–F141. doi:10.1152/ajprenal.00400.2009
Meng, H., Qu, Z., Xiao, Z., Kong, B., Yang, H., Shuai, W., et al. (2025). Ubiquitin-specific protease 38 modulates atrial fibrillation susceptibility in chronic kidney disease via STRAP stabilization and activation of TGF-β/SMAD signaling. Mol. Med. 31, 238. doi:10.1186/s10020-025-01296-1
Meyer-Schwesinger, C. (2019). The ubiquitin-proteasome system in kidney physiology and disease. Nat. Rev. Nephrol. 15, 393–411. doi:10.1038/s41581-019-0148-1
Miguel, V., Shaw, I. W., and Kramann, R. (2025). Metabolism at the crossroads of inflammation and fibrosis in chronic kidney disease. Nat. Rev. Nephrol. 21, 39–56. doi:10.1038/s41581-024-00889-z
Miljuš, G., Penezić, A., Pažitná, L., Gligorijević, N., Baralić, M., Vilotić, A., et al. (2024). Glycosylation and characterization of human transferrin in an end-stage kidney disease. Int. J. Mol. Sci. 25, 4625. doi:10.3390/ijms25094625
Mohamed, R. H., Abdelrahim, D. S., Hay, N. H. A., Fawzy, N. M., M, D. K. M., Yehia, D. A. Y., et al. (2024). The role of protein prenylation inhibition through targeting FPPS by zoledronic acid in the prevention of renal fibrosis in rats. Sci. Rep. 14, 18283. doi:10.1038/s41598-024-68303-z
Mu, B., Zeng, Y., Luo, L., and Wang, K. (2024). Oxidative stress-mediated protein sulfenylation in human diseases: past, present, and future. Redox Biol. 76, 103332. doi:10.1016/j.redox.2024.103332
Ni, J. Y., Wang, X., Xie, H. Y., Yang, N. H., Li, J. Y., Sun, X. A., et al. (2023). Deubiquitinating enzyme USP11 promotes renal tubular cell senescence and fibrosis via inhibiting the ubiquitin degradation of TGF-β receptor II. Acta Pharmacol. Sin. 44, 584–595. doi:10.1038/s41401-022-00977-5
Nishima, N., and Tanaka, S. (2024). Lactate: a missing link between metabolism and inflammation in CKD progression? Kidney Int. 106, 183–185. doi:10.1016/j.kint.2024.05.016
Noels, H., Jankowski, V., Schunk, S. J., Vanholder, R., Kalim, S., and Jankowski, J. (2024). Post-translational modifications in kidney diseases and associated cardiovascular risk. Nat. Rev. Nephrol. 20, 495–512. doi:10.1038/s41581-024-00837-x
Nogueira, A., Vala, H., Vasconcelos-Nóbrega, C., Faustino-Rocha, A. I., Pires, C. A., Colaço, A., et al. (2017). Long-term treatment with chaethomellic acid A reduces glomerulosclerosis and arteriolosclerosis in a rat model of chronic kidney disease. Biomed. Pharmacother. 96, 489–496. doi:10.1016/j.biopha.2017.09.137
Paik, W. K., Paik, D. C., and Kim, S. (2007). Historical review: the field of protein methylation. Trends Biochem. Sci. 32, 146–152. doi:10.1016/j.tibs.2007.01.006
Pan, S., Yuan, T., Xia, Y., Yu, W., Zhou, X., and Cheng, F. (2024). Role of histone modifications in kidney fibrosis. Med. Kaunas. 60, 888. doi:10.3390/medicina60060888
Pedrosa, R., Villar, V. A., Pascua, A. M., Simão, S., Hopfer, U., Jose, P. A., et al. (2008). H2O2 stimulation of the Cl-/HCO3- exchanger by angiotensin II and angiotensin II type 1 receptor distribution in membrane microdomains. Hypertension 51, 1332–1338. doi:10.1161/hypertensionaha.107.102434
Pei, S., and Piao, H. L. (2024). Exploring protein S-Palmitoylation: Mechanisms, detection, and strategies for inhibitor discovery. ACS Chem. Biol. 19, 1868–1882. doi:10.1021/acschembio.4c00110
Peng, Z., Huang, X., Pan, Y., Li, W., Hu, H., Chen, X., et al. (2025). USP22 promotes angiotensin II-induced podocyte injury by deubiquitinating and stabilizing HMGB1. Cell Signal 131, 111771. doi:10.1016/j.cellsig.2025.111771
Politis, P. K., and Charonis, A. S. (2022). Calreticulin in renal fibrosis: a short review. J. Cell Mol. Med. 26, 5949–5954. doi:10.1111/jcmm.17627
Qi, W., Keenan, H. A., Li, Q., Ishikado, A., Kannt, A., Sadowski, T., et al. (2017). Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat. Med. 23, 753–762. doi:10.1038/nm.4328
Qian, H., Ding, Y., Deng, X., Huang, W., Li, Z., Liu, F., et al. (2023). Synthesis-accessibility-oriented design of c-Jun N-terminal kinase 1 inhibitor. Eur. J. Med. Chem. 256, 115442. doi:10.1016/j.ejmech.2023.115442
Qiao, J., Tan, Y., Liu, H., Yang, B., Zhang, Q., Liu, Q., et al. (2024). Histone H3K18 and ezrin lactylation promote renal dysfunction in sepsis-associated Acute Kidney injury. Adv. Sci. (Weinh) 11, e2307216. doi:10.1002/advs.202307216
Rai, R., Verma, S. K., Kim, D., Ramirez, V., Lux, E., Li, C., et al. (2017). A novel acetyltransferase p300 inhibitor ameliorates hypertension-associated cardio-renal fibrosis. Epigenetics 12, 1004–1013. doi:10.1080/15592294.2017.1370173
Rodríguez-Peña, A. B., Fuentes-Calvo, I., Docherty, N. G., Arévalo, M., Grande, M. T., Eleno, N., et al. (2014). Effect of angiotensin II and small GTPase Ras signaling pathway inhibition on early renal changes in a murine model of obstructive nephropathy. Biomed. Res. Int. 2014, 124902. doi:10.1155/2014/124902
Romagnani, P., Remuzzi, G., Glassock, R., Levin, A., Jager, K. J., Tonelli, M., et al. (2017). Chronic kidney disease. Nat. Rev. Dis. Prim. 3, 17088. doi:10.1038/nrdp.2017.88
Romagnani, P., Agarwal, R., Chan, J. C. N., Levin, A., Kalyesubula, R., Karam, S., et al. (2025). Chronic kidney disease. Nat. Rev. Dis. Prim. 11, 8. doi:10.1038/s41572-024-00589-9
Roza, P., Divya, H., and Biswa Prasun, C. (2023). Recent histone deacetylase inhibitors in cancer therapy. Cancer 129, 3372–3380. doi:10.1002/cncr.34974
Šakić, Z., Atić, A., Potočki, S., and Bašić-Jukić, N. (2024). Sphingolipids and chronic kidney disease. J. Clin. Med. 13, 5050. doi:10.3390/jcm13175050
Santiago-Hernandez, A., Martin-Lorenzo, M., Gómez-Serrano, M., Lopez, J. A., Martin-Blazquez, A., Vellosillo, P., et al. (2024). The urinary glycopeptide profile differentiates early cardiorenal risk in subjects not meeting criteria for chronic kidney disease. Int. J. Mol. Sci. 25, 7005. doi:10.3390/ijms25137005
Sasaki, K., Doi, S., Nakashima, A., Irifuku, T., Yamada, K., Kokoroishi, K., et al. (2016). Inhibition of SET domain-containing lysine methyltransferase 7/9 ameliorates renal fibrosis. J. Am. Soc. Nephrol. 27, 203–215. doi:10.1681/asn.2014090850
Sawa-Aihara, A., Hattori, K., Nagao, G., Yamada, Y., and Ishida, T. (2023). Potential efficacy of proteasome inhibitor, Delanzomib, for the treatment of renal fibrosis. Biol. Pharm. Bull. 46, 279–285. doi:10.1248/bpb.b22-00713
Schnee, P., Pleiss, J., and Jeltsch, A. (2024). Approaching the catalytic mechanism of protein lysine methyltransferases by biochemical and simulation techniques. Crit. Rev. Biochem. Mol. Biol. 59, 20–68. doi:10.1080/10409238.2024.2318547
Seto, E., and Yoshida, M. (2014). Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6, a018713. doi:10.1101/cshperspect.a018713
Shao, Y., Zhang, W., Du, D., Yu, Y., Li, Q., and Peng, X. (2022). Ubiquitin-like protein FAT10 promotes renal fibrosis by stabilizing USP7 to prolong CHK1-mediated G2/M arrest in renal tubular epithelial cells. Aging (Albany NY) 14, 7527–7546. doi:10.18632/aging.204301
Sharpe, C. C., Dockrell, M. E., Scott, R., Noor, M. I., Cowsert, L. M., Monia, B. P., et al. (1999). Evidence of a role for Ki-Ras in the stimulated proliferation of renal fibroblasts. J. Am. Soc. Nephrol. 10, 1186–1192. doi:10.1681/asn.V1061186
Shen, N., Lin, H., Wu, T., Wang, D., Wang, W., Xie, H., et al. (2013). Inhibition of TGF-β1-receptor posttranslational core fucosylation attenuates rat renal interstitial fibrosis. Kidney Int. 84, 64–77. doi:10.1038/ki.2013.82
Shen, F., Hou, X., Li, T., Yu, J., Chen, H., Liu, N., et al. (2022). Pharmacological and genetic inhibition of HDAC4 alleviates renal injury and fibrosis in mice. Front. Pharmacol. 13, 929334. doi:10.3389/fphar.2022.929334
Sheng, Z., Wang, X., Ma, Y., Zhang, D., Yang, Y., Zhang, P., et al. (2019). MS-based strategies for identification of protein SUMOylation modification. Electrophoresis 40, 2877–2887. doi:10.1002/elps.201900100
Sheng, Z., Zhu, J., Deng, Y. N., Gao, S., and Liang, S. (2021). SUMOylation modification-mediated cell death. Open Biol. 11, 210050. doi:10.1098/rsob.210050
Shi, Y., Jia, B., Xu, W., Li, W., Liu, T., Liu, P., et al. (2017). Chidamide in relapsed or refractory peripheral T cell lymphoma: a multicenter real-world study in China. J. Hematol. Oncol. 10, 69. doi:10.1186/s13045-017-0439-6
Shi, Y., Tao, M., Chen, H., Ma, X., Wang, Y., Hu, Y., et al. (2022). Ubiquitin-specific protease 11 promotes partial epithelial-to-mesenchymal transition by deubiquitinating the epidermal growth factor receptor during kidney fibrosis. Kidney Int. 103, 544–564. doi:10.1016/j.kint.2022.11.027
Shi, Y., Tao, M., Chen, H., Ma, X., Wang, Y., Hu, Y., et al. (2023). Ubiquitin-specific protease 11 promotes partial epithelial-to-mesenchymal transition by deubiquitinating the epidermal growth factor receptor during kidney fibrosis. Kidney Int. 103, 544–564. doi:10.1016/j.kint.2023.11.027
Silva-Aguiar, R. P., Bezerra, N. C. F., Lucena, M. C., Sirtoli, G. M., Sudo, R. T., Zapata-Sudo, G., et al. (2018). O-GlcNAcylation reduces proximal tubule protein reabsorption and promotes proteinuria in spontaneously hypertensive rats. J. Biol. Chem. 293, 12749–12758. doi:10.1074/jbc.RA118.001746
Singh, R., Rathore, A. S., Dilnashin, H., Keshri, P. K., Gupta, N. K., Prakash, S. A. S., et al. (2024). HAT and HDAC: enzyme with contradictory action in neurodegenerative diseases. Mol. Neurobiol. 61, 9110–9124. doi:10.1007/s12035-024-04115-6
Sternberg, C., Armstrong, A., Pili, R., Ng, S., Huddart, R., Agarwal, N., et al. (2016). Randomized, Double-Blind, placebo-controlled phase III Study of tasquinimod in men with metastatic castration-resistant prostate cancer. J. Clin. Oncol. 34, 2636–2643. doi:10.1200/jco.2016.66.9697
Stinghen, A. E., Massy, Z. A., Vlassara, H., Striker, G. E., and Boullier, A. (2016). Uremic toxicity of advanced glycation end products in CKD. J. Am. Soc. Nephrol. 27, 354–370. doi:10.1681/asn.2014101047
Su, H., Wan, Q., Tian, X. J., He, F. F., Gao, P., Tang, H., et al. (2015). MAD2B contributes to podocyte injury of diabetic nephropathy via inducing cyclin B1 and Skp2 accumulation. Am. J. Physiol. Ren. Physiol. 308, F728–F736. doi:10.1152/ajprenal.00409.2014
Su, H., Liu, B., Chen, H., Zhang, T., Huang, T., Liu, Y., et al. (2022). LncRNA ANRIL mediates endothelial dysfunction through BDNF downregulation in chronic kidney disease. Cell Death Dis. 13, 661. doi:10.1038/s41419-022-05068-1
Sun, G., Reddy, M. A., Yuan, H., Lanting, L., Kato, M., and Natarajan, R. (2010). Epigenetic histone methylation modulates fibrotic gene expression. J. Am. Soc. Nephrol. 21, 2069–2080. doi:10.1681/asn.2010060633
Sun, X., Chen, S., Zhao, Y., Wu, T., Zhao, Z., Luo, W., et al. (2024). OTUD6A in tubular epithelial cells mediates angiotensin II-induced kidney injury by targeting STAT3. Am. J. Physiol. Cell Physiol. 326, C400–c413. doi:10.1152/ajpcell.00394.2023
Tabaczar, S., Czogalla, A., Podkalicka, J., Biernatowska, A., and Sikorski, A. F. (2017). Protein palmitoylation: palmitoyltransferases and their specificity. Exp. Biol. Med. (Maywood) 242, 1150–1157. doi:10.1177/1535370217707732
Tabata, T., Kokura, K., Ten Dijke, P., and Ishii, S. (2009). Ski co-repressor complexes maintain the basal repressed state of the TGF-beta target gene, SMAD7, via HDAC3 and PRMT5. Genes cells. 14, 17–28. doi:10.1111/j.1365-2443.2008.01246.x
Tan, M., Luo, H., Lee, S., Jin, F., Yang, J. S., Montellier, E., et al. (2011). Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028. doi:10.1016/j.cell.2011.08.008
Tan, R. Z., Jia, J., Li, T., Wang, L., and Kantawong, F. (2024). A systematic review of epigenetic interplay in kidney diseases: crosstalk between long noncoding RNAs and methylation, acetylation of chromatin and histone. Biomed. Pharmacother. 176, 116922. doi:10.1016/j.biopha.2024.116922
Tan, R. Z., Bai, Q. X., Jia, L. H., Wang, Y. B., Li, T., Lin, J. Y., et al. (2025). Epigenetic regulation of macrophage function in kidney disease: new perspective on the interaction between epigenetics and immune modulation. Biomed. Pharmacother. 183, 117842. doi:10.1016/j.biopha.2025.117842
Tang, M., and Kalim, S. (2022). Avenues for post-translational protein modification prevention and therapy. Mol. Asp. Med. 86, 101083. doi:10.1016/j.mam.2022.101083
Tang, Y., Li, Y., Yang, X., Lu, T., Wang, X., Li, Z., et al. (2025). Intestinal metabolite TMAO promotes CKD progression by stimulating macrophage M2 polarization through histone H4 lysine 12 lactylation. Cell Death Differ. doi:10.1038/s41418-025-01554-z
Tian, E., Wang, S., Zhang, L., Zhang, Y., Malicdan, M. C., Mao, Y., et al. (2019). Galnt11 regulates kidney function by glycosylating the endocytosis receptor megalin to modulate ligand binding. Proc. Natl. Acad. Sci. U. S. A. 116, 25196–25202. doi:10.1073/pnas.1909573116
Vaz de Castro, P. A. S., Amaral, A. A., Almeida, M. G., Selvaskandan, H., Barratt, J., and Simões, E. S. A. C. (2024). Examining the association between serum galactose-deficient IgA1 and primary IgA nephropathy: a systematic review and meta-analysis. J. Nephrol. 37, 2099–2112. doi:10.1007/s40620-023-01874-8
Villeneuve, L. M., Reddy, M. A., Lanting, L. L., Wang, M., Meng, L., and Natarajan, R. (2008). Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc. Natl. Acad. Sci. U. S. A. 105, 9047–9052. doi:10.1073/pnas.0803623105
Wan, J., Hu, M., Jiang, Z., Liu, D., Pan, S., Zhou, S., et al. (2021). Lysine acetylation in the proteome of renal tubular epithelial cells in diabetic nephropathy. Front. Genet. 12, 767135. doi:10.3389/fgene.2021.767135
Wang, N., and Zhang, C. (2024). Oxidative stress: a culprit in the progression of diabetic kidney disease. Antioxidants (Basel) 13, 455. doi:10.3390/antiox13040455
Wang, J., Pan, J., Li, H., Long, J., Fang, F., Chen, J., et al. (2018). lncRNA ZEB1-AS1 was suppressed by p53 for renal fibrosis in diabetic nephropathy. Mol. Ther. Nucleic Acids 12, 741–750. doi:10.1016/j.omtn.2018.07.012
Wang, M., Hu, J., Yan, L., Yang, Y., He, M., Wu, M., et al. (2019). High glucose-induced ubiquitination of G6PD leads to the injury of podocytes. Faseb J. 33, 6296–6310. doi:10.1096/fj.201801921R
Wang, M., Dai, W., Ke, Z., and Li, Y. (2020a). Functional roles of E3 ubiquitin ligases in gastric cancer. Oncol. Lett. 20, 22. doi:10.3892/ol.2020.11883
Wang, Y., Zuo, B., Wang, N., Li, S., Liu, C., and Sun, D. (2020b). Calcium dobesilate mediates renal interstitial fibrosis and delay renal peritubular capillary loss through Sirt1/p53 signaling pathway. Biomed. Pharmacother. 132, 110798. doi:10.1016/j.biopha.2020.110798
Wang, B., Dai, T., Sun, W., Wei, Y., Ren, J., Zhang, L., et al. (2021a). Protein N-myristoylation: functions and mechanisms in control of innate immunity. Cell Mol. Immunol. 18, 878–888. doi:10.1038/s41423-021-00663-2
Wang, B. S., Ma, X. F., Zhang, C. Y., Li, Y. X., Liu, X. Z., and Hu, C. Q. (2021b). Astragaloside IV improves angiogenesis and promotes wound healing in diabetic rats via the activation of the SUMOylation pathway. Biomed. Environ. Sci. 34, 124–129. doi:10.3967/bes2021.018
Wang, S., Osgood, A. O., and Chatterjee, A. (2022). Uncovering post-translational modification-associated protein-protein interactions. Curr. Opin. Struct. Biol. 74, 102352. doi:10.1016/j.sbi.2022.102352
Wang, X., Xie, X., Ni, J. Y., Li, J. Y., Sun, X. A., Xie, H. Y., et al. (2024a). USP11 promotes renal tubular cell pyroptosis and fibrosis in UUO mice via inhibiting KLF4 ubiquitin degradation. Acta Pharmacol. Sin. 46, 159–170. doi:10.1038/s41401-024-01363-z
Wang, Y., Li, H., Jiang, S., Fu, D., Lu, X., Lu, M., et al. (2024b). The glycolytic enzyme PFKFB3 drives kidney fibrosis through promoting histone lactylation-mediated NF-κB family activation. Kidney Int. 106, 226–240. doi:10.1016/j.kint.2024.04.016
Wang, S., Zhang, B., Wang, Y., Lan, Q., Lv, L., Xiao, T., et al. (2025). Acetylation-regulated DUSP1 deficiency contributes to renal fibrosis progression. Theranostics 15, 3781–3796. doi:10.7150/thno.108992
Wehbi, G. J., Zimpelmann, J., Carey, R. M., Levine, D. Z., and Burns, K. D. (2001). Early streptozotocin-diabetes mellitus downregulates rat kidney AT2 receptors. Am. J. Physiol. Ren. Physiol. 280, F254–F265. doi:10.1152/ajprenal.2001.280.2.F254
Wei, H., Wang, B., Miyagi, M., She, Y., Gopalan, B., Huang, D. B., et al. (2013). PRMT5 dimethylates R30 of the p65 subunit to activate NF-κB. Proc. Natl. Acad. Sci. U. S. A. 110, 13516–13521. doi:10.1073/pnas.1311784110
White, J., Derheimer, F. A., Jensen-Pergakes, K., O'Connell, S., Sharma, S., Spiegel, N., et al. (2024). Histone lysine acetyltransferase inhibitors: an emerging class of drugs for cancer therapy. Trends Pharmacol. Sci. 45, 243–254. doi:10.1016/j.tips.2024.01.010
Woo, H., Oh, J., Cho, Y. J., Oh, G. T., Kim, S. Y., Dan, K., et al. (2024). N-terminal acetylation of Set1-COMPASS fine-tunes H3K4 methylation patterns. Sci. Adv. 10, eadl6280. doi:10.1126/sciadv.adl6280
Wu, W., and Huang, C. (2023). SUMOylation and DeSUMOylation: prospective therapeutic targets in cancer. Life Sci. 332, 122085. doi:10.1016/j.lfs.2023.122085
Wu, Q., Schapira, M., Arrowsmith, C. H., and Barsyte-Lovejoy, D. (2021). Protein arginine methylation: from enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov. 20, 509–530. doi:10.1038/s41573-021-00159-8
Wu, Q. P., Vang, S., Zhou, J. Q., Krick, S., Barnes, J. W., and Sanders, Y. Y. (2024). O-GlcNAc regulates anti-fibrotic genes in lung fibroblasts through EZH2. J. Cell Mol. Med. 28, e18191. doi:10.1111/jcmm.18191
Wusiman, R., Haimiti, S., Abuduaini, H., Yang, M., Wang, Y., Gu, M., et al. (2025). Increased SUMO-activating enzyme subunit 1 promotes glycolysis and fibrotic phenotype of diabetic nephropathy. Biochem. Pharmacol. 237, 116920. doi:10.1016/j.bcp.2025.116920
Xiang, T., Wang, X., Huang, S., Zhou, K., Fei, S., Zhou, B., et al. (2024). Inhibition of PKM2 by shikonin impedes TGF-β1 expression by repressing histone lactylation to alleviate renal fibrosis. Phytomedicine 136, 156324. doi:10.1016/j.phymed.2024.156324
Xiang, T., Wang, X., Huang, S., Zhou, K., Fei, S., Zhou, B., et al. (2025). Inhibition of PKM2 by shikonin impedes TGF-β1 expression by repressing histone lactylation to alleviate renal fibrosis. Phytomedicine 136, 156324. doi:10.1016/j.phymed.2025.156324
Xiao, Q., Tang, Y., Xia, J., Luo, H., Yu, M., Chen, S., et al. (2022). Ubiquitin-specific protease 47 is associated with vascular calcification in chronic kidney disease by regulating osteogenic transdifferentiation of vascular smooth muscle cells. Ren. Fail 44, 752–766. doi:10.1080/0886022x.2022.2072337
Xie, J. Y., Ju, J., Zhou, P., Chen, H., Wang, S. C., Wang, K., et al. (2024). The mechanisms, regulations, and functions of histone lysine crotonylation. Cell Death Discov. 10, 66. doi:10.1038/s41420-024-01830-w
Xiong, C., Guan, Y., Zhou, X., Liu, L., Zhuang, M. A., Zhang, W., et al. (2019). Selective inhibition of class IIa histone deacetylases alleviates renal fibrosis. Faseb J. 33, 8249–8262. doi:10.1096/fj.201801067RR
Xiong, C., Chen, H., Su, B., Zhang, L., Hu, J., Wang, Q., et al. (2025a). PRMT1-mediated BRD4 arginine methylation and phosphorylation promote partial epithelial-mesenchymal transformation and renal fibrosis. Faseb J. 39, e70293. doi:10.1096/fj.202401838R
Xiong, Z., Hu, X., Wang, R., Li, C., Cheng, H., Zhao, W., et al. (2025b). Jingtian granule alleviates adenine-induced renal fibrosis in mice through SIRT3-Mediated deacetylation of P53. Front. Pharmacol. 16, 1526414. doi:10.3389/fphar.2025.1526414
Xu, T. H., Sheng, Z., Li, Y., Qiu, X., Tian, B., and Yao, L. (2020). OGT knockdown counteracts high phosphate-induced vascular calcification in chronic kidney disease through autophagy activation by downregulating YAP. Life Sci. 261, 118121. doi:10.1016/j.lfs.2020.118121
Xu, X., Qin, Z., Zhang, C., Mi, X., Zhang, C., Zhou, F., et al. (2023). TRIM29 promotes podocyte pyroptosis in diabetic nephropathy through the NF-kB/NLRP3 inflammasome pathway. Cell Biol. Int. 47, 1126–1135. doi:10.1002/cbin.12006
Yang, S., Yang, G., Wang, X., Xiang, J., Kang, L., and Liang, Z. (2023). SIRT2 alleviated renal fibrosis by deacetylating SMAD2 and SMAD3 in renal tubular epithelial cells. Cell Death Dis. 14, 646. doi:10.1038/s41419-023-06169-1
Yang, Y., Ren, S., Xue, J., Dong, W., He, W., Luo, J., et al. (2024). DeSUMOylation of RBMX regulates exosomal sorting of cargo to promote renal tubulointerstitial fibrosis in diabetic kidney disease. J. Adv. Res. 74, 175–189. doi:10.1016/j.jare.2024.09.021
Yang, L., Chen, Y., Guo, F., Wang, B., Ying, Z., Kuang, Y., et al. (2025). Transcription factor ATF3 aggravates kidney fibrosis by maintaining the state of histone H3 lysine 27 acetylation. Chin. Med. J. Engl. doi:10.1097/cm9.0000000000003425
Ye, W., Zheng, Y., Zhang, S., Yan, L., Cheng, H., and Wu, M. (2016). Oxamate improves glycemic control and insulin sensitivity via inhibition of tissue lactate production in db/db mice. PLOS ONE 11, e0150303. doi:10.1371/journal.pone.0150303
Yimamu, Y., Ohtani, A., Takei, Y., Furuichi, A., Kamei, Y., Yamanaka-Okumura, H., et al. (2022). 25-hydroxyvitamin D-1α-hydroxylase (CYP27B1) induces ectopic calcification. J. Clin. Biochem. Nutr. 71, 103–111. doi:10.3164/jcbn.22-16
Ying, S., Liu, L., Luo, C., Liu, Y., Zhao, C., Ge, W., et al. (2023). Sublytic C5b-9 induces TIMP3 expression by glomerular mesangial cells via TRAF6-dependent KLF5 K63-linked ubiquitination in rat Thy-1 nephritis. Int. Immunopharmacol. 124, 110970. doi:10.1016/j.intimp.2023.110970
You, H., Zhang, H., Jin, X., and Yan, Z. (2024). Dysregulation of ubiquitination modification in renal cell carcinoma. Front. Genet. 15, 1453191. doi:10.3389/fgene.2024.1453191
Yu, A., Zhao, J., Yadav, S. P. S., Molitoris, B. A., Wagner, M. C., and Mechref, Y. (2021a). Changes in the expression of renal brush border membrane N-Glycome in model rats with chronic kidney diseases. Biomolecules 11, 1677. doi:10.3390/biom11111677
Yu, A., Zhao, J., Zhong, J., Wang, J., Yadav, S. P. S., Molitoris, B. A., et al. (2021b). Altered O-glycomes of renal brush-border membrane in model rats with chronic kidney diseases. Biomolecules 11, 1560. doi:10.3390/biom11111560
Yu, C., Xiong, C., Tang, J., Hou, X., Liu, N., Bayliss, G., et al. (2021c). Histone demethylase JMJD3 protects against renal fibrosis by suppressing TGFβ and notch signaling and preserving PTEN expression. Theranostics 11, 2706–2721. doi:10.7150/thno.48679
Yu, L., Bian, X., Zhang, C., Wu, Z., Huang, N., Yang, J., et al. (2022). Ginkgolic acid improves bleomycin-induced pulmonary fibrosis by inhibiting SMAD4 SUMOylation. Oxid. Med. Cell Longev. 2022, 8002566. doi:10.1155/2022/8002566
Zeng, H., Li, D., Dong, J., Zhou, X., Ou, M., Xue, W., et al. (2023). Qualitative proteome-wide lysine crotonylation profiling reveals protein modification alteration in the leukocyte extravasation pathway in systemic lupus erythematosus. ACS Omega 8, 44905–44919. doi:10.1021/acsomega.3c06293
Zeniya, M., Mori, T., Yui, N., Nomura, N., Mandai, S., Isobe, K., et al. (2017). The proteasome inhibitor bortezomib attenuates renal fibrosis in mice via the suppression of TGF-β1. Sci. Rep. 7, 13086. doi:10.1038/s41598-017-13486-x
Zhan, Q., Liu, H., Zhao, M., and Huang, H. (2025). M2 macrophages-derived exosomes inhibited podocyte pyroptosis via lncRNA AFAP1-AS1/EZH2 axis. Clin. Exp. Pharmacol. Physiol. 52, e70056. doi:10.1111/1440-1681.70056
Zhang, M. M., and Hang, H. C. (2017). Protein S-palmitoylation in cellular differentiation. Biochem. Soc. Trans. 45, 275–285. doi:10.1042/bst20160236
Zhang, Z., Tan, M., Xie, Z., Dai, L., Chen, Y., and Zhao, Y. (2011). Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7, 58–63. doi:10.1038/nchembio.495
Zhang, X., Wu, J., Wu, C., Chen, W., Lin, R., Zhou, Y., et al. (2018). The LINC01138 interacts with PRMT5 to promote SREBP1-mediated lipid desaturation and cell growth in clear cell renal cell carcinoma. Biochem. Biophys. Res. Commun. 507, 337–342. doi:10.1016/j.bbrc.2018.11.036
Zhang, L., Chen, L., Gao, C., Chen, E., Lightle, A. R., Foulke, L., et al. (2020a). Loss of Histone H3 K79 methyltransferase Dot1l facilitates kidney fibrosis by upregulating endothelin 1 through Histone deacetylase 2. J. Am. Soc. Nephrol. 31, 337–349. doi:10.1681/asn.2019070739
Zhang, Y., Zou, J., Tolbert, E., Zhao, T. C., Bayliss, G., and Zhuang, S. (2020b). Identification of histone deacetylase 8 as a novel therapeutic target for renal fibrosis. Faseb J. 34, 7295–7310. doi:10.1096/fj.201903254R
Zhang, Y., He, L., Tu, M., Huang, M., Chen, Y., Pan, D., et al. (2021a). The ameliorative effect of terpinen-4-ol on ER stress-induced vascular calcification depends on SIRT1-mediated regulation of PERK acetylation. Pharmacol. Res. 170, 105629. doi:10.1016/j.phrs.2021.105629
Zhang, Y., Wen, P., Luo, J., Ding, H., Cao, H., He, W., et al. (2021b). Sirtuin 3 regulates mitochondrial protein acetylation and metabolism in tubular epithelial cells during renal fibrosis. Cell Death Dis. 12, 847. doi:10.1038/s41419-021-04134-4
Zhang, Y., Zheng, S., Mao, Y., Cao, W., Zhao, L., Wu, C., et al. (2021c). Systems analysis of plasma IgG intact N-glycopeptides from patients with chronic kidney diseases via EThcD-sceHCD-MS/MS. Analyst 146, 7274–7283. doi:10.1039/d1an01657a
Zhang, C., Guan, Y., Zou, J., Yang, X., Bayliss, G., and Zhuang, S. (2022). Histone methyltransferase MLL1 drives renal tubular cell apoptosis by p53-dependent repression of E-cadherin during cisplatin-induced acute kidney injury. Cell Death Dis. 13, 770. doi:10.1038/s41419-022-05104-0
Zhang, T., Zhang, Y., Xu, H., Lan, J., Feng, Z., Huang, R., et al. (2023). LINC00355 mediates CTNNBIP1 promoter methylation and promotes endoplasmic reticulum stress-induced podocyte injury in diabetic nephropathy. Antioxid. Redox Signal 39, 225–240. doi:10.1089/ars.2021.0227
Zhang, X., Wang, S., Chong, N., Chen, D., Shu, J., Sun, J., et al. (2024). GDF-15 alleviates diabetic nephropathy via inhibiting NEDD4L-mediated IKK/NF-κB signalling pathways. Int. Immunopharmacol. 128, 111427. doi:10.1016/j.intimp.2023.111427
Zhao, X., Wang, S., He, X., Wei, W., and Huang, K. (2024a). Quercetin prevents the USP22-Snail1 signaling pathway to ameliorate diabetic tubulointerstitial fibrosis. Food Funct. 15, 11990–12006. doi:10.1039/d4fo03564j
Zhao, Y., Fan, S., Zhu, H., Zhao, Q., Fang, Z., Xu, D., et al. (2024b). Podocyte OTUD5 alleviates diabetic kidney disease through deubiquitinating TAK1 and reducing podocyte inflammation and injury. Nat. Commun. 15, 5441. doi:10.1038/s41467-024-49854-1
Zhao, Y., Zhang, Y., Meng, B., Luo, M., Li, G., Liu, F., et al. (2024c). A novel integrated pipeline for site-specific quantification of N-glycosylation. Phenomics 4, 213–226. doi:10.1007/s43657-023-00150-w
Zhou, X., Gao, C., Huang, W., Yang, M., Chen, G., Jiang, L., et al. (2014). High glucose induces sumoylation of Smad4 via SUMO2/3 in mesangial cells. Biomed. Res. Int. 2014, 782625. doi:10.1155/2014/782625
Zhou, X., Zang, X., Ponnusamy, M., Masucci, M. V., Tolbert, E., Gong, R., et al. (2016). Enhancer of zeste homolog 2 inhibition attenuates renal fibrosis by maintaining Smad7 and phosphatase and tensin homolog expression. J. Am. Soc. Nephrol. 27, 2092–2108. doi:10.1681/asn.2015040457
Zhou, X., Chen, H., Hu, Y., Ma, X., Li, J., Shi, Y., et al. (2023). Enhancer of zeste homolog 2 promotes renal fibrosis after acute kidney injury by inducing epithelial-mesenchymal transition and activation of M2 macrophage polarization. Cell Death Dis. 14, 253. doi:10.1038/s41419-023-05782-4
Zhou, G., Zhang, C., Peng, H., Su, X., Huang, Q., Zhao, Z., et al. (2024a). PRMT3 methylates HIF-1α to enhance the vascular calcification induced by chronic kidney disease. Mol. Med. 30, 8. doi:10.1186/s10020-023-00759-7
Zhou, H., Deng, N., Li, Y., Hu, X., Yu, X., Jia, S., et al. (2024b). Distinctive tumorigenic significance and innovative oncology targets of SUMOylation. Theranostics 14, 3127–3149. doi:10.7150/thno.97162
Zhu, Y., Cui, H., Lv, J., Liang, H., Zheng, Y., Wang, S., et al. (2019). AT1 and AT2 receptors modulate renal tubular cell necroptosis in angiotensin II-infused renal injury mice. Sci. Rep. 9, 19450. doi:10.1038/s41598-019-55550-8
Zhu, Q., Dong, H., Bukhari, A. A., Zhao, A., Li, M., Sun, Y., et al. (2020a). HUWE1 promotes EGFR ubiquitination and degradation to protect against renal tubulointerstitial fibrosis. Faseb J. 34, 4591–4601. doi:10.1096/fj.201902751R
Zhu, Y., Yu, C., and Zhuang, S. (2020b). Protein arginine methyltransferase 1 mediates renal fibroblast activation and fibrogenesis through activation of Smad3 signaling. Am. J. Physiol. Ren. Physiol. 318, F375–f387. doi:10.1152/ajprenal.00487.2019
Zong, Z., Ren, J., Yang, B., Zhang, L., and Zhou, F. (2025). Emerging roles of lysine lactyltransferases and lactylation. Nat. Cell Biol. 27, 563–574. doi:10.1038/s41556-025-01635-8
Keywords: enzymatic post-translational modifications, chronic kidney disease, therapeutictargets, methylation, acetylation, lactylation
Citation: Wei M, Lin J, Zeng Y, Wang X, Wen J, Wang J, Zou W, Tu K, Liu M and Li J (2025) Enzymatic post-translational modifications of proteins in chronic kidney disease: mechanisms, regulation, and clinical significance. Front. Pharmacol. 16:1678812. doi: 10.3389/fphar.2025.1678812
Received: 03 August 2025; Accepted: 25 September 2025;
Published: 08 October 2025.
Edited by:
Blythe D. Shepard, Georgetown University Medical Center, United StatesReviewed by:
Hewang Lee, George Washington University, United StatesXiumei Wu, Third Affiliated Hospital of Sun Yat-sen University, China
Copyright © 2025 Wei, Lin, Zeng, Wang, Wen, Wang, Zou, Tu, Liu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Menghua Liu, bGl1bWVuZ2h1YUBzbXUuZWR1LmNu; Juan Li, bGlqdWFuMTBAc211LmVkdS5jbg==
†These authors have contributed equally to this work