



- 1 Department of Psychiatry and Neurobehavioral Sciences, University of Virginia, Charlottesville, VA, USA
- 2 Institute of Biological Science and Bioengineering, Beijing Jiaotong University, Beijing, China
- 3 Department of Public Health Sciences, University of Virginia, Charlottesville, VA, USA
- 4 Department of Otolaryngology, ACT Center for Tobacco Treatment, Education and Research, University of Mississippi Medical Center, Jackson, MS, USA
We previously reported that catechol-O-methyltransferase (COMT) is significantly associated with nicotine dependence (ND) in humans. In this study, we examined whether there exists any difference in the extent of methylation of CpG dinucleotides in the promoter region of COMT in smokers and non-smokers by analyzing the methylation status of cytosines at 33 CpG sites through direct sequencing of bisulfite-treated DNA (N = 50 per group). The cytosine was methylated at 13 of 33 CpG sites, and two of these sites showed significant differences between smokers and matched non-smoker controls. Specifically, in the −193 CpG site, the degree of methylation was 19.1% in smokers and 13.2% in non-smokers (P < 0.01). This finding was confirmed by methylation-specific PCR using an additional 100 smoker and 100 non-smoker control samples, which showed the degree of methylation to be 22.2% in smokers and 18.3% in non-smokers (P < 0.01). For the −39 CpG site, the degree of methylation was 9.2% in smokers, whereas no methylation was found in non-smoker controls. Together, our findings provide the first molecular explanation at the epigenetic level for the association of ND with methylation of the COMT promoter, implying that methylation plays a role in smoking dependence.
Introduction
Tobacco use is the leading cause of preventable death throughout the world. In the United States alone, smoking causes approximately 438,000 premature deaths and $167 billion in health-related costs annually (Mokdad et al., 2004 ). Numerous earlier studies demonstrated that nicotine is the primary addictive substance in tobacco smoke, stimulating the release of dopamine from neurons in the ventral tegmental area (Stolerman and Jarvis, 1995 ; USDHHS, 2000 ; WHO, 2002 ). Furthermore, twin and family studies reveal that nicotine dependence (ND) is a complex disorder determined by both genetic and environmental factors, as well as interactions between genes or between genes and the environment (Carmelli et al., 1992 ; Heath et al., 1999 ; Kendler et al., 2000 ; Li, 2003 ; Li et al., 2003 ; Swan et al., 2003 ; Swan and Lessov, 2004 ). Although numerous studies have shown that genetic variants contribute to the etiology of ND (Li, 2008 ; Li and Burmeister, 2009 ), the complex interplay between genetic and environmental factors is largely unknown.
It has become increasingly clear that environmental factors can influence epigenetic mechanisms such as DNA methylation, histone modification, and chromatin restructuring, leading to changes in gene expression (Tsankova et al., 2007 ). Such changes can be especially problematic in individuals with genetic susceptibilities to specific diseases (Abdolmaleky et al., 2004 ). Methylation of CpG islands in the promoter region of a gene has been suggested to be a primary epigenetic modification pattern in humans (Jones and Takai, 2001 ) and a critical mechanism for regulating gene expression and facilitating adaptation to environmental stressors (Abdolmaleky et al., 2004 ). Changes in methylation patterns play an important role in various human diseases, such as cancer (Feinberg and Tycko, 2004 ) and psychiatric disorders (Murphy et al., 2005 ; Abdolmaleky et al., 2006 ). On the other hand, there have been only a few studies of methylation in humans involved in substance abuse (Bonsch et al., 2004 , 2005 ; Philibert et al., 2008a ,b ; Biermann et al., 2009 ; Hillemacher et al., 2009 ; Nielsen et al., 2009 ). For example, Philibert et al. (2008a) found that the methylation status of the promoter region of monoamine oxidase A (MAOA) was significantly associated with lifetime symptom counts for ND and alcoholism in women, a conclusion they reached after performing quantitative methylation analysis of DNA extracted from lymphoblasts derived from 191 subjects participating in the Iowa Adoption Studies (IAS). In another study with this sample, the same research group found that CpG methylation of the serotonin transporter gene was higher in female than male subjects (Philibert et al., 2008b ). By direct sequencing of bisulfite-treated DNA obtained from lymphocytes of 194 former heroin addicts stabilized on methadone maintenance and of 134 control subjects, Nielsen et al. (2009) found that the percent methylation at two of 16 CpG sites in the promoter region of OPM1 was significantly associated with heroin addition. Because both of the sites are located in potential Sp1 transcriptional factor-binding sites, it has been hypothesized that methylation of these CpG sites leads to reduced OPM1 expression in the lymphocytes of these former heroin addicts. Considering the role that dopaminergic neurotransmission plays in the genesis and maintenance of drug dependence, Hillemacher et al. (2009) examined promoter specific methylation of the dopamine transporter (DAT) gene and found that subjects with alcohol dependent showed significant hypermethylation in the DAT promoter.
Previously, we reported that the genetic variants of catechol-O-methyltransferase (COMT) were significantly associated with ND in smokers (Beuten et al., 2006 ). Further, two recent investigations revealed a strong association between the COMT Val108/158Met genotype and smoking cessation or other smoking behaviors (Colilla et al., 2005 ; Johnstone et al., 2007 ). The enzyme encoded by this gene catalyzes the transfer of a methyl group from S-adenosyl-methionine to the hydroxyl group of catecholamines, including the neurotransmitters dopamine, epinephrine, and norepinephrine, or of the catechol estrogen (Weinshilboum et al., 1999 ). Also, COMT plays a critical role in the degradation and inactivation of extraneuronal dopamine and may be involved in a variety of mental disorders (Akil et al., 2003 ; Chen et al., 2004 ).
The COMT has two promoters, P1 and P2, which control transcription of different mRNAs. The membrane-bound COMT (MB-COMT), encoded in mRNA transcribed from the P2 promoter, is expressed predominantly in brain neurons (Tenhunen et al., 1994 ). Hypermethylation of CpG islands in the promoter of MB-COMT correlates with hypoexpression of COMT in endometrial cancer (Sasaki et al., 2003 ), and a recent independent study showed this methylation to be a significant risk factor for schizophrenia and bipolar disorder (Abdolmaleky et al., 2006 ). Together, these findings imply that MB-COMT is a good candidate for the study of DNA methylation in determining vulnerability to ND. The present study was performed to determine differences in the methylation of the MB-COMT promoter in patients with ND and age- and sex-matched non-smokers by detecting methylation of cytosines at 33 CpG sites in the promoter region of the gene through sodium bisulfite modification using DNA sequencing and methylation-specific PCR.
Materials and Methods
Study Participants
All participants were selected from the database developed for a large family-based genetic study of ND, called the Mid-South Tobacco Family (MSTF) study, in which every participant provided informed consent. The study protocol and all other related forms/procedures were approved by all participating Institutional Review Boards. A detailed description of the MSTF study is provided in previous publications from this group (Li et al., 2006 , 2007 , 2008 ). Briefly, we requested each participant to have no (1) history of using abused substances other than nicotine and alcohol over the past 12 months; (2) a history of any serious mental disorder; or (3) a blood/genetic relationship between smokers and non-smoker controls. Additional selection criteria for smokers were that they had to be at least 18 years of age, to have smoked for at least the last 5 years, and to have consumed an average of 20 cigarettes per day for the last 12 months. Non-smoker controls were subject to the following additional selection criteria: (1) smoked no more than 100 cigarettes in their lifetimes; (2) engaged in no regular use of any other tobacco products (e.g., cigars, smokeless tobacco) or nicotine replacement products (e.g., nicotine gum, nicotine patches); (3) had not used any tobacco or nicotine replacement products in the last year; and (4) had an expired alveolar CO < 8.0 ppm.
In the current study, 150 smokers and 150 non-smoker controls of African-American origin were selected, with an equal number of men and women in each group. The average age was 45.5 ± 5.86 (SD) years for the smoker group and 44.6 ± 6.59 years for the non-smoker control group. The average number of cigarettes smoked per day, Heaviness of Smoking Index (HSI), and Fagerstroőm Test for ND (FTND) (Heatherton et al., 1991 ) of smokers were 24.6 ± 14.7, 4.64 ± 1.4, and 7.47 ± 3.15, respectively.
Preparation of Genomic DNA and Bisulfite Modification
Genomic DNA was isolated from whole blood samples of each participant using the DNA Maxi kit from Qiagen, Inc. (Valencia, CA, USA) according to the manufacturer’s protocol. About 500 ng of DNA was treated with sodium bisulfite using the EpiTect Bisulfite Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s protocol. The bisulfite-treated DNA was eluted in 30 µl of EB buffer.
Bisulfite Sequencing and Methylation Analysis
Primers for amplifying the CpG islands in the promoter region of MB-COMT from bisulfite-treated DNA were designed with Methyl Primer Express version 1.0 (Applied Biosystems, Foster City, CA, USA). Amplification was performed with 1 µl of bisulfite-treated DNA; 0.4 µM of each primer (primer A: 5′-TGGGATATTAGTTTTGGGAGAT-3′ and primer B: 5′-TCCCAATATTCCACCCTAAATCTA-3′); 0.2 mM each of dATP, dCTP, dGTP, and dTTP; 1.5 mM MgCl2; and 1.0 unit of Platinum Taq DNA Polymerase (Invitrogen) in a total volume of 25 µl. The amplification conditions consisted of 4 min at 95°C; 5 cycles of 30 s at 95°C, 90 s at 56°C, and 120 s at 72°C; then 25 cycles of 30 s at 95°C, 90 s at 56°C, and 90 s at 72°C; followed by a final elongation step at 72°C for 4 min. A second round of amplification was performed using 2 µl of the first amplified product with primer A and the nested primer C with the sequence 5′-AACAACCCTAACTACCCCAAAA-3′). The amplification conditions for the second round consisted of 4 min at 95°C; 5 cycles of 30 s at 95°C, 90 s at 52°C, and 120 s at 72°C; then 25 cycles of 30 s at 95°C, 90 s at 52°C, and 90 s at 72°C; followed by a final elongation step at 72°C for 4 min. After the second round of amplification, the DNA fragments were recovered and sequenced using the ABI 3730 XL sequencer.
Quantitative methylation rates were estimated from sequence trace files using ESME software (v. 3.2.1) from Epigenomics AG (Berlin, Germany), which involved appropriate quality control, normalization of signals, correction for incomplete bisulfite convention, and mapping of positions in the trace file to CpGs in reference sequences as described by Lewin et al. (2004) . The ESME software was run on a virtual machine (VMware player on Windows XP) running Red Hat Enterprise Linux 4.
Methylation-Specific PCR
After sodium bisulfite conversion, genomic DNA was amplified with primers A and C using the procedure described above for the first round of amplification, except that 15 instead of 25 cycles were used. The amplification products were detected with fluorescence-based, real-time quantitative PCR according to the manual of the TaqMan® Gene Expression Assay (Applied Biosystems). Briefly, real-time PCR was performed in a 96-well optical microplate in a final volume of 20 µl consisting of 10 µl of TaqMan® Universal PCR Master Mix, No AmpErase® UNG (uracil-N-glycosylase), 2 µl of amplified products, an additional 2.5 U of AmpliTaq Gold (Perkin Elmer), 2.5 µM each of the primers 193Fwd and 193Rev, and 150 nM each of the fluorescently labeled probes 193Met and 193Unmet (the sequences of these primers are given below). Serial dilutions of a control sample were included in each plate to generate a standard curve. Initial denaturation at 95°C for 10 min was followed by 40 cycles of denaturation at 95°C for 15 s and annealing and extension at 60°C for 1 min (ABI Prism, 7500 Sequence Detection System). All samples were measured in duplicate, and an average of the two duplicates was used for statistical analysis. The methylation percentage of each sample was compared with a standard curve according to the approach described by Zeschnigk et al. (2004) .
To generate a standard curve, different ratios of methylated and unmethylated target sequences were prepared. The unmethylated sequence was produced using an amplified DNA fragment ensured to be unmethylated by bisulfite sequencing; it was cloned using the TA Cloning Kit (Invitrogen), yielding the PCR193UM plasmid. For the methylated target sequence, plasmid PCR193M was generated from the PCR193UM plasmid with the target nucleotide “T” being changed to “C” using a site-directed mutagenesis kit purchased from Stratagene Inc. (La Jolla, CA, USA). The following methylated/unmethylated ratios were prepared by mixing appropriate target sequences: 0/100, 20/80, 40/60, 60/40, 80/20, and 100/0.
The TaqMan® probes and primers were designed to amplify the target CpG site detected by bisulfite sequencing. The primer sequences are: 193Fwd: 5′-AGATTAGATTAAGAGGTTGGTATGTGGAT-3′ and 193Rev: 5′- CAACAATAACCAAAATATCCCCATAA-3′. The probe sequences are: 193Met: 5′-VIC-TTTTGCGTGGGTATTT-MGB-3′ and 193Unmet: 5′-6FAM- CCACACAAAAATATCC-MGB-3′.
Statistical Analysis
Statistical analysis and plotting of data were performed with GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA). The MB-COMT methylation is given as percentage of methylation of each CpG site. A significant level of α = 0.05 or less was considered significant. Comparisons were made using Student’s t-test on Systat software (Systat Software Inc., Chicago, IL, USA), and all tests were two-tailed. To meet the assumption of normality, we did a logarithmic transformation to the base e for the percentage of methylation at each CpG site.
Results
Methylation Status of 33 CpG Sites in the Promoter Region of MB-COMT
Using the program provided on the website www.urogene.org/methprimer , we predicted a CpG island in the promoter region of MB-COMT (i.e., within about 560 nucleotides from the transcription start site) under the definition island size >100, GC% >50.0%, and Obs/Exp >0.6 (Gardiner-Garden and Frommer, 1987 ). Figure 1 shows the sequence of the MB-COMT promoter from nucleotides −256 to +51 relative to the transcription start site. This DNA fragment contains potential binding motifs for transcription factors AP-2, Sp1, Ets-1, and NF-D (Tenhunen et al., 1994 ). In the current study, 33 CpG dinucleotides were investigated; they are located at nucleotides −211, −195, −193, −179, −161, −158, −156, −154, −142, −131, −127, −125, −123, −112, −110, −104, −99, −79, −74, −72, −69, −67, −62, −55, −51, −46, −39, −35, −29, −23, −8, −3, and +4.
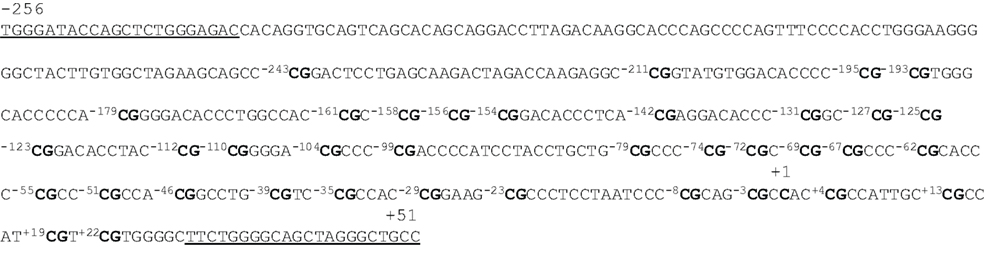
Figure 1. Sequence of the MB-COMT promoter. This DNA fragment is located from nucleotides −256 to +51, and the cytosine shown in bold marked +1 is the 5′ end of the encoded mRNA. The underlined sequences mark the location of the nested PCR primers for bisulfite sequencing. The cytosines at 33 CpG sites are numbered −243 ∼ +22 with CpGs shown in bold.
By direct bisulfite sequencing of the PCR product from sodium bisulfate-modified genomic DNA, we examined the methylation states of all 33 CpG sites in 50 smokers and 50 non-smoker controls. We found that 13 cytosines of the 33 CpG dinucleotides were methylated in at least some of the samples. This pattern was read as “N” by the sequence reader or showed “C” and “T” peaks to different degrees.
Differential Methylation at the −193 and −39 CpG Sites in Smokers and Non-Smoker Controls
On the basis of direct bisulfite sequencing results from 48 smoker and 45 non-smoker control samples from which we have generated reliable sequence information with certainty, we found no significant difference in the mean methylation of all the 13 methylated CpG sites between the two groups. However, two sites showed significantly different methylation extents between the smoker and non-smoker samples (Figure 2 ). At the −193 CpG site, the level of methylation was 19.1% in smokers and 13.2% in non-smokers (P < 0.01). And at the −39 CpG site, the level of methylation was 9.2% in smokers, whereas no methylation was found in non-smoker controls.
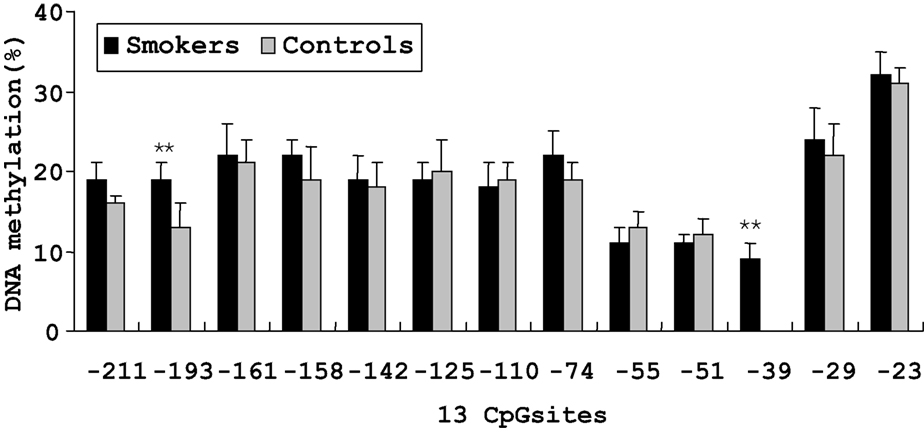
Figure 2. Percent methylation of 13 CpG sites by bisulfite sequencing in the promoter region of MB-COMT in smoker and non-smoker control samples. **P < 0.01. Error bar represents SEM.
To validate the significance of these methylation differences, we examined an additional 100 DNA samples plus the original 50 samples used for bisulfite sequencing employing real-time methylation-specific PCR. Compared with the standard curve, the average extent of methylation at the −193 CpG site was 22.2% (ranging from 12.1% to 39.4%) for the smoker group and 18.3% (ranging from 11.1% to 31.1%) for the non-smoker control group. When the methylation ratios were logarithmic-transformed to the base e, the smokers showed a significantly higher methylation rate than controls (P < 0.01; Figure 3 ). As for the −39 CpG site, although we examined different TaqMan® probes and primers designed specifically for this site, we found no probes or primers that worked effectively. Therefore, real-time methylation-specific PCR could not be performed.
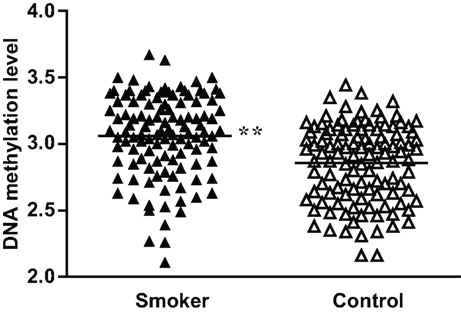
Figure 3. Percent methylation of the −193 CpG site by methylation-specific PCR in MB-COMT promoter in smoker and non-smoker control samples. To meet the assumption of normality, methylation rates were logarithmic-transformed to the base e. **P < 0.01.
Discussion
In this study, we investigated the methylation patterns of cytosines at 33 CpG islands in the promoter region of MB-COMT using bisulfite sequencing. Although no significant difference was detected between smokers and non-smoker controls in the mean methylation level of all the sites, a significantly higher extent of methylation was found in smokers at the −193 and −39 CpG sites. Moreover, the hypermethylation at −193 was confirmed by another methylation detection approach and in more DNA samples. To our knowledge, no studies have investigated the methylation status of MB-COMT in smoking-related phenotypes. Thus, our findings provide the first molecular explanation at the epigenetic level for a direct relation between the risk of ND and methylation alterations of MB-COMT.
Previously, on the basis of a family-based genetic association study, we found that allelic variants of COMT are significantly associated with ND (Beuten et al., 2006 ); and in the present study, hypermethylation at two CpG sites in the promoter region of COMT was found in smokers compared with age- and sex-matched non-smokers, suggesting both genetic and epigenetic processes play a role in the association of COMT with ND. Given that several potential binding motifs for transcription factors are contained in the flanking sequence of the two CpG sites in the MB-COMT promoter, the hypermethylation of the two specific CpG sites may be associated with a change in expression of this gene, generally reducing its expression (Ngo et al., 1996 ). This implies that in our samples, low expression of MB-COMT correlates with a higher risk of ND, which is consistent with the results of our previous genetic association study, where we found that low enzyme activity correlated with higher vulnerability to ND (Beuten et al., 2006 ). Therefore, individuals with low expression of COMT may experience longer-lasting and more abundant dopamine release in the brain, increasing the magnitude or duration of the reward derived from cigarette smoking and the risk of becoming nicotine dependent. Furthermore, the reward processes of neurotransmitters are mediated primarily by dopaminergic pathways (Kalivas, 1993 ). Recently, significant hypermethylation of the DAT promoter was found in patients with alcohol dependence compared with healthy controls, suggesting that hypermethylation of the DAT promoter may play an important role in dopaminergic neurotransmission and is associated with decreased alcohol craving (Hillemacher et al., 2009 ).
In the current study, two methods (i.e., direct bisulfite sequencing and methylation-specific PCR) were used to detect the methylation of MB-COMT. Although our bioinformatical analysis predicted that CpG islands exist in the promoter region of the gene, it cannot predict which one is indeed methylated. Direct bisulfite sequencing can be used for detailed analysis of DNA methylation and inform us of the status of methylation of every CpG site, but the use of this method is commonly limited by the number of samples that can be assayed, as many labor-intensive steps are involved in the process. On the other hand, methylation-specific PCR is more specific, sensitive, and only need small amounts of template DNA and allows rapid analysis of large samples. However, the primers and probes have to be designed specifically on the basis of the status of methylation known from genomic sequencing, which can be challenging in some cases (Eads et al., 2000 ; Zeschnigk et al., 2004 ). For example, in the current study, we could not perform the methylation-specific PCR specifically for the −39 CpG site although we had tested various combinations of different TaqMan® probes and primers designed specifically for the site. Compared with the aforementioned two approaches, the pyrosequencing technique has some advantageous, as it combines the capability for direct quantitative sequencing with reproducibility, speed, and ease of use (Dupont et al., 2004 ). However, the pyrosequencing has its own limitation as well such as it can only read the length of approximately 150 bases (Tost and Gut, 2007 ), which makes it less suitable for an investigation where a large DNA fragment is requested to be examined such as the case reported here. Based on these comparisons, we adopted direct bisulfite sequencing and methylation-specific PCR in this investigation. Through direct genome sequencing analysis of a small number of samples, we detected the methylation status of all CpG sites and identified two sites (i.e., −193 and −39) that appeared to be significantly different between smokers and non-smoker controls. Following identification of these two CpG sites, we analyzed more samples using methylation-specific PCR with the goal of confirming our direct sequencing finding. Unfortunately, significant methylation rate was verified with methylation-specific PCR only at −193 site, thus the significant methylation status at −39 site remains to be further verified in the future with other techniques in the same or different samples.
There are two limitations of the present study. First, the DNA was extracted from the lymphocytes of peripheral blood, and it is possible that the methylation status does not represent exactly what happens in the brain, a key organ for the development of ND. Although we acknowledge it is important to investigate methylation status in DNA extracted from brain tissue, this does not imply that investigation of the methylation status in lymphocytes is less important. This is because: (1) the blood cells, which are more readily obtained, have been valuable biological resources for many genetic studies at various molecular levels, including methylation status; and (2) genome-wide expression profiling studies reveal a high similarity of expression pattern in the cells of the brain and the peripheral blood (Gladkevich et al., 2004 ). Nevertheless, this is an issue faced by almost all brain researchers, as brain tissue is not readily available from human research subjects. Thus, whether our detected methylation signatures of COMT gene in the blood can be replicated in the brain remains to be verified in postmortem tissue. Second, we were not able to determine whether these methylation changes lead to different expression of genes between smokers and non-smokers. To answer this question would require a new study in which we can collect both DNA and RNA simultaneously from each subject. Unfortunately, we do not have RNA extracted from the blood of these subjects as we did for DNA sample, because the samples were collected several years ago and processed for our genetic studies on ND. Although this is indeed an essential question, it is beyond of scope of the current study. A new study designed specifically to test this hypothesis is needed in the future.
In summary, we identified two hypermethylation sites in the promoter region of MB-COMT in smokers compared with non-smoker controls. This indicates that methylation in the promoter region of the gene contributes at least partly to our previously observed association of COMT with ND. Given that DNA methylation is an important factor in regulating gene expression, we hypothesize that increased methylation in the promoter region of MB-COMT in smokers would lead to low level of expression of the genes in smokers compared to non-smokers. Unfortunately, we could not test this hypothesis, as we have no RNA samples available from these subjects. Thus, a study designed specifically to test this hypothesis is warranted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by National Institutes of Health grants DA-12844 and DA-13783 to Ming D. Li. We are grateful for the invaluable contributions of clinical information and blood by all participants in the genetic study, as well as the dedicated work of many research staff at the clinical sites. We also thank Drs. David Nielsen and Mary Jeanne Kreek of the Rockefeller University for their help in methylation analysis of sequencing data using ESME software. Finally, we thank Dr. Joseph Cubells and Dr. Alicia Smith of Emory University, and Dr. Robert Philibert of University of Iowa for their critical and valuable comments and suggestions on the paper.
References
Abdolmaleky, H. M., Cheng, K. H., Faraone, S. V., Wilcox, M., Glatt, S. J., Gao, F., Smith, C. L., Shafa, R., Aeali, B., Carnevale, J., Pan, H., Papageorgis, P., Ponte, J. F., Sivaraman, V., Tsuang, M. T., and Thiagalingam, S. (2006). Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum. Mol. Genet. 15, 3132–3145.
Abdolmaleky, H. M., Smith, C. L., Faraone, S. V., Shafa, R., Stone, W., Glatt, S. J., and Tsuang, M. T. (2004). Methylomics in psychiatry: modulation of gene-environment interactions may be through DNA methylation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 127B, 51–59.
Akil, M., Kolachana, B. S., Rothmond, D. A., Hyde, T. M., Weinberger, D. R., and Kleinman, J. E. (2003). Catechol-O-methyltransferase genotype and dopamine regulation in the human brain. J. Neurosci. 23, 2008–2013.
Beuten, J., Payne, T. J., Ma, J. Z., and Li, M. D. (2006). Significant association of catechol-O-methyltransferase (COMT) haplotypes with nicotine dependence in male and female smokers of two ethnic populations. Neuropsychopharmacology 31, 675–684.
Biermann, T., Reulbach, U., Lenz, B., Frieling, H., Muschler, M., Hillemacher, T., Kornhuber, J., and Bleich, S. (2009). N-methyl-D-aspartate 2b receptor subtype (NR2B) promoter methylation in patients during alcohol withdrawal. J. Neural Transm. 116, 615–622.
Bonsch, D., Lenz, B., Kornhuber, J., and Bleich, S. (2005). DNA hypermethylation of the alpha synuclein promoter in patients with alcoholism. Neuroreport 16, 167–170.
Bonsch, D., Lenz, B., Reulbach, U., Kornhuber, J., and Bleich, S. (2004). Homocysteine associated genomic DNA hypermethylation in patients with chronic alcoholism. J. Neural Transm. 111, 1611–1616.
Carmelli, D., Swan, G. E., Robinette, D., and Fabsitz, R. (1992). Genetic influence on smoking – a study of male twins. N. Engl. J. Med. 327, 829–833.
Chen, J., Lipska, B. K., Halim, N., Ma, Q. D., Matsumoto, M., Melhem, S., Kolachana, B. S., Hyde, T. M., Herman, M. M., Apud, J., Egan, M. F., Kleinman, J. E., and Weinberger, D. R. (2004). Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am. J. Hum. Genet. 75, 807–821.
Colilla, S., Lerman, C., Shields, P. G., Jepson, C., Rukstalis, M., Berlin, J., DeMichele, A., Bunin, G., Strom, B. L., and Rebbeck, T. R. (2005). Association of catechol-O-methyltransferase with smoking cessation in two independent studies of women. Pharmacogenet. Genomics 15, 393–398.
Dupont, J. M., Tost, J., Jammes, H., and Gut, I. G. (2004). De novo quantitative bisulfite sequencing using the pyrosequencing technology. Anal. Biochem. 333, 119–127.
Eads, C. A., Danenberg, K. D., Kawakami, K., Saltz, L. B., Blake, C., Shibata, D., Danenberg, P. V., and Laird, P. W. (2000). MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 28, E32.
Feinberg, A. P., and Tycko, B. (2004). The history of cancer epigenetics. Nat. Rev. Cancer 4, 143–153.
Gardiner-Garden, M., and Frommer, M. (1987). CpG islands in vertebrate genomes. J. Mol. Biol. 196, 261–282.
Gladkevich, A., Kauffman, H. F., and Korf, J. (2004). Lymphocytes as a neural probe: potential for studying psychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 28, 559–576.
Heath, A. C., Kirk, K. M., Meyer, J. M., and Martin, N. G. (1999). Genetic and social determinants of initiation and age at onset of smoking in Australian twins. Behav. Genet. 29, 395–407.
Heatherton, T. F., Kozlowski, L. T., Frecker, R. C., and Fagerstrom, K. O. (1991). The fagerstrom test for nicotine dependence: a revision of the fagerstrom tolerance questionnaire. Br. J. Addict. 86, 1119–1127.
Hillemacher, T., Frieling, H., Hartl, T., Wilhelm, J., Kornhuber, J., and Bleich, S. (2009). Promoter specific methylation of the dopamine transporter gene is altered in alcohol dependence and associated with craving. J. Psychiatr. Res. 43, 388–392.
Johnstone, E. C., Elliot, K. M., David, S. P., Murphy, M. F., Walton, R. T., and Munafo, M. R. (2007). Association of COMT Val108/158Met genotype with smoking cessation in a nicotine replacement therapy randomized trial. Cancer Epidemiol. Biomarkers Prev. 16, 1065–1069.
Jones, P. A., and Takai, D. (2001). The role of DNA methylation in mammalian epigenetics. Science 293, 1068–1070.
Kalivas, P. W. (1993). Neurotransmitter regulation of dopamine neurons in the ventral tegmental area. Brain Res. Brain Res. Rev. 18, 75–11.3
Kendler, K. S., Thornton, L. M., and Pedersen, N. L. (2000). Tobacco consumption in Swedish twins reared apart and reared together. Arch. Gen. Psychiatry 57, 886–892.
Lewin, J., Schmitt, A. O., Adorjan, P., Hildmann, T., and Piepenbrock, C. (2004). Quantitative DNA methylation analysis based on four-dye trace data from direct sequencing of PCR amplificates. Bioinformatics 20, 3005–3012.
Li, M. D. (2003). The genetics of smoking related behavior: a brief review. Am. J. Med. Sci. 326, 168–173.
Li, M. D. (2008). Identifying susceptibility loci for nicotine dependence: 2008 update based on recent genome-wide linkage analyses. Hum. Genet. 123, 119–131.
Li, M. D., and Burmeister, M. (2009). New insights into the genetics of addiction. Nat. Rev. Genet. 10, 225–231.
Li, M. D., Cheng, R., Ma, J. Z., and Swan, G. E. (2003). A meta-analysis of estimated genetic and environmental effects on smoking behavior in male and female adult twins. Addiction 98, 23–31.
Li, M. D., Ma, J. Z., Payne, T. J., Lou, X. Y., Zhang, D., Dupont, R. T., and Elston, R. C. (2008). Genome-wide linkage scan for nicotine dependence in European Americans and its converging results with African Americans in the Mid–South Tobacco Family sample. Mol. Psychiatry 13, 407–416.
Li, M. D., Payne, T. J., Ma, J. Z., Lou, X. Y., Zhang, D., Dupont, R. T., Crews, K. M., Somes, G., Williams, N. J., and Elston, R. C. (2006). A genomewide search finds major susceptibility Loci for nicotine dependence on chromosome 10 in african americans. Am. J. Hum. Genet. 79, 745–751.
Li, M. D., Sun, D., Lou, X. Y., Beuten, J., Payne, T. J., and Ma, J. Z. (2007). Linkage and association studies in African- and Caucasian-American populations demonstrate that SHC3 is a novel susceptibility locus for nicotine dependence. Mol. Psychiatry 12, 462–473.
Mokdad, A. H., Marks, J. S., Stroup, D. F., and Gerberding, J. L. (2004). Actual causes of death in the United States, 2000. JAMA 291, 1238–1245.
Murphy, B. C., O’Reilly, R. L., and Singh, S. M. (2005). Site-specific cytosine methylation in S-COMT promoter in 31 brain regions with implications for studies involving schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 133B, 37–42.
Ngo, V., Gourdji, D., and Laverriere, J. N. (1996). Site-specific methylation of the rat prolactin and growth hormone promoters correlates with gene expression. Mol. Cell. Biol. 16, 3245–3254.
Nielsen, D. A., Yuferov, V., Hamon, S., Jackson, C., Ho, A., Ott, J., and Kreek, M. J. (2009). Increased OPRM1 DNA methylation in lymphocytes of methadone-maintained former heroin addicts. Neuropsychopharmacology 34, 867–873.
Philibert, R. A., Gunter, T. D., Beach, S. R., Brody, G. H., and Madan, A. (2008a). MAOA methylation is associated with nicotine and alcohol dependence in women. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 565–570.
Philibert, R. A., Sandhu, H., Hollenbeck, N., Gunter, T., Adams, W., and Madan, A. (2008b). The relationship of 5HTT (SLC6A4) methylation and genotype on mRNA expression and liability to major depression and alcohol dependence in subjects from the Iowa Adoption Studies. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 543–549.
Sasaki, M., Kaneuchi, M., Sakuragi, N., and Dahiya, R. (2003). Multiple promoters of catechol-O-methyltransferase gene are selectively inactivated by CpG hypermethylation in endometrial cancer. Cancer Res. 63, 3101–3106.
Stolerman, I. P., and Jarvis, M. J. (1995). The scientific case that nicotine is addictive. Psychopharmacology (Berl.) 117, 2–10; discussion 14–20.
Swan, G. E., Hudmon, K. S., Jack, L. M., Hemberger, K., Carmelli, D., Khroyan, T. V., Ring, H. Z., Hops, H., Andrews, J. A., Tildesley, E., McBride, D., Benowitz, N., Webster, C., Wilhelmsen, K. C., Feiler, H. S., Koenig, B., Caron, L., Illes, J., and Cheng, L. S. (2003). Environmental and genetic determinants of tobacco use: methodology for a multidisciplinary, longitudinal family-based investigation. Cancer Epidemiol. Biomarkers Prev. 12, 994–1005.
Swan, G. E., and Lessov, C. N. (2004). Gene-environment interaction in nicotine addiction: the need for a large-scale, collaborative effort. Subst. Use. Misuse 39, 2083–2085.
Tenhunen, J., Salminen, M., Lundstrom, K., Kiviluoto, T., Savolainen, R., and Ulmanen, I. (1994). Genomic organization of the human catechol O-methyltransferase gene and its expression from two distinct promoters. Eur. J. Biochem. 223, 1049–1059.
Tost, J., and Gut, I. G. (2007). DNA methylation analysis by pyrosequencing. Nat. Protoc. 2, 2265–2275.
Tsankova, N., Renthal, W., Kumar, A., and Nestler, E. J. (2007). Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 8, 355–367.
USDHHS. (2000). Reducing Tobacco Use: A Report of the Surgeon General. Atlanta, Georgia: US Department of Health & Human Services, Center for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion.
Weinshilboum, R. M., Otterness, D. M., and Szumlanski, C. L. (1999). Methylation pharmacogenetics: catechol O-methyltransferase, thiopurine methyltransferase, and histamine N-methyltransferase. Annu. Rev. Pharmacol. Toxicol. 39, 19–52.
Keywords: methylation, nicotine, tobacco smoking, promoter, MB-COMT, CpG site
Citation: Xu Q, Ma JZ, Payne TJ and Li MD (2010) Determination of methylated CpG sites in the promoter region of catechol-O-methyltransferase (COMT) and their involvement in the etiology of tobacco smoking. Front. Psychiatry 1:16. doi: 10.3389/fpsyt.2010.00016
This article was submitted to Frontiers in Molecular Psychiatry, a specialty of Frontiers in Psychiatry.
Received: 17 February 2010;
Paper pending published: 22 April 2010;
Accepted: 24 May 2010;
Published online: 10 June 2010
Edited by:
Joseph F. Cubells, Emory University, USAReviewed by:
Alicia K. Smith, Emory University, USARobert Philibert, The University of Iowa, USA
Copyright: © 2010 Xu, Ma, Payne and Li. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Ming D. Li, Department of Psychiatry and Neurobehavioral Sciences, University of Virginia, 1670 Discovery Drive, Suite 110, Charlottesville, VA 22911, USA. e-mail:bWluZ19saUB2aXJnaW5pYS5lZHU=