



- Medical Research Council Centre for Neurodegeneration Research, Institute of Psychiatry, Department of Neuroscience, King’s College London, London, UK
Alzheimer’s disease (AD) is characterized by the extracellular deposition of β-amyloid in senile plaques, the intraneuronal accumulation of hyperphosphorylated tau aggregates as neurofibrillary tangles, and progressive neuronal loss leading to the onset of dementia. Increasing evidence suggests that neuroinflammatory processes contribute to the progression of AD. Minocycline is a semi-synthetic tetracycline derivative commonly used in the treatment of acne. Many studies have revealed that minocycline also has potent anti-inflammatory actions that are neuroprotective in rodent models of Huntington’s disease, Parkinson’s disease and motor neuron disease. Recently, we demonstrated that minocycline reduces the development of abnormal tau species in the htau mouse model of Alzheimer’s disease. We have now extended these findings by examining the impact of minocycline on inflammatory processes in htau mice. Immunohistochemical analysis revealed that minocycline treatment resulted in fewer activated astrocytes in several cortical regions of htau mice, but did not affect astrocytosis in the hippocampus. We found htau mice have significantly elevated amounts of several cortical pro-inflammatory cytokines. In addition, we find that minocycline treatment significantly reduced the amounts of several inflammatory factors, including monocyte chemoattractant proteins 1 and 5, interleukins -6 and -10, eotaxin, and I-309. Furthermore, the reduced amounts of these cytokines significantly correlated with the amount of tau phosphorylated at Ser396/404 in the cortex of htau mice. These results may reveal new cytokine targets of minocycline that could be associated with its inhibition of tau pathology development in vivo. It is possible that further investigation of the role of these cytokines in neurodegenerative processes may identify novel therapeutic targets for Alzheimer’s disease and related disorders.
Introduction
Tauopathies are a group of neurodegenerative disorders that account for the most common forms of dementia, and include Alzheimer’s disease (AD) and frontotemporal dementia with Parkinsonism linked to tau mutations on chromosome 17 (FTDP-17T). Tauopathies are characterized by the deposition of hyperphosphorylated aggregates of the microtubule-associated protein tau in neurons and glia in the central nervous system (CNS). The characteristic tau pathology in these diseases results from abnormal processing of tau, a predominantly neuronal protein, whose main functions include polymerization of tubulin, stabilization of the microtubule cytoskeleton, and regulation of axonal transport (Buée et al., 2000; Gallo et al., 2007; Hanger et al., 2009a,b).
In most tauopathies, prolonged and widespread activation of astrocytes and microglia is apparent in postmortem brain (Salmina, 2009; Mandrekar-Colucci and Landreth, 2010). This characteristic neuroinflammation, while not necessarily causative, is increasingly regarded as an important modulator of disease progression. In AD brain, increased release of several pro-inflammatory mediators from activated glia has been reported. These factors include IL-(interleukin)-1β (Griffin et al., 1995), IL-6 (Huell et al., 1995), tumor necrosis factor (TNF)-α (Dickson et al., 1993), macrophage inflammatory protein (MIP)-1β (Xia et al., 1998), IL-18 (Ojala et al., 2009), S100β (Van Eldik and Griffin, 1994), and monocyte chemoattractant protein (MCP-1) (Ishizuka et al., 1997). Indeed, the amount of MCP-1 in brain tissue has recently been identified as a major predictor of AD (Sokolova et al., 2009). The severity of neuroinflammation is reported to correlate with the extent of brain atrophy (Cagnin et al., 2001) and cognitive decline (Parachikova et al., 2007) observed in AD patients.
While most studies have concentrated on the relationship between inflammation and amyloid pathology in AD, increasing evidence indicates that tau pathology is also associated with neuroinflammation. Substantial reactive astrocytosis and microgliosis is evident in many transgenic mice over-expressing wild-type or mutant human tau (reviewed in Noble et al., 2010). For example, the pattern of activated glia correlates closely with the distribution and density of NFTs in P301S transgenic mice (Yoshiyama et al., 2007). In addition, in transgenic mice that develop both tau and amyloid pathology (3 × Tg-AD line), lipopolysaccharide-induced activation of glia exacerbates tau, but not amyloid pathology (Kitazawa et al., 2005). Furthermore, recent work has shown that tau is also a target of non-steroidal anti-inflammatory drugs, with reduced tau phosphorylation and memory impairment observed following treatment of 3 × Tg-AD mice with ibuprofen (McKee et al., 2008).
Minocycline is a tetracycline-derived antibiotic with proven neuroinflammatory properties (Familian et al., 2006; Noble et al., 2009b) that has been shown to slow disease onset and confer neuroprotection in a number of mouse models of CNS diseases, including Parkinson’s disease (Du et al., 2001) Huntingdon’s disease (Chen et al., 2000; Wang et al., 2003) amyotrophic lateral sclerosis (Kriz et al., 2002; Zhu et al., 2002), multiple sclerosis (Popovic et al., 2002), spinal cord injury (SCI) (Lee et al., 2003; Stirling et al., 2004; Teng et al., 2004), and traumatic brain injury (Sanchez Mejia et al., 2001). Furthermore, recent work has demonstrated that minocycline treatment affects behavioral impairments and plaque formation in APP transgenic mice (Seabrook et al., 2006; Cuello et al., 2010), inhibits neuronal death and attenuates learning and memory deficits following administration of Aβ to rats (Ryu et al., 2006; Choi et al., 2007), and reduces cognitive deficits in the 3 × Tg-AD mouse line (Parachikova et al., 2010). Although many of the beneficial effects of minocycline are attributed to its inhibitory actions on inflammatory and/or apoptotic cell death pathways, the precise targets by which minocycline protects from neuronal death have not yet been identified.
In a recent study we found that administration of minocycline reduced the development of disease-associated tau species in the htau mouse model of tauopathy (Noble et al., 2009a). Htau mice develop age-dependent tau pathology in the hippocampus and cortex (Andorfer et al., 2003, 2005), with the resulting neurodegeneration preceded by deficits in object recognition and spatial memory (Polydoro et al., 2009). Here, we have extended these previous findings by examining the impact of minocycline treatment on neuroinflammatory processes in htau mice. We find that minocycline treatment of relatively young htau mice reduces cortical, but not hippocampal astrocytosis. In addition, using unbiased methods, we have identified several pro-inflammatory mediators whose cortical protein amounts are significantly elevated in htau mice compared to wild-type controls, and that are significantly altered following minocycline treatment. Furthermore, the cortical amounts of selected cytokines correlate strongly with the amount of tau phosphorylated at Ser396/404 in the cortex of htau mice. Thus, this work has identified novel cytokine targets of minocycline that may be closely associated with the development of tau pathology in a mouse model of tauopathy. It is possible that identifying the role of these cytokines in tau-associated neurodegeneration may reveal novel therapeutic targets for AD and related neurodegenerative tauopathies.
Materials and Methods
Animals and Treatment Protocol
Tau transgenic mice (htau line) were obtained from Jackson Laboratories (Bar Habor, Maine, USA). Htau mice (Andorfer et al., 2003) were originally generated by crossing 8c mice that express the entire wild-type human gene (Duff et al., 2000), with tau knock-out (KO) mice that have a targeted disruption of exon 1 of tau (Tucker et al., 2001). For this study 3- to 4-month-old htau mice, and wild-type controls on an identical genetic background, were used. Htau mice received intraperitoneal injections of 10 mg/kg minocycline hydrochloride (Sigma) in phosphate buffered saline (PBS) daily for 14 days. According to the methods of the Friedlander group (Zhu et al., 2002; Wang et al., 2003; Zhang et al., 2003), minocycline was freshly prepared each day. Control htau mice received only sterile NaCl (vehicle) for comparison. Mice were sacrificed by cervical dislocation 2 h after the final treatment. One hemisphere was immediately snap-frozen on dry ice for biochemical analysis, while the other was immersion fixed in 4% (w/v) paraformaldehyde (PFA) in PBS.
Animals were maintained and treated in accordance with the Animals (Scientific Procedures) Act, 1986, following approval by the local ethical review committee.
Immunohistochemistry
Hemibrains were fixed in 4% (v/v) PFA overnight at 4°C, washed in PBS and stored at 4°C in PBS containing 0.1% (w/v) sodium azide. Prior to sectioning, hemispheres were immersed in cryoprotectant solution (30% (w/v) sucrose in 50 mM tris-buffered saline [TBS]) for 48 h. Thick coronal sections (40-μm) were then cut on a Leitz 1321 freezing microtome. Sections were collected in 30% (v/v) ethylene glycol, 15% (w/v) sucrose, 0.05% (w/v) sodium azide in 50 mM TBS, and stored at 4°C prior to histological processing.
A one in six series of sections was used for immunohistochemistry with an antibody directed against glial fibrillary acidic protein (GFAP) (DAKO Ltd.) according to a standard protocol (Pontikis et al., 2005). Briefly, endogenous peroxidase activity was quenched by incubating sections in 1% (v/v) H2O2 in TBS for 15 min, followed by washing. After blocking in 15% (v/v) normal serum for 2 h, sections were incubated overnight with the GFAP antibody (1/5000). A biotinylated anti-rabbit IgG secondary antibody (Vector Laboratories) was used in combination with an avidin–biotin peroxidase complex (Vectastain Elite ABC Kit, Vector Laboratories) for antigen detection. Immunoreactivity was visualized using a 0.05% (w/v) DAB solution, with the staining times carefully controlled with all batches of sections stained at the same time. Sections were mounted on gelatin-chrome alum coated glass slides prior to clearing through a graded series of alcohol.
Quantitative Analysis of Immunohistochemical Staining
The intensity of GFAP labeling was quantified using a semi-automated thresholding image analysis system (Pontikis et al., 2004). This analysis was performed blind to treatment. Briefly, 40 non-overlapping images were captured, on four consecutive sections, through distinct brain regions. All RGB images were captured via a live video camera (JVC, 3CCD, KY-F55B), mounted onto a Zeiss Axioplan microscope using a ×40 objective. Images were subsequently analyzed using Image-Pro Plus 4.0 (Media Cybernetics) image analysis software. Data were plotted graphically as the mean percentage area of immunoreactivity per field ± SEM for each region.
Cytokine Antibody Arrays
Frozen cortical tissue was homogenized in lysis buffer (50 mM TBS [pH 7.4], 2 mM ethylene glycol tetraacetic acid (EGTA), 1 mM phenylmethylsulphonylfluoride (PMSF), 1 mM sodium orthovanadate, 1 mM sodium fluoride, and protease inhibitor cocktail) using a mechanical homogenizer (Polytron; Fisher Scientific Ltd., Loughborough, UK) at 100 mg/ml (w/v). Homogenates were centrifuged at 20,000 g av for 20 min at 4°C, and supernatants were collected. Protein concentrations were determined using a BCA protein assay kit (Pierce Endogen), and 10 μg protein from each sample was analyzed by Cytokine Array, as described below.
Mouse Cytokine Array Panels were used according the manufacturers instructions (R&D Systems, Inc.). Briefly, cortical lysates were mixed with a cocktail of biotinylated detection antibodies prior to incubation at 4°C with the array membranes. Following washing, streptavidin-HRP was applied for 30 min at ambient temperature. Immunoreactivity was then visualized using enhanced chemiluminescence reagent (GE Healthcare). Densitometric analysis was then performed.
Statistical Analysis
Immunohistochemical data and Cytokine Arrays were analyzed using students unpaired t-tests (Graphpad Prism 5.0 Software). Correlation coefficients and significance were determined by Spearman’s Rho non-parametric correlation analysis (SPPS 15.0 for Windows). Differences were considered statistically significant when p < 0.05.
Results
Minocycline Treatment Reduces Cortical Astrocytosis in Htau Mice
Increased inflammatory responses are characteristic of many transgenic mouse models of AD that develop tau pathology, with activated astrocytes and microglia often accumulating around neurons containing highly phosphorylated tau (Schindowski et al., 2006).
To investigate the impact of minocycline treatment on astrocytosis in the CNS of htau mice, brain sections were immunolabeled using a primary antibody directed against GFAP. Changes in GFAP immunoreactivity in specific brain regions were first assessed qualitatively. In general, GFAP labeling revealed activated astrocytes throughout the hippocampus, piriform, primary motor, and somatosensory cortices of htau mice (Figure 1). Within neocortical regions, GFAP-positive astrocytes were almost exclusively present in laminae I, II, and VI.
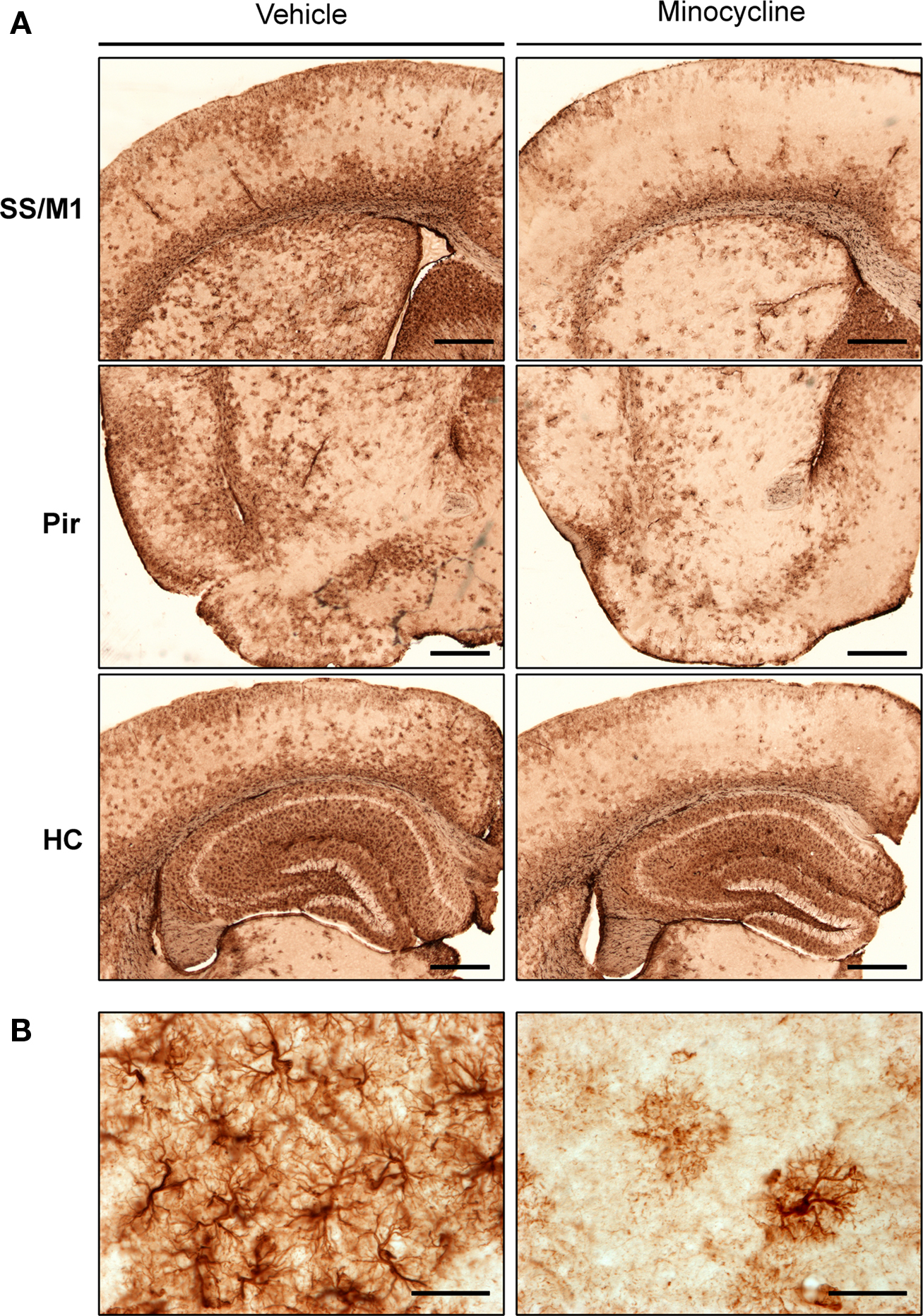
Figure 1. GFAP labeling in minocycline- and vehicle-treated htau mice. Representative images of (A) GFAP immunoreactive astrocytes in the somatosensory and primary motor cortices (SS and M1), piriform cortex (Pir), and hippocampus (HC) of 3- to 4-month-old vehicle- and minocycline-treated htau mice. Scale bars are 500 μm (B) Protoplasmic GFAP labeling in the somatosensory cortex. Scale bars are 40 μm. n = 3.
Qualitative assessment suggested that minocycline treatment resulted in reduced GFAP immunoreactivity in cortical regions, when compared to vehicle-treated htau controls. In contrast, there was no apparent difference in the intensity of hippocampal GFAP immunoreactivity between minocycline- and vehicle-treated htau mice. Higher magnification images revealed a protoplasmic appearance of GFAP immunoreactive astrocytes in both young vehicle- and minocycline- treated htau mice, and there was some evidence of GFAP-positive astrocytes with hypertrophied cell bodies and thickened processes in vehicle-treated mice only (Figure 1B). Indeed, in vehicle-treated htau mice considerable overlapping of neighboring astrocyte processes was observed, which is indicative of severe diffuse astrogliosis as defined by Sofroniew and Vinters (2010).
To quantitatively assess the intensity of GFAP staining in minocycline- and vehicle-treated htau mice, a thresholding image analysis system was used. The barrel field of the somatosensory cortex (S1BF) was selected for this analysis since this region is easily identified, and is representative of general cortical GFAP labeling. The barrel field of the somatosensory cortex refers to discrete multineuronal units called “barrels” that are located in lamina IV of the somatosensory cortex (Rice and Van der Loos, 1977). However, when used for volumetric analysis, the term S1BF, applies to all laminae of the cortex in this region.
Thresholding image analysis of GFAP immunoreactivity revealed a decrease in the intensity of GFAP labeling in the S1BF of minocycline-treated htau mice, when compared to the vehicle-treated controls (mean 37.6% ± 12.6% vs mean 24.4% ± 1.8%, respectively, Figure 2). However this difference was not statistically significant, most likely due to the relatively large variability in GFAP immunoreactivity within the vehicle-treated group. However, the variation within sample groups was significantly reduced by minocycline treatment (p < 0.05), which likely indicates a stabilizing effect of minocycline on astrocyte activation.
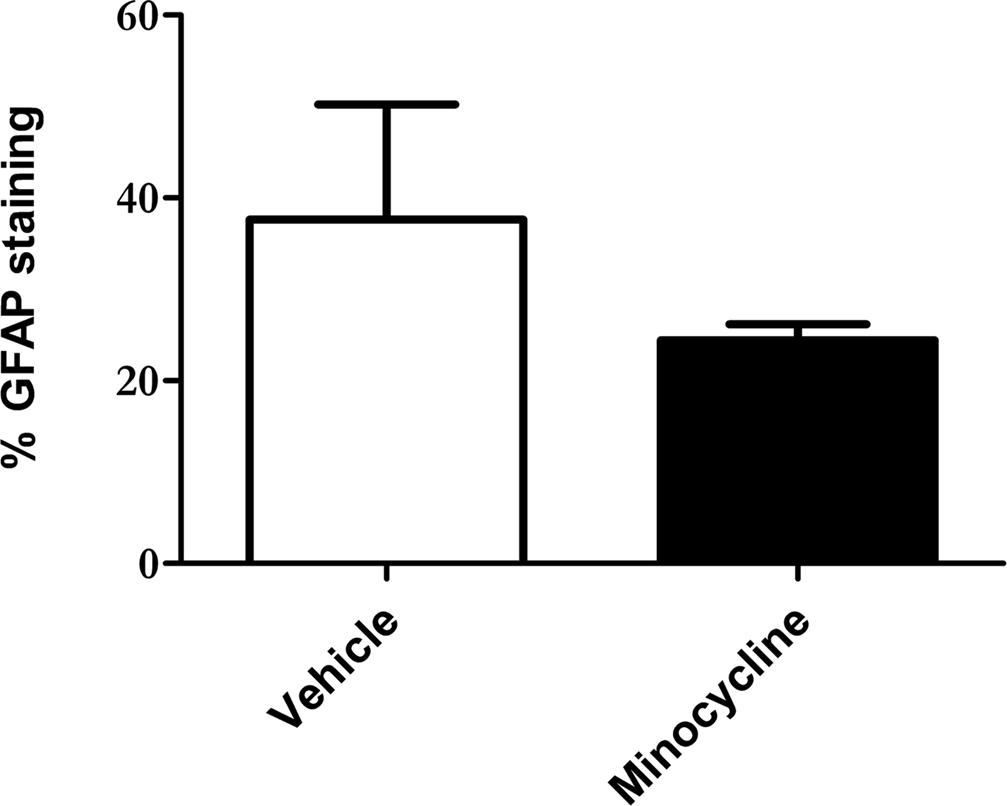
Figure 2. Minocycline treatment reduces GFAP immunoreactivity in the somatosensory cortex of 3- to 4-month-old htau mice. Bar chart shows GFAP labeling as a % of total section area. Values represent mean ± SEM. n = 3.
Htau Mice Have Elevated Amounts of Several Cortical Pro-Inflammatory Cytokines
We next examined cortical lysates from htau and wild-type mice to determine if htau mice display increased neuroinflammatory responses in comparison to wild-type controls. To achieve this, cytokine antibody arrays were used to assess the amount of cytokines present in cortical lysates from these mice. These arrays were selected since they allow unbiased and simultaneous measurement of the amounts of 40 mouse cytokines and chemokines. Positive controls within each array enable standardization between individual membranes allowing comparisons to be made between different samples in independent experiments. Many of the pro-inflammatory factors included in these arrays have been implicated in neuroinflammatory responses.
The results of this work indicated that htau mice have increased cortical amounts of several cytokines when compared to wild-type controls. In particular htau mice displayed increased amounts of complement (C)5a, granulocyte-colony stimulating factor (G-CSF), I-309, soluble inter-cellular adhesion molecule (sICAM)-1, interferon (IFN)-γ, interleukin (IL)-1β, IL-1ra, IL-2, IL-4, IL-7, IL-10, IL-12, IL-13, IL-16, Il1-7, IL-23, IL-27, monokine induced by IFN-γ (MIG), MIP-1α, MIP-1β, MIP-2, stromal cell-derived factor (SDF)-1, TNFα and triggering receptor expressed on myeloid cells (TREM)-1, relative to wild-type controls (p < 0.05 for all, Figure 3).
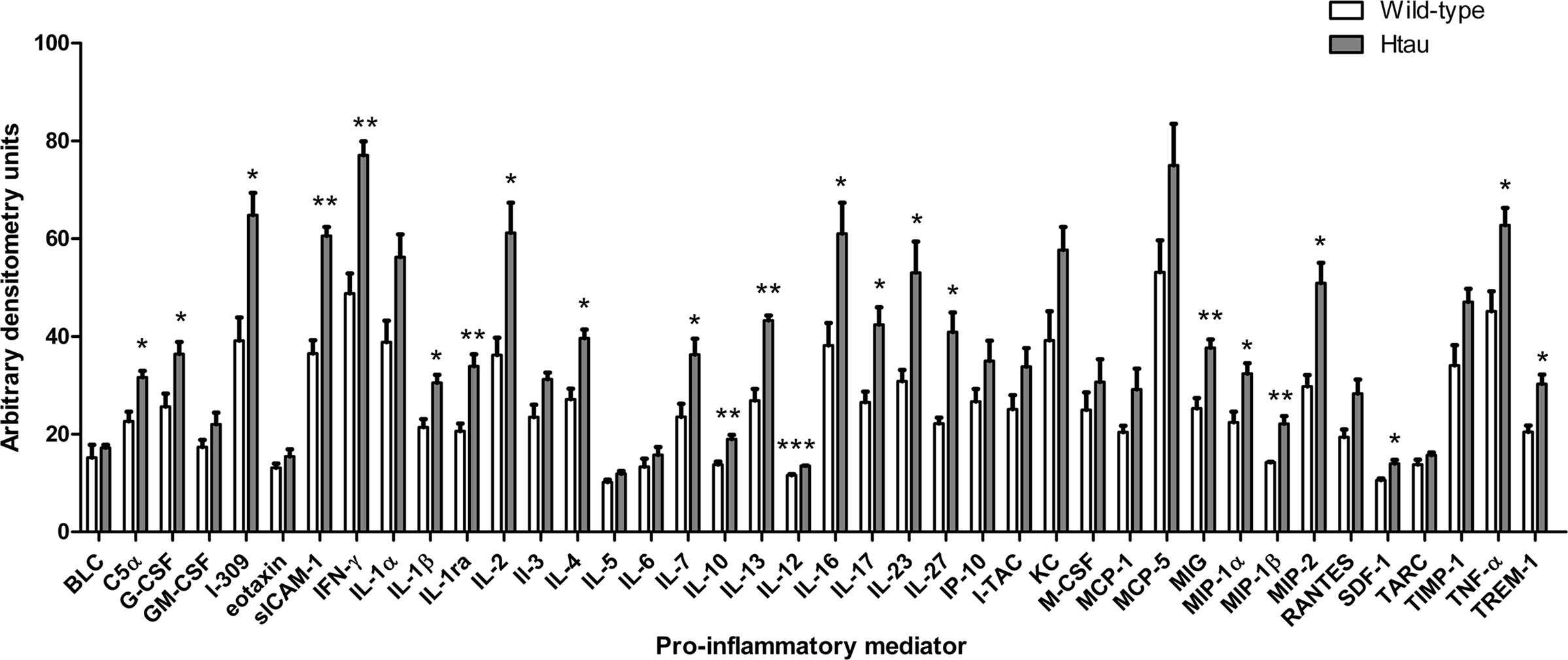
Figure 3. Cytokine amounts in the cortex of htau and wild-type mice. Bar chart shows cytokine protein amounts in arbitrary densitometry units. Values represent mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001. n = 3.
Minocycline Treatment Reduces the Protein Amounts of Several Inflammatory mediators in the Cortex of Htau Mice
Since immunohistochemical analysis revealed that minocycline treatment reduces astrocyte activation in htau mice, we were interested in the effect of this treatment on the cortical abundance of pro-inflammatory mediators. Astrocytes are known to release a variety of pro- and anti-inflammatory mediators in response to stimulation, including IL-1, IL-6, IL-8, IL-10, IP-10, MCP-1, TGF-β, TNF-α, and RANTES (Nair et al., 2008), many of which are known to be upregulated in AD brain (Cameron and Landreth, 2010; Galasko and Montine, 2010). The secretion of these cytokines from activated astrocytes is believed to drive specific immune responses, attract T cells, and enhance the migration of lymphocytes across the blood–brain barrier (Nair et al., 2008).
Cytokine arrays were used to measure differences in cortical cytokine amounts in minocycline- and vehicle-treated htau mice. The results of this experiment revealed that minocycline treatment resulted in significantly decreased cortical amounts of several inflammatory mediators in the cortex of htau mice (Figure 4), including GM-CSF (p < 0.05), I-309 (p < 0.001), eotaxin (p < 0.001), IL-6 (p < 0.01), IL-10 (p < 0.01), macrophage-colony stimulating factor (M-CSF) (p < 0.01), MCP-1 (p < 0.01), MCP-5 (p < 0.01), and TREM-1 (p < 0.05). Of these cytokines, we have shown that the amounts of I-309, IL-10 and TREM-1 are increased in htau mice relative to wild-type controls. In addition, minocycline treatment resulted in significantly increased cortical amounts of IL-17 (p < 0.01) and IL-1β (p < 0.05) in htau cortex.
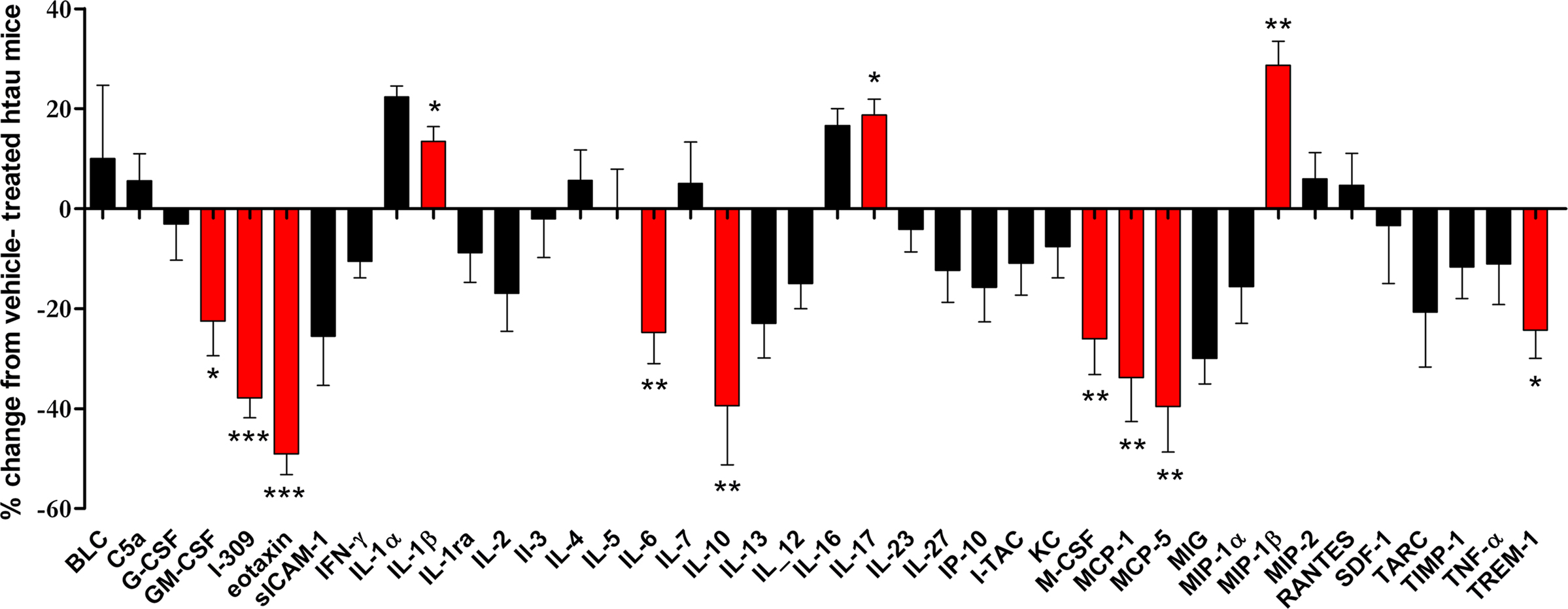
Figure 4. The effect of minocycline treatment on protein amounts of inflammatory mediators in the cortex of 3- to 4-month-old htau mice. Bar chart shows the protein amounts of cytokines in the cortex of minocycline-treated as a percentage of those in vehicle-treated htau mice. Values represent mean ± SEM, *p < 0.05, **p < 0.01, *** p < 0.001. n = 4.
The Amount of Selected Inflammatory Cytokines Correlates Strongly with Tau Phosphorylation
We have previously shown that tau phosphorylation at the PHF1 epitope, Ser396/404, is significantly reduced following minocycline treatment of 3- to 4-month-old htau mice (Noble et al., 2009a). To extend these findings, we next correlated the amounts of cytokines measured in the cortex of these animals, with amounts of pSer396/404 tau (Table 1). We found that there were significant positive correlations between the amount of tau phosphorylated at Ser396/404 and GM-CSF, I-309, eotaxin, IL-6, IL-10, M-CSF, MCP-1, MCP-5, and TREM-1. In addition, there was a significant negative correlation between tau phosphorylation at the PHF1 site and IL-1β, IL-17, and MIP-1β, cytokines we found to be increased by minocycline treatment. Finally, there was a significant correlation between MIG amounts and tau phosphorylation at Ser396/404, although the amounts of this cytokine were not significantly affected by minocycline treatment.
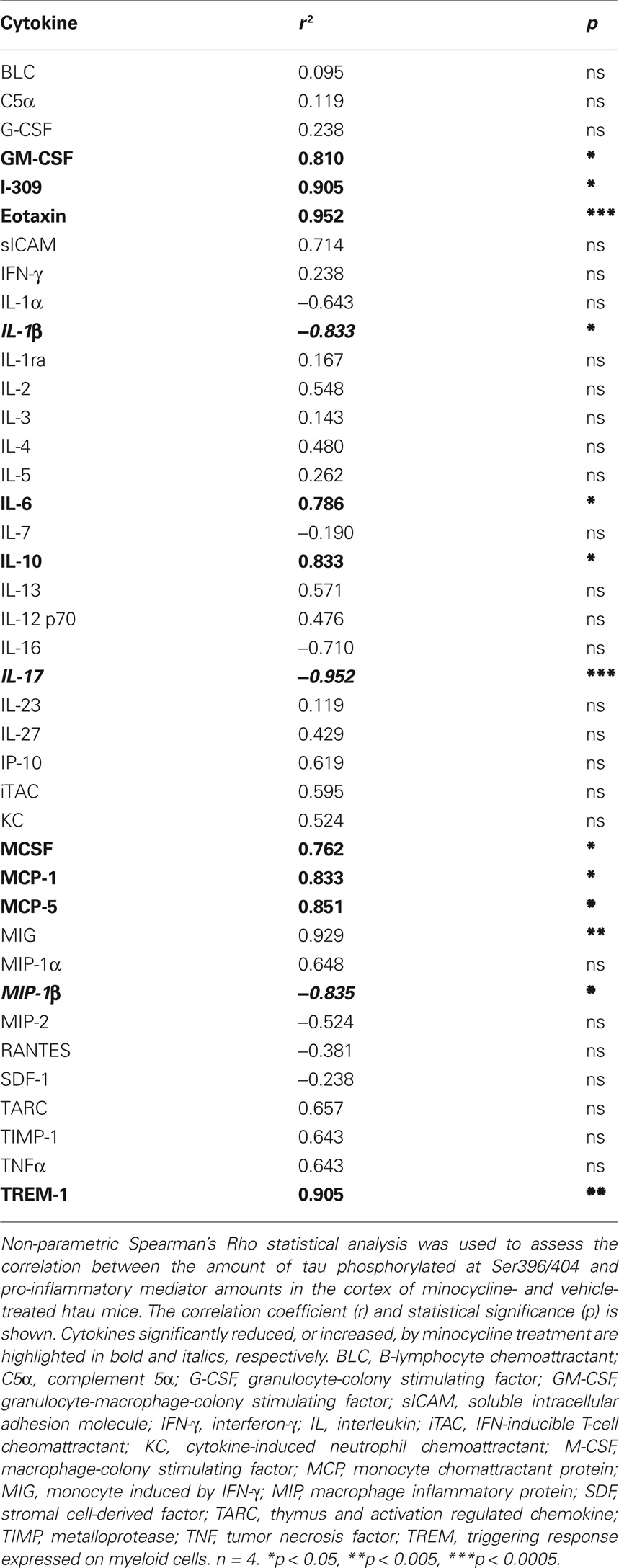
Table 1. Correlation between cortical amounts of pSer396/404 tau and cytokines.
Discussion
Here, we have shown that htau mice have an increased inflammatory response, and also that minocycline treatment inhibits a subset of the cytokines upregulated in this transgenic mouse model of tauopathy. In minocycline-treated mice there was an appreciable decrease in astrocytosis in all cortical regions, but no effect on the intensity of GFAP labeling in the hippocampus. The reason for a lack of an effect of minocycline on astrocytosis within the hippocampus is not understood, but may reflect the relatively high level of GFAP expression evident in this brain region, even in neurologically normal control mice. In general, there was abundant labeling of activated astrocytes in the neocortex, piriform cortex and hippocampus of htau mice. It has been shown previously in tau transgenic mice that glial activation accompanies the development of tau pathology (Bellucci et al., 2004; Yoshiyama et al., 2007). This might suggest that astrocyte activation precedes the development of tau pathology in htau mice, and indeed this has been suggested in other mouse models of tauopathy (Schindowski et al., 2006).
Astrocytes are known to release a variety of factors when activated in vitro and in vivo, including complement factors, iNOS, COX-2, and various interleukins (Akiyama et al., 2000a,b). A recent review summarizes inflammatory responses in various transgenic mouse models of AD (Schwab et al., 2009). Most studies so far have concentrated on transgenic models of AD with plaque, but not tangle, pathology and as a result there is limited literature relating to the inflammatory profile of tau transgenic mice. However, in mice over-expressing P301S mutant human tau, iNOS, COX-2, and IL-1β positive cells have been identified in affected brain regions (Bellucci et al., 2004). Furthermore, increased amounts of TNFα, IL-12, RANTES, IL-5, IL-4, and decreased amounts of eotaxin, KC, MIP-1α, and IL-2, have been found in 3 × Tg-AD mice compared to non-transgenic mice (Parachikova et al., 2010).
Here, we showed that htau mice have increased amounts of C5a, G-CSF, I-309, sICAM-1, IFN-γ, IL-1β, -2, -4, -7, -10, -12, -13, -27, MIG, MIP-1α, MIP-1β, MIP-2, SD)-1, TNFα, and TREM-1, relative to wild-type controls. Thus, TNF-α, IL-12, and IL-4 are upregulated in both htau and 3 × Tg-AD mice, while MIP-1α and IL-2 are decreased in 3 × Tg-AD mice, but increased in the htau line, relative to their respective controls. The effect of minocycline treatment on cytokine levels in the cortex of young htau mice was then assessed, since we have previously demonstrated that minocycline significantly reduces the development of abnormal tau species in these mice (Noble et al., 2009a). A number of cytokines were found to be significantly decreased in response to minocycline treatment, including GM-CSF, M-CSF, I-309, eotaxin, MCP-1, MCP-5, IL-6, and TREM-1.
GM-CSF is a hematopoietic cytokine, produced by macrophages, astrocytes and neurons (McGeer et al., 1996). Together, with M-CSF, GM-CSF stimulates microglial proliferation in vivo (Giulian and Ingeman, 1988). Increased amounts of both GM-CSF (Tarkowski et al., 2001) and M-CSF (Laske et al., 2010) have been found in the CSF of AD patients compared to control cases, and interestingly, GM-CSF levels correlate with the abundance of tau proteins in CSF (Tarkowski et al., 2001). In addition, I-309 has recently been identified as a novel biomarker for AD and mild cognitive impairment (Hu et al., 2010), whilst the serum amounts of eotaxin are known to be increased in AD patients (Choi et al., 2008). Furthermore, CNS amounts of the predominantly astrocyte-derived cytokine MCP-1, are considered a major predictor of AD (Sokolova et al., 2009), and IL-6 co-localizes with plaque-associated microglia (Bauer et al., 1991; Huell et al., 1995). To date, there is no known link between MCP-5 or TREM-1 and AD or related tauopathies.
Minocycline treatment of htau mice also significantly increased the cortical amounts of two inflammatory mediators, IL-17 and Il-1β. IL-17 functions in a synergistic manner to IL-6, and IL-17 protein amounts are reported to be increased in the brains of transgenic AD mice (Jin et al., 2008). IL-1β co-localizes with amyloid plaques in AD brain, and minocycline treatment has previously been shown to suppress secretion of IL-1β from Aβ-treated microglia in culture (Seabrook et al., 2006). The reason for the apparent discrepancy in the effect of minocycline treatment on IL-1β in amyloid and tau models of AD is not clear, but is worthy of further investigation.
Indeed, a recent paper by Parachikova using the 3 × Tg-AD model has raised several discrepancies between our findings, and those that they have reported. While, we detected reduced tau phosphorylation at Ser202 and Ser396/404 in htau mice treated with minocycline (Noble et al., 2009a), in 3 × Tg-AD mice, minocycline treatment did not affect tau phosphorylation at Ser199/202, reduced phosphorylation at Thr212/Ser214, and increased tau phosphorylation at Ser396/400/403/404 (Parachikova et al., 2010). Furthermore, while both Parachikova et al. (2010) and ourselves both found reductions in GFAP in minocycline-treated mice, these authors detected decreased amounts of TNFα and RANTES, two cytokines not affected here. Furthermore, here we show a significant decrease in eotaxin amounts, while Parachikova et al. (2010) report that this cytokine is increased by minocycline treatment of 3 × Tg-AD mice. There are differences in the administration method utilized by these studies (chow vs intraperitoneal injection), the method used to detect cytokine amounts (Bio-plex multiplex analysis vs Antibody cytokine arrays) and in the antibodies used to examine tau phosphorylation, thus the reasons for these discrepancies are not clear. However, it is likely that the distinct findings of these studies may likely be related to the elevated Aβ present in the 3 × Tg-AD line, which may induce distinct inflammatory responses and intracellular signaling pathways to those present in the htau mouse line. In support of this, recent work from Mastrangelo et al. (2009) report that administration of IFN-γ to 3 × Tg-AD mice results in an attenuation of tau phosphorylation, but increased Aβ pathology. Thus, it is possible that individual pro-inflammatory cytokines may be (i) differentially regulated by Aβ and tau, and (ii) exert opposing effects on these two important proteins.
In summary, we have demonstrated that minocycline treatment of htau mice reduces the activation of cortical astrocytes, and lowers the cortex amounts of several pro-inflammatory mediators implicated in AD. The results of this work have identified novel CNS cytokine targets of minocycline, including GM-CSF, M-CSF, I-309, MCP-5, and TREM-1. We have previously shown that minocycline treatment reduced the amount of aggregated tau in the cortex of young htau mice (Noble et al., 2009a), and it is possible that the data presented here may suggest that several, previously unreported cytokines, could be associated with the development of tau pathology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in the authors’ laboratories is supported by funding from the Medical Research Council, Alzheimer’s Society, and the Alzheimer’s Research Trust.
References
Akiyama, H., Arai, T., Kondo, H., Tanno, E., Haga, C., and Ikeda, K. (2000a). Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis. Assoc. Disord. 14(Suppl. 1), S47–S53.
Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G. M., Cooper, N. R., Eikelenboom, P., Emmerling, M., Fiebich, B. L., Finch, C. E., Frautschy, S., Griffin, W. S., Hampel, H., Hull, M., Landreth, G., Lue, L., Mrak, R., Mackenzie, I. R., McGeer, P. L., O’Banion, M. K., Pachter, J., Pasinetti, G., Plata-Salaman, C., Rogers, J., Rydel, R., Shen, Y., Streit, W., Strohmeyer, R., Tooyoma, I., Van Muiswinkel, F. L., Veerhuis, R., Walker, D., Webster, S., Wegrzyniak, B., Wenk, G., and Wyss-Coray, T. (2000b). Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421.
Andorfer, C., Acker, C. M., Kress, Y., Hof, P. R., Duff, K., and Davies, P. (2005). Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 25, 5446–5454.
Andorfer, C., Kress, Y., Espinoza, M., de Silva, R., Tucker, K. L., Barde, Y. A., Duff, K., and Davies, P. (2003). Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 86, 582–590.
Bauer, J., Strauss, S. Volk, B., and Berger, M. (1991). IL-6-mediated events in Alzheimer’s disease pathology. Immunol. Today 12, 422.
Bellucci, A., Westwood, A. J., Ingram, E., Casamenti, F., Goedert, M., and Spillantini, M. G. (2004). Induction of inflammatory mediators and microglial activation in mice transgenic for mutant human P301S tau protein. Am. J. Pathol. 165, 1643–1652.
Buée, L., Bussière, T., Buée-Scherrer, V., Delacourte, A., and Hof, P. R. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 33, 95–130.
Cagnin, A., Brooks, D. J., Kennedy, A. M., Gunn, R. N., Myers, R., Turkheimer, F. E., Jones, T., and Banati, R. B. (2001). In vivo measurement of activated microglia in dementia. Lancet 358, 461–467.
Cameron, B., and Landreth, G. E. (2010). Inflammation, microglia, and Alzheimer’s disease. Neurobiol. Dis. 37, 503–509.
Chen, M., Ona, V. O., Li, M., Ferrante, R. J., Fink, K. B., Zhu, S., Bian, J., Guo, L., Farrell, L. A., Hersch, S. M., Hobbs, W., Vonsattel, J. P., Cha, J. H., and Friedlander, R. M. (2000). Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 6, 797–801.
Choi, C., Jeong, J. H., Jang, J. S., Choi, K., Lee, J., Kwon, J., Choi, K. G., Lee, J. S., and Kang, S. W. (2008). Multiplex analysis of cytokines in the serum and cerebrospinal fluid of patients with Alzheimer’s disease by color-coded bead technology. J. Clin. Neurol. 4, 84–88.
Choi, Y., Kim, H. S., Shin, K. Y., Kim, E. M., Kim, M., Kim, H. S., Park, C. H., Jeong, Y. H., Yoo, J., Lee, J. P., Chang, K. A., Kim, S., and Suh, Y. H. (2007). Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer’s disease models. Neuropsychopharmacology. 32, 2393–2404.
Cuello, A. C., Ferretti, M. T., Leon, W. C., Iulita, M. F., Melis, T., Ducatenzeiler, A., Bruno, M. A., and Canneva, F. (2010). Early-stage inflammation and experimental therapy in transgenic models of the Alzheimer-like amyloid pathology. Neurodegener. Dis. 7, 96–98.
Dickson, D. W., Lee, S. C., Mattiace, L. A., Yen, S. H., and Brosnan, C. (1993). Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia 7, 75–83.
Du, Y., Ma, Z., Lin, S., Dodel, R. C., Gao, F., Bales, K. R., Triarhou, L. C., Chernet, E., Perry, K. W., Nelson, D. L., Luecke, S., Phebus, L. A., Bymaster, F. P., and Paul, S. M. (2001). Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 98, 14669–14674.
Duff, K., Knight, H., Refolo, L. M., Sanders, S., Yu, X., Picciano, M., Malester, B., Hutton, M., Adamson, J., Goedert, M., Burki, K., and Davies, P. (2000). Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol. Dis. 7, 87–98.
Familian, A., Boshuizen, R. S., Eikelenboom, P., and Veerhuis, R. (2006). Inhibitory effect of minocycline on amyloid beta fibril formation and human microglial activation. Glia 53, 233–240.
Galasko, D., and Montine, T. J. (2010). Biomarkers of oxidative damage and inflammation in Alzheimer’s disease. Biomark. Med. 4, 27–36.
Gallo, J. M., and Noble, W., and Martin, T. R. (2007). RNA and protein-dependent mechanisms in tauopathies: consequences for therapeutic strategies. Cell. Mol. Life Sci. 64, 1701–1714.
Giulian, D., and Ingeman, J. E. (1988). Colony-stimulating factors as promoters of ameboid microglia. J. Neurosci. 8, 4707–4717.
Griffin, W. S., Sheng, J. G., Roberts, G. W., and Mrak, R. E. (1995). Interleukin-1 expression in different plaque types in Alzheimer’s disease: significance in plaque evolution. J. Neuropathol. Exp. Neurol. 54, 276–281.
Hanger, D. P., Anderton, B. H., and Noble, W. (2009a). Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends. Mol. Med. 15, 112–119.
Hanger, D. P., Seereeram, A., and Noble, W. (2009b). Mediators of tau phosphorylation in the pathogenesis of Alzheimer’s disease. Expert Rev. Neurother. 9, 1647–1666.
Hu, W. T., Chen-Plotkin, A., Arnold, S. E., Grossman, M., Clark, C. M., Shaw, L. M., Pickering, E., Kuhn, M., Chen, Y., McCluskey, L., Elman, L., Karlawish, J., Hurtig, H. I., Siderowf, A., Lee, V. M., Soares, H., and Trojanowski, J. Q. (2010). Novel CSF biomarkers for Alzheimer’s disease and mild cognitive impairment. Acta Neuropathol. 119, 669–678.
Huell, M., Strauss, S., Volk, B., Berger, M., and Bauer, J. (1995). Interleukin-6 is present in early stages of plaque formation and is restricted to the brains of Alzheimer’s disease patients. Acta Neuropathol. 89, 544–551.
Ishizuka, K., Kimura, T., Igata-yi, R., Katsuragi, S., Takamatsu, J., and Miyakawa, T. (1997). Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer’s disease. Psychiatry Clin. Neurosci. 51, 135–138.
Jin, J. J., Kim, H. D., Maxwell, J. A., Li, L., and Fukichi, K. (2008). Toll-like receptor 4-dependent upregulation of cytokines in a transgenic model of Alzheimer’s disease. J. Neuroinflamm. 5, 23.
Kitazawa, M., Oddo, S., Yamasaki, T. R., Green, K. N., and Laferla, F. M. (2005). Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 25, 8843–8853.
Kriz, J., Nguyen, M. D., and Julien, J. P. (2002). Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 10, 268–278.
Laske, C., Stransky, E., Hoffmann, N., Maetzler, W., Straten, G., Eschweiler, G. W., and Leyhe, T. (2010). Macrophage colony-stimulating factor (M-CSF) in plasma and CSF of patients with mild cognitive impairment and Alzheimer’s disease. Curr. Alzheimer Res. 7, 409–414.
Lee, S. M., Yune, T. Y., Kim, S. J., Park, D. W., Lee, Y. K., Kim, Y. C., Oh, Y. J., Markelonis, G. J., and Oh, T. H. (2003). Minocycline reduces cell death and improves functional recovery after traumatic spinal cord injury in the rat. J. Neurotrauma 20, 1017–1027.
Mandrekar-Colucci, S., and Landreth, G. E. (2010). Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord Drug Targets 9, 156–167.
Mastrangelo, M. A., Sudol, K. L., Narrow, W. C., and Bowers, W. J. (2009). Interferon-{gamma} differentially affects Alzheimer’s disease pathologies and induces neurogenesis in triple transgenic-AD mice. Am. J. Pathol. 175, 2076–2088.
McGeer, P. L., Schulzer, M., and McGeer, E. G. (1996). Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 47, 425–432.
McKee, A. C., Carreras, I., Hossain, L., Ryu, H., Klein, W. L., Oddo, S., Laferla, F. M., Jenkins, B. G., Kowall, N. W., and Dedeoglu, A. (2008). Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res. 1207, 225–236.
Nair, A., Frederick, T. J., and Miller, S. D. (2008). Astrocytes in multiple sclerosis: a product of their environment. Cell. Mol. Life Sci. 65, 2702–2720.
Noble, W., Garwood, C. J., Stephenson, J., Kinsey, A. M., Hanger, D. P., and Anderton, B. H. (2009a). Minocycline reduces the development of abnormal tau species in models of Alzheimer’s disease. FASEB J. 23, 739–750.
Noble, W., Garwood, C. J., and Hanger, D. P. (2009b). Minocycline as a potential therapeutic agent in neurodegenerative disorders characterised by protein misfolding. Prion 3, 78–83.
Noble, W., Hanger, D. P., and Gallo, J. M. (2010). Transgenic mouse models of tauopathy in drug discovery. CNS Neurol. Disord. Drug Targets 9, 403–428.
Ojala, J., Alafuzoff, I., Herukka, S. K., van Groen, T., Tanila H., and Pirttila, T. (2009). Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol. Aging 30, 198–209.
Parachikova, A., Agadjanyan, M. G., Cribbs, D. H., Blurton-Jones, M., Perreau, V., Rogers, J., Beach, T. G., and Cotman, C. W. (2007). Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol. Aging 28, 1821–1833.
Parachikova, A., Visilevko, V., Cribbs, D. H., Laferla, F. M., and Green, K. N. (2010). Reductions in amyloid-beta-derived neuroinflammation, with minocycline, restore cognition but do not significantly affect tau hyperphosphorylation. J. Alzheimers Dis. 21, 527–542.
Polydoro, M., Acker, C. M., Duff, K., Castillo, P. E., and Davies, P. (2009). Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J. Neurosci. 29, 10741–10749.
Pontikis, C. C., Cella, C. V., Parihar, N., Lim, M. J., Chakrabarti, S., Mitchison, H. M., Mobley, W. C., Rezaie, P., Pearce, D. A., and Cooper, J. D. (2004). Late onset neurodegeneration in the Cln3−/− mouse model of juvenile neuronal ceroid lipofuscinosis is preceded by low level glial activation. Brain Res. 1023, 231–242.
Pontikis, C. C., Cotman, S. L., MacDonald, M. E., and Cooper, J. D. (2005). Thalamocortical neuron loss and localized astrocytosis in the Cln3Deltaex7/8 knock-in mouse model of Batten disease. Neurobiol. Dis. 20, 823–836.
Popovic, N., Schubart, A., Goetz, B. D., Zhang, S. C., Linington, C., and Duncan, I. D. (2002). Inhibition of autoimmune encephalomyelitis by a tetracycline. Ann. Neurol. 51, 215–223.
Rice, F. L., and Van der Loos, H. (1977). Development of the barrels and barrel field in the somatosensory cortex of the mouse. J. Comp. Neurol. 171, 545–560.
Ryu, J. K., and McLarnon, J. G. (2006). Minocycline or iNOS inhibition block 3-nitrotyrosine increases and blood–brain barrier leakiness in amyloid beta-peptide-injected rat hippocampus. Exp. Neurol. 198, 552–557. [Epub 2006 February 9].
Salmina, A. B. (2009). Neuron-glia interactions as therapeutic targets in neurodegeneration. J. Alzheimers Dis. 16, 485–502.
Sanchez Mejia, R. O., Ona, V. O., Li, M., and Friedlander, R. M. (2001). Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery 48, 1393–1399.
Schindowski, K., Bretteville, A., Leroy, K., Begard, S., Brion, J. P., Hamdane, M., and Buee, L. (2006). Alzheimer’s disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am. J. Pathol. 169, 599–616.
Schwab, C., Klegeris, A., and McGeer, P. L. (2009). Inflammation in transgenic mouse models of neurodegenerative disorders. Biochim. Biophys. Acta. 1802, 889–902.
Seabrook, T. J., Jiang, L., Maier, M., and Lemere, C. A. (2006). Minocycline affects microglia activation, Abeta deposition, and behavior in APP-tg mice. Glia 53, 776–782.
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35.
Sokolova, A., Hill, M. D., Rahimi, F., Warden, L. A., Halliday, G. M., and Shepherd, C. E. (2009). Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer’s disease. Brain Pathol. 19, 392–398.
Stirling, D. P., Khodarahmi, K., Liu, J., McPhail, L. T., McBride, C. B., Steeves, J. D., Ramer, M. S., and Tetzlaff, W. (2004). Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J. Neurosci. 24, 2182–2190.
Tarkowski, E., Wallin, A., Regland, B., Blennow, K., and Tarkowski, A. (2001). Local and systemic GM-CSF increase in Alzheimer’s disease and vascular dementia. Acta Neurol. Scand. 103, 166–174.
Teng, Y. D., Choi, H., Onario, R. C., Zhu, S., Desilets, F. C., Lan, S., Woodard, E. J., Snyder, E. Y., Eichler, M. E., and Friedlander, R. M. (2004). Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc. Natl. Acad. Sci. U.S.A. 101, 3071–3076.
Tucker, K. L., Meyer, M., and Barde, Y. A. (2001). Neurotrophins are required for nerve growth during development. Nat. Neurosci. 4, 29–37.
Van Eldik, L. J., and Griffin, W. S. (1994). S100 beta expression in Alzheimer’s disease: relation to neuropathology in brain regions. Biochim. Biophys. Acta 1223, 398–403.
Wang, X., Zhu, S., Drozda, M., Zhang, W., Stavrovskaya, I. G., Cattaneo, E., Ferrante, R. J., Kristal, B. S., and Friedlander, R. M. (2003). Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 100, 10483–10487.
Xia, M. Q., Qin, S. X., Wu, L. J., Mackay, C. R., and Hyman, B. T. (1998). Immunohistochemical study of the beta-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer’s disease brains. Am. J. Pathol. 153, 31–37.
Yoshiyama, Y., Higuchi, M., Zhang, B., Huang, S. M., Iwata, N., Saido, T. C., Maeda, J., Suhara, T., Trojanowski, J. Q., and Lee, V. M. (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351.
Zhang, W., Narayanan, M., and Friedlander, R. M. (2003). Additive neuroprotective effects of minocycline with creatine in a mouse model of ALS. Ann. Neurol. 53, 267–270.
Zhu, S., Stavrovskaya, I. G., Drozda, M., Kim, B. Y., Ona, V., Li, M., Sarang, S., Liu, A. S., Hartley, D. M., Wu, D. C., Gullans, S., Ferrante, R. J., Przedborski, S., Kristal, B. S., and Friedlander, R. M. (2002). Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 417, 74–78.
Keywords: tau, inflammation, minocycline, Alzheimer’s disease, cytokine, immunohistochemistry, array
Citation: Garwood CJ, Cooper JD, Hanger DP and Noble W (2010) Anti-inflammatory impact of minocycline in a mouse model of tauopathy. Front. Psychiatry 1:136. doi: 10.3389/fpsyt.2010.00136
Received: 25 June 2010;
Paper pending published: 13 July 2010;
Accepted: 06 September 2010;
Published online: 12 October 2010.
Edited by:
Selina Wray, University College London, UKReviewed by:
Luc Buee, Institut National de la Santé et de la Recherche Médicale, FranceJesus Avila, Centro de Biología Molecular Severo Ochoa Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid, Spain
Copyright: © 2010 Garwood, Cooper, Hanger and Noble. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Wendy Noble, Medical Research Council Centre for Neurodegeneration Research, Institute of Psychiatry, Department of Neuroscience, King’s College London, PO37, De Crespigny Park, London, UK. e-mail:d2VuZHkubm9ibGVAa2NsLmFjLnVr