

- Department of Molecular Neuroscience, University College London Institute of Neurology, London, UK
Parkinson’s disease (PD) represents a major challenge for health care systems around the world: it is the most common degenerative movement disorder of old age, affecting over 100,000 people in the UK alone (Schrag et al., 2000). Despite the remarkable success of treatments directed at potentiating or replacing dopamine within the brain, which can relieve symptoms for over a decade, PD remains an incurable and invariably fatal disorder. As such, efforts to understand the processes that lead to cell death in the brains of patients with PD are a priority for neurodegenerative researchers. A great deal of progress has been made in this regard by taking advantage of advances in genetics, initially by the identification of genes responsible for rare Mendelian forms of PD (outlined in Table 1), and more recently by applying genome wide association studies (GWAS) to the sporadic form of the disease (Hardy et al., 2009). Several such GWAS have now been carried out, with a meta-analysis currently under way. Using over 6000 cases and 10,000 controls, two of these studies have identified variation at a number of loci as being associated with an increased risk of disease (Satake et al., 2009; Simon-Sanchez et al., 2009). Three genes stand out as candidates from these studies – the SNCA gene, coding for α-synuclein, the LRRK2 gene, coding for leucine rich repeat kinase 2, and MAPT, coding for the microtubule-associated protein tau. Mutations at all three of these loci have been associated with Mendelian forms of disease presenting with the clinical syndrome of Parkinsonism, however only SNCA and LRRK2 have been previously associated with pathologically defined PD (Hardy et al., 2009). Point mutations in α-synuclein, along with gene multiplication events, result in autosomal dominant PD, often with a significant dementia component. In addition to this, α-synuclein is the principle component of the main pathological hallmark of idiopathic PD, the Lewy body, making it an unsurprising hit in the GWAS (Spillantini et al., 1997). Mutations in LRRK2 are the most common genetic cause of PD, and so again made this gene a likely candidate as a susceptibility locus for the sporadic form of disease (Kumari and Tan, 2009). More surprising, perhaps, was the identification of tau as a susceptibility factor for Parkinson’s. In this review we will outline the role of tau in neurodegeneration and in different forms of Parkinsonism, and speculate as to what the functional basis of the association between MAPT and PD might be.
Tau Protein and its Role in Neurodegeneration
The microtubule-associated protein tau is a predominantly neuronal protein whose major function is to bind to and stabilize microtubules. In the adult human CNS, tau exists as six protein isoforms which differ by the presence of either 0, 1, or 2 N-terminal inserts and either three (3R) or four (4R) microtubule binding repeats located at the C terminus of the protein (Goedert et al., 1989; Andreadis et al., 1992). Tau binds to and stabilizes microtubules via its C-terminal repeats in a process regulated by phosphorylation, where the phosphorylation of tau at specific residues favors its detachment from the microtubules (Lindwall and Cole, 1984).
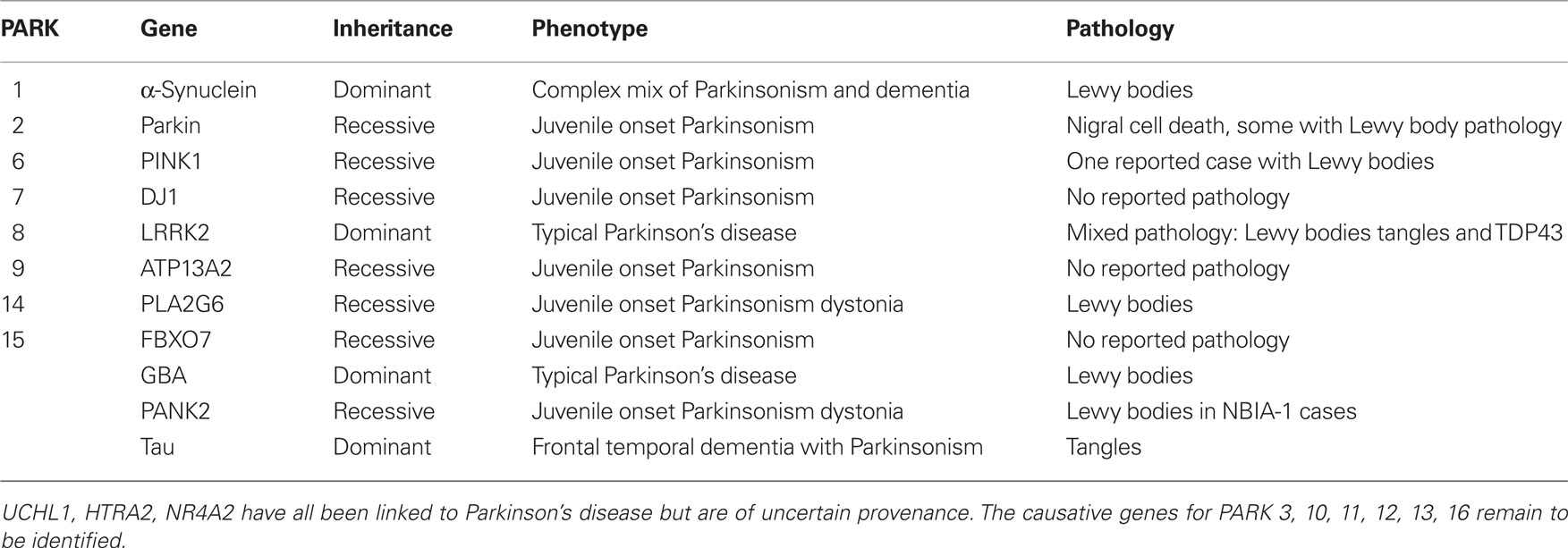
Table 1. Genes linked to autosomal forms of Parkinsonism.
In neurodegenerative disease, hyperphosphorylated, insoluble aggregates of tau are observed in neurons and glia of affected brain regions. Such disorders are collectively named the tauopathies, the most well known of which is Alzheimer’s disease (AD), where neurofibrillary tangles (NFTs) comprised of hyperphosphorylated, insoluble tau are one of the defining pathological features of the disease, alongside extracellular plaques composed of Aβ peptide (Grundke-Iqbal et al., 1986). The discovery of familial AD mutations that increase Aβ production led to the development of the amyloid cascade hypothesis, where tau pathology sits downstream of Aβ in the pathological cascade (Hardy and Allsop, 1991; Hardy and Higgins, 1992). For many years afterward, there was debate as to whether tau pathology was critical for disease pathogenesis, or was just a non-specific marker of disease. Several studies have shown that tau is necessary for Aβ-mediated toxicity, for example primary neuronal cultures from tau knockout mice are resistant to Aβ, and crossing APP transgenic mice with tau knockout mice resulted in a reduction of Aβ induced deficits (Rapoport et al., 2002; Roberson et al., 2007). The presence of NFTs correlates highly with the degree of dementia in AD suggesting a central role for tau in neuronal demise (Braak and Braak, 1991). This, combined with the discovery of mutations in the tau gene (MAPT) that cause frontotemporal dementia (discussed further below), have made understanding the links between tau and neuronal death an area of intense research.
Tau is subject to heavy post-translational modification, and hyperphosphorylation, truncation, and glycosylation have all been linked to disrupting tau function and promoting tau aggregation. This in turn leads to disruption in the microtubule network and impaired axonal transport, eventually causing synaptic and neuronal degeneration. It is beyond the scope of this review to discuss post-translational modifications to tau and their potential roles in neurodegeneration, but a comprehensive overview is given in (Iqbal et al., 2009).
Beyond AD, other tauopathies include progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17; Bird et al., 1999; Gasparini et al., 2007). In addition to being characterized by tau deposition, mutations, or common variation within MAPT has been linked to these disorders and is discussed further below.
The Genetics of MAPT in Neurodegeneration
The confirmation that tau dysfunction is sufficient to cause neurodegeneration came in 1998 with the discovery of mutations in MAPT that cause frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17; Hutton et al., 1998; Poorkaj et al., 1998). More than 40 tau mutations have been identified to date and these can be split into two groups: coding mutations that affect the microtubule binding activity of tau and/or its propensity to aggregate, and mutations that alter the splicing of exon 10 of MAPT (Hasegawa et al., 1998; Jiang et al., 2003). Exon 10 codes for the fourth microtubule binding repeat seen in 4R tau isoforms, and the majority of splice mutations increase exon 10 inclusion, thereby leading to an increase in 4R tau levels. In the adult human CNS the ratio of 3R:4R tau is approximately one, which appears to be critical for proper neuronal function since mutations that disrupt this ratio are sufficient to cause disease. A disruption in tau splicing is also seen in the sporadic disorders PSP and CBD, which are characterized pathologically by selective deposition of 4R tau isoforms.
As mutations in MAPT cause autosomal dominant disease, examining how common variation at the MAPT locus can predispose to sporadic tauopathy has been a major focus of research. The genetic architecture of the MAPT locus is complex. The tau gene is located within the largest known block of linkage disequilibrium in the human genome, spanning 1.8 Mb, where the two major haplotypes are termed H1 and H2 (Stefansson et al., 2005). Homozygosity for the H1 haplotype has been shown to be strongly associated with increased risk of developing PSP and CBD in several populations (Conrad et al., 1997; Baker et al., 1999; Houlden et al., 2001; Pittman et al., 2004, 2006). Further dissection of the MAPT locus has revealed H2 to be largely invariant, whereas H1 can be divided into the subhaplotypes H1a, H1b, and H1c (Pittman et al., 2004, 2006). Of these, it is the H1c haplotype which exhibits the strongest association with PSP and CBD (Pittman et al., 2004, 2006).
The strongest association of the H1 haplotype with PSP and CBD has been mapped to the promoter region of MAPT (Myers et al., 2007), and as both the H1 and H2 haplotypes produce tau protein with the same coding sequence, it is logical to hypothesize that they may influence disease risk by altering expression levels or splicing of tau. This concept, where increased levels of normal sequence proteins are sufficient to cause neurodegeneration, has been described previously, for example duplications and triplications of SNCA cause PD, and AD-type pathology is seen in Down’s syndrome where triplication of the APP gene occurs (Hardy, 2005). Tau levels, in particular the 4R isoforms, were found to be higher in post-mortem tissue from H1 homozygotes than H1/H2 heterozygotes and H2 homozygotes (Myers et al., 2007). In vitro reporter assays have shown the H1 promoter region to exhibit increased transcriptional activity when compared to H2 and allele-specific expression assays in H1/H2 heterozygote neuroblastoma have shown that 4R tau isoform production is highest from the H1 allele (Caffrey et al., 2006). These experiments provide a functional basis for the association of the MAPT H1 haplotype with disease.
Tau in Parkinsonism
In addition to the mutations in tau linked to FTD, there are a number of examples of disorders where tau deposition is linked to a movement disorder phenotype. It is important to note that the clinical syndrome of parkinsonism is distinct from PD, which refers to the pathologically defined disorder, and also that there are many other causes of parkinsonism in addition to those associated with tau (Hughes et al., 1992). There are two historical examples of tau being involved in epidemic Parkinsonism: post-encephalitic Parkinsonism and the Parkinsonism/dementia complex (PDC) of Guam (Gibbs and Gajdusek, 1972; Wong et al., 1996). The former is Parkinsonism found subsequent to cases of von Economos encephalitis, which has a temporal if not explicitly causative link to the 1918 Influenza epidemic (Casals et al., 1998). The disease was characterized by a state of akinetic mutism that proved to be responsive to l-DOPA (immortalized by Oliver Sachs in his novel Awakenings). Crucially, patients with post-encephalitic Parkinsonism have extensive tangle pathology in their brains, although the connection between this pathology and von Economo’s encephalitis is unclear. Tau is a prominent pathological component of the PDC of Guam, part of a group of tangle disorders with differing phenotypes that were, for a period, a leading cause of death amongst the Chamorros people of Guam (Plato et al., 2003). The causative insult of the tangle disorders found amongst the Chamorros is unknown, but again there is a link between a syndrome involving Parkinsonism and tau pathology.
A syndrome with a Parkinsonism component where the causative insult has been identified is dementia pugilistica, a disorder presented by patients who have been the recipient of repeated head trauma - most famously exemplified by Muhammed Ali, the noted pugilist. Again, this is a disorder with a complex phenotype, combining marked cognitive decline with a movement disorder component (Roberts et al., 1990; Schmidt et al., 2001). It is also notable that head trauma is a significant risk factor for AD, suggesting a causative link between head trauma and tauopathies (Graves et al., 1990). The insight that these links yields, however, is limited by the fact that we still do not know whether the tau accumulation is deleterious or protective to the damaged cells in these cases.
Tau in Parkinson’s Disease
So far, we have discussed the links between tau and the clinical presentation of parkinsonism, rather than PD, which must be pathologically defined by the presence of Lewy bodies containing α-synuclein. However, the association of tau mutations with the clinical syndrome of Parkinsonism in FTDP-17 prompted several groups to analyze common variation at the MAPT locus as a risk factor for PD. An association between homozygosity of the H1 haplotype and increased PD risk has been described by several groups, although this was contradicted by other studies (Farrer et al., 2002; Kwok et al., 2004; Skipper et al., 2004; Vandrovcova et al., 2009). However, the recent GWAS study identifying MAPT as a risk factor for PD confirm the candidate gene association studies using an unbiased experimental approach (Simon-Sanchez et al., 2009). A separate GWAS in a Japanese cohort confirmed the associations of SNCA and LRRK2, but failed to identify variation at MAPT as a risk factor for PD (Satake et al., 2009). The H2 haplotype is Caucasian in origin and is absent from Japanese populations (Evans et al., 2004), suggesting that the lack of association in the Japanese cohort is due to population heterogeneity at the MAPT locus, rather than lack of reproducibility. The lack of tau pathology in idiopathic PD makes the genetic link puzzling, especially as MAPT has not been identified as a risk factor in GWAS of AD, where NFT pathology is extensive (Lambert et al., 2009). Why should variation in tau be a risk factor for a disease where tau pathology is not consistently observed, but not influence disease risk where there is tau deposition is prevalent?
Explaining the Link vetween MAPT and PD
Although there are many intriguing links between the clinical presentation of Parkinsonism and tau, how relevant these are to PD is unclear, and understanding the functional basis for the genetic link between tau and pathologically defined PD will be a key area of research for the future.
From a genetic perspective, fine mapping of the PD risk haplotype has shown that, although the subhaplotype associated with PD it is part of the H1 clade, it is a novel haplotype that is associated with PD and not the same that is associated with PSP and CBD (Tobin et al., 2008). However, increased 4R tau has been detected in PD brain suggesting that the common mechanism of increased expression underlies the association of MAPT with PD, PSP, and CBD (Tobin et al., 2008).
As variation at MAPT influences PD risk, where tangles are not consistently found, but has only a moderate influence on AD risk, where NFTs are a defining pathological feature (Pittman et al., 2006), one could hypothesize that it is some pre-tangle, toxic oligomer species that is responsible for tau-mediated neurodegeneration. It is possible that NFTs are a marker of damage already done rather than the primary effector of neurodegeneration. Several lines of evidence have recently emerged to support this idea. In AD, tau pathology correlates strongly with degree of dementia and the extent of neuronal loss, however there is evidence to suggest some neurons die without tangle formation (Gomez-Isla et al., 1997). Additionally, some FTDP-17 patients with pathogenic mutations in MAPT have relatively few tangles at post-mortem despite widespread neuronal loss (Bird et al., 1999). Neurodegeneration occurs in a Drosophila model of tauopathy without NFT formation, although this was more pronounced in flies expressing R406W mutant tau than flies expressing wild-type tau (Wittmann et al., 2001). In a transgenic mouse model overexpressing non-mutant, human tau, the presence of tau filaments did not correlate with cell death (Andorfer et al., 2005). Inducible transgenic mice expressing the P301L FTDP-17 tau mutation continue to accumulate tangles when transgene expression is switched off, despite a recovery in cognitive function and stabilization of neuronal numbers, providing compelling evidence for the dissociation between NFT formation and neurotoxicity (Santacruz et al., 2005). Further work in this model identified several tau multimeric species, which were better correlated with neuronal loss and behavioral decline than tau tangles (Berger et al., 2007). Equivalent species were isolated from human FTD post-mortem brain tissue, suggestive of the existence of a toxic tau oligomer (Berger et al., 2007). These data point to the existence of a toxic tau species that is yet to be identified. These toxic species may not be readily detectable, and would constitute an “invisible assassin,” Macavity like character in PD, hitting neurons in the substantia nigra but leaving no pawmarks (Eliot, 1939).
Interactions between MAPT, Snca, and LRRK2
Further insight into the role of tau as a risk factor in PD can be gained from understanding how MAPT interacts with the two other major genetic influences on PD risk: SNCA and LRRK2. The known interactions between these three proteins are summarized below and in Figure 1.
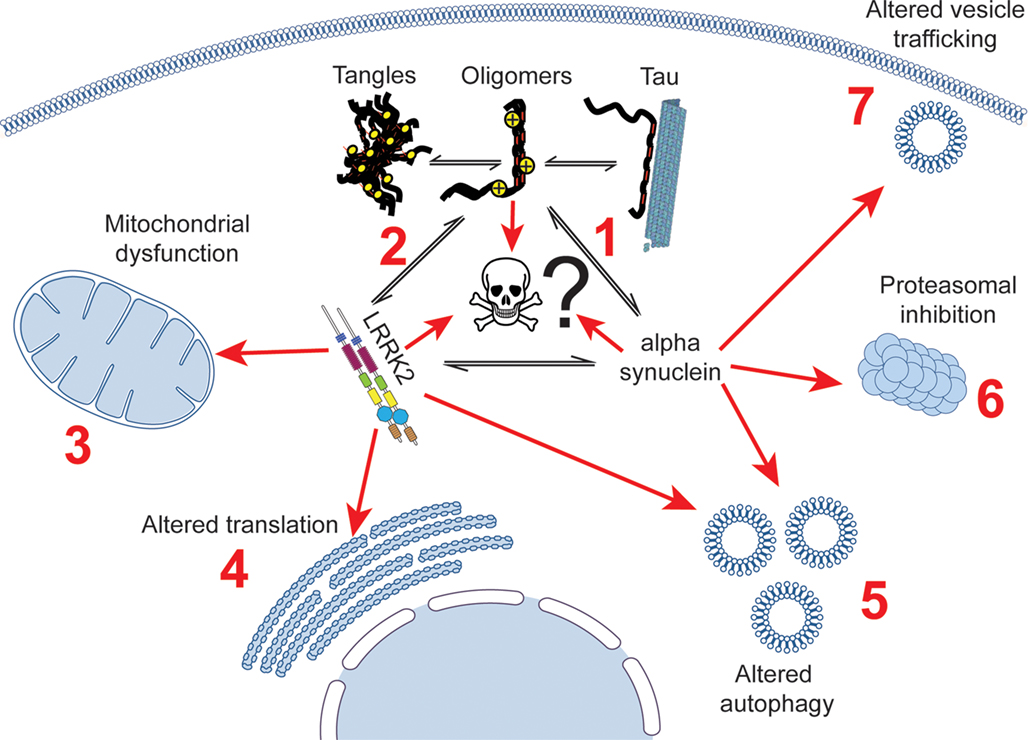
Figure 1. Potential interactions linking tau to cell death in Parkinson’s disease patients, showing established links between tau, LRRK2, α-synuclein and cellular dysfunction. Tau has been shown to alter the aggregation of α-synuclein (1), and has been described as being aggregated in mouse models of LRRK2 disease (2). A putative cellular interaction between tau, α-synuclein and lrrk2 could impact on a large number of cellular processes that all three proteins have been suggested to be involved in, including mitochondrial dysfunction (3), impaired translation (4), altered autophagy (5), proteasomal dysfunction (6), and altered vesicle trafficking (7).
Tau and α-synuclein are both found as insoluble aggregates in the tauopathies and synucleinopathies respectively. Several studies have demonstrated the co-occurrence of tau and α-synuclein pathologies in the same brain at post-mortem. A high proportion of AD cases display synuclein positive Lewy bodies in addition to plaques and tangles, and α-synuclein itself was originally isolated as the None Amyloid Component of Plaques or NACP (Ueda et al., 1993). Lewy bodies were detected in the amygdala of 60% of familial AD cases as well as 50% of Down’s syndrome cases who had concomitant AD (Lippa et al., 1998, 1999). Subsequently, it has been shown that the majority of sporadic AD cases also display some Lewy body pathology (Hamilton, 2000). A family with early-onset dementia characterized pathologically by the widespread appearance of both Lewy Bodies and NFTs, but not amyloid deposits has also been described (Clarimon et al., 2009). Additionally, although the α-synuclein positive Lewy Body is the pathological hallmark of PD, there have been several reports describing the presence of tau tangles in PD brain at post-mortem and tangle pathology in PD is observed at a higher frequency than in controls (Joachim et al., 1987; Bancher et al., 1993). One study found tangles in the SNPc of 78% of PD patients, and the presence of NFTs was correlated with gait impairment, but not other symptoms (Schneider et al., 2006). This is in contrast to AD, where tangles are found in mainly cortical regions and not in the SNPc (Braak and Braak, 1991). In PSP and CBD, tangle pathology is most predominant in the brainstem, and double immunofluorescence has shown that tau and synuclein epitopes colocalize in brainstem Lewy bodies of PD, although tau immunoreactivity was only observed in a minority of LB (Arima et al., 1999). Finally, post-mortem examination of an individual with the rare A53T mutation in α-synuclein revealed widespread tau pathology in addition to α-synuclein pathology (Duda et al., 2002). The co-occurrence of α-synuclein and tau pathology is higher than would be expected from chance alone; suggestive of a functional relationship between these two proteins and their deposition.
In PD, variation at the SNCA locus has been shown to act synergistically with variation at MAPT to increase disease risk, i.e., the increase in risk when carrying both risk variants is greater than the additive risk of carrying each variant in isolation (Goris et al., 2007). In vitro work has shown that α-synuclein and tau are able to influence each other’s polymerization, and diffuse tau pathology is observed in transgenic mice expressing the α-synuclein A53T mutation (Giasson et al., 2003). Furthermore, transgenic mice expressing the A30P α-synuclein mutation display increased tau phosphorylation, although interestingly this is not detected in the same neurons where synuclein pathology is found (Frasier et al., 2005). A recent report crossed a triple-transgenic mouse model of AD (expressing mutations in APP, PSEN1, and MAPT) with a SNCA transgenic, and demonstrated that the resultant mice exhibited more rapid cognitive decline than the triple transgenics alone (Clinton et al., 2010). As previously observed by Frasier and colleagues, tau and α-synuclein pathology did not colocalize in the same neurons. Intriguingly, in the synuclein-only transgenics the majority of pathology is observed in the brainstem, but in the combined transgenics the α-synuclein pathology was predominantly cortical (Clinton et al., 2010). This suggests an extremely tight relationship between tau and α-synuclein levels that is not understood at present. Hyperphosphorylation of tau is a common feature in all of the tauopathies, and α-synuclein has been shown to mediate tau phosphorylation at the pathological epitope S396, and increased phosphorylation at this site is observed in PD (Muntane et al., 2008; Duka et al., 2009).
Another intriguing genetic link between Parkinsonism and tau is provided by one of the other genes highlighted by the GWA studies: LRRK2. The autosomal dominant form of PD linked to mutations in LRRK2 is characterized by pleomorphic pathology, including Lewy bodies, Lewy neurites and, significantly, tau tangles (Zimprich et al., 2004). There does not seem to be a direct genotype/pathology correlation between specific mutations in LRRK2 and deposition of a specific type of pathology, indeed patients within the same kindred possessing the same mutation can present with different pathologies (Cookson et al., 2008). Evidence for a functional link between LRRK2 and tau has been forthcoming from several model systems. Tau positive axonal swellings have been observed in rat neuronal cultures overexpressing a fragment of LRRK2 and a recent mouse model of LRRK2 PD expressing human LRRK2 containing the R1441G mutation displayed extensive tangle pathology (MacLeod et al., 2006).
As tau is hyperphosphorylated in neurodegeneration, the obvious hypothesis is that tau could be a substrate for LRRK2. However, to date, the direct phosphorylation of tau by LRRK2 has not been reported. This does not rule out a functional link between the two proteins: LRRK2 may influence tau phosphorylation by modulating the activity of other major tau kinases, such as GSK3beta and cdk5. Were this to be the case, it may be that even in cases without explicit tau pathology there may be altered phosphorylation of tau. It will be interesting to see if levels of tau pathology are enhanced in tau and LRRK2 double transgenics. It will also be interesting to understand if MAPT risk haplotypes are able to act synergistically with LRRK2 variation to influence disease risk.
Summary
Recent GWAS have provided compelling evidence that variation within MAPT is a major risk factor for the development of sporadic PD. The lack of tau pathology in sporadic PD makes this link puzzling at first glance: however, pathological and functional studies have provided us with some hints as to how MAPT may interact with the other PD risk loci, SNCA, and LRRK2, to enhance PD risk. In the light of research into Alzheimer’s, where a role for tau has been explored for several decades and yet we still do not understand the link between the pathology that we observe in the post-mortem brain and the underlying process that leads to cell death, it is perhaps naive to expect that we should immediately comprehend how tau is impacting on the disease process in PD. That said, a thorough understanding of the links between MAPT, SNCA, and LRRK2 will be crucial to understand the etiology of PD, which in turn is critical for the development of novel therapies to help those who are afflicted by this disorder. The genetic dissection of PD continues apace: we now need to develop the tools and approaches to dissect the cell biology of this disease, and the highlighting of tau in this process can only help us.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank the Alzheimer’s Research Trust, the Brain Research Trust, Parkinson’s UK, and the Michael J. Fox Foundation for their generous support.
References
Andorfer, C., Acker, C. M., Kress, Y., Hof, P. R., Duff, K., and Davies, P. (2005). Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 25, 5446–5454.
Andreadis, A., Brown, W. M., and Kosik, K. S. (1992). Structure and novel exons of the human tau gene. Biochemistry 31, 10626–10633.
Arima, K., Hirai, S., Sunohara, N., Aoto, K., Izumiyama, Y., Ueda, K., Ikeda, K., and Kawai, M. (1999). Cellular co-localization of phosphorylated tau- and NACP/alpha-synuclein-epitopes in Lewy bodies in sporadic Parkinson’s disease and in dementia with Lewy bodies. Brain Res. 843, 53–61.
Baker, M., Litvan, I., Houlden, H., Adamson, J., Dickson, D., Perez-Tur, J., Hardy, J., Lynch, T., Bigio, E., and Hutton, M. (1999). Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum. Mol. Genet. 8, 711–715.
Bancher, C., Braak, H., Fischer, P., and Jellinger, K. A. (1993). Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer’s and Parkinson’s disease patients. Neurosci. Lett. 162, 179–182.
Berger, Z., Roder, H., Hanna, A., Carlson, A., Rangachari, V., Yue, M., Wszolek, Z., Ashe, K., Knight, J., Dickson, D., Andorfer, C., Rosenberry, T. L., Lewis, J., Hutton, M., and Janus, C. (2007). Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J. Neurosci. 27, 3650–3662.
Bird, T. D., Nochlin, D., Poorkaj, P., Cherrier, M., Kaye, J., Payami, H., Peskind, E., Lampe, T. H., Nemens, E., Boyer, P. J., and Schellenberg, G. D. (1999). A clinical pathological comparison of three families with frontotemporal dementia and identical mutations in the tau gene (P301L). Brain 122(Pt 4), 741–756.
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259.
Caffrey, T. M., Joachim, C., Paracchini, S., Esiri, M. M., and Wade-Martins, R. (2006). Haplotype-specific expression of exon 10 at the human MAPT locus. Hum. Mol. Genet. 15, 3529–3537.
Casals, J., Elizan, T. S., and Yahr, M. D. (1998). Postencephalitic parkinsonism–a review. J. Neural Transm. 105, 645–676.
Clarimon, J., Molina-Porcel, L., Gomez-Isla, T., Blesa, R., Guardia-Laguarta, C., Gonzalez-Neira, A., Estorch, M., Ma, G. J., Barraquer, L., Roig, C., Ferrer, I., and Lleo, A. (2009). Early-onset familial Lewy body dementia with extensive tauopathy: a clinical, genetic, and neuropathological study. J. Neuropathol. Exp. Neurol. 68, 73–82.
Clinton, L. K., Blurton-Jones, M., Myczek, K., Trojanowski, J. Q., and LaFerla, F. M. (2010). Synergistic interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J. Neurosci. 30, 7281–7289.
Conrad, C., Andreadis, A., Trojanowski, J. Q., Dickson, D. W., Kang, D., Chen, X., Wiederholt, W., Hansen, L., Masliah, E., Thal, L. J., Katzman, R., Xia, Y., and Saitoh, T. (1997). Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann. Neurol. 41, 277–281.
Cookson, M. R., Hardy, J., and Lewis, P. A. (2008). Genetic neuropathology of Parkinson’s disease. Int. J. Clin. Exp. Pathol. 1, 217–231.
Duda, J. E., Giasson, B. I., Mabon, M. E., Miller, D. C., Golbe, L. I., Lee, V. M., and Trojanowski, J. Q. (2002). Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol. 104, 7–11.
Duka, T., Duka, V., Joyce, J. N., and Sidhu, A. (2009). Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J. 23, 2820–2830.
Evans, W., Fung, H. C., Steele, J., Eerola, J., Tienari, P., Pittman, A., Silva, R., Myers, A., Vrieze, F. W., Singleton, A., and Hardy, J. (2004). The tau H2 haplotype is almost exclusively Caucasian in origin. Neurosci. Lett. 369, 183–185.
Farrer, M., Skipper, L., Berg, M., Bisceglio, G., Hanson, M., Hardy, J., Adam, A., Gwinn-Hardy, K., and Aasly, J. (2002). The tau H1 haplotype is associated with Parkinson’s disease in the Norwegian population. Neurosci. Lett. 322, 83–86.
Frasier, M., Walzer, M., McCarthy, L., Magnuson, D., Lee, J. M., Haas, C., Kahle, P., and Wolozin, B. (2005). Tau phosphorylation increases in symptomatic mice overexpressing A30P alpha-synuclein. Exp. Neurol. 192, 274–287.
Gasparini, L., Terni, B., and Spillantini, M. G. (2007). Frontotemporal dementia with tau pathology. Neurodegener. Dis. 4, 236–253.
Giasson, B. I., Forman, M. S., Higuchi, M., Golbe, L. I., Graves, C. L., Kotzbauer, P. T., Trojanowski, J. Q., and Lee, V. M. (2003). Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300, 636–640.
Gibbs, C. J. Jr., and Gajdusek, D. C. (1972). Amyotrophic lateral sclerosis, Parkinson’s disease, and the amyotrophic lateral sclerosis-Parkinsonism dementia complex on Guam: a review and summary of attempts to demonstrate infection as the aetiology. J. Clin. Pathol. Suppl. (R. Coll. Pathol.) 6, 132–140.
Goedert, M., Spillantini, M. G., Potier, M. C., Ulrich, J., and Crowther, R. A. (1989). Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 8, 393–399.
Gomez-Isla, T., Hollister, R., West, H., Mui, S., Growdon, J. H., Petersen, R. C., Parisi, J. E., and Hyman, B. T. (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 41, 17–24.
Goris, A., Williams-Gray, C. H., Clark, G. R., Foltynie, T., Lewis, S. J., Brown, J., Ban, M., Spillantini, M. G., Compston, A., Burn, D. J., Chinnery, P. F., Barker, R. A., and Sawcer, S. J. (2007). Tau and alpha-synuclein in susceptibility to, and dementia in, Parkinson’s disease. Ann. Neurol. 62, 145–153.
Graves, A. B., White, E., Koepsell, T. D., Reifler, B. V., van, B. G., Larson, E. B., and Raskind, M. (1990). The association between head trauma and Alzheimer’s disease. Am. J. Epidemiol. 131, 491–501.
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917.
Hamilton, R. L. (2000). Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 10, 378–384.
Hardy, J. (2005). Expression of normal sequence pathogenic proteins for neurodegenerative disease contributes to disease risk: “permissive templating” as a general mechanism underlying neurodegeneration. Biochem. Soc. Trans. 33, 578–581.
Hardy, J., and Allsop, D. (1991). Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 12, 383–388.
Hardy, J., Lewis, P., Revesz, T., Lees, A., and Paisan-Ruiz, C. (2009). The genetics of Parkinson’s syndromes: a critical review. Curr. Opin. Genet. Dev. 19, 254–265.
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185.
Hasegawa, M., Smith, M. J., and Goedert, M. (1998). Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett. 437, 207–210.
Houlden, H., Baker, M., Morris, H. R., MacDonald, N., Pickering-Brown, S., Adamson, J., Lees, A. J., Rossor, M. N., Quinn, N. P., Kertesz, A., Khan, M. N., Hardy, J., Lantos, P. L., St George-Hyslop, P., Munoz, D. G., Mann, D., Lang, A. E., Bergeron, C., Bigio, E. H., Litvan, I., Bhatia, K. P., Dickson, D., Wood, N. W., and Hutton, M. (2001). Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology 56, 1702–1706.
Hughes, A. J., Daniel, S. E., Kilford, L., and Lees, A. J. (1992). Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatr. 55, 181–184.
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., Pickering-Brown, S., Chakraverty, S., Isaacs, A., Grover, A., Hackett, J., Adamson, J., Lincoln, S., Dickson, D., Davies, P., Petersen, R. C., Stevens, M., de, G. E., Wauters, E., van, B. J., Hillebrand, M., Joosse, M., Kwon, J. M., Nowotny, P., Che, L. K., Norton, J., Morris, J. C., Reed, L. A., Trojanowski, J., Basun, H., Lannfelt, L., Neystat, M., Fahn, S., Dark, F., Tannenberg, T., Dodd, P. R., Hayward, N., Kwok, J. B., Schofield, P. R., Andreadis, A., Snowden, J., Craufurd, D., Neary, D., Owen, F., Oostra, B. A., Hardy, J., Goate, A., van, S. J., Mann, D., Lynch, T., and Heutink, P. (1998). Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705.
Iqbal, K., Liu, F., Gong, C. X., Alonso, A. C., and Grundke-Iqbal, I. (2009). Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 118, 53–69.
Jiang, Z., Tang, H., Havlioglu, N., Zhang, X., Stamm, S., Yan, R., and Wu, J. Y. (2003). Mutations in tau gene exon 10 associated with FTDP-17 alter the activity of an exonic splicing enhancer to interact with Tra2 beta. J. Biol. Chem. 278, 18997–19007.
Joachim, C. L., Morris, J. H., Kosik, K. S., and Selkoe, D. J. (1987). Tau antisera recognize neurofibrillary tangles in a range of neurodegenerative disorders. Ann. Neurol. 22, 514–520.
Kumari, U., and Tan, E. K. (2009). LRRK2 in Parkinson’s disease: genetic and clinical studies from patients. FEBS J. 276, 6455–6463.
Kwok, J. B., Teber, E. T., Loy, C., Hallupp, M., Nicholson, G., Mellick, G. D., Buchanan, D. D., Silburn, P. A., and Schofield, P. R. (2004). Tau haplotypes regulate transcription and are associated with Parkinson’s disease. Ann. Neurol. 55, 329–334.
Lambert, J. C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., Combarros, O., Zelenika, D., Bullido, M. J., Tavernier, B., Letenneur, L., Bettens, K., Berr, C., Pasquier, F., Fievet, N., Barberger-Gateau, P., Engelborghs, S., De, D. P., Mateo, I., Franck, A., Helisalmi, S., Porcellini, E., Hanon, O., de Pancorbo, M. M., Lendon, C., Dufouil, C., Jaillard, C., Leveillard, T., Alvarez, V., Bosco, P., Mancuso, M., Panza, F., Nacmias, B., Bossu, P., Piccardi, P., Annoni, G., Seripa, D., Galimberti, D., Hannequin, D., Licastro, F., Soininen, H., Ritchie, K., Blanche, H., Dartigues, J. F., Tzourio, C., Gut, I., Van, B. C., Alperovitch, A., Lathrop, M., and Amouyel, P. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1099.
Lindwall, G., and Cole, R. D. (1984). Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 259, 5301–5305.
Lippa, C. F., Fujiwara, H., Mann, D. M., Giasson, B., Baba, M., Schmidt, M. L., Nee, L. E., O’Connell, B., Pollen, D. A., St George-Hyslop, P., Ghetti, B., Nochlin, D., Bird, T. D., Cairns, N. J., Lee, V. M., Iwatsubo, T., and Trojanowski, J. Q. (1998). Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am. J. Pathol. 153, 1365–1370.
Lippa, C. F., Schmidt, M. L., Lee, V. M., and Trojanowski, J. Q. (1999). Antibodies to alpha-synuclein detect Lewy bodies in many Down’s syndrome brains with Alzheimer’s disease. Ann. Neurol. 45, 353–357.
MacLeod, D., Dowman, J., Hammond, R., Leete, T., Inoue, K., and Abeliovich, A. (2006). The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52, 587–593.
Muntane, G., Dalfo, E., Martinez, A., and Ferrer, I. (2008). Phosphorylation of tau and alpha-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer’s disease, and in Parkinson’s disease and related alpha-synucleinopathies. Neuroscience 152, 913–923.
Myers, A. J., Pittman, A. M., Zhao, A. S., Rohrer, K., Kaleem, M., Marlowe, L., Lees, A., Leung, D., McKeith, I. G., Perry, R. H., Morris, C. M., Trojanowski, J. Q., Clark, C., Karlawish, J., Arnold, S., Forman, M. S., Van, D., de, V. S. R., and Hardy, J. (2007). The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol. Dis. 25, 561–570.
Pittman, A. M., Fung, H. C., and de, S. R. (2006). Untangling the tau gene association with neurodegenerative disorders. Hum. Mol. Genet. 15, R188–R195.
Pittman, A. M., Myers, A. J., Duckworth, J., Bryden, L., Hanson, M., Abou-Sleiman, P., Wood, N. W., Hardy, J., Lees, A., and de, S. R. (2004). The structure of the tau haplotype in controls and in progressive supranuclear palsy. Hum. Mol. Genet. 13, 1267–1274.
Plato, C. C., Garruto, R. M., Galasko, D., Craig, U. K., Plato, M., Gamst, A., Torres, J. M., and Wiederholt, W. (2003). Amyotrophic lateral sclerosis and parkinsonism-dementia complex of Guam: changing incidence rates during the past 60 years. Am. J. Epidemiol. 157, 149–157.
Poorkaj, P., Bird, T. D., Wijsman, E., Nemens, E., Garruto, R. M., Anderson, L., Andreadis, A., Wiederholt, W. C., Raskind, M., and Schellenberg, G. D. (1998). Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 43, 815–825.
Rapoport, M., Dawson, H. N., Binder, L. I., Vitek, M. P., and Ferreira, A. (2002). Tau is essential to beta -amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 99, 6364–6369.
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., Gerstein, H., Yu, G. Q., and Mucke, L. (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754.
Roberts, G. W., Allsop, D., and Bruton, C. (1990). The occult aftermath of boxing. J. Neurol. Neurosurg. Psychiatr. 53, 373–378.
Santacruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., Guimaraes, A., DeTure, M., Ramsden, M., McGowan, E., Forster, C., Yue, M., Orne, J., Janus, C., Mariash, A., Kuskowski, M., Hyman, B., Hutton, M., and Ashe, K. H. (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481.
Satake, W., Nakabayashi, Y. Mizuta, I., Hirota, Y., Ito, C., Kubo, M., Kawaguchi, T., Tsunoda, T., Watanabe, M., Takeda, A., Tomiyama, H., Nakashima, K., Hasegawa, K., Obata, F., Yoshikawa, T., Kawakami, H., Sakoda, S., Yamamoto, M., Hattori, N., Murata, M., Nakamura, Y., and Toda, T. (2009). Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 41, 1303–1307.
Schmidt, M. L., Zhukareva, V., Newell, K. L., Lee, V. M., and Trojanowski, J. Q. (2001). Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer’s disease. Acta Neuropathol. 101, 518–524.
Schneider, J. A., Li, J. L., Li, Y., Wilson, R. S., Kordower, J. H., and Bennett, D. A. (2006). Substantia nigra tangles are related to gait impairment in older persons. Ann. Neurol. 59, 166–173.
Schrag, A., Ben-Shlomo, Y., and Quinn, N. P. (2000). Cross sectional prevalence survey of idiopathic Parkinson’s disease and Parkinsonism in London. BMJ 321, 21–22.
Simon-Sanchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., Paisan-Ruiz, C., Lichtner, P., Scholz, S. W., Hernandez, D. G., Kruger, R., Federoff, M., Klein, C., Goate, A., Perlmutter, J., Bonin, M., Nalls, M. A., Illig, T., Gieger, C., Houlden, H., Steffens, M., Okun, M. S., Racette, B. A., Cookson, M. R., Foote, K. D., Fernandez, H. H., Traynor, B. J., Schreiber, S., Arepalli, S., Zonozi, R., Gwinn, K., van der Brug, M., Lopez, G., Chanock, S. J., Schatzkin, A., Park, Y., Hollenbeck, A., Gao, J., Huang, X., Wood, N. W., Lorenz, D., Deuschl, G., Chen, H., Riess, O., Hardy, J. A., Singleton, A. B., and Gasser, T. (2009). Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 41, 1308–1312.
Skipper, L., Wilkes, K., Toft, M., Baker, M., Lincoln, S., Hulihan, M., Ross, O. A., Hutton, M., Aasly, J., and Farrer, M. (2004). Linkage disequilibrium and association of MAPT H1 in Parkinson disease. Am. J. Hum. Genet. 75, 669–677.
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840.
Stefansson, H., Helgason, A., Thorleifsson, G., Steinthorsdottir, V., Masson, G., Barnard, J., Baker, A., Jonasdottir, A., Ingason, A., Gudnadottir, V. G., Desnica, N., Hicks, A., Gylfason, A., Gudbjartsson, D. F., Jonsdottir, G. M., Sainz, J., Agnarsson, K., Birgisdottir, B., Ghosh, S., Olafsdottir, A., Cazier, J. B., Kristjansson, K., Frigge, M. L., Thorgeirsson, T. E., Gulcher, J. R., Kong, A., and Stefansson, K. (2005). A common inversion under selection in Europeans. Nat. Genet. 37, 129–137.
Tobin, J. E., Latourelle, J. C., Lew, M. F., Klein, C., Suchowersky, O., Shill, H. A., Golbe, L. I., Mark, M. H., Growdon, J. H., Wooten, G. F., Racette, B. A., Perlmutter, J. S., Watts, R., Guttman, M., Baker, K. B., Goldwurm, S., Pezzoli, G., Singer, C., Saint-Hilaire, M. H., Hendricks, A. E., Williamson, S., Nagle, M. W., Wilk, J. B., Massood, T., Laramie, J. M., DeStefano, A. L., Litvan, I., Nicholson, G., Corbett, A., Isaacson, S., Burn, D. J., Chinnery, P. F., Pramstaller, P. P., Sherman, S., Al-hinti, J., Drasby, E., Nance, M., Moller, A. T., Ostergaard, K., Roxburgh, R., Snow, B., Slevin, J. T., Cambi, F., Gusella, J. F., and Myers, R. H. (2008). Haplotypes and gene expression implicate the MAPT region for Parkinson disease: the GenePD Study. Neurology 71, 28–34.
Ueda, K., Fukushima, H., Masliah, E., Xia, Y., Iwai, A., Yoshimoto, M., Otero, D. A., Kondo, J., Ihara, Y., and Saitoh, T. (1993). Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 11282–11286.
Vandrovcova, J., Pittman, A. M., Malzer, E., Abou-Sleiman, P. M., Lees, A. J., Wood, N. W., and de, S. R. (2009). Association of MAPT haplotype-tagging SNPs with sporadic Parkinson’s disease. Neurobiol. Aging 30, 1477–1482.
Wittmann, C. W., Wszolek, M. F., Shulman, J. M., Salvaterra, P. M., Lewis, J., Hutton, M., and Feany, M. B. (2001). Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293, 711–714.
Wong, K. T., Allen, I. V., McQuaid, S., and McConnell, R. (1996). An immunohistochemical study of neurofibrillary tangle formation in post-encephalitic Parkinsonism. Clin. Neuropathol. 15, 22–25.
Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., Kachergus, J., Hulihan, M., Uitti, R. J., Calne, D. B., Stoessl, A. J., Pfeiffer, R. F., Patenge, N., Carbajal, I. C., Vieregge, P., Asmus, F., Muller-Myhsok, B., Dickson, D. W., Meitinger, T., Strom, T. M., Wszolek, Z. K., and Gasser, T. (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607.
Keywords: genome wide association study, Parkinson’s disease, synuclein, tau
Citation: Wray S and Lewis PA (2010) A tangled web – tau and sporadic Parkinson’s disease. Front. Psychiatry 1:150. doi: 10.3389/fpsyt.2010.00150
Received: 01 July 2010;
Accepted: 03 December 2010;
Published online: 27 December 2010.
Edited by:
Mark R. Cookson, National Institutes of Health, USAReviewed by:
Isidro Ferrer, University of Barcelona, SpainCheng-Xin Gong, The City University of New York, USA
Leonard Petrucelli, Mayo Clinic, USA
Copyright: © 2010 Wray and Lewis. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Selina Wray and Patrick A. Lewis, Department of Molecular Neuroscience, University College London Institute of Neurology, Queen Square, London WC1N 3BG, UK. e-mail:cy53cmF5QGlvbi51Y2wuYWMudWs=;cC5sZXdpc0Bpb24udWNsLmFjLnVr