 Bernhard J. Mitterauer1
Bernhard J. Mitterauer1 Birgitta Kofler-Westergren2*
Birgitta Kofler-Westergren2*- 1 Volitronics – Institute for Basic Research, Psychopathology and Brain Philosophy, Wals/Salzburg, Austria
- 2 Forensic Neuropsychiatry, University of Salzburg, Salzburg, Austria
A model of glial–neuronal interactions is proposed that could be explanatory for the demyelination identified in brains with schizophrenia. It is based on two hypotheses: (1) that glia–neuron systems are functionally viable and important for normal brain function, and (2) that disruption of this postulated function disturbs the glial categorization function, as shown by formal analysis. According to this model, in schizophrenia receptors on astrocytes in glial–neuronal synaptic units are not functional, loosing their modulatory influence on synaptic neurotransmission. Hence, an unconstrained neurotransmission flux occurs that hyperactivates the axon and floods the cognate receptors of neurotransmitters on oligodendrocytes. The excess of neurotransmitters may have a toxic effect on oligodendrocytes and myelin, causing demyelination. In parallel, an increasing impairment of axons may disconnect neuronal networks. It is formally shown how oligodendrocytes normally categorize axonic information processing via their processes. Demyelination decomposes the oligodendrocyte–axonic system making it incapable to generate categories of information. This incoherence may be responsible for symptoms of disorganization in schizophrenia, such as thought disorder, inappropriate affect and incommunicable motor behavior. In parallel, the loss of oligodendrocytes affects gap junctions in the panglial syncytium, presumably responsible for memory impairment in schizophrenia.
Introduction and Hypothesis
The findings of white matter abnormalities in brains with schizophrenia (Davis et al., 2003; Di et al., 2009; Kyriakopoulos et al., 2009) suggest to focus on glial–neuronal interactions in schizophrenia research. Moreover, a deeper insight into the pathophysiology of this severe disorder is dependent on the underlying brain model from which the abnormalities are deduced.
To start out, the model of glial–neuronal interactions based on pertinent experimental results (Mitterauer, 1998, 2010b; Mitterauer and Kopp, 2003) is presented. The disorders deduced from this model that may explain the pathophysiology of schizophrenia focus indeed on oligodendrocyte–axonic interactions, but information processing in glial–neuronal synaptic units and their networks must be regarded as well. The present hypothesis is as follows: non-functional astrocytic receptors may cause an unconstrained neurotransmitter flux in the synapse which in turn hyperexcites the axon. Such hyperactivation of the axon may exert an excess of neurotransmitters with a toxic effect on the oligodendrocytes and their myelin sheaths, leading to demyelination. In this manner, the oligodendrocyte–axonic system increasingly decomposes (disconnects) which may lead to the symptoms of incoherence in schizophrenia. In addition, the loss of oligodendrocytes may also result in a loss of gap junctions in the panglial syncytium, causing memory impairment.
Brain Model of Glial–Neuronal Interactions
Description of the Networks
The proposed biological brain model is based on glial–neuronal interactions (Mitterauer, 1998, 2007). The nervous tissue of the brain consists of the neuronal system (neurons, axons, dendrites) and the glial system (astrocytes, oligodendrocytes with myelin sheaths enfolding axons, radial glia, and microglia). Experimental results are inspiring a major reexamination of the role of glia in the regulation of neural integration in the central nervous system (Kettenmann and Ransom, 2005; Halassa and Haydon, 2010). Figure 1 shows a schematic diagram of the glial–neuronal interaction: two astrocytes (Ac1,2) are shown in this very simple model, whereby in each case only one neuron (N1,2) belonging to an astrocyte is taken into consideration. Halassa et al. (2007) identified how a single astrocyte contacts only four to eight neurons, but 300–600 synapses via its processes. The glial network (syncytium) consists in this schema of two astrocytes and two oligodendrocytes (Oc1,2) interconnected via gap junctions (g.j.). The neuronal system shows two neurons (N1,2) with two afferent axons (Axi,j) and two afferent axodendritic synapses (Sai,j), two efferent axons (Ax1,2) with myelin sheaths (Ms) and a node of Ranvier (N.R.), as well as two dendro-dendritic synapses (Sd1,2,3,4) with the corresponding dendrites (D1,D2;D3D4).
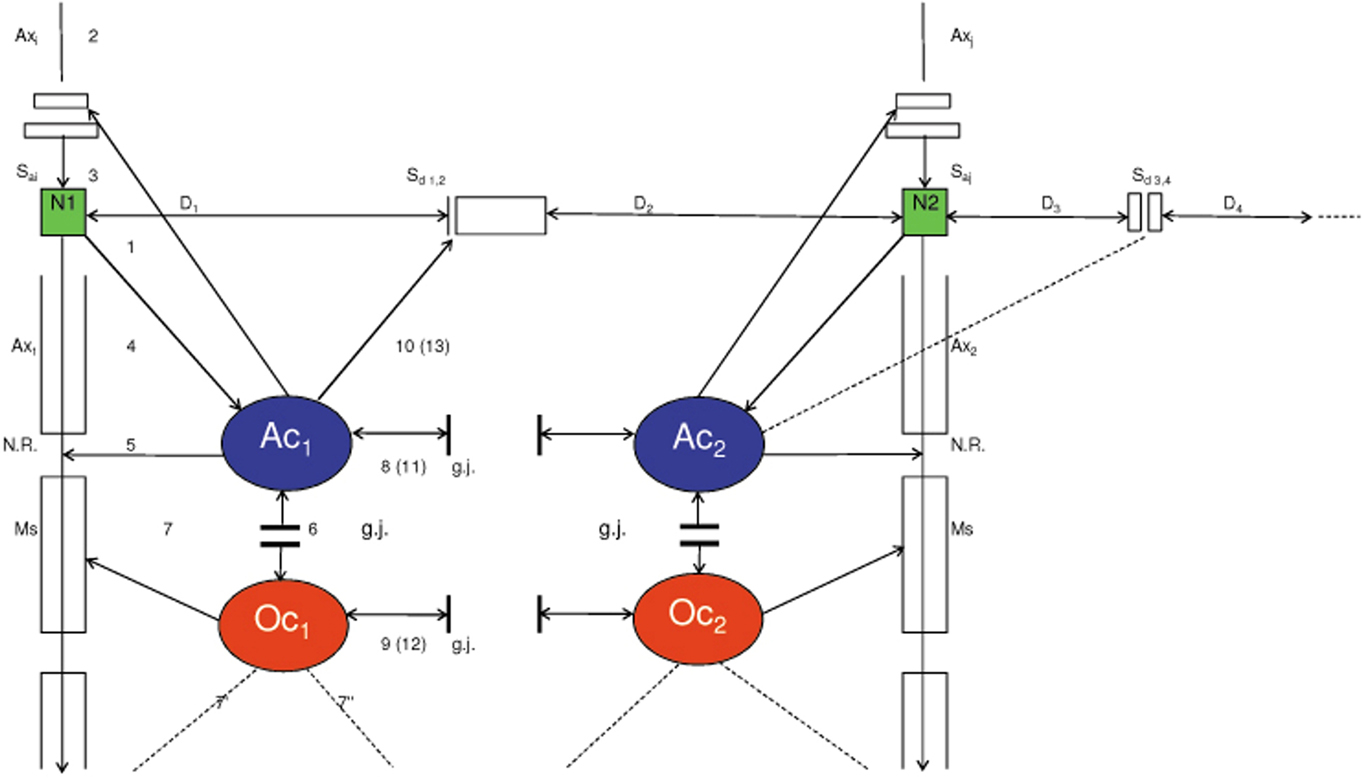
Figure 1. Schematic diagram of the glial–neuronal interaction. Two astrocytes (Ac1, 2) are shown in this very simple model, whereby in each case only one neuron belonging to an astrocyte is taken into consideration. The glial network (syncytium) consists of two astrocytes and two oligodendrocytes (Oc1, 2) belonging to them. Gap junctions (g.j.) exist between the astrocytes and the oligodendrocytes. The neuronal system shows two neurons (N1, 2) with two afferent axons (Axi, j) and two afferent axodendritic synapses (Sai, j), two efferent axons (Ax1, 2) with myelin sheaths (Ms) and a node of Ranvier (N.R.), as well as two dendro-dendritic synapses (Sd1,2,3,4) with the corresponding dendrites (D1, D2, D3, D4).
Glial–Neuronal Synaptic Units
Although a true understanding of how the astrocyte, the dominant glial cell type, interacts with neurons is still missing, several models have been published. Here, the focus is on a modified model proposed by Newman (2005). Figure 2 depicts a schematic diagram of possible glial–neuronal interactions at a glutamatergic glial–neuronal synaptic unit. Release of glutamate (GLU) from the presynaptic terminal activates glial receptors (glR) and postsynaptic receptors (poR) (1) (for the sake of clarity only one receptor is shown). The occupancy of glR evokes a Ca2+ increase (2) and the release of glutamate from the astrocyte. Glutamate excitation of presynaptic receptors (prR) (3) modulates glutamate release while activation of postsynaptic receptors (4) directly depolarizes the postsynapse. Activation of the astrocyte also elicits the release of adenosine-triphosphate (ATP), which depolarizes the postsynaptic neuron (5) and inhibits the presynaptic terminal (6) via occupancy of the cognate receptors (Haydon and Carmignoto, 2006). Hence, glia may exert a temporal boundary-setting function in synaptic information processing (Mitterauer, 1998).
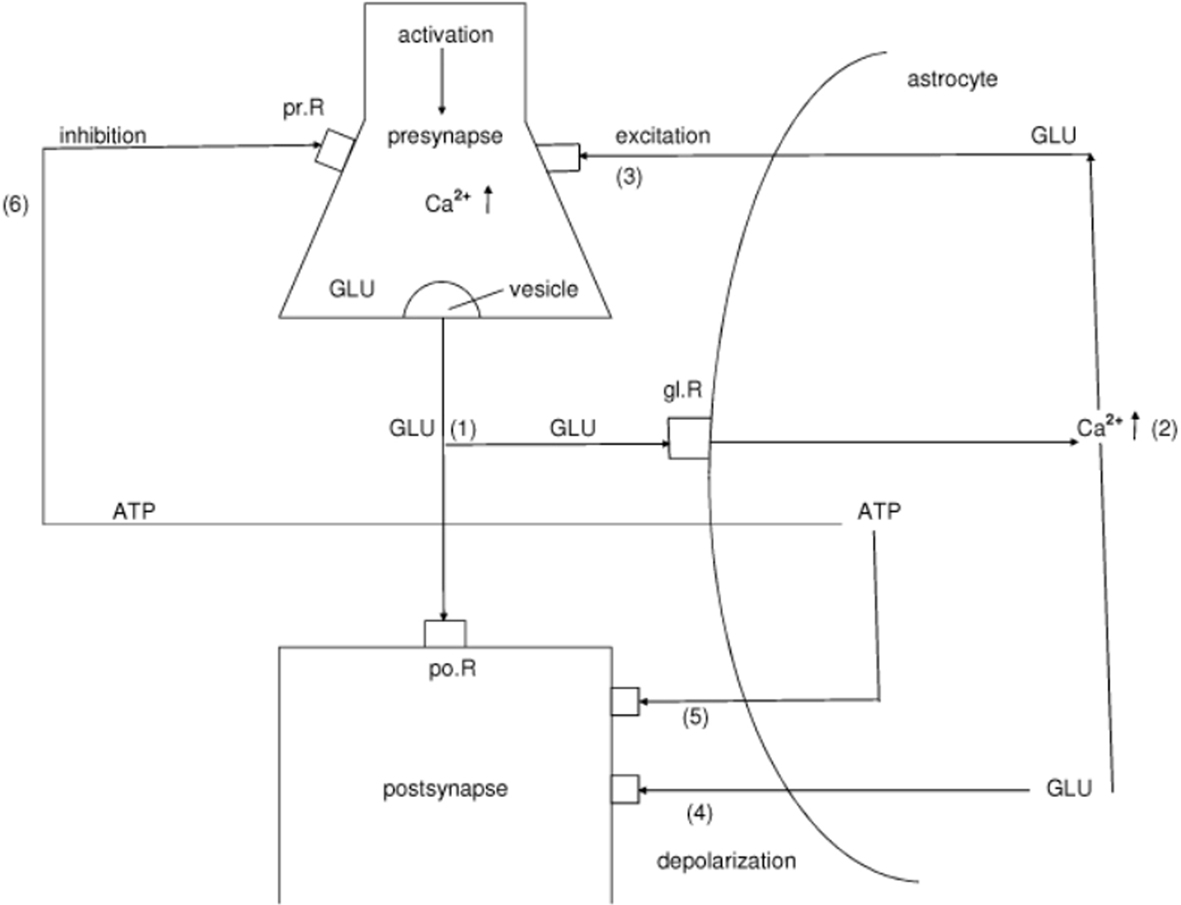
Figure 2. Schematic diagram of possible glial–neuronal interactions of the glutamatergic tripartite synapse (modified after Newman, 2005). Release of glutamate (GLU) from the presynapse activates (arrows) glial receptors (glR) and postsynaptic receptors (poR) (1), (for the sake of clarity only one receptor is shown). The occupancy of glR evokes a Ca2+ increase (2) and the release of GLU from the astrocyte. GLU excitation of presynaptic receptors (prR) (3) modulates GLU-release, while activation of prR (4) directly depolarizes the postsynapse. Activation of the astrocyte also elicits the release of adenosine-triphosphate (ATP), which depolarizes the postsynapse (5) and inhibits the presynapse (6) via occupancy of the cognate receptors.
Outline of the Glial Syncytium
Since the astrocytic syncytium may influence synaptic information processing, its structure and possible function must also be described. Figure 3 shows a diagrammatic schema depicting an astrocytic syncytium composed of two astrocytes (Ac1, Ac2) interconnected via gap junctions (g.j.). Each astrocyte contacts four synapses (Sy) with four different qualities (a, b, c, d) building an astrocytic-neuronal compartment. A quality is defined as the specific neurotransmitter type that mainly operates in synaptic neurotransmission, say glutamate (a), acetylcholine (b), serotonin (c), and dopamine (d). The gap junctions consist of the four identified astrocytic connexins Cx43, Cx30, Cx26, and Cx45, forming homotypic and heterotypic gap junction channels (for the sake of clarity not shown in the figure).
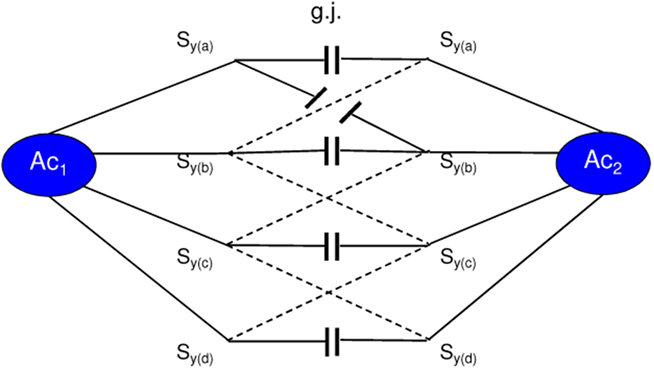
Figure 3. Diagrammatic schema of an astrocytic syncytium. Two astrocytes (Ac1, Ac2) are interconnected via gap junctions (g.j.). Each astrocyte contacts four synapses (Sy) with four different qualities (a, b, c, d) building two astrocytic-neuronal compartments. Since these two compartments are interconnected via gap junctions, a network is generated, called syncytium.
Moreover, astrocytes are also connected with oligodendrocytes via gap junctions (g.j.); one speaks of a panglial syncytium (Rash, 2010). Figure 4 outlines a panglial syncytium consisting of four astrocytes and four oligodendrocytes interconnected by gap junctions. Importantly, astrocytes may activate or inhibit the axonal information flux on the nodes of Ranvier (N.R.). In addition to “traditional” myelin formation, oligodendrocytes may influence synaptic regulation and signaling of the nodes of Ranvier (DeFelipe et al., 2010). There is growing experimental evidence that oligodendrocytes co-determine axonal information processing via myelin sheaths, by ions and transmitters. Since oligodendrocytes are interconnected with astrocytes via gap junctions, the processes in the astrocytic syncytium may determine the function of the oligodendrocytes (Fields, 2009).
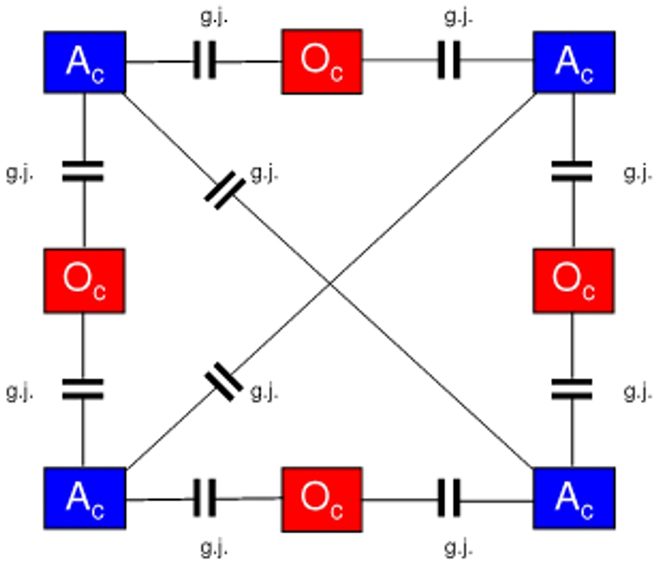
Figure 4. Outline of a panglial syncytium. Four astrocytes (Ac) and four oligodendrocytes (Oc) are interconnected via gap junctions (g.j.). The network of gap junctions between astrocytes is called astrocytic syncytium. If oligodendrocytes are included, the network forms a panglial syncytium.
Astrocyte Domain Organization
In all mammals, protoplasmic astrocytes are organized into spatially non-overlapping domains that encompass both neurons and vasculature. An astrocyte domain defines a contiguous cohort of synapses that interacts exclusively with a single astrocyte. One can also speak of a glial–neuronal compartment. Synapses within a particular territory are thereby linked via a shared astrocyte partner, independent of a neuronal networking (Oberheim et al., 2006). It is experimentally verified that astrocytes can express almost all receptors for important transmitter systems (Kettenmann and Steinhäuser, 2005). In certain cases, individual astroglial cells express as many as five different receptor systems linked to Ca2+ mobilization (McCarthy and Salm, 1991). Each astrocyte territory represents an island made up of many thousands of synapses (about 140000 in the hippocampal region of the brain, for instance), whose activity is controlled by that astrocyte (Santello and Volterra, 2010). On the average, human astrocytes extend 40 large processes radially and symmetrically in all directions from the soma so that each astrocyte supports and modulates the function of roughly two million synapses in the cerebral cortex (Oberheim et al., 2006). Astrocytic receptors are mainly located on the endfeet of the processes. Here, we apparently deal with a high combinational complexity of astrocyte–synaptic interactions.
Oligodendrocyte–Axonic Interactions
For normal brain function it is essential that signals pass rapidly between neurons. Oligodendrocytes play an important role in assuring fast neuronal signaling in the central nervous system. By covering neuronal axons with myelin which decreases the effective axonal membrane capacity, they reduce the charge needed to depolarize the axon and hence allow the action potential to travel much faster by saltatory conduction from one node of Ranvier to the next.
During development, oligodendrocytes are generated from precursor cells. These initially differentiate into immature cells that put out processes seeking axons to myelinate, and eventually they form mature cells with parallel processes myelinating up to 30 different axons. The production of myelinated axons requires a precise matching of the number of oligodendrocytes generated to the length of axons to be myelinated. This may be regulated in part by neurotransmitter receptors activated by substances released by active axons. Such interactions may also be important for maintaining the myelination of mature axons. Thus, neurotransmitter receptors play an important role in the life and death of oligodendrocytes (Káradóttir and Attwell, 2007).
Impulse activity in axons affects the development of oligodendrocytes, and thus myelination. Some of the effects of impulse activity inhibit myelination and some stimulate it. Here, the question arises as to how oligodendrocytes know which axons are electrically active. Three mechanisms have been identified that regulate myelination or the development of myelin forming glia in response to electrical stimulation of axons in vitro (Fields, 2010). Specific frequencies of electrical impulses control the amount of L1CAM present on unmyelinated axons, a cell adhesion molecule that is necessary for myelination (Stevens et al., 1998). The neurotransmitter adenosine 5’-triphosphate (ATP) is released from axons and activates receptors on astrocytes, causing them to release the leukemia inhibitory factor that stimulates myelination by mature oligodendrocytes (Ishibashi et al., 2006). Adenosine derived from hydrolysis of released ATP promotes oligodendrocyte precursor cell development and thus increases myelination (Stevens et al., 2002). Moreover, a non-synaptic mechanism for ATP release from axons has also been identified (Fields and Ni, 2010).
Combinational Analysis of Axonal Properties via the Processes of Oliogodendrocytes
Generally speaking, the oligodendrocyte–axonic system embodies a structure. The elements of this structure are n axons interconnected with m processes of oligodendrocytes that envelop axons by their myelin sheaths. Supposing that each axon conducts information of a special property, the processes of oligodendrocytes may categorize a set of axonic properties. A property is defined as an essential quality common to all members of a class. The logical definition of a category is any of the various basic concepts into which all knowledge can be classified. The structure of the oligodendrocyte–axonic system can be analyzed on three levels of complexity (Guenther, 1971):
(1) Protoanalysis: How many different properties are there? This corresponds to cardinality.
(2) Deuteroanalysis: How are the properties distributed? This corresponds to distribution.
(3) Tritoanalysis: Where are the individual properties located? This corresponds to the position.
Now, let us concentrate on the formalism underlying the highest level of complexity (tritoanalysis) and take oligodendrocytes with four processes as an example. If the oligodendrocyte–axonic system uses four processes, we must qualitatively count to four in order to achieve all combinational possibilities. Table 1 shows the qualitative counting from one to four (Thomas, 1985). On the left from the vertical line the numbers of the processes of oligodendrocytes (1…4) are enumerated. On the right from that line the categories of all possible oligodendrocyte–axonic structures are counted. In the first case only one process of an oligodendrocyte is available. This can merely connect one axon with property one (1) or another with property two (2). Oligodendrocytes with two processes are able to connect two axons with the same (11) or with different (12) properties. In the case of three processes, the position of the processes becomes relevant. In order to refer to all possible positions of the axonic properties, already five categories are generated. Applying this rule to four processes, we must count to 15 (1111…1234). Each number designates a specific category (brackets) with increasing complexity. Such qualitative counting or categorization follows the sequence of the Bell numbers. Bell numbers B(n) are generated according to the formula
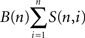

Table 1. Qualitative counting in categories (n = 4) shown on an oligodendrocyte-axonic system.
(S(n, i) are the Stirling numbers of the second kind). Table 2 shows the first 10 Bell numbers that rapidly increase. Taken this integer sequence, oligodendrocytes with 10 processes generate a combinational structure with 115975 categories. Here one is seduced to speculate if this high potential of the brain to categorize information could be an inexhaustible source of creativity.
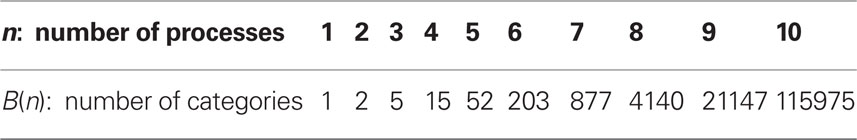
Table 2. The Bell number B(n) indicates how many categories n processes of oligodendrocytes can generate.
Figure 5 gives an example how oligodendrocytes with one to four processes connect 10 axonic lines with four different properties (1…4). Each oligodendrocyte is characterized with the category that its processes generate. From the left to the right examples of one to four processes (according to Table 1) are given. Together, the capacity of the oligodendrocyte–axonic system to generate categories may lead to a coherent information processing. Coherence in cognition means that we are able to abstract, generalize, and extract meaning from different properties of information. This capacity can be severely disturbed in schizophrenia.
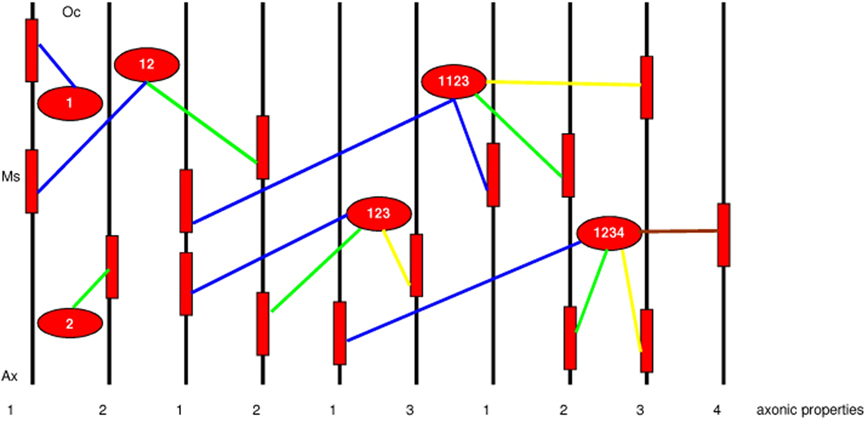
Figure 5. Example of an oligodendrocyte-axonic structure according to the rule of categorization (Table 1). Oligodendrocytes (Oc) and myelin sheaths (Ms) are represented in red color. Ten axons (black) with different or equal properties of information processing (1…4) are shown. Each oligodendrocyte is characterized with the category that its processes generate. From the left to the right examples of one to four processes (according to Table 1) are given. Blue colored processes connect axonic property 1, green property 2, yellow property 3, and brown property 4.
It may be controversial to assign information processing and topical properties to axons, especially if the brain is placed into conceptual framework of non-linear dynamics (Werner, 2007). However, in considering the myriads of various receptors within the brain and on the sensory receptor sheaths, structural specificity and topical precision may be very relevant for information processing. Take for example the identified specific molecular quality of olfactory receptors (Axel, 1995; Mori et al., 1999). Another argument is visual perception where the concept of retinotopy supports a topological organization of information processing (Cowan and Friedman, 1995). Furthermore, there are various types of neurons in the brain that exert specific function in the sense of special purpose computers as, e.g., identified in the visual cortex (Hubel and Wiesel, 1968). If a neuron or sensory receptor transfers their information via axons to other systems in the brain, then these axons may not only conduct currents, but also specific properties of information.
Categorization is fundamental to our perception and understanding of the environment (Grinband et al., 2006; Husain et al., 2006). Many recent studies have examined the neural basis of category learning. Behavioral neuroscience results suggest that both the prefrontal cortex and the basal ganglia play important category-learning roles; neurons that develop category-specific firing properties are found in both regions and lesions to both areas cause category-learning deficits (Ashby and Spiering, 2004). However, present research on categorization function in the brain is based on the neuronal system and not referring to the glial system.
Tying Formalism and Brain-Biological Substrates Together
(1) The boundary-setting function (compartmentalization) of glia could be the brain-biological substrate of the tritogrammatical categorization process.
(2) A tritogram consisting of places with a numeral located at each place is a certain glial structure.
(3) Categories are specific types of brain operations and/or information processed or produced in the brain.
(4) The categorization process corresponds to glial–neuronal interactions. We also refer to this interaction as compartmentalization, i.e., the way oligodendrocytes divide groups of axons into both spatially and temporarily limited functional units.
(5) Astrocytes may also categorize a set of synapses into domains via their processes, formalizable as tritograms.
(6) If processes of oligodendrocytes are impaired or lost, their categorization function is disrupted.
Synaptic Model of Unconstrained Neurotransmitter Flux in Schizophrenia
Since we hypothesize that an unconstrained neurotransmitter flux in schizophrenia may represent a major pathophysiological mechanism responsible for the impairment of oligodendrocyte–axonic interactions, it must be shown how this disorder of glial–neuronal synaptic units may arise.
If the glR are totally non-functional and therefore cannot be occupied by neurotransmitters, the system is unbalanced. As in Figure 6 depicted, the glR are non-functional (crosses) and cannot be occupied by neurotransmitters (NT), so that the activation of the gliotransmitters (GT) is impossible. Hence, they cannot negatively feedback to the receptors on the presynapse (prR). As a consequence, the glia lose their inhibitory or boundary-setting function and the neural transmitter flux is unconstrained, as the flux of thought on the phenomenological level.
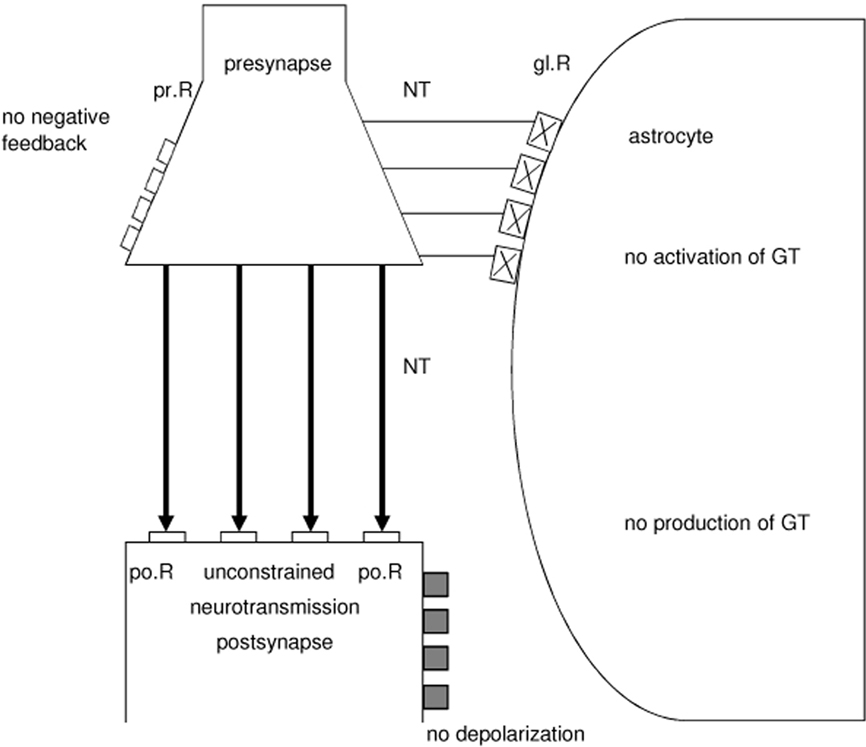
Figure 6. Model of an unbalanced tripartite synapse that may cause schizophrenia. Non-functional glial receptors (glR), depicted by crosses, cannot be occupied by neurotransmitters (NT). Since the activation and production of gliotransmitters (GT) is not possible, glia do not negatively feed back to the presynaptic receptors (prR) and cannot depolarize the postsynaptic neuron. This severe synaptic disturbance leads to an unconstrained neurotransmission (fat arrows).
The brain theory presented here is essentially based on the experimentally supported hypothesis that the glial system in its interaction with the neuronal system generates glial–neuronal compartments in the sense of specific functional units or operational domains (Mitterauer, 1998; Mitterauer and Kopp, 2003). The interactional structure of an astrocyte with n-neurons can be defined as an elementary compartment of nerve cells. By simultaneously activating and deactivating neurotransmission in all of the synapses enveloped by an astrocyte, the astrocyte calcium wave may coordinate synapses into synchronously firing groups (Antanitus, 1998; Fellin et al., 2006), interpretable as harmonization (Mitterauer, 2001).
A loss of the glial boundary-setting function caused by unbalanced tripartite synapses is depicted in Figure 7. The astrocytes (Aci; Acj) of compartment x and compartment y have non-functional glR (asterisks), so that glia cannot influence neuronal information processing. This genetically determined disturbance results in a compartmentless neuronal network displayed as a group of eight neurons with 28 connecting lines (according to the formula [n2 − n]/2). Such a brain is unable to structure the environmental information. One may argue that a glial determination of neuronal networks into functional units is not necessary because the neuronal system is compartmentalized per se (Rall, 1995). However, according to our view, there is a qualitative difference between the purely neuronal compartments and the glia-determined compartments. Neuronal compartments may be merely functional for information processing, whereas glial–neuronal compartments may in addition have an information-structuring potency that we need for recognizing the qualitative differences between objects and individuals in our environment. That capacity may be lost in schizophrenic patients. Therefore, one can also speak of a loss of conceptual boundaries in schizophrenia.
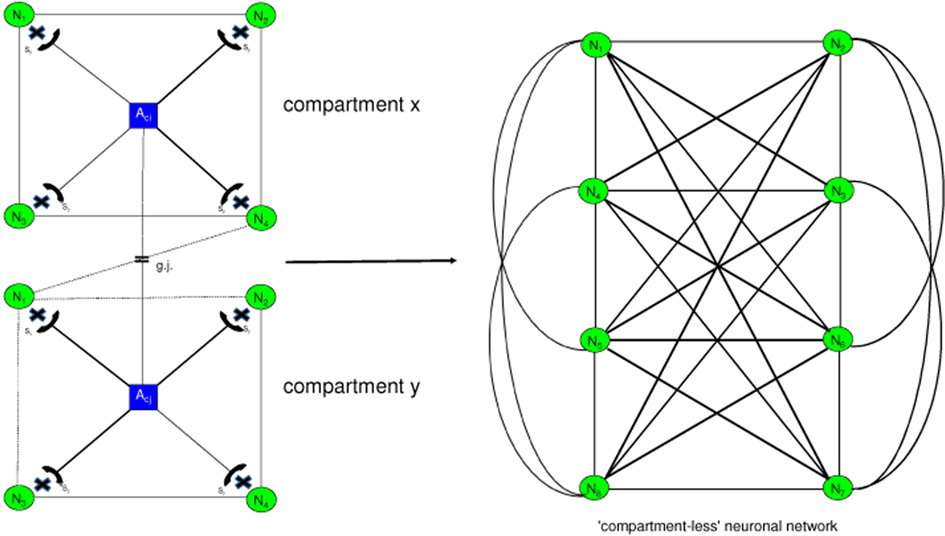
Figure 7. Loss of glial boundary-setting functions. Generalization of neuronal information processing. In the glial–neuronal compartments (x, y) the astrocytes (Aci, j) are unable to modulate any of the neurons (N1,…, N4), since they cannot influence the neurotransmission in the synapses (asterisks). This loss of glial boundary-setting results in a “compartmentless” neuronal network in which all neurons are interconnected (Mitterauer, 2003).
From an ontological point of view, delusions are the consequence of the loss of boundaries between the self and the others (non-selves). Here, the self is defined as a living system capable of self-observation (Mitterauer and Pritz, 1978). One could also say that our brain embodies a distinct ontological locus of self-observation. Everything taking place in the brains of schizophrenic patients is reality because they cannot differentiate between their inner world and the outer world. Therefore, they cannot see ontological differences between the selves and the non-selves. This loss of ontological boundaries may lead to a delusional misinterpretation of reality.
Hallucinations may be caused by the same disorder. However, the perception systems are phenomenologically affected. A schizophrenic who hears the voice of a person in his head is absolutely convinced that this person is really speaking to him. The loss of ontological boundaries or inner/outer confusion shows its phenomenological manifestation in the auditory system. Such a disorder can also occur in other sensory systems.
If the loss of boundaries affects the motor system in the brain, the symptomatology is called catatonia. A state of catatonic agitation in which a disinhibited discharge of nearly all motor systems occurs is an expression of motor generalization with raging and screaming as behavioral components. One could also say that the brain’s inability to constrain information processing among motor modules appears in catatonic phenomena. Hence, the catatonic type of schizophrenia represents a serious disorder of motor behavior. Typical symptoms are excessive motor activity and motoric immobility (stupor). Both phenomena appear to be purposeless and not influenced by external stimuli. In such a catatonic state, a patient is unable to communicate. He or she cannot see the other. Everything that happens takes place in the brain of the patient.
Affective flattening is regarded as a negative schizophrenic symptom (Arajärvi et al., 2006). This symptom can also be explained as a loss of boundary setting. The different affective or emotional qualities cannot be produced within the brain, and the communication of feelings is disturbed as a result (Holden, 2003).
The Neurotransmitter Hypothesis in View of Non-Functional Astrocytic Receptors
There are a number of theories of schizophrenia that implicate aberrant neurotransmission systems. The neurotransmitter hypothesis comprises dopamine, glutamate, serotonin, noradrenaline, acetylcholine, and gamma-aminobutyric acid (GABA). The classical dopamine hypothesis of schizophrenia postulates a hyperactivity of dopaminergic transmission at the D2 receptor in the mesencephalic projections to the limbic striatum (Carlsson, 2003). The dopamine hypothesis has received support from post-mortem and positron emission tomography (PET) indications of increased dopamine D2 receptor levels in the brains of schizophrenic patients. Furthermore, in drug-free patients a greater release of dopamine from presynaptic terminals was shown as compared to normal control subjects (Laruelle et al., 1996; Breier et al., 1997; Abi-Dargham et al., 1998). On the contrary, the functional activity of dopamine may be decreased in the neocortex of schizophrenia, which could at least partially be associated with negative symptoms (Okubo et al., 1997).
The glutamate hypothesis proposes that abnormal glutamatergic function underlies schizophrenia (Kristiansen et al., 2007). NMD-R hypofunction (a subtype of glutamate receptors) in the cortical association pathways could be responsible for a variety of cognitive and other symptoms (Pilowsky et al., 2005). However, studies of NMDA receptor numbers in post-mortem schizophrenic brains are inconsistent. The numbers of those receptors have been investigated in the prefrontal cortex, temporal cortex, hippocampus, and striatum. Although increases and decreases of receptors have been found, normal amounts have been counted as well. Importantly, the existence of anatomical and functional interrelationships between dopamine and glutamate systems in the central nervous system suggests that inhibition of the NMDA-R would influence dopamine transmission. For example, NMDR antagonists decrease corticofugal inhibition of subcortical dopamine neurons, thereby enhancing the firing rate of dopamine neurons (Seeman, 2009).
The GABA hypothesis focuses on a defect in neurotransmission involving GABA, especially in the frontal cortex of brains with schizophrenia limited to the parvalbumin-class of GABAergic interneurons (Blum and Mann, 2002). Post-mortem findings have then formed the basis for theories of schizophrenia based on defective neural synchrony (Ford and Mathalon, 2008). Interestingly, neonatal lesions of the ventral hippocampus elicit alterations in the GABAergic system, leading to functional consequences on brain excitability, lending support to the idea that GABAergic systems could be involved in the pathophysiology of schizophrenia (Francois et al., 2009).
The serotonin hypothesis of schiozophrenia is formed by several observations. Serotonin receptors are involved in the psychotomimetic and psychotogenic properties of hallucinogens. The number of cortical 5-HT2A and 5-HT1A receptors is altered in schizophrenic brains. These receptors play a role in the therapeutic and/or side-effect profiles of atypical antipsychotics. 5-HT2A receptor-mediated activation of the prefrontal cortex may be impaired in some schizophrenics. Meltzer and Nash (1991) marshalled a range of evidence from preclinical studies to argue that the therapeutic superiority of clozapine was due to its simultaneous antagonism of dopamine, 5-HT2 and 5-HT3 receptors. Hence, serotoninergic and dopaminergic systems are interdependent and may be simultaneously affected in schizophrenia.
Noradrenaline was another early neurochemical candidate in schizophrenia. Stein and Wise (1971) argued that progressive damage to a noradrenergic reward pathway might cause the negative symptoms and long-term downhill course of schizophrenia. Subsequently, this transmitter has been marginalized, although interest has been kept alive by some researchers (Yamamoto and Hornykiewicz, 2004).
According to Raedler et al. (2007) cholinergic neurons of the central nervous system are also involved in schizophrenia. However, it is not entirely clear whether a functional excess or deficiency is postulated.
To many researchers it seems unimaginable that there is no neurochemical disturbance in schizophrenia. Yet, after 50 years of well-diversified investigation, an adequate neurochemical theory of schizophrenia seems as elusive as ever (McKenna, 2010). The reason may be that the current neurotransmitter hypothesis of schizophrenia does not refer to the glial variable in synaptic neurotransmission (Mitterauer, 2003, 2005, 2010b). As already described, in glial–neuronal synaptic units astrocytes may have a temporal boundary-setting function by temporarily turning off synaptic information transmission (Mitterauer, 1998). Moreover, based on a logic of balance, divergent synaptic dysfunctions can be unified (Mitterauer, 2005, 2010a).
Balance, Imbalance, and Unbalance between Neurotransmitters and Astrocytic Receptors
The interaction between neurotransmitters and astrocytic receptors can be system-theoretically interpreted (Guenther, 1963) as balanced, underbalanced, overbalanced or even unbalanced. The proposition is that the operations of a living system are balanced if the number of variables and values are equal. From a biocybernetic point of view, a variable is something that retains its own identity while capable of changing its state in the sense of material realization (Krippendorff, 1986). Here, we interpret the amount of neurotransmitters as values and the number of astrocytic receptors as variables. Figure 8 shows balance, underbalance, overbalance, and unbalance between neurotransmitters and their cognate astrocytic receptors. If the concentration of neurotransmitters for occupancy of astrocytic receptors is appropriate, the synaptic system is in balance (a). In the case of an excess of astrocytic receptors, the amount of neurotransmitters is too small, leading to an underbalanced synaptic system (b). This state may cause depression (Mitterauer, 2004, 2010a). If the number of astrocytic receptors is low in relation to a normal neurotransmitter concentration, the synaptic system is overbalanced (c). This state may cause mania (Mitterauer, 2004, 2010a). Supposing that astrocytic receptors do not function at all, the system is totally unbalanced (d). Such a synaptic state may be responsible for the pathophysiology of schizophrenia.
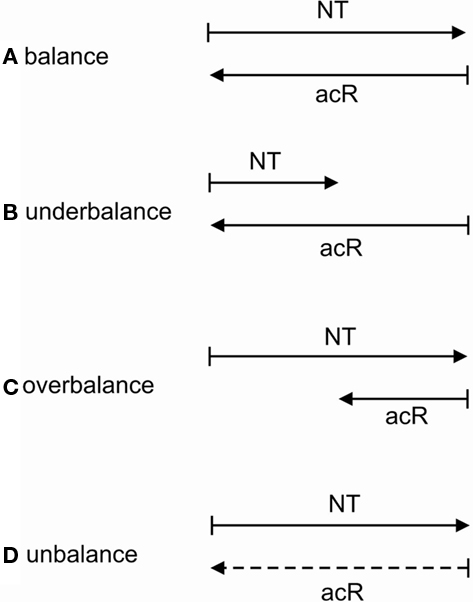
Figure 8. Balance, underbalance, overbalance, and unbalance between neurotransmitters (NT) and astrocytic receptors (acR). Here, NT are interpreted as values and acR as variables. In the case where the amount of NT and acR is equal, the synaptic system is balanced (A). If the number of acR surpasses the amount of NT available, the system is underbalanced (B). In the inverse situation, the synaptic glial–neuronal interaction is overbalanced (C). An unbalanced synaptic neurotransmission occurs if acR are non-functional (dashed line) for occupancy with NT (D).
Therefore, a functional excess or deficiency in the neuronal components of synapses may not be decisive for the pathophysiology of schizophrenia, but rather non-functional astrocytic receptors may essentially cause the disorder. Accordingly, the loss of the glial temporal boundary-setting function leads to an unconstrained synaptic neurotransmission in various degrees of the amount of neurotransmitters. This mechanism may unify seemingly contradictory findings of the neurotransmitter hypothesis.
Unconstrained Neurotransmission may Cause Demyelination
Generally, in pathological conditions neurotransmitters can be released excessively, damaging the cells they normally act on. Since oligodendrocytes have receptors for the various transmitters, neurotransmitter excess can cause demyelination (Káradóttir and Attwell, 2007). Already in 1977 a study was published documenting severe breakdown of myelin in dogs injected with myelin (Saakov et al., 1977). In the gray matter of the brain the death of neurons in pathological conditions is often caused by a rise of extracellular glutamate concentration activating NMDA receptors and causing an excessive rise of Ca2+. Glutamate can also damage white matter oligodendrocytes. Concentrations of glutamate which alone are not toxic sensitize oligodendrocytes to subsequent complementary attack that inserts membrane attack complexes into the oligodendrocyte, allowing a toxic Ca2+ influx to occur (Alberdi et al., 2006).
In line with the presented synaptic model of the pathophysiology of schizophrenia, an unconstrained flux of neurotransmitters occurs. This may hold for all the various neurotransmitter types. This unconstrained flux of neurotransmitters may affect oligodendrocytes either by flooding of their cognate receptors on oligodendrocytes, exerting a toxic Ca2+ influx, or via a hyperactivation of axons with an excess of axonic ATP production and a consequent toxic effect on oligodendrocytes (Figure 9). In addition, ATP released from axons cannot activate astrocytic receptors, since they do not function. Therefore, a stimulation of myelination by mature oligodendrocytes is not possible. These pathological mechanisms may cause demyelination, as observed in brains with schizophrenia (Skelly et al., 2008; Takahashi et al., 2010). Note, although decreased expression of oligodendrocyte-related genes has been identified (Höstad et al., 2009), it seems implausible that genetics alone could account for demyelination in schizophrenia (Fields, 2009).
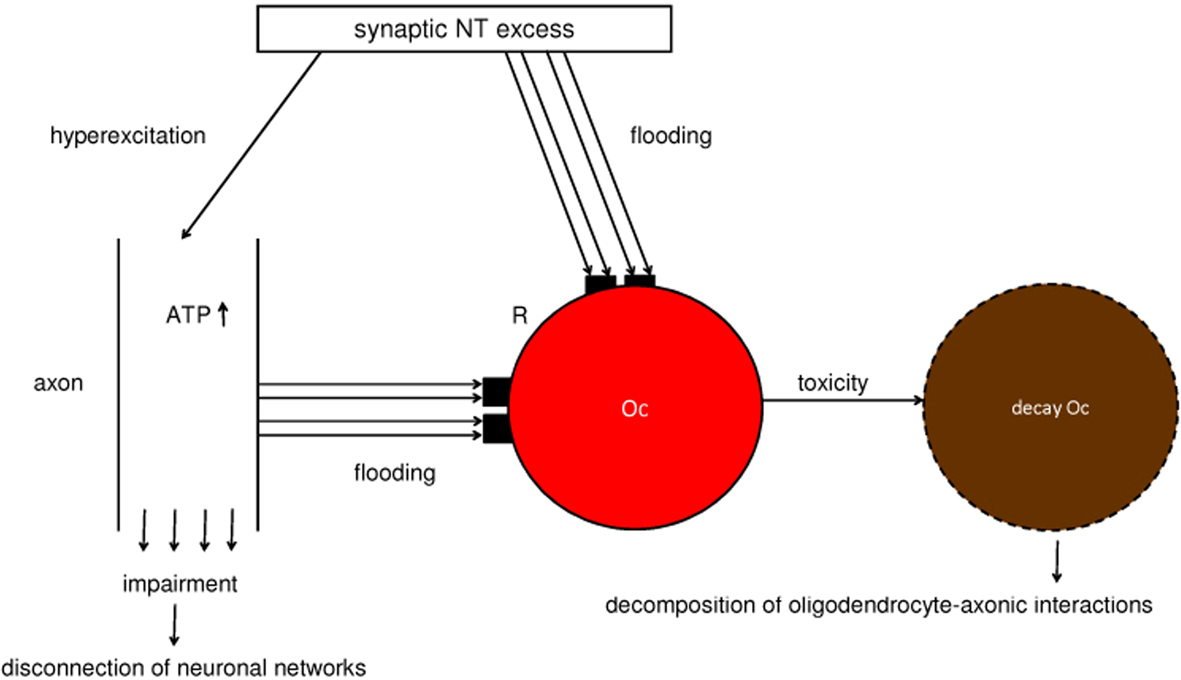
Figure 9. Schematic diagram of the excess of neurotransmitters that may cause a loss of oligodendrocytes and decomposition or disconnection of oligodendrocyte–axonic interactions and neuronal networks. A synaptic neurotransmitter (NT) excess hyperexcites the axon and floods the cognate receptors (R) on the oligodendrocyte (Oc). In parallel, a non-synaptic ATP excess occurs, also flooding R. This flooding of NT and ATP exerts a toxic effect on the Oc, leading to its decay. These mechanisms may be responsible for the decomposition of oligodendrocyte–axonic interactions and the disconnection of neuronal networks.
Moreover, the permanent hyperactivation of axons may also impair them so that neuronal networks disconnect. This may represent a possible mechanism that could at least in part be responsible for the disconnection of neuronal networks identified in brains with schizophrenia (Höstad et al., 2009).
Loss of Oligodendrocytes may Cause a Decay of Gap Junctions in the Panglial Syncytium Responsible for Memory Impairment
Since gap junctions between astrocytes and oligodendrocytes are heterotypic, composed of special astrocytic connexins and different oligodendrocytic connexins, a loss of oligodendrocytes disrupts astrocyte-oligodendrocytic gap junctions in the panglial syncytium, leading to a “leaky” syncytium (Figure 10).
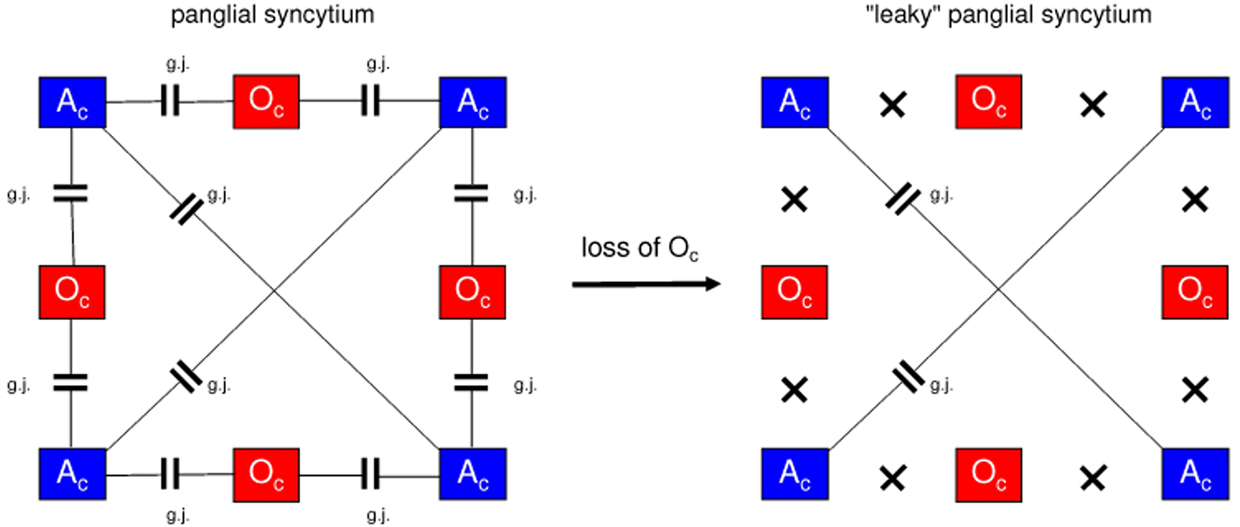
Figure 10. Loss of oligodendrocytes disconnects the panglial syncytium, causing memory impairment in schizophrenia. On the left, a panglial syncytium composed of astrocytes (Ac) and oligodendrocytes (Oc) interconnected via gap junctions (g.j.) is outlined (Figure 4). A loss of Oc may cause a decay of Oc-Ac gap junctions (crosses). Such a “leaky” panglial syncytium may be responsible for memory impairment.
Gap junctions form plaques that may embody memory structures (Robertson, 2002). Therefore, a loss of gap junctions may impair memory. In addition, genetic or (and) exogenous disorders can affect gap junctional plaque formation which may be the case in schizophrenia. In this context one could speak of a syncytiopathy in schizophrenia (Mitterauer, 2009). However, memory impairment may also be caused by a disorganization of neuronal networks (Wolf et al., 2008).
Schizophrenic Symptoms Presumably Caused by Incoherence in the Oligodendrocyte-Axonic System
The research criteria for alternative dimensional descriptors for schizophrenia (American Psychiatric Association, 1998) differentiate between a psychotic (hallucinations, delusions) dimension, a disorganized dimension and a negative (deficit) dimension of schizophrenia. The disorganized dimension describes the degree to which disorganized speech, disorganized behavior or inappropriate affect have been present. Here, we will focus on the disorganized domain and attempt to deduce this kind of schizophrenic symptomatology from the incoherence of brain functions that may be caused by decomposed oligodendrocyte–axonic interactions.
First, let us discuss thought disorder, the main symptom of cognitive disorganization. This appears in incoherent words, termed neologisms, and in incoherent sentences (word salad). Formal testing of patients rated as incoherent shows that they are producing words that are unusual or inappropriate in context. In verbal fluency tasks patients with incoherence produce unusual and deviant words. This means that they produce words that are very unusual for the category (e.g., “aardvark” for “animals”) or words that are not in the category prescribed (e.g., “ginger” as a “fruit”). In word association tasks with ambiguous words patients produced associations to the less likely meaning (Frith, 1999). For example, to the word pair tree-bark, an incoherent patient might give the response “dog” (Allen, 1983). These symptoms represent incoherence of thought that may be caused by decomposed oligodendrocyte–axonic interactions, if the cognitive systems are affected.
The same mechanism may be responsible for the typical motoric symptoms of schizophrenia. According to Frith (1999), disorders of action associated with schizophrenia fall into three categories. First, there is poverty of action; no action is produced. Second, there is perseveration, in which the same action is produced inappropriately. Third, actions occur that are inappropriate to the context. Here we deal with symptoms of the catatonic type of schizophrenia. Let us focus on excessive, purposeless motor activity and inappropriate motor behavior like stereotypy and mannerisms (Table 3). These symptoms may also be caused by incoherence in the motoric pathways in the sense of a decomposed oligodendrocyte–axonic system. In other words: motor programs cannot be composed into categories necessary for effective and communicative behavior. Finally, if the emotional systems of the brain are disordered by the same mechanism proposed, then the generation of affects is incoherent. We speak of inappropriate affects defined as a disharmony between the emotional feeling tone and the idea, thought or speech accompanying it. A flat affect is a typical negative symptom of schizophrenia. These patients are apparently unable to produce any emotional quality (Table 3). The incapacity of the oligodendrocyte–axonic system to generate categories for coherent and comprehensive emotional communication may be responsible for these symptoms.

Table 3. Schizophrenic symptoms possibly caused by incoherence in the oligodendrocyte–axonic system.
However, one may argue that multiple sclerosis is also based on a demyelinating disorder of the brain where oligodendroglia is decreased or lost, comparable to the mechanism that is hypothesized for symptoms of incoherence in schizophrenia. In addition, patients with multiple sclerosis show not only the typical motoric symptoms, but can also suffer from cognitive impairments, affective disorders, and even psychotic symptoms (Honer et al., 1987; Beatty, 1993). First of all, although the pathophysiology of schizophrenia is as yet unknown, there is a consensus that the core symptoms (delusions and hallucinations) of schizophrenia may be basically caused by a disorder in the synaptic information processing. In glial–neuronal synaptic units an imbalance in the sense of an unconstrained information flux may be responsible for the core symptoms of schizophrenia (Mitterauer, 2003, 2005, 2010a). Based on that synaptic pathophysiological approach, it is here attempted to hypothesize that a decomposition of the oligodendrocyte–axonic system may cause symptoms of incoherence in schizophrenia. Most importantly, according to the presented model of the pathophysiology of schizophrenia, the astrocyte–neuronal interaction in synapses must primarily be disordered, which is not the case in multiple sclerosis.
Conclusion
Figure 11 summarizes the possible pathophysiology of glial–neuronal interactions in brains with schizophrenia as proposed in this paper. A synaptic imbalance of neurotransmission caused by non-functional astrocytic receptors, leads to an unconstrained neurotransmitter flux that hyperexcites axons and may affect oligodendrocytes directly. In parallel, a non-synaptic ATP excess arises. The unconstrained flux of synaptic and non-synaptic neurotransmitters exerts a toxic effect on the oligodendrocytes and their myelin, leading to demyelination. Whereas the synaptic imbalance described may mainly be responsible for psychotic symptoms, demyelination has a double effect: on the one hand, a decomposition of oligodendrocyte–axonic interaction occurs, responsible for symptoms of disorganization or incoherence. On the other hand, the panglial syncytium is affected. Such a “leaky” syncytium may cause memory impairment and negative symptoms.
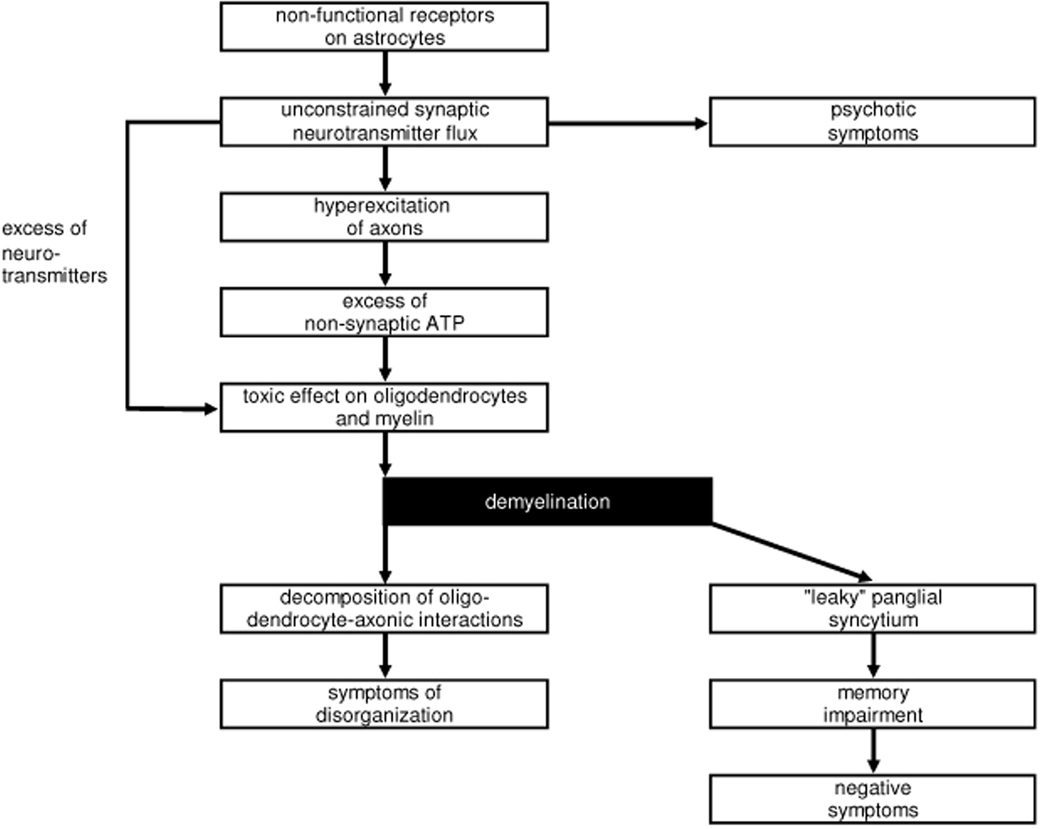
Figure 11. Schematic diagram of impaired glial-neuronal interactions that may be responsible for the main symptoms of schizphrenia (see text).
Importantly, all macroglial cells (astrocytes and oligodendrocytes) with their syncytia must be considered in their interactions with the neuronal system with regard to research into the pathophysiology of schizophrenia. Therefore, abnormalities of white matter in brains with schizophrenia may be mainly responsible for symptoms of incoherence and not for the whole schizophrenic syndrome.
Although pertinent treatment strategies are not the topic of the present paper, a concluding remark seems to be necessary. First of all, if antipsychotic drugs reduce the synaptic neurotransmission flux, they certainly influence not only psychotic symptoms but may also inhibit demyelination. Unfortunately, antipsychotic substances cannot influence or improve the functional impairment of astrocytic receptors, the core disorder in our model of the pathophysiology of schizophrenia. Here, the substitution of astrocytic receptor proteins is imaginable.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abi-Dargham, A., Gil, R., Krystal, J., Baldwin, R. M., Seibyl, J. P., Bowers, M., van Dyck, C. H., Charney, D. S., Innis, R. B., and Laruelle, M. (1998). Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am. J. Psychiatry 155, 761–767.
Alberdi, E., Sánchez-Gómez, M. V., Torre, I., Domercq, M., Pérez-Samartín, A., Pérez-Cerdá, F., and Matute, C. (2006). Activation of kainate receptors sensitizes oligodendrocytes to complement attack. J. Neurosci. 26, 3220–3228.
Allen, H. A. (1983). Do positive and negative language symptom subtypes of schizophrenia show qualitative differences in language production? Psychol. Med. 13, 787–797.
American Psychiatric Association. (1998). Diagnostic and Statistical Manual of Mental Disorders. Washington: American Psychiatric Association.
Antanitus, D. S. (1998). A theory of cortical neuron-astrocyte interaction. Neuroscientist 4, 154–159.
Arajärvi, R., Varilo, T., and Suvisaari, J. (2006). Affective flattening and alogia associate with familial form of schizophrenia. Psychiatry Res 141, 161–172.
Ashby, F. G., and Spiering, B. J. (2004). The neurobiology of category learning. Behav. Cognit. Neurosci. Rev. 3, 101–113.
Beatty, W. W. (1993). Cognitive and emotional disturbances in multiple sclerosis. Neurol. Clin. 11, 189–204.
Blum, B. P., and Mann, J. J. (2002). The GABAergic system in schizophrenia. Int. J. Neuropsychopharmacol. 5, 159–179.
Breier, A., Su, T. P., Saunders, R., Carson, R. E., Kolachana, B. S., de Bartolomeis, A., Weinberger, D. R., Weisenfeld, N., Malhotra, A. K., Eckelman, W. C., and Pickar, D. (1997). Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc. Natl. Acad. Sci. U.S.A. 94, 2569–2574.
Carlsson, A. (2003). “Historical aspects and future directions,” in Dopamine in the Pathophysiology and Treatment of Schizophrenia, eds S. Kapur and Y. Lecrubier (London: Martin Dunitz), 1–13.
Cowan, J. D., and Friedman, A. E. (1995). “Development and regeneration of eye-brain maps”, in The Handbook of Brain Theory and Neural Networks, ed. M. A. Arbib (Cambridge, MA: MIT Press), 295–299.
Davis, K. L., Stewart, D. G., Friedman, J. I., Buchsbaum, M., Harvey, P. D., Hof, P. R., Buxbaum, J., and Haroutunian, V. (2003). White matter changes in schizophrenia. Arch. Gen. Psychiatry 60, 443–456.
DeFelipe, J., Fields, R. D., Hof, P. R., Höistad, M., Kostovic, I., Meyer, G., and Rockland, K. S. (2010). Cortical white matter: beyond the pale remarks, main conclusions and discussion. Front. Neuroanat. 4:4. doi: 10.3389/neuro.05.004.2010
Di, X., Chan, R. C., and Gong, Q. (2009). White matter reduction in patients with schizophrenia as revealed by voxel-based morphometry: an activation likelihood estimation meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 33, 1390–1394.
Fellin, T., Pascual, O., and Haydon, P. G. (2006). Astrocytes coordinate synaptic networks: balanced excitation and inhibition. Physiology 21, 208–215.
Fields, R. D., and Ni, Y. (2010). Nonsynaptic communication through ATP release from volume-activated anion channels in axons. Sci. Signal. 3, ra 73.
Ford, J. M., and Mathalon, D. H. (2008). Neural synchrony in schizophrenia. Schizophr. Bull. 34, 904–906.
Francois, J., Ferrandon, A., Koning, E., Angst, M. J., Sandner, G., and Nehlig, A. (2009). Selective reorganization of GABAergic transmission in neonatal ventral hippocampal-lesioned rats. Int. J. Neuropsychopharmacol. 12, 1097–1110.
Grinband, J., Hirsch, J., and Perrera, V. P. (2006). A neural representation of categorization uncertainty in the human brain. Neuron 49, 757–763.
Halassa, M. M., Fellin, T., Takano, H., Dong, J., and Haydon, P. G. (2007). Synaptic islands defined by the territory of a single astrocyte. J. Neurosci. 27, 6473–6477.
Halassa, M. M., and Haydon, P. G. (2010). Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu. Rev. Physiol. 72, 335–355.
Haydon, P. G., and Carmignoto, G. (2006). Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 86, 1009–1031.
Honer, W. G., Hurwitz, T., Li, D. K., Palmer, M., and Paty, D. W. (1987). Temporal lobe involvement in multiple sclerosis patients with psychiatric disorders. Arch. Neurol. 44, 187–190.
Höistad, M., Segal, D., Takahashi, N., Sakurai, T., Buxbaum, J. D., and Hof, P. R. (2009). Linking white and grey matter in schizophrenia: oligodendrocyte and neuron pathology in the prefrontal cortex. Front. Neuroanat. 3:9. doi: 10.3389/neuro.05.009.2009
Hubel, D. H., and Wiesel, T. N. (1968). Receptive fields and functional architecture of monkey striate cortex. J. Physiol. 195, 215–244.
Husain, F. T., Fromm, S. J., Pursley, R. H., Hosey, L. A., Braun, A. R., and Horwitz, B. (2006). Neural basis of categorization of simple speech and nonspeech sounds. Hum. Brain Mapp. 27, 636–651.
Ishibashi, T., Dakin, K. A., Stevens, B., Lee, P. R., Kozlov, S. V., Stewart, C. L., and Fields, R. D. (2006). Astrocytes promote myelination in response to electrical impulses. Neuron 49, 823–832.
Káradóttir, R., and Attwell, D. (2007). Neurotransmitter receptors in the life and death of oligodendrocytes. Neuroscience 145, 1426–1438.
Kettenmann, H., and Steinhäuser, C. (2005). “Receptors for neurotransmitters and hormones”, in Neuroglia, eds H. Kettenmann and B. R. Ransom (Oxford: Oxford University Press), 131–145.
Krippendorff, K. (1986). A Dictionary of Cybernetics. The Annenberg School of Communications, Philadelphia; University of Pennsylvania.
Kristiansen, L. V., Huerta, I., Beneyto, M., and Meador-Woodruff, J. H. (2007). NMDA receptors and schizophrenia. Curr. Opin. Pharmacol. 7, 48–55.
Kyriakopoulos, M., Perez-Iglesias, R., Woolley, J. B., Kanaan, R. A., Vyas, N. S., Barker, G. J., Frangou, S., and McGuire, P. K. (2009). Effect of age at onset of schizophrenia on white matter abnormalities. Br. J. Psychiatry 195, 346–353.
Laruelle, M., Abi-Dargham, A., van Dyck, C. H., Gil, R., D’Souza, C. D., Erdos, J., McCance, E., Rosenblatt, W., Fingado, C., Zoghbi, S. S., Baldwin, R. M., Seibyl, J. P., Krystal, J. H., Charney, D. S., and Innis, R. B. (1996). Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug free schizophrenic subjects. Proc. Natl. Acad. Sci. U.S.A. 93, 9235–9240.
McCarthy, K. D., and Salm, A. K. (1991). Pharmacologically-distinct subsets of astroglia can be identified by their calcium response to neuroligands. Neuroscience 2/3, 325–333.
McKenna, P. J. (2010). “Neurochemical theories of schizophrenia,” in Therapeutic Strategies in Schizophrenia, eds A. M. Mortimer and P. J. McKenna (Oxford: Clinical Publishing), 173–191.
Meltzer, H. Y., and Nash, J. F. (1991). Effects of antipsychotic drugs on serotonin receptors. Pharmacol. Rev. 21, 106S-115S.
Mitterauer, B. (2003). The loss of self-boundaries: towards a neuromolecular theory of schizophrenia. BioSystems 72, 209–215.
Mitterauer, B. (2004). Imbalance of glial-neuronal interaction in synapses: a possible mechanism of the pathophysiology of bipolar disorder. Neuroscientist 10, 199–208.
Mitterauer, B. (2005). Nonfunctional glial proteins in tripartite synapses: a pathophysiological model of schizophrenia. Neuroscientist 11, 192–198.
Mitterauer, B. (2009). Loss of function of glial gap junctions may cause severe cognitive impairments in schizophrenia. Med. Hypotheses 73, 393–397.
Mitterauer, B. (2010b). Many realities: outline of a brain philosophy based on glial-neuronal interactions. J. Intell. Syst. 19, 1–29.
Mitterauer, B., and Kopp, C. (2003). The self-composing brain: towards a glial-neuronal brain theory. Brain Cogn. 51, 357–367.
Mitterauer, B., and Pritz, W. F. (1978). The concept of the self: a theory of self-observation. Int. Rev. Psychoanal. 5, 179–188.
Mitterauer, B. J. (1998). An interdisciplinary approach towards a theory of consciousness. BioSystems 45, 99–121.
Mitterauer, B. J. (2007). Where and how could intentional programs be generated in the brain? A hypothetical model based on glial-neuronal interactions. BioSystems 88, 101–112.
Mori, K., Nagao, H., and Yoshihara, Y. (1999). The olfactory bulb: coding and processing of odor molecular information. Science 286, 711–715.
Newman, E. A. (2005). “Glia and synaptic transmission”, in Neuroglia, eds H. Kettenmann and B. R. Ransom (Oxford: Oxford University Press), 355–366.
Oberheim, N. A., Wang, X., Goldman, S., and Nedergaard, M. (2006). Astrocytic complexity distinguishes the human brain. Trends Neurosci. 29, 547–553.
Okubo, Y., Suhara, T., Suzuki, K., Kobayashi, K., Inoue, O., Terasaki, O., Someya, Y., Sassa, T., Sudo, Y., Matsushima, E., Iyo, M., Tateno, Y., and Toru, M. (1997). Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature 385, 634–636.
Pilowsky, L. S., Bressan, R. A., Stone, J. M., Erlandsson, K., Mulligan, R. S., Krystal, J. H., and Ell, P. J. (2005). First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol. Psychiatry 11, 118–119.
Raedler, T. J., Bymaster, F. P., Tandon, R., Copolov, D., and Dean, B. (2007). Towards a muscarinic hypothesis of schizophrenia. Mol. Psychiatry 12, 232–246.
Rall, W. (1995). “Theoretical significance of dendritic trees for neuronal input-output relations,” in The Theoretical Foundation of Dendrite Function, eds I. Segev, J. Rinzel, and G. M. Shephard (Cambridge: MIT Press), 122–146.
Rash, J. E. (2010). Molecular disruptions of the panglial syncytium block potassium siphoning and axonal saltatory conduction: pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience 168, 982–1008.
Robertson, J. M. (2002). The astrocentric hypothesis: proposed role of astrocytes in consciousness and memory formation. J. Physiol. Paris 96, 251–255.
Saakov, B. A., Khoruzhaya, T. A., and Bardakhchyan, E. A. (1977). Ultrastructural mechanisms of serotonin demyelination. Biulleten Eksperimental noi Biologii I Meditsiny 83, 606–610.
Seeman, P. (2009). Glutamate and dopamine components in schizophrenia. J. Psychiatry Neurosci. 34, 143–149.
Skelly, L. R., Calhoun, V., Meda, S. A., Kim, J., Mathalon, D. H., and Pearlson, G. D. (2008). Diffusion tensor imaging in schizophrenia: relationship to symptoms. Schizophr. Res. 98, 157–162.
Stein, L., and Wise, C. D. (1971). Possible etiology of schizophrenia: progressive damage to the noradrenergic reward system by 6-hydroxydopamine. Science 171, 1032–1036.
Stevens, B., Porta, S., Haak, L. L., Gallo, V., and Fields, R. D. (2002). Adenosine, a neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron 36, 855–868.
Stevens, B., Tanner, S., and Fields, R. D. (1998). Control of myelination by specific patterns of neural impulses. J. Neurosci. 18, 9303–9311.
Takahashi, N., Sakurai, T., Davis, K. L., and Buxbaum, J. D. (2010). Linking oligodendrocyte and myelin dysfunction to neurocircuitry abnormalities in schizophrenia. Prog. Neurobiol. 93, 13–24.
Thomas, G. G. (1985). Introduction to kenogrammatics, Proceedings of the 13th Winter School on Abstract Analysis, Section of Topology. Rendiconti del Circolo Matematico di Palermo 11, 113–123.
Werner, G. (2007). Perspectives on the neuroscience of cognition and consciusness. BioSystems 87, 82–95.
Wolf, R. C., Höse, A., Frasch, K., Walter, H., and Vasic, N. (2008). Volumetric abnormalities with cognitive deficits in patients with schizophrenia. Eur. Psychiatry 23, 541–548.
Keywords: glial–neuronal interactions, demyelination, schizophrenia, synaptic imbalance
Citation: Mitterauer BJ and Kofler-Westergren B (2011) Possible effects of synaptic imbalances on oligodendrocyte–axonic interactions in schizophrenia: a hypothetical model. Front. Psychiatry 2:15. doi:10.3389/fpsyt.2011.00015
Received: 17 January 2011;
Accepted: 28 March 2011;
Published online: 12 April 2011.
Edited by:
Miles A. Whittington, Newcastle University, UKReviewed by:
Daniel J. Lodge, University of Texas Health Science Center in San Antonio, USAGerhard Werner, University of Texas at Austin, USA
Copyright: © 2011 Mitterauer and Kofler-Westergren. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Birgitta Kofler-Westergren, Forensic Neuropsychiatry, University of Salzburg, Ignaz-Harrer Strasse 79, A-5020 Salzburg, Austria. e-mail:YmlyZ2l0dGEua29mbGVyLXdlc3RlcmdyZW5Ac2JnLmFjLmF0