

- 1 Department of Pharmacology, Bowles Center for Alcohol Studies, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
- 2 Department of Psychiatry, Bowles Center for Alcohol Studies, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
Repeated drug use/abuse amplifies psychopathology, progressively reducing frontal lobe behavioral control, and cognitive flexibility while simultaneously increasing limbic temporal lobe negative emotionality. The period of adolescence is a neurodevelopmental stage characterized by poor behavioral control as well as strong limbic reward and thrill seeking. Repeated drug abuse and/or stress during this stage increase the risk of addiction and elevate activator innate immune signaling in the brain. Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is a key glial transcription factor that regulates proinflammatory chemokines, cytokines, oxidases, proteases, and other innate immune genes. Induction of innate brain immune gene expression (e.g., NF-κB) facilitates negative affect, depression-like behaviors, and inhibits hippocampal neurogenesis. In addition, innate immune gene induction alters cortical neurotransmission consistent with loss of behavioral control. Studies with anti-oxidant, anti-inflammatory, and anti-depressant drugs as well as opiate antagonists link persistent innate immune gene expression to key behavioral components of addiction, e.g., negative affect-anxiety and loss of frontal–cortical behavioral control. This review suggests that persistent and progressive changes in innate immune gene expression contribute to the development of addiction. Innate immune genes may represent a novel new target for addiction therapy.
Introduction
Chronic use of alcohol, stimulants, and/or opiates leads to progressive changes in brain and behavior. Addiction is the continued use of a drug despite harm, e.g., a loss of behavioral control over drug use. The frontal cortex regulates decision making and other executive functions, such as motivation, planning, goal setting, and inhibition of impulses. In contrast, the amygdala, hippocampus, and other limbic structures contribute to emotion, emotional learning, and mood. Persistent use of alcohol and other drugs of abuse result in changes to neurobiology that culminate in a loss of attention, poor decision making, increased impulsivity, and anxious urgency that promotes the progressive loss of behavioral control over drug use. Although it is well accepted that drug intoxication changes neurochemistry ultimately leading to altered behavior, the importance of persistent drug-induced changes in the brain that underlie persistent harmful behaviors has only recently been appreciated. Indeed, drug dependence and addiction involves a disruption of the normal balance between self-control mechanisms and emotional needs. Across drugs of addiction, the progression from abuse to addiction involves increased drug wanting, negative emotional urgency, and diminished behavioral control (Jentsch and Taylor, 1999; Robinson and Berridge, 2003). Frontal lobe executive function involves the ability to recognize future consequences, choose between good and bad actions (or better and best), override and suppress unacceptable social responses, and determine similarities and differences between things or events. This cortical region also plays an important role in retaining long-term memories associated with emotions derived from the brain’s limbic system. These emotions are then modified via the frontal cortex to generally fit societal norms important for individual integration into society. In addition, the frontal lobes also inhibit impulsivity, making predictions that adjust behavior to current rewards when environment changes (Schoenbaum and Shaham, 2008). Thus, a key element of the behavioral pathology of addiction and substance dependence centers on the loss of frontal–cortical executive behavioral control, increased impulsivity, reduced behavioral flexibility, and a mounting limbic anxiety and urgency.
Adolescent Brain Development and Addiction
Adolescence: A Unique Period of Development
Adolescence is a critical developmental period that encompasses the transition from childhood to adulthood. It is best defined by characteristic behaviors that include high levels of risk-taking, increased exploration, novelty and sensation seeking, social interaction, high activity, and play behaviors that likely promote the acquisition of skills necessary for maturation and independence (Spear, 2000; Ernst et al., 2009). These behaviors are suggested to facilitate the adolescents’ development of social skills necessary to gain independence from their family or become senior adults in their group. In rodents, increased social interactions help guide their food choices (Galef, 1977) and other adult actions, such as sexual and aggressive behaviors (see e.g., Fagen, 1976; Smith, 1982). Unfortunately, the increased incidence of novelty/sensation-seeking behaviors during adolescence are also strong predictors of drug and alcohol use (Baumrind, 1987; Andrucci et al., 1989; Wills et al., 1994; Faden, 2006). Indeed, the adolescent brain is in a unique state of transition as it undergoes both progressive and regressive changes providing a biological basis for unique adolescent behaviors and the associated changes in these behaviors during maturation to adulthood. Human magnetic resonance imaging (MRI) studies have demonstrated an inverted U-shape change in gray matter volume during the adolescent period, with pre-adolescent increases followed by post-adolescent reductions (Giedd et al., 1999; Giedd, 2004). At the cellular level, these changes correspond with a marked overproduction of axons and synapses during early puberty, but rapid pruning in later adolescence (Giedd et al., 1999; Andersen et al., 2000; Andersen and Teicher, 2004). Although the exact mechanisms underlying such synaptic changes are not well understood, it is speculated that such remodeling is the biological basis of developmental plasticity wherein the neurological circuits are effectively shaped to adapt to environmental needs leading to mature adult behavior. Such a period of remodeling could also make the adolescent brain more vulnerable to external insults and other psychiatric disorders.
The prefrontal cortex (PFC) and the limbic system, which includes the hippocampus, amygdala, nucleus accumbens (NAcc), and the hypothalamus, undergo prominent reorganization during adolescence. Indeed, absolute PFC gray matter volumes decline in humans (Sowell et al., 1999, 2001) as well as in rats (van Eden et al., 1990) during adolescence. Similarly, a substantial loss of synapses, especially excitatory glutamatergic inputs to the PFC, occur during the adolescent period in humans and non-human primates (Huttenlocher, 1984; Zecevic et al., 1989). In contrast to such adolescent-associated pruning, dopaminergic, and serotonergic inputs to the PFC increase to peak levels well above those observed earlier or later in life (Kalsbeek et al., 1988; Rosenberg and Lewis, 1994). In a similar fashion, cholinergic innervation of the PFC also increases at this time point, ultimately reaching mature levels in rats (Gould et al., 1991) and humans (Kostovic, 1990). Within the hippocampus, the exuberant outgrowth of excitatory axon collaterals and synapses during youth are morphologically remodeled, and branches within dendritic arbors are pruned during this period of maturation (Swann et al., 1999 #3027). Similarly, significant dendritic pruning and synaptic regression also occurs in the medial amygdala (Zehr et al., 2006), NAcc (Teicher et al., 1995; Tarazi et al., 1998b), and hypothalamus (Choi and Kellogg, 1992; Choi et al., 1997). Although most synaptic pruning is likely glutamatergic, dopaminergic receptor expression peaks in early adolescence at postnatal day (P) 28 followed by a one-third reduction of receptors between P35 and P60 (Tarazi et al., 1998a). In terms of hypothalamic function, adolescent rats often exhibit more prolonged stress-induced increases in cortisol than adults (Walker et al., 2001). In addition, rats at P28 evidence less stress-induced Fos-like immunoreactivity in cortical and amygdaloid nuclei than adult rats (Kellogg et al., 1998), but higher novelty-induced Fos activation in the hippocampus during this period (Waters et al., 1997). Thus, significant maturation of the cortical and limbic systems characterizes the adolescent period of development.
Behavioral studies have demonstrated that performance on tasks involving inhibitory control, decision making, and processing speed continues to develop during adolescence. During this developmental stage, selective attention, working memory, and problem solving skills consistently improve as frontal–cortical synaptic pruning and myelination progress (Blakemore and Choudhury, 2006). Similarly, executive inhibitory control improves from adolescence through to adulthood. Studies measuring behavioral inhibition on a Go–No-Go task and functional MRI data reveal greater activation of dorsolateral frontal and orbitofrontal cortices in children than adolescence, and greater activation during adolescence than adults with the adults showing the lowest dorsolateral, but equal orbitofrontal activation and greater inhibitory control performance (Casey et al., 1997; Tamm et al., 2002). These studies support the concept that the immature brain, with excess synapses, possesses more extensive, and less efficient frontal activation and lower performance than adults that have a more efficient frontal cortex that results in more focused, lower overall activation and faster reaction times and better performance (Blakemore and Choudhury, 2006). Taken together, these studies suggest that remodeling of the cortex during the developmental transition from youth to adolescence to adulthood has functional implications for the adult stages of life.
Neurogenic Processes in the Adolescent Brain
Although neurogenesis is primarily an early developmental process with most neurons generated during the prenatal and early postnatal periods, it continues throughout adulthood in discrete brain regions, including the forebrain subventricular zone and subgranular zone of the hippocampal dentate gyrus. The generation and functional integration of nascent neurons into preexisting adult neural circuits is believed to enable the hippocampus to adapt to novel and more complex situations (Kempermann, 2002). Indeed, the contribution of hippocampal neurogenesis to learning and memory (Shors et al., 2001) as well as mood and affective state (Malberg et al., 2000) is supported by many studies. Adolescent neurogenesis, and its role in brain remodeling and unique adolescent behaviors, has to date not been investigated. Studies indicate that adolescent animals have higher levels of hippocampal neurogenesis (He and Crews, 2007), but that neurogenesis in the adolescent brain is very sensitive to alcohol-induced degeneration (Crews et al., 2006a). Thus, disruption of the neurogenic process by drugs and alcohol use during adolescence might produce long-lasting changes that persist into adulthood.
Binge Drinking during Critical Periods in Cortical Development might Lead to Lifelong Changes in Executive Function
The effect of alcohol on the adolescent brain is different from those observed in adulthood. Adolescents are less sensitive to the sedative effects of alcohol (Silveri and Spear, 1998), which allows them to binge drink. However, they are more vulnerable to alcohol-induced neurotoxicity (Monti et al., 2005; Crews et al., 2007). The increased sensitivity of the adolescent brain to alcohol-induced toxicity (Peleg-Oren et al., 2009), coupled with the dynamic synaptic remodeling that characterizes this stage, might strengthen the learning components of heavy drinking behaviors and perpetuate the loss of important self-control and goal setting components of the maturing brain’s executive centers. Indeed, studies of adolescent individuals with alcohol use disorder have demonstrated smaller prefrontal gray and white matter volumes than age-matched controls. These lower PFC volumes, in turn, correlate with a higher maximum number of drinks per drinking episode (De Bellis et al., 2005). Furthermore, binge ethanol exposure during adolescence reduces D1 and D2 receptors in the frontal cortex while simultaneously increased histone acetylation in the frontal cortex and limbic system (Pascual et al., 2009). Thus, it is likely that both genetics and environment (heavy drinking) contribute to the development of an alcohol use disorder and lower PFC volumes in adolescents. Studies of social drinkers have found that the heaviest binge drinkers have more negative moods and performed worse on executive function tasks (Townshend and Duka, 2003; Weissenborn and Duka, 2003). Furthermore, alcoholics report more fear in facial expressions and animal studies have suggested these alterations in fear response are the result of alcohol-induced deficits in associative learning (Duka et al., 2004). Additional studies have demonstrated perseverative relearning deficits following a rat model of binge drinking that relates to damage of the association cortex (Obernier et al., 2002). However, none of these studies directly reveal a critical period during adolescence when executive function is liable to disruption by ethanol. In contrast, other work on the deleterious effects of ethanol on critical periods involving of visual cortical development, coupled with ethanol-induced cortical neurotoxicity and ethanol-induced alterations in executive function, support the theory that disruption of frontal–cortical development and executive function maturation occurs in adolescent alcohol abusers. It is plausible that adolescent alcohol abuse might disrupt impulse inhibition, attention, and motivation thereby promoting adult alcohol dependence and underlie the high risk of lifetime alcohol dependence found among those who begin drinking as adolescents. In total, the evidence does support a link between adolescent alcohol abuse during a critical period of executive function maturation and an increased risk of lifetime alcohol dependence and perhaps other psychopathologies.
Drugs and Stress Induce Innate Immune Genes through Activation of NF-κB Transcription
Neuroimmune signaling contributes to enteric, sensory, and endocrine hypothalamic–pituitary–adrenal (HPA) responses to external and internal environmental factors. Monocytes and tissue specific monocytes, such as brain microglia, are key cells involved in neuroimmune signaling. These cells are regularly generated from bone marrow stem cells where they migrate to blood, and under normal states, replenish tissue resident macrophages and dendritic cells, including brain microglia. Monocytes have multiple stages of activation that represent a progressive cascade of innate immune gene activation (Graeber, 2010). Monocyte responses regulate cellular movement to sites of tissue damage, secretion of chemokine signals to other cells, secretion of proinflammatory cytokines, proteases, and “danger” signaling molecules, and increased expression of Toll-like receptors (TLRs), oxidases [nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, cyclooxygenase (COX), and inducible nitric oxide synthases (iNOS)], and other innate immune molecules that increase across a spectrum of activation states that range from proinflammatory to trophic. Microglia have a low threshold of activation with initial states of activation secreting signaling molecules, slight morphological changes, upregulation of major histocompatibility complex (MHC) and TLR proteins, and activation of synaptic stripping. In contrast, highly activated microglia progress to mitosis, proliferation, and phagocytic oxidative bursts that oxidize and engulf waste (Graeber, 2010). Under healthy conditions, microglia as well as monocytes in the peripheral sensory nerves and endocrine organs contributes to the integration of sensory systems aimed at maintaining health. However, stress, alcohol, and other addictive drugs as well as sensory and hormonal signals activate the oxidation sensitive transcription factor NF-κB that is highly expressed in microglia (see Figure 1).
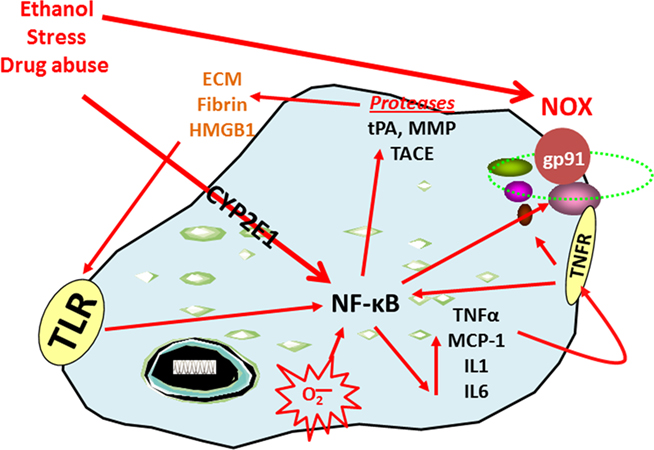
Figure 1. Nuclear factor kappa-light-chain-enhancer of activated B cells transcription increases the expression of chemokines, cytokines, oxidases, and proteases. The transcription factor NF-κB is involved in the induction of innate immune genes (Ghosh and Hayden, 2008). Stimuli such as stress, drugs of abuse, peptides, chemokines, cytokines, reactive oxygen species (ROS), ultraviolet irradiation, bacteria, viruses, trauma, and other factors all increase NF-κB-DNA binding and transcription. Reactive oxygen species resulting from oxidases such as NADPH-oxidase or ethanol metabolism by CYP2E1 increase NF-κB transcription of NOX2phox, a key NOX catalytic subunit (Cao et al., 2005) that produces ROS (Qin et al., 2008). Loops of activation also occur through induction of genes that stimulate further NF-κB activation leading to autocrine and paracrine amplification and persistent signals. Cytokines and chemokines, such as TNFα, IL1β, IL6, and MCP-1 as well as their receptors (TNFR in figure), are also induced resulting in amplification loops. Toll-like receptors are increased by ethanol (Dolganiuc et al., 2006; Alfonso-Loeches et al., 2010) as are other damage-associated molecular pattern receptors and there agonists resulting in the formation of positive activation loops (Garg et al., 2010). Toll-like receptors and HMGB1 interact to create another activation-amplification loop. Persistent and repeated activation occurs through positive cycles of activation. These loops spread innate immune signaling across the brain causing altered neurocircuitry and neurobiology. Figure abbreviations: CYP2E1, cytochrome P450 2E1; ECM, extracellular matrix; EtOH, ethanol; gp91, NADPH-oxidase flavocytochrome b components; HMGB1, high-mobility group box 1; IL-1β, interleukin-1 beta; IL1, interleukin-1; LPS, lipopolysaccharide; MMP, matrix metalloproteinase; MCP-1, monocyte chemoattractant protein-1; NOX, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; TACE, TNFα converting enzyme; TLR, toll-like receptor; TNFα, tissue necrosis factor-alpha; tPA, tissue plasminogen activator.
The transcription factor NF-κB is involved in the induction of innate immune genes in microglia and other monocyte-like cells in the periphery. Stimuli such as stress, cytokines, oxidative free radicals, ultraviolet irradiation, bacterial or viral antigens, and many other signaling molecules increase NF-κB-DNA binding and transcription of many genes, particularly chemokines, cytokines, oxidases, and proteases. Our laboratory has previously demonstrated that ethanol increases NF-κB–DNA binding in the brain in vivo (Crews et al., 2006b) and in vitro in hippocampal–entorhinal cortex slice cultures (Zou and Crews, 2006). Furthermore, work from our laboratory and others indicate that ethanol also increases the transcription of NF-κB target genes, including chemokine monocyte chemoattractant protein-1 (MCP-1, CCL2; He and Crews, 2008), proinflammatory cytokines [tumor necrosis factor-α (TNFα), Interleukin (IL)-1β, and IL-6], proinflammatory oxidases [iNOS (Zou and Crews, 2010), cyclooxygenase (COX; Knapp and Crews, 1999), and NOX (Qin et al., 2008)], and proteases (TACE and tPA; Zou and Crews, 2010). Similarly, stress increases the expression of NF-κB (Madrigal et al., 2001), cytokines, prostaglandin E2, and COX-2 levels (Madrigal et al., 2003) in the brain. In addition, chronic stress causes the reversal of acute glucocorticoid anti-inflammatory responses to proinflammatory NF-κB activation in the cortex (Munhoz et al., 2010). Similarly, all addictive drugs cause chronic elevations of basal glucocorticoids (Armario, 2010) that likely contribute to activation of brain NF-κB. Thus, activation of NF-κB by stress and drugs of abuse is a common molecular mechanism involving innate immune gene induction that is consistent with a stress-drug synergy culminating in progressive increases in loss of behavioral control and addiction.
Astrocytes and microglia show morphological changes in response to exposure to drugs of abuse. Using both in vitro and in vivo models through a series of elegant studies, Guerri and colleagues have established that chronic ethanol treatment induces astroglial activation and astrogliosis in the brain as indicated by marked upregulation of glial fibrillary acidic protein immunoreactivity along with hypertrophic astrocytes (Alfonso-Loeches et al., 2010). In addition to the altered astrocyte morphology, microglia also evidence increased expression of TLRs, which are both NF-κB target genes and activators of NF-κB transcription. Recently, TLR4 was discovered to contribute to persistent innate immune gene induction following ethanol exposure. Indeed, chronic ethanol exposure produces upregulation and activation of TLR4-glial NF-κB signaling that contributes to alcohol-induced neurodegeneration (Alfonso-Loeches et al., 2010). Similarly, acute ethanol exposure disrupts membrane lipid rafts thereby activating TLR4 signaling to NF-κB as well as increased expression of TLR4 (Blanco et al., 2008). Indomethacin, an anti-inflammatory drug, reduces chronic intermittent ethanol induction of brain innate immune genes (iNOS and COX-2) in astrocytes and reduces markers of cell death and behavioral dysfunction (Pascual et al., 2007). Innate immune activation resulting from oxidized phospholipids (Yang et al., 2010), and/or release of damage-associated molecular pattern danger sensing molecules such as high-mobility group box 1 (Garg et al., 2010), activate TLR and other signals that contribute to innate immune gene induction (Huang et al., 2010). Loops of NF-κB activation likely vary across individuals and exposure to specific addictive drugs. However, all addictive drugs activate NF-κB transcription across the development of addiction (Russo et al., 2009; Loftis et al., 2010). In an extensive series of studies, repeated bouts of moderate ethanol consumption and/or stress or innate immune activator exposure increased negative affect and anxiety in rats. The finding that the effects of ethanol and stress on brain function can be mimicked by injection of the chemokine MCP-1, the cytokine TNFα, or lipopolysaccharide (LPS), is consistent with the notion that stress and ethanol act through induction of innate immune genes to progressively increase negative affect (Breese et al., 2008). Thus, NF-κB transcription of innate immune genes in the brain occurs during exposure to ethanol and other addictive drugs as well as stress promoting vicious cycles of NF-κB induction of innate immune genes that culminate in changes to neurocircuitry and neurobiology.
In addition to alcohol, opiates are known to be addicting drugs, and endogenous opioid receptors and agonists clearly contribute to the neurobiology of addiction (Koob and Volkow, 2010). In contrast, opiate antagonists are used to treat both alcohol and opiate addiction. Interestingly, a potential mechanism of opiate antagonists appears to involve blockade of innate immune gene activation. Indeed, studies have found that opiate antagonists blunt LPS inherent immune responses (Liu et al., 2000b) and protect dopaminergic neurons via inhibition of microglial activation and reduced NOX formation of reactive oxygen species (Liu et al., 2000a; Qin et al., 2005). Other studies have demonstrated that opiate antagonists block TLR4 activation of innate immune transcription, which is a site of action in innate immune loops (Hutchinson et al., 2008, 2010). Thus, opiate antagonist therapy might exert some of its beneficial effects through blockade of innate immune gene induction. However, the progressive nature of innate immune gene induction and addictive behaviors suggest that therapeutic treatments aimed at reducing the induction of these genes would be more advantageous at preventing than reversing addiction.
Innate Immune Activation is Involved in Ethanol Drinking, Depression-Like Behavior, and Addiction
Numerous studies have investigated the neurobiological consequences of addiction. Our laboratory demonstrated that MCP-1 (CCL2), a key chemokine induced by chronic ethanol treatment in mice known to regulate ethanol consumptive behavior, is upregulated in post-mortem human alcoholic brains (see Figure 2). Neuroanatomical assessment of MCP-1 protein levels as well as histological assessment of microglia from alcoholic human brains indicate increased levels in the ventral tegmental area, substantia nigra, hippocampus, and amygdala relative to healthy control subjects (He and Crews, 2008). In another post-mortem human alcoholic brain study, Okvist et al. (2007) reported increased NF-κB nuclear binding of p50 subunits with 479 NF-κB driven genes being generally upregulated in the frontal cortex, but not motor cortex. In addition, a human gene expression analysis conducted on post-mortem tissue (Liu et al., 2006) revealed altered expression of a group of cell adhesion genes that is consistent with altered extracellular membrane components and innate immune activation. Thus, studies of human alcoholic brain are consistent with the hypothesis that drug addiction activates brain innate immune gene expression.
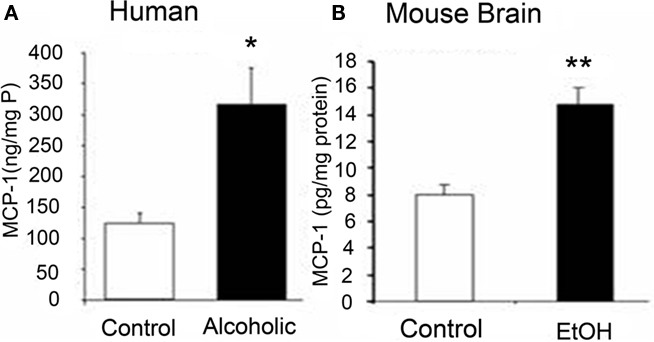
Figure 2. Comparisons of increased levels of CCL2 in post-mortem human alcoholic brain and mouse brain following chronic ethanol treatment. Shown is data from different studies within our laboratory illustrating increased levels of the chemokine MCP-1 (CCL2) in humans and mice following ethanol exposure. (A) MCP-1 protein levels from human hippocampal homogenate measured using ELISA. Increased MCP-1 levels were also found in ventral tegmental area, substantia nigra, and amygdala (see He and Crews, 2008). (B) Levels of MCP-1 in mouse brain increased following chronic ethanol treatment. Mice (C57Bl/6) treated with 10 daily doses of ethanol (5.0 g/kg. i.g.) and brain MCP-1 levels were determined 24 h after the last ethanol administration (see Qin et al., 2008 for details). These studies indicate that ethanol upregulates the innate immune chemokine MCP-1 in post-mortem human alcoholic and mouse brain samples, which is consistent with ethanol activation of innate immune genes.
The induction of innate immune gene expression is known to alter behavior. Perhaps the most serious consequence of cancer treatment with proinflammatory interferon and interleukin is the development of severe depression that requires treatment with anti-depressant medication (O’Connor et al., 2007). Several other studies have linked negative affect and depression to innate immune activation (see e.g., Kelley and Dantzer, 2011). For instance, bacterial endotoxin induces sickness behavior and negative affect across multiple species. Indeed, Eisenberger et al. (2010) recently demonstrated that infusions of LPS into healthy humans reduced reward responses and increased depressed mood. Similarly, cycles of drug abuse, stress, and other environmental changes amplify anxiety and negative affect. Interestingly, animal studies determining the genetic basis of behavior find that innate immune genes increase alcohol drinking behavior. For example, gene expression studies of genetically paired rats and mice that differ primarily in their preference for ethanol consumption find that NF-κB, its regulatory proteins, and many innate immune genes are central to high ethanol drinking behaviors (Mulligan et al., 2006). Furthermore, beta-2 microglobulin (β2M), which is a NF-κB target gene involved in MHC immune signaling (Pahl, 1999) evidenced the largest increase in high ethanol preferring brain transcriptomes (Mulligan et al., 2006). In addition, work from Blednov et al. (2005, 2011b) have provided interesting and novel data supporting the hypothesis that innate immune genes regulate ethanol drinking behavior. Across multiple strains of transgenic mice with innate immune gene deletion, these animals universally drink significantly less ethanol than matched controls across multiple ethanol drinking paradigms. Recently, Blednov et al. (2011a) discovered that innate immune activation through LPS can cause long-lasting increases in ethanol drinking. Indeed, strains of mice show varied innate immune responses to LPS that correspond to increases in the consumption of ethanol. Furthermore, a single injection of LPS is capable of producing a delayed, but long-lasting increase in ethanol consumption even in strains of high drinking mice. Similarly, a single LPS treatment induces persistent increases in brain innate immune gene expression (Qin et al., 2007). Taken together, these findings are consistent with genetic regulation of brain innate immune gene expression contributing to risk for alcoholism, alcohol drinking (both preference and quantity), and behavioral sensitivity to alcohol across multiple species.
A significant body of evidence supports the hypothesis that innate immune gene induction in the brain results in negative affect and depression-like behavior (Raison et al., 2009). Patients with major depressive disorder evidence increased blood inflammatory markers, and anti-depressant therapy is associated with a reduction of these markers. In addition to increased innate immune gene expression, human depression involves structural changes in the hippocampus as multiple studies have demonstrated decreased hippocampal volume in patients with depression (see e.g., Videbech and Ravnkilde, 2004). These findings are consistent with depression-associated diminution of hippocampal neurogenesis and anti-depressant-induced increases in neurogenesis, hippocampal volume in humans, and reversal of depressive symptomology (Dranovsky and Hen, 2006). The reductions of adult hippocampal neurogenesis may underlie depression and provide an index of mood and negative affect that allow for molecular studies. Indeed, both alcoholism and depression may be mediated by changes in adult hippocampal neurogenesis (Crews and Nixon, 2003; Koo et al., 2011). Similarly, stress, multiple addictive drugs, and other factors that precipitate depression also reduce neurogenesis (Tanapat et al., 2001; Malberg and Duman, 2003; Gregus et al., 2005). Many of the factors that reduce neurogenesis also increase depression-like behaviors (Johnson et al., 2006; see Figure 3). Recent research has revealed that activation of NF-κB is necessary for stress-induced inhibition of neurogenesis and induction of depression-like behaviors (Koo and Duman, 2008), such as the social defeat model of depression (Christoffel et al., 2011). In addition, anti-depressant efficacy in rodent behavioral models is dependent upon hippocampal neurogenesis (Santarelli et al., 2003). In animal studies, endotoxin-induced increases in innate immune genes reduce neurogenesis and increase depression-like behavior (Kelley and Dantzer, 2011). Immune activation includes induction of microglial tryptophan metabolism that could reduce serotonin thereby contributing to depression (Kelley and Dantzer, 2011). TLRs are necessary components of both ethanol neurotoxicity (Alfonso-Loeches et al., 2010) and innate immune-induced depressive behavior and reduction of neurogenesis (Kelley and Dantzer, 2011). We have found that chronic ethanol increases brain innate immune genes, reduces brain neurogenesis, and increases depression-like behavior. In addition, mice self-administering ethanol in a chronic heavy drinking model evidenced depression-like behavior during abstinence that was associated with reduced neurogenesis (Stevenson et al., 2009). Ethanol-induced loss of neurogenesis parallels the onset of depression-like behavior, which is reversed via anti-depressant treatment. Similarly, stress-induced IL-1β reduces neurogenesis causing depression-like behaviors (Koo and Duman, 2008). Inhibition of neurogenesis is also associated with negative affect and depression, which are key elements in the neurobiology of addiction. Thus, neurogenesis reflects mood, with reduced neurogenesis associated with innate immune gene induction, drug-induced negative affect, and depression-like behavior.
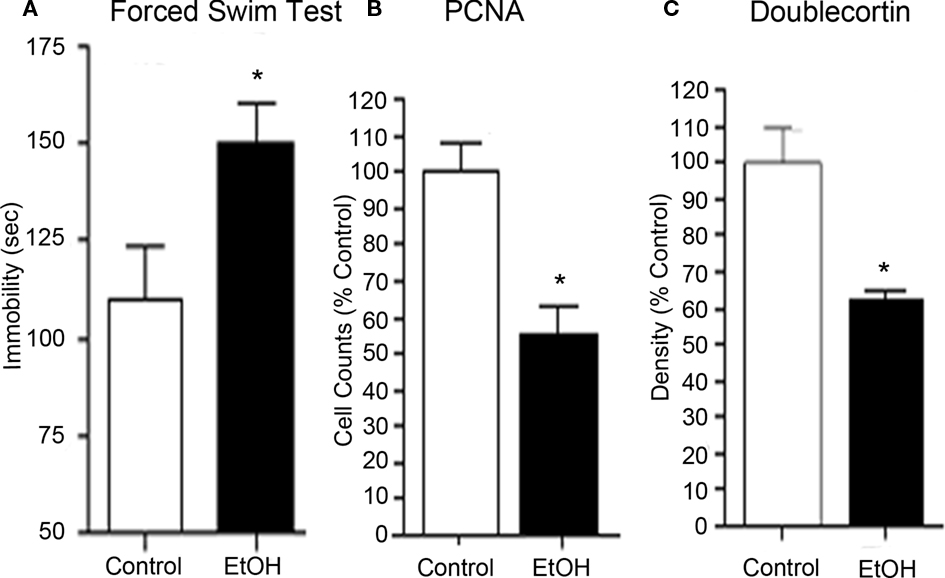
Figure 3. Chronic ethanol self-administration induces depression-like behavior and inhibits hippocampal neurogenesis. C57BL/6J mice self-administered either ethanol (10% v/v) or water for 28 days. (A) Abstinence-induced increase in immobility (seconds) on the forced swim test provides an index of depression-like behavior. Abstinence from chronic ethanol consumption resulted in increased negative affect. (B) Ethanol self-administration decreased PCNA, a marker of cell proliferation, in the neurogenic region of the hippocampal dentate gyrus. (C) Ethanol self-administration decreased doublecortin expression, a marker of neurogenesis, in the dentate gyrus. Reduced progenitor cell proliferation and neurogenesis is associated with increased depression-like behavior. Furthermore, these studies are consistent with the research suggesting that decreased hippocampal neurogenesis is linked to depression. Finally, desipramine treatment, an anti-depressant, reversed both the reduced hippocampal neurogenesis and the depression-like behavior in abstinent mice (see Stevenson et al., 2009).
Innate immune gene activation in the brain persists for long periods (Qin et al., 2007, 2008), consistent with the persistence of addiction. This persistent nature is likely amplified in the adolescent brain (Spear, 2000) because of their increased ethanol consumption (Silveri and Spear, 1998) and greater vulnerability to the neurotoxic effects of alcohol (Monti et al., 2005; Crews et al., 2007). As such, chronic intermittent ethanol exposure during adolescence increases COX-2 and iNOS expression as well as apoptotic cell death in the neocortex and hippocampus (see Figure 4). Importantly, and relevant to the potential involvement of innate immune gene induction in adolescent binge drinking, administration of indomethacin attenuates the behavioral dysfunction associated with adolescent intermittent ethanol in early adulthood (Pascual et al., 2007).
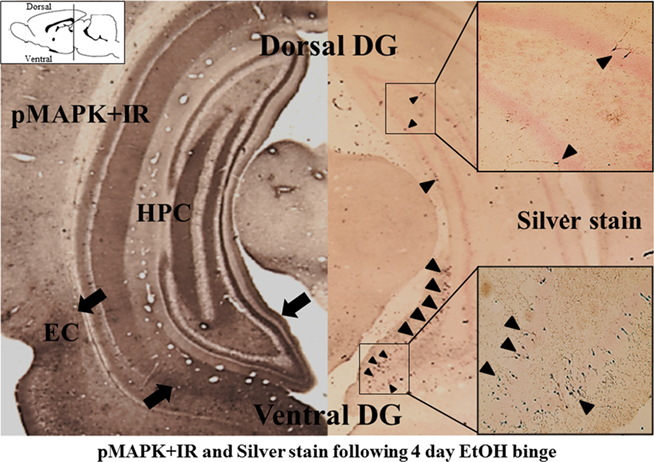
Figure 4. Ventral hippocampus shows greater activation (pMAPK + IR) and neurotoxicity (Silver stain) following binge ethanol treatment. Coronal sections of rats exposed to a 4-day binge ethanol model are shown [approximately −5.80 mm from bregma, adapted from Crews et. al. (2006a; The position of the coronal histological sections are depicted in the sagittal diagram in the upper left corner. The histological coronal sections show both dorsal (upper) and ventral (lower) dentate gyrus of hippocampus)]. Left: Mitogen-activated protein kinase (MAPK) is a family of kinases activated by phosphorylation. Phosphorylated MAPK (pMAPK) provides an index of kinase activation. Note the lower-ventral dentate gyrus, hippocampus, and entorhinal cortex (indicated by black arrows) contains more pMAPK + IR than the upper-dorsal sections consistent with greater activation of ventral hippocampus. Right: Silver stain identifies dying neurons (see Crews et. al., 2006a). Note that the ventral hippocampus contains many more silver stained neurons (black arrowheads) compared to the ventral hippocampus. Boxes on the right show higher magnification of silver stained dorsal and ventral hippocampus. These findings are consistent with ethanol causing greater emotional ventral hippocampus activation (pMAPK + IR) and cell death (silver stain).
Maturation of the frontal cortex during adolescence is paralleled by the development of behavioral control (Ernst et al., 2009). Adolescence is a recognized risk period for the initiation of drug experimentation and addiction due to the vulnerability of the developing frontal cortex (Crews et al., 2007). Other studies have suggested that genetic factors linked to a hyperglutamatergic state might contribute to alcoholism (Spanagel et al., 2005) and ethanol-induced NF-κB activation to increased extracellular glutamate (Ward et al., 2009). Innate immune gene induction results in hyperexcitability in the spinal cord related to neuropathic pain (Graeber, 2010) and in the hippocampus related to seizures (Maroso et al., 2010). Similarly, hyperexcitability in the frontal cortex results in loss of cognitive flexibility creating addiction-like behavior (Gruber et al., 2010). In elegant studies by Kaliva and colleagues have established that cocaine and stimulant addiction are related to a hyperglutamatergic states due to alterations of the cortical glutamate transporters (Reissner and Kalivas, 2010). Studies of both human cocaine and alcohol addicts have revealed dysfunctional decision making on tasks involving delayed reward for more value and reversal learning tasks that probe cognitive flexibility and frontal lobe function (Bechara et al., 2002). Thus, frontal–cortical hyperexcitability due to innate immune gene induction likely contributes to the neurobiology of addiction.
Frontal–cortical dysfunction is often investigated using reversal learning tasks. During reversal learning, expected outcomes are incorrect requiring flexible behavior in response to outcomes that do not match those predicted by the preceding cues (Stalnaker et al., 2009). In behavioral studies, this learning paradigm mimics the inability of drug addicted individuals to learn new healthy behaviors. Thus, proper frontal cortex function is needed to weigh the value of decisions and is important when new learning and/or behavior is necessary. Our laboratory found that models of binge ethanol drinking induces persistent deficits in reversal learning in rats (Obernier et al., 2002) and in adult mice following a model of adolescent binge drinking (Coleman et al., 2011). Other studies have demonstrated that rats with previous experience, either with self-administration of cocaine or with passive cocaine injections, are abnormally slow to learn reversals even though they learn initial contingencies at a normal rate (Schoenbaum et al., 2004; Calu et al., 2007). Furthermore, lesion of the frontal cortex produce reversal learning deficits similar in nature to chronic drug abuse-induced deficits (Schoenbaum et al., 2006). In addition, frontal cortex dysfunction results in perseveration and repetition of previously learned behaviors due to failure to associate new information (e.g., negative consequences) into decision making. Thus, innate immune gene induction disrupts frontal–cortical functions leading to loss of behavioral control. Similarly, limbic negative affect is promoted by innate immune gene induction. Together, the loss of behavioral control and increased limbic drive due to innate immune gene induction is consistent with innate immune gene induction culminating in the neurobiology of addiction.
Conclusion
The neurobiology of addiction is complex (see Figure 5) and high rates of co-morbid depression psychopathology suggest common overlapping molecular changes in the brain (Grant and Dawson, 1998). Drug-induced induction of brain innate immune genes was initially thought to reflect drug-induced neurodegeneration. However, more recent studies suggest that increased glutamate hyperexcitability in the frontal cortex occurs as well as increased sensitivity to excitotoxicity. Recent research supports a role for innate immune gene induction in altered neurotransmission and neurocircuitry that contribute to the dysfunctional behaviors associated with addiction. Indeed, increased innate immune gene expression in increasingly associated with the molecular mechanisms underlying negative affect, anxiety, and depression that are known to increase in the addicted brain. The recent discovery that chronic glucocorticoids, elevated by stress and/or drug abuse, promote NF-κB proinflammatory transcription in the frontal cortex support a common molecular mechanism of drug abuse and stress promoting common changes in neurobiology that parallel the progressive and persistent psychopathology of addiction. Increased innate immune gene expression in post-mortem brain tissue of addicted individuals’ mimic findings in pre-clinical studies of drug- and stress-induced activation of brain NF-κB transcription and the prolonged psychopathology of chronic addiction. Stimuli that activate brain innate immune gene expression independent of any addictive drug experience promote addiction-like behaviors and increase drug consumption. Human genetic studies on alcohol dependence have also found innate immune gene polymorphisms that are associated with risk for alcoholism. A promising recent discovery that anti-opiate drugs used to treat addiction also disrupt innate immune gene induction suggests new and unanticipated mechanisms of action. Taken together, these findings support innate immune gene induction as a key mechanism causing addiction. This new mechanism includes many new and novel targets for addiction, depression, and other psychopathology. It is hoped that the discoveries of the role of innate immune genes in addiction will lead to improved prevention and/or therapy for addiction.
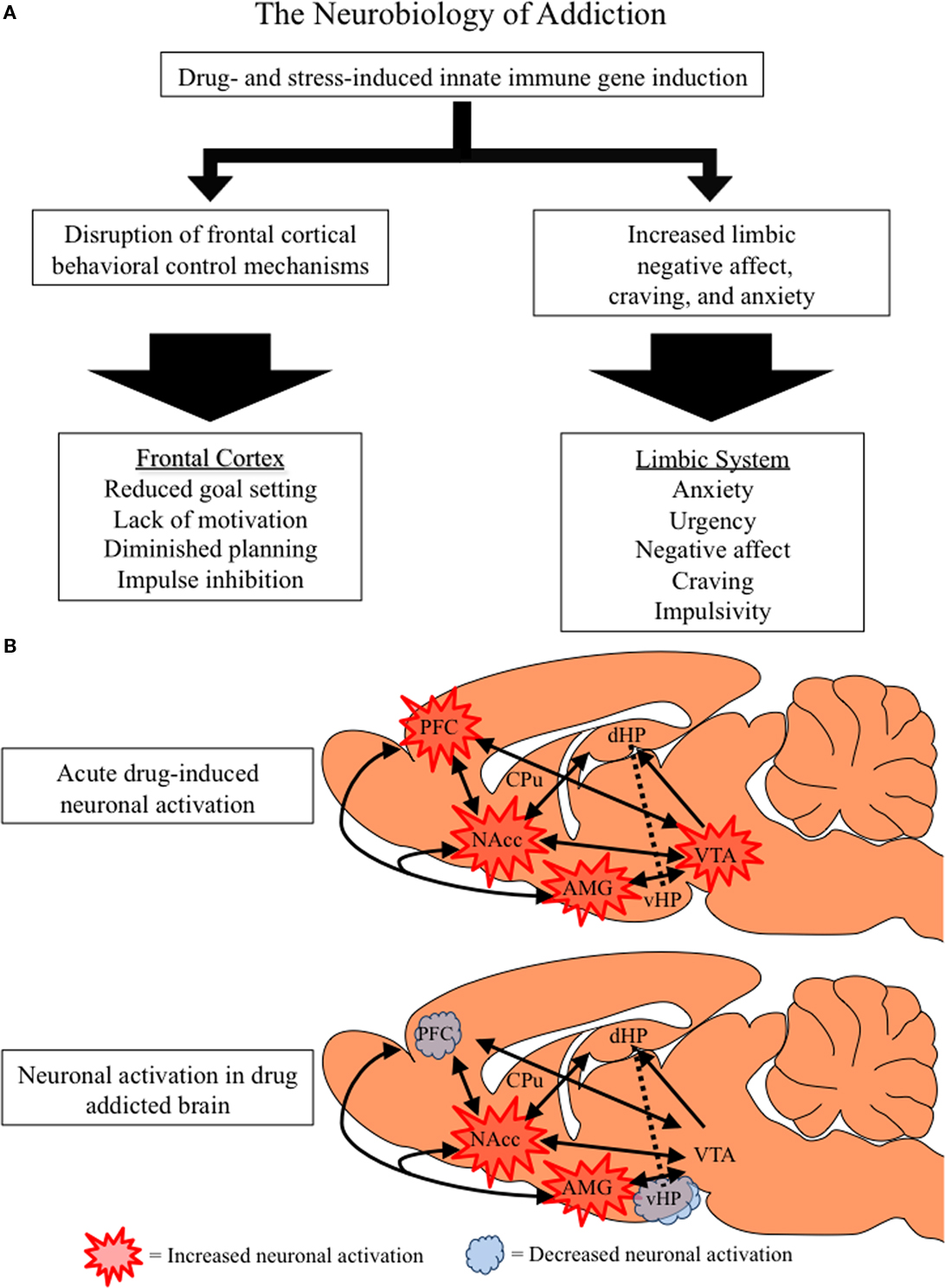
Figure 5. The neurobiology of addiction. (A) Flow chart distinguishing the frontal–cortical and limbic changes associated with drug addiction. Both stress and drug abuse activate innate immune gene expression, which increases limbic activation and disrupts frontal–cortical function. (B) A simplified schematic of the frontal–cortical and limbic circuitry that contributes to addictive behavior. Depicted is a rat brain with internal structures highlighted and accompanying projections (as indicated by black arrows). The frontal–cortical areas include the medial prefrontal, anterior cingulate, and orbitofrontal cortices, and are involved in attention, goal setting, planning, and impulse control (Schoenbaum et al., 2006; Schoenbaum and Shaham, 2008). The limbic circuitry, comprising the nucleus accumbens (NAcc), amygdala (AMG), hippocampus (HPC), and ventral tegmental area (VTA), is involved in emotion, learning, and memory. Acute drug abuse activates frontal–cortical attention mechanisms, prompting limbic learning. Similarly, innate immune gene induction in the brain leads to PFC hyperexcitability (Zou and Crews 2005; Crews et al., 2006a) that inactivates frontal–cortical regulation of limbic structures (Gruber et al., 2010). Innate immune gene induction in limbic regions increases negative affect and depression-like behaviors prompting further drug abuse and self-medication. The harmful consequences of prolonged alcohol, opiate and stimulant drug dependence result in diminished activation of frontal–cortical circuits leading to a loss of attention and poor decision combined with increasing urgency and negative affect motivating persistent drug taking behaviors. Decreased ventral hippocampal activation likely contributes to frontal hyperexcitability and loss of cognitive flexibility (Gruber et al., 2010). Thus, an inactivated PFC, loss of behavioral flexibility, and increasing limbic negative emotion characterizes the drug-addicted brain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Authors wish to acknowledge support from the Bowles Center for Alcohol Studies, School of Medicine, University of North Carolina and the National Institutes of Health, National Institute on Alcoholism and Alcohol Abuse, AA020023, AA020024, AA020022, AA019767, AA11605, and AA007573.
References
Alfonso-Loeches, S., Pascual-Lucas, M., Blanco, A. M., Sanchez-Vera, I., and Guerri, C. (2010). Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J. Neurosci. 30, 8285–8295.
Andersen, S. L., and Teicher, M. H. (2004). Delayed effects of early stress on hippocampal development. Neuropsychopharmacology 29, 1988–1993.
Andersen, S. L., Thompson, A. T., Rutstein, M., Hostetter, J. C., and Teicher, M. H. (2000). Dopamine receptor pruning in prefrontal cortex during the periadolescent period in rats. Synapse 37, 167–169.
Andrucci, G. L., Archer, R. P., Pancoast, D. L., and Gordon, R. A. (1989). The relationship of MMPI and sensation seeking scales to adolescent drug use. J. Pers. Assess. 53, 253–266.
Armario, A. (2010). Activation of the hypothalamic–pituitary–adrenal axis by addictive drugs: different pathways, common outcome. Trends Pharmacol. Sci. 31, 318–325.
Baumrind, D. (1987). “A developmental perspective on adolescent risk taking in contemporary America,” in Adolescent Social Behavior and Health, ed. C. E. Irwin Jr. (San Francisco, CA: Jossey-Bass), 93–125.
Bechara, A., Dolan, S., and Hindes, A. (2002). Decision-making and addiction (part II): myopia for the future or hypersensitivity to reward? Neuropsychologia 40, 1690–1705.
Blakemore, S. J., and Choudhury, S. (2006). Development of the adolescent brain: implications for executive function and social cognition. J. Child Psychol. Psychiatry 47, 296–312.
Blanco, A. M., Perez-Arago, A., Fernandez-Lizarbe, S., and Guerri, C. (2008). Ethanol mimics ligand-mediated activation and endocytosis of IL-1RI/TLR4 receptors via lipid rafts caveolae in astroglial cells. J. Neurochem. 106, 625–639.
Blednov, Y. A., Benavidez, J. M., Geil, C., Perra, S., Morikawa, H., and Harris, R. A. (2011a). Activation of neuroinflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav. Immun. doi: 10.1016/j.bbi.2011.01.008. [Epub ahead of print].
Blednov, Y. A., Ponomarev, I., Geil, C., Bergeson, S., Koob, G. F., and Harris, R. A. (2011b). Neuroimmune regulation of alcohol consumption: behavioral validation of genes obtained from genomic studies. Addict. Biol. doi: 10.1111/j.1369-1600.2010.00284.x. [Epub ahead of print].
Blednov, Y. A., Bergeson, S. E., Walker, D., Ferreira, V. M., Kuziel, W. A., and Harris, R. A. (2005). Perturbation of chemokine networks by gene deletion alters the reinforcing actions of ethanol. Behav. Brain Res. 165, 110–125.
Breese, G. R., Knapp, D. J., Overstreet, D. H., Navarro, M., Wills, T. A., and Angel, R. A. (2008). Repeated lipopolysaccharide (LPS) or cytokine treatments sensitize ethanol withdrawal-induced anxiety-like behavior. Neuropsychopharmacology 33, 867–876.
Calu, D. J., Roesch, M. R., Stalnaker, T. A., and Schoenbaum, G. (2007). Associative encoding in posterior piriform cortex during odor discrimination and reversal learning. Cereb. Cortex 17, 1342–1349.
Cao, Q., Mak, K. M., and Lieber, C. S. (2005). Cytochrome P4502E1 primes macrophages to increase TNF-alpha production in response to lipopolysaccharide. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G95–G107.
Casey, B. J., Trainor, R. J., Orendi, J. L., Schubert, A. B., Nystrom, L. E., Giedd, J. N., Castellanos, F. X., Haxby, J. V., Noll, D. C., Cohen, J. D., Forman, S. D., Dahl, R. E., and Rapoport, J. L. (1997). A developmental functional MRI study of prefrontal activation during performance of a go–no-go task. J. Cogn. Neurosci. 9, 835–847.
Choi, S., and Kellogg, C. K. (1992). Norepinephrine utilization in the hypothalamus of the male rat during adolescent development. Dev. Neurosci. 14, 369–376.
Choi, S., Weisberg, S. N., and Kellogg, C. K. (1997). Control of endogenous norepinephrine release in the hypothalamus of male rats changes over adolescent development. Brain Res. Dev. Brain Res. 98, 134–141.
Christoffel, D. J., Golden, S. A., Dumitriu, D., Robinson, A. J., Janssen, W. G., Ahn, H. F., Krishnan, V., Reyes, C. M., Han, M. H., Ables, J. L., Eisch, A. J., Dietz, D. M., Ferguson, D., Neve, R. L., Greengard, P., Kim, Y., Morrison, J. H., and Russo, S. J. (2011). IkB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J. Neurosci. 31, 314–321.
Coleman, L., Jun, H., Joohwi, L., Styner, M., and Crews, F. T. (2011). Adolescent binge drinking alters adult brain neurotransmitter gene expression, behavior, brain regional volumes, and neurochemistry in mice. Alcohol. Clin. Exp. Res. 35, 671–688.
Crews, F. T., He, J., and Hodge, C. (2007). Adolescent cortical development: a critical period of vulnerability for addiction. Pharmacol. Biochem. Behav. 86, 189–199.
Crews, F. T., Mdzinarishvili, A., Kim, D., He, J., and Nixon, K. (2006a). Neurogenesis in adolescent brain is potently inhibited by ethanol. Neuroscience 137, 437–445.
Crews, F. T., Nixon, K., Kim, D., Joseph, J., Shukitt-Hale, B., Qin, L., and Zou, J. (2006b). BHT blocks NF-kappaB activation and ethanol-induced brain damage. Alcohol. Clin. Exp. Res. 30, 1938–1949.
Crews, F. T., and Nixon, K. (2003). Alcohol, neural stem cells, and adult neurogenesis. Alcohol Res. Health 27, 197–204.
De Bellis, M. D., Narasimhan, A., Thatcher, D. L., Keshavan, M. S., Soloff, P., and Clark, D. B. (2005). Prefrontal cortex, thalamus, and cerebellar volumes in adolescents and young adults with adolescent-onset alcohol use disorders and comorbid mental disorders. Alcohol. Clin. Exp. Res. 29, 1590–1600.
Dolganiuc, A., Bakis, G., Kodys, K., Mandrekar, P., and Szabo, G. (2006). Acute ethanol treatment modulates toll-like receptor-4 association with lipid rafts. Alcohol. Clin. Exp. Res. 30, 76–85.
Dranovsky, A., and Hen, R. (2006). Hippocampal neurogenesis: regulation by stress and antidepressants. Biol. Psychiatry 59, 1136–1143.
Duka, T., Gentry, J., Malcolm, R., Ripley, T. L., Borlikova, G., Stephens, D. N., Veatch, L. M., Becker, H. C., and Crews, F. T. (2004). Consequences of multiple withdrawals from alcohol. Alcohol. Clin. Exp. Res. 28, 233–246.
Eisenberger, N. I., Berkman, E. T., Inagaki, T. K., Rameson, L. T., Mashal, N. M., and Irwin, M. R. (2010). Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol. Psychiatry 68, 748–754.
Ernst, M., Romeo, R. D., and Andersen, S. L. (2009). Neurobiology of the development of motivated behaviors in adolescence: a window into a neural systems model. Pharmacol. Biochem. Behav. 93, 199–211.
Faden, V. B. (2006). Trends in initiation of alcohol use in the United States 1975 to 2003. Alcohol. Clin. Exp. Res. 30, 1011–1022.
Fagen, R. M. (1976). “Exercise, play, and physical training in animals,” in Perspectives in Ethology, Vol. 2, eds P. P. G. Bateson and P. M. Klopfer (New York, NY: Plenum Press), 189–219.
Galef, B. G. Jr. (1977). “Mechanisms for the social transmission of food preferences from adult to weanling rats,” in Learning Mechanisms in Food Selection, eds. L. M. Barker, M. Best, and M. Domjan (Waco, TX: Baylor University Press), 123–148.
Garg, A. D., Nowis, D., Golab, J., Vandenabeele, P., Krysko, D. V., and Agostinis, P. (2010). Immunogenic cell death, DAMPs, and anticancer therapeutics: an emerging amalgamation. Biochim. Biophys. Acta 1805, 53–71.
Ghosh, S., and Hayden, M. S. (2008). New regulators of NF-kappaB in inflammation. Nat. Rev. Immunol. 8, 837–848.
Giedd, J. N. (2004). Structural magnetic resonance imaging of the adolescent brain. Ann. N. Y. Acad. Sci. 1021, 77–85.
Giedd, J. N., Blumenthal, J., Jeffries, N. O., Castellanos, F. X., Liu, H., Zijdenbos, A., Paus, T., Evans, A. C., and Rapoport, J. L. (1999). Brain development during childhood and adolescence: a longitudinal MRI study. Nat. Neurosci. 2, 861–863.
Gould, E., Woolf, N. J., and Butcher, L. L. (1991). Postnatal development of cholinergic neurons in the rat: I. Forebrain. Brain Res. Bull. 27, 767–789.
Grant, B. F., and Dawson, D. A. (1998). Age of onset of drug use and its association with DSM-IV drug abuse and dependence: results from the National Longitudinal Alcohol Epidemiologic Survey. J. Subst. Abuse 10, 163–173.
Gregus, A., Wintink, A. J., Davis, A. C., and Kalynchuk, L. E. (2005). Effect of repeated corticosterone injections and restraint stress on anxiety and depression-like behavior in male rats. Behav. Brain Res. 156, 105–114.
Gruber, A. J., Calhoun, Gwendolyn, G., Shusterman, I., Schoenbaum, G., Roesch, M. R., and O’Donnell, P. (2010). More is less: a disinhibited prefrontal cortex impairs cognitive flexibility. J. Neurosci. 30, 17102–17110.
He, J., and Crews, F. T. (2007). Neurogenesis decreases during brain maturation from adolescence to adulthood. Pharmacol. Biochem. Behav. 86, 327–333.
He, J., and Crews, F. T. (2008). Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp. Neurol. 210, 349–358.
Huang, W., Tang, Y., and Li, L. (2010). HMGB1, a potent proinflammatory cytokine in sepsis. Cytokine 51, 119–126.
Hutchinson, M. R., Zhang, Y., Brown, K., Coats, B. D., Shridhar, M., Sholar, P. W., Patel, S. J., Crysdale, N. Y., Harrison, J. A., Maier, S. F., Rice, K. C., and Watkins, L. R. (2008). Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4). Eur. J. Neurosci. 28, 20–29.
Hutchinson, M. R., Zhang, Y., Shridhar, M., Evans, J. H., Buchanan, M. M., Zhao, T. X., Slivka, P. F., Coats, B. D., Rezvani, N., Wieseler, J., Hughes, T. S., Landgraf, K. E., Chan, S., Fong, S., Phipps, S., Falke, J. J., Leinwand, L. A., Maier, S. F., Yin, H., Rice, K. C., and Watkins, L. R. (2010). Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav. Immun. 24, 83–95.
Huttenlocher, P. R. (1984). Synapse elimination and plasticity in developing human cerebral cortex. Am. J. Ment. Defic. 88, 488–496.
Jentsch, J. D., and Taylor, J. R. (1999). Impulsivity resulting from frontostriatal dysfunction in drug abuse: implications for the control of behavior by reward-related stimuli. Psychopharmacology (Berl.) 146, 373–390.
Johnson, S. A., Fournier, N. M., and Kalynchuk, L. E. (2006). Effect of different doses of corticosterone on depression-like behavior and HPA axis responses to a novel stressor. Behav. Brain Res. 168, 280–288.
Kalsbeek, A., Voorn, P., Buijs, R. M., Pool, C. W., and Uylings, H. B. (1988). Development of the dopaminergic innervation in the prefrontal cortex of the rat. J. Comp. Neurol. 269, 58–72.
Kelley, K. W., and Dantzer, R. (2011). Alcoholism and inflammation: neuroimmunology of behavioral and mood disorders. Brain Behav. Immun. doi: 10.1016/j.bbi.2010.12.013. [Epub ahead of print].
Kellogg, C. K., Awatramani, G. B., and Piekut, D. T. (1998). Adolescent development alters stressor-induced Fos immunoreactivity in rat brain. Neuroscience 83, 681–689.
Kempermann, G. (2002). Why new neurons? Possible functions for adult hippocampal neurogenesis. J. Neurosci. 22, 635–638.
Knapp, D. J., and Crews, F. T. (1999). Induction of cyclooxygenase-2 in brain during acute and chronic ethanol treatment and ethanol withdrawal. Alcohol. Clin. Exp. Res. 23, 633–643.
Koo, J. W., and Duman, R. S. (2008). IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc. Natl. Acad. Sci. U.S.A. 105, 751–756.
Koo, J. W., Russo, S. J., Ferguson, D., Nestler, E. J., and Duman, R. S. (2011). Nuclear factor-kappa B is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc. Natl. Acad. Sci. U.S.A. 107, 2669–2674.
Koob, G. F., and Volkow, N. D. (2010). Neurocircuitry of addiction. Neuropsychopharmacology 35, 217–238.
Kostovic, I. (1990). Structural and histochemical reorganization of the human prefrontal cortex during perinatal and postnatal life. Prog. Brain Res. 85, 223–240.
Liu, B., Du, L., and Hong, J. S. (2000a). Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J. Pharmacol. Exp. Ther. 293, 607–617.
Liu, B., Jiang, J. W., Wilson, B. C., Du, L., Yang, S. N., Wang, J. Y., Wu, G. C., Cao, X. D., and Hong, J. S. (2000b). Systemic infusion of naloxone reduces degeneration of rat substantia nigral dopaminergic neurons induced by intranigral injection of lipopolysaccharide. J. Pharmacol. Exp. Ther. 295, 125–132.
Liu, J., Lewohl, J. M., Harris, R. A., Iyer, V. R., Dodd, P. R., Randall, P. K., and Mayfield, R. D. (2006). Patterns of gene expression in the frontal cortex discriminate alcoholic from nonalcoholic individuals. Neuropsychopharmacology 31, 1574–1582.
Loftis, J. M., Choi, D., Hoffman, W., and Huckans, M. S. (2010). Methamphetamine causes persistent immune dysregulation: a cross-species, translational report. Neurotox. Res. doi: 10.1007/s12640-010-9223-x. [Epub ahead of print].
Madrigal, B., Puebla, P., Caballero, E., Pelaez, R., Gravalos, D. G., and Medarde, M. (2001). Synthesis of imidazo[4,5-d]oxazolo[3,4-a]pyridines. New heterocyclic analogues of lignans. Arch. Pharm. (Weinheim) 334, 177–179.
Madrigal, J. L., Garcia-Bueno, B., Moro, M. A., Lizasoain, I., Lorenzo, P., and Leza, J. C. (2003). Relationship between cyclooxygenase-2 and nitric oxide synthase-2 in rat cortex after stress. Eur. J. Neurosci. 18, 1701–1705.
Malberg, J. E., and Duman, R. S. (2003). Cell proliferation in adult hippocampus is decreased by inescapable stress: reversal by fluoxetine treatment. Neuropsychopharmacology 28, 1562–1571.
Malberg, J. E., Eisch, A. J., Nestler, E. J., and Duman, R. S. (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 20, 9104–9110.
Maroso, M., Balosso, S., Ravizza, T., Liu, J., Aronica, E., Iyer, A. M., Rossetti, C., Moteni, M., Casalgrandi, M., Manfredi, A. A., Bianchi, M. E., and Vezzani, A. (2010). Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 16, 413–419.
Monti, P. M., Miranda, R. Jr., Nixon, K., Sher, K. J., Swartzwelder, H. S., Tapert, S. F., White, A., and Crews, F. T. (2005). Adolescence: booze, brains, and behavior. Alcohol. Clin. Exp. Res. 29, 207–220.
Mulligan, M. K., Ponomarev, I., Hitzemann, R. J., Belknap, J. K., Tabakoff, B., Harris, R. A., Crabbe, J. C., Blednov, Y. A., Grahame, N. J., Phillips, T. J., Finn, D. A., Hoffman, P. L., Iyer, V. R., Koob, G. F., and Bergeson, S. E. (2006). Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc. Natl. Acad. Sci. U.S.A. 103, 6368–6373.
Munhoz, C. D., Sorrells, S. F., Caso, J. R., Scavone, C., and Sapolsky, R. M. (2010). Glucocorticoids exacerbate lipopolysaccharide-induced signaling in the frontal cortex and hippocampus in a dose-dependent manner. J. Neurosci. 30, 13690–13698.
Obernier, J. A., White, A. M., Swartzwelder, H. S., and Crews, F. T. (2002). Cognitive deficits and CNS damage after a 4-day binge ethanol exposure in rats. Pharmacol. Biochem. Behav. 72, 521–532.
O’Connor, M. F., Irwin, M. R., Seldon, J., Kwan, L., and Ganz, P. A. (2007). Pro-inflammatory cytokines and depression in a familial cancer registry. Psychooncology 16, 499–501.
Okvist, A., Johansson, S., Kuzmin, A., Bazov, I., Merino-Martinez, R., Ponomarev, I., Mayfield, R. D., Harris, R. A., Sheedy, D., Garrick, T., Harper, C., Hurd, Y. L., Terenius, L., Ekstrom, T. J., Bakalkin, G., and Yakovleva, T. (2007). Neuroadaptations in human chronic alcoholics: dysregulation of the NF-kappaB system. PLoS ONE 2, e930. doi: 10.1371/journal.pone.0000930
Pahl, H. L. (1999). Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18, 6853–6866.
Pascual, M., Blanco, A. M., Cauli, O., Minarro, J., and Guerri, C. (2007). Intermittent ethanol exposure induces inflammatory brain damage and causes long-term behavioural alterations in adolescent rats. Eur. J. Neurosci. 25, 541–550.
Pascual, M., Boix, J., Felipo, V., and Guerri, C. (2009). Repeated alcohol administration during adolescence causes changes in the mesolimbic dopaminergic and glutamatergic systems and promotes alcohol intake in the adult rat. J. Neurochem. 108, 920–931.
Peleg-Oren, N., Saint-Jean, G., Cardenas, G. A., Tammara, H., and Pierre, C. (2009). Drinking alcohol before age 13 and negative outcomes in late adolescence. Alcohol. Clin. Exp. Res. 3, 1966–1972.
Qin, L., Block, M. L., Liu, Y., Bienstock, R. J., Pei, Z., Zhang, W., Wu, X., Wilson, B., Burka, T., and Hong, J. (2005). Microglial NADPH oxidase is a novel target for femtomolar neuroprotection against oxidative stress. FASEB J. 19, 550–557.
Qin, L., He, J., Hanes, R. N., Pluzarev, O., Hong, J. S., and Crews, F. T. (2008). Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J. Neuroinflammation 5, 10.
Qin, L., Wu, X., Block, M. L., Liu, Y., Breese, G. R., Hong, J. S., Knapp, D. J., and Crews, F. T. (2007). Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55, 453–462.
Raison, C. L., Borisov, A. S., Majer, M., Drake, D. F., Pagnoni, G., Woolwine, B. J., Vogt, G. J., Massung, B., and Miller, A. H. (2009). Activation of central nervous system inflammatory pathways by interferon-alpha: relationship to monoamines and depression. Biol. Psychiatry 65, 296–303.
Reissner, K. J., and Kalivas, P. W. (2010). Using glutamate homeostasis as a target for treating addictive disorders. Behav. Pharmacol. 21, 514–522.
Rosenberg, D. R., and Lewis, D. A. (1994). Changes in the dopaminergic innervation of monkey prefrontal cortex during late postnatal development: a tyrosine hydroxylase immunohistochemical study. Biol. Psychiatry 36, 272–277.
Russo, S. J., Mazei-Robison, M. S., Ables, J. L., and Nestler, E. J. (2009). Neurotrophic factors and structural plasticity in addiction. Neuropharmacology 56, 73–82.
Santarelli, L., Saxe, M., Gross, C., Surget, A., Battaglia, F., Dulawa, S., Weisstaub, N., Lee, J., Duman, R., Arancio, O., Belzung, C., and Hen, R. (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301, 805–809.
Schoenbaum, G., Roesch, M. R., and Stalnaker, T. A. (2006). Orbitofrontal cortex, decision-making, and drug addiction. Trends Neurosci. 29, 116–124.
Schoenbaum, G., Saddoris, M. P., Ramus, S. J., Shaham, Y., and Setlow, B. (2004). Cocaine-experienced rats exhibit learning deficits in a task sensitive to orbitofrontal cortex lesions. Eur. J. Neurosci. 19, 1997–2002.
Schoenbaum, G., and Shaham, Y. (2008). The role of orbitofrontal cortex in drug addiction: a review of preclinical studies. Biol. Psychiatry 63, 256–262.
Shors, T. J., Miesegaes, G., Beylin, A., Zhao, M., Rydel, T., and Gould, E. (2001). Neurogenesis in the adult in involved in the formation of trace memories. Nature 410, 372–376.
Silveri, M. M., and Spear, L. P. (1998). Decreased sensitivity to the hypnotic effects of ethanol early in ontogeny. Alcohol. Clin. Exp. Res. 22, 670–676.
Smith, P. K. (1982). Does play matter? Functional and evolutionary aspects of animal and human play. Behav. Brain Sci. 5, 139–184.
Sowell, E. R., Thompson, P. M., Holmes, C. J., Jernigan, T. L., and Toga, A. W. (1999). In vivo evidence for post-adolescent brain maturation in frontal and striatal regions. Nat. Neurosci. 2, 859–861.
Sowell, E. R., Thompson, P. M., Tessner, K. D., and Toga, A. W. (2001). Mapping continued brain growth and gray matter density reduction in dorsal frontal cortex: inverse relationships during postadolescent brain maturation. J. Neurosci. 22, 8819–8829.
Spanagel, R., Rosenwasser, A. M., Schumann, G., and Sarker, D. K. (2005). Alcohol consumption and the body’s biological clock. Alcohol. Clin. Exp. Res. 29, 1550–1557.
Spear, L. P. (2000). The adolescent brain and age-related behavioral manifestations. Neurosci. Biobehav. Rev. 24, 417–463.
Stalnaker, T. A., Takahashi, Y., Roesch, M. R., and Schoenbaum, G. (2009). Neural substrates of cognitive inflexibility after chronic cocaine exposure. Neuropharmacology 56, 63–72.
Stevenson, J. R., Schroeder, J. P., Nixon, K., Besheer, J., Crews, F. T., and Hodge, C. W. (2009). Abstinence following alcohol drinking produces depression-like behavior and reduced hippocampal neurogenesis in mice. Neuropsychopharmacology 34, 1209–1222.
Swann, J. W., Pierson, M. G., Smith, K. L., and Lee, C. L. (1999). Developmental neuroplasticity: roles in early life seizures and chronic epilepsy. Adv. Neurol. 79, 203–216.
Tamm, L., Menon, V., and Reiss, A. L. (2002). Maturation of brain function associated with response inhibition. J. Am. Acad. Child Adolesc. Psychiatry 41, 1231–1238.
Tanapat, P., Hastings, N. B., Rydel, T. A., Galea, L. A., and Gould, E. (2001). Exposure to fox odor inhibits cell proliferation in the hippocampus of adult rats via an adrenal hormone-dependent mechanism. J. Comp. Neurol. 437, 496–504.
Tarazi, F. I., Tomasini, E. C., and Baldessarini, R. J. (1998a). Postnatal development of dopamine D4-like receptors in rat forebrain regions: comparison with D2-like receptors. Brain Res. Dev. Brain Res. 110, 227–233.
Tarazi, F. I., Tomasini, E. C., and Baldessarini, R. J. (1998b). Postnatal development of dopamine and serotonin transporters in rat caudate-putamen and nucleus accumbens septi. Neurosci. Lett. 254, 21–24.
Teicher, M. H., Andersen, S. L., and Hostetter, J. C. Jr. (1995). Evidence for dopamine receptor pruning between adolescence and adulthood in striatum but not nucleus accumbens. Brain Res. Dev. Brain Res. 89, 167–172.
Townshend, J. M., and Duka, T. (2003). Mixed emotions: alcoholics’ impairments in the recognition of specific emotional facial expressions. Neuropsychologia 41, 773–782.
van Eden, C. G., Kros, J. M., and Uylings, H. B. (1990). The development of the rat prefrontal cortex. Its size and development of connections with thalamus, spinal cord and other cortical areas. Prog. Brain Res. 85, 169–183.
Videbech, P., and Ravnkilde, B. (2004). Hippocampal volume and depression: meta-analysis of MRI studies. Am. J. Psychiatry 161, 1957–1966.
Walker, E. F., Walder, D. J., and Reynolds, F. (2001). Developmental changes in cortisol secretion in normal and at-risk youth. Dev. Psychopathol. 13, 721–732.
Ward, R. J., Colivicchi, M. A., Allen, R., Schol, F., Lallemand, F., de Witte, P., Ballini, C., Corte, L. D., and Dexter, D. (2009). Neuroinflammation induced in the hippocampus of “binge drinking” rats may be mediated by elevated extracellular glutamate content. J. Neurochem. 111, 1119–1128.
Waters, N. S., Klintsova, A. Y., and Foster, T. C. (1997). Insensitivity of the hippocampus to environmental stimulation during postnatal development. J. Neurosci. 17, 7967–7973.
Weissenborn, R., and Duka, T. (2003). Acute alcohol effects on cognition function in social drinkers: their relationship to drinking habits. Psychopharmacology (Berl.) 165, 306–312.
Wills, T. A., Vaccaro, D., and McNamara, G. (1994). Novelty seeking, risk taking, and related constructs as predictors of adolescent substance use: an application of Cloninger’s theory. J. Subst. Abuse 6, 1–20.
Yang, L., Seifert, A., Wu, D., Wang, X., Rankovic, V., Schroder, H., Brandenburg, L. O., Hollt, V., and Koch, T. (2010). Role of phospholipase D2/phosphatidic acid signal transduction in micro- and delta-opioid receptor endocytosis. Mol. Pharmacol. 78, 105–113.
Zecevic, N., Bourgeois, J. P., and Rakic, P. (1989). Changes in synaptic density in motor cortex of rhesus monkey during fetal and postnatal life. Brain Res. Dev. Brain Res. 50, 11–32.
Zehr, J. L., Todd, B. J., Schulz, K. M., McCarthy, M. M., and Sisk, C. L. (2006). Dendritic pruning of the medial amygdala during pubertal development of the Syrian hamster. J. Neurobiol. 66, 578–590.
Zou, J. Y., and Crews, F. T. (2005). TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: Neuroprotection by NF kappa B inhibition. Brain Res. 1034, 11–24.
Zou, J., and Crews, F. T. (2006). CREB and NF-kappaB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cell. Mol. Neurobiol. 26, 385–405.
Keywords: addiction, alcoholism, chemokines, microglia, neurogenesis
Citation: Crews FT and Vetreno RP (2011) Addiction, adolescence, and innate immune gene induction. Front. Psychiatry 2:19. doi: 10.3389/fpsyt.2011.00019
Received: 10 March 2011; Paper pending published: 27 March 2011;
Accepted: 11 April 2011; Published online: 27 April 2011.
Edited by:
Josephine Johns, University of North Carolina at Chapel Hill, USAReviewed by:
Cynthia J. M. Kane, University of Arkansas for Medical Sciences, USAConsuelo Guerri, Research Center Principe Felipe, Spain
Copyright: © 2011 Crews and Vetreno. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Fulton T. Crews, Bowles Center for Alcohol Studies, The School of Medicine, The University of North Carolina at Chapel Hill, CB #7178, 1021 Thurston-Bowles Building, Chapel Hill, NC 27599-71780, USA. e-mail:ZnVsdG9uX2NyZXdzQG1lZC51bmMuZWR1