
 Bojana Zupan
Bojana Zupan
- Department of Pharmacology, Weill Medical College of Cornell University, New York, NY, USA
Risk factors for psychiatric disorders have traditionally been classified as genetic or environmental. Risk (candidate) genes, although typically possessing small effects, represent a clear starting point to elucidate downstream cellular/molecular pathways of disease. Environmental effects, especially during development, can also lead to altered behavior and increased risk for disease. An important environmental factor is the mother, demonstrated by the negative effects elicited by maternal gestational stress and altered maternal care. These maternal effects can also have a genetic basis (e.g., maternal genetic variability and mutations). The focus of this review is “maternal genotype effects” that influence the emotional development of the offspring resulting in life-long psychiatric disease-like phenotypes. We have recently found that genetic inactivation of the serotonin 1A receptor (5-HT1AR) and the fmr1 gene (encoding the fragile X mental retardation protein) in mouse dams results in psychiatric disease-like phenotypes in their genetically unaffected offspring. 5-HT1AR deficiency in dams results in anxiety and increased stress responsiveness in their offspring. Offspring of 5-HT1AR deficient dams display altered development of the hippocampus, which could be linked to their anxiety-like phenotype. Maternal inactivation of fmr1, like its inactivation in the offspring, results in a hyperactivity-like condition and is associated with receptor alterations in the striatum. These data indicate a high sensitivity of the offspring to maternal mutations and suggest that maternal genotype effects can increase the impact of genetic risk factors in a population by increasing the risk of the genetically normal offspring as well as by enhancing the effects of offspring mutations.
Maternal Genotype Effects Can Contribute to the High Heritability of Psychiatric Diseases
A large number of studies indicate that the pre/postnatal maternal environment can increase the offspring’s risk for psychopathology. Although some of these maternal effects may have a genetic basis or are at least influenced by genes, only a few examples of “maternal genotype effects” are currently known/confirmed in humans and in animal models (Doolin et al., 2002; Rouse and Azen, 2004). A classical example is maternal phenylketonuria (high blood phenylalanine levels due to the lack of phenylalanine hydroxylase) that leads to mental retardation, seizures, microcephaly, and growth retardation in the offspring. Also, maternal mutations in methionine biosynthesis (G allele of methionine synthase A2756G and methionine synthase reductase A66G polymorphisms) can lead to spina bifida in the offspring. Reports also indicate that maternal genotype effects contribute not only to neurological disorders but also to common psychiatric conditions such as autism and attention deficit hyperactivity disorder (ADHD). Glutathione S-transferases (GST), a protective factor against reactive oxygen species, is expressed in multiple forms and polymorphic variants. Maternal transmission disequilibrium tests showed that the Val allele of the Ile105Val (A313G) polymorphism in GSTP1 (a pi class GST) is overtransmitted to mothers of autistic children (Williams et al., 2007). The variation at position 105 affects thermostability and catalytic activity suggesting that a change in enzyme activity in mothers during pregnancy may be related to the increased likelihood of autism in their children. By using a similar approach, maternal (but not offspring or paternal) TPH1 (tryptophan hydroxylase) mutations have been shown to increase the risk for ADHD (Halmoy et al., 2011). There are additional examples supporting the effect of maternal mutations on gestational development, but they are all based on statistical data from relatively small populations that will need to be replicated (Johnson, 2003). On the other hand, the list of maternal genes that affect offspring development could be expanded to include those that have an effect on maternal behavior prepartum and during early postnatal life. Experimental data in animals clearly show a significant impact of maternal care on the development of psychiatric disease-like conditions (Meaney, 2001). Also, clinical studies indicate that maternal anxiety, depression, and ADHD increase the risk of psychopathology (Halligan et al., 2004, 2007; Murray and Johnston, 2006; Davis and Tremont, 2007; Van den Bergh et al., 2008; Yehuda et al., 2008; Figueiredo and Costa, 2009; Brummelte and Galea, 2010; Murray et al., 2010). Although it is possible that these postnatal maternal effects may also have a genetic component, no maternal mutations have been identified in either human or animal models.
Since most association studies do not include the mother’s genotype, it is likely that the prevalence of maternal genotype effects is greatly underestimated in pedigrees and in populations. Indeed, maternal genotype effects could significantly contribute to the high heritability of common disorders including psychiatric diseases. It is striking that despite the high heritability in these disorders (proportion of variability in a population attributable to genetic variation among individuals), only a small percentage of the risk can be explained by identified genetic variants, a phenomenon often referred to as missing heritability (Manolio et al., 2009). Since they are apparent at the pedigree and population level but not detectable at the genetic level, maternal genotype effects could be one of the mechanisms that explain “missing heritability.” Therefore, maternal genotype effects may have major significance in understanding and treating common diseases, including psychiatric conditions discussed in this review.
Animal Models Can Help Identify Mechanisms Underlying Maternal Genotype Effects
Although an altered prenatal environment due to maternal genetic variability has been shown as a risk factor in psychiatric diseases such as autism and ADHD (Williams et al., 2007; Halmoy et al., 2011), the underlying disease mechanisms are difficult to identify because of the limitations associated with human studies. Animal (rodent) models have been invaluable tools in studying diseases, especially at the mechanistic level. Many rat and mouse lines are considered inbred – or lacking genetic differences between individuals – and therefore genetic mutations/modifications in the dams are not likely modified by between-subject genetic background variability. In addition to the highly controllable genetic background, rodent models allow for high degree of control of environmental factors. This control is of critical importance because environmental influences could interfere with the maternal genotype effect. Lastly, the advantages of rodent models include the availability of methods to determine if a particular behavioral change in the offspring is dependent on prenatal and/or postnatal parental or maternal environment. Cross-fostering, a procedure in which rodent pups are placed with a foster mother within the first postnatal day, can be used to determine whether a phenotype requires a postnatal maternal contribution, which may be either behavioral or physiological, transmitted through maternal milk. The advantages of animal models are clearly demonstrated in a work that explored the effect of a maternal mutation in Peg-3 (paternally expressed gene 3). The maternal allele of this gene is imprinted and silenced and therefore the offspring of mutant mothers and wild-type (WT) fathers are essentially WT (expressing the paternal allele) allowing to study the maternal genotype effect without the interference of or interaction with the mutation in the offspring. The offspring of mutant mothers displayed increased neophobia and decreased exploration, although these effects were seen only in females (Champagne et al., 2009). Furthermore, an association was found between the offspring behavioral abnormalities and the reduced postpartum maternal care of Peg-3 KO dams (Curley et al., 2008).
To demonstrate the feasibility of mouse models in studying maternal genotype effects and their mechanisms, we present our research on two maternal mutations that alter specific behavioral phenotypes and correlated cellular or pharmacologic properties in the genetically unaffected offspring.
Maternal Serotonin 1A Receptor and Offspring Anxiety
The serotonin (5-HT1A) receptor has been implicated in anxiety and depression through receptor binding and pharmacological studies (Drevets et al., 1999; Lemonde et al., 2003; Strobel et al., 2003; Neumeister et al., 2004). The 5-HT1A receptor knockout mouse model demonstrates increased anxiety-related behavior in several behavioral assays, on multiple genetic backgrounds (Heisler et al., 1998; Parks et al., 1998; Ramboz et al., 1998). However, the interpretation of the anxiety phenotype of 5-HT1A receptor KO mice is complicated by the fact that following a typical heterozygote × heterozygote (H × H) breeding, the offspring is exposed to a receptor deficient maternal environment. Considering the association between the 5-HT1A receptor and depression (Van den Hove et al., 2006; Spinelli et al., 2010), including postpartum depression (Moses-Kolko et al., 2008), and the importance of maternal care on the normal emotional development of the offspring, the maternal receptor genotype itself may modulate anxiety-like behavior in the WT offspring and/or could interact with the null allele in the KO offspring, making the dissociation between the maternal and offspring genotype effects on offspring anxiety levels difficult. By studying maternal–offspring 5-HT1A receptor genotype interactions, Weller et al. (2003) found reduced adult anxiety when the behavior of the H offspring of KO dams were compared to that of the WT dams suggesting an anxiolytic-like effect of the maternal KO alleles on offspring behavior. Since the offspring themselves were receptor deficient, the effect of maternal receptor deficiency as a single factor on offspring behavior could not be determined.
We recently showed that partial or complete 5-HT1A receptor deficiency in Swiss Webster (SW) mouse dams can cause increased anxiety-related behavior and enhanced stress reactivity in their offspring, independently of offspring genotype (Gleason et al., 2010; Figure 1) Genetically WT offspring of 5-HT1A receptor deficient mice displayed increased anxiety-related behavior in the elevated plus maze. This phenotype was found to require prenatal maternal 5-HT1A receptor deficiency, as mice which developed from WT embryos implanted into 5-HT1A receptor deficient mothers and raised by either 5-HT1A deficient or WT mothers after birth displayed increased anxiety-related behavior (Figure 1). This phenotype was also shown to be dependent on strain background (Gleason et al., 2010). In contrast, increased anxiety-related behavior in the open field, a less stressful assay for unconditioned anxiety, was found to be dependent on offspring 5-HT1A receptor genotype. Finally, we demonstrated that maternal 5-HT1A receptor deficiency leads to reduced immobility time in the Porsolt Forced Swim Test, which can be interpreted as a lack of normal coping skills and an increase in reactivity to inescapable stress. The development of this phenotype is independent of offspring genotype and requires both prenatal and postnatal maternal receptor deficit, as shown by embryo transfer and cross-fostering experiments.
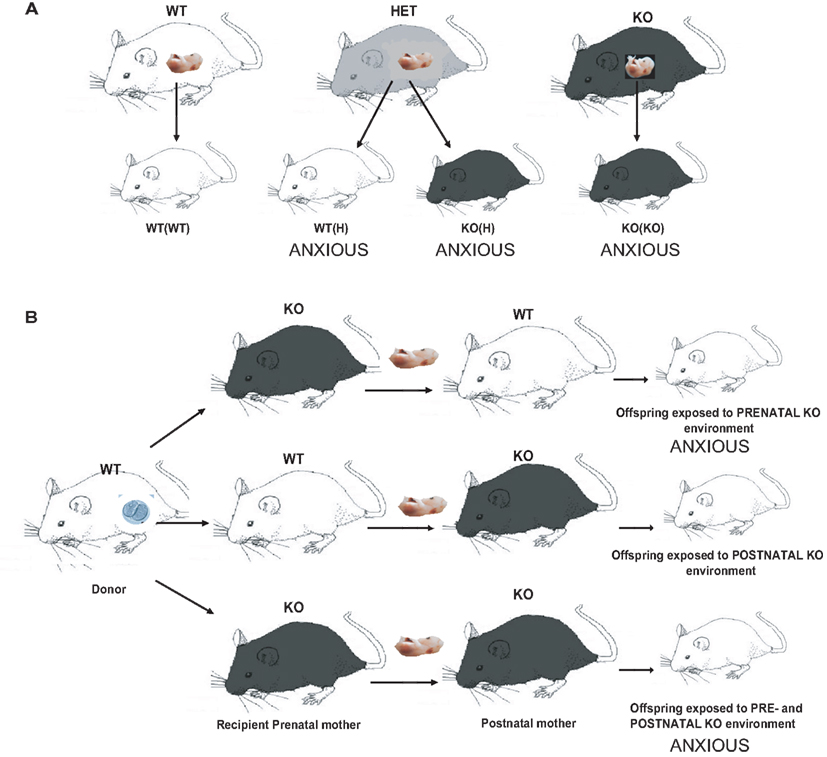
Figure 1. Diagram showing breeding and embryonic cross-fostering schemes for the 5-HT1A receptor deficient line. (A) WT mice are bred together to generate WT(WT) control animals with neither a maternal nor an offspring 5-HT1A receptor effect. Homozygous 5-HT1A receptor knockout mice are bred together to generate KO(KO) mice with both a maternal and an offspring 5-HT1A receptor genotype effect, which display increased anxiety-related behavior in the elevated plus maze. WT and 5-HT1A receptor homozygous knockout mice are bred together to generate 5-HT1A receptor heterozygotes, which are intercrossed to generate both WT(H) and KO(H) mice, both of which also display increased anxiety-related behavior. WT(H) mice have a maternal but not offspring 5-HT1A receptor genotype effect, while KO(H) mice have both a maternal and offspring 5-HT1A receptor genotype effect. (B) WT embryos (indicated by the oocyte in the donor) are implanted into WT and 5-HT1A receptor homozygous KO pseudopregnant mothers. Within 24 h after birth, pups are cross-fostered to WT or 5-HT1A receptor homozygous KO mothers, resulting in offspring raised with a prenatal, postnatal, or combined pre and postnatal maternal KO environment. Mice with either a prenatal or combined pre and postnatal maternal 5-HT1A receptor genotype effect exhibit increased anxiety-related behavior.
In addition, we examined early postnatal development of the ventral dentate gyrus of the hippocampus in mice exposed to maternal 5-HT1A receptor deficiency, because the ventral hippocampus and this time period have been linked to the development of anxiety (Gross et al., 2002; Bannerman et al., 2003). We identified several developmental changes that correlate with later life anxiety-related behavior, including an increased volume of the ventral granule cell layer (GCL) during the first postnatal week, which normalized by the age of 4 weeks (in the absence of changes in the number of proliferating cells) and a maturational delay of developing neurons in the ventral GCL at postnatal day 7. To provide a possible link between the developmental delay in the ventral dentate gyrus and the maternal 5-HT1A receptor mediated anxiety-related behavior, we examined the effect of inactivating a candidate gene involved in neuronal precursor maturation, p16Ink4a. We found that deletion of this gene phenocopies both the ventral GCL volume increase in the first postnatal week, and the increased anxiety-related behavior in the elevated plus maze. These data indicate that the maternal 5-HT1A receptor deficit alters hippocampal development in the offspring, and that these developmental changes could contribute to the increased anxiety-like behavior in the elevated plus maze (Figure 2).
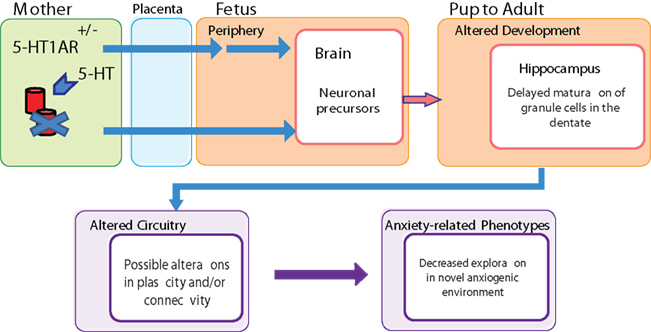
Figure 2. Schematic showing how prenatal maternal 5-HT1A receptor deficiency may lead to alterations in offspring hippocampal development and increased anxiety-related behavior. 5-HT1A receptor deficiency in the mouse mother is signaled across the placenta during embryonic development and transmuted to the developing brain. This prenatal reprogramming results in delayed maturation in the early postnatal dentate gyrus, which may contribute to changes in hippocampal network formation and ultimately to increased anxiety-related behavior.
It is important to note that the offspring were more sensitive to the maternal than to their own receptor gene dosage as a partial maternal receptor deficit was sufficient to elicit a full anxiety phenotype while a strong anxiety phenotype developed only in the homozygote knockout offspring (in the absence of maternal effect). The significance of this finding is that while a complete loss of the receptor has not been observed in humans, a 40–50% reduction in receptor binding, associated with stress and psychiatric disease, is relatively common (Lesch et al., 1992; Lopez et al., 1998; Drevets et al., 1999, 2007; Mann, 1999; Arango et al., 2001; Moses-Kolko et al., 2008). Therefore, a maternal 5-HT1AR deficit could be more relevant than an offspring deficit to human anxiety.
Maternal Fragile X Mental Retardation Protein and Offspring Hyperactivity
Our second mouse model of human disease is produced by the inactivation of the fmr1 gene encoding the fragile X mental retardation protein (FMRP). This model reproduces key behavioral features of the human fragile X syndrome (FXS; Consortium TD-BFX, 1994). FMRP is an RNA binding protein that affects multiple stages of RNA translation and is involved in the regulation of both local and global protein synthesis (Kao et al., 2010). Mice lacking Fmrp exhibit a number of FXS-like phenotypes including locomotor hyperactivity, sensory hyper-reactivity, cognitive defect, and macroorchidism (D’Hooge et al., 1997; Peier et al., 2000; Chen and Toth, 2001; Spencer et al., 2005; Yun et al., 2006). In addition, Fmrp deficient mice reproduce some of the autistic-like behaviors seen in FXS. Interestingly, the manifestation of autistic-like behaviors is genetic background dependent, consistent with the observation that autistic behaviors in FXS vary considerably, presumably as a result of genetic modifiers (Spencer et al., 2011).
Although FMRP has been identified as the singular cause of FXS, a typical Mendelian disease, we found that the maternal Fmrp deficit can contribute to the development of some of the disease-associated phenotypes. Specifically, we found that the genetically unaffected adult male offspring of heterozygote fmr1 KO dams displayed increased constitutive locomotor activity and that the combination of maternal and offspring genotype effects in the FXS mouse model had an additive effect on locomotor activity (Zupan and Toth, 2008a). This finding suggests that even a partial deficit in Fmrp in the dam has long-term effects on offspring behavior and is sufficient to induce a disease-like phenotype. Other FXS-associated phenotypes such as macroorchidism and sensory hypersensitivity were unaffected by the maternal genotype and present only if fmr1 was mutated in the offspring.
Locomotor activity is regulated in part by the activity of mesolimbic dopamine neurons originating in the ventral tegmental area and projecting to the ventral striatum (Koob and Swerdlow, 1988; Szczypka et al., 2001). Hyperactivity has been linked to low tonic dopamine activity promoted by D2 autoreceptors and high phasic dopamine neurotransmission (Grace, 2001). Activation of D2 autoreceptors, by reducing the amount of DA released into the synapse (presynaptically) and reducing the excitability of the DA neurons (in the somatodendritic compartment), inhibits locomotor activity in rodents (Starke et al., 1989; Cory-Slechta et al., 1996; Usiello et al., 2000; Figure 3). When we probed the dopamine system using quinpirole at a D2 autoreceptor preferring dose, we found that the hyperactive offspring of FMRP deficient dams, regardless of their own genotype, had attenuated behavioral responses to quinpirole. This indicates a functional downregulation of the D2 autoreceptor that can explain or contribute to the hyperactivity phenotype (Figure 4). While the functional D2 receptor downregulation was not affected by the offspring genotype, the KO offspring of heterozygote fmr1 KO dams had a higher level of hyperactivity than their WT offspring indicating that only the maternally induced component of the hyperactivity may be explained by the downregulation of presynaptic D2 receptors. In addition to the D2 autoreceptors, the presynaptic GABAB receptors also inhibit DA release while receptors located somatodendritically reduce the firing rate of DA neurons (Engberg et al., 1993; Smolders et al., 1995; Madden and Johnson, 1998; Labouebe et al., 2007; Figure 3). Administration of the GABAB agonist baclofen at doses that had no sedative effects resulted in a more prominent reduction in locomotor activity in the offspring of fmr1 heterozygote KO dams as compared to offspring of WT dams (Zupan and Toth, 2008b; Figure 4). Again, this change was maternal but not offspring genotype dependent. This indicated a maternal genotype-dependent sensitization of the GABAB receptor in the offspring that may compensate for the hyperactivity related to the D2 receptor downregulation. Taken together, these data indicate that the maternal genotype effect can be linked not only to behavioral alterations but also to neurochemical changes which can ultimately help elucidate the underlying mechanisms.

Figure 3. Illustration of dopamine D2 autoreceptors (orange) and GABAB receptors (green) on somatodendritic and presynaptic sites of a mesolimbic dopamine neuron. Maternal fmr1 insufficiency resulted in reduced D2 autoreceptor and enhanced GABAB receptor function in adult male offspring as measured by the locomotor response following quinpirole and baclofen administration, respectively.
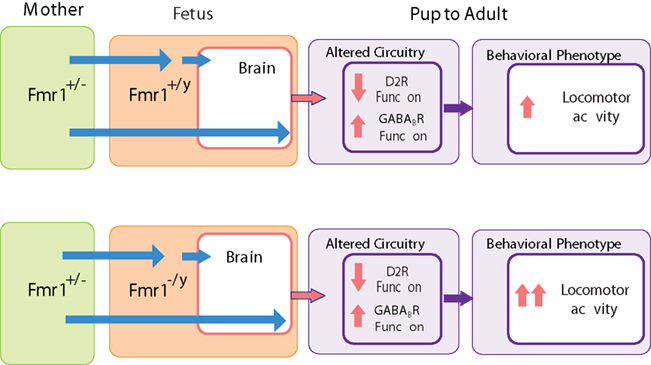
Figure 4. Maternal and offspring fmr1 genotype effects on locomotor activity. Locomotor activity is increased in genetically unaffected animals due to maternal FMRP deficit, and this behavioral effect is further enhanced by the offspring’s own lack of FMRP. Maternal FMRP deficit increases GABAB receptor and decreases D2 autoreceptor function and this effect is not further modified by the offspring’s own fmr1 expression suggesting that these neurochemical changes are specifically associated with the maternal deficit in FMRP.
Fmrp has been directly linked to FXS, as the sole cause of this Mendelian disorder. However, FMRP, at least in the mouse model, may have an additional function as its partial or complete deficit in the mother results in hyperactivity in the offspring that can be further increased by the offspring’s own mutation. This raises the possibility that in affected sons of mothers with full fmr1 mutation some of the behavioral phenotypes may be caused by a non-genetic mechanism related to the mother’s mutation and that genetically non-affected sons may also acquire some vulnerability to mental disorder. Interestingly, FMRP levels were found to be reduced in conditions unrelated to FXS such as autism, schizophrenia, bipolar disorder, and major depressive disorder (Fatemi and Folsom, 2011; Fatemi et al., 2010). Because the offspring are highly sensitive to even a partial reduction in FMRP, we speculate that these non-FXS conditions, via maternal effects, can expand the impact of FMRP in the human population.
Possible Mediators of Maternal Genotype Effects
Maternal effects, unrelated to the genetic transfer of any genetic variation or mutation, are in principle either behavioral, in particular during the postnatal period, or are related to maternal substances that reach or signal to the developing brain during pre and/or postnatal life. First we discuss maternal genotype effects which are likely mediated by the behavioral interaction between mother and infant. Then we will discuss potential mediators of the maternal genotype effect that act during prenatal but also postnatal life.
Influence of Maternal Care and Behavior on the Offspring
Several genes have been identified as being required for normal maternal care, and mutations in these genes lead to phenotypes including impaired pup retrieval, failure of pups to thrive, and increased pup mortality. Some of these maternal mutations lead to severe impairment of maternal behavior and subsequent reductions in pup survival or impaired somatic development, and thus are not directly relevant to maternal genotype related psychiatric conditions. Nevertheless, we briefly summarize these genes below because they could provide insights to mother–infant interaction mechanisms. FosB null mouse dams exhibit impaired nurturing behavior (time crouching over pups, failure to maintain pups in a huddle), and impaired pup retrieval (Brown et al., 1996; Kuroda et al., 2008). CREBαδ null mouse dams also exhibit impaired pup retrieval, and CREBαδ heterozygous pups fail to thrive (Jin et al., 2005). Dams carrying a null allele of Peg-3 show reduced nurturing behavior toward their offspring and associated reductions in offspring survival, as well as reduced lactation, which was correlated with a reduction in the number of oxytocin neurons in the hypothalamus (Li et al., 1999). In a follow-up study, Curley et al. (2004, 2008) examined the effect of Peg-3 maternal and/or offspring deficiency on offspring development, and demonstrated that pups born to WT mothers who inherited a mutant Peg-3 allele exhibit reduced suckling activity and weight gain, and that their mothers ate less during pregnancy, indicating impaired fetal–maternal signaling. Peg-3 mutant mothers of WT pups also failed to increase caloric intake during pregnancy and had reduced milk let-down, while their offspring gained weight less rapidly and entered puberty later. When both mother and pup were mutant for Peg-3, these abnormalities were additive, and led to substantially increased pup mortality. Other genes implicated in maternal behavior include Mest/Peg1 (Lefebvre et al., 1998), the prolactin receptor gene (Lucas et al., 1998), the corticotropin-releasing factor I gene (Gammie et al., 2007), Pet-1, encoding a serotonergic transcription factor (Lerch-Haner et al., 2008), the brain vasopressin gene (Bosch and Neumann, 2008), the oxytocin and the oxytocin receptor genes (Takayanagi et al., 2005), and CD38 (Jin et al., 2007).
As discussed earlier, thus far there have been few animal models in which specific maternal mutations were found to contribute to offspring behavioral changes that are reminiscent of psychiatric-like conditions. Instead, research has focused mostly on strain differences, innate variability within rodent strains, or rodents selectively bred for behavioral traits. For instance, there is a line of Wistar rats selectively bred for anxiety (high anxiety behavior and low anxiety behavior rats; Liebsch et al., 1998), in which high anxiety rats have been shown to display reduced levels of maternal behavior relative to low anxiety rats (Kessler et al., 2011). However, these rat lines are bred to maximize phenotypic variability, and as such have a large number of genetic differences, making it difficult to determine which genetic changes are relevant to the observed behavioral changes.
Meaney and colleagues have compiled an extensive body of work demonstrating how innate variability in maternal behavior in Long–Evans rats can lead to altered offspring behavior. A large cohort of rats was phenotyped for natural variations in maternal behavior, and two subpopulations were identified, the 10% displaying the most licking–grooming and arched back nursing (High LG–ABN), as well as the 10% displaying the lowest frequency of these behaviors (Low LG–ABN; Liu et al., 1997). The offspring of high LG–ABN rats were found to display reduced fearfulness, reduced hypothalamic–pituitary–adrenal (HPA) axis responses to stress, and decreased startle response, as well as increased hippocampal glucocorticoid receptor expression. When rats were cross-fostered at birth, their adult behavioral phenotypes and their own maternal behavior resembled that of their foster mothers, indicating a postnatal and transgenerational effect (Francis et al., 1999). Again, the selection procedure segregated two different populations that likely differ in multiple genetic mutations or polymorphisms. The variability in maternal behavior was found to selectively alter the epigenome of the offspring by differential methylation of the glucocorticoid receptor promoter during the first postnatal week, an effect which persisted into adulthood and was reversible through cross-fostering (Weaver et al., 2004). The group differences in epigenetic modification, glucocorticoid receptor expression, and HPA responses to stress could be removed through central infusion of a histone deacetylase inhibitor into the adult offspring (Weaver et al., 2004).
A similar approach was also used with mouse strains showing genetic and phenotypic heterogeneity. For instance, the C57/Bl6 (B6) mouse displays greater licking/grooming behavior, reduced anxiety-related behavior, increased learning ability in the Morris water maze (MWM), and increased pre-pulse inhibition (PPI) of acoustic startle, relative to the BalbC. When BalbC zygotes were implanted into B6 hosts and raised by B6 foster mothers dams during both the prenatal and postnatal period, elevated plus maze, open field, and MWM behavior were shown to be no different from that of B6 mice raised by B6 mothers (Francis et al., 2003). Carola et al. (2008) used the maternal care variability in this strains to create mice that are genetically identical (B6/BalbC heterozygotes), but nonetheless display different maternal behavior based on maternal strain. They then utilized B6/BalbC heterozygote dams with either B6 or BalbC like behavior to study the effect of maternal behavior on offspring behavior. These data, although revealing no maternal genes, again indicate the importance of the maternal genotype on the behavior of genetically unrelated offspring producing psychiatric disease-like conditions.
Maternal Cytokines
Maternal cytokines have been proposed as potential mediators that signal across the placenta to the developing embryo, either directly, or through induction of fetal cytokines. Although we have no direct evidence that the maternal genotype effects in the 5-HT1A receptor and Fmrp deficient mouse lines or in other models of maternal genotype effects would be mediated by cytokines, these molecules are plausible candidates because they can cross the placenta and blood–brain barrier and because cytokine receptors are abundant in the brain, providing a means to convey the maternal genotype effects to developing neurons. Both the 5-HT1A receptor and FMRP are expressed in immune cells and could alter their functions, including cytokine production and secretion. Indeed, the 5-HT1A receptor is prominently expressed in the immune system and we and others have shown that 5-HT, via the 5-HT1A receptor, has chemotactic activity for eosinophils and mast cells (Boehme et al., 2004; Kushnir-Sukhov et al., 2006). Regarding FMRP, significant differences in the plasma levels of a number of cytokines, including IL-1alpha, were reported between FXS individuals and controls (Ashwood et al., 2011).
The evidence for the ability of cytokines to alter offspring brain development and function comes from studies in which the maternal immune system is challenged, resulting in offspring behavioral abnormalities (Smith et al., 2007; Patterson, 2009). Maternal infection and inflammation during pregnancy have long been implicated as potential predisposing factors to offspring psychiatric disorders, including schizophrenia and autism. For instance, retrospective studies examining maternal medical records have shown that maternal infection increases the risk of schizophrenia in offspring (2× for respiratory infection, 8× for influenza infection; Brown et al., 1996; Byrne et al., 2007). Schizophrenia in offspring has also been associated with elevated cytokines and anti-influenza antibodies in archived maternal serum (Brown et al., 2004a,b). A Danish registry study recently showed that maternal hospital admission due to either viral infection in the first trimester or bacterial infection in the second trimester was significantly associated with the diagnosis of Autism Spectrum Disorder in offspring (Atladottir et al., 2010). Animal models that have been used to test the link between maternal inflammation and offspring psychiatric-like phenotypes include administration of lipopolysaccharide (LPS), polyinosinic polycytidylic acid [Poly(I:C)] or antibodies, or direct exposure to pathogens such as influenza, all of which induce an immune response in the dam, which may directly or via the induction of fetal cytokines have profound consequences for the developing fetus (reviewed in Boksa, 2010).
Poly(I:C) is a synthetic dsRNA that acts through the toll-like receptor (TLR) 3 (Cunningham et al., 2007). The injection of Poly(I:C) into rodent dams during gestation has been shown to lead to a panoply of schizophrenia-related behavioral abnormalities in their offspring including deficits in PPI, social interaction, latent inhibition, working memory, and novel object exploration (Patterson, 2009). In addition, maternal Poly(I:C) exposure induces a range of histological and structural changes in systems relevant to schizophrenia, including increased GABAA receptor α2 immunoreactivity and dopamine hyperfunction, ventricular enlargement, reduced NMDA receptor expression in the hippocampus, and reduced dopamine D1 and D2 receptor expression in the prefrontal cortex (Patterson, 2009). These neurochemical changes may underlie the behavioral abnormalities. Extending the Poly(I:C) model of maternal immune activation, Abazyan et al. (2010)exposed an mhDISC1 mutant mouse to Poly(I:C) at gestational day 9 (GD9), which increased the anxiety-related behaviors, depression-like responses, and altered social behaviors in adults as compared to mutant mice of uninfected dams. Polymorphisms in the DISC1 gene have been linked to increased risk of several psychiatric diseases, including schizophrenia, major depression, bipolar disorder, autism, and Asperger’s syndrome (Millar et al., 2000, 2001; Blackwood et al., 2001; Kilpinen et al., 2008) and DISC1 mutant animals display a range of cellular and behavioral phenotypes relevant to schizophrenia (Clapcote et al., 2007; Hikida et al., 2007; Kvajo et al., 2008; Pletnikov et al., 2008; Shen et al., 2008). Importantly, Poly(I:C) treatment altered cytokine levels in the fetal brain in a genotype-dependent manner indicating that offspring mutations can increase the maternal effect presumably via the induction of fetal cytokines. Tuberous sclerosis (TSC) is a genetic condition in which 40–50% of affected individuals display autism spectrum disorder (Ehninger et al., 2010). These authors exposed a mouse mutant for Tsc2 to gestational immune activation, and demonstrated that this mutation increases in utero mortality after Poly(I:C) injection, and that the combined genetic and environmental factors lead to abnormalities in social interaction in surviving offspring (Ehninger et al., 2010).
Conclusion
The high heritability of psychiatric disorders is likely explained by a number of mechanisms including the inheritance of rare alleles with high penetrance and various combinations of low penetrance alleles. Here we describe another mechanism that could also significantly contribute to the heritability of these disorders. Specifically, here we show that maternal mutations alone or in combination with offspring mutations can result in anxiety and hyperactivity in the offspring. The importance of maternal genotype effects is twofold. First, it can increase the severity of the phenotype caused by the offspring genotype. Second, maternal genotype effects can produce disease-like phenotypes in genetically unaffected individuals. This finding expands the population vulnerable to psychiatric disease. Since conventional association studies do not include the maternal genome, the identification of maternal genotype effects is difficult. Efforts to incorporate parental genomes into large scale genome-wide association studies (Kong et al., 2009), could eventually help to estimate the overall impact of maternal genotypes in increasing risk for mental disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abazyan, B., Nomura, J., Kannan, G., Ishizuka, K., Tamashiro, K. L., Nucifora, F., Pogorelov, V., Ladenheim, B., Yang, C., Krasnova, I. N., Cadet, J. L., Pardo, C., Mori, S., Kamiya, A., Vogel, M. W., Sawa, A., Ross, C. A., and Pletnikov, M. V. (2010). Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol. Psychiatry 68, 1172–1181.
Arango, V., Underwood, M. D., Boldrini, M., Tamir, H., Kassir, S. A., Hsiung, S., Chen, J. J., and Mann, J. J. (2001). Serotonin 1A receptors, serotonin transporter binding and serotonin transporter mRNA expression in the brainstem of depressed suicide victims. Neuropsychopharmacology 25, 892–903.
Ashwood, P., Nguyen, D. V., Hessl, D., Hagerman, R. J., and Tassone, F. (2011). Plasma cytokine profiles in Fragile X subjects: is there a role for cytokines in the pathogenesis? Brain Behav. Immun. 24, 898–902.
Atladottir, H. O., Thorsen, P., Schendel, D. E., Ostergaard, L., Lemcke, S., and Parner, E. T. (2010). Association of hospitalization for infection in childhood with diagnosis of autism spectrum disorders: a Danish cohort study. Arch. Pediatr. Adolesc. Med. 164, 470–477.
Bannerman, D. M., Grubb, M., Deacon, R. M., Yee, B. K., Feldon, J., and Rawlins, J. N. (2003). Ventral hippocampal lesions affect anxiety but not spatial learning. Behav. Brain Res. 139, 197–213.
Blackwood, D. H., Fordyce, A., Walker, M. T., St Clair, D. M., Porteous, D. J., and Muir, W. J. (2001). Schizophrenia and affective disorders – cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am. J. Hum. Genet. 69, 428–433.
Boehme, S. A., Lio, F. M., Sikora, L., Pandit, T. S., Lavrador, K., Rao, S. P., and Sriramarao, P. (2004). Cutting edge: serotonin is a chemotactic factor for eosinophils and functions additively with eotaxin. J. Immunol. 173, 3599–3603.
Boksa, P. (2010). Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav. Immun. 24, 881–897.
Bosch, O. J., and Neumann, I. D. (2008). Brain vasopressin is an important regulator of maternal behavior independent of dams’ trait anxiety. Proc. Natl. Acad. Sci. U.S.A. 105, 17139–17144.
Brown, A. S., Begg, M. D., Gravenstein, S., Schaefer, C. A., Wyatt, R. J., Bresnahan, M., Babulas, V. P., and Susser, E. S. (2004a). Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch. Gen. Psychiatry 61, 774–780.
Brown, A. S., Hooton, J., Schaefer, C. A., Zhang, H., Petkova, E., Babulas, V., Perrin, M., Gorman, J. M., and Susser, E. S. (2004b). Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am. J. Psychiatry 161, 889–895.
Brown, J. R., Ye, H., Bronson, R. T., Dikkes, P., and Greenberg, M. E. (1996). A defect in nurturing in mice lacking the immediate early gene fosB. Cell 86, 297–309.
Brummelte, S., and Galea, L. A. (2010). Depression during pregnancy and postpartum: contribution of stress and ovarian hormones. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 766–776.
Byrne, M., Agerbo, E., Bennedsen, B., Eaton, W. W., and Mortensen, P. B. (2007). Obstetric conditions and risk of first admission with schizophrenia: a Danish national register based study. Schizophr. Res. 97, 51–59.
Carola, V., Frazzetto, G., Pascucci, T., Audero, E., Puglisi-Allegra, S., Cabib, S., Lesch, K. P., and Gross, C. (2008). Identifying molecular substrates in a mouse model of the serotonin transporter × environment risk factor for anxiety and depression. Biol. Psychiatry 63, 840–846.
Champagne, F. A., Curley, J. P., Swaney, W. T., Hasen, N. S., and Keverne, E. B. (2009). Paternal influence on female behavior: the role of Peg3 in exploration, olfaction, and neuroendocrine regulation of maternal behavior of female mice. Behav. Neurosci. 123, 469–480.
Chen, L., and Toth, M. (2001). Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience 103, 1043–1050.
Clapcote, S. J., Lipina, T. V., Millar, J. K., Mackie, S., Christie, S., Ogawa, F., Lerch, J. P., Trimble, K., Uchiyama, M., Sakuraba, Y., Kaneda, H., Shiroishi, T., Houslay, M. D., Henkelman, R. M., Sled, J. G., Gondo, Y., Porteous, D. J., and Roder, J. C. (2007). Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 54, 387–402.
Consortium TD-BFX. (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 78, 23–33.
Cory-Slechta, D. A., Zuch, C. L., and Fox, R. A. (1996). Comparison of the stimulus properties of a pre- vs. a putative postsynaptic dose of quinpirole. Pharmacol. Biochem. Behav. 55, 423–432.
Cunningham, C., Campion, S., Teeling, J., Felton, L., and Perry, V. H. (2007). The sickness behaviour and CNS inflammatory mediator profile induced by systemic challenge of mice with synthetic double-stranded RNA (poly I:C). Brain Behav. Immun. 21, 490–502.
Curley, J. P., Barton, S., Surani, A., and Keverne, E. B. (2004). Coadaptation in mother and infant regulated by a paternally expressed imprinted gene. Proc. Biol. Sci. 271, 1303–1309.
Curley, J. P., Champagne, F. A., Bateson, P., Keverne, E. B., and Curley, K. O. Jr. (2008). Transgenerational effects of impaired maternal care on behaviour of offspring and grandoffspring. Anim. Behav. 75, 1551–1561.
Davis, J. D., and Tremont, G. (2007). Neuropsychiatric aspects of hypothyroidism and treatment reversibility. Minerva Endocrinol. 32, 49–65.
D’Hooge, R., Nagels, G., Franck, F., Bakker, C. E., Reyniers, E., Storm, K., Kooy, R. F., Oostra, B. A., Willems, P. J., and De Deyn, P. P. (1997). Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience 76, 367–376.
Doolin, M. T., Barbaux, S., McDonnell, M., Hoess, K., Whitehead, A. S., and Mitchell, L. E. (2002). Maternal genetic effects, exerted by genes involved in homocysteine remethylation, influence the risk of spina bifida. Am. J. Hum. Genet. 71, 1222–1226.
Drevets, W. C., Frank, E., Price, J. C., Kupfer, D. J., Holt, D., Greer, P. J., Huang, Y., Gautier, C., and Mathis, C. (1999). PET imaging of serotonin 1A receptor binding in depression. Biol. Psychiatry 46, 1375–1387.
Drevets, W. C., Thase, M. E., Moses-Kolko, E. L., Price, J., Frank, E., Kupfer, D. J., and Mathis, C. (2007). Serotonin-1A receptor imaging in recurrent depression: replication and literature review. Nucl. Med. Biol. 34, 865–877.
Ehninger, D., Sano, Y., de Vries, P. J., Dies, K., Franz, D., Geschwind, D. H., Kaur, M., Lee, Y. S., Li, W., Lowe, J. K., Nakagawa, J. A., Sahin, M., Smith, K., Whittemore, V., and Silva, A. J. (2010). Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Mol. Psychiatry, [Epub ahead of print].
Engberg, G., Kling-Petersen, T., and Nissbrandt, H. (1993). GABAB-receptor activation alters the firing pattern of dopamine neurons in the rat substantia nigra. Synapse 15, 229–238.
Fatemi, S. H., and Folsom, T. D. (2011). The role of fragile X mental retardation protein in major mental disorders. Neuropharmacology 60, 1221–1226.
Fatemi, S. H., Kneeland, R. E., Liesch, S. B., and Folsom, T. D. (2010). Fragile X mental retardation protein levels are decreased in major psychiatric disorders. Schizophr. Res. 124, 246–247.
Figueiredo, B., and Costa, R. (2009). Mother’s stress, mood and emotional involvement with the infant: 3 months before and 3 months after childbirth. Arch. Womens Ment. Health 12, 143–153.
Francis, D., Diorio, J., Liu, D., and Meaney, M. J. (1999). Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science 286, 1155–1158.
Francis, D. D., Szegda, K., Campbell, G., Martin, W. D., and Insel, T. R. (2003). Epigenetic sources of behavioral differences in mice. Nat. Neurosci. 6, 445–446.
Gammie, S. C., Bethea, E. D., and Stevenson, S. A. (2007). Altered maternal profiles in corticotropin-releasing factor receptor 1 deficient mice. BMC Neurosci. 8, 17. doi: 10.1186/1471-2202-8-17
Gleason, G., Liu, B., Bruening, S., Zupan, B., Auerbach, A., Mark, W., Oh, J. E., Gal-Toth, J., Lee, F., and Toth, M. (2010). The serotonin1A receptor gene as a genetic and prenatal maternal environmental factor in anxiety. Proc. Natl. Acad. Sci. U.S.A. 107, 7592–7597.
Grace, A. (2001). “Psychostimulant actions on dopamine and limbic system function: relevance to the pathophysiology and treatment of ADHD,” in Stimulant Drugs and ADHD: Basic and Clinical Neuroscience, eds M. V. Solanto, A. F. T. Arnsten, and F. X. Castellanos (New York: Oxford University Press), 134–155.
Gross, C., Zhuang, X., Stark, K., Ramboz, S., Oosting, R., Kirby, L., Santarelli, L., Beck, S., and Hen, R. (2002). Serotonin1A receptor acts during development to establish normal anxiety-like behaviour in the adult. Nature 416, 396–400.
Halligan, S. L., Herbert, J., Goodyer, I., and Murray, L. (2007). Disturbances in morning cortisol secretion in association with maternal postnatal depression predict subsequent depressive symptomatology in adolescents. Biol. Psychiatry 62, 40–46.
Halligan, S. L., Herbert, J., Goodyer, I. M., and Murray, L. (2004). Exposure to postnatal depression predicts elevated cortisol in adolescent offspring. Biol. Psychiatry 55, 376–381.
Halmoy, A., Johansson, S., Winge, I., McKinney, J. A., Knappskog, P. M., and Haavik, J. (2011). Attention-deficit/hyperactivity disorder symptoms in offspring of mothers with impaired serotonin production. Arch. Gen. Psychiatry 67, 1033–1043.
Heisler, L. K., Chu, H. M., Brennan, T. J., Danao, J. A., Bajwa, P., Parsons, L. H., and Tecott, L. H. (1998). Elevated anxiety and antidepressant-like responses in serotonin 5-HT1A receptor mutant mice. Proc. Natl. Acad. Sci. U.S.A. 95, 15049–15054.
Hikida, T., Jaaro-Peled, H., Seshadri, S., Oishi, K., Hookway, C., Kong, S., Wu, D., Xue, R., Andrade, M., Tankou, S., Mori, S., Gallagher, M., Ishizuka, K., Pletnikov, M., Kida, S., and Sawa, A. (2007). Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc. Natl. Acad. Sci. U.S.A. 104, 14501–14506.
Jin, D., Liu, H. X., Hirai, H., Torashima, T., Nagai, T., Lopatina, O., Shnayder, N. A., Yamada, K., Noda, M., Seike, T., Fujita, K., Takasawa, S., Yokoyama, S., Koizumi, K., Shiraishi, Y., Tanaka, S., Hashii, M., Yoshihara, T., Higashida, K., Islam, M. S., Yamada, N., Hayashi, K., Noguchi, N., Kato, I., Okamoto, H., Matsushima, A., Salmina, A., Munesue, T., Shimizu, N., Mochida, S., Asano, M., and Higashida, H. (2007). CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 446, 41–45.
Jin, S. H., Blendy, J. A., and Thomas, S. A. (2005). Cyclic AMP response element-binding protein is required for normal maternal nurturing behavior. Neuroscience 133, 647–655.
Kao, D. I., Aldridge, G. M., Weiler, I. J., and Greenough, W. T. (2010). Altered mRNA transport, docking, and protein translation in neurons lacking fragile X mental retardation protein. Proc. Natl. Acad. Sci. U.S.A. 107, 15601–15606.
Kessler, M. S., Bosch, O. J., Bunck, M., Landgraf, R., and Neumann, I. D. (2011). Maternal care differs in mice bred for high vs. low trait anxiety: impact of brain vasopressin and cross-fostering. Soc. Neurosci. 6, 156–168.
Kilpinen, H., Ylisaukko-Oja, T., Hennah, W., Palo, O. M., Varilo, T., Vanhala, R., Nieminen-von Wendt, T., von Wendt, L., Paunio, T., and Peltonen, L. (2008). Association of DISC1 with autism and Asperger syndrome. Mol. Psychiatry 13, 187–196.
Kong, A., Steinthorsdottir, V., Masson, G., Thorleifsson, G., Sulem, P., Besenbacher, S., Jonasdottir, A., Sigurdsson, A., Kristinsson, K. T., Jonasdottir, A., Frigge, M. L., Gylfason, A., Olason, P. I., Gudjonsson, S. A., Sverrisson, S., Stacey, S. N., Sigurgeirsson, B., Benediktsdottir, K. R., Sigurdsson, H., Jonsson, T., Benediktsson, R., Olafsson, J. H., Johannsson, O. T., Hreidarsson, A. B., Sigurdsson, G., Ferguson-Smith, A. C., Gudbjartsson, D. F., Thorsteinsdottir, U., and Stefansson, K. (2009). Parental origin of sequence variants associated with complex diseases. Nature 462, 868–874.
Koob, G. F., and Swerdlow, N. R. (1988). The functional output of the mesolimbic dopamine system. Ann. N. Y. Acad. Sci. 537, 216–227.
Kuroda, K. O., Meaney, M. J., Uetani, N., and Kato, T. (2008). Neurobehavioral basis of the impaired nurturing in mice lacking the immediate early gene FosB. Brain Res. 1211, 57–71.
Kushnir-Sukhov, N. M., Gilfillan, A. M., Coleman, J. W., Brown, J. M., Bruening, S., Toth, M., and Metcalfe, D. D. (2006). 5-hydroxytryptamine induces mast cell adhesion and migration. J. Immunol. 177, 6422–6432.
Kvajo, M., McKellar, H., Arguello, P. A., Drew, L. J., Moore, H., MacDermott, A. B., Karayiorgou, M., and Gogos, J. A. (2008). A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc. Natl. Acad. Sci. U.S.A. 105, 7076–7081.
Labouebe, G., Lomazzi, M., Cruz, H. G., Creton, C., Lujan, R., Li, M., Yanagawa, Y., Obata, K., Watanabe, M., Wickman, K., Boyer, S. B., Slesinger, P. A., and Luscher, C. (2007). RGS2 modulates coupling between GABAB receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nat. Neurosci. 10, 1559–1568.
Lefebvre, L., Viville, S., Barton, S. C., Ishino, F., Keverne, E. B., and Surani, M. A. (1998). Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat. Genet. 20, 163–169.
Lemonde, S., Turecki, G., Bakish, D., Du, L., Hrdina, P. D., Bown, C. D., Sequeira, A., Kushwaha, N., Morris, S. J., Basak, A., Ou, X. M., and Albert, P. R. (2003). Impaired repression at a 5-hydroxytryptamine 1A receptor gene polymorphism associated with major depression and suicide. J. Neurosci. 23, 8788–8799.
Lerch-Haner, J. K., Frierson, D., Crawford, L. K., Beck, S. G., and Deneris, E. S. (2008). Serotonergic transcriptional programming determines maternal behavior and offspring survival. Nat. Neurosci. 11, 1001–1003.
Lesch, K. P., Wiesmann, M., Hoh, A., Muller, T., Disselkamp-Tietze, J., Osterheider, M., and Schulte, H. M. (1992). 5-HT1A receptor-effector system responsivity in panic disorder. Psychopharmacology (Berl.) 106, 111–117.
Li, L., Keverne, E. B., Aparicio, S. A., Ishino, F., Barton, S. C., and Surani, M. A. (1999). Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science 284, 330–333.
Liebsch, G., Montkowski, A., Holsboer, F., and Landgraf, R. (1998). Behavioural profiles of two Wistar rat lines selectively bred for high or low anxiety-related behaviour. Behav. Brain Res. 94, 301–310.
Liu, D., Diorio, J., Tannenbaum, B., Caldji, C., Francis, D., Freedman, A., Sharma, S., Pearson, D., Plotsky, P. M., and Meaney, M. J. (1997). Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science 277, 1659–1662.
Lopez, J. F., Chalmers, D. T., Little, K. Y., and Watson, S. J. (1998). A.E. Bennett Research Award. Regulation of serotonin1A, glucocorticoid, and mineralocorticoid receptor in rat and human hippocampus: implications for the neurobiology of depression. Biol. Psychiatry 43, 547–573.
Lucas, B. K., Ormandy, C. J., Binart, N., Bridges, R. S., and Kelly, P. A. (1998). Null mutation of the prolactin receptor gene produces a defect in maternal behavior. Endocrinology 139, 4102–4107.
Madden, T. E., and Johnson, S. W. (1998). Gamma-hydroxybutyrate is a GABAB receptor agonist that increases a potassium conductance in rat ventral tegmental dopamine neurons. J. Pharmacol. Exp. Ther. 287, 261–265.
Mann, J. J. (1999). Role of the serotonergic system in the pathogenesis of major depression and suicidal behavior. Neuropsychopharmacology 21, 99S–105S.
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., McCarthy, M. I., Ramos, E. M., Cardon, L. R., Chakravarti, A., Cho, J. H., Guttmacher, A. E., Kong, A., Kruglyak, L., Mardis, E., Rotimi, C. N., Slatkin, M., Valle, D., Whittemore, A. S., Boehnke, M., Clark, A. G., Eichler, E. E., Gibson, G., Haines, J. L., Mackay, T. F., McCarroll, S. A., and Visscher, P. M. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753.
Meaney, M. J. (2001). Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu. Rev. Neurosci. 24, 1161–1192.
Millar, J. K., Christie, S., Anderson, S., Lawson, D., Hsiao-Wei Loh, D., Devon, R. S., Arveiler, B., Muir, W. J., Blackwood, D. H., and Porteous, D. J. (2001). Genomic structure and localisation within a linkage hotspot of disrupted in Schizophrenia 1, a gene disrupted by a translocation segregating with schizophrenia. Mol. Psychiatry 6, 173–178.
Millar, J. K., Wilson-Annan, J. C., Anderson, S., Christie, S., Taylor, M. S., Semple, C. A., Devon, R. S., St Clair, D. M., Muir, W. J., Blackwood, D. H., and Porteous, D. J. (2000). Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 9, 1415–1423.
Moses-Kolko, E. L., Wisner, K. L., Price, J. C., Berga, S. L., Drevets, W. C., Hanusa, B. H., Loucks, T. L., and Meltzer, C. C. (2008). Serotonin 1A receptor reductions in postpartum depression: a positron emission tomography study. Fertil. Steril. 89, 685–692.
Murray, C., and Johnston, C. (2006). Parenting in mothers with and without attention-deficit/hyperactivity disorder. J. Abnorm. Psychol. 115, 52–61.
Murray, L., Halligan, S. L., Goodyer, I., and Herbert, J. (2010). Disturbances in early parenting of depressed mothers and cortisol secretion in offspring: a preliminary study. J. Affect. Disord. 122, 218–223.
Neumeister, A., Bain, E., Nugent, A. C., Carson, R. E., Bonne, O., Luckenbaugh, D. A., Eckelman, W., Herscovitch, P., Charney, D. S., and Drevets, W. C. (2004). Reduced serotonin type 1A receptor binding in panic disorder. J. Neurosci. 24, 589–591.
Parks, C. L., Robinson, P. S., Sibille, E., Shenk, T., and Toth, M. (1998). Increased anxiety of mice lacking the serotonin1A receptor. Proc. Natl. Acad. Sci. U.S.A. 95, 10734–10739.
Patterson, P. H. (2009). Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav. Brain Res. 204, 313–321.
Peier, A. M., McIlwain, K. L., Kenneson, A., Warren, S. T., Paylor, R., and Nelson, D. L. (2000). (Over)correction of FMR1 deficiency with YAC transgenics: behavioral and physical features. Hum. Mol. Genet. 9, 1145–1159.
Pletnikov, M. V., Ayhan, Y., Nikolskaia, O., Xu, Y., Ovanesov, M. V., Huang, H., Mori, S., Moran, T. H., and Ross, C. A. (2008). Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol. Psychiatry 13, 173–186.
Ramboz, S., Oosting, R., Amara, D. A., Kung, H. F., Blier, P., Mendelsohn, M., Mann, J. J., Brunner, D., and Hen, R. (1998). Serotonin receptor 1A knockout: an animal model of anxiety-related disorder. Proc. Natl. Acad. Sci. U.S.A. 95, 14476–14481.
Rouse, B., and Azen, C. (2004). Effect of high maternal blood phenylalanine on offspring congenital anomalies and developmental outcome at ages 4 and 6 years: the importance of strict dietary control preconception and throughout pregnancy. J. Pediatr. 144, 235–239.
Shen, S., Lang, B., Nakamoto, C., Zhang, F., Pu, J., Kuan, S. L., Chatzi, C., He, S., Mackie, I., Brandon, N. J., Marquis, K. L., Day, M., Hurko, O., McCaig, C. D., Riedel, G., and St Clair, D. (2008). Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. J. Neurosci. 28, 10893–10904.
Smith, S. E., Li, J., Garbett, K., Mirnics, K., and Patterson, P. H. (2007). Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 27, 10695–10702.
Smolders, I., De Klippel, N., Sarre, S., Ebinger, G., and Michotte, Y. (1995). Tonic GABA-ergic modulation of striatal dopamine release studied by in vivo microdialysis in the freely moving rat. Eur. J. Pharmacol. 284, 83–91.
Spencer, C. M., Alekseyenko, O., Hamilton, S. M., Thomas, A. M., Serysheva, E., Yuva-Paylor, L. A., and Paylor, R. (2011). Modifying behavioral phenotypes in Fmr1KO mice: genetic background differences reveal autistic-like responses. Autism Res. 4, 40–56.
Spencer, C. M., Alekseyenko, O., Serysheva, E., Yuva-Paylor, L. A., and Paylor, R. (2005). Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes Brain Behav. 4, 420–430.
Spinelli, S., Chefer, S., Carson, R. E., Jagoda, E., Lang, L., Heilig, M., Barr, C. S., Suomi, S. J., Higley, J. D., and Stein, E. A. (2010). Effects of early-life stress on serotonin(1A) receptors in juvenile Rhesus monkeys measured by positron emission tomography. Biol. Psychiatry 67, 1146–1153.
Starke, K., Gothert, M., and Kilbinger, H. (1989). Modulation of neurotransmitter release by presynaptic autoreceptors. Physiol. Rev. 69, 864–989.
Strobel, A., Gutknecht, L., Rothe, C., Reif, A., Mossner, R., Zeng, Y., Brocke, B., and Lesch, K. P. (2003). Allelic variation in 5-HT1A receptor expression is associated with anxiety- and depression-related personality traits. J. Neural Transm. 110, 1445–1453.
Szczypka, M. S., Kwok, K., Brot, M. D., Marck, B. T., Matsumoto, A. M., Donahue, B. A., and Palmiter, R. D. (2001). Dopamine production in the caudate putamen restores feeding in dopamine-deficient mice. Neuron 30, 819–828.
Takayanagi, Y., Yoshida, M., Bielsky, I. F., Ross, H. E., Kawamata, M., Onaka, T., Yanagisawa, T., Kimura, T., Matzuk, M. M., Young, L. J., and Nishimori, K. (2005). Pervasive social deficits, but normal parturition, in oxytocin receptor-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 102, 16096–16101.
Usiello, A., Baik, J. H., Rouge-Pont, F., Picetti, R., Dierich, A., LeMeur, M., Piazza, P. V., and Borrelli, E. (2000). Distinct functions of the two isoforms of dopamine D2 receptors. Nature 408, 199–203.
Van den Bergh, B. R., Van Calster, B., Smits, T., Van Huffel, S., and Lagae, L. (2008). Antenatal maternal anxiety is related to HPA-axis dysregulation and self-reported depressive symptoms in adolescence: a prospective study on the fetal origins of depressed mood. Neuropsychopharmacology 33, 536–545.
Van den Hove, D. L., Lauder, J. M., Scheepens, A., Prickaerts, J., Blanco, C. E., and Steinbusch, H. W. (2006). Prenatal stress in the rat alters 5-HT1A receptor binding in the ventral hippocampus. Brain Res. 1090, 29–34.
Weaver, I. C., Cervoni, N., Champagne, F. A., D’Alessio, A. C., Sharma, S., Seckl, J. R., Dymov, S., Szyf, M., and Meaney, M. J. (2004). Epigenetic programming by maternal behavior. Nat. Neurosci. 7, 847–854.
Weller, A., Leguisamo, A. C., Towns, L., Ramboz, S., Bagiella, E., Hofer, M., Hen, R., and Brunner, D. (2003). Maternal effects in infant and adult phenotypes of 5HT1A and 5HT1B receptor knockout mice. Dev. Psychobiol. 42, 194–205.
Williams, T. A., Mars, A. E., Buyske, S. G., Stenroos, E. S., Wang, R., Factura-Santiago, M. F., Lambert, G. H., and Johnson, W. G. (2007). Risk of autistic disorder in affected offspring of mothers with a glutathione S-transferase P1 haplotype. Arch. Pediatr. Adolesc. Med. 161, 356–361.
Yehuda, R., Bell, A., Bierer, L. M., and Schmeidler, J. (2008). Maternal, not paternal, PTSD is related to increased risk for PTSD in offspring of Holocaust survivors. J. Psychiatr. Res. 42, 1104–1111.
Yun, S. W., Platholi, J., Flaherty, M. S., Fu, W., Kottmann, A. H., and Toth, M. (2006). Fmrp is required for the establishment of the startle response during the critical period of auditory development. Brain Res. 1110, 159–165.
Zupan, B., and Toth, M. (2008a). Wild-type male offspring of fmr-1(+/−) mothers exhibit characteristics of the fragile X phenotype. Neuropsychopharmacology 33, 2667–2675.
Keywords: anxiety, fragile X, transgenerational, epigenetic, maternal
Citation: Gleason G, Zupan B and Toth M (2011) Maternal genetic mutations as gestational and early life influences in producing psychiatric disease-like phenotypes in mice. Front. Psychiatry 2:25. doi: 10.3389/fpsyt.2011.00025
Received: 20 February 2011;
Accepted: 26 April 2011;
Published online: 11 May 2011.
Edited by:
Sheryl Moy, University of North Carolina, USAReviewed by:
Frances A. Champagne, Columbia University, USAPaul H. Patterson, The Patterson Lab, USA
Copyright: © 2011 Gleason, Zupan and Toth. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Miklos Toth, Weill Medical College of Cornell University, 1300 York Avenue, New York, NY, USA. e-mail:bXRvdGhAbWVkLmNvcm5lbGwuZWR1