
 Steven B. Harrod
Steven B. Harrod- Behavioral Neuroscience Program, Department of Psychology, University of South Carolina, Columbia, SC, USA
One clue regarding the basis of cocaine-induced deficits in attentional processing is provided by the clinical findings of changes in the infants’ startle response; observations buttressed by neurophysiological evidence of alterations in brainstem transmission time. Using the IV route of administration and doses that mimic the peak arterial levels of cocaine use in humans, the present study examined the effects of prenatal cocaine on auditory information processing via tests of the auditory startle response (ASR), habituation, and prepulse inhibition (PPI) in the offspring. Nulliparous Long–Evans female rats, implanted with an IV access port prior to breeding, were administered saline, 0.5, 1.0, or 3.0 mg/kg/injection of cocaine HCL (COC) from gestation day (GD) 8–20 (1×/day-GD8–14, 2×/day-GD15–20). COC had no significant effects on maternal/litter parameters or growth of the offspring. At 18–20 days of age, one male and one female, randomly selected from each litter displayed an increased ASR (>30% for males at 1.0 mg/kg and >30% for females at 3.0 mg/kg). When reassessed in adulthood (D90–100), a linear dose–response increase was noted on response amplitude. At both test ages, within-session habituation was retarded by prenatal cocaine treatment. Testing the females in diestrus vs. estrus did not alter the results. Prenatal cocaine altered the PPI response function across interstimulus interval and induced significant sex-dependent changes in response latency. Idazoxan, an α2-adrenergic receptor antagonist, significantly enhanced the ASR, but less enhancement was noted with increasing doses of prenatal cocaine. Thus, in utero exposure to cocaine, when delivered via a protocol designed to capture prominent features of recreational usage, causes persistent, if not permanent, alterations in auditory information processing, and suggests dysfunction of the central noradrenergic circuitry modulating, if not mediating, these responses.
Introduction
Although a variety of data sources suggest that the years of 1979–1985 marked the height of the past epidemic of cocaine abuse in the U.S., the extraordinary levels of cocaine availability, increased purity, and the facility of smoking “crack” continue to captivate a significant segment of our population. The population of young female drug users entering childbearing age is a major concern. In 2009, an estimated 21.8 million Americans aged 12 or older were current (past month) illicit drug users, meaning they had used an illicit drug during the month prior to the survey interview (Substance Abuse and Mental Health Services Administration, 2010). This estimate represents 8.7% of the population aged 12 years old or older. Regarding cocaine usage, there are 1.6 million current cocaine users aged 12 or older, comprising 0.7% of the population (Johnston et al., 2010). These estimates are similar to the number and rate in 2008 (1.9 million or 0.7%). Clearly, illicit drug abuse, including cocaine/crack use, among young women of childbearing age remains a significant societal concern, placing future generations at risk. Comprehending the effects of prenatal drug exposure, such as cocaine/crack, on behavioral and neural development remains extremely important.
Drug use around the world is not distributed evenly and is not simply related to drug policy, as countries with more strict user-level drug policies did not have lower levels of use than countries with more liberal policies. Of the 17 countries participating in the World Health Organization’s (WHO’s) World Mental Health (WMH) Survey Initiative, the highest lifetime cumulative incidence of cocaine was in the United States (16.2%), followed by New Zealand, Spain, Colombia, and Mexico (range of 4.0–4.3%; Degenhardt et al., 2008). A more recent review of countries around the world has suggested high lifetime prevalence use of cocaine in Argentina (7.9%), Italy (6.6%), United Kingdom (6.5%), Chile (5.9%), and Ireland 5.3%; (Degenhardt et al., 2011). Thus, the implication of cocaine abuse for future generations appears to be far more than a U.S. health issue.
Comprehensive literature reviews providing ∼15 and ∼20 year perspectives on preschool (<6 years of age, Frank et al., 2001) and school-aged (>6 years of age, Ackerman et al., 2010) prenatal cocaine-exposed children, respectively, have failed to definitively identify any unique constellation of effects of prenatal cocaine exposure. Many findings once thought to be specific effects of in utero cocaine exposure are correlated with other factors, including prenatal exposure to tobacco, marijuana, or alcohol, and/or a host of social/environmental factors, such as poverty, caregiver education, placement stability, and quality of child-caregiver relationships that are known to affect a child’s development. For example, among children aged 6 years or younger, across four domains (physical growth; cognition; language skills; motor skills) Frank et al. (2001) concluded there was no convincing evidence that prenatal cocaine exposure is associated with developmental toxic effects that are different in severity, scope, or kind from the sequelae of multiple other risk factors. A similar recent reaffirmation noted that studies of school-aged children have shown no long-term direct effects of prenatal cocaine exposure on children’s physical growth, developmental test scores, or language outcomes (Ackerman et al., 2010).
Nevertheless, the most consistently suggested, and perhaps the most prominent, fetal cocaine effect reported to date is that of an enduring attentional dysfunction in preschool and school-aged children. Problems with attentional processes have been documented over the past 15 years with several different paradigms, including computer-controlled continuous performance tasks (Richardson et al., 1996; Heffelfinger et al., 1997, 2002; Leech et al., 1999; Bandstra et al., 2001; Noland et al., 2005; Savage et al., 2005; Accornero et al., 2007; Chiriboga et al., 2009; Carmody et al., 2011). Given the myriad of correlated factors that often characterizes this population, as discussed above, preclinical studies are important in defining and characterizing the behavioral and neural basis(es) of alterations in attentional processes. Consistent alterations in “attentionally sensitive” neurobehavioral paradigms have been reported in rodents when maternal cocaine was administered via the intravenous route and at relatively low “recreational” doses (Mactutus, 1999; Bayer et al., 2000, 2002; Garavan et al., 2000; Morgan et al., 2002; Gendle et al., 2003, 2004a,1996; Foltz et al., 2004).
One clue as to the basis for the cocaine-induced deficits in attention is provided by the clinical findings of changes in the infant’s startle response as well as in neurophysiological evidence of alterations in brainstem transmission time. Initial clinical studies suggested an altered startle reactivity to a variety of stimuli (e.g., Chasnoff et al., 1985, 1989; Griffith, 1988) and impairments in auditory information processing, characterized by impaired habituation in infancy (Potter et al., 2000). Although ordinal measurement of Brazelton Behavioral Assessment Scale remains rather subjective and is a caveat of the earlier studies, reflex modification procedures also reported cocaine-exposed infants were more reactive (glabellar reflex) and more responsive to auditory stimuli, as indicated by an increased blink when the tone accompanied the tap (Anday et al., 1989). Significantly depressed startle reactivity has also been reported in prenatal cocaine-exposed children at 54 months (Mayes et al., 1998). Although there are neither striking nor consistent data across the various rodent models to corroborate the clinical in utero cocaine effects on the infant startle response or its plasticity (Foss and Riley, 1988, 1991a, b; Dow-Edwards and Hughes, 1995; Vorhees et al., 1995, 2000; Hughes et al., 1996; Overstreet et al., 2000), to the best of our knowledge, there is no preclinical data available with the more clinically relevant intravenous route of cocaine administration.
Neurophysiologically based clinical studies have consistently reported alterations in auditory brainstem responses (ABR, or also called brainstem auditory-evoked responses) in prenatal cocaine-exposed infants (Shih et al., 1988; Salamy et al., 1990; Cone-Wesson and Spingarn, 1993; Lester et al., 2003; Tan-Laxa et al., 2004). Prolonged interpeak latencies of the ABR of infants prenatally cocaine-exposed was reported relative to non-exposed controls (Shih et al., 1988) and replicated in both term and low-birth weight infants exposed in utero to cocaine (Salamy et al., 1990) as well as in infants matched for birth weight and conceptual age (Cone-Wesson and Spingarn, 1993). No increased incidence of hearing impairment was found in these early clinical studies. Further, the one report that failed to find effects of prenatal cocaine exposure on the ABR used relatively low intensity stimuli (30 dB) as the aim of that study was to ascertain if there were hearing deficits consequent to prenatal cocaine exposure (Carzoli et al., 1991). Tan-Laxa et al. (2004) reported abnormal interpeak latencies in prenatal cocaine-exposed neonates, compared with non-exposed controls, confirmed with meconium drug analysis. Lester et al. (2003) confirmed and expanded the effects of prenatal cocaine on prolonging ABR interpeak latencies at 1 month of age, and did so in a large sample in which covariate adjustments were made (other drugs, gestational age, social class); the main effects were attributable to heavy maternal cocaine use. Prolonged interpeak latencies, particularly as a function of increasing stimulation rates, may indicate that neural recovery time is slower for prenatal cocaine-exposed infants, such as attributable to neurotransmitter depletion or dysfunction. Prolongation of the I–V latency, which provides information regarding the integrity of the eighth cranial nerve to the auditory brain stem, may indicate delayed brainstem auditory system development (Salamy, 1984; Krumholz et al., 1985). In a rodent model, prolongation of the interpeak latencies was also observed as a function of prenatal cocaine exposure, but only at the highest dose (100 mg/kg/day, subcutaneous), only at 35 days of age (not at 6–10 months of age), and only at the highest stimulus intensity (Church and Overbeck, 1990). The consistent ABR evidence with in utero cocaine-exposed infants, in conjunction with the limited animal model data, collectively suggests an ontogenetic impairment in auditory information processing.
Auditory processing disorders are commonly thought to be the basis of some language problems. Rate of processing of brief, rapidly presented, non-linguistic auditory stimuli in infancy has been shown to the single best predictor of subsequent language development (Benasich and Tallal, 2002). Although the effects of prenatal cocaine on preschool language development are commonly referred to as equivocal and/or subtle (e.g., Singer et al., 2001; Beeghly et al., 2006; Dinehart et al., 2009), it is of particular note that significant positive associations of language decrements with prenatal cocaine are reported in covariate-controlled prospective studies by Singer’s group (Singer et al., 2001; Lewis et al., 2004, 2007, 2011) and by Bandstra’s group (Bandstra et al., 2002, 2004, 2011; Morrow et al., 2003, 2004). Not surprisingly, other large covariate-controlled prospective studies failed to find such effects, as documented by Hurt and colleagues (Hurt et al., 1997, 2009; Betancourt et al., 2011), the cohort studied by Frank and colleagues (Frank et al., 2005; Beeghly et al., 2006) and by others (Delaney-Black et al., 2000; Kilbride et al., 2000, 2006). One possible mitigating factor in the disparate outcomes on language development within the larger prospective longitudinal studies is that of retention rate. Clearly, retention has been high among those studies that have reported positive associations of prenatal cocaine with language decrements (e.g., 89% of infant cohort assessed at 3 years, Morrow et al., 2004; 79% of the original cohort available for follow up at 3, 5, and 7 years, Bandstra et al., 2004; 77% of the original cohort through 10 years, Lewis et al., 2011). Although it is tempting to argue that the absence of an effect of prenatal cocaine on language may reflect low retention rates, e.g., 55%, this lowest rate was across 20 years (Betancourt et al., 2011). Perhaps most importantly, to the best of our knowledge, none of the large scale prospective studies have reported any differential effect of prenatal cocaine on retention rates. Collectively, these studies suggest that if an effect of prenatal cocaine on language is detected in a study cohort, a stable language decrement will likely be observed through to adolescence.
Because of these clinical and preclinical reports of prenatal cocaine-induced alterations in attentional processes and auditory information processing (with implications for receptive language development), and that the regulation of sensory processing serves as a gating mechanism to optimize attention, the present paper sought to investigate potential prenatal cocaine-induced alterations in auditory information processing in a rodent model within auditory startle, habituation, and reflex modification paradigms. The ASR and its plasticity [habituation, prepulse inhibition (PPI)] have much to recommend it for drug abuse studies, including its rapidity, its objectivity, its sensitivity, and its suitability for use in both humans and a variety of laboratory animals (Ison, 1984). Accordingly, the goals of the present study were fivefold. We sought to determine if prenatal cocaine, administered via the clinically relevant IV route and with doses that mimic the peak arterial levels of cocaine used by humans (cf. Evans et al., 1996; Booze et al., 1997): (1) alters the development of the ASR in preweanling rats, (2) has a long-term effect on the ASR and its habituation, (3) produces any differential effect on the ASR as a function of the sex and/or stage of the estrous cycle of the offspring, (4) alters the preattentive process of sensory gating as indexed by PPI, and (5) exerts its effects on the ASR via alterations in the noradrenergic system.
Materials and Methods
Animals
Nulliparous female Long–Evans rats were obtained from Harlan Sprague-Dawley (Indianapolis, IN, USA) at 11–12 weeks of age. The animals were pair-housed and given ad libitum access to both food (Purina Rat Chow) and water for 2 weeks prior to the beginning of the experiment. The animals were maintained according to NIH guidelines in AAALAC-accredited facilities. The animal facility was maintained at 21 ± 2°C, 50 ± 10% relative humidity, and had a 12-h light: 12-h dark cycle with lights on at 0700 h (EST). The protocols for the use of rats in this research were approved by the university IACUC.
Surgery
Catheterization of the female rats with a vascular access port was performed as previously described in detail (Mactutus et al., 1994). Briefly, animals were anesthetized with a mixture of ketamine (100 mg/kg/ml) and xylazine (3.3 mg/kg/ml); this anesthetic mixture was chosen over pentobarbital because of the potent effect of the latter agent on cytochrome P450 activity (LaBella and Queen, 1993; Loch et al., 1995). A sterile Intracath IV catheter (22 ga., Becton/Dickinson and Co., Franklin Lakes, NJ, USA) with a Luer lock injection cap (Medex Inc., Carlsbad, CA, USA) was tunneled under the skin from the back (between the shoulder blades) and inserted into the jugular vein approximately 3 mm in the direction of the right atria. Two sutures were placed on either side of the catheter to hold the catheter in place. Both the cap and the catheter were implanted dorsally under the skin in a subcutaneous pouch. Following the surgery, animals were observed periodically and returned to the vivarium upon recovery from the anesthesia. The catheters were flushed daily with approximately 0.2 ml of 2.5% heparinized saline and the animals were observed for any signs of discomfort or behavioral distress. Body weights were recorded prior to the surgery and daily throughout recovery.
Animal Mating
At 4–7 days following surgery, the females were group-housed with males overnight for breeding. Vaginal cytology and the presence of sperm were checked daily in the females. A sperm positive female was considered pregnant at gestation day (GD)0 and individually housed in plastic cages with wood-chip bedding throughout pregnancy and lactation.
Experimental Design/Drug Treatment
The pregnant catheterized female Long–Evans rats were randomly assigned to one of four treatment groups (ns = 12–13) and received 0.0 (saline), 0.5, 1.0, or 3.0 mg/kg of cocaine hydrochloride (Sigma, St. Louis, MO, USA). Cocaine and saline injections were administered in a volume of 1 ml/kg, via the intravenous access port, once per day from GD8–14 and twice per day from GD15–20. Weights of dams were taken daily. Day of birth was considered postnatal day 0 (D0). Pups were culled to four males and four females per liter. Pup body weights were obtained on D1 and thereafter at weekly intervals.
The IV injection procedure was chosen because it mimics the rapidly peaking pharmacokinetic profile following inhalation or IV injection of cocaine in humans (Isenschmid et al., 1992). The 3.0-mg/kg dose produces peak arterial plasma levels that are similar to those reported for humans IV administered 32 mg of cocaine (Evans et al., 1996; Booze et al., 1997). The acute heart rate and blood pressure responses in the late gestation pregnant rat are similar to those produced in a variety of other species (Mactutus et al., 2000). Under experimental conditions, this dose is self-administered by “users” multiple times in a 2.5-h session (Fischman and Schuster, 1982), and thus represents a low or recreational dose, highly relevant to the clinical situation being modeled. The lower doses (0.5 and 1.0 mg/kg) of cocaine in the male rat also produced peak arterial levels that reasonably approximated the peak arterial levels in humans IV administered 8 or 16 mg of cocaine (Evans et al., 1996; Booze et al., 1997). This regimen (route, dose, and rate) produces no evidence of overt maternal or fetal toxicity, no maternal seizure activity, no effect on maternal weight, and no effect on offspring growth or mortality (e.g., Mactutus et al., 1994; Mactutus, 1999; Foltz et al., 2004). Furthermore, this IV injection procedure does not reduce food intake of dams even when utilizing a cocaine dose as high as 6 mg/kg (Robinson et al., 1994), precluding the need for pair-fed controls.
Apparatus
The commercially available startle apparatus (SR-Lab Startle Reflex System, San Diego Instruments, Inc.) was used, but with a 10-cm thick double-walled, 81-cm × 81-cm × 116-cm isolation cabinet (external dimensions; Industrial Acoustic Company, INC., Bronx, NY, USA), rather than the 1.91-cm thick ABS plastic cabinet offered with this system. The high-frequency loudspeaker of the SR-Lab system (Radio Shack model #40-1278B, frequency range of 5–16 kHz) was mounted inside the chamber, 30 cm above the perspex test cylinder, for delivery of the auditory stimuli. The perspex test cylinder varied in size with the age of the animal: 3.8 cm internal diameter (ID) for the weanling rats and 8.9 cm ID for the adult rats. The perspex test cylinder was positioned on a 12.5-cm × 20-cm perspex platform, centrally located within the test chamber. A piezoelectric accelerometer, permanently affixed to the base of each perspex cylinder, converted the deflection of the perspex cylinder produced by the animal’s startle response to an analog signal. All accelerometer signals were then digitized (12 bit A–D) and saved to a hard disk on a Pentium class computer. Auditory stimulus intensities were measured and calibrated with a sound level meter (Extech Instruments, Waltham, MA, USA). Response sensitivities were calibrated using the SR-LAB startle calibration system.
General Procedures
For all paradigms, each animal was tested individually in the dark. All white noise stimuli were passed as broad-band through the range possible by the horn tweeter (5–16 kHz). This broad-band white noise spans the peak sensitivity of the audiogram of the Long–Evans rat at 8 kHz (Heffner et al., 1994). For all paradigms, the dependent measures collected were peak response amplitude within a 100-ms sampling window, average response amplitude across the 100-ms sampling window, and response latency. Within the constraints of the software, response latency represents the time from the onset of the startle stimulus to the time of the peak response, rather than the more traditional definition of latency, which would be until the onset of the startle response. The error introduced by this protocol is estimated to be approximately 2–3 ms, accounting for roughly 10% of the error in response latency values.
Preweaning ASR and habituation
At 18–20 days of age, one male and one female were randomly selected for a preweaning assessment. The ASR was tested, for approximately 11 min, using the following parameters: 5 min adaptation, 70 dB(A) background, followed by 36 startle trials, 100 dB(A) white noise stimulus of 20 ms duration, and a fixed 10-s intertrial interval (ITI). The startle stimulus intensity was 120 dB(A) at 2.5 cm from the speaker, but 100 dB(A) measured inside the perspex test cylinder. Habituation across the 36 trials was examined in six-trial blocks.
Adult ASR and habituation
The adult assessment (D90+) followed the parameters as described for ASR for each animal’s preweaning assessment.
Adult PPI
The animals were tested again approximately 14 days later, but with the PPI protocol. All rats were tested for approximately 18 min. Only the 3.0-mg/kg prenatal cocaine and vehicle control groups were tested in PPI. The females were tested in diestrus vs. estrus to assess any potential influence of estrous cycle. Under PPI, the animals received a 5-min adaptation period, followed by six single startle only white noise stimuli [100 dB(A), 5–16 kHz], 20 ms duration, as habituation trials with a fixed ITI of 10 s, and then 36 PPI trials [interstimulus intervals (ISIs) of 0, 8, 40, 80, 120, or 4000 ms] with a variable ITI of 20 s (range of 15–25 s), assigned by a Latin-square design. The prepulse stimulus intensity was an 85-dB(A) white noise stimulus (5–16 kHz) with a duration of 20 ms. The incorporation of a range of ISIs was fundamental to establishment of a relatively precise and defined response function, and consequently, a more accurate assessment of response inhibition. Similar ISIs have been employed in studies of the ontogeny of PPI (Parisi and Ison, 1979, 1981) as well as alterations in PPI as a function of developmental drug or toxin exposure (Ison, 1984; Fitting et al., 2006a,b,c; Lacy et al., 2011); these intervals produced clear, definable response inhibition and response latency curves. Given the constraints of the software regarding the use of any ISI intervals shorter than the prepulse stimulus duration, the ISI represented the time from the offset of the prepulse stimulus to the onset of the startle stimulus.
Adult idazoxan–ASR and habituation
The final testing of ASR of each animal occurred at approximately 120 days of age. Each animal received a subcutaneous injection of 0.5 mg/kg idazoxan (Sigma, St. Louis, MO, USA), an α2-adrenergic receptor antagonist, immediately prior to testing. A 10-min adaptation period was the only variation to the ASR parameters stated above; a procedure implemented to permit reasonable absorption and distribution of the drug.
Statistical Analysis
Weight gains of dams during their gestation period were converted to a percentage of weekly and total weight gain. The litter parameters of gestation length, number of male and female pups per litter, and mean D1 body weights of male and female pups were compared based on litter means. All data were analyzed by ANOVA techniques (Statistical Solutions Ltd, 2009). Log10 transformations of the peak amplitude response were employed, as necessary to help provide data that were consistent with the normality assumption of ANOVA. For repeated measures factors, either orthogonal decompositions were used for those variables that classically violate compound symmetry assumptions (e.g., trials) or the Greenhouse–Geisser df correction factor was used (Greenhouse and Geisser, 1959). Tests of simple mains effects and specific linear contrasts were also used to evaluate dose-dependent and trial-dependent effects of the cocaine treatment, respectively (Winer, 1971). An α level of p ≤ 0.05 was the significance level set for rejection of the null hypothesis.
Results
Offspring Growth
Prenatal IV cocaine, administered from GD8–20, had no adverse effect on any of the maternal or litter parameters measured, including maternal weight gain during pregnancy, gestation length, birth weight of male or females pups, or number of male or female pups (Table 1; Fs < 1.8). No latent effects of prenatal cocaine on adult body weight were apparent (Table 2; Fs < 1.0).
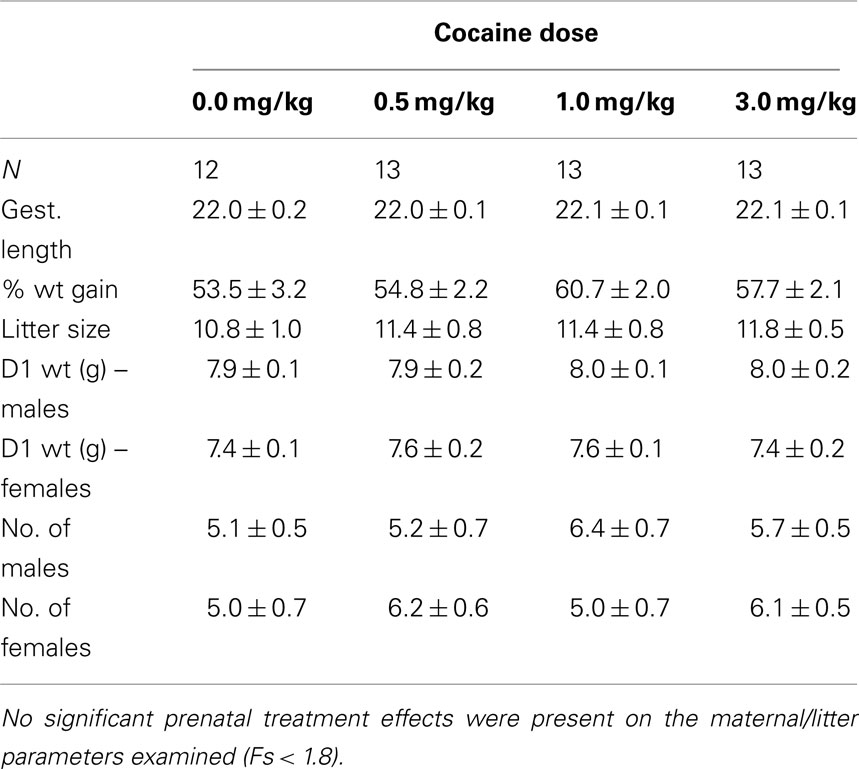
Table 1. Prenatal treatment (mean ± SEM).
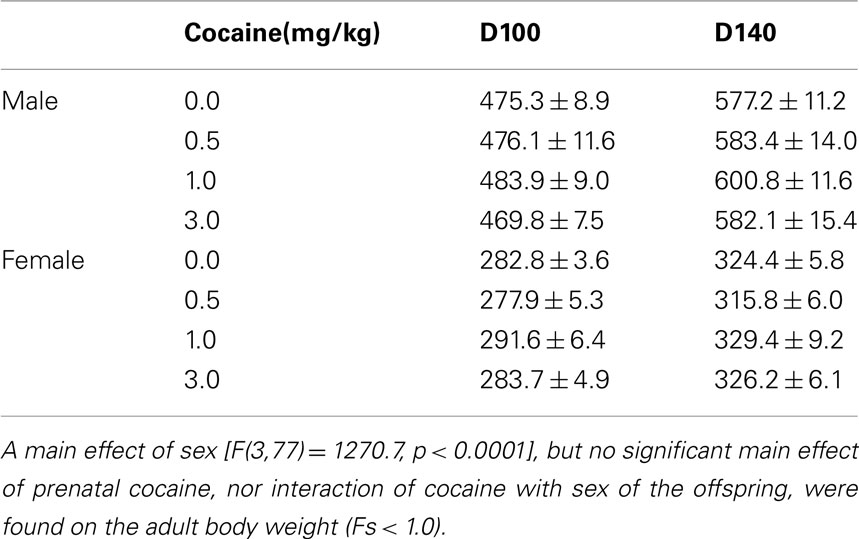
Table 2. Adult body weights (g) (mean ± SEM).
ASR-Preweanling
The peak amplitude measure of the ASR in the preweanling rats is portrayed in Figure 1 (36-trial mean) and Figure 2 (habituation across six-trial blocks). Most importantly, specific assessment of dose–response functions indicated a significant prenatal cocaine dose by offspring sex interaction [quadratic dose by sex, F(1,78) = 5.95, p ≤ 0.017]. Response amplitude was increased >18% for M > F (0.5 mg/kg), >30% for M > F (1 mg/kg) and >30% for F > M (3 mg/kg). The best fit quadratic function for males accounted for 91% of the variance (r2 = 0.91), whereas the best fit quadratic fit for females accounted for 94% of the variance(r2 = 0.94). A comparable pattern of results was seen with the average response amplitude measure, with a significant prenatal cocaine dose by offspring sex interaction [quadratic dose by sex, F(1,78) = 5.0, p ≤ 0.028]; these redundant data are not illustrated. There were no statistically significant effects detectable on average response latency. The mean (±SEM) response latency across all pups was 30.6 ± 0.3 ms; all treatment/sex group means were within the range of 29.1–32.7 ms (data not illustrated).
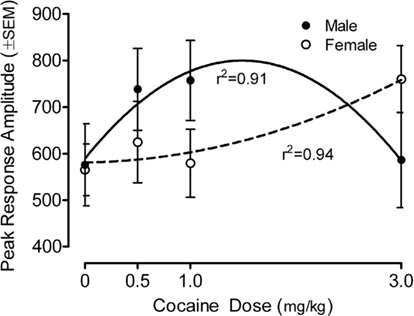
Figure 1. Mean (±SEM) peak amplitude of the ASR in the preweanling rats illustrates the quadratic cocaine dose by offspring sex interaction [F(1,78) = 5.95, p ≤ 0.017]. For the male offspring the best fit quadratic function accounted for 91% of the variance across dose; for the female offspring the best fit quadratic function accounted for 94% of the variance across dose. A similar finding of a quadratic cocaine dose by offspring sex interaction with the average amplitude measure was found [F(1,78) = 5.0, p ≤ 0.028]; those data are not shown).
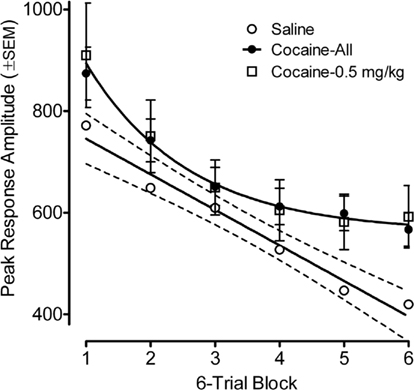
Figure 2. Mean (±SEM) peak amplitude of the ASR in the preweanling rats, illustrated as a function of six-trial blocks, depicts the retarding of within-session habituation by prenatal IV cocaine. Comparison of the average prenatal cocaine group habituation with that for the saline group found a prenatal treatment by six-trial block interaction [F(1,78) = 2.55, pGG ≤ 0.045], with significant group differences apparent on trial blocks five and six [F(1,82) = 5.8, p ≤ 0.018 and F(1,82) = 5.1, p ≤ 0.027, respectively]. The best fit linear regression for the habituation curve of the saline group accounted for 97% of the variance; the best fit function for the average cocaine habituation was a quadratic that accounted for >99% of the variance. A similar pattern of differences is also illustrated when comparing the low 0.5 mg/kg cocaine dose vs. saline.
Habituation of the ASR across six-trial blocks revealed a prominent linear decrease [F(1,78) = 106.5, p ≤ 0.001] in peak response amplitude across trials with the contribution of a quadratic component [F(1,78) = 10.4, p ≤ 0.002]. A comparison of the average prenatal cocaine group habituation vs. that for the saline group revealed a significant six-trial block by prenatal treatment interaction [F(1,78) = 2.55, pGG ≤ 0.045], with significant group differences apparent on trial blocks five and six [F(1,82) = 5.8, p ≤ 0.018 and F(1,82) = 5.1, p ≤ 0.027, respectively]. The best fit linear regression for the habituation curve of the saline group accounted for 97% of the variance; the best fit function for the average cocaine habituation was a quadratic that accounted for 99.6% of the variance. A comparison of the lowest 0.5 mg/kg cocaine dose group vs. the saline group found a very similar pattern, although the three-way interaction of six-trial block, prenatal cocaine treatment, and offspring sex was statistically significant [F(5,190) = 3.12, pGG ≤0.021]. The linear six-trial block by prenatal treatment interaction was prominent in males [F(1,38) = 10.10, p ≤ 0.003], but was not evident in females. Nevertheless, significant treatment group differences (0.5 mg/kg cocaine vs. saline) were again suggested on trial blocks five and six [F(1,38) = 3.91, p ≤ 0.055 and F(1,38) = 5.0, p ≤ 0.031, respectively].
ASR-Adult
The peak amplitude measure of the ASR in the adult offspring is portrayed in Figure 3 (36-trial mean) and Figure 4 (habituation across six-trial blocks). The peak amplitude of the ASR in the adults displayed an overall effect of prenatal dose of cocaine [F(3,80) = 6.0, p ≤ 0.001], but neither of sex of the offspring nor the interaction of dose and sex approached significance (ps > 0.10). Trend analyses indicated the prenatal treatment factor displayed a prominent linear dose–response effect [F(1,80) = 14.5, p ≤ 0.001] with a maximal facilitation of >75%.
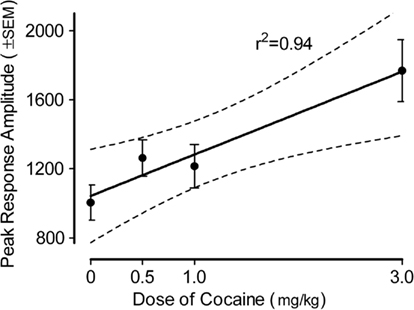
Figure 3. Mean (±SEM) peak amplitude of the ASR in the offspring as adults. The effect of prenatal dose of cocaine [F(3,80) = 6.0, p ≤ 0.001] with a prominent linear dose–response component [F(1,80) = 14.5, p ≤ 0.001] accounted for 84% of the dose group variance. A similar prominent linear dose–response effect [F(1,80) = 10.9, p ≤ 0.0015] was noted on the average amplitude measure (data not shown).
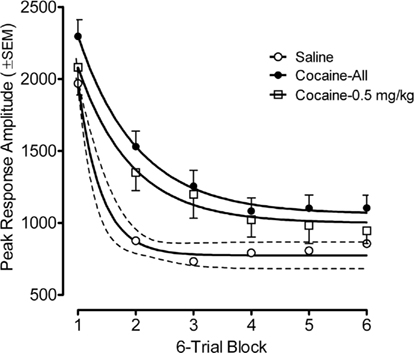
Figure 4. Mean (±SEM) peak amplitude of the ASR in the offspring as adults shown as a function of six-trial blocks. The best fit function for the habituation curves of the saline group, the average cocaine group, and the 0.5-mg/kg cocaine group were each quadratic fits that accounted for >99% of the variance; Examination of habituation across all prenatal treatment dose groups revealed significant group differences were apparent on trial blocks one through six, inclusive [Fs(3,80) ≥ 2.7, p ≤ 0.05]. Thus, within-session habituation of the ASR in adulthood was also retarded by prenatal IV cocaine.
The average amplitude of the ASR in the offspring as adults also indicated an augmented overall effect of prenatal dose of cocaine [F(3,80) = 4.8, p ≤ 0.004], but neither sex of the offspring nor the interaction of dose and sex approached significance (ps > 0.10). Trend analyses indicated the prenatal treatment factor displayed a prominent linear dose–response effect [F(1,80) = 11.1, p ≤ 0.001] with a maximal facilitation of ∼85%; this redundant data is not illustrated.
There was no overall effect of prenatal dose of cocaine on peak response latency of the ASR in the offspring as adults [F(3,80) = 1.5, p > 0.10], sex of the offspring nor on the interaction of dose and sex [Fs < 1.0]. Further, trend analyses failed to detect any systematic relation among the treatment dose groups. The mean (±SEM) response latency across all rats was 29.1 ± 0.6 ms; all treatment group means were within the range of 28.1–31.2 ms (data not illustrated). The numeric change as a function of treatment ranged from between −3 and +8% relative to vehicle controls.
Habituation of the ASR across six-trial blocks revealed both prominent linear and quadratic components to the decrease in peak response amplitude across trials [F(1,80) = 135.6, p ≤ 0.001 and F(1,80) = 83.2, p ≤ 0.001, respectively]. The best fit function for the habituation curve of the saline group was a quadratic fit that accounted for >99% of the variance; similarly, the best fit function for the average cocaine habituation was a quadratic accounting for >99% of the variance; these curves were significantly different (p < 0.001). The best fit function for the 0.5-mg/kg cocaine group also accounted for >99% of the variance. Examination of habituation across all prenatal treatment dose groups revealed significant group differences were apparent on trial blocks one through six, inclusive [F(3,80) = 2.8, p ≤ 0.045, F(3,80) = 7.5, p ≤ 0.001, F(3,80) = 5.6, p ≤ 0.002, F(3,80) = 2.9, p ≤ 0.039, F(3,80) = 2.8, p ≤ 0.048, F(3,80) = 2.7, p ≤ 0.05]. A comparison of the average prenatal cocaine group habituation vs. that for the saline group revealed an overall prenatal cocaine effect [F(1,84) = 5.9, p ≤ 0.018] with significant group differences apparent on trial blocks two and three [F(1,84) = 10.2, p ≤ 0.002 and F(1,84) = 6.2, p ≤ 0.014, respectively]. A comparison of the lowest 0.5 mg/kg cocaine dose group vs. the saline group found a very similar pattern; significant treatment group differences (0.5 mg/kg cocaine vs. saline) were again suggested on trial blocks two and three [F(1,40) = 7.9, p ≤ 0.008 and F(1,40) = 5.3, p ≤ 0.026, respectively].
Estrous Cycle
Comparison of the peak response amplitude (mean ± SEM: 1227 ± 166.4 vs. 1355 ± 176.3), average response amplitude (280 ± 26.1 vs. 320 ± 30.1) and response latency (28.3 ± 0.76 vs. 29.7 ± 0.88) in the adult females as a function of stage of the estrous cycle (diestrus vs. estrus, respectively) demonstrated that there was no significant effect on any of the dependent measures [Fs(1,20) < 1.1].
Prepulse Inhibition
An overall effect of the prenatal dose of cocaine was detected on peak amplitude on the habituation trials (six-trial average) of the PPI protocol [F(1,34) = 11.2, p ≤ 0.002], but neither sex of the offspring nor the interaction of dose and sex were significant (ps > 0.10). A marked facilitation was observed in both the males (65%) and females (55%) that had prenatally received the 3.0-mg/kg dose of cocaine. The average amplitude measure displayed a similar prenatal cocaine effect [F(1,34) = 11.3 p ≤ 0.002], with pronounced facilitation in both males (99%) and female (82%).
The PPI peak response amplitude data from the adult offspring is illustrated in Figure 5, across the ISI function. The peak amplitude on the PPI trials as a function of the ISI (six-trial average/ISI) showed that the main effects of prenatal treatment and sex, as well as the interaction of treatment and sex, were not significant. The ISI function was best described by a quadratic fit [F(1,34) = 138.1 p ≤ 0.001 and F(1,34) = 75.2, p ≤ 0.001, peak and average amplitude measures, respectively]. Of greater interest, however, a significant prenatal treatment effect by prepulse ISI was observed [F(5,170) = 4.0, pGG ≤ 0.038, with the suggestion of linear and quadratic components F(1,34) = 6.4, p ≤ 0.016 and F(1,34) = 3.8, p ≤ 0.059, respectively]. The average amplitude measure similarly confirmed a significant prenatal treatment by prepulse ISI interaction [F(5,170) = 4.7, pGG ≤ 0.026], with linear and quadratic components [F(1,34) = 5.6, p ≤ 0.024, and F(1,34) = 4.6, p ≤ 0.039, respectively]. Specific examination of the prepulse trials (ISIs 4–120) noted differential modulation of inhibition by ISI as a function of prenatal cocaine treatment. The characteristic quadratic function displayed by the saline control group account for 88% of the variance whereas a linear function accounted for 67% of the variance for the prenatal cocaine group. The different functional characterization of the inhibition curve further demonstrated a shift in the peak of inhibition from 40 to 120 ms. The evidence for an effect of prenatal cocaine treatment on the control trials (0 and 4000 ms ISI intervals) was suggested, but not consistent [peak amplitude, F(1,34) = 3.4, p ≤ 0.072 vs. average amplitude, F(1,34) = 4.2, p ≤ 0.048]. The calculation of a percent PPI measure, to take into account any such difference in the control trials, failed to detect any overall difference in magnitude of inhibition (prenatal saline, 87.2% vs. prenatal cocaine, 87.7%). Thus, although prenatal cocaine exposure did not alter the derived percent PPI measure, the assessment of inhibition across ISI displayed a functional alteration in modulation of inhibition by ISI, in other words, an alteration in sensorimotor gating.
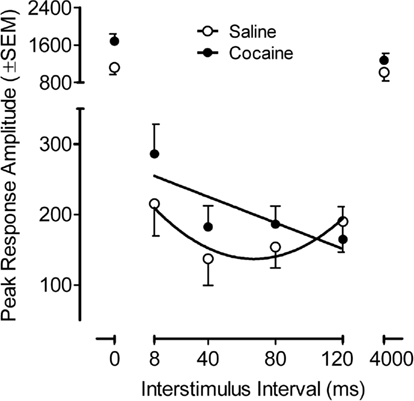
Figure 5. Mean (±SEM) peak amplitude on the prepulse inhibition trials as a function of the ISI (six-trial average/ISI). A significant prenatal treatment effect by prepulse ISI was observed [F(5,170) = 4.0, pGG ≤ 0.038]. The characteristic quadratic function across ISIs 8–120, accounted for 88% of the variance in the control group. In contrast the progressive linear change in inhibition across ISI for the 3.0-mg/kg prenatal cocaine group accounted for 67% of the variance. A shift in the peak of inhibition from 40 to 120 ms is also apparent.
The response latency measure for the PPI paradigm with the adult offspring is portrayed in Figure 6. An examination of response latencies across the inhibition trials (8–120 ms), where the functional alterations in response inhibition was noted above, confirmed there was a pronounced effect of ISI on response latency [F(3,102) = 20.5, p ≤ 0.001, with a prominent linear component, F(1,34) = 44.4, p ≤ 0.001], but ISI did not interact with prenatal treatment. Specifically, all groups generally displayed shorter latencies with the shorter ISIs. Nevertheless, a significant offspring sex effect [F(1,34 = 4.5, p ≤ 0.041] as well as a prenatal treatment by offspring sex interaction [F(1,34) = 5.2, p ≤ 0.030] was observed on the inhibition trials (ISIs of 8–120 ms). An examination of this interaction revealed that there was a significant sex difference in response latency for the saline control group [F(1,34) = 9.2, p ≤ 0.005], which was not present in the prenatal cocaine-treated adults [F < 1.0]. From an alternative view, there was a significant prenatal treatment effect in the male adult offspring [F(1,34) = 6.6, p ≤ 0.015], but not in the female adult offspring [F < 1.0]. The locus of these group differences appeared to be the attributable to the difference in response latency at the 40-ms ISI [F(1,34) = 8.0, p ≤ 0.008].
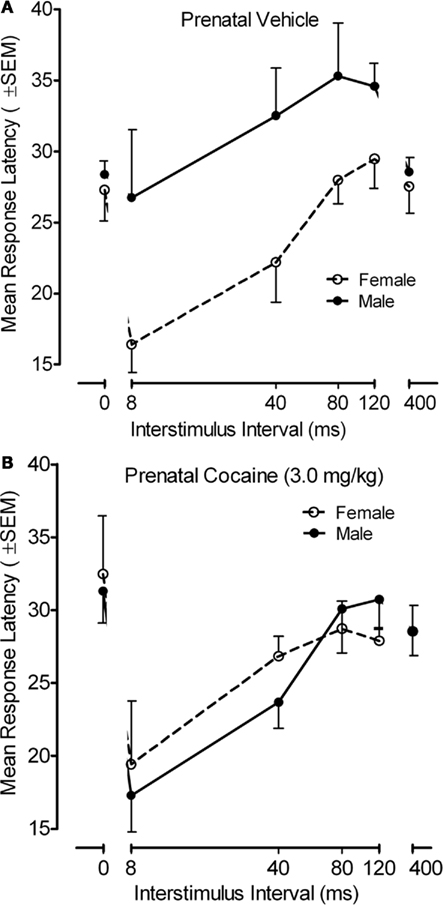
Figure 6. (A) Mean (±SEM) response latency in as a function of ISI and sex of the offspring for the prenatal (A) vehicle- and (B) cocaine-treated animals. A prenatal treatment by offspring sex interaction [F(1,34) = 5.2, p ≤ 0.030] was observed on the inhibition trials (ISIs of 8–120 ms). The significant sex difference in response latency for the saline control group [F(1,34) = 9.2, p ≤ 0.005] was not present in the prenatal cocaine-treated adults [F < 1.0]. The most marked treatment group difference in response latency occurred at the 40-ms ISI [F(1,34) = 8.0, p ≤ 0.008].
Idazoxan
The percent change in the peak amplitude of the ASR following idazoxan administration in the adult offspring is displayed as a function of prenatal cocaine dose in Figure 7. There was a significant prenatal treatment effect [F(3,79) = 3.1, p ≤ 0.031] which was characterized by a prominent linear trend [F(1,79) = 8.9, p ≤ 0.004]. Although an effect of offspring sex approached significance [F(1,79) = 3.6, p ≤ 0.062], there was little evidence for an interaction of prenatal cocaine dose and sex of the offspring was [F(1,79) = 1.3]. An evaluation of the percent change in the average amplitude of the ASR confirmed the major finding of a differential sensitivity to idazoxan as a linear function of prenatal cocaine dose [F(1,79) = 4.2, p ≤ 0.043]. The evidence for idazoxan-induced changes in response latency [F(3,79) = 3.5, p < 0.019] displayed a prominent quadratic component across increasing prenatal cocaine dose [F(1,79) = 9.3. p < 0.003] having no obvious association with the response amplitude effects of idazoxan. The response latency data (mean ± SEM), across increasing prenatal cocaine dose, were 3.0 ± 3.1, 8.7 ± 3.7, 6.4 ± 4.5, and −10.3 ± 7.0%, respectively.
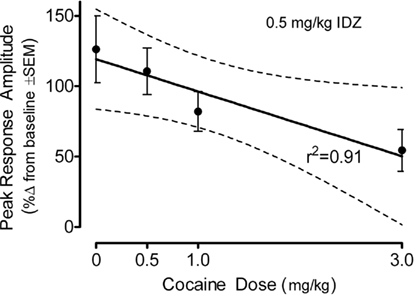
Figure 7. Mean (±SEM) percent change in the peak amplitude of the ASR following idazoxan (0.5 mg/kg) administration in the adult offspring shown as a function of prenatal cocaine dose. There was a significant prenatal treatment effect [F(3,79) = 3.1, p ≤ 0.03] which was characterized by a prominent linear trend [F(1,79) = 8.9, p ≤ 0.004]. A similar linear trend was shown by the average amplitude response data [F(1,79) = 4.2, p ≤ 0.043] (not shown).
Discussion
Administration of cocaine to pregnant rats from the time of implantation until parturition produced significant alterations in auditory information processing in the Long–Evans rat within auditory startle, habituation, and reflex modification paradigms. Three prominent effects observed were: (1) the magnitude of the ASR, as indexed by waveform amplitude, was significantly increased as a function of prenatal dose of cocaine; (2) decreased rates of within-session habituation were observed at even the lowest maternally administered dose; and (3) differential sensitivity to PPI was noted as a function of prenatal cocaine with manipulation of the ISI function; this differential sensitivity of the preattentive process of sensorimotor gating was observed for both response amplitude and response latency measures. Perhaps more importantly, (4) significant alterations in each of the three paradigms were found in adulthood, regardless of whether or not there was evidence available for the persistence, as opposed to a latent emergence, of the effects. Further, (5) an alteration in the function of the noradrenergic system in adulthood was suggested by the prenatal cocaine dose-dependent attenuation of the ability of 0.5 mg/kg idazoxan to enhance the ASR. Finally, these alterations in auditory information processing were observed with cocaine being delivered by the clinically relevant IV route and at physiologically relevant doses; conditions under which no adverse effects on maternal/litter parameters were significantly altered.
Early clinical data suggested an altered startle reactivity to a variety of stimuli (e.g., Chasnoff et al., 1985, 1989; Griffith, 1988) may be observed in prenatal cocaine-exposed infants. Subsequent experimental data reported increased responsiveness to auditory stimuli (Anday et al., 1989); but depressed startle reactivity has also been noted (Mayes et al., 1998). The increased ASR response presently reported during the preweaning period (∼30%) and in adulthood (>75%) was particularly notable in light of the relatively modest, if any, changes available in the literature with rodent models (Foss and Riley 1988,1991a,b; Dow-Edwards and Hughes, 1995; Vorhees et al., 1995, 2000; Hughes et al., 1996). In the initial, preliminary study, the ASR of prenatal cocaine rats (60 mg/kg/day, peroral (PO) route, GD14–21) was enhanced at 90, but not at 60, days of age (Foss and Riley, 1988). However, a very systematic replication study by the same laboratory, with the addition of pair-fed controls, and a second set of rats administered cocaine via the subcutaneous (SC) route (40 mg/kg/day, GD8–21) failed to find any significant changes in the ASR, in startle habituation, or in reflex modification tests that could be attributed to prenatal exposure to cocaine (Foss and Riley, 1991a). Not surprisingly, prenatal cocaine during a more restricted period of gestation (GD14–21, SC route) also failed to significantly affect the peak ASR in adult rats (Foss and Riley, 1991b). Repeated daily dosing (five doses/day × 20 mg/kg/dose) on GD7–12 or GD13–18, failed to affect the ASR on D50–52 (Vorhees et al., 1995). In the one “positive” peer-reviewed prenatal study, cocaine administered intragastric (IG; GD8–22) failed to affect the peak ASR at 60 days of age, although there was a small, statistically significant decrease in the ASR of females (Hughes et al., 1996). Even the neonatal rat model, promoted to more specifically mimic fetal brain exposure to cocaine during the third trimester of human pregnancy (Dobbing and Sands, 1979), has provided, at best, equivocal results (Dow-Edwards and Hughes, 1995; Vorhees et al., 2000). Comparing D1–10 vs. D11–20 treatment with cocaine revealed no significant alteration in the ASR on an initial test at 60–65 days of age (Dow-Edwards and Hughes, 1995). In the “positive” study, neonatal cocaine-treated rats (60 mg/kg, SC, PND 1–10) showed a significantly augmented (∼30%) peak ASR at D50–52; however, an analysis of covariance adjusting for significant cocaine-induced decreases in body weight, reduced the augmentation to a statistically non-significant level (Vorhees et al., 2000). Clearly, there are neither striking nor consistent positive data within the extant rodent models to corroborate the clinical in utero cocaine effects on the infant ASR.
Dose-dependent alterations in the ASR characterized by prenatal cocaine treatment were apparent during both the preweaning period and in adulthood. The early alterations in the ASR were differently expressed dependent upon the sex of the offspring. Prenatal cocaine-exposed males were most adversely affected by the low and middle doses of cocaine, as reflected in a quadratic dose–response fit, whereas the females were most adversely affected by the high-dose of cocaine, as reflected in a growth curve fit. In adulthood, a linear dose–response function, independent of offspring sex, captured 94% of the variance in ASR amplitude. The presence of a sex-dependent effect of cocaine well before puberty, but not in adulthood, on the same dependent measure suggests the likelihood of an organizational effect on the brain (Gorski et al., 1975). The consequences of such an organizational effect are not readily revealed as a function of ASR testing of the adult females in estrus vs. diestrus vs. males. Nevertheless, as discussed in detail below, the pronounced prenatal cocaine by sex interaction on response latency in the PPI paradigm is wholly consistent with such an effect on early brain development.
Deficits in auditory information processing, characterized by impaired habituation in prenatal cocaine-exposed neonates, were reported using habituation and recovery of head-turning toward an auditory stimulus (Potter et al., 2000). The response pattern of the prenatal cocaine-exposed infants was consistent with a slower speed on auditory information processing. Cocaine-exposed newborns showed inferior performance on the habituation cluster of the Brazelton scale, requiring more trials than controls to habituate to auditory (as well as visual and tactile) stimuli (Eisen et al., 1991). The presently reported alterations in within-session habituation detected during the preweaning period as well as in adulthood were also especially notable in light of the paucity of such information in the preclinical literature. No differences were seen as a function of prenatal cocaine in rate of habituation to a pulsing tone in assessment of the heart rate orienting response in preweanling rats (Heyser et al., 1994). Impaired between-session habituation at 60–65 days of age was found following neonatal cocaine exposure on D1–10; ASR amplitude was higher throughout session 2 than session 1, however, the polynomial fit across startle trials did not differentiate the prenatal treatments (Dow-Edwards and Hughes, 1995).
The use of the relatively short ITI, to facilitate within-session habituation, was able to differentiate habituation between prenatal cocaine vs. vehicle controls in the preweaning period. The linear habituation in controls contrasts sharply with the best fit quadratic function in prenatal cocaine animals, reflecting their failure to show progressive habituation throughout the ASR session. In adulthood a similar differentiation of within-session habituation was apparent. The very rapid decrease in response amplitude to a plateau after the first six-trial block in controls is markedly distinct from the gradual curvilinear process observed with the prenatal cocaine-exposed adults. Perhaps of greater note, at both test ages, the overall cocaine effect was readily apparent with the lowest administered dose. It will be of great interest to ascertain the generality of this impaired habituation with tactile startling stimuli, such as with an air-puff.
Reflex modification procedures have much to recommend it for drug abuse studies in both humans and a variety of laboratory animals (Ison, 1984). The preattentive processes underlying reflex modification procedures, such as PPI, are presumed to protect encoding by gating out other stimulation that occurs in close temporal proximity to the initial stimulus; i.e., attenuated PPI would suggest less efficient gating mechanisms (Hoffman and Ison, 1980; Ison and Hoffman, 1983). In the only clinical reflex modification study of which we are aware, prenatal cocaine-exposed infants displayed an exaggerated glabellar reflex when the tap was accompanied by a tone (Anday et al., 1989). Although more reflex modification studies are available in the preclinical literature, they have consistently failed to find significant alterations in PPI.
Presently, the differential sensitivity to PPI observed as a function of prenatal cocaine was revealed by systematic manipulation of the ISI function; this differential sensitivity of the preattentive process of sensorimotor gating was observed for both response amplitude and response latency measures. It is of interest that the commonly reported metric for response amplitude, percentage PPI, was not altered by the prenatal cocaine treatment when tested in adulthood. However, the incorporation of a reasonably complete ISI function was sensitive to revealing alterations in sensorimotor gating. In contrast to the characteristic curvilinear function relating PPI to ISI, as seen with other stimulants or toxic proteins (e.g., Ison, 1984; Fitting et al., 2006a,b,c; Lacy et al., 2011), the prenatal cocaine-exposed adults displayed a progressive increase in PPI as the ISI increased throughout the range examined (8–120 ms). Thus, the temporal process of sensorimotor gating, as revealed by manipulation of ISI, appeared adversely affected by the prenatal cocaine treatment.
Alterations in response latencies during PPI were striking, particularly in light of the alterations in the brainstem evoked potential alteration reported in human infants (Shih et al., 1988; Salamy et al., 1990; Cone-Wesson and Spingarn, 1993; Lester et al., 2003; Tan-Laxa et al., 2004). The response latencies on PPI latency trials of the saline animals demonstrated a clear sex-dependent pattern whereas those of the prenatal IV cocaine-exposed males and females failed to display any significant variation as a function of sex. Again, of the limited prior rodent studies, either no significant alterations were observed in prenatal cocaine-exposed animals on any measure of PPI (Foss and Riley, 1991a; Vorhees et al., 1995, 2000), or a small, albeit statistically significant increase in the magnitude of PPI was reported compared to controls (relative difference of 8%) at 50–60 days of age (30 mg/kg/day cocaine on GD1–20; Hughes et al., 1996).
One factor that differed between the present and prior studies is that the cocaine was delivered to the dam by different routes of administration, IV vs. SC or PO. IV injection is one of the most clinically relevant routes; stimulant abuse liability is a function of the rapidity with which the drug reaches the brain (e.g., Russell and Feyerabend, 1978; Henningfield and Keenan, 1993; Abreu et al., 2001); a consequence not unexpected given that rate of IV drug delivery is well-established to increase maximum arterial drug concentration (Gibaldi, 1991). Furthermore, IV maternal administration of cocaine not only results in significant fetal plasma levels of cocaine (3 mg/kg – 300 ng/ml; 6 mg/kg – 500 ng/ml – Robinson et al., 1994), but also an appreciable tissue uptake of cocaine by fetal brain (at least under a repeated daily dosing regimen; Robinson et al., 1994). Moreover, unlike the SC and PO routes of administration (Spear et al., 1989a; Dow-Edwards, 1990), the IV route is associated with a rapid elimination of cocaine from fetal brain (Robinson et al., 1994). Thus, the fetus is rapidly and transiently exposed to significant amounts of cocaine using the IV dosing model.
The pharmacokinetic profile of near instantaneous distribution and short half-life for IV cocaine in rats closely mimics that observed in humans following inhalation or IV injection of cocaine (Evans et al., 1996; Booze et al., 1997). In contrast, the pharmacokinetics of cocaine delivered by the SC route involves a prolonged absorption process as well as a protracted elimination half-life (Collins et al., 1999). Second, the doses of cocaine employed in the current study are not only 1–2 orders of magnitude less than that employed with SC or PO dosing, but are known to produce arterial plasma levels of cocaine comparable to that of humans provided recreational doses of cocaine (Evans et al., 1996). Furthermore, the IV model precludes the potential confounds of cocaine-induced necrotic lesions characteristic of other routes of administration (e.g., intranasal, SC, Bruckner et al., 1982) and the possibility of extraplacental fetal absorption of cocaine (SC, Lipton et al., 1998). Third, the animals in the present study were tested beginning at 90 days of age vs. 50–60 used in many of the prior studies. Although there have been clear ontogenetic drifts in physiological profiles, at least historically, vaginal opening of LE female rats occurred at approximately D72 with maximum fertility achieved at D100–D300 (Farris, 1949); it may be prudent to refrain from labeling 50 to 60-day-old LE rats as adults.
Pretreatment with an acute low dose of idazoxan at 120 days of age significantly enhanced the magnitude of the ASR, but as the dose of prenatal IV cocaine increased, less enhancement of the ASR was observed. The enhancement of the ASR by idazoxan, an α2-adrenergic receptor antagonist, was expected based upon the well-characterized anatomy and pharmacology of the ASR (Davis, 1984; Yeomans and Frankland, 1996). Idazoxan may affect noradrenergic activity via pre-synaptic and/or postsynaptic actions, since α2-receptors (the drug’s binding site) are localized both pre- and post-synaptically (Nicholas et al., 1996; Docherty, 1998). Idazoxan blockade of pre-synaptic α2-receptors increases NE release due to a reduction in NE-mediated negative feedback, whereas idazoxan’s blockade of postsynaptic α2-receptors antagonizes the effects of released NE at synapses with post-junctional α2-receptors. The marked stimulation of the ASR in control animals (>225%) is consistent with a pre-synaptic “stimulatory” effect and an increase in LC firing and release of NE (Freedman and Aghajanian, 1984; Dennis et al., 1987). Moreover, previous work suggests that idazoxan specifically altered selective attention in a distraction task that also revealed altered selective attention in rats exposed to cocaine prenatally (Bunsey and Strupp, 1995; Bayer et al., 2002). Prenatal IV cocaine exposure results in a persistent alteration in forebrain NE systems as indicated by alterations in receptor density in adolescent (D35; Booze et al., 2006) and adult (D395) rats (Ferris et al., 2007). The sex-dependent nature of these alterations in receptor proteins is of note, particularly given the striking sex-dependent alterations in PPI response latencies, as discussed above. Collectively, the above findings suggest that the enduring changes produced by prenatal cocaine in the ASR and its plasticity may be due to underlying changes in the ceuruleocortical NE system. These data are important as they suggest that the increased reactivity seen in the prenatal cocaine-exposed animals was attributable to dysfunction in the descending NE system.
Specific effects of idazoxan in a distraction task have been previously shown in adult offspring, following maternal IV cocaine, without similarly affecting vigilance implicating endogenous norepinephrine influences on distractibility and/or selective attention (Bayer et al., 2002). The pattern of results and the specificity of the IDZ dose effect rules out non-specific alterations in performance (Bunsey and Strupp, 1995). Given that the noradrenergic system appears to be involved in the focusing of attention, possibly by attenuating distraction caused by irrelevant stimuli (Coull, 1994; Aston-Jones and Cohen, 2005), these data suggest alterations in the ascending noradrenergic system provide the basis for this “distracting” effect.
Traditionally, the actions of cocaine have been attributed to non-selective inhibition of catecholaminergic neurotransmitter reuptake systems. However, emerging evidence supports the view that cocaine may have non-traditional mechanisms for its effects on neuron function (Snow et al., 2001, 2004; Dey et al., 2006). In both in vitro and in vivo studies noradrenergic neurons of the locus coeruleus were exposed to cocaine at a physiologically relevant concentration, and for the in vivo studies via the clinically relevant IV route of administration (Evans et al., 1996; Booze et al., 1997). Following 7 days of in vitro administration, cocaine decreased cell survival as well as neurite elongation in comparison to vehicle controls (Snow et al., 2001, 2004). In subsequent studies, cocaine exposure in vitro induced apoptosis in fetal LC neurons putatively regulated by Bax, via activation of caspases and their downstream target proteins (Dey et al., 2006; Dey and Snow, 2007). The results were obtained from 7 days of cocaine administration (in vitro); a longer term exposure might augment the adverse neuronal effects. These data suggest a decreased ability of LC neurons to network with target cells through both ascending (attention/distraction task with idazoxan challenge, Bayer et al., 2002) and descending (present auditory startle and idazoxan challenge studies) NE pathways, where damage to the NE cell bodies of the LC might provide a unifying mechanism underlying these widespread effects.
Importantly, our evidence for an alteration in the cocaine-exposed offspring’s processing of novel auditory information was detected in the absence of any support for the contribution of indirect effects on maternal growth and nutrition. The IV administration of cocaine to pregnant rats during the last 2 weeks of gestation produced no detectable evidence of toxicity in either the dams or the offspring. A similar failure to find evidence of maternal or fetal toxicity attributable to cocaine, when delivered via the IV route, was previously reported (Mactutus et al., 1994; Mactutus, 1999; Foltz et al., 2004). This negative finding is also consonant with several other studies in rats, mice, and rabbits, all of which have employed IV cocaine doses greater than or equal to those employed here (Mehanny et al., 1991; Kunko et al., 1993; Robinson et al., 1994; Murphy et al., 1997). Together, these observations argue that the IV route of exposure does not require the use of pair-fed nutritional controls as, unlike the SC (Church et al., 1988; Spear et al., 1989b) or PO (Dow-Edwards et al., 1989; Hutchings et al., 1989) models, there appear to be no detectable effects on pregnancy weight gain, putatively the most sensitive routine measure of maternal health. Thus, it is tempting to speculate that the effects of maternal IV cocaine may satisfy the conditions of a pure neurobehavioral teratogen. Nevertheless, higher IV doses of cocaine, as typically delivered to pregnant rabbits (4 mg/kg 2× day, Harvey, 2004) causes a loss of approximately half of the kits within the first postnatal week (Murphy et al., 1997); no such effects have ever been reported with the 3-mg/kg 2× dose as employed by others that use the IV rabbit model (e.g., Stanwood and Levitt, 2007).
Despite the growing and converging evidence for a primary effect of prenatal IV cocaine on the LC and noradrenergic system, alterations in dopaminergic systems may also be contributing to the pattern of observed results, i.e., idazoxan may conceivably be altering norepinephrine-modulated release of dopamine from the ventral tegmental area and/or substantia nigra (Gresch et al., 1995; Devoto et al., 2004). Nevertheless, such an effect would remain consistent with a primary effect of prenatal IV cocaine on the noradrenergic system. When a direct comparison of potential noradrenergic vs. dopaminergic system alterations has been made, dissociations of the effects on prenatal IV cocaine on noradrenergic vs. dopaminergic systems are observed, e.g., on neurite outgrowth (Dey et al., 2006) and apoptotic signaling (Dey et al., 2007). Future studies that manipulate the timing of gestational cocaine exposure may permit further dissociation of the effects on noradrenergic vs. dopaminergic systems.
Finally, while there is very good reason to believe that the reported alterations are attributable to direct effects of cocaine on the developing fetus, one must acknowledge the possibility that there was also some contribution of altered maternal care that may have contributed to the alterations noted in the offspring, i.e., the offspring were not fostered to non-exposed dams at birth. However, we have purposely used a low dose of cocaine in our research program to preclude effects on maternal nutrition and pregnancy weight gain, and on fetal growth. We have never been interested in a high-dose model which would confound nutritional effects with other direct effects of cocaine. Across the studies of the past 15 years in which we have performed research with prenatal cocaine, we have never seen an adverse effect of prenatal cocaine on offspring growth (often touted as one of the most sensitive variables one can measure) implicating any impairment in maternal care. Further, in light of the half-life of cocaine in the rat when delivered via the IV route (Booze et al., 1997), one would not expect the presence of cocaine in the maternal compartment on the day the animals gave birth. Finally, the dosage regimen employed has no detectable effects on maternal retrieval in newborn pups (unpublished observations), again suggesting the low dosages of cocaine employed with the IV route are not consistent with any impairment in maternal care.
In sum, the ASR and its plasticity (habituation, PPI) were sensitive to revealing teratogenic effects of cocaine when delivered by the clinically relevant IV route and at physiologically relevant doses. These conditions produced no detectable adverse effects on maternal/litter parameters. Significant alterations in each of the three paradigms were found in adulthood, regardless of whether or not there was evidence available for the persistence, as opposed to a latent emergence, of the effects. Functional alteration of the noradrenergic system in adulthood was suggested by the prenatal cocaine dose-dependent attenuation of the ability of 0.5 mg/kg idazoxan to enhance the ASR. Thus, in utero exposure to cocaine, when delivered via a protocol designed to capture prominent features of recreational usage, causes persistent, if not permanent, alterations in auditory information processing, and suggests dysfunction of the central noradrenergic circuitry modulating, if not mediating, these responses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported, in part, by grants from the National Institutes of Health: National Institute on Drug Abuse, DA 009160 (Charles F. Mactutus), DA013137 (Rosemarie M. Booze), and DA 021287 (Steven B. Harrod), and National Institute of Child Health and Human Development, HD 043680 (Charles F. Mactutus). The authors gratefully acknowledge the efforts of M. A. Welch for her technical assistance in the conduct of this research. Graduate students Lauren L. Hord and Landhing M. Moran contributed equally to the research; their authorship position was determined by a coin toss. The authors also gratefully acknowledge the input of the referees for their comments and suggestions during the review process.
References
Abreu, M. E., Bigelow, G. E., Fleisher, L., and Walsh, S. L. (2001). Effect of intravenous injection speed on responses to cocaine and hydromorphone in humans. Psychopharmacology (Berl.) 154, 76–84.
Accornero, V. H., Amado, A. J., Morrow, C. E., Xue, L., Anthony, J. C., and Bandstra, E. S. (2007). Impact of prenatal cocaine exposure on attention and response inhibition as assessed by continuous performance tests. J. Dev. Behav. Pediatr. 28, 195–205.
Ackerman, J. P., Riggins, T., and Black, M. M. (2010). A review of the effects of prenatal cocaine exposure among school-aged children. Pediatrics 125, 554–565.
Anday, E., Cohen, M., Kelley, N., and Leitner, D. (1989). Effect of in utero cocaine exposure on startle and its modification. Dev. Pharmacol. Ther. 12, 137–145.
Aston-Jones, G., and Cohen, J. D. (2005). An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu. Rev. Neurosci. 28, 403–450.
Bandstra, E. S., Morrow, C. E., Accornero, V. H., Mansoor, E., Xue, L., and Anthony, J. C. (2011). Estimated effects of in utero cocaine exposure on language development through early adolescence. Neurotoxicol. Teratol. 33, 25–35.
Bandstra, E. S., Vogel, A. L., Morrow, C. E., Xue, L., and Anthony, J. C. (2004). Severity of prenatal cocaine exposure and child language functioning through age seven years: a longitudinal latent growth curve analysis. Subst. Use. Misuse 39, 25–59.
Bandstra, E. S., Morrow, C. E., Vogel, A. L., Fifer, R. C., Ofir, A. Y., Dausa, A. T., Xue, L., and Anthony, J. C. (2002). Longitudinal influence of prenatal cocaine exposure on child language functioning. Neurotoxicol. Teratol. 24, 297–308.
Bandstra, E. S., Morrow, C. E., Anthony, J. C., Accornero, V. H., and Fried, P. A. (2001). Longitudinal investigation of task persistence and sustained attention in children with prenatal cocaine exposure. Neurotoxicol. Teratol. 23, 545–559.
Bayer, L. E., Brown, A., Mactutus, C. F., Booze, R. M., and Strupp, B. J. (2000). Prenatal cocaine exposure increases sensitivity to the attentional effects of the dopamine D1 agonist SKF81297. J. Neurosci. 20, 8902–8908.
Bayer, L. E., Kakumanu, S., Mactutus, C. F., Booze, R. M., and Strupp, B. J. (2002). Prenatal cocaine exposure alters sensitivity to the effects of idazoxan in a distraction task. Behav. Brain Res. 133, 185–196.
Beeghly, M., Martin, B., Rose-Jacobs, R., Cabral, H., Heeren, T., Augustyn, M., Bellinger, D., and Frank, D. A. (2006). Prenatal cocaine exposure and children’s language functioning at 6 and 9.5 years: moderating effects of child age, birthweight, and gender. J. Pediatr. Psychol. 31, 98–115.
Benasich, A. A., and Tallal, P. (2002). Infant discrimination of rapid auditory cues predicts later language impairment. Behav. Brain Res. 136, 31–49.
Betancourt, L. M., Yang, W., Brodsky, N. L., Gallagher, P. R., Malmud, E. K., Giannetta, J. M., Farah, M. J., and Hurt, H. (2011). Adolescents with and without gestational cocaine exposure: longitudinal analysis of inhibitory control, memory and receptive language. Neurotoxicol. Teratol. 33, 36–46.
Booze, R. M., Lehner, A. F., Wallace, D. R., Welch, M. A., and Mactutus, C. F. (1997). Dose-response cocaine pharmacokinetics and metabolite profile following intravenous administration and arterial sampling in unanesthesized, freely moving male rats. Neurotoxicol. Teratol. 19, 7–15.
Booze, R. M., Wallace, D. R., Silvers, J. M., Strupp, B. J., Snow, D. M., and Mactutus, C. F. (2006). Prenatal cocaine exposure alters alpha2 receptor expression in adolescent rats. BMC Neurosci. 7, 33. doi: 10.1186/1471-2202-7-33
Bruckner, J. V., Jiang, W. D., Ho, B. T., and Levy, B. M. (1982). Histopathological evaluation of cocaine-induced skin lesions in the rat. J. Cutan. Pathol. 9, 83–95.
Bunsey, M. D., and Strupp, B. J. (1995). Specific effects of idazoxan in a distraction task: evidence that endogenous norepinephrine plays a role in selective attention in rats. Behav. Neurosci. 109, 903–911.
Carmody, D. P., Bennett, D. S., and Lewis, M. (2011). The effects of prenatal cocaine exposure and gender on inhibitory control and attention. Neurotoxicol. Teratol. 33, 61–68.
Carzoli, R. P., Murphy, S. P., Hammer-Knisely, J., and Houy, J. (1991). Evaluation of auditory brain-stem response in full-term infants of cocaine-abusing mothers. Am. J. Dis. Child. 145, 1013–1016.
Chasnoff, I. J., Burns, W. J., Schnoll, S. H., and Burns, K. A. (1985). Cocaine use in pregnancy. N. Engl. J. Med. 313, 666–669.
Chasnoff, I. J., Griffith, D. R., MacGregor, S., Dirkes, K., and Burns, K. A. (1989). Temporal patterns of cocaine use in pregnancy. Perinatal outcome. J. Am. Med. Assoc. 261, 1741–1744.
Chiriboga, C. A., Starr, D., Kuhn, L., and Wasserman, G. A. (2009). Prenatal cocaine exposure and prolonged focus attention. Poor infant information processing ability or precocious maturation of attentional systems? Dev. Neurosci. 31, 149–158.
Church, M. W., Dintcheff, B. A., and Gessner, P. K. (1988). Dose dependent consequences of cocaine on pregnancy outcome in the Long–Evans rat. Neurotoxicol. Teratol. 10, 51–58.
Church, M. W., and Overbeck, G. W. (1990). Prenatal cocaine exposure in the Long-Evans rat: III. Developmental effects on the brainstem auditory-evoked potential. Neurotoxicol. Teratol. 12, 345–351.
Collins, L. M., Pahl, J. A., and Meyer, J. S. (1999). Distribution of cocaine and metabolites in the pregnant rat and fetus in a chronic subcutaneous injection model. Neurotoxicol. Teratol. 21, 639–646.
Cone-Wesson, B., and Spingarn, A. (1993). Effects of maternal cocaine abuse on neonatal auditory brainstem responses. Am. J. Audiol. 2, 48–54.
Coull, J. T. (1994). Pharmacological manipulation of the α2-noradrenergic system. Effects on cognition. Drugs Aging 5, 116–126.
Davis, M. (1984). “The mammalian startle response,” in Neural Mechanisms of Startle Behavior, ed. R. C. Eaton (New York, NY: Plenum Press), 287–351.
Degenhardt, L., Bucello, C., Calabria, B., Nelson, P., Roberts, A., Hall, W., Lynskey, M., Wiessing, L., and The GBD Illicit Drug Use Writing Group. (2011). What data are available on the extent of illicit drug use and dependence globally? Results of four systematic reviews. Drug Alcohol Depend. doi: 10.1016/j.drugalcdep.2010.11.032. [Epub ahead of print].
Degenhardt, L., Chiu, W. T., Sampson, N., Kessler, R. C., Anthony, J. C., Angermeyer, M., Bruffaerts, R., de Girolamo, G., Gureje, O., Huang, Y., Karam, A., Kostyuchenko, S., Lepine, J. P., Mora, M. E., Neumark, Y., Ormel, J. H., Pinto-Meza, A., Posada-Villa, J., Stein, D. J., Takeshima, T., and Wells, J. E. (2008). Toward a global view of alcohol, tobacco, cannabis, and cocaine use: findings from the WHO World Mental Health Surveys. PLoS Med. 5, e141. doi: 10.1371/journal.pmed.0050141
Delaney-Black, V., Covington, C., Templin, T., Kershaw, T., Nordstrom-Klee, B., Ager, J., Clark, N., Surendran, A., Martier, S., and Sokol, R. J. (2000). Expressive language development of children exposed to cocaine prenatally: literature review and report of a prospective cohort study. J. Commun. Disord. 33, 463–480.
Dennis, T., L’Heureux, R., Carter, C., and Scatton, B. (1987). Presynaptic alpha-2 adrenoceptors play a major role in the effects of idazoxan on cortical noradrenaline release (as measured by in vivo dialysis) in the rat. J. Pharmacol. Exp. Ther. 241, 642–649.
Devoto, P., Flore, G., Pira, L., Longu, G., and Gessa, G. L. (2004). Alpha2-adrenoceptor mediated co-release of dopamine and noradrenaline from noradrenergic neurons in the cerebral cortex. J. Neurochem. 88, 1003–1009.
Dey, S., Mactutus, C. F., Booze, R. M., and Snow, D. M. (2006). Specificity of prenatal cocaine on inhibition of locus coeruleus neurite outgrowth. Neuroscience 139, 899–907.
Dey, S., Mactutus, C. F., Booze, R. M., and Snow, D. M. (2007). Cocaine exposure in vitro induces apoptosis in fetal locus coeruleus neurons by altering the Bax/Bcl-2 ratio and through caspase-3 apoptotic signaling. Neuroscience 144, 509–521.
Dey, S., and Snow, D. M. (2007). Cocaine exposure in vitro induces apoptosis in fetal locus coeruleus neurons through TNF-alpha-mediated induction of Bax and phosphorylated c-Jun NH(2)-terminal kinase. J. Neurochem. 103, 542–556.
Dinehart, L. H. B., Kaiser, M. Y., and Hughes, C. (2009). Language delay and elicitation intervention in children born cocaine exposed: a pilot study. J. Dev. Phys. Disabil. 21, 9–22.
Dobbing, J., and Sands, J. (1979). Comparative aspects of the brain growth spurt. Early Hum. Dev. 3, 79–83.
Docherty, J. R. (1998). Subtypes of functional alpha1- and alpha2-adrenoceptors. Eur. J. Pharmacol. 361, 1–15.
Dow-Edwards, D. L., Fico, T. A., Osman, M., Gamagaris, Z., and Hutchings, D. E. (1989). Comparison of oral and subcutaneous routes of cocaine administration on behavior, plasma drug concentration and toxicity in female rats. Pharmacol. Biochem. Behav. 33, 167–173.
Dow-Edwards, D. (1990). Fetal and maternal cocaine levels peak rapidly following intragastric administration in the rat. J. Subst. Abuse 2, 427–437.
Dow-Edwards, D., and Hughes, H. (1995). Adult reactivity in rats exposed to cocaine during two early postnatal periods. Neurotoxicol. Teratol. 17, 553–557.
Eisen, L. N., Field, T. M., Bandstra, E. S., Roberts, J. P., Morrow, C., Larson, S. K., and Steele, B. M. (1991). Perinatal cocaine effects on neonatal stress behavior and performance on the Brazelton Scale. Pediatrics 88, 477–480.
Evans, S. M., Cone, E. J., and Henningfeld, J. E. (1996). Arterial and venous cocaine plasma concentrations in humans: relationship to route of administration, cardiovascular effects and subjective effects. J. Pharmacol. Exp. Ther. 279, 1345–1356.
Farris, E. J. (1949). “Breeding of the rat,” in The Rat in Laboratory Investigation, 2nd edn. eds E. J. Farris, and J. Q. Jr. Griffith (New York, NY: Hafner Publishing Co.), 1–18.
Ferris, M. J., Mactutus, C. F., Silvers, J. M., Hasselrot, U., Beaudin, S. A., Strupp, B. J., and Booze, R. M. (2007). Sex mediates dopamine and adrenergic receptor expression in adult rats exposed prenatally to cocaine. Int. J. Dev. Neurosci. 25, 445–454.
Fischman, M. W., and Schuster, C. R. (1982). Cocaine self-administration in humans. Fed. Proc. 41, 241–246.
Fitting, S., Booze, R. M., Hasselrot, U., and Mactutus, C. F. (2006a). Intrahippocampal injections of Tat: effects on prepulse inhibition of the auditory startle response in adult male rats. Pharmacol. Biochem. Behav. 84, 189–196.
Fitting, S., Booze, R. M., and Mactutus, C. F. (2006b). Neonatal intrahippocampal glycoprotein 120 injection: the role of dopaminergic alterations in prepulse inhibition in adult rats. J. Pharmacol. Exp. Ther. 318, 1352–1358.
Fitting, S., Booze, R. M., and Mactutus, C. F. (2006c). Neonatal hippocampal Tat injections: developmental effects on prepulse inhibition (PPI) of the auditory startle response. Int. J. Dev. Neurosci. 24, 275–283.
Foltz, T. L., Snow, D. M., Strupp, B. J., Booze, R. M., and Mactutus, C. F. (2004). Prenatal intravenous cocaine and the heart rate-orienting response: a dose-response study. Int. J. Dev. Neurosci. 22, 285–296.
Foss, J. A., and Riley, E. (1988). Behavioral evaluation of animals exposed prenatally to cocaine. Teratology 37, 517.
Foss, J. A., and Riley, E. (1991a). Elicitation and modification of the acoustic startle reflex in animals prenatally exposed to cocaine. Neurotoxicol. Teratol. 13, 541–546.
Foss, J. A., and Riley, E. (1991b). Failure of acute cocaine administration to differentially affect acoustic startle and activity in rats prenatally exposed to cocaine. Neurotoxicol. Teratol. 13, 547–551.
Frank, D. A., Augustyn, M., Knight, W. G., Pell, T., and Zuckerman, B. (2001). Growth, development, and behavior in early childhood following prenatal cocaine exposure: a systematic review. J. Am. Med. Assoc. 285, 1613–1625.
Frank, D. A., Rose-Jacobs, R., Beeghly, M., Wilbur, M., Bellinger, D., and Cabral, H. (2005). Level of prenatal cocaine exposure and 48-month IQ: importance of preschool enrichment. Neurotoxicol. Teratol. 27, 15–28.
Freedman, J. E., and Aghajanian, G. K. (1984). Idazoxan (RX 781094) selectively antagonizes alpha 2-adrenoceptors on rat central neurons. Eur. J. Pharmacol. 105, 265–272.
Garavan, H., Morgan, R. E., Mactutus, C. F., Levitsky, D. A., Booze, R. M., and Strupp, B. J. (2000). Prenatal cocaine exposure impairs selective attention: evidence from serial reversal and extradimensional shift tasks. Behav. Neurosci. 114, 725–738.
Gendle, M. H., Strawderman, M. S., Mactutus, C. F., Booze, R. M., Levitsky, D. A., and Strupp, B. J. (2003). Impaired sustained attention and altered reactivity to errors in an animal model of prenatal cocaine exposure. Brain Res. Dev. Brain Res. 147, 85–96.
Gendle, M. H., White, T. L., Strawderman, M., Mactutus, C. F., Booze, R. M., Levitsky, D. A., and Strupp, B. J. (2004a). Enduring effects of prenatal cocaine exposure on selective attention and reactivity to errors: evidence from an animal model. Behav. Neurosci. 118, 290–297.
Gendle, M. H., Strawderman, M. S., Mactutus, C. F., Booze, R. M., Levitsky, D. A., and Strupp, B. J. (2004b). Prenatal cocaine exposure does not alter working memory in adult rats. Neurotoxicol. Teratol. 26, 319–329.
Gibaldi, M. (1991). Biopharmaceutics and Clinical Pharmacokinetics, 4th Edn. Philadelphia, PA: Lea & Febiger.
Gorski, R. A., Mennin, S. P., and Kubo, K. (1975). The neural and hormonal bases of the reproductive cycle of the rat. Adv. Exp. Med. Biol. 54, 115–153.
Greenhouse, S. W., and Geisser, S. (1959). On methods in the analysis of profile data. Psychometrika 24, 95–112.
Gresch, P. J., Sved, A. F., Zigmond, M. J., and Finlay, J. M. (1995). Local influence of endogenous norepinephrine on extracellular dopamine in rat medial prefrontal cortex. J. Neurochem. 65, 111–116.
Griffith, D. R. (1988). “The effects of perinatal cocaine exposure on infant neurobehavior and early maternal-infant interactions,” in Drugs, Alcohol, Pregnancy and Parenting, ed. I. J. Chasnoff (Dordrecht: Kluwer Academic Publishers), 105–113.
Harvey, J. A. (2004). Cocaine effects on the developing brain: current status. Neurosci. Biobehav. Rev. 27, 751–764.
Heffelfinger, A., Craft, S., and Shyken, J. (1997). Visual attention in children with prenatal cocaine exposure. J. Int. Neuropsychol. Soc. 3, 237–245.
Heffelfinger, A. K., Craft, S., White, D. A., and Shyken, J. (2002). Visual attention in preschool children prenatally exposed to cocaine: implications for behavioral regulation. J. Int. Neuropsychol. Soc. 8, 12–21.
Heffner, H. E., Heffner, R. S., Contos, C., and Ott, T. (1994). Audiogram of the hooded Norway rat. Hear. Res. 73, 244–247.
Henningfield, J., and Keenan, R. (1993). Nicotine delivery kinetics and abuse liability. J. Consult. Clin. Psychol. 61, 743–750.
Heyser, C. J., McKinzie, D. L., Athalie, F., Spear, N. E., and Spear, L. P. (1994). Effects of prenatal exposure to cocaine on heart rate and nonassociative learning and retention in infant rats. Teratology 49, 470–478.
Hoffman, H. S., and Ison, J. R. (1980). Reflex modification in the domain of startle: I. Some empirical findings and their implications for how the nervous system processes sensory input. Psychol. Rev. 87, 175–189.
Hughes, H. E., Donohue, L. M., and Dow-Edwards, D. L. (1996). Prenatal cocaine exposure affects the acoustic startle response in adult rats. Behav. Brain Res. 75, 83–90.
Hurt, H., Betancourt, L. M., Malmud, E. K., Shera, D. M., Giannetta, J. M., Brodsky, N. L., and Farah, M. J. (2009). Children with and without gestational cocaine exposure: a neurocognitive systems analysis. Neurotoxicol. Teratol. 31, 334–341.
Hurt, H., Malmud, E., Betancourt, L., Brodsky, N. L., and Giannetta, J. (1997). A prospective evaluation of early language development in children with in utero cocaine exposure and in control subjects. J. Pediatr. 130, 310–312.
Hutchings, D. E., Fico, T. A., and Dow-Edwards, D. L. (1989). Prenatal cocaine: maternal toxicity, fetal effects and locomotor activity in rat offspring. Neurotoxicol. Teratol. 11, 65–69.
Isenschmid, D. S., Fischman, M. W., Foltin, R. W., and Caplan, Y. H. (1992). Concentration of cocaine and metabolites in plasma of humans following intravenous administration and smoking of cocaine. J. Anal. Toxicol. 16, 311–314.
Ison, J. R. (1984). Reflex modification as an objective test for sensory processing following toxicant exposure. Neurobehav. Toxicol. Teratol. 6, 437–445.
Ison, J. R., and Hoffman, H. S. (1983). Reflex modification in the domain of startle: II. The anomalous history of a robust and ubiquitous phenomenon. Psychol. Bull. 94, 3–17.
Johnston, L. D., O’Malley, P. M., Bachman, J. G., and Schulenberg, J. E. (2010). Monitoring the Future National Survey Results on Drug Use, 1975-2009. Volume I: Secondary School Students (NIH Publication No. 10-7584). Bethesda, MD: National Institute on Drug Abuse, 734.
Kilbride, H., Castor, C., Hoffman, E., and Fuger, K. L. (2000). Thirty-six-month outcome of prenatal cocaine exposure for term or near-term infants: impact of early case management. J. Dev. Behav. Pediatr. 21, 19–26.
Kilbride, H. W., Castor, C. A., and Fuger, K. L. (2006). School-age outcome of children with prenatal cocaine exposure following early case management. J. Dev. Behav. Pediatr. 27, 181–187.
Krumholz, A., Felix, J. K., Goldstein, P. J., and McKenzie, E. (1985). Maturation of the brain-stem auditory evoked potential in premature infants. Electroencephalogr. Clin. Neurophysiol. 62, 124–134.
Kunko, P. M., Moyer, D., and Robinson, S. E. (1993). Intravenous gestational cocaine in rats: effects on offspring development and weanling behavior. Neurotoxicol. Teratol. 15, 335–344.
LaBella, F. S., and Queen, G. (1993). General anesthetics inhibit cytochrome P450 monoxygenases and arachidonic acid metabolism. Can. J. Physiol. Pharmacol. 71, 48–53.
Lacy, R. T., Mactutus, C. F., and Harrod, S. B. (2011). Prenatal IV nicotine exposure produces a sex difference in sensorimotor gating of the auditory startle reflex in adult rats. Int. J. Dev. Neurosci. 29, 153–161.
Leech, S. L., Richardson, G. A., Goldschmidt, L., and Day, N. L. (1999). Prenatal substance exposure: effects on attention and impulsivity of 6-year-olds. Neurotoxicol. Teratol. 21, 109–118.
Lester, B. M., Lagasse, L., Seifer, R., Tronick, E. Z., Bauer, C. R., Shankaran, S., Bada, H. S., Wright, L. L., Smeriglio, V. L., Liu, J., Finnegan, L. P., and Maza, P. L. (2003). The Maternal Lifestyle Study (MLS): effects of prenatal cocaine and/or opiate exposure on auditory brain response at one month. J. Pediatr. 142, 279–285.
Lewis, B. A., Kirchner, H. L., Short, E. J., Minnes, S., Weishampel, P., Satayathum, S., and Singer, L. T. (2007). Prenatal cocaine and tobacco effects on children’s language trajectories. Pediatrics 120, 78–85.
Lewis, B. A., Minnes, S., Short, E. J., Weishampel, P., Satayathum, S., Min, M. O., Nelson, S., and Singer, L. T. (2011). The effects of prenatal cocaine on language development at 10 years of age. Neurotoxicol. Teratol. 33, 17–24.
Lewis, B. A., Singer, L. T., Short, E. J., Minnes, S., Arendt, R., Weishampel, P., Klein, N., and Min, M. O. (2004). Four-year language outcomes of children exposed to cocaine in utero. Neurotoxicol. Teratol. 26, 617–627.
Lipton, J. W., Robie, H. C., Ling, Z., Weese-Mayer, D. E., and Carvey, P. M. (1998). The magnitude of brain dopamine depletion from prenatal cocaine exposure is a function of uterine position. Neurotoxicol. Teratol. 20, 373–382.
Loch, J. M., Potter, J., and Bachmann, K. A. (1995). The influence of anesthetic agents on rat hepatic cytochromes P450 in vivo. Pharmacology 50, 146–153.
Mactutus, C. F. (1999). Prenatal intravenous cocaine adversely affects attentional processing in preweanling rats. Neurotoxicol. Teratol. 21, 539–550.
Mactutus, C. F., Booze, R. M., and Dowell, R. T. (2000). The influence of route of administration on the acute cardiovascular effects of cocaine in conscious unrestrained pregnant rats. Neurotoxicol. Teratol. 22, 357–368.
Mactutus, C. F., Herman, A. S., and Booze, R. M. (1994). Chronic intravenous model for studies of drug abuse in the pregnant and/or group-housed rat: an initial study with cocaine. Neurotoxicol. Teratol. 16, 183–191.
Mayes, L. C., Grillon, C., Granger, R., and Schottenfeld, R. (1998). Regulation of arousal and attention in preschool children exposed to cocaine prenatally. Ann. N. Y. Acad. Sci. 846, 126–143.
Mehanny, S. Z., Abdel-Rahman, M. S., and Ahmed, Y. Y. (1991). Teratogenic effect of cocaine and diazepam in CF1 mice. Teratology 43, 11–17.
Morgan, R. E., Garavan, H. P., Mactutus, C. F., Levitsky, D. A., Booze, R. M., and Strupp, B. J. (2002). Enduring effects of prenatal cocaine exposure on attention and reaction to errors. Behav. Neurosci. 116, 624–633.
Morrow, C. E., Bandstra, E. S., Anthony, J. C., Ofir, A. Y., Xue, L., and Reyes, M. B. (2003). Influence of prenatal cocaine exposure on early language development: longitudinal findings from four months to three years of age. J. Dev. Behav. Pediatr. 24, 39–50.
Morrow, C. E., Vogel, A. L., Anthony, J. C., Ofir, A. Y., Dausa, A. T., and Bandstra, E. S. (2004). Expressive and receptive language functioning in preschool children with prenatal cocaine exposure. J. Pediatr. Psychol. 7, 543–554.
Murphy, E. H., Fischer, I., Friedman, E., Grayson, D., Jones, L., Levitt, P., O’Brien-Jenkins, A., Wang, H. Y., and Wang, X. H. (1997). Cocaine administration in pregnant rabbits alters cortical structure and function in their progeny in the absence of maternal seizures. Exp. Brain Res. 114, 433–441.
Nicholas, A. P., Hökfelt, T., and Pieribone, V. A. (1996). The distribution and significance of CNS adrenoceptors examined with in situ hybridization. Trends Pharmacol. Sci. 17, 245–255.
Noland, J. S., Singer, L. T., Short, E. J., Minnes, S., Arendt, R. E., Kirchner, H. L., and Bearer, C. (2005). Prenatal drug exposure and selective attention in preschoolers. Neurotoxicol. Teratol. 27, 429–438.
Overstreet, D. H., Moy, S. S., Lubin, D. A., Gause, L. R., Lieberman, J. A., and Johns, J. M. (2000). Enduring effects of prenatal cocaine administration on emotional behavior in rats. Physiol. Behav. 70, 149–156.
Parisi, T., and Ison, J. R. (1979). Development of the acoustic startle response in the rat: ontogenetic changes in the magnitude of inhibition by prepulse stimulation. Dev. Psychobiol. 12, 219–230.
Parisi, T., and Ison, J. R. (1981). Ontogeny of control over the acoustic startle reflex by visual prestimulation in the rat. Dev. Psychobiol. 14, 311–316.
Potter, S. M., Zelazo, P. R., Stack, D. M., and Papageorgiou, A. N. (2000). Adverse effects of fetal cocaine exposure on neonatal auditory information processing. Pediatrics 105, E40.
Richardson, G. A., Conroy, M. L., and Day, N. L. (1996). Prenatal cocaine exposure: effects on the development of school-age children. Neurotoxicol. Teratol. 18, 627–634.
Robinson, S. E., Enters, E. K., Jackson, G. F., Chinchilli, V. M., Maher, J. R., McDowell, K. P., Allen, H. M., and Guo, H. (1994). Maternal and fetal brain and plasma levels of cocaine and benzoylecgonine after acute or chronic maternal intravenous administration of cocaine. J. Pharmacol. Exp. Ther. 271, 1234–1239.
Russell, M. A., and Feyerabend, C. (1978). Cigarette smoking: a dependence on high-nicotine boli. Drug Metab. Rev. 8, 29–57.
Salamy, A. (1984). Maturation of the auditory brainstem response from birth through early childhood. J. Clin. Neurophysiol. 1, 293–329.
Salamy, A., Eldredge, L., Anderson, J., and Bull, D. (1990). Brain-stem transmission time in infants exposed to cocaine in utero. J. Pediatr. 117, 627–629.
Savage, J., Brodsky, N. L., Malmud, E., Giannetta, J. M., and Hurt, H. (2005). Attentional functioning and impulse control in cocaine-exposed and control children at age ten years. J. Dev. Behav. Pediatr. 26, 42–47.
Shih, L., Cone-Wesson, B., and Reddix, B. (1988). Effects of maternal cocaine abuse on the neonatal auditory system. Int. J. Pediatr. Otorhinolaryngol. 15, 245–251.
Singer, L. T., Arendt, R., Minnes, S., Salvator, A., Siegel, A. C., and Lewis, B. A. (2001). Developing language skills of cocaine-exposed infants. Pediatrics 107, 1057–1064.
Snow, D. M., Carman, H. M., Smith, J. D., Booze, R. M., Welch, M. A., and Mactutus, C. F. (2004). Cocaine-induced inhibition of process outgrowth in locus coeruleus neurons: role of gestational exposure period and offspring sex. Int. J. Dev. Neurosci. 22, 297–308.
Snow, D. M., Smith, J. D., Booze, R. M., Welch, M. A., and Mactutus, C. F. (2001). Cocaine decreases cell survival and inhibits neurite extension of rat locus coeruleus neurons. Neurotoxicol. Teratol. 23, 225–234.
Spear, L. P., Frambes, N. A., and Kirstein, C. L. (1989a). Fetal and maternal brain and plasma levels of cocaine and benzoylecgonine following chronic subcutaneous administration of cocaine during gestation in rats. Psychopharmacology (Berl.) 97, 427–431.
Spear, L. P., Kirstein, C. L., Bell, J., Yoottanasumpun, V., Greenbaum, R., Oshea, J., Hoffmann, H., and Spear, N. E. (1989b). Effects of prenatal cocaine exposure on behavior during the early postnatal period. Neurotoxicol. Teratol. 11, 57–63.
Stanwood, G. D., and Levitt, P. (2007). Prenatal exposure to cocaine produces unique developmental and long-term adaptive changes in dopamine D1 receptor activity and subcellular distribution. J. Neurosci. 27, 152–157.
Substance Abuse and Mental Health Services Administration. (2010). Results from the 2009 National Survey on Drug Use and Health: Volume I. Summary of National Findings (Office of Applied Studies, NSDUH Series H-38A, HHS Publication No. SMA 10-4586 Findings). Rockville, MD.
Tan-Laxa, M. A., Sison-Switala, C., Rintelman, W., and Ostrea, E. M. Jr. (2004). Abnormal auditory brainstem response among infants with prenatal cocaine exposure. Pediatrics 113, 357–360.
Vorhees, C., Wood, S., Morford, L., Reed, T., Moran, M., Pu, C., and Cappon, G. (2000). Evaluation of neonatal exposure to cocaine on learning, activity, startle, scent marking, immobility, and plasma cocaine concentrations. Neurotoxicol. Teratol. 22, 255–265.
Vorhees, C. V., Reed, T. M., Acuff-Smith, K. D., Schilling, M. A., Cappon, G. D., Fisher, J. E., and Pu, C. (1995). Long-term learning deficits and changes in unlearned behaviors following in utero exposure to multiple daily doses of cocaine during different exposure periods and maternal plasma cocaine concentrations. Neurotoxicol. Teratol. 17, 253–264.
Keywords: prenatal cocaine, intravenous administration, auditory startle, habituation, prepulse inhibition, dose–response, norepinephrine, idazoxan
Citation: Mactutus CF, Harrod SB, Hord LL, Moran LM and Booze RM (2011) Prenatal IV cocaine: alterations in auditory information processing. Front. Psychiatry 2:38. doi: 10.3389/fpsyt.2011.00038
Received: 04 March 2011;
Paper pending published: 21 March 2011;
Accepted: 11 June 2011;
Published online: 28 June 2011.
Edited by:
Josephine Johns, University of North Carolina at Chapel Hill, USAReviewed by:
Charles Heyser, Franklin and Marshall College, USALynn Singer, Case Western Reserve University, USA
Copyright: © 2011 Mactutus, Harrod, Hord, Moran and Booze. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Charles F. Mactutus, Behavioral Neuroscience Program, Department of Psychology, University of South Carolina, Columbia, SC 29208-0001, USA. e-mail:bWFjdHV0dXNAbWFpbGJveC5zYy5lZHU=