
- Academic Clinical Psychiatry, Department of Neuroscience, University of Sheffield, Sheffield, UK
Schizophrenia is a disorder with a large number of clinical, neurobiological, and cognitive manifestations, none of which is invariably present. However it appears to be a single nosological entity. This article considers the likely characteristics of a pathology capable of such diverse consequences. It is argued that both deficit and psychotic symptoms can be manifestations of a single pathology. A general model of psychosis is proposed in which the informational sensitivity or responsivity of a network (“hodological resonance”) becomes so high that it activates spontaneously, to produce a hallucination, if it is in sensory cortex, or another psychotic symptom if it is elsewhere. It is argued that this can come about because of high levels of modulation such as those assumed present in affective psychosis, or because of high levels of baseline resonance, such as those expected in deafferentation syndromes associated with hallucinations, for example, Charles Bonnet. It is further proposed that schizophrenia results from a process (probably neurodevelopmental) causing widespread increases of variance in baseline resonance; consequently some networks possess high baseline resonance and become susceptible to spontaneous activation. Deficit symptoms might result from the presence of networks with increased activation thresholds. This hodological variance model is explored in terms of schizo-affective disorder, transient psychotic symptoms, diathesis-stress models, mechanisms of antipsychotic pharmacotherapy and persistence of genes predisposing to schizophrenia. Predictions and implications of the model are discussed. In particular it suggests a need for more research into psychotic states and for more single case-based studies in schizophrenia.
Introduction
Despite the effort that has gone into the study of schizophrenia over the last century or so, and the discovery of a large number of abnormalities, progress in understanding the nature of this disease has been limited. In particular, we have so far failed to formulate a theoretical framework which is coherent, comprehensive, and compelling. This in turn has left much of schizophrenia research as an exercise in cataloging the abnormalities associated with the disorder, in the hope that this will shed light on its fundamental nature. Paradoxically, the number and variety of abnormalities described have seemed to make it ever harder to formulate an all-encompassing hypothetical model.
This article first examines the claim that schizophrenia is a single nosological entity, then explores the nature of the disease’s essence with reference to the diversity of its manifestations, both within an individual, and between individuals. The nature of the disease is also discussed in relation to evidence for genetic pleiotropy (the potential of predisposing genes to manifest in different ways). Some contemporary models of schizophrenia are briefly reviewed and their limitations discussed.
Following this, a hypothetical general model of psychosis is described, and a hypothetical pathology is described for schizophrenia, consistent with the more general model. This is compared with a hypothetical mechanism for affective psychosis, and the nature of a hybrid schizo-affective disorder is explored. The model is discussed in relation to the diathesis-stress model, relapse, and treatment, the continuum of psychosis, and the persistence of schizophrenia genes in the population. Finally, the model is discussed in terms of its predictions, implications for schizophrenia research, and its limitations.
The Nosological Integrity of Schizophrenia
We must first ask whether the difficulty in understanding schizophrenia arises because we are attempting to understand a mixture of different disorders. By this I mean disorders which are different in etiology and pathology, and share only clinical similarities. Schizophrenia is etiologically heterogeneous in that genetic risk is thought to be conferred by a large number of genes of modest effect (International Schizophrenia Consortium, 2009) and some rare copy number variants (chromosomal deletions or multiple copies) of greater effect (Manolio et al., 2009). There are also several putative environmental etiological risk factors none of which appear to confer odds ratios above 5 (MacDonald and Schulz, 2009). Therefore it appears that there are multiple potential causative factors, but none appears likely to be responsible for a large proportion of the disorder.
Schizophrenia is also clinically heterogeneous for instance in terms of psychiatric symptomatology (Kendler et al., 1994), course (Huber, 1997), and neuropsychological function (Kremen et al., 2004). Moreover some symptom clusters are strongly correlated in MZ twin pairs (Cardno et al., 2001), and there is evidence that some susceptibility genes are associated with particular symptom dimensions (Fanous et al., 2005; McClay et al., 2006). However there are reasons to suppose that the factors producing clinical heterogeneity are largely distinct from those contributing to susceptibility to the disease. Genes associated with susceptibility have in the main not been shown to be associated with particular clinical features (Kendler et al., 2000; Fanous and Kendler, 2005); and symptom correlations are modest for sibs (Rietkerk et al., 2008), suggesting that the susceptibility genes shared by the sib-pair are not potent symptom-modifiers. The search for pathoplastic (symptom modifying) genes has now begun (Fanous et al., 2007) which will allow further scrutiny of this issue. Even considering those identified genetic factors associated with both susceptibility to the disease and to particular symptoms, symptom associations are not very strong, and are not always replicated (Bergen et al., 2010). Moreover their modest effect size in relation to the disease suggests that they act in conjunction with other (generic) risk factors to produce the disease.
At present therefore the evidence suggests that genetic susceptibility to schizophrenia is associated with susceptibility to all clinical forms of the disorder, and so schizophrenia cannot be genetically parsed into pathologically distinct disorders. Instead, we might think of an hourglass shape with a range of etiological factors at the top, which at some point converge on a common pathology, and diversify again into a range of manifestations, in the same way that, for instance, atherosclerosis has diverse causes, which converge to produce the characteristic pathology of the vascular lesions, which in turn produce diverse consequences. Although the etiological mechanisms are incompletely understood, the common pathology of atherosclerosis has been characterized. In the case of schizophrenia, our challenge is to identify the conceptual level and nature of the common pathology.
A Neuroanatomical Perspective
While genetics provided the earliest and strongest evidence of biological etiology, neuroanatomy, and neuroimaging provided early and compelling evidence of the biological manifestations of the disorder. And just as genetic models have had to become more complex, models of pathology in schizophrenia have begun to move away from simple consideration of a single neuroanatomical locus or neurotransmitter to explore more widespread patterns of pathology (Lewis, 2000). Honea et al. (2005) performed a meta-analysis of 15 studies of brain morphology in schizophrenia using voxel-based morphometry. They found 32 brain regions which showed regional volume deficits on at least three of the studies. Only two regions (the left superior temporal gyrus and the left middle temporal gyrus) were reported as abnormal in more than 50% of the studies (8/15 and 9/15 respectively). This broad spread suggests heterogeneity in the anatomical location of brain pathology. Moreover, the fact that no anatomical region can be shown to be invariably involved suggests that schizophrenia is not definable in terms of a dysfunction of a particular neuroanatomical structure.
Dysconnectivity models (e.g., Bullmore et al., 1997; Friston, 1998) have more capacity to accommodate the contribution of lesions at different sites, although a rather widespread network would need to be invoked to accommodate the number of brain regions in which abnormalities have been reported to be associated with the disease. However, although a network model can explain how different patterns of lesions can manifest as the same disorder, it does not explain the nature or the anatomical variability of the lesions, and has little to say about how and why such a rich variety of clinical manifestations can be produced by disruption of the same network. There is another possibility: that the disease manifests as a characteristic lesion which can occur at various different neuroanatomical locations or within different networks, and this idea is developed later.
Pleiotropy and Schizophrenia
If many of the genes predisposing to schizophrenia have little effect on the pattern of individual symptoms (as discussed above), this suggests a degree of pleiotropy; in other words, the same genetic predisposition may manifest in more than one fashion, depending on other genetic and environmental factors. This is known to be the case for at least one of the genes predisposing to schizophrenia, Neuregulin 1 (Harrison and Law, 2006).
Further evidence can be gleaned from patterns of inheritance, such as that seen in the inheritance of smooth pursuit eye movement disorder (SPEM) associated with schizophrenia. Abnormalities of SPEM are more common in both schizophrenia patients and their relatives compared to the general population, suggesting that it is a genetic disorder linked to schizophrenia susceptibility (Holzman, 2000; Lee and Williams, 2000). However, even patients with normal SPEM have families showing increased rates of SPEM abnormalities (Matthysse et al., 1986), suggesting that the same genetic factor(s) may manifest as either SPEM disorder or schizophrenia or both. Sustained attention, as measured by a Continuous Performance Test, has recently been shown to display a similar pattern of inheritance (Birkett et al., 2007). Therefore multiple lines of evidence suggest that schizophrenia is the result of pleiotropic gene expression.
The Essence of Schizophrenia
Earlier in this article, the pathogenesis of schizophrenia was compared to an hourglass. In pleiotropy, we have a model of the way in which a single pathology can produce a variety of manifestations. One of the chief difficulties in the elucidation of schizophrenia is the heterogeneity of the causes and manifestations of the disorder. Crucially therefore we must ask what is the common essence, the neck of the hourglass.
Let us consider the proposition that there is a single neuropathology associated with schizophrenia which may be manifest in a variety of different neuroanatomical locations, and it is the location and degree of the pathology which determines the particular symptoms experienced by an individual. In support of this idea, there is evidence suggesting a degree of anatomical specificity for particular symptoms both in terms of structural (Marsh et al., 1997; Spencer et al., 2007) and functional anatomy (Ffytche et al., 1998; Shergill et al., 2001).
Nevertheless, there are difficulties with this view. From a clinical standpoint, schizophrenia is most strikingly characterized by hallucinations, delusions, and thought disorder. These symptoms are state-dependent. They may remit spontaneously, or in response to antipsychotic medication. In contrast, deficit or “negative” symptoms, and cognitive impairments, tend to be more stable over time (Reichenberg et al., 2005) and show less response to antipsychotics. These differences seem unlikely to be explainable on the basis of the anatomical site of lesions alone. Therefore it seems that at least two kinds of symptoms are involved.
However, this does not necessarily imply at least two kinds of pathology. For instance coarse brain damage may result in loss of normal function, and also lead to epileptic seizures (new, abnormal function). It is possible that the psychotic symptoms represent abnormal neuronal activity which arises from the brain pathology characteristic of schizophrenia, whereas deficit symptoms represent the disturbance to normal brain function caused by this pathology. This account has the advantage of simplicity, but does not explain why factor analysis of symptoms has repeatedly shown that psychomotor poverty symptoms are independent of psychotic symptoms. (Reichenberg et al., 2005).
As discussed above, the evidence suggests that the same genes predispose to both types of symptoms, and the co-existence of psychotic and deficit symptoms as part of the same syndrome implies an association within the general population. Therefore at some level there must be a unifying pathology.
We can resolve this paradox if we suppose that in some parts of the brain, the aberrant “state” activity is clinically more conspicuous, while the loss of normal function is relatively silent; in other parts of the brain the opposite relationship pertains, causing the apparent dissociation on the basis of the neuroanatomical distribution of pathology within the individual.
The normal function of prefrontal and frontal cortex is to generate and regulate action, therefore loss of that function is conspicuous, and aberrant activity in this part of the brain is relatively inconspicuous. This is because, although it might be expected to manifest as abnormal actions and ideas, these will be conflated with, and perhaps masked by, the abnormal action and ideas which come about as a result of abnormal sensory and cognitive information, due to dysfunction in temporal lobes and elsewhere. In contrast, in an area such as the sensory cortex where output is normally highly dependent on sensory input, additional aberrant activity (in the absence of sensory input) will be manifest as hallucinations, and likely to be more conspicuous than a degree of impairment in normal function.
According to this view, then, the deficit symptoms are rather less specific than the psychotic symptoms because if one supposes that normal brain function is at or near optimum, then there must be a multiplicity of ways in which that normal function could be impaired, unless the impairment is quite circumscribed, which, in the case of schizophrenia, it is not (Heinrichs and Zakzanis, 1998). Although the specificity of psychotic symptoms has recently been challenged by suggestions of a continuum of psychosis shading into normality (Verdoux and van Os, 2002), nevertheless a clinical diagnosis of schizophrenia can invariably be made with a higher degree of certainty on the basis of psychotic symptoms than on the basis of even the most exhaustive profile of psychomotor poverty and cognitive performance results. Consistent with this view, a DSM-IV-TR (American Psychiatric Association, 2000) diagnosis of schizophrenia cannot be made on the basis of negative symptoms alone. Therefore a promising strategy for understanding schizophrenia is the search for a general mechanism which manifests in different neuroanatomical locations or networks, to produce the characteristic psychotic symptoms of schizophrenia.
Mechanisms of Psychotic Symptoms in Schizophrenia-Contemporary Ideas
Contemporary hypotheses explaining psychotic symptoms in schizophrenia can be considered to fall into two broad schools. The first explains psychotic symptoms as deriving from the abnormal interpretation of physiological processes, so that internal cues such as thoughts (manifest as verbal imagery), are mis-labeled as hallucinations. Crow (2000) has argued that this may happen because of the loss of the normal deictic frame of reference (where the meaning of some words such as “I” and “you” is determined by the context; in these examples, by the identities of the speaker and the person addressed). Crow argues that this loss is caused by linguistic dysfunction, which is, in turn, due to disturbance of normal cerebral asymmetry. Feinberg, Frith, and others (Feinberg, 1982, 1978; Feinberg and Guazzelli, 1999; Frith et al., 2000; Johns et al., 2001) have instead argued that a mis-labeling of internal percepts as external occurs because of the loss of normal monitoring of self-generated action. It has also been argued that this mechanism can explain the symptom of “alien control” (Frith and Done, 1989).
In contrast, the other school regards psychotic symptoms seen in schizophrenia as resulting from pathological brain activity, which may, for example, take the form of auditory hallucinations generated by abnormal activity in the auditory cortex (Hoffman and McGlashan, 1997) perhaps caused by “parasitic foci,” where some parts of the brain are prone to produce the same output regardless of input (Hoffman and Dobscha, 1989). Thus an auditory hallucination may be judged as ego alien because the abnormal activity is not under voluntary control, and identified as occurring in external space because the same parts of the cortex are activated which respond to external sounds (Hunter et al., 2003; Hunter, 2004). A more recent theoretical approach hypothesises that abnormal perception arises from abnormal sensory predictions within a Bayesian framework; perhaps due to abnormally strong top-down predictions in combination with a noisy bottom-up sensory signal (Corlett et al., 2009), and that faulty error-correction within a similar Bayesian framework can explain delusional beliefs (Fletcher and Frith, 2009).
Several studies have demonstrated a deficit in self monitoring in schizophrenia which is consistent with the first explanation (e.g., Johns et al., 2001; Stirling et al., 2001) however there is also experimental evidence against this hypothesis: Goldberg et al. (1997) showed that in a situation where hallucinated subjects might be expected to benefit from impaired self monitoring, they actually performed worse than controls. Moreover, there are theoretical problems with this account. First, it requires us to believe that auditory hallucinations reflect the thought content of the psychotic individual. This seems more plausible in the case of the individual with psychotic depression who is hearing mood-congruent derogatory hallucinations in the second person, than in the case of an individual with schizophrenia who is hearing two or more hallucinated voices discussing him in the third person, in a manner which is not mood congruent; and indeed the inner speech of those with schizophrenia appears to be no different from the general population (Langdon et al., 2009). Second, misattribution theories require the presence of a stream of normal self-generated action or imagery, which is then available for misattribution. While this is clearly the case for physical movement or thought, it is more doubtful when applied to, say, olfaction, or vision. Third, it does not explain why some thoughts would be experienced as auditory hallucinations, while others would be experienced as ego-alien thoughts. Thought echo, in which correctly attributed thoughts are later experienced as hallucinations, is harder still to explain if hallucinations are misattributed thoughts. Fourth, it can offer no explanation for delusions, other than those which concern the presence of the hallucination (or made action) itself (e.g., a running commentary generating the delusion of being spied on). Delusions related to the content of the hallucination (e.g., the hallucination “we are coming to kill you” generating the corresponding persecutory delusion) would need to be regarded as primary delusions, since, according to this model, the belief would need to precede the hallucination containing it. In fact, there are several other major features of psychosis, including thought-disordered speech, incongruent affect, and delusions of reference, which seem difficult to explain on the basis of faulty attribution of self-generated content.
In contrast, if hallucinations result from aberrant activity in sensory cortex, we would not expect the content to reflect normal thought. Nor does this explanation require a stream of imagery or volition in each affected modality. We can explain the difference between the genesis of ego-alien thought and auditory hallucinations on the basis of the anatomical location and physiological function of the particular cortex involved (Hoffman and McGlashan, 1993).
Moreover there is evidence directly linking abnormal cortical activity with hallucinations. It is well established that stimulation of cortical areas may evince hallucinations (Perot and Penfield, 1960). Recent work includes stimulation of visual cortex to produce complex hallucinations (Schulz et al., 2007). Areas of visual cortex activated during hallucinations have been shown to correlate with their normal functions (Ffytche et al., 1998; Ffytche, 2008). Primary visual cortex activity has been associated with color-word synesthesia (Aleman et al., 2001). There is also evidence to show that auditory hallucinations are associated with anomalous activity of auditory sensory cortex (Shergill et al., 2001; van de Ven et al., 2005; Marti-Bonmati et al., 2007).
This mechanism can also explain other psychotic phenomena. Many aspects of the normal working of the mind involve internal percepts; mental elements such as emotions, memories, associations, models, and beliefs, any of which could potentially be the subject of anomalous activity. For instance, a delusion of reference could be explained as abnormal spontaneous activity resulting in a tag of “personal salience” being anomalously attached to a percept. Many delusions could be classified as secondary to other phenomena, especially the content of auditory hallucinations. Primary delusions could be due to activation of abnormal semantic content. Therefore it seems reasonable to prefer the “abnormal activity” hypothesis as having more explanatory power in terms of schizophrenia symptomatology.
Hoffmann and colleagues (Hoffman and Dobscha, 1989; Hoffman and McGlashan, 1997) have shown how aberrant spontaneous activity could be generated through excessive synaptic pruning. This model can explain the distribution of age of onset of schizophrenia which corresponds with a period of synaptic pruning (Huttenlocher, 1979; Feinberg, 1982). However the models discussed above do not offer an account of the relationship between the mechanism generating hallucinations in schizophrenia, and that generating hallucinations in other disorders. This is particularly important in relation to affective psychoses, which clinically and etiologically may lie on a continuum with schizophrenia (Crow, 1995; Craddock et al., 2006; Cheniaux et al., 2008). A further difficulty is that existing models do not explain the degree of heterogeneity discussed above. The following Sections (Resonance, Hodology, and the Brain and Psychosis) will present a general model of hallucinations and psychosis. A mechanism specific for schizophrenia (as a special case of the general model) is proposed in Section “Schizophrenia” which is further developed and discussed in the remainder of this paper.
Resonance, Hodology, and the Brain
Resonance and Hallucinations
Most people will be familiar with the “howl” that is produced when a microphone is pointed toward loudspeakers carrying its amplified output. This howling is often termed “feedback,” because the output is being fed back into the system. However, it might be more accurate to term this phenomenon “resonance” because the frequency produced by the howl is not random, but represents the natural resonant frequency of the circuit including the acoustic space. This is the frequency whose energy is most efficiently absorbed (and therefore propagated) by the system. Therefore in distinction to a simple positive feedback loop, in which the output is an upwardly moving scalar, this resonant frequency loop has imposed its own characteristic signal onto the system. Another example is the filter of an electronic musical synthesizer, in which the degree of resonance (at the filtering frequency) can be increased to the point at which it self-oscillates, and produces a sound even when there is no apparent input from the dedicated oscillators.
These are examples of systems whose normal function is to process signal, but which under certain circumstances instead generate it. This is of interest, because it has been argued above that the same is true of the way in which schizophrenia causes sensory and other cortex to behave. Below I shall develop the argument that psychotic symptoms are caused by a mechanism conceptually related to resonance.
In classical resonance theory (Crowell, 2008), resonance describes a property possessed by all objects. The degree of resonance is a measure of the time it takes for oscillation at each frequency to decay by a fixed amount (the decay being exponential), and is highest at the object’s natural frequency. At equilibrium, the amount of energy being lost from the system is equal to the amount being fed in. Normally, objects lose energy quickly and therefore do not tend to oscillate noticeably even when subject to vibrational energy. However, when resonance is high, it is possible to cause that object to vibrate at (or near) its natural frequency by supplying energy, and this is the principle by which all pitched acoustic musical instruments work. Different pitches are produced by changing the instrument’s natural frequency. The cases of microphone howl and filter self-oscillation cited above occur when resonance becomes so high that even the very small vibrational energies which occur as background noise are sufficient to drive the system into noticeable oscillation (a condition I shall call hyper-resonance).
Hodological Resonance
It should be emphasized that this hypothesis does not suggest that classical resonance is involved in schizophrenia (which would produce oscillations of an epileptiform nature). Rather, it is suggested that neural networks such as those in the brain, possess (and indeed rely on) a quality analogous to classical resonance, in the information domain. In this (typical connectionist) model, excitation of a given node will feed informational “energy” into a network whose conformation is determined by the strength of the connections between nodes, as modified by the particular modulations in effect at that moment.
Recently Ffytche and Catani have drawn attention to the importance of hodology in relation to psychotic phenomena (Ffytche and Catani, 2005; Ffytche, 2008). Hodology is a general term meaning the study of pathways, and, as applied to neuroscience, to the study of neuronal pathways. I shall use the term “hodological” to signify considerations of the conformation of neural connections. Although the term is often used in connection with longer-range connections, I am including short-range local connections within the meaning.
It is hypothesized that the degree to which information is propagated from one node to another connected node is based on four groups of factors
(1) Long-term neurodevelopmental factors
(2) Medium term neuroplastic factors, such as axonal sprouting, neuroreceptor up- or down-regulation
(3) Short term neuromodulational factors determined by excitatory and inhibitory afferents (the weighting of neuromodulatory afferents will also be affected by factors 1 and 2)
(4) The sum of any collateral indirect pathways (factors 1, 2, and 3 will affect these too).
In this account the nodes are assumed to be at the level of neuronal microcircuitry. Several interlinked nodes may be considered as a network with its own profile of sensitivity to particular afferent impulses, in other words the circumstances under which that given network will activate; or (more precisely) the relationship between various informational inflows and modulations, and the degree of response. The degree of sensitivity may be regarded as analogous to resonance in respect of a particular network. Individual nodes will be involved in many overlapping networks. I will use the term “hodological resonance” to mean the neuroanatomical and neurocognitive profile of neuronal network sensitivity across the brain in relation to different kinds of input.
The point at which a network moves from being “primed” to being “activated” may be regarded as a phase transition and so may be associated with a threshold (the notion of an “activation threshold” simplifies discussion but does not materially affect the principles discussed here; alternatively, if “primed” and “activated” are not qualitatively different states but arbitrary points on a scale of activation, discussion of movement of the activation threshold should be taken to mean movement of the input/response curve left or right).
Examples and Evidence
To illustrate this concept of hodological resonance, consider the following everyday example. A woman is speaking on a hands-free telephone. She is earnestly trying to explain something delicate to the listener. Although her listener cannot see her, she gesticulates as she speaks as if these actions were an inseparable part of the communication process. What is going on? In this model, the resonance of a wide range of gesticulations is being increased by the desire to communicate, the subjective importance of the material, its emotional content, and the perceived risk of being misunderstood. Under these conditions, volitional communicative intent giving rise to each verbal clause now also triggers a corresponding gesture which is appropriate to that clause, even though not to the circumstances of the communication. To complete the illustration, we should note that it would be possible for the woman to suppress these gesticulations, but that this would be effortful (which is why she allows herself to continue despite the absurdity). The suppression would correspond to a process of damping.
Thus modulational states (which may be regarded as setting the context for the input event) can determine that a particular network is in a state of heightened sensitivity or resonance, meaning that it is more likely to be triggered by an input. The activation threshold in relation to a given event will depend on the baseline responsivity of the network to that event, and on any modulation acting on the network. Evidence to support this general proposition comes from several sources. First, there is a large literature on priming (for example, see McNamara, 2005), which has been demonstrated in several different forms such as semantic priming, negative priming, and repetition priming. Second, there is neuroimaging evidence for the facilitatory effect of top-down attentional modulation on sensory cortical activation. Woodruff et al. (1996), showed that when auditory and visual stimuli are presented simultaneously, the response of auditory or visual cortex was increased by the subject attending to the corresponding sensory modality. It is also widely accepted that non-conscious contextual modulation may modify sensory perception and cognition (e.g., a hungry man will notice the smell of food more strongly, and subjects fasted for 24 h suffer more Stroop-interference from food-related words (Lavy and Hout, 1993)). Third, one of the leading theories of speech production (Dell, 1986; Dell and O’Seaghdha, 1991) suggests that speech is produced as activation spreads across nodes representing concepts, words, morphemes, and phonemes. The most activated nodes at each level influence activations at lower levels, which finally determine the contents of speech. In fact connectionist networks feature prominently in currently dominant models of speech production (Levelt, 1999). A fourth line of evidence is provided by the McGurk–MacDonald effect (McGurk and MacDonald, 1976). McGurk and MacDonald (1976) showed that visual cues can alter speech perception. An audio recording of the syllable “ba” dubbed onto a video of a speaker saying “ga” is usually perceived as the syllable “da.” The illusion may come about because the audio input activates networks for “ba” and, to a lesser extent, “da,” while the visual input increases resonance of networks representing “ga” and, to a lesser extent, “da.” Thus the syllable “da” is heard because visual cues potentiate auditory processing of that syllable over the true auditory perception. Finally, this kind of connectionist model explains the pattern of interference effects revealed using the Stroop task (Williams et al., 1996).
Therefore just as the resonance of an acoustic space will tend to selectively emphasize the resonant frequencies of the sounds played within it, so informational resonance within the brain will determine the resources allocated to particular aspects of external and internal information.
Psychosis, Hodological Variance, and Schizophrenia
Psychosis
It has been argued above that anomalous cortical activity can explain psychotic symptoms better than a “faulty monitoring” hypothesis. A central hypothesis proposed here is that anomalous activity (resulting perhaps in a hallucination) will occur if the sensitivity or resonance of a network rises to the point where it will begin to show significant activation even though there is little or no input (which I will call hyper-resonance).
Several lines of evidence support the hypothesis that psychotic symptoms are associated with lower activation thresholds (consistent with increased resonance). A recent meta-analysis of studies of semantic priming in schizophrenia (Pomarol-Clotet et al., 2008) concludes that it is increased in schizophrenia patients with thought disorder. Oliveri and Calvo (2003) showed that for users of the drug “ecstasy,” the phosphene threshold for transcranial magnetic stimulation (TMS) to the visual cortex (i.e., the level at which the subject begins to experience visual phenomena) was lower in those with visual hallucinations. Sensory deafferentation, typically associated with upregulation (Zis and Fibiger, 1975; Burke, 2002), is known to be associated with tactile (Kooijman et al., 2000), auditory (Griffiths, 2000), and visual (Ffytche, 2005) hallucinations.
These examples illustrate that this hypothetical hodological hyper-resonance mechanism is consistent with psychotic symptoms caused by a variety of disorders. As a further example, let us consider affective disorder. The chief characteristic of hallucinations and delusions associated with affective disorder is that their content is congruent with the mood, usually carrying a high emotional charge. Likewise, the mood is profoundly disturbed. It is proposed here that the extreme mood causes neuromodulation outside the normal physiological range, leading to hyper-resonance in networks representing constructs with a high matching emotional valence.
Schizophrenia
What of schizophrenia itself? It was noted above that there is considerable clinical and neuroanatomical heterogeneity in schizophrenia, whilst the genetic predisposition for the disorder is largely independent of this variation, suggesting genetic pleiotropy. Moreover, the available evidence seems most consistent with a polygenic transmission (International Schizophrenia Consortium, 2009) and involvement of the cortical microcircuit (Harrison and Weinberger, 2005) although there is also evidence for the involvement of more widespread networks including deep brain structures (Silbersweig et al., 1995; Ffytche, 2008).
Polygenic transmission implies a quantitative aspect to the predisposition. Pleiotropy suggests this then may manifest in many ways. A parsimonious model would be that the genetic predisposition contributes pathology which varies in neuroanatomical distribution between individuals, giving rise to a composite distribution of pathology within functional networks. If the pathology in question results in altered hodological resonance within a network, it becomes possible to reconcile a specific pathology for schizophrenia with a more general pathological mechanism for psychotic symptoms.
It is proposed that genetic predisposition to schizophrenia is expressed as a neurodevelopmental process resulting directly or indirectly in an increased variation (between networks) of network excitability. This might result from a reduction in buffering to stochastic (random) events during neurodevelopment, consistent with evidence of developmental instability associated with the disease (Murphy and Owen, 1996; Yeo et al., 1999). The unevenness (or its effect) is exacerbated following maturational synaptic elimination, perhaps because pruning produces networks with fewer connections, which therefore have a larger variance in connectivity/resonance; and results in some networks with low activation thresholds, which can become hyper-resonant under certain conditions of neuroplasticity and neuromodulation.
This model (subsequently referred to as the hodological variance model) can provide some explanation for bizarre and complex phenomena such as auditory hallucinations consisting of a commentary on a patient’s actions. In those susceptible, networks with a semantic connection to the current context are primed by that context, and those networks which already have a lowered threshold (because of the disease) activate producing the hallucination. Compare this with the telephone call described above, where each gesticulation is appropriate to the context of that moment’s utterance.
Furthermore, variable connection and modulation strengths will lead to some networks which are over-damped, and so have increased activation thresholds. This will be especially critical for networks involved in the initiation of action not driven by environmental stimuli (such as those in the prefrontal cortex), where self-activation is necessary. Therefore this hypothetical mechanism suggests another explanation for negative symptoms, more specific to schizophrenia than that discussed earlier. The heterogeneous clinical picture produced by the disorder can be explained in terms of the degree of hodological variance within the individual (corresponding to general severity), and the anatomical distribution of (and balance between) hyper-resonant and hypo-resonant networks.
The model can accommodate a continuum between schizophrenia and affective psychosis. This paper does not attempt to impute a mechanism for affective psychosis beyond providing an account of how profound affective disturbance may give rise to psychotic symptoms. However it is possible to see how an equal mixture of schizophrenic and affective pathology could give rise to a distribution of psychotic symptoms, most of which would involve contributions from both pathologies. These symptoms would arise in networks which had lowered activation thresholds (due to schizophrenia pathology), and additionally carry some affective valence (i.e., the network activation threshold would be lower in the context of the corresponding affect). These networks could then become hyper-resonant under conditions of high affective modulation. Because of this double mechanism, the affective disturbance would not need to be as profound as in a pure affective psychosis; neither would the networks involved need to carry the same degree of emotional valence; in other words, the “mixed” psychotic symptoms could be less mood congruent.
Figures 1A,B illustrate these concepts. Figure 1A shows notional intra-individual distributions for brain network resonance (at baseline; i.e., in the absence of modulation) and illustrates the excess of networks with low and high resonance hypothesized in schizophrenia. Figure 1B (which shares the same x-axis) shows a notional hyper-resonance threshold, to the right of which the network is activated as if in response to an input, even in the absence of any input. Greater degrees of network base resonance require less modulation in order to become hyper-resonant. The second boundary represents the maximal physiological modulation for a network. Values above this are assumed to exist in affective and schizo-affective disorders, and may cause hyper-resonance in a population of networks. The third boundary, between affective psychosis and schizo-affective disorder represents the level of network base resonance which is assumed to be uncommon in normal individuals (Figure 1A). In principle, schizo-affective disorder may also produce hyper-resonance across these boundaries (i.e., mood-congruent hyper-modulation of normal networks or physiological modulation of networks with high baseline resonance), in other words it may also generate psychotic symptoms like schizophrenia or affective psychosis if it is sufficiently severe. In practice these diagnostic distinctions will be dimensional rather than categorical.
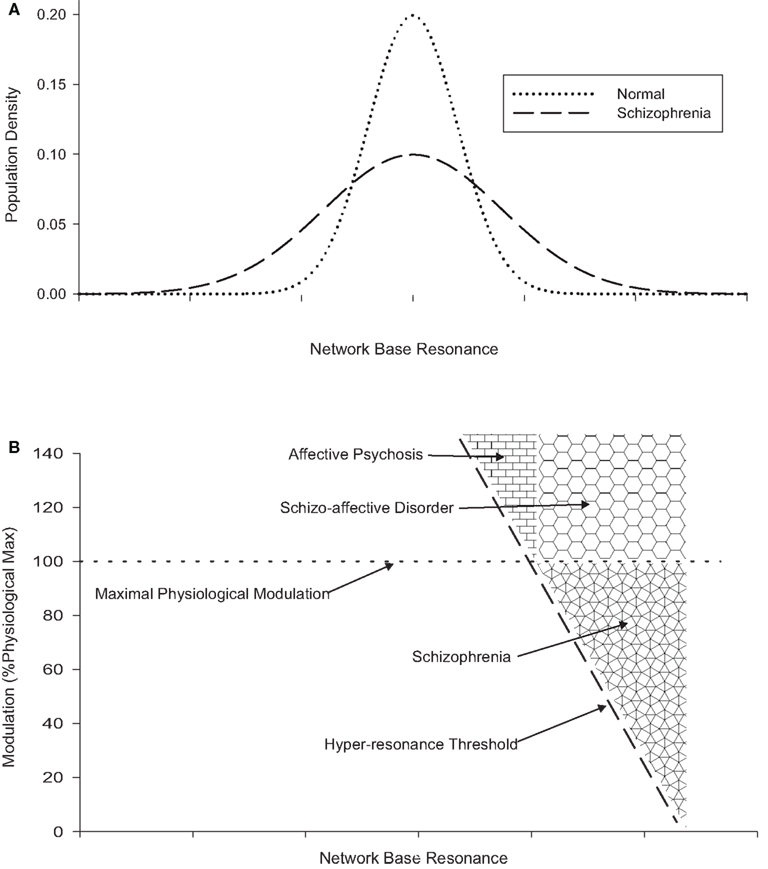
Figure 1. (A) (Top) showing the effect of greater hodological variance on network base resonance in schizophrenia. (B) (Bottom, using same x-axis) showing the relationship between network base resonance, modulation, and hyper-resonance, and the relationship between schizophrenia, schizo-affective disorder, and affective psychosis.
Schizophrenia at the Cellular and Biochemical Level
It has been argued above that consideration of evidence from a variety of sources including genetics, phenomenology, cognitive psychology, linguistics, neuroimaging, and epidemiology provide reasons to consider an unevenness of network excitability as the core pathology of schizophrenia. It might reasonably be asked how this proposed pathology is related to the various abnormalities reported in schizophrenia at the cellular and biochemical level. However, although the question is important, there are major obstacles in providing an answer. The first is, simply, the limits of our current understanding of brain function, which make the emergent effects of particular small-scale abnormalities very difficult to predict. Next, there are several plausible mechanisms which might result in an increase in hodological variance; it might arise from static variance in connections between the components, from decompensation in some neuroplastic process or a defect in a compensatory process. Increased variance might also arise from dynamic instability in function, for instance, through altered function of cross-modulatory components. Finally the interconnected nature of the brain often makes it difficult to say whether a particular finding it is part of the primary pathology, a consequence of primary pathology or whether it results from a compensatory process (Lewis and Gonzalez-Burgos, 2008). At the present time the scale of the complexity appear to render the problem intractable; therefore it appears difficult to resist the conclusion that currently, at least, small-scale findings neither lend support to, nor cast doubt on this model.
The Diathesis – Stress Model and the Disease State
The above account of schizo-affective disorder illustrates interplay of affect and schizophrenic pathology similar to that which may characterize the transition from predisposition (or remission) to active illness. It is known that a stressful life-event or life situation may precipitate the illness in a predisposed individual (Day et al., 1987). These conditions may induce strong affect, especially anxiety, and feelings of being under threat. Here it is hypothesized that affective modulation may cause predisposed networks to enter a state of hyper-resonance. Importantly, the networks receiving strong modulation under conditions of anxiety and perceived threat are likely to be those concerned with the detection, monitoring, and appraisal of external threats; for instance networks responsible for detecting hostile gaze, or other external reference, those concerned with hostile comment or malicious intent. A similar argument (that expectation shapes perception) is made in Bayesian models of psychosis (Corlett et al., 2009). Therefore in a susceptible individual, stress may trigger a state of positive feedback, where vigilance toward external threat produces an aberrant perception of external threat, resulting in increasing levels of affective arousal and progressive involvement of more networks.
This is illustrated in Figure 2. Thus this model can explain both how stress may trigger the onset of illness, and how the illness can become a self-perpetuating state. It also explains why the onset of illness in the absence of a sudden precipitant is a poor prognostic sign; in this scenario less modulation has been required to produce the hyper-resonant state, implying higher baseline resonance.
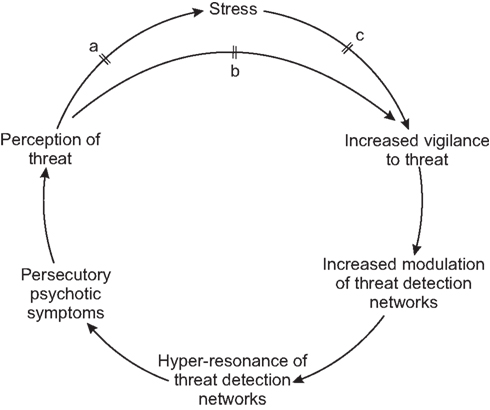
Figure 2. Schema showing external precipitation/exacerbation of psychosis in schizophrenia, maintenance of psychosis, and hypothetical mechanism of action of antipsychotics at points a, b, and c.
The Mechanism of Antipsychotic Pharmacotherapy
In order to understand how antipsychotic medication fits this model, let us first consider some of the key facts. Pharmacotherapy brings about symptomatic control rather than cure. It is far more effective at controlling the psychotic symptoms than the negative symptoms. Nevertheless, there are individuals for whom no amount of medication will fully control the psychotic symptoms. Reduction of dopaminergic neurotransmission appears central to the mechanism, but there is a lag of several weeks between that blockade and maximum therapeutic response.
Recent considerations of the role of dopamine in relation to schizophrenia have highlighted its role in the attribution of salience, particularly in relation to stimuli involved in reward, punishment, and motivation (Howes and Kapur, 2009). Courvoisier’s classic experiment (Courvoisier, 1956) showed that rats trained to associate the sound of a bell with a subsequent electric shock, would no longer run away at the sound of the bell after being treated with chlorpromazine, although they still responded to the shock itself. It can be seen from Figure 2, that if the salience of potentially threatening perceptions is diminished, then not only will they cause less stress (and antipsychotics are known to have an anxiolytic action; Quality Assurance Project, 1985; Depping et al., 2010), but also the required action (vigilance toward potential threats) becomes less pressing, allowing a reversal of the cycle (described in relation to stress) to occur. This reversal takes some time because it is mediated via the emotional and cognitive systems involved in the appraisal of threat.
Therefore it can be seen that within this model, dopaminergic blockade acts by diminishing modulation, perhaps even to sub-physiological levels; and that, rather than effecting a correction of the core pathology, it provides a means to ameliorate the psychotic symptoms. This model would predict no positive impact of dopamine blockade on negative symptoms caused directly by the disease, though it would allow an impact on any symptoms secondary to, or exacerbated by, the psychotic symptoms. On the other hand, some negative symptoms might be exacerbated through this mechanism. Some individuals more severely affected by the disease would have networks whose high baseline resonance caused them to continue to be hyper-resonant even under conditions of much reduced modulation brought about by maximal dopaminergic blockade. Dopamine’s hypothetical role in affecting modulational states via the attribution of salience may also explain the efficacy of antipsychotics in other psychotic states.
Psychotic symptoms in normal individuals
It can be seen that the model predicts that there will be a population of individuals in whom peaks of modulation may produce transient psychotic symptoms, which do not become self-sustaining as described above, because the population density of the more resonant networks is too low. Hunter et al. (2006) studied normal subjects and found episodes of spontaneous activity in speech-sensitive cortex during silence. These episodes did not correspond to the subjective experience of hallucinations, perhaps because the degree of activation was insufficient. The existence of sub-threshold hallucination-like activity in non-symptomatic individuals is consistent with the notion of a continuum and is predicted by the model described above.
It should be noted that, while there may indeed be a symptomatic continuum between psychotic and normal individuals, the distribution within the general population does not appear to be either Gaussian or exponential. In other words, if we consider the fraction of the population who, untreated, would experience auditory hallucinations every 8 h or less, we do not see the percentage of the population affected increase rapidly as we halve or quarter the threshold for the frequency of the psychotic symptoms. This is difficult to explain if we consider psychosis-proneness to be a single factor, but can be explained in this model as being due to the interplay of baseline resonance and the modulation threshold for hyper-resonance. If we consider the total population of networks within the general population of individuals, the model seems to suggest that, as we consider more common networks with lower baseline resonance (which need higher degrees of modulation to produce psychotic symptoms), the statistical likelihood of a modulation great enough to drive the network into hyper-resonance decreases faster than the population density of such networks increases. In the particular case of hallucinations relating to bereavement of long-term partners, both modulation and resonance are likely to be involved. Networks which have been used to responding to characteristics such as the deceased’s form and voice are likely to be in a state akin to denervation supersensitivity, while simultaneously strong feelings of longing for this person will provide modulation of these networks.
The Persistence of Schizophrenia Genes and Species-Specificity of Schizophrenia
An important issue is the persistence of genes predisposing to schizophrenia which has a negative impact on reproductive fitness. Indeed Crow (2000) puts this question at the heart of his hypothetical model, arguing that schizophrenia is the price that Homo Sapiens pays for language, in other words, that it is an inevitable side-effect of the evolutionary brain changes which have made speech possible and characterize the species. Many commentators have suggested that alleles predisposing to schizophrenia persist because those with an intermediate genetic loading have some advantage (“balancing selection”) such as increased creativity (Nettle and Clegg, 2006). Others argue that the most likely explanation is that these genes are not advantageous in any way, but the persistence of schizophrenia represents a balance between the selection against the genes, and their generation through mutation (Keller and Miller, 2006).
Consider first the mutation-selection balance hypothesis. It is perhaps to be preferred for its greater simplicity, and is compatible with the hypothesis presented here. However, to my knowledge, no (natural) analog of schizophrenia has been found in other animals, and, at least in the case of mice, might have been expected to have been discovered and exploited, had it existed with anything approaching the frequency of schizophrenia. Therefore there is good reason to suspect a degree of specificity for the primate brain, if not the human brain. A potential weakness of the mutation-selection balance model is its failure to account for this specificity. One attractive explanation is that the loci involved are newer, and specific to humans or their close relatives. However, most of the loci for schizophrenia candidate genes identified so far are present within the mouse genome. Another possibility, suggested by Burns (2004) is that some novel aspect of the human brain causes old loci to manifest in a novel way. If this is the case, the mutation-selection balance hypothesis is compatible with the HR hypothesis as far as it goes, but in turn raises the question of what it is about the human brain that unlocks the possibility of schizophrenia.
Let us now consider the balancing selection hypothesis. One of the main objections to this hypothesis has been the lack of evidence of a clear advantage for relatives of those with schizophrenia, either in terms of overall fertility (Haukka et al., 2003) or other qualities such as creativity (Waddell, 1998). However, in order to exclude a balancing selection advantage in a polygenic trait we should need to look in the region of the modal point for gene loading for that trait, where the selection advantage needed to offset the effect of schizophrenia is likely to be smaller than for the few percent of the population representing close relatives of those with the disease. It is possible that the closest relatives may have a larger than optimal dose of genetic loading (Hoffman et al., 2004). Since it is not currently possible to identify the population nearer the modal loading, any such balancing advantage is for the time being untestable. Therefore we should ask if there are any theoretical reasons why some advantage might be conferred through the mechanisms hypothesized above.
According to these mechanisms we should see an increase in inter-individual and intra-individual variability in brain function across cognitive domains in the population with modal genetic loading, compared to those with no loading. Some networks would have a diminished threshold for activation (corresponding to cognitive domains more easily generating interest and attention) while others would have an increased threshold. This could plausibly predispose individuals to focus interest and specialize in some cognitive domain, and to increase the chances that a population will include an individual excelling in any particular domain, at the expense of diminishing the numbers of individuals having all-round interests. Such a mechanism could be particularly advantageous for Homo Sapiens because the existence of language facilitates the transfer of knowledge; consequently in many domains, all can enjoy the insights of those with the greatest experience and expertise. In other words, just as hemispheric specialization reduces duplication to leverage cognitive power within the brain, cognitive diversity could leverage cognitive power within a community. Thus the evolutionary relationship between language and schizophrenia posited by Crow might be reformulated as follows: “schizophrenia is the price Homo Sapiens pays for cognitive diversity; language makes the price worth paying.” This aspect of the hypothesis could be explored by comparing cognitive diversity within H. Sapiens with that of other mammals.
Therefore this hypothesis is compatible with both mutation-selection balance and balancing selection explanations for the persistence of schizophrenia genes, but offers more explanatory power for species-specificity in the latter.
Implications and Predictions of the Model
Hodological variance itself may be beyond our current capability to observe directly, because of the potentially small scale, and the distributed and intermeshed nature of the networks involved. A number of indirect consequences would be predicted, including increased inter-subject variation (and perhaps also intra-subject variation) in the functional neuroanatomy associated with a given task (although once again this might be on a small scale). We might also expect to see an increase in neuroanatomical and cognitive variation, as well as the well-documented shifts in means. Hodological resonance is potentially observable and, as argued above, may account for the increase in semantic priming seen in thought-disordered patients with schizophrenia.
In the context of our current research capability, perhaps the most important implication of this model is the need for more research using single-case studies, because of the variability of pathology between individuals, and the risk that important results will be masked within group means. Although a start has been made using traditional neuropsychology (Shallice et al., 1991; Chan et al., 2006) this needs to be extended using techniques such as non-invasive structural and functional neuroimaging, experimental psychology, and neurophysiological techniques. The emphasis should be on functional and structural localization of areas of abnormality within the individual, with progressive characterization of the abnormalities using a wide range of techniques. The objective should be to attempt to characterize the nature of lesions separately from the consequences of their neuroanatomical location and distribution.
This article highlights the greater specificity of psychotic symptoms and therefore suggests the need for greater research emphasis on psychotic symptoms. The model suggests the need for more data on neurocognitive abnormalities associated with particular psychotic states (such as auditory hallucinations), and to distinguish these from those associated with the corresponding psychotic traits. In particular, there should be further work using priming and other functional paradigms in those currently subject to psychotic symptoms. However a potential difficulty in schizophrenia is that many networks may have unchanged or increased as well as lowered activation thresholds.
There is a need for more qualitative research into the phenomenological psychopathology of schizophrenia. In particular, there is a need for further consideration (in the light of modern knowledge) of the systems likely to be involved in each type of symptom in order to test the hypothesis that all symptoms cannot be parsimoniously explained by lesions in a small number of systems/anatomical locations. This work has also begun (Nayani and David, 1996; Woodruff, 2004) but requires elaboration. There is also a need for further study of precipitating and relieving factors for psychotic symptoms. These approaches may be particularly fruitful in conjunction with the case-study approach discussed above.
The endophenotype strategy (Gottesman and Gould, 2003) proposed that neuropsychological and other brain abnormalities seen in close relatives of those with schizophrenia might represent genetically and pathologically simpler entities more amenable to study than the full disorder. The hodological variance hypothesis suggests that this strategy will not prove to be fertile because endophenotypes will be found to result from pleiotropy of a common genetic predisposition (rather than a genetically simpler subset of that predisposition). Specifically, the model predicts that each abnormality associated with the schizophrenia endophenotype will be increased in relatives of schizophrenia patients without that abnormality. This was already implied by the evidence of apparent separation of genetic susceptibility and pathoplastic factors discussed earlier. This model adds weight by showing how a single pathology can plausibly give rise to a wide variety of manifestations consistent with findings in schizophrenia. Nevertheless, research on genetically predisposed individuals unaffected by the disease will continue to be valuable because we know that any abnormalities present are manifestations of the disease predisposition and not due to consequences of the disease or its treatment. A related prediction is that genetic pathoplastic factors will not account for more than a minority of the variance in pathological form, as much of this variance would derive ultimately from environmental and stochastic neurodevelopmental factors.
Summary and Appraisal of the Hodological Variance Model
The model described here suggests that in schizophrenia, many genes of small effect together with copy number variants and environmental factors, contribute to a neurodevelopmental diathesis. Genetic factors may be mediated through impaired developmental buffering and the diathesis is characterized by an increase in the variability of activation and/or inhibition strengths of neuronal inputs and modulation. This in turn leads to an increased variability in neuronal networks, which is exacerbated through maturational dendritic pruning. This leads to both psychotic and deficit symptoms, the former through the phenomenon of hodological hyper-resonance, and the latter through what might be termed hypo-resonance. The increased variance is assumed to be widespread and can therefore give rise to abnormal activation in a wide range of networks, and so can produce bizarre (Schneiderian) hallucinations, misperceptions, and delusions. The account shows how this hyper-resonance can be considered a general mechanism for psychotic phenomena, which is produced through a different pathway (excessive modulation) in affective disorder, and illustrates how the two pathways would interact additively or synergistically to give schizo-affective disorder. The model can explain observations relating to the onset of schizophrenia, its relationship to stress, and its response to antipsychotic medication.
The guiding principle employed during development of this model was to achieve an optimum ratio between parsimony and explanatory power. A particular objective was to address some of the apparent paradoxes of schizophrenia: to present a single class of pathology which is compatible with considerable heterogeneity including asymptomatic and alternative (intermediate) phenotypes; to explain how this could give rise to negative and positive symptoms as different dimensions, how some individuals with schizophrenia can appear cognitively within normal limits (Chan et al., 2006), how patterns of onset and treatment effects could be explained, how the genes could persist by conferring a balancing selection advantage, and how the pathology could fit within a spectrum of psychosis alongside schizo-affective and affective psychosis, and within a continuum of psychosis shading into the psychotic-like experiences of otherwise apparently healthy individuals.
Therefore these theoretical difficulties were used to constrain and test the model. In the process it was necessary to develop sub-hypotheses to demonstrate explanatory power and provide a context. In developing a model of schizophrenia it was necessary to invoke a more general model of psychotic symptoms (hodological resonance). However, although these hypotheses interlock, they are not all interdependent. For instance, two different (though not mutually exclusive) hypotheses were proposed to explain negative symptoms. The model was left unconstrained except where the evidence seemed to compel the constraint. The hodological variance mechanism would still function as described if the variance only applied to (for instance) neuromodulatory inputs, or to particular classes of networks. Indeed, although the model was described above in terms of static variance, it could equally apply to mechanisms producing increased dynamic variance, or dynamic instability. There is some evidence that this is present in schizophrenia – for instance there is evidence that most if not all the deficit in reaction time seen in schizophrenia can be accounted for by greater variability in performance, as opposed to any impairment of best performance (Birkett et al., 2007) and schizophrenia seems to be associated with more intra-individual variation than affective disorders (Schwartz et al., 1989; Kaiser et al., 2008). Some readers may feel that this lack of specification amounts to vagueness and diminishes the value of the model. However, as the scientist and philosopher Charles Peirce pointed out, the use of more general hypotheses may hasten scientific progress. Using the parlor game “twenty questions” to explore the efficiency of hypothesis-generation, he wrote “twenty skillful hypotheses will ascertain what two hundred thousand stupid ones might fail to do. The secret of the business lies in the caution which breaks a hypothesis up into its smallest logical components, and only risks them one at a time” (Peirce, 1998, p. 109). In other words, when nothing is known about the subject, testing the general hypothesis “It is alive” is more likely to yield useful information than the more specific hypothesis “It is a horse.”
Perhaps the most important potential criticism of this model is the present lack of direct evidence for increased variability in network excitability, which is after all at the center of the hypothesis. Testing this directly is most likely beyond our current capability. However, this current lack of detection-capability should not be seen as a problem for the model; indeed it is to be expected. By definition the core abnormality in schizophrenia should be present around 100% of the time in affected individuals, and completely absent in unaffected individuals (subject to misdiagnosis and limitations in our detection of the abnormality). In terms of quantitative traits, it would need to represent the most extreme 0.7% or so of the population, or a minimum of 2.4 SD beyond the mean, with lower values always present in unaffected individuals. If such an abnormality had been demonstrated, there would be little debate about the core pathology of schizophrenia; conversely all hypotheses built around abnormalities with less evidential support than this must all be considered at best, incomplete. Hence it may be supposed that the core abnormality is so far undetected, perhaps because it is currently beyond our capacity to do so. Accordingly, there is some virtue in approaching the problem from the theoretical direction described above, which complements the traditional more data-driven approach.
In addition to the detailed hypotheses presented here, consideration of the model raises and illustrates potential difficulties with current strategies of genetic and neuroanatomical reductionism, and suggests the need for new emphasis on the study of individuals, of variation, and of phenomenology informed by modern cognitive science.
It is not claimed that every aspect of the pathology of schizophrenia has been explained (for instance, no attempt has been made to explain the disease’s differential effects on the morphology of neuroanatomical structures), nor that every aspect discussed has been fully specified and detailed. The dynamic aspects in terms of neuroplastic processes of compensation and decompensation have been left largely unexplored. The “nodes” described in this connectionist account are much simpler than the neurohistological reality, and therefore the model does not easily lend itself to biochemical or histological predictions. This is primarily a neurocognitive connectionist, rather than anatomical, neurochemical, or physiological account, and as such it is possible to see areas of potential compatibility with other hypothetical models which use a different frame of reference. Nevertheless it makes specific predictions not suggested by other models. There is no doubt that the hodological variance and resonance models would benefit from further elaboration and refinement. However I hope the reader will find that the ideas presented here have achieved that favorable ratio between parsimony and explanatory power, and therefore represent a good basis for further study.
Conclusion
An accurate model of schizophrenia faces a number of challenges. It should be consistent with a broader model of psychosis, while highlighting the distinct mechanistic features peculiar to schizophrenia, and relating these to the corresponding characteristic clinical features. It should be consistent with the intermediate forms of psychosis (such as schizo-affective disorder) seen in clinical practice. It should explain the extensive clinical variation seen in schizophrenia, either by explaining variation within a single disorder, or by providing a practical rationale for parsing it into disorders with distinct pathologies. It should account for both deficit and psychotic symptoms, and should be consistent with what is known of the etiology and natural history of the disorder. Finally, for a single disorder, the core pathology should be conceptually simple (i.e., a mechanism which cannot be further parsed into independent pathologies).
In this account I have shown how in increase in the variation of strength of brain connectivity (hodological variance) can satisfy these tests and can therefore be considered as a candidate for the core pathology of schizophrenia. By its nature, an abnormality in intra-individual variation does not easily yield up its secrets to experimental designs based on comparison of group means; therefore in the study of schizophrenia there is a need for more single-case studies and for experimental designs based on exploration of intra-individual variance, to compliment more traditional research designs.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I am very grateful to Professor Tim Crow, Dr. Alastair Cardno, Dr. Tom Farrow, Professor Peter Woodruff, Dr. Mike Hunter, Dr. Kwang-Hyuk Lee, and the staff of ScanLab, University of Sheffield for helpful comments on earlier drafts. I would also like to thank the reviewers whose thoughtful comments and suggestions have helped me to improve the manuscript.
References
Aleman, A., Rutten, G. J., Sitskoorn, M. M., Dautzenberg, G., and Ramsey, N. F. (2001). Activation of striate cortex in the absence of visual stimulation: an fMRI study of synesthesia. Neuroreport 12, 2827–2830.
American Psychiatric Association. (2000). Diagnostic and Statistical Manual of Mental Disorders, 4th Edn (Text Revision). Washington, DC: American Psychiatric Association.
Bergen, S. E., Fanous, A. H., Kuo, P.-H., Wormley, B. K., O’Neill, F. A., Walsh, D., Riley, B. P., and Kendler, K. S. (2010). No association of dysbindin with symptom factors of schizophrenia in an Irish case-control sample. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 700–705.
Birkett, P., Sigmundsson, T., Sharma, T., Toulopoulou, T., Griffiths, T. D., Reveley, A., and Murray, R. (2007). Reaction time and sustained attention in schizophrenia and its genetic predisposition. Schizophr. Res. 95, 76–85.
Bullmore, E. T., Frangou, S., and Murray, R. M. (1997). The dysplastic net hypothesis: an integration of developmental and dysconnectivity theories of schizophrenia. Schizophr. Res. 28, 143–156.
Burke, W. (2002). The neural basis of Charles Bonnet hallucinations: a hypothesis. J. Neurol. Neurosurg. Psychiatr. 73, 535–541.
Burns, J. K. (2004). An evolutionary theory of schizophrenia: cortical connectivity, metarepresentation, and the social brain. Behav. Brain Sci. 27, 831–855; discussion 855–885.
Cardno, A. G., Sham, P. C., Murray, R. M., and McGuffin, P. (2001). Twin study of symptom dimensions in psychoses. Br. J. Psychiatry 179, 39–45.
Chan, R. C., Chen, E. Y., Cheung, E. F., Chen, R. Y., and Cheung, H. K. (2006). The components of executive functioning in a cohort of patients with chronic schizophrenia: a multiple single-case study design. Schizophr. Res. 81, 173–189.
Cheniaux, E., Landeira-Fernandez, J., Lessa Telles, L., Lessa, J. L., Dias, A., Duncan, T., and Versiani, M. (2008). Does schizoaffective disorder really exist? A systematic review of the studies that compared schizoaffective disorder with schizophrenia or mood disorders. J. Affect. Disord. 106, 209–217.
Corlett, P. R., Frith, C. D., and Fletcher, P. C. (2009). From drugs to deprivation: a Bayesian framework for understanding models of psychosis. Psychopharmacology (Berl.) 206, 515–530.
Courvoisier, S. (1956). Pharmacodynamic basis for the use of chlorpromazine in psychiatry. J. Clin. Exp. Psychopathol. 17, 25–37.
Craddock, N., O’Donovan, M. C., and Owen, M. J. (2006). Genes for schizophrenia and bipolar disorder? Implications for psychiatric nosology. Schizophr. Bull. 32, 9–16.
Crow, T. J. (1995). A continuum of psychosis, one human gene, and not much else – the case for homogeneity. Schizophr. Res. 17, 135–145.
Crow, T. J. (2000). Schizophrenia as the price that homo sapiens pays for language: a resolution of the central paradox in the origin of the species. Brain Res. Rev. 31, 118–129.
Day, R., Nielsen, J. A., Korten, A., Ernberg, G., Dube, K. C., Gebhart, J., Jablensky, A., Leon, C., Marsella, A., Olatawura, M., Sartorius, N., Stromgren, E., Takahashi, R., Wig, N., and Wynne, L. C. (1987). Stressful life events preceding the acute onset of schizophrenia: a cross-national study from the World Health Organization. Cult. Med. Psychiatry 11, 123–205.
Dell, G. S. (1986). A spreading-activation theory of retrieval in sentence production. Psychol. Rev. 93, 283–321.
Dell, G. S., and O’Seaghdha, P. G. (1991). Mediated and convergent lexical priming in language production: a comment on Levelt et al. (1991). Psychol. Rev. 98, 604–614.
Depping, A. M., Komossa, K., Kissling, W., and Leucht, S. (2010). Second-generation antipsychotics for anxiety disorders. Cochrane Database Syst. Rev. doi: 10.1002/14651858.CD008120.pub2
Fanous, A. H., and Kendler, K. S. (2005). Genetic heterogeneity, modifier genes, and quantitative phenotypes in psychiatric illness: searching for a framework. Mol. Psychiatry 10, 6–13.
Fanous, A. H., Neale, M. C., Webb, B. T., Straub, R. E., Amdur, R. L., O’Neill, F. A., Walsh, D., Riley, B. P., and Kendler, K. S. (2007). A genome-wide scan for modifier loci in schizophrenia. Am. J. Med. Genet. B 144, 589–595.
Fanous, A. H., van den Oord, E. J., Riley, B. P., Aggen, S. H., Neale, M. C., O’Neill, F. A., Walsh, D., and Kendler, K. S. (2005). Relationship between a high-risk haplotype in the DTNBP1 (dysbindin) gene and clinical features of schizophrenia. Am. J. Psychiatry 162, 1824–1832.
Feinberg, I. (1978). Efference copy and corollary discharge: implications for thinking and its disorders. Schizophr. Bull. 4, 636–640.
Feinberg, I. (1982). Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J. Psychiatr. Res. 17, 319–334.
Feinberg, I., and Guazzelli, M. (1999). Schizophrenia – a disorder of the corollary discharge systems that integrate the motor systems of thought with the sensory systems of consciousness. Br. J. Psychiatry 174, 196–204.
Ffytche, D. H. (2005). Visual hallucinations and the Charles Bonnet syndrome. Curr. Psychiatry Rep. 7, 168–179.
Ffytche, D. H., and Catani, M. (2005). Beyond localization: from hodology to function. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 767–779.
Ffytche, D. H., Howard, R. J., Brammer, M. J., David, A., Woodruff, P., and Williams, S. (1998). The anatomy of conscious vision: an fMRI study of visual hallucinations. Nat. Neurosci. 1, 738–742.
Fletcher, P. C., and Frith, C. D. (2009). Perceiving is believing: a Bayesian approach to explaining the positive symptoms of schizophrenia. Nat. Rev. Neurosci. 10, 48–58.
Frith, C. D., Blakemore, S., and Wolpert, D. M. (2000). Explaining the symptoms of schizophrenia: abnormalities in the awareness of action. Brain Res. Brain Res. Rev. 31, 357–363.
Frith, C. D., and Done, D. J. (1989). Experiences of alien control in schizophrenia reflect a disorder in the central monitoring of action. Psychol. Med. 19, 359–363.
Goldberg, T. E., Gold, J. M., Coppola, R., and Weinberger, D. R. (1997). Unnatural practices, unspeakable actions: a study of delayed auditory feedback in schizophrenia. Am. J. Psychiatry 154, 858–860.
Gottesman, I. I., and Gould, T. D. (2003). The endophenotype concept in psychiatry: etymology and strategic intentions. Am. J. Psychiatry 160, 636–645.
Griffiths, T. D. (2000). Musical hallucinosis in acquired deafness. Phenomenology and brain substrate. Brain 123(Pt 10), 2065–2076.
Harrison, P. J., and Law, A. J. (2006). Neuregulin 1 and schizophrenia: genetics, gene expression, and neurobiology. Biol. Psychiatry 60, 132–140.
Harrison, P. J., and Weinberger, D. R. (2005). Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol. Psychiatry 10, 40–68.
Haukka, J., Suvisaari, J., and Lonnqvist, J. (2003). Fertility of patients with schizophrenia, their siblings, and the general population: a cohort study from 1950 to 1959 in Finland. Am. J. Psychiatry 160, 460–463.
Heinrichs, D. W., and Zakzanis, K. K. (1998). Neurocognitive deficit in schizophrenia: a quantititative review of the evidence. Neuropsychology 12, 426–445.
Hoffman, R. E., and Dobscha, S. K. (1989). Cortical pruning and the development of schizophrenia: a computer model. Schizophr. Bull. 15, 477–490.
Hoffman, R. E., Hampson, M., Varanko, M., and McGlashan, T. H. (2004). Auditory hallucinations, network connectivity and schizophrenia. Behav. Brain Sci. 27, 860–861.
Hoffman, R. E., and McGlashan, T. H. (1993). Parallel distributed processing and the emergence of schizophrenic symptoms. Schizophr. Bull. 19, 119–140.
Hoffman, R. E., and McGlashan, T. H. (1997). Synaptic elimination, neurodevelopment, and the mechanism of hallucinated “voices” in schizophrenia. Am. J. Psychiatry 154, 1683–1689.
Holzman, P. S. (2000). Eye movements and the search for the essence of schizophrenia. Brain Res. Brain Res. Rev. 31, 350–356.
Honea, R., Crow, T. J., Passingham, D., and Mackay, C. E. (2005). Regional deficits in brain volume in schizophrenia: a meta-analysis of voxel-based morphometry studies. Am. J. Psychiatry 162, 2233–2245.
Howes, O. D., and Kapur, S. (2009). The dopamine hypothesis of schizophrenia: version III – the final common pathway. Schizophr. Bull. 35, 549–562.
Hunter, M. D. (2004). Locating voices in space: a perceptual model for auditory hallucinations? Cogn. Neuropsychiatry 9, 93–105.
Hunter, M. D., Eickhoff, S. B., Miller, T. W., Farrow, T. F., Wilkinson, I. D., and Woodruff, P. W. (2006). Neural activity in speech-sensitive auditory cortex during silence. Proc. Natl. Acad. Sci. U.S.A. 103, 189–194.
Hunter, M. D., Griffiths, T. D., Farrow, T. F. D., Zheng, Y., Wilkinson, I. D., Hegde, N., Woods, W., Spence, S. A., and Woodruff, P. W. R. (2003). A neural basis for the perception of voices in external auditory space. Brain 126, 161–169.
Huttenlocher, P. R. (1979). Synaptic density in human frontal cortex – developmental changes and effects of aging. Brain Res. 163, 195–205.
International Schizophrenia Consortium. (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752.
Johns, L. C., Rossell, S., Frith, C., Ahmad, F., Hemsley, D., Kuipers, E., and McGuire, P. K. (2001). Verbal self-monitoring and auditory verbal hallucinations in patients with schizophrenia. Psychol. Med. 31, 705–715.
Kaiser, S., Roth, A., Rentrop, M., Friederich, H.-C., Bender, S., and Weisbrod, M. (2008). Intra-individual reaction time variability in schizophrenia, depression and borderline personality disorder. Brain Cogn. 66, 73–82.
Keller, M. C., and Miller, G. (2006). Resolving the paradox of common, harmful, heritable mental disorders: which evolutionary genetic models work best? Behav. Brain Sci. 29, 385–404; discussion 405–452.
Kendler, K. S., McGuire, M., Gruenberg, A. M., and Walsh, D. (1994). Clinical heterogeneity in schizophrenia and the pattern of psychopathology in relatives: results from an epidemiologically based family study. Acta Psychiatr. Scand. 89, 294–300.
Kendler, K. S., Myers, J. M., O’Neill, F. A., Martin, R., Murphy, B., MacLean, C. J., Walsh, D., and Straub, R. E. (2000). Clinical features of schizophrenia and linkage to chromosomes 5q, 6p, 8p, and 10p in the Irish Study of High-Density Schizophrenia Families. Am. J. Psychiatry 157, 402–408.
Kooijman, C. M., Dijkstra, P. U., Geertzen, J. H., Elzinga, A., and van der Schans, C. P. (2000). Phantom pain and phantom sensations in upper limb amputees: an epidemiological study. Pain 87, 33–41.
Kremen, W. S., Seidman, L. J., Faraone, S. V., Toomey, R., and Tsuang, M. T. (2004). Heterogeneity of schizophrenia: a study of individual neuropsychological profiles. Schizophr. Res. 71, 307–321.
Langdon, R., Jones, S. R., Connaughton, E., and Fernyhough, C. (2009). The phenomenology of inner speech: comparison of schizophrenia patients with auditory verbal hallucinations and healthy controls. Psychol. Med. 39, 655–663.
Lavy, E. H., and Hout, M. v. d. (1993). Attentional bias for appetitive cues:effects of fasting on normal subjects. Behav. Res. Ther. 31, 297–310.
Lee, K.-H., and Williams, L. M. (2000). Eye movement dysfunction as a biological marker of risk for schizophrenia. Aust. N. Z. J. Psychiatry 34, S91–S100.
Lewis, D. A. (2000). Distributed disturbances in brain structure and function in schizophrenia. Am. J. Psychiatry 157, 1–2.
Lewis, D. A., and Gonzalez-Burgos, G. (2008). Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology 33, 141–165.
MacDonald, A. W., and Schulz, S. C. (2009). What we know: findings that every theory of schizophrenia should explain. Schizophr. Bull. 35, 493–508.
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., McCarthy, M. I., Ramos, E. M., Cardon, L. R., Chakravarti, A., Cho, J. H., Guttmacher, A. E., Kong, A., Kruglyak, L., Mardis, E., Rotimi, C. N., Slatkin, M., Valle, D., Whittemore, A. S., Boehnke, M., Clark, A. G., Eichler, E. E., Gibson, G., Haines, J. L., Mackay, T. F. C., McCarroll, S. A., and Visscher, P. M. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753.
Marsh, L., Harris, D., Lim, K. O., Beal, M., Hoff, A. L., Minn, K., Csernansky, J. G., DeMent, S., Faustman, W. O., Sullivan, E. V., and Pfefferbaum, A. (1997). Structural magnetic resonance imaging abnormalities in men with severe chronic schizophrenia and an early age at clinical onset. Arch. Gen. Psychiatry 54, 1104–1112.
Marti-Bonmati, L., Lull, J. J., Garcia-Marti, G., Aguilar, E. J., Moratal-Perez, D., Poyatos, C., Robles, M., and Sanjuan, J. (2007). Chronic auditory hallucinations in schizophrenic patients: MR analysis of the coincidence between functional and morphologic abnormalities. Radiology 244, 549–556.
Matthysse, S., Holzman, P. S., and Lange, K. (1986). The genetic transmission of schizophrenia: application of Mendelian latent structure analysis to eye tracking dysfunctions in schizophrenia and affective disorder. J. Psychiatr. Res. 20, 57–67.
McClay, J. L., Fanous, A., van den Oord, E. J., Webb, B. T., Walsh, D., O’Neill, F. A., Kendler, K. S., and Chen, X. (2006). Catechol-O-methyltransferase and the clinical features of psychosis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141, 935–938.
Murphy, K. C., and Owen, M. J. (1996). Minor physical anomalies and their relationship to the aetiology of schizophrenia. Br. J. Psychiatry 168, 139–142.
Nayani, T. N., and David, A. S. (1996). The auditory hallucination: a phenomenological survey. Psychol. Med. 26, 177–189.
Nettle, D., and Clegg, H. (2006). Schizotypy, creativity and mating success in humans. Proc. Biol. Sci. 273, 611–615.
Oliveri, M., and Calvo, G. (2003). Increased visual cortical excitability in ecstasy users: a transcranial magnetic stimulation study. J. Neurol. Neurosurg. Psychiatr. 74, 1136–1138.
Peirce, C. S. (1998). The Essential Peirce – Vol. 2 Selected Philosophical Writings (1893–1913). Bloomington: Indiana University Press.
Perot, P., and Penfield, W. (1960). Hallucinations of past experience and experiential responses to stimulation of temporal cortex. Trans. Am. Neurol. Assoc. 85, 80–84.
Pomarol-Clotet, E., Oh, T. M., Laws, K. R., and McKenna, P. J. (2008). Semantic priming in schizophrenia: systematic review and meta-analysis. Br. J. Psychiatry 192, 92–97.
Quality Assurance Project. (1985). Treatment outlines for the management of anxiety states. Aust. N. Z. J. Psychiatry 19, 138–151.
Reichenberg, A., Rieckmann, N., and Harvey, P. D. (2005). Stability in schizophrenia symptoms over time: findings from the Mount Sinai Pilgrim Psychiatric Center Longitudinal Study. J. Abnorm. Psychol. 114, 363–372.
Rietkerk, T., Boks, M. P., Sommer, I. E., Liddle, P. F., Ophoff, R. A., and Kahn, R. S. (2008). The genetics of symptom dimensions of schizophrenia: review and meta-analysis. Schizophr. Res. 102, 197–205.
Schulz, R., Woermann, F. G., and Ebner, A. (2007). When written words become moving pictures: complex visual hallucinations on stimulation of the lateral occipital lobe. Epilepsy Behav. 11, 147–151.
Schwartz, F., Carr, A. C., Munich, R. L., Glauber, S., Lesser, B., and Murray, J. (1989). Reaction time impairment in schizophrenia and affective illness: the role of attention. Biol. Psychiatry 25, 540–548.
Shallice, T., Burgess, P. W., and Frith, C. D. (1991). Can the neuropsychological case-study approach be applied to schizophrenia? Psychol. Med. 21, 661–673.
Shergill, S. S., Cameron, L. A., Brammer, M. J., Williams, S. C. R., Murray, R. M., and McGuire, P. K. (2001). Modality specific neural correlates of auditory and somatic hallucinations. J. Neurol. Neurosurg. Psychiatr. 71, 688–690.
Silbersweig, D. A., Stern, E., Frith, C., Cahill, C., Holmes, A., Grootoonk, S., Seaward, J., McKenna, P., Chua, S. E., Schnorr, L., Jones, T., and Frackowiak, R. S. J. (1995). A functional neuroanatomy of hallucinations in schizophrenia. Nature 378, 176–179.
Spencer, M. D., Moorhead, T. W., McIntosh, A. M., Stanfield, A. C., Muir, W. J., Hoare, P., Owens, D. G., Lawrie, S. M., and Johnstone, E. C. (2007). Grey matter correlates of early psychotic symptoms in adolescents at enhanced risk of psychosis: a voxel-based study. Neuroimage 35, 1181–1191.
Stirling, J. D., Hellewell, J. S. E., and Ndlovu, D. (2001). Self-monitoring dysfunction and the positive symptoms of schizophrenia. Psychopathology 34, 198–202.
van de Ven, V. G., Formisano, E., Roder, C. H., Prvulovic, D., Bittner, R. A., Dietz, M. G., Hubl, D., Dierks, T., Federspiel, A., Esposito, F., Di Salle, F., Jansma, B., Goebel, R., and Linden, D. E. (2005). The spatiotemporal pattern of auditory cortical responses during verbal hallucinations. Neuroimage 27, 644–655.
Verdoux, H., and van Os, J. (2002). Psychotic symptoms in non-clinical populations and the continuum of psychosis. Schizophr. Res. 54, 59–65.
Williams, J. M. G., Mathews, A., and MacLeod, C. (1996). The emotional Stroop task and psychopathology. Psychol. Bull. 120, 3–24.
Woodruff, P. W. (2004). Auditory hallucinations: insights and questions from neuroimaging. Cogn. Neuropsychiatry 9, 73–91.
Woodruff, P. W., Benson, R. R., Bandettini, P. A., Kwong, K. K., Howard, R. J., Talavage, T., Belliveau, J., and Rosen, B. R. (1996). Modulation of auditory and visual cortex by selective attention is modality-dependent. Neuroreport 7, 1909–1913.
Yeo, R. A., Gangestad, S. W., Edgar, C., and Thoma, R. (1999). The evolutionary genetic underpinnings of schizophrenia: the developmental instability model. Schizophr. Res. 39, 197–206.
Keywords: schizophrenia, disease-model, hallucination, pleiotropy, psychosis, connectivity, priming, hodology
Citation: Birkett PBL (2011) Hodological resonance, hodological variance, psychosis, and schizophrenia: a hypothetical model. Front. Psychiatry 2:46. doi: 10.3389/fpsyt.2011.00046
Received: 08 November 2010;
Accepted: 08 July 2011;
Published online: 25 July 2011.
Edited by:
Bernat Kocsis, Harvard Medical School, USAReviewed by:
Vaibhav A. Diwadkar, Wayne State University School of Medicine, USAArmando D’Agostino, University of Milan, Italy
Copyright: © 2011 Birkett. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Paul Brian Lawrie Birkett, Academic Clinical Psychiatry, Department of Neuroscience, University of Sheffield, Longley Centre, Norwood Grange Drive, Sheffield S5 7JT, UK. e-mail:cC5iLmJpcmtldHRAc2hlZmZpZWxkLmFjLnVr