
 Yan Deng2
Yan Deng2
- 1 Department of Physiology and Neuroscience, New York University School of Medicine, New York, NY, USA
- 2 Skirball Institute of Biomolecular Medicine, New York University School of Medicine, New York, NY, USA
- 3 Department of Psychiatry, New York University School of Medicine, New York, NY, USA
Recent studies have shown that immunotherapy clears amyloid beta (Aβ) plaques and reduces Aβ levels in mouse models of Alzheimer’s disease (AD), as well as in AD patients. Tangle pathology is also relevant for the neurodegeneration in AD, and our studies have shown that active immunization with an AD related phospho-tau peptide reduces aggregated tau within the brain and slows the progression of tauopathy-induced behavioral impairments. Thus, clearance of neurofibrillary tangles and/or their precursors may reduce synaptic and neuronal loss associated with AD and other tauopathies. So far the mechanisms involved in antibody-mediated clearance of tau pathology are yet to be elucidated. In this study we have used a mouse brain slice model to examine the uptake and localization of FITC labeled anti-tau antibodies. Confocal microscopy analysis showed that the FITC labeled anti-tau antibody co-stained with phosphorylated tau, had a perinuclear appearance and co-localized with markers of the endosomal/lysosomal pathway. Additionally, tau and FITC–IgG were found together in an enriched lysosome fraction. In summary, antibody-mediated clearance of intracellular tau aggregates appears to occur via the lysosomal pathway.
Introduction
The presence of intracellular neurofibrillary tangles (NFT’s) composed of hyperphosphorylated tau is a characteristic feature of Alzheimer’s disease (AD) and other tauopathies. The causative role of tau pathology in neurodegeneration has been unequivocally proven with the identification of tau mutations in a range of disorders termed frontotemporal dementia and Parkinsonism linked to chromosome 17 (FTDP-17; Hutton et al., 1998; Poorkaj et al., 1998). Therapeutic approaches targeting tau pathology have concentrated on reducing its level of phosphorylation by modifying tau kinase and phosphatase activities (Gong et al., 2010; Medina and Avila, 2010; Piedrahita et al., 2010). A novel approach developed by our group is to use immunomodulation to clear tau pathology, akin to what has been tried with reducing amyloid beta (Aβ) load in transgenic mice and in recent clinical trials (Schenk et al., 1999; Sigurdsson et al., 2001; Lemere and Masliah, 2010). We have shown in two models with tangle pathology that active or passive immunization targeting a tauopathy related phospho-tau epitope, reduces tau aggregates and slows the progression of tangle related motor deficits (Asuni et al., 2007; Boutajangout et al., 2011) and prevents cognitive impairments (Boutajangout et al., 2010).
A natural follow up of these studies is to investigate the cellular mechanisms involved in this phenomenon. The most likely route for clearance of tau aggregates would appear to be the endosomal–lysosomal pathway (Asuni et al., 2007). Previously, it had been shown that lysosomal tau is detected in AD and control brains (Ikeda et al., 1998), and pathological changes in the lysosomal pathway in AD are well established (Nixon et al., 2005). More recently, lysosomal processing has been shown to influence tau aggregation and clearance in an inducible tauopathy cell model (Wang et al., 2009). Moreover, using immuno electron microscopy techniques, Meeker et al. (1987) showed that antibodies could be detected within lysosomes. Thus, we propose that the antibody-mediated removal of tau aggregates is facilitated by clearance through the endosomal–lysosomal pathway (Sigurdsson, 2008, 2009).
Further support for the validity of this view comes from a study using a mouse model of Parkinson’s disease which was immunized with α-synuclein or its antibodies, and showed clearance of α-synuclein aggregates, most likely via lysosomal pathways (Masliah et al., 2005, 2011) Additionally, Tampellini et al. (2007) showed that anti-Aβ antibodies could clear Aβ aggregates via the endosomal–lysosomal pathway in a neuronal cell culture system. More recently, decreased levels of the lysosomal proteases, cathepsin D and L, were observed in tangle mice receiving tau immunotherapy (Boimel et al., 2010), which perhaps may be a consequence of diminished tau pathology.
Another established mechanism for the clearance of cellular proteins is via the ubiquitin proteasome system (UPS). The UPS is considered to be mainly involved in the degradation of short lived, misfolded, and truncated proteins (Pickart, 2004). It has been known for some time that ubiquitin could be detected with NFT’s (Mori et al., 1987), and in AD brain tissue, impaired proteasome peptidase activity has been noted (Keller et al., 2000). Thus, impairment of the UPS has been proposed to be a contributing factor for the accumulation of misfolded proteins in several neurodegenerative diseases including AD and Parkinson’s disease.
Similar to other soluble proteins, normal tau, and certain species of non-aggregated hyperphosphorylated tau are likely degraded by the ubiquitin proteosome system (Petrucelli and Dawson, 2004). However, upon aggregation or under pathological conditions, clearance through the autophagy endosomal–lysosomal system should be favored. Indeed, prevalent lysosomal and autophagic vesicles have been detected by ultrastructural analysis in the JNPL3 tangle mouse model (Lin et al., 2003), as well as in neuronal cultures that express various tau mutations (Lim et al., 2001).
Herein, we studied the uptake and localization of FITC labeled anti-tau antibodies in an organotypic brain slice model derived from adult homozygous JNPL3 mice with established tau pathology. Fixed slices were sectioned and stained with antibodies against cellular organelle markers such as early and late endosomes, and lysosomes, as well as antibodies that recognize pathological and phosphorylated tau. Additionally, biochemical studies examining antibody compartmentalization were performed.
Materials and Methods
Acute Brain Slice Model and FITC–IgG Treatment
Brain slices were prepared from adult (15–26 months old) homozygous JNPL3 mice (Taconic; Lewis et al., 2000) as described by Gong et al. (2001), allowed to stabilize at room temperature for 1 h and incubated with FITC–IgG for 2 h at 35°C with intermittent oxygenation. The IgG was purified from an immunized JNPL3 mouse with a high antibody titer against our phos-tau immunogen (Tau 379–408[pSer396, 404]; Asuni et al., 2007). FITC conjugation was performed with the FluoroTag™ FITC Conjugation Kit (Sigma) as per the manufacturer’s protocol. Slices were either fixed in periodate–lysine–paraformaldehyde buffer (PLP) for immunohistochemical analysis or frozen for biochemical analysis.
Immunohistochemistry
Fixed slices were sectioned at 40 μm and were stained with the tau antibodies CP13 (pSer202 epitope) and MC1 (conformational epitope). Tau antibodies CP13 and MC1 were kindly provided by Prof. P. Davies, AECOM, New York. Sections were also stained with antibodies against lysosomal-associated membrane protein 2 (LAMP2; Abl93; Chen et al., 1985) and Rab5, markers of lysosomes and early endosomes respectively, kindly provided by Dr. P. Mathews, Nathan Kline Institute, Orangeburg. All sections were stained with appropriate secondary antibodies conjugated to Texas Red (Molecular Probes), counterstained with the nuclear marker Hoechst 33342 (Molecular Probes) and visualized by confocal microscopy (Zeiss LSM710, Zeiss Plan-Apochromat 63×/1.40 Oil DIC objective).
Lysosome Preparation
Frozen slices were homogenized and an enriched lysosome fraction was prepared as per the Lysosome Enrichment Kit for Tissue and Cultured Cells (Pierce). Briefly, brain slices were homogenized in 1× v/w Lysosome Enrichment Reagent A containing protease inhibitors. An equal volume of Lysosome Enrichment Reagent B containing protease inhibitors was then added and the tube mixed by inverting. Samples were centrifuged at 500 × g for 10 min at 4°C. The supernatant obtained was brought to a final concentration of 15% OptiPrep™ Media, overlaid on top of a discontinuous density gradient and spun at 145,000 × g for 2 h at 4°C. Post ultracentrifugation, 400 μl fractions were removed from around and including the top visible band that had formed in the gradient of the ultracentrifuge tube and processed for removal of lysosomes as indicated in the manufacturer’s protocol (Pierce). Lysosome pellets were resuspended in 2× STOP buffer for western blot analysis. The enriched lysosome preparation was repeated multiple times (n = 6).
Western Blot
Enriched lysosome pellet fractions were resuspended in 2× STOP buffer (0.25 M Tris–HCl, pH 7.5, 2% SDS, 25 mM dithiothreitol, 5 mM EDTA, 5 mM EGTA, 10% glycerol, and 0.01% bromophenol blue), boiled and loaded onto 8 or 10% polyacrylamide gels. Membranes were probed with antibodies to LAMP2 (Abl93; Chen et al., 1985, gift from Dr. P. Mathews), IgG–FITC (200-032-037 Jackson Immunoresearch), or total tau (DAKO A0024). The blots were incubated with the appropriate peroxidase-conjugated goat anti-rat, anti-rabbit, or anti-mouse IgG (Jackson Immunoresearch). Immunoreactive bands were visualized and analyzed by enhanced chemiluminescence reagent (Thermo Scientific) using a Fujifilm LAS4000 imaging system and the Multi Gage software (Fujifilm Life Science, USA). Immunoblotting for LAMP2, IgG–FITC, and total tau was performed four separate times.
Results
Organotypic brain slices were prepared and used to determine the localization of our FITC labeled phos-tau antibody that had been added to the slice culture. Slices were co-stained with antibodies CP13, which recognizes tau phosphorylated at the Ser202 site (Weaver et al., 2000), and MC1, which detects a pathological conformation of tau that is present in AD brain (Jicha et al., 1997), and a Texas Red conjugated secondary antibody. Confocal microscopy images showed extensive but partial co-localization between FITC–IgG and the tau antibodies CP13 and MC1 (Figure 1). Furthermore, we determined that the FITC–IgG was completely associated with cellular markers of lysosomes, LAMP2, and early endosomes, Rab5, with perinuclear vesicles as the main areas of co-staining. In wild type mice, limited non-specific FITC–IgG binding was observed (Figure 1 bottom panel).
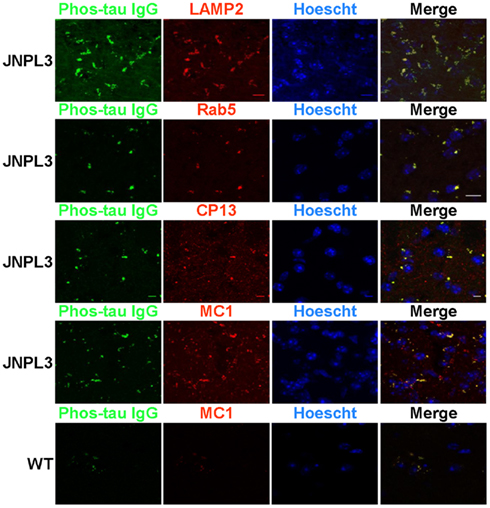
Figure 1. FITC labeled IgG from a high titer mouse co-localizes with phosphorylated and pathological tau within the endosomal/lysosomal system. Confocal microscope images of brain slice sections from JNPL3 transgenic mice and WT (bottom panel) mice. Slices were incubated with FITC–IgG from a high titer Tau 379–408[pSer396, 404] immunized mouse (green) and after sectioning co-stained with an antibody to LAMP2 (red), a marker of late endosomes/lysosomes, and an antibody to Rab5 (red), a marker of early endosomes and the nuclear stain Hoechst 33342. The merged images indicate areas of co-localization between FITC–IgG and endosomes and lysosomes (orange/yellow), mostly in perinuclear areas. Slices were also co-stained with an antibody to MC1 (red), which recognizes a disease related conformational tau epitope and CP13 (red) which recognizes tau pSer202. The merged images indicate areas of co-localization between FITC–IgG and pathological tau (yellow), mostly in perinuclear areas. Minimal staining was observed in the WT mouse. Scale bar = 10 μm.
A higher magnification confocal microscope image of brain slices from a JNPL3 mouse incubated with FITC labeled phos-tau antibody and co-stained with LAMP2 clearly shows the perinuclear regions of co-staining. The delineated shape of the cell has a neuronal morphology and indicates that neurons are capable of taking up our FITC labeled phos-tau antibody (Figure 2).
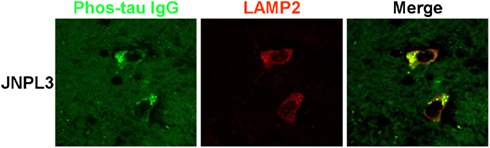
Figure 2. Neuronal co-localization of FITC–IgG and LAMP2. High magnification confocal microscope images of brain slice sections from a JNPL3 transgenic mouse. Brain slices were incubated with FITC–IgG from a high titer Tau 379–408[pSer396, 404] immunized mouse (green) and after sectioning co-stained with an antibody to LAMP2 (red), which is a marker of late endosomes and lysosomes. The merged image indicates areas of co-localization (green/yellow) between FITC–IgG and late endosomes/lysosomes, mainly in perinuclear areas. The neuronal morphology is clearly delineated by the regions of staining.
To examine antibody compartmentalization, we next prepared an enriched lysosome fraction from our FITC–IgG treated JNPL3 brain slices. Fractions were removed post separation and analyzed by western blot. Figure 3 indicates enriched lysosome fractions obtained from two separate mice, A and B respectively. Fractions (400 μl) were obtained from around and inclusive of the top visible band in the ultracentrifuge tube. Immunoblotting with an antibody to LAMP2, a marker of lysosomes, showed LAMP2 immunoreactivity in fractions A2, B1, B2, and B3. Fraction A1 is most likely only OptiPrep™ media without any lysosomes. Mouse kidney lysates were also run as a positive control. The kidney lysate is a whole tissue homogenate rather than a lysosome fraction (Figure 3). Immunostaining of the enriched lysosome fractions with an anti-FITC–IgG antibody showed more intense reactivity in fractions A2 and B1, indicating that our FITC conjugated antibody was taken up by the brain slices and present in the lysosome preparations (Figure 3). To test if the lysosome fractions also contained tau, the FITC–IgG blot was reprobed with an antibody to total tau (DAKO A0024). Lysosome fractions A2, B1, B2, and B3 were all positive for tau. Fraction A1 was weakly positive for tau, probably due to the fraction not being completely pure. And mouse kidney lysates were also tau positive since tau is known to be present in non-neuronal tissue (Gu et al., 1996; Figure 3).
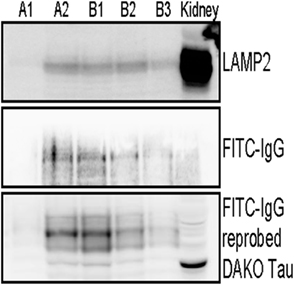
Figure 3. FITC labeled IgG from a high titer mouse and tau are present in enriched lysosome fractions. Immunoblotting analysis of enriched lysosome fractions from two separate JNPL3 mice brain preparations is shown, A and B respectively. Brain slices (400 μm) were incubated with FITC–IgG from a high titer Tau 379–408[pSer396, 404] immunized mouse for 2 h. Post treatment slices were processed to obtain an enriched lysosome fraction. Several fractions were obtained and the presence of lysosomes was confirmed in LAMP2 positive fractions A2, B1, B2, and B3. Mouse kidney lysate was used as a positive control. The kidney control is not a purified kidney preparation but a whole organ lysate, hence it has more background staining when compared to the enriched lysosome fractions from brain. Lysosome fractions were also immunoblotted for FITC–IgG. Fractions A2, B1, B2, and B3 were positive for FITC–IgG. To test if the lysosome fractions also contained tau, the FITC–IgG blot was reprobed with an antibody to total tau (DAKO A0024). Lysosome fractions A2, B1, B2, B3, and kidney lysate, all were positive for tau. Importantly, most of the FITC–IgG was detected in fractions with the highest levels of tau (A2 and B1). Overall, these findings are in accordance with the histological findings.
JNPL3 mice overexpress mutant tau (P301L) and develop extensive tau pathology (Lewis et al., 2000). A potential route involved in the clearance of pathological tau is the endosomal–lysosomal pathway, hence we next examined if markers of the lysosomal pathway were upregulated in these animals. Enriched lysosome fractions were prepared from wild type and JNPL3 brain slices. The presence of lysosomes was confirmed in LAMP2 positive fractions from both wild type and JNPL3 mice (Figure 4). The transgenic mice appeared to have more lysosomes than wild type mice. Additionally, immunoblotting of lysosome fractions with the total tau antibody showed the presence of tau in both wild type and transgenic mice, but as expected lysosomes from the latter group had much more tau than lysosomes obtained from wild type brain (Figure 4).
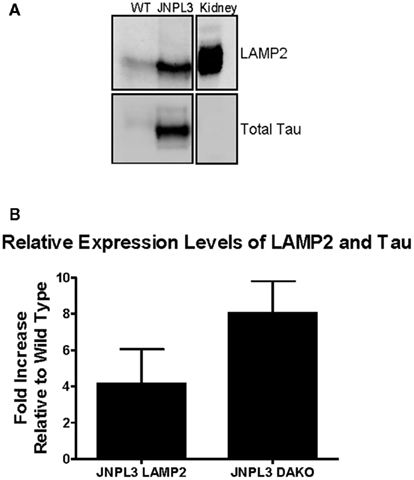
Figure 4. Non-immunized JNPL3 mice have more lysosomes than WT mice and these contain tau. Brain slices (400 μm) were processed to obtain an enriched lysosome fraction. (A) Representative immunoblot showing the presence of lysosomes (LAMP2) and tau (DAKO A0024) was confirmed in fractions from both WT and JNPL3 mice. Mouse kidney lysate was used as a positive control. (B) Quantification of immunoblots indicated that non-immunized JNPL3 mice had fourfold more lysosomes than WT mice, with corresponding eightfold higher amounts of tau (n = 3 separate experiments).
Discussion
The idea of using immunotherapy to treat neurodegenerative disorders such as AD is gaining more momentum. Several active and passive immunotherapy approaches are under investigation in clinical trials with the aim of accelerating Aβ clearance from the brain of AD patients. The most advanced of these immunological approaches is bapineuzumab, composed of a humanized anti-Aβ monoclonal antibody, which is being tested in a Phase III trial (Frisardi et al., 2010).
To date the mechanism by which immunomodulation can result in a clearance of tau pathology has not been elucidated. In fact a major caveat to accepting the validity of immunotherapy has been the notion that since the brain is an immunoprivileged site, it is not amenable to a vaccine based treatment. With particular respect to tau immunotherapy, it did not seem logical to many that antibodies could access intracellular proteins such as tau. The rationale for and potential mechanisms involved in antibody-mediated clearance of tau aggregates has been outlined in (Sigurdsson, 2008, 2009). Briefly, active or passive immunization targeting pathological tau should promote the clearance of tau aggregates and aid in restoring or saving neuronal function. To facilitate this, antibodies must first enter the CNS. In fact antibody entry into the CNS has been shown under normal conditions (Nerenberg and Prasad, 1975), and also antibodies can be actively transported via adsorptive mediated transcytosis (Zlokovic et al., 1990; Bickel, 1995). Additionally, the blood brain barrier (BBB) is known to be compromised in neurodegenerative diseases such as AD (Zlokovic, 1997), and we have detected increased permeability of the BBB in JNPL3 mice with advanced tau pathology (Asuni et al., 2007). It is also well established that neurons have several receptors that can bind IgG, which can then be internalized via receptor-mediated endocytosis (Sigurdsson, 2008, 2009). The antibodies may also diffuse through damaged membranes. Importantly, we previously reported that the same FITC–IgG preparation entered neurons within the brain and bound to pathological tau following injection into the carotid artery (Asuni et al., 2007). Once the tau antibodies have entered the brain and are internalized the most likely method of clearance of phosphorylated tau oligomers/aggregates would be via the endosomal-autophagy-lysosomal pathways. This is based on evidence from ultrastructural analysis of JNPL3 mice which has shown the presence of axonal spheroids containing filamentous tau as well as autophagic and lysosomal vacuoles (Lin et al., 2003), and the fact that antibodies have also been detected within lysosomes (Meeker et al., 1987).
The data from the present study shows that in a JNPL3 mouse brain slice model system, purified IgG from a mouse immunized with the phos-tau peptide (Tau 379–408[pSer396,404] is able to co-localize with tau species that are hyperphosphorylated and have a pathological conformation – CP13 and MC1 positive. There was no discernable MC1 or purified IgG immunostaining in wild type brain slices, indicating that our phos-tau immunized mice had developed antibodies to pathological tau species and the antibodies were preferentially internalized in neurons with tau aggregates. The brain slice model provides a unique system to help analyze antibody uptake, given the fact that we have circumvented the requirement for antibodies to cross the BBB.
The most likely mechanism for antibody-mediated clearance of large and probably long lived tau aggregates is via the endosomal–autophagy–lysosomal pathways, rather than the UPS (Sigurdsson, 2009). Thus when we stained JNPL3 brain slices with antibodies to Rab5 and LAMP2, markers of early endosomes and lysosomes, we clearly saw that our labeled purified IgG did in fact co-localize with endosomal and lysosomal organelles. We are currently exploring the potential involvement of autophagic pathways.
Proteasome activity has been found to be decreased in AD brain tissue (Keller et al., 2000). A potential reason for this was reported by Keck and colleagues. They showed that the inhibition of the endogenous proteasome system in AD brain tissue was due to the binding of PHF tau to the 20S core proteasome (Keck et al., 2003). We would expect the capacity of the proteasomal degradation pathway has likely been exceeded in the aged JNPL3 mice with extensive tau pathology, and the endosomal–autophagy–lysosomal pathways should be the preferred mechanism for removing tau aggregates, similar to what has been reported in an inducible tauopathy cell model (Wang et al., 2010). Also, the aggregates are too large to enter the proteasome (Sigurdsson, 2009). However, it is interesting in this context that high affinity antibody receptor on ubiquitin E4 ligase (TRIM21) has recently been identified (Mallery et al., 2010). This opens up the possibility that tau antibodies may facilitate clearance of soluble misfolded tau, which may be particularly relevant under prophylactic conditions to prevent further aggregation.
Our immunostaining data supports our idea that the clearance of tau aggregates can proceed via the endosomal–lysosomal system. This is also supported by ultrastructural analysis of two transgenic mouse lines. In the JNPL3 mouse model, electron microscopy analysis has shown lysosomal and autophagic vesicles to be associated with tau filaments (Lin et al., 2003), and in a transgenic mouse expressing four microtubule binding repeats and three FTDP-17 mutations, there was evidence of increased numbers of morphologically abnormal lysosomes (Lim et al., 2001). Additional evidence for the involvement of the endosomal–lysosomal pathway was seen in the biochemical preparations of enriched lysosomes from JNPL3 mouse slices treated with purified and FITC labeled IgG. Enriched lysosome fractions were positive for FITC–IgG as well as tau protein, and as anticipated, JNPL3 mice had more lysosomes containing tau than wild type mice. It is unlikely that FITC labeled IgG would be present in other enriched organelle fractions such as Golgi or mitochondrial fractions. There are no reports of IgG being detected in these fractions in contrast to lysosomes (Meeker et al., 1987). However, these are important controls and should be examined in future studies.
Recently, two studies reported that tau pathology could spread through different brain regions. Frost et al. (2009) showed that cultured neuronal cells could take up aggregated tau and these extracellular tau aggregates could induce fibril formation intracellularly. Additionally, they showed that the aggregated tau could be transmitted between co-cultured cells (Frost et al., 2009). How endocytosed extracellular aggregates can promote fibrillization of a cytoplasmic protein is not known. But we can envision that tau antibody treatment could readily bind to extracellular aggregates and promote their clearance in a manner similar to what has been proposed for antibodies targeting Aβ (Sigurdsson, 2009), and thus prevent the propagation of misfolded tau through the brain. Just as Frost and colleagues predicted, Clavaguera et al. (2009) demonstrated that injecting brain extract from a mouse with the FTDP-17 P301S tau mutation into the brain of a transgenic mouse expressing unmutated human tau, led to the formation of tau filaments and the spreading of pathology from one brain region to another. Again, we can only speculate at the mechanism responsible for the spreading of tau pathology. Mutant tau could potentially be acting as a seeding or nucleating factor for aggregates. Treatment with tau antibodies would again be beneficial as they could disrupt this event and so prevent the spread of tau pathology between different brain regions. The spread of amyloidosis within the affected organ is likely to be similar for all amyloid diseases (Sigurdsson et al., 2002). Hence, both tau and Aβ can be targeted with immunotherapy regardless of whether the main pool of aggregates is intra or extracellular.
Overall, based on the findings from our brain slice system we can confirm that the endosomal–lysosomal pathway is involved in antibody-mediated clearance of tau aggregates.
Conflict of Interest Statement
Patent pending on tau immunotherapy.
Acknowledgments
Supported by NIH grants AG032611, AG020197, the Alzheimer’s Association and the Alzheimer’s Drug Discovery Foundation/Association for Frontotemporal Degeneration. Funding for the Zeiss LSM710 confocal microscope was provided by grant NCRR S10 RR023704-01A1. We are extremely grateful to Prof. Peter Davies, Albert Einstein College of Medicine, New York, for providing the tau antibodies CP13 and MC1, and Dr. Paul Mathews, Nathan Kline Institute, Orangeburg, for providing LAMP2 and Rab5 antibodies.
References
Asuni, A. A., Boutajangout, A., Quartermain, D., and Sigurdsson, E. M. (2007). Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J. Neurosci. 27, 9115–9129.
Bickel, U. (1995). Antibody delivery through the blood-brain barrier. Adv. Drug Deliv. Rev. 15, 53–72.
Boimel, M., Grigoriadis, N., Lourbopoulos, A., Haber, E., Abramsky, O., and Rosenmann, H. (2010). Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp. Neurol. 224, 472–485.
Boutajangout, A., Ingadottir, J., Davies, P., and Sigurdsson, E. M. (2011). Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J. Neurochem. 118, 658–667.
Boutajangout, A., Quartermain, D., and Sigurdsson, E. M. (2010). Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J. Neurosci. 30, 16559–16566.
Chen, J. W., Murphy, T. L., Willingham, M. C., Pastan, I., and August, J. T. (1985). Identification of two lysosomal membrane glycoproteins. J. Cell Biol. 101, 85–95.
Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., Fraser, G., Stalder, A. K., Beibel, M., Staufenbiel, M., Jucker, M., Goedert, M., and Tolnay, M. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913.
Frisardi, V., Solfrizzi, V., Imbimbo, P. B., Capurso, C., D’introno, A., Colacicco, A. M., Vendemiale, G., Seripa, D., Pilotto, A., Capurso, A., and Panza, F. (2010). Towards disease-modifying treatment of Alzheimer’s disease: drugs targeting beta-amyloid. Curr. Alzheimer Res. 7, 40–55.
Frost, B., Jacks, R. L., and Diamond, M. I. (2009). Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 284, 12845–12852.
Gong, C. X., Grundke-Iqbal, I., and Iqbal, K. (2010). Targeting tau protein in Alzheimer’s disease. Drugs Aging 27, 351–365.
Gong, C. X., Lidsky, T., Wegiel, J., Grundke-Iqbal, I., and Iqbal, K. (2001). Metabolically active rat brain slices as a model to study the regulation of protein phosphorylation in mammalian brain. Brain Res. Brain Res. Protoc. 6, 134–140.
Gu, Y., Oyama, F., and Ihara, Y. (1996). Tau is widely expressed in rat tissues. J. Neurochem. 67, 1235–1244.
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., Pickering-Brown, S., Chakraverty, S., Isaacs, A., Grover, A., Hackett, J., Adamson, J., Lincoln, S., Dickson, D., Davies, P., Petersen, R. C., Stevens, M., De Graaff, E., Wauters, E., Van Baren, J., Hillebrand, M., Joosse, M., Kwon, J. M., Nowotny, P., Che, L. K., Norton, J., Morris, J. C., Reed, L. A., Trojanowski, J., Basun, H., Lannfelt, L., Neystat, M., Fahn, S., Dark, F., Tannenberg, T., Dodd, P. R., Hayward, N., Kwok, J. B., Schofield, P. R., Andreadis, A., Snowden, J., Craufurd, D., Neary, D., Owen, F., Oostra, B. A., Hardy, J., Goate, A., van Swieten, J., Mann, D., Lynch, T., and Heutink, P. (1998). Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705.
Ikeda, K., Akiyama, H., Arai, T., Kondo, H., Haga, C., Iritani, S., and Tsuchiya, K. (1998). Alz-50/Gallyas-positive lysosome-like intraneuronal granules in Alzheimer’s disease and control brains. Neurosci. Lett. 258, 113–116.
Jicha, G. A., Bowser, R., Kazam, I. G., and Davies, P. (1997). Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J. Neurosci. Res. 48, 128–132.
Keck, S., Nitsch, R., Grune, T., and Ullrich, O. (2003). Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 85, 115–122.
Keller, J. N., Hanni, K. B., and Markesbery, W. R. (2000). Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 75, 436–439.
Lemere, C. A., and Masliah, E. (2010). Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat. Rev. Neurol. 6, 108–119.
Lewis, J., Mcgowan, E., Rockwood, J., Melrose, H., Nacharaju, P., Van Slegtenhorst, M., Gwinn-Hardy, K., Paul Murphy, M., Baker, M., Yu, X., Duff, K., Hardy, J., Corral, A., Lin, W. L., Yen, S. H., Dickson, D. W., Davies, P., and Hutton, M. (2000). Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 25, 402–405.
Lim, F., Hernandez, F., Lucas, J. J., Gomez-Ramos, P., Moran, M. A., and Avila, J. (2001). FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and Tau filaments in forebrain. Mol. Cell. Neurosci. 18, 702–714.
Lin, W. L., Lewis, J., Yen, S. H., Hutton, M., and Dickson, D. W. (2003). Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J. Neurocytol. 32, 1091–1105.
Mallery, D. L., Mcewan, W. A., Bidgood, S. R., Towers, G. J., Johnson, C. M., and James, L. C. (2010). Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. U.S.A. 107, 19985–19990.
Masliah, E., Rockenstein, E., Adame, A., Alford, M., Crews, L., Hashimoto, M., Seubert, P., Lee, M., Goldstein, J., Chilcote, T., Games, D., and Schenk, D. (2005). Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 46, 857–868.
Masliah, E., Rockenstein, E., Mante, M., Crews, L., Spencer, B., Adame, A., Patrick, C., Trejo, M., Ubhi, K., Rohn, T. T., Mueller-Steiner, S., Seubert, P., Barbour, R., Mcconlogue, L., Buttini, M., Games, D., and Schenk, D. (2011). Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of lewy body disease. PLoS ONE 6, e19338. doi: 10.1371/journal.pone.0019338
Medina, M., and Avila, J. (2010). Glycogen synthase kinase-3 (GSK-3) inhibitors for the treatment of Alzheimer’s disease. Curr. Pharm. Des. 16, 2790–2798.
Meeker, M. L., Meeker, R. B., and Hayward, J. N. (1987). Accumulation of circulating endogenous and exogenous immunoglobulins by hypothalamic magnocellular neurons. Brain Res. 423, 45–55.
Mori, H., Kondo, J., and Ihara, Y. (1987). Ubiquitin is a component of paired helical filaments in Alzheimer’s disease. Science 235, 1641–1644.
Nerenberg, S. T., and Prasad, R. (1975). Radioimmunoassays for Ig classes G, A, M, D, and E in spinal fluids: normal values of different age groups. J. Lab. Clin. Med. 86, 887–898.
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., and Cuervo, A. M. (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122.
Petrucelli, L., and Dawson, T. M. (2004). Mechanism of neurodegenerative disease: role of the ubiquitin proteasome system. Ann. Med. 36, 315–320.
Piedrahita, D., Hernandez, I., Lopez-Tobon, A., Fedorov, D., Obara, B., Manjunath, B. S., Boudreau, R. L., Davidson, B., Laferla, F., Gallego-Gomez, J. C., Kosik, K. S., and Cardona-Gomez, G. P. (2010). Silencing of CDK5 reduces neurofibrillary tangles in transgenic Alzheimer’s mice. J. Neurosci. 30, 13966–13976.
Poorkaj, P., Bird, T. D., Wijsman, E., Nemens, E., Garruto, R. M., Anderson, L., Andreadis, A., Wiederholt, W. C., Raskind, M., and Schellenberg, G. D. (1998). Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 43, 815–825. [published erratum appears in Ann. Neurol. 1998 September; 44, 428].
Schenk, D., Barbour, R., Dunn, W., Gordon, G., Grajeda, H., Guido, T., Hu, K., Huang, J., Johnson-Wood, K., Khan, K., Kholodenko, D., Lee, M., Liao, Z., Lieberburg, I., Motter, R., Mutter, L., Soriano, F., Shopp, G., Vasquez, N., Vandevert, C., Walker, S., Wogulis, M., Yednock, T., Games, D., and Seubert, P. (1999). Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177.
Sigurdsson, E., Wisniewski, T., and Frangione, B. (2002). Infectivity of amyloid diseases. Trends Mol. Med. 8, 411.
Sigurdsson, E. M. (2008). Immunotherapy targeting pathological tau protein in Alzheimer’s disease and related tauopathies. J. Alzheimers Dis. 15, 157–168.
Sigurdsson, E. M. (2009). Tau-focused immunotherapy for Alzheimer’s disease and related tauopathies. Curr. Alzheimer Res. 6, 446–450.
Sigurdsson, E. M., Scholtzova, H., Mehta, P. D., Frangione, B., and Wisniewski, T. (2001). Immunization with a nontoxic/nonfibrillar amyloid-beta homologous peptide reduces Alzheimer’s disease-associated pathology in transgenic mice. Am. J. Pathol. 159, 439–447.
Tampellini, D., Magrane, J., Takahashi, R. H., Li, F., Lin, M. T., Almeida, C. G., and Gouras, G. K. (2007). Internalized antibodies to the Abeta domain of APP reduce neuronal Abeta and protect against synaptic alterations. J. Biol. Chem. 282, 18895–18906.
Wang, Y., Kruger, U., Mandelkow, E., and Mandelkow, E. M. (2010). Generation of tau aggregates and clearance by autophagy in an inducible cell model of tauopathy. Neurodegener. Dis. 7, 103–107.
Wang, Y., Martinez-Vicente, M., Kruger, U., Kaushik, S., Wong, E., Mandelkow, E. M., Cuervo, A. M., and Mandelkow, E. (2009). Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet. 18, 4153–4170.
Weaver, C. L., Espinoza, M., Kress, Y., and Davies, P. (2000). Conformational change as one of the earliest alterations of tau in Alzheimer’s disease. Neurobiol. Aging 21, 719–727.
Zlokovic, B. (1997). Can blood-brain barrier play a role in the development of cerebral amyloidosis and Alzheimer’s disease pathology. Neurobiol. Dis. 4, 23–26.
Keywords: tau, antibodies, immunotherapy, slice culture, endosomes, lysosomes
Citation: Krishnamurthy PK, Deng Y and Sigurdsson EM (2011) Mechanistic studies of antibody-mediated clearance of tau aggregates using an ex vivo brain slice model. Front. Psychiatry 2:59. doi: 10.3389/fpsyt.2011.00059
Received: 03 June 2011; Accepted: 06 October 2011;
Published online: 21 October 2011.
Edited by:
Ottavio Arancio, Columbia University, USAReviewed by:
Emmanuel Planel, Centre Hospitalier de lÕUniversitŽ Laval, CanadaAlejandra Alonso, The College of Staten Island, CUNY, USA
Gunnar K. Gouras, Weill Cornell Medical College, USA
Cynthia Lemere, Harvard University, USA
Copyright: © 2011 Krishnamurthy, Deng and Sigurdsson. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Einar M. Sigurdsson, Department of Physiology and Neuroscience, New York University School of Medicine, 550 First Avenue, MSB 459, New York, NY 10016, USA. e-mail:ZWluYXIuc2lndXJkc3NvbkBueXVtYy5vcmc=