 Armin P. Piehler1,2†
Armin P. Piehler1,2† Mustafa Özcürümez3† and Wolfgang E. Kaminski4*
Mustafa Özcürümez3† and Wolfgang E. Kaminski4*- 1 Fürst Medical Laboratory, Oslo, Norway
- 2 Department of Medical Biochemistry, Oslo University Hospital, Oslo, Norway
- 3 Bioscientia Institut für Medizinische Diagnostik GmbH, Medizinisches Versorgungszentrum Berlin, Berlin, Germany
- 4 Institute for Clinical Chemistry, Medical Faculty Mannheim, University of Heidelberg, Mannheim, Germany
The A-subclass of ATP-binding cassette (ABC) transporters comprises 12 structurally related members of the evolutionarily highly conserved superfamily of ABC transporters. ABCA transporters represent a subgroup of “full-size” multispan transporters of which several members have been shown to mediate the transport of a variety of physiologic lipid compounds across membrane barriers. The importance of ABCA transporters in human disease is documented by the observations that so far four members of this protein family (ABCA1, ABCA3, ABCA4, ABCA12) have been causatively linked to monogenetic disorders including familial high-density lipoprotein deficiency, neonatal surfactant deficiency, degenerative retinopathies, and congenital keratinization disorders. Recent research also point to a significant contribution of several A-subfamily ABC transporters to neurodegenerative diseases, in particular Alzheimer’s disease (AD). This review will give a summary of our current knowledge of the A-subclass of ABC transporters with a special focus on brain lipid homeostasis and their involvement in AD.
ABC Transporters
The superfamily of ATP-binding cassette (ABC) transporters constitutes a large group of highly conserved, multispan transmembrane transport proteins which can be found in all organisms from bacteria to man (Higgins, 1992; Holland et al., 2002). In most species, ABC transporters are abundantly represented (Linton and Higgins, 1998). In the human genome, 48 functional, protein-coding ABC transporter genes (Dean, 2005), and several ABC pseudogenes (Piehler et al., 2006, 2008) have been identified. All ABC proteins share a common global configuration and are composed of four specific core domains; two highly conserved ABCs and two transmembrane domains (TMDs; Figure 1; Higgins et al., 1986; Linton and Higgins, 1998). These four domains can be encoded in a single polypeptide chain, which is then called a “full-size” transporter (Figure 1). Alternatively, two homo- or heterodimers (“half-size” transporters), each containing one ABC and one TMD, attach to form the functional transporter (Dean, 2005). Based on phylogenetic analyses of the ABCs, human ABC transporters are divided into seven subfamilies, denoted ABC A–G (Dean, 2002). ABC transporters are primary active transporters using the energy from ATP-binding and -hydrolysis to translocate a wide variety of substrates including lipids, ions, sugars, peptides, amino acids, carbohydrates, vitamins, steroid hormones (Higgins, 1992; Holland et al., 2002), and xenobiotics, such as anticancer drugs (Gottesman et al., 2002). On a subcellular level, ABC transporters are located in eukaryotes in the plasma membrane and in the lipid membranes of the Golgi apparatus, endosomes, multivesicular bodies, endoplasmic reticulum, peroxisomes, and mitochondria (Kaminski et al., 2006). The function of ABC transporters in a broad range of physiological systems is also reflected by the fact that mutations in several ABC proteins result in monogenetic disorders affecting diverse physiological systems (Wenzel et al., 2007).
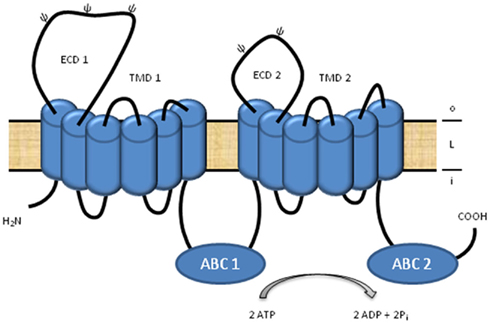
Figure 1. Schematic view of an A-subclass ABC transporter. A-subclass ABC transporters are full-size transporters and consist of two transmembrane domains (TMD), which anchor the transporter into a lipid membrane (L), and two ATP-binding cassettes (ABC), at which two molecules ATP are bound and hydrolyzed to support the energy for substrate translocation between the inside (i) and outside (o) of a cell or compartment. A special feature of the A-subfamily members is the unusually large, first extracellular domain (ECD) of each TMD.
Outstanding experts in the field have recently contributed with reviews of single members or subsets of ABC transporters covering diverse aspects of ABC transporters and their role in health and disease, including brain lipid metabolism and Alzheimer’s disease (AD; Kim et al., 2008; Mack et al., 2008; Hirsch-Reinshagen et al., 2009; Koldamova et al., 2010). In the present review, we focus on the A-subfamily of ABC transporters and their role in brain lipid transport and neurodegeneration.
The A-Subclass of ABC Transporters
In 1994, in an effort to clone members of the ABC transporter superfamily, Luciani et al. (1994) identified two novel ABC transporter genes, named ABC1 (ABCA1) and ABC2 (ABCA2), in close proximity to each other on chromosome 9. Due to their structural features that clearly set them apart from other ABC transporters, Luciani et al. suggested that these transporters define a novel subgroup of ABC transporters, the A-subfamily of ABC transporters. Altogether, the human ABCA subfamily comprises 12 protein-coding genes, ABCA1–ABCA13, with “ABCA11” representing a transcribed pseudogene (Kaminski et al., 2006; Piehler et al., 2008). All ABCA proteins are full-size transporters (Tusnady et al., 2006).
At least three characteristics define the members of the A-subfamily of ABC transporters: (i) the A-subfamily contains the largest members of the ABC transporter superfamily. According to their predicted primary structure, ABCA transporters are polypeptides ranging from 1543 aa (ABCA10) to 5058 aa (ABCA13) in size with a calculated molecular weight between 176 and 576 kDa (Kaminski et al., 2006). (ii) The extracellular domain between the first and second membrane spanning helix of each predicted TMD is particularly large in this subfamily (Kaminski et al., 2006). (iii) A highly hydrophobic domain in ABCA transporters interrupts the cytosolic linker that connects the two halves of an ABC transporter (Bungert et al., 2001; Vulevic et al., 2001; Peelman et al., 2003; Albrecht and Viturro, 2007). Most ABCA transporters show a broad tissue-specificity, including expression in brain (Kaminski et al., 2006), beside ABCA4, which is mainly expressed in the eye (Allikmets et al., 1997), and ABCA13 with detectable expression in only a small variety of tissues (Prades et al., 2002; Table 1).
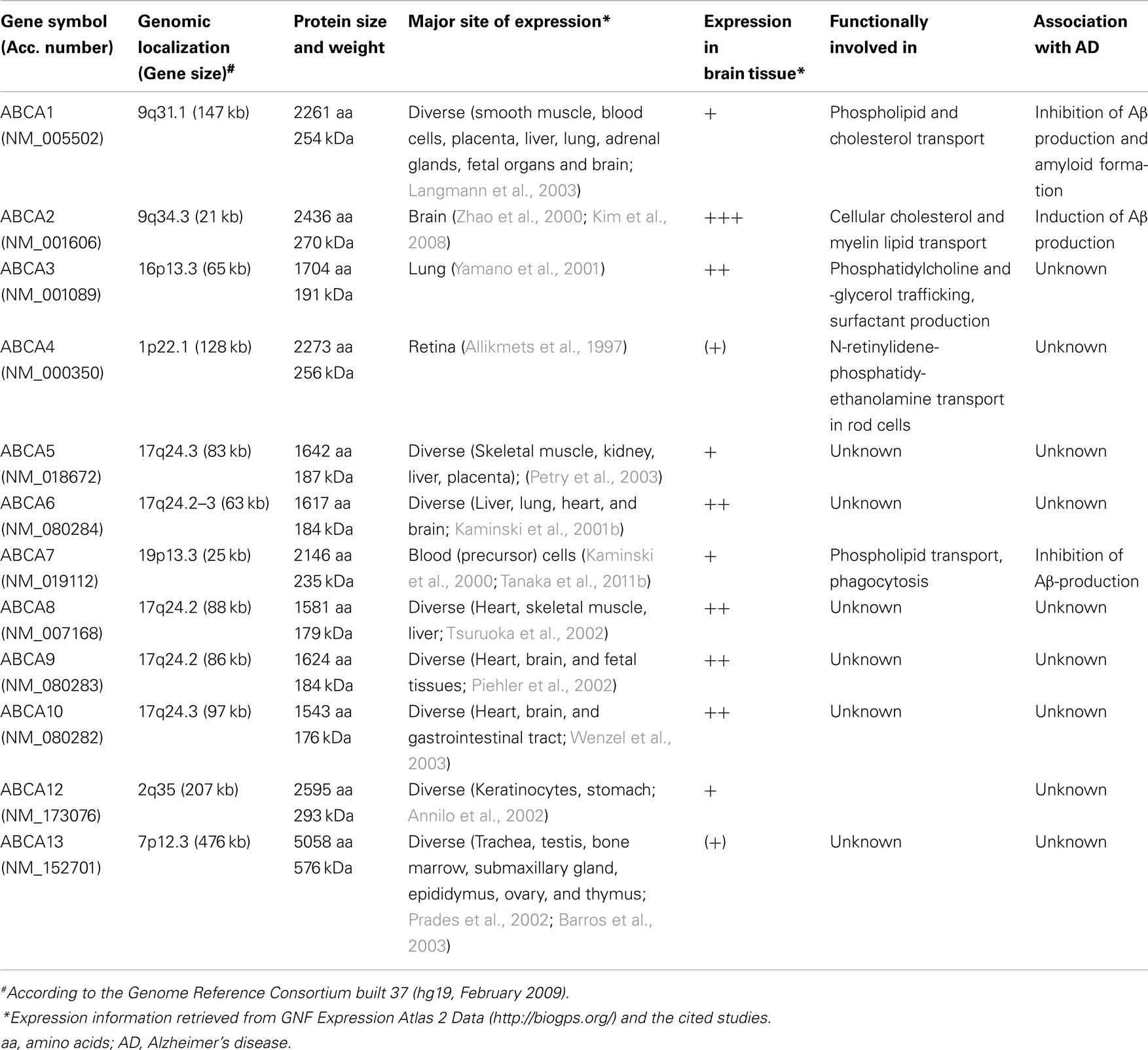
Table 1. Biological characteristics of A-subfamily ABC transporters.
Table 1 lists some of the biological properties of the members of the ABCA subfamily.
Phylogenetic analyses suggest that all ABCA subfamily transporters have evolved from a common ancestor gene (Kaminski et al., 2006) and can be further divided into two subgroups. The so-called “ABCA6-like” transporters comprise ABCA5, ABCA6, ABCA8, ABCA9, and ABCA10 and represent subgroup I (Piehler et al., 2002). These transporters form a compact gene cluster on chromosome 17q24 and are characterized by a strikingly high, mutual amino acid sequence identity and a significantly smaller size (between 1543 aa and 1642 aa) compared to the other ABCA proteins (Arnould et al., 2001; Piehler et al., 2002; Kaminski et al., 2006). The remaining seven ABCA transporters (ABCA1, ABCA2, ABCA3, ABCA4, ABCA7, ABCA12, and ABCA13) are included in subgroup II of the ABCA transporter subfamily and dispersed on six chromosomes (Dean, 2002).
The A-subfamily of ABC transporters has gained special focus over the last years as mutations in four members were identified as the underlying causes of monogenetic diseases in humans, including mutations in ABCA1 [high-density lipoprotein (HDL)-deficiency/Tangier disease (TD)], ABCA3 (neonatal surfactant deficiency), ABCA4 (several forms of autosomal recessive macular dystrophies), and ABCA12 (two forms of hereditary keratinization disorders; Kaminski et al., 2006). The phenotypes of these loss-of-function mutations have resulted in valuable information about the physiological function of ABCA transporters and identified them as lipid transporters. Over the last years, evidence has accumulated that members of the A-subfamily of ABC transporters are also involved in more complex diseases like atherosclerosis (ABCA1), pediatric interstitial lung diseases (ABCA3), age-related macular degeneration (ABCA4), and AD (ABCA1, ABCA2, ABCA7; Kaminski et al., 2006).
A-Subfamily ABC Transporters in Brain Lipid Homeostasis and Neurodegeneration
ABCA1
Molecular properties of ABCA1
ABCA1 is the prototypic member of the A-subfamily of ABC transporters. It has been extensively studied since its identification in 1994. In particular, the discovery in 1999 that mutations in this ABCA subfamily transporter compromise cellular cholesterol export has attracted a lot of interest from researchers worldwide. The role of ABCA1 in cholesterol transport, HDL particle formation and atherosclerosis has been reviewed previously (Kaminski et al., 2006; Wenzel et al., 2007; Kang et al., 2010; Nagao et al., 2011b), and detailed reports on the implication of ABCA1 in AD and neurodegeneration have been published recently (Kim et al., 2008; Hirsch-Reinshagen et al., 2009; Koldamova et al., 2010).
The gene encoding ABCA1 is located on chromosome 9q31.1 and comprises 50 exons which span 147 kb DNA. The 254 kDa protein of ABCA1 consists of 2261 amino acids and displays the typical structure of a full-size ABC transporter with two ATP-bindings cassettes and two TMDs. ABCA1 shows a broad expression pattern with highest expression levels in smooth muscle, whole blood, placenta, liver, lung, adrenal glands, fetal organs, and brain (Langmann et al., 1999; Su et al., 2004; Kim et al., 2008). On the subcellular level, ABCA1 is present in the plasma membrane, the Golgi compartment, and in lysosomes (Neufeld et al., 2001; Tanaka et al., 2003a).
ABCA1 and HDL metabolism
First clues for the involvement of ABCA1 in HDL metabolism came from our own studies demonstrating that ABCA1 expression is up-regulated by cholesterol uptake and down-regulated during HDL-mediated cholesterol efflux in human macrophages (Langmann et al., 1999). The identification of mutations in ABCA1 causing HDL-deficiency syndromes, including TD, confirmed the role of ABCA1 as a key regulator in cellular HDL metabolism (Bodzioch et al., 1999; Brooks-Wilson et al., 1999; Lawn et al., 1999; Rust et al., 1999). To date, more than 80 mutations within the ABCA1 gene have been described (for an overview see The Human Gene Mutation Database at http://www.hgmd.org). In TD patients, cholesterol accumulates in peripheral tissues like tonsils, spleen, liver, and the artery wall leading to premature atherosclerosis and hepatosplenomegaly. This accumulation is due to an impaired efflux of cellular cholesterol to its extracellular acceptor apolipoprotein A-I (apoA-I), the main apolipoprotein in HDL particles, by ABCA1 leading to a lack of HDL in the patient’s plasma (Bodzioch et al., 1999; Brooks-Wilson et al., 1999; Lawn et al., 1999; Rust et al., 1999). Further studies documented that both hepatic and intestinal ABCA1 contributes to HDL metabolism and that the liver is the predominant source of plasma HDL (Orso et al., 2000; Basso et al., 2003; Wellington et al., 2003; Timmins et al., 2005; Brunham et al., 2006).
Despite extensive efforts, the molecular mechanisms by which ABCA1 modulates the cellular cholesterol efflux and HDL biogenesis are still unclear. A current model suggests that the initial step is binding of small amounts of apoA-I, the main protein component of HDL particles, to the large, first extracellular domain of ABCA1 that leads to stabilization and activation of the transporter. Activation of ABCA1 triggers increased translocation of phospholipids from the cytosolic to the exofacial leaflet of the plasma membrane which subsequently bends and reveals high affinity binding sites for apoA-I in so-called exovesiculated lipid domains. Membrane bound apoA-I then passively accepts membrane phospholipids and cholesterol to form discoidal HDL particles (Vedhachalam et al., 2007a,b, 2010; Kang et al., 2010; Nagao et al., 2011a,b). An alternative model postulates that apoA-I is internalized after binding to ABCA1 and the resulting apoA-I/ABCA1 complex is then targeted to late endosomes where apoA-I binds lipids. These apolipoprotein–lipid complexes are subsequently released to the extracellular space by exocytosis (Oram, 2008; Yvan-Charvet et al., 2010). Evidence has accumulated to suggest that both mechanisms of ABCA1-dependent apoA-I loading with lipids exist. However, there is still an ongoing controversy as to which one is the dominant mechanism in HDL biogenesis.
ABCA1 in the CNS and alzheimer’s disease
The expression of ABCA1 in the brain was already noted by Luciani et al. (1994) describing the identification and cloning of ABCA1 from embryonic mouse brain RNA. Subsequent studies reported highest expression of ABCA1 in the brain regions of the olfactory bulb, hippocampus, cerebellar cortex, choroid plexus, and in germinal regions of embryonic and early postnatal brains (Fukumoto et al., 2002; Tachikawa et al., 2005). Experiments on isolated brain cells showed expression of ABCA1 in neurons, astrocytes, microglia, and oligodendrocytes, with highest expression levels in neurons and microglia (Koldamova et al., 2003; Kim et al., 2006). ABCA1 expression in brain capillary endothelial cells has also been reported (Panzenboeck et al., 2002; Ohtsuki et al., 2004).
Several observations led to the investigation of a potential link between ABCA1 and apoE: (i) ABCA1 is a major regulator of HDL biogenesis outside the CNS by facilitating cholesterol and phospholipid loading onto apoA-I (Kaminski et al., 2006).(ii) ApoE is the most abundant apolipoprotein in the CNS and present in HDL-like particles in the cerebrospinal fluid (Hayashi, 2011). (iii) The major apoE isoforms (apoE2, apoE3, and apoE4) are highly associated with disposition to and onset of AD (Hirsch-Reinshagen et al., 2009). (iv) Membrane cholesterol is known to regulate processing of amyloid precursor protein (APP) and generation of beta-amyloid (Aβ)-fibrils (Simons et al., 1998; Puglielli et al., 2003). Initial clues for an interdependency between apoE and ABCA1 came from experiments showing that Abca1 expression induced by liver X receptor (LXR) and retinoid X receptor (RXR) agonists increases efflux of cholesterol to lipid-free apoA-I and apoE3 in isolated mouse neurons, astrocytes, and microglia (Koldamova et al., 2003). Despite the fact that apoE is mainly secreted from astrocytes (Hayashi, 2011), further studies identified ABCA1-dependent loading of apoE with cholesterol as a mechanism by which cholesterol is targeted to neurons (Kim et al., 2007). Importantly, ABCA1 also appears to enhance the reverse process, the removal of cholesterol from neurons. Studies in ABCA1−/− mice finally documented the central role of ABCA1 in apoE lipidation and that intact ABCA1 is required for normal CNS apoE concentrations (Hirsch-Reinshagen et al., 2004; Wahrle et al., 2004). These studies demonstrated that ABCA1−/− mice had significantly reduced (about 80% reduction) apoE levels in the brain, CSF, and plasma. In addition, they revealed that the observed apoE reduction is specific as levels of apoJ, another major apolipoprotein in the brain, were unchanged (Hirsch-Reinshagen et al., 2004; Wahrle et al., 2004). Moreover, endogenous apoE particles that are secreted from ABCA1-deficient glia showed poor lipidation (Hirsch-Reinshagen et al., 2004; Wahrle et al., 2004). Interestingly, several independent groups were able to demonstrate that these poorly lipidated apoE particles enhance Aβ deposition in offsprings from Abca1−/− mice crossbred with four different murine models of AD (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005; Wahrle et al., 2005). Unexpectedly, the Abca1 null mutant offsprings exhibited increased Aβ immunoreactivity and amyloid load demonstrating that the poorly lipidated apoE particles from ABCA1-deficient glia enhance beta-amyloid deposition. None of these studies, however, found evidence for a significant contribution of Abca1 deficiency to APP processing or Aβ production in vivo strongly suggesting that Abca1 enhances amyloid formation indirectly via facilitation of apoE lipidation. Conversely, ABCA1 overexpression studies revealed that robust (>6-fold over endogenous expression) but not weak overexpression (about 50%) of ABCA1 results in decreased amyloid deposition (Hirsch-Reinshagen et al., 2007; Wahrle et al., 2008). Based on the findings that ABCA1 depletion results in increased amyloid deposition and ABCA1 induction shows a reciprocal effect and the fact that APP processing or Aβ production is not influenced by Abca1 depletion in vivo, the concept of the ABCA1–apoE pathway of Aβ clearance has been put forward. In this model, nascent apoE particles are secreted from astrocytes and microglia and are subsequently lipidated by ABCA1 forming discoidal lipid–apoE complexes. In a second step, these are further loaded with lipids by ABCG1 (and likely other ABC transporters) to form spherical, mature apoE-containing lipid particles present in the CNS. Finally, mature apoE–lipid particles bind Aβ and facilitate the cellular uptake and clearance of Aβ via a functional apoE-receptor (Figure 2; Hirsch-Reinshagen et al., 2009).
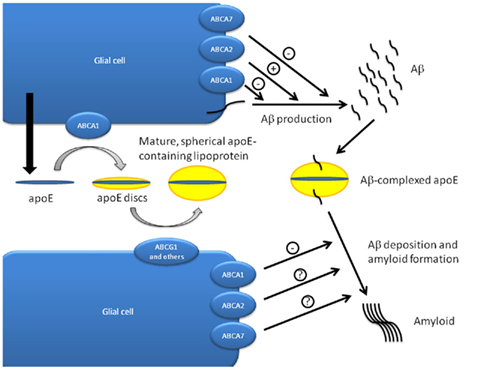
Figure 2. ABC transporters in apoE lipidation and Aβ metabolism. The ABCA1-apoE pathway of Aβ clearance hypothesis postulates that nascent apoE particles secreted from glial cells are initially lipidated by ABCA1 to form discoidal apoE-lipid particles. Further maturation and lipidation of these complexes by ABCG1 and presumably other ABC transporters finally results in mature, spherical apoE-containing lipoproteins. Both ABCA transporters and apoE have been implicated in the production, deposition, and clearance of Aβ. Knock-down and overexpression studies indicate that ABCA2 promotes Aβ production which is inhibited by ABCA1 and ABCA7. Amyloid formation is inhibited by ABCA1, and ApoE is required for Aβ deposition. ApoE also facilitates the cellular uptake of Aβ via apoE receptors and has a negative effect on Aβ clearance across the blood–brain barrier (Figure modified after Hirsch-Reinshagen et al., 2009; Hayashi, 2011).
Of note, a most recent study reports that transient expression of ABCA1 TD mutants in CHO cells stably expressing APP reduces Aβ production to a similar degree as wild-type ABCA1 but independently of cholesterol efflux (Kim et al., 2011). These findings, which challenge the proposed model of Aβ clearance via ABCA1-lipidated apoE, rather suggest a direct effect of the ABCA1 protein on Aβ production and amyloid formation.
Although the molecular mechanisms by which ABCA1 impacts Aβ production and amyloid deposition is not fully understood, a substantial body of evidence has accumulated during the past years to suggest critical roles of ABCA1 in the development of AD. In light of this, ABCA1 may be a promising candidate for therapeutical interventions aiming at the prevention and treatment of AD.
Association studies of ABCA1 and alzheimer’s disease
Beside these efforts to explore the functional implication of ABCA1 in amyloid deposition and development of AD, 11 studies have thus far investigated associations between single nucleotide polymorphisms (SNP) in the ABCA1 gene and the development of AD in various populations. Among the allelic variants identified in the ABCA1 gene, the SNPs rs2230806 (R219K), rs4149313 (I883M), and rs2230808 (R1587K) have been most extensively studied. At this point, however, the data from available association studies are inconclusive. This sobering resume is likely the result of major variations in the genetic and ethnic background of the cohorts investigated and varying experimental power of the studies. The 219K allele of the coding SNP rs2230806, for example, has been associated with both predisposition to AD (Rodriguez-Rodriguez et al., 2007; Sundar et al., 2007) and the opposite, a protective effect for the development of AD (Wollmer et al., 2003; Katzov et al., 2004; Wang and Jia, 2007; Wavrant-De et al., 2007; Reynolds et al., 2009). Other studies were unable to identify an association between common ABCA1 sequence variants and AD or delayed onset of AD (Li et al., 2004; Kolsch et al., 2006; Shibata et al., 2006; Wahrle et al., 2007). Although an anti-Alzheimer effect of the ABCA1 219K variant cannot be excluded at this point, further studies that rely on stringent design and strictly defined cohorts are necessary to explore the potential association between ABCA1 allelic variants and AD.
ABCA2
Molecular properties of ABCA2
Originally, ABCA2 was co-identified with ABCA1 in mice and is the second member of the A-subfamily of ABC transporters (Luciani et al., 1994). The human gene comprises 48 exons, is located on chromosome 9q34 and shows an extreme compact structure within a genomic region of only 21 kb (Kaminski et al., 2001a; Vulevic et al., 2001; Ile et al., 2004). The ABCA2 full-length cDNA is 7.3 kb in size and codes for a 270 kDa protein consisting of 2436 aa (Kaminski et al., 2001a; Vulevic et al., 2001). ABCA2 is mainly expressed in brain and nervous tissues with particularly high expression in cells facilitating myelin production in the brain (oligodendrocytes) and peripheral nerves (Schwann cells), respectively (Luciani et al., 1994; Zhao et al., 2000; Vulevic et al., 2001; Zhou et al., 2001, 2002; Langmann et al., 2003; Tanaka et al., 2003b; Su et al., 2004; Wang et al., 2005; Kim et al., 2006; Saito et al., 2007; Wahrle et al., 2008). Based on comparison of ABCA2 expression in astrocytic tumors and oligodendrogliomas, and in schwannomas and healthy Schwann cells, respectively, ABCA2 has also been proposed as a molecular marker for oligodendrogliomas and vestibular schwannomas (Wang et al., 2005; Soichi et al., 2007). However, ABCA2 has also been detected in a variety of other brain cells including neurons and brain endothelial cells (Ohtsuki et al., 2004; Broccardo et al., 2006; Kim et al., 2006; Warren et al., 2009; Shawahna et al., 2011), and other tissues outside the CNS including monocytes, macrophages, T-cells, thyroid gland, kidney, liver, thymus, heart, ovary, lung, and various tumor cells, respectively (Zhao et al., 2000; Kaminski et al., 2001a; Vulevic et al., 2001; Zhou et al., 2001, 2002; Ile et al., 2004; Broccardo et al., 2006). On a subcellular level, both electron microscopy and immunocolocalization experiments indicate a localization of ABCA2 around lysosomes and in the Golgi apparatus (Vulevic et al., 2001; Zhou et al., 2001, 2002). Ile et al. (2004) identified a novel 5′-localized exon of ABCA2 encoding an alternative N-terminus. Using Laser scanning confocal microscopy, the authors found that this novel isoform also co-localizes with lysosome-associated proteins-1 and -2 (LAMP1 and 2) and shows highest expression in peripheral blood leukocytes. In addition to this alternative exon 1-transcript, ABCA2 is transcribed at least into five alternative transcripts which are differentially expressed in different brain regions (Piehler et al., unpublished data).
ABCA2 and lipid transport in the CNS
Although the intracellular localization of ABCA2 is in agreement with the finding that ABCA2 overexpressing HEK293 cells do not stimulate cholesterol efflux to apoA-I, apoE, or apoE disks (Kim et al., 2007), several observations suggest a role of ABCA2 in brain cholesterol metabolism: (i) the putative promoter sequence of ABCA2 contains multiple potential transcription factor binding sites for neural cell differentiation (Kaminski et al., 2001a). (ii) ABCA2 transcription displays cholesterol–responsive regulation (Kaminski et al., 2001a; Davis Jr. et al., 2004; Davis, Jr., 2011) and (iii) ABCA2 is coordinately expressed with other sterol-dependent genes (Davis Jr. et al., 2004). (iv) ABCA2 overexpression in CHO cells leads to an increase in LDL receptor expression and other genes involved in cholesterol metabolism (Davis Jr. et al., 2004). (v) This results in a reduction of LDL-derived free cholesterol esterification and an accumulation of un-esterified LDL-cholesterol in the endosomal/lysosomal pathway (Davis Jr. et al., 2004), which is part of the processing of APP to Aβ. A recent study by Davis using N2a neuroblastoma cells, however, reported a decrease of the LDL receptor in response to ABCA2 overexpression and thus a decrease in the uptake of a fluorescent LDL analog (Davis, Jr., 2011). In this work, the author also shows that ABCA2 overexpression leads to a reduction in total, free- and esterified cholesterol levels in the neuronal cells overexpressing ABCA2 (Davis, Jr., 2011). In addition, a decrease in plasma membrane cholesterol was noted, but not in other organelles or lipid raft compartments. Moreover, de novo cholesterol synthesis or trafficking of cholesterol to the plasma membrane or the endoplasmic reticulum were unaffected (Davis, Jr., 2011). Together, these results clearly indicate a regulatory role of ABCA2 cholesterol metabolism within the cell.
Next to the studies documenting highest expression of ABCA2 in oligodendrocytes and Schwann cells, which facilitate myelination of neurons in the CNS and the peripheral nervous system, several experiments point to a role of ABCA2 in myelin lipid transport in addition to cholesterol homeostasis. Analysis of maturing central and peripheral nervous tissues revealed that temporal and spatial expression of ABCA2 was closely correlated with that of myelin sheath-associated proteins (Zhou et al., 2002; Tanaka et al., 2003b). To date, two independent groups have reported the generation of Abca2 deficient mice. In both studies, Abca2-null mice phenotypically displayed reduced body weight and an obvious distinct tremor of their limbs and were reported to be easily startled (Mack et al., 2007; Sakai et al., 2007a). In the study by Mack and colleagues, Abca2−/− mice exhibited ultrastructurally abnormal myelin sheathes with increased myelin sheath thickness in the spinal cord and a reduced periodicity of the myelin membrane both in the spinal cord and cerebrum. In contrast, no apparent change in total, esterified or free plasma cholesterol, or in total CNS tissue lipid composition (ceramide, sphingosine, or sphingomyelin species) were observed in the Abca2 deficient mice. Because female Abca2-null mice had a lower body weight compared to their male littermates, the authors suggest a hormone-dependent role of Abca2 in neurological development (Mack et al., 2007). Sakai et al. (2007a) observed no abnormalities in the cytoarchitectonic or compact myelin structure in their Abca2 knock-out mice, but significant differences in lipid concentrations of both total brain tissue and myelin fractions compared to wild-type animals. From 4 to 64 weeks of age, Abca2-null mice brains exhibited an accumulation of gangliosides along with reduced sphingomyelin, and an accumulation of cerebrosides and sulfatides at 64 weeks of age. Analysis of the brain of Abca2 knock-out mice revealed reduced sphingomyelin and a significant increase of the major ganglioside GM1. The latter finding is of particular interest as it has been shown that raised levels of gangliosides in brain tissue induce beta-amyloid fibril formation (Yanagisawa, 2007). In conclusion, functional studies from the past years corroborate an involvement of ABCA2 in brain lipid metabolism. However, further work is required to define in detail the molecular involvement of ABCA2 in neuronal cholesterol homeostasis and myelin lipid metabolism.
ABCA2 in alzheimer’s disease
Most recently, functional studies indicate a link between ABCA2 and the central molecular process in AD: beta-amyloid production. Using amplified differential gene expression, Chen et al. (2004) showed that overexpression of ABCA2 results in upregulation of genes commonly associated with oxidative stress and the pathogenesis of AD, including seladin-1, amyloid b (A4) precursor protein, vimentin, LDL receptor-related protein 3, Slc23a1, and calsarcin-1. Using confocal microscopy, the authors showed that increased ABCA2 levels impact the expression of Aβ and APP, and that ABCA2 co-localizes with both Aβ and APP in discrete intracellular vesicles that also stained positively for the endolysosome markers LAMP1 and LAMP2 (Chen et al., 2004). Further evidence for ABCA2 as a key regulator of APP metabolism has recently been provided by Davis showing that overexpression of ABCA2 in neuronal N2a cells increased both the transcription of APP and the amount of APP holoprotein, and promoted amyloidogenic processing of APP (Davis, Jr., 2011). These findings are in line with a most recent report demonstrating that knock-down/knock-out of ABCA2 in mammalian cells in vitro, in Drosophila melanogaster and in mice results in reduced production of Aβ (Michaki et al., 2011). In this study, ABCA2 depletion led to a relative shift from β- to α-secretase dependent cleavage of APP resulting in increased levels of APPsα and a relative decrease in APPsβ production. This observation computes well with the results by Davis reporting enhanced β-secretase cleavage of APP at the alternative cleavage site Glu11 (β′-site; Davis, Jr., 2011) which is increasingly used alternatively to the canonical Asp1 amino acid site (β-site) under conditions of β-secretase excess (Fluhrer et al., 2002; Liu et al., 2002). As ABCA2 is a likely regulator of cellular homeostasis and β-secretase acts within the detergent resistant domains (DRMs) of membranes (Wahrle et al., 2002; Ehehalt et al., 2003; Abad-Rodriguez et al., 2004; Vetrivel et al., 2004), depletion of ABCA2 may change DRM stabilization and thus the relative amount of APP processed by α- and β-secretase. Of note, Mickaki et al. also found in their study that ABCA2 depletion leads to a reduction of APP processing by γ-secretase. This was due altered γ-secretase complex formation which in turn was caused by aberrant glycosylation of Nicastrin, one of the components of γ-secretase (Michaki et al., 2011). Interestingly, the reduction of γ-secretase cleavage of APP by ABCA2 depletion occurred in a substrate-selective manner since processing of Notch, another important substrate of γ-secretase, was unaffected (Michaki et al., 2011).
Association studies of ABCA2 and alzheimer’s disease
In an effort to explore a potential association between genetic variation within ABCA2 and AD, two groups reported a strong correlation between a synonymous SNP (rs908832) and early-onset and sporadic AD in a Caucasian and a Western European population, respectively (Mace et al., 2005; Wollmer et al., 2006). In one study, however, the association was ethnicity-dependent and not present in a Southern European and an Asian population (Wollmer et al., 2006). In the same study, the authors reported an association of this SNP with cholesterol levels in the cerebrospinal fluid, a known determinant of AD (Wollmer et al., 2006). In contrast, Minster et al. (2008) could not to confirm the observed association of rs908832 with early onset AD (EOAD) or sporadic AD. Despite the rather limited cohort size of the EOAD cohort (137 EOAD patients, 1006 controls), the study had a power of >90% to detect the odd-ratios reported by Mace et al. and Wollmer et al. respectively. At this point, more work is required to establish whether a clinically relevant association does exist between AD and genetic ABCA2 variants.
Taken together, evidence has accumulated through the past years to suggest important roles of ABCA2 in intracellular brain cholesterol homeostasis and APP processing. Although the molecular basis of the interdependency of ABCA2 and cholesterol traffic and Alzheimer disease are currently not fully understood, the knowledge of altered secretase activity due to changes in ABCA2 activity and cholesterol homeostasis may represent a starting point for developing therapeutical approaches to AD.
ABCA7
Molecular properties of ABCA7
ABCA7 is a 220 kDa A-subfamily ABC transporter that exhibits highest sequence similarity to ABCA1 (54%) and ABCA4 (49%; Kaminski et al., 2000, 2006; Broccardo et al., 2001). Initially, ABCA7 was cloned from human macrophages (Kaminski et al., 2000), and shows highest expression in cells from the myelo-lymphatic lineage comprising both mature and blood cell precursors (Kaminski et al., 2000; Sasaki et al., 2003; Tanaka et al., 2011b). However, expression of ABCA7 in other tissues has also been reported, including keratinocytes and brain (Kaminski et al., 2000; Kielar et al., 2003; Kim et al., 2006; Sakai et al., 2007a). Specifically, in situ hybridization experiments revealed the presence of Abca7 mRNA throughout the brain of adult mice with highest expression in the densely packed neurons of the hippocampus (Kim et al., 2005), and RT-qPCR analysis of the different brain cell types and several cell lines derived from the CNS showed highest ABCA7 transcription levels in microglia (Kim et al., 2006). On the subcellular level, the localization of ABCA7 remains still unclear. Whereas initial experiments in HEK293 cells indicated the presence of ABCA7 in the plasma membrane (Wang et al., 2003), a subsequent study could not confirm this localization by immunofluorescence microscopy but suggested an intracellular presence in peritoneal mouse macrophages (Linsel-Nitschke et al., 2005). Several studies have thus far reported both intracellular detection of ABCA7 and its localization in the plasma membrane (Ikeda et al., 2003; Sasaki et al., 2003; Abe-Dohmae et al., 2004; Iwamoto et al., 2006). The putative presence of ABCA7 both in the cell and on the cell surface may be explained by the observation that ABCA7 is transcribed into several alternative variants (Ikeda et al., 2003; Kaminski et al., 2006).
The elusive function of ABCA7
Despite its identification more than 10 years ago, the definite molecular function of ABCA7 still needs to be elucidated. Several lines of evidence point to a role in cellular lipid transport: (i) ABCA7 shares highest sequence identity with ABCA1, the main molecule for cholesterol export to apoA-I to produce HDL particles (Kaminski et al., 2006), (ii) ABCA7 is up-regulated upon cholesterol load of macrophages and down-regulated upon cholesterol efflux from these cells (Kaminski et al., 2000), (iii) the ABCA7 promoter contains sterol responsive elements and is regulated by SREPBs (Iwamoto et al., 2006) and (iv) other A-subfamily members, of which the substrate has been identified, serve as transporters of lipids (ABCA1, ABCA3, ABCA4, ABCA12; Kaminski et al., 2006). Results from studies investigating apoA-I mediated release of phospholipids and cholesterol by ABCA7 under different conditions, however, are conflicting. Whereas some reports suggest that cells overexpressing ABCA7 exhibit increased apolipoprotein-mediated efflux of both phospholipids and cholesterol (Ikeda et al., 2003; Abe-Dohmae et al., 2004; Hayashi et al., 2005), data from other experiments could not confirm that ABCA7 facilitates the release of cholesterol from cells (Wang et al., 2003; Abe-Dohmae et al., 2004; Linsel-Nitschke et al., 2005). The situation is complicated by the observations that peritoneal macrophages treated with ABCA7 siRNA and from heterozygous ABCA7+/− mice showed unaffected apoA-I mediated cellular cholesterol and phospholipid release (Linsel-Nitschke et al., 2005). Moreover, Abca7 null mice, which have been reported to be lethal by one group (Jehle et al., 2006), showed unchanged cholesterol and phospholipid efflux levels in bone marrow-derived macrophages investigated by another group (Kim et al., 2005). Abca7 knock-out mice showed no obvious phenotypic abnormalities, but surprisingly exhibited a gender dependent reduction in serum total cholesterol, HDL concentration and visceral fat (Kim et al., 2005). At this point, further work is required to establish how and to which degree ABCA7 contributes to human lipid trafficking.
ABCA7 in CNS lipid transport and alzheimer’s disease
Based on the notion that ABCA7 is expressed in the brain and potentially functions as a lipid transporter, Chan et al. (2008) investigated whether ABCA7 regulates cholesterol efflux to apoE, the major apolipoprotein in the brain, and whether it influences beta-amyloid production. They found that overexpression of ABCA7 stimulated cholesterol efflux to discoidal apoE-lipid complexes independent of the apoE isoform. Lipid-free apoE did not accept cholesterol suggesting that apoE disks present in the CNS may function as acceptors for ABCA7-mediated cholesterol efflux. Moreover, the authors noted that ABCA7 significantly inhibited beta-amyloid secretion from human amyloid precursor protein expressing cells. Using fluorogenic substrates and GFP-tagged APP they demonstrated that the observed inhibition was due to an apparent retention of APP in a perinuclear location rather than an inhibitory effect on α-, β-, or γ-secretase activity (Chan et al., 2008).
Along with the sustained efforts to elucidate the role of ABCA7 in cellular lipid transport, evidence has accumulated during the past years that links another potential function of this transporter to neurodegenerative disease. Recent reports indicate a central role of ABCA7 in phagocytosis and the engulfment of apoptotic cells (Iwamoto et al., 2006; Jehle et al., 2006; Tanaka et al., 2010, 2011a,b). In addition to the finding that ABCA7 mRNA and protein is up-regulated during phagocytosis via the SREBP2 pathway (Iwamoto et al., 2006), these studies showed that knock-down of ABCA7 results in a decreased phagocytic activity (Iwamoto et al., 2006) and that heterozygous ABCA7+/− mice exhibit a defective clearance of apoptotic cells (Jehle et al., 2006). Both the phagocytic rate and the expression of ABCA7 are increased in ABCA1-deficient fibroblasts (Bared et al., 2004; Iwamoto et al., 2006). In contrast, the phagocytic activity is reduced in the peritoneal cavity of ABCA7−/− mice compared to wild-type animals (Tanaka et al., 2010). These studies also indicate that apoA-I and apoA-II both stabilize and increase surface ABCA7 and thus increases the phagocytic rate rather than contribute to lipid efflux from the cell. This stimulation of phagocytosis was sustained in ABCA1 siRNA treated J774 macrophages and in the peritoneal macrophages from ABCA1−/− mice (Tanaka et al., 2010). Another study by the same group also demonstrated that statin treatment of J774 cells induces ABCA7 expression by its cholesterol-lowering effect and simultaneously enhances phagocytosis. This increase in phagocytic activity was abolished by ABCA7 siRNA treatment of the statin stimulated cells (Tanaka et al., 2011a).
The finding that ABCA7 promotes Fc receptor-independent phagocytosis is of particular interest taking into account that human microglia cells contribute to the phagocytic removal of apoptotic debris from the brain (Stolzing and Grune, 2004; Napoli and Neumann, 2009) and given that among brain cells microglia displays highest ABCA7 expression. Based on this, it is tempting to speculate that microglial ABCA7 has an active role in the Fc receptor-independent phagocytic uptake of debris and plaques generated in the process of neurodegenerative disorders.
Moreover, the recent demonstration of a T cell receptor (TCR)-based machinery for specific immune recognition in macrophages that functions as a modulator of phagocytic capacity (Beham et al., 2011) raises the intriguing possibility of a functional link between ABCA7 and the novel variable macrophages immune system. It will be most challenging to investigate whether and to which degree an interdependencies exists between ABCA7 and the macrophage TCR-like immunoreceptors in the pathogenesis of macrophage-dependent neurodegenerative diseases such as multiple sclerosis and HIV dementia.
Association studies of ABCA7 and alzheimer’s disease
Next to functional studies, independent evidence for roles of ABCA7 in brain lipid homeostasis and neurodegeneration comes from a recent genome-wide association study (GWAS) and re-analysis of combined GWAS datasets which identified a candidate locus for AD in the ABCA7 gene (Jones et al., 2010; Hollingworth et al., 2011). The hot spot, which is defined by a common genetic variant in the ABCA7 gene (rs3764650) located in intron 13, exhibited a strong association with this disease. Also a non-synonymous SNP in ABCA7 exon 32 (rs3752246) which showed the highest linkage disequilibrium with rs3764650 was found to be significantly associated with LOAD (Hollingworth et al., 2011). However, as rs3752246 encodes a glycine to alanine substitution at position 1527 of the protein, which is regarded as a benign change, this SNP is unlikely to be the relevant functional variant. The authors also excluded an association between rs3764650 and the expression of ABCA7 analyzing data from two expression quantitative trait loci (eQTL) datasets. Although these studies present compelling evidence for an association of ABCA7 with AD, the genetic mechanisms that define a causative interrelationship still await elucidation.
Other A-subfamily ABC Transporters
Beside the above ABC transporters, other A-subfamily members are expressed only to a limited degree in brain and nervous tissues. Anecdotic reports on potential functions of these are rare, but mainly point to an involvement in cellular lipid metabolism. Future detailed studies will help to determine whether these A-subfamily transporters are implicated in brain lipid homeostasis and the pathophysiology of neurodegenerative processes. The following subchapter provides a brief synopsis of our current knowledge on additional members of the A-subfamily of ABC transporters.
ABCA3
ABCA3 is mainly expressed in the human lung alveolar type II cells on the limiting membrane of lamellar bodies, which represent the storage form of lung surfactant. Mutations in ABCA3 have been shown to cause fatal surfactant deficiency in full-term newborns (Shulenin et al., 2004) and are associated with milder forms of interstitial lung disease as well (Bullard et al., 2005; Kunig et al., 2007; Karjalainen et al., 2008; Yokota et al., 2008). The phenotype of surfactant deficiency caused by mutations in ABCA3 has been confirmed in ABCA3 knock-out mice by several groups (Ban et al., 2007; Cheong et al., 2007; Fitzgerald et al., 2007; Hammel et al., 2007). Although the exact molecular contribution of ABCA3 to surfactant production is still unknown, an essential role of this transporter in lipid transport, in particular in phosphatidylcholine and phosphatidylglycerol trafficking, during lamellar body formation has been established. Expression of ABCA3 has also been detected in total brain tissue (Fitzgerald et al., 2007; Stahlman et al., 2007), oligodendrocytes, neurons, astrocytes, and microglia (Kim et al., 2006), a tissue expression pattern that is similar to that of ABCA2 in the developing mouse brain (Tachikawa et al., 2005). Given the expression of ABCA3 in the brain and the fact that phosphatidylcholine and phophatidylglycerol are the lipid compounds mainly affected in the ABCA3-defective lung, it appears realistic to assume roles of ABCA3 in the transport of these lipids in the brain. However, studies investigating this function are currently unavailable. In detail examination of ABCA3 deficient mouse brains will help to gain further insight into a possible role of ABCA3 in brain lipid homeostasis.
ABCA4
ABCA4 is one of the best characterized A-subclass ABC transporters. Already in 1997 it was shown that mutations in this retina-specific transporter, also termed ABCR, cause Stargadt disease. This degenerative eye-disease is characterized by progressive impairment of central vision early in life, presence of lipofuscin deposits in the central retina, and bilateral photoreceptor atrophy in the macula (Allikmets et al., 1997; Molday, 2007). Also the rare, but more severe cone-rod dystrophy type 3 and retinitis pigmentosa type 19 have been shown to be caused by mutations in ABCA4 (Cremers et al., 1998; Martinez-Mir et al., 1998; Maugeri et al., 2000). Extensive studies of this transporter strongly suggest that ABCA4 facilitates the export of the phospholipid N-retinylidenephosphatidylethanolamine, a byproduct of the visual cycle, from the lumen to the cytoplasm of disk membranes in rod cells (Sun et al., 1999; Weng et al., 1999; Ahn et al., 2000; Mata et al., 2000; Beharry et al., 2004; Molday, 2007). Depletion of ABCA4 activity results in accumulation of retinoids in disk membranes and subsequent retinal pigment epithelial cell death and neurodegeneration of photoreceptors, a process that ultimately leads to severe vision loss in affected individuals (Weng et al., 1999; Mata et al., 2000; Molday, 2007). ABCA4 is also expressed outside the retina in rat choroid plexus epithelial cells and human brain capillary endothelial cells (Ohtsuki et al., 2004; Bhongsatiern et al., 2005; Tachikawa et al., 2005) suggesting a role of this ABC protein in blood-cerebrospinal fluid barrier function. Because ABCA4 dysfunction leads to accumulation of retinoids in the retinal pigment epithelium, it may be worthwhile investigating whether ABCA4 present in the choroid plexus is involved in the regulation of retinoid concentrations in the CNS which are associated with motor neuron disease and AD (Maden, 2007).
ABCA6-like transporters
Only little is known about the function of the subgroup of ABCA6-like transporters which form a compact gene cluster located on chr 17q24.2-3. This cluster comprises the transporters ABCA5, ABCA6, ABCA8, ABCA9, and ABCA10, respectively. Although all ABCA6-like transporters are expressed at detectable levels in the brain (Nagase et al., 1998; Arnould et al., 2001; Kaminski et al., 2001b, 2006; Piehler et al., 2002; Tsuruoka et al., 2002; Langmann et al., 2003; Wenzel et al., 2003, 2007; Kim et al., 2006) and it is likely that they are involved in lipid transport processes, their potential implication in human brain lipid homeostasis and neurodegeneration remains purely speculative at this point.
Reports on ABCA5 expression and function are anecdotic and the sites of expression appear to be highly diverse. Interestingly, ABCA5 expression has been documented mainly in the context of epithelial malignancies including prostate cancer (Hu et al., 2007), adenocarcinoma (Ohtsuki et al., 2007), melanomas (Heimerl et al., 2007), esophageal carcinoma (Huang et al., 2009), mesothelioma (Shukla et al., 2010), and oral squamosa cell carcinoma (Cha et al., 2011), respectively. Other studies report high ABCA5 expression in the brain, lung, heart, thyroid gland, and testis (Kubo et al., 2005; Petry et al., 2006). Upregulation of ABCA5 in human brain microvascular endothelial cells derived from the blood–brain barrier endothelium in response to tacrolimus treatment has also been reported (Quezada et al., 2008). On the subcellular level, ABCA5 localizes to lysosomes and late endosomes, and Abca5−/− mice exhibit exophthalmos and a collapse of the thyroid gland (Kubo et al., 2005). In adulthood, these animals develop a dilated cardiomyopathy-like heart and die due to cardiac insufficiency (Kubo et al., 2005). Despite its expression in the brain and its potential involvement in cholesterol metabolism (Ye et al., 2008, 2010), it appears unlikely that ABCA5 plays a crucial role in brain lipid homeostasis since overt abnormalities are not observed in the brain of ABCA5 deficient mice (Kubo et al., 2005).
ABCA6, ABCA9, and ABCA10 are also expressed at detectable levels in the brain and regulated during monocyte differentiation and cholesterol transport in human macrophages (Kaminski et al., 2001b, 2006; Piehler et al., 2002; Wenzel et al., 2003; Albrecht and Viturro, 2007) suggesting roles of these transporters in lipid transport. However, definitive evidence supporting an implication of these highly homologous transporters in brain lipid transport is still outstanding.
Initial studies have indicated a role of ABCA8 in drug transport with a substrate specificity close to that of ABCC2 which is located at the blood–brain barrier (Tsuruoka et al., 2002). Moreover, Abca8a transcription is induced in the mouse liver upon acute digoxin intoxication (Wakaumi et al., 2005). A recent study by Reppe et al. (2010) also suggests an association between ABCA8 and bone mineral density in postmenopausal Caucasian women.
ABCA12
ABCA12 was co-identified with ABCA13 in 2002 and mapped to the locus linked to lamellar ichthyosis on chromosome 2q34 (Parmentier et al., 1999; Annilo et al., 2002). Subsequent studies revealed that mutations in this transporter cause three related hereditary skin diseases: lamellar ichthyosis type 2 (Lefevre et al., 2003), non-bullous congenital ichthyosiform erythroderma (Natsuga et al., 2007; Akiyama et al., 2008) and harlequin ichthyosis (HI; Akiyama et al., 2005; Kelsell et al., 2005; Akiyama, 2006). All three entities represent hereditary dyskeratinization disorders, but present with different clinical severity. Whereas lamellar ichthyosis type 2 is a relatively mild form of dyskeratinization with skin desquamation over the whole body and large, pigmented scales (Lefevre et al., 2003), HI presents with a distorting phenotype in which the patient’s body is covered with an enormous horny shell with deep fissures leading to water loss, electrolyte abnormalities, severe infections, and mostly death within the first days after birth (Unamuno et al., 1987; Akiyama et al., 2005; Akiyama, 2006). Analysis of the mutations identified in the different dyskeratinization entities suggests that the severity of the phenotype correlates with the remaining activity of the mutated ABCA12 protein product. The molecular correlate of these dyskeratinization disorders is the absence or malformation of so-called lamellar granules (LG; Akiyama et al., 2005; Kelsell et al., 2005) which physiologically contribute to assemble the skin barrier by extruding their lipid content into the extracellular space during the keratinization process (Wertz, 2000). Since mutations in ABCA12 result in an abnormal distribution pattern of glucosylceramide, a major lipid component of LG (Kelsell et al., 2005; Akiyama et al., 2008), and ABCA12 ultrastructurally co-localizes with glucosylceramide to LG on their way from the Golgi apparatus to the cell periphery (Sakai et al., 2007b), it is realistic to assume an essential role of ABCA12 in intracellular sphingolipid transport processes (Akiyama, 2011). Of note, expression of ABCA12 has also been reported in fetal brain (Annilo et al., 2002). It remains to be established, however, whether ABCA12 exerts functions in the regulation of the sphingolipid metabolism in the developing brain.
ABCA13
ABCA13 is the largest known ABC transporter with a gene size of 450 kb comprising 62 exons which code for a 576-kDa polypeptide of 5058 aa and with an exceptionally large extracellular domain of more than 3500 aa (Prades et al., 2002; Barros et al., 2003). In normal tissues, highest mRNA expression was found in human trachea, testis, bone marrow, submaxillary gland, epididymis, ovary, and thymus (Prades et al., 2002; Barros et al., 2003). High expression in the brain tumor cell line SNB-19 has also been reported (Prades et al., 2002). The molecular function of ABCA13 is currently unknown. Intriguingly, a recent report points to an association between genetic variants of ABCA13 and schizophrenia, bipolar disorder, and depression (Knight et al., 2009). In this study, the population attributable risk of the identified mutations was 2.2% for schizophrenia and 4.0% for bipolar disorder. However, a more recent report failed to confirm this observation (Dwyer et al., 2011).
Non A-Subfamily ABC Transporters
Next to the A-subfamily ABC transporters reviewed above, several studies point to roles of ABC transporters outside this subfamily in brain lipid homeostasis and neurodegeneration (Kim et al., 2008). ABCB1, also known as MDR1 and P-glycoprotein, is one of the best characterized ABC transporters. It functions mainly as an exporter of amphipathic molecules such as anticancer drugs that confer multidrug resistance to tumor cells (Sarkadi et al., 2006). Transport of lipids (phosphatidylcholine and ethanolamine, glycosylceramide, and sphingomyelin) by ABCB1 has also been reported (van Helvoort et al., 1996). Despite its overall low expression in human brain (Langmann et al., 2003), its presence and functional implication in the blood–brain barrier have been well documented (Cordon-Cardo et al., 1989; Tatsuta et al., 1992; Tishler et al., 1995; Seetharaman et al., 1998; ElAli and Hermann, 2011; Vogelgesang et al., 2011). Importantly, recent studies indicate a central role of ABCB1 in the efflux of Aβ from the brain (Lam et al., 2001; Vogelgesang et al., 2004, 2011; Cirrito et al., 2005; Kuhnke et al., 2007; Hartz et al., 2010) which is reduced by approximately 30% in AD patients compared to healthy individuals (Mawuenyega et al., 2010). In addition to ABCB1, additional studies strongly suggest the involvement of the ABC proteins ABCA1, ABCC1, and ABCG2, respectively, in the export of Aβ (Xiong et al., 2009; Koldamova et al., 2010; Krohn et al., 2011). Finally, another G-subfamily ABC transporter, designated ABCG1, has been implicated in the processing of APP to generate Aβ peptides (Sarkadi et al., 2006; Kim et al. (2007). Each of these transporters may represent a potential pharmaceutical target for therapeutic interventions aiming at the reduction of Aβ accumulation and the prevention of AD progression.
Conclusion
ATP-binding cassette transporters constitute an evolutionary ancient group of large proteins which mediate the transmembrane transport of a diverse spectrum of substrates. Specifically, the members of the recently identified A-subfamily of ABC transporters serve as key regulators of cellular lipid transport processes. Given that the central nervous system, next to adipose tissue, represents the second major lipid rich area in higher organisms, it is reasonable to assume that A-subfamily ABC transporters are of critical importance for the integrity of the CNS. In fact, recent evidence links A-subfamily transporters and also other members of the ABC protein family to the maintenance of brain lipid homeostasis and neurodegenerative diseases. In particular, the transporters ABCA1, ABCA2, and ABCA7 are promising research targets that bear significant therapeutic potential for the treatment of neurodegenerative disease. Whereas ABCA7 appears to inhibit Aβ production, a positive effect on Aβ production has been reported for ABCA2. Moreover, available evidence suggests that ABCA1 acts as a suppressor of Aβ production and deposition. Recent experiments involving ABCA1 inducing LXR agonists have shown promising, antiamyloidogenic effects and thus highlight the importance of ACBA1 as a promising drug target in combating AD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abad-Rodriguez, J., Ledesma, M. D., Craessaerts, K., Perga, S., Medina, M., Delacourte, A., Dingwall, C., De, S. B., and Dotti, C. G. (2004). Neuronal membrane cholesterol loss enhances amyloid peptide generation. J. Cell Biol. 167, 953–960.
Abe-Dohmae, S., Ikeda, Y., Matsuo, M., Hayashi, M., Okuhira, K., Ueda, K., and Yokoyama, S. (2004). Human ABCA7 supports apolipoprotein-mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J. Biol. Chem. 279, 604–611.
Ahn, J., Wong, J. T., and Molday, R. S. (2000). The effect of lipid environment and retinoids on the ATPase activity of ABCR, the photoreceptor ABC transporter responsible for Stargardt macular dystrophy. J. Biol. Chem. 275, 20399–20405.
Akiyama, M. (2006). Pathomechanisms of harlequin ichthyosis and ABCA transporters in human diseases. Arch. Dermatol. 142, 914–918.
Akiyama, M. (2011). The roles of ABCA12 in keratinocyte differentiation and lipid barrier formation in the epidermis. Dermatoendocrinol. 3, 107–112.
Akiyama, M., Sakai, K., Hatamochi, A., Yamazaki, S., McMillan, J. R., and Shimizu, H. (2008). Novel compound heterozygous nonsense and missense ABCA12 mutations lead to nonbullous congenital ichthyosiform erythroderma. Br. J. Dermatol. 158, 864–867.
Akiyama, M., Sugiyama-Nakagiri, Y., Sakai, K., McMillan, J. R., Goto, M., Arita, K., Tsuji-Abe, Y., Tabata, N., Matsuoka, K., Sasaki, R., Sawamura, D., and Shimizu, H. (2005). Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J. Clin. Invest. 115, 1777–1784.
Albrecht, C., and Viturro, E. (2007). The ABCA subfamily – gene and protein structures, functions and associated hereditary diseases. Pflugers Arch. 453, 581–589.
Allikmets, R., Singh, N., Sun, H., Shroyer, N. F., Hutchinson, A., Chidambaram, A., Gerrard, B., Baird, L., Stauffer, D., Peiffer, A., Rattner, A., Smallwood, P., Li, Y., Anderson, K. L., Lewis, R. A., Nathans, J., Leppert, M., Dean, M., and Lupski, J. R. (1997). A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 15, 236–246.
Annilo, T., Shulenin, S., Chen, Z. Q., Arnould, I., Prades, C., Lemoine, C., Maintoux-Larois, C., Devaud, C., Dean, M., Denefle, P., and Rosier, M. (2002). Identification and characterization of a novel ABCA subfamily member, ABCA12, located in the lamellar ichthyosis region on 2q34. Cytogenet. Genome Res. 98, 169–176.
Arnould, I., Schriml, L. M., Prades, C., Lachtermacher-Triunfol, M., Schneider, T., Maintoux, C., Lemoine, C., Debono, D., Devaud, C., Naudin, L., Bauché, S., Annat, M., Annilo, T., Allikmets, R., Gold, B., Denèfle, P., Rosier, M., and Dean, M. (2001). Identifying and characterizing a five-gene cluster of ATP-binding cassette transporters mapping to human chromosome 17q24: a new subgroup within the ABCA subfamily. GeneScreen 1, 157–164.
Ban, N., Matsumura, Y., Sakai, H., Takanezawa, Y., Sasaki, M., Arai, H., and Inagaki, N. (2007). ABCA3 as a lipid transporter in pulmonary surfactant biogenesis. J. Biol. Chem. 282, 9628–9634.
Bared, S. M., Buechler, C., Boettcher, A., Dayoub, R., Sigruener, A., Grandl, M., Rudolph, C., Dada, A., and Schmitz, G. (2004). Association of ABCA1 with syntaxin 13 and flotillin-1 and enhanced phagocytosis in tangier cells. Mol. Biol. Cell 15, 5399–5407.
Barros, S. A., Tennant, R. W., and Cannon, R. E. (2003). Molecular structure and characterization of a novel murine ABC transporter, Abca13. Gene 307, 191–200.
Basso, F., Freeman, L., Knapper, C. L., Remaley, A., Stonik, J., Neufeld, E. B., Tansey, T., Amar, M. J., Fruchart-Najib, J., Duverger, N., Santamarina-Fojo, S., and Brewer, H. B. Jr. (2003). Role of the hepatic ABCA1 transporter in modulating intrahepatic cholesterol and plasma HDL cholesterol concentrations. J. Lipid Res. 44, 296–302.
Beham, A. W., Puellmann, K., Laird, R., Fuchs, T., Streich, R., Breysach, C., Raddatz, D., Oniga, S., Peccerella, T., Findeisen, P., Kzhyshkowska, J., Gratchev, A., Schweyer, S., Saunders, B., Wessels, J. T., Mobius, W., Keane, J., Becker, H., Ganser, A., Neumaier, M., and Kaminski, W. E. (2011). A TNF-regulated recombinatorial macrophage immune receptor implicated in granuloma formation in tuberculosis. PLoS. Pathog. 7, e1002375. doi:10.1371/journal.ppat.1002375
Beharry, S., Zhong, M., and Molday, R. S. (2004). N-retinylidene-phosphatidylethanolamine is the preferred retinoid substrate for the photoreceptor-specific ABC transporter ABCA4 (ABCR). J. Biol. Chem. 279, 53972–53979.
Bhongsatiern, J., Ohtsuki, S., Tachikawa, M., Hori, S., and Terasaki, T. (2005). Retinal-specific ATP-binding cassette transporter (ABCR/ABCA4) is expressed at the choroid plexus in rat brain. J. Neurochem. 92, 1277–1280.
Bodzioch, M., Orso, E., Klucken, J., Langmann, T., Bottcher, A., Diederich, W., Drobnik, W., Barlage, S., Buchler, C., Porsch-Ozcurumez, M., Kaminski, W. E., Hahmann, H. W., Oette, K., Rothe, G., Aslanidis, C., Lackner, K. J., and Schmitz, G. (1999). The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 22, 347–351.
Broccardo, C., Nieoullon, V., Amin, R., Masmejean, F., Carta, S., Tassi, S., Pophillat, M., Rubartelli, A., Pierres, M., Rougon, G., Nieoullon, A., Chazal, G., and Chimini, G. (2006). ABCA2 is a marker of neural progenitors and neuronal subsets in the adult rodent brain. J. Neurochem. 97, 345–355.
Broccardo, C., Osorio, J., Luciani, M. F., Schriml, L. M., Prades, C., Shulenin, S., Arnould, I., Naudin, L., Lafargue, C., Rosier, M., Jordan, B., Mattei, M. G., Dean, M., Denefle, P., and Chimini, G. (2001). Comparative analysis of the promoter structure and genomic organization of the human and mouse ABCA7 gene encoding a novel ABCA transporter. Cytogenet. Cell Genet. 92, 264–270.
Brooks-Wilson, A., Marcil, M., Clee, S. M., Zhang, L. H., Roomp, K., van, D. M., Yu, L., Brewer, C., Collins, J. A., Molhuizen, H. O., Loubser, O., Ouelette, B. F., Fichter, K., shbourne-Excoffon, K. J., Sensen, C. W., Scherer, S., Mott, S., Denis, M., Martindale, D., Frohlich, J., Morgan, K., Koop, B., Pimstone, S., Kastelein, J. J., Genest, J. Jr., and Hayden, M. R. (1999). Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 22, 336–345.
Brunham, L. R., Kruit, J. K., Iqbal, J., Fievet, C., Timmins, J. M., Pape, T. D., Coburn, B. A., Bissada, N., Staels, B., Groen, A. K., Hussain, M. M., Parks, J. S., Kuipers, F., and Hayden, M. R. (2006). Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J. Clin. Invest. 116, 1052–1062.
Bullard, J. E., Wert, S. E., Whitsett, J. A., Dean, M., and Nogee, L. M. (2005). ABCA3 mutations associated with pediatric interstitial lung disease. Am. J. Respir. Crit. Care Med. 172, 1026–1031.
Bungert, S., Molday, L. L., and Molday, R. S. (2001). Membrane topology of the ATP binding cassette transporter ABCR and its relationship to ABC1 and related ABCA transporters: identification of N-linked glycosylation sites. J. Biol. Chem. 276, 23539–23546.
Cha, J. D., Kim, H. J., and Cha, I. H. (2011). Genetic alterations in oral squamous cell carcinoma progression detected by combining array-based comparative genomic hybridization and multiplex ligation-dependent probe amplification. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 111, 594–607.
Chan, S. L., Kim, W. S., Kwok, J. B., Hill, A. F., Cappai, R., Rye, K. A., and Garner, B. (2008). ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J. Neurochem. 106, 793–804.
Chen, Z. J., Vulevic, B., Ile, K. E., Soulika, A., Davis, W. Jr., Reiner, P. B., Connop, B. P., Nathwani, P., Trojanowski, J. Q., and Tew, K. D. (2004). Association of ABCA2 expression with determinants of Alzheimer’s disease. FASEB J. 18, 1129–1131.
Cheong, N., Zhang, H., Madesh, M., Zhao, M., Yu, K., Dodia, C., Fisher, A. B., Savani, R. C., and Shuman, H. (2007). ABCA3 is critical for lamellar body biogenesis in vivo. J. Biol. Chem. 282, 23811–23817.
Cirrito, J. R., Deane, R., Fagan, A. M., Spinner, M. L., Parsadanian, M., Finn, M. B., Jiang, H., Prior, J. L., Sagare, A., Bales, K. R., Paul, S. M., Zlokovic, B. V., Piwnica-Worms, D., and Holtzman, D. M. (2005). P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Invest. 115, 3285–3290.
Cordon-Cardo, C., O’Brien, J. P., Casals, D., Rittman-Grauer, L., Biedler, J. L., Melamed, M. R., and Bertino, J. R. (1989). Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. U.S.A. 86, 695–698.
Cremers, F. P., van de Pol, D. J., van, D. M., den Hollander, A. I., van Haren, F. J., Knoers, N. V., Tijmes, N., Bergen, A. A., Rohrschneider, K., Blankenagel, A., Pinckers, A. J., Deutman, A. F., and Hoyng, C. B. (1998). Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum. Mol. Genet. 7, 355–362.
Davis, W. Jr. (2010). The ATP-binding cassette transporter-2 (ABCA2) increases endogenous amyloid precursor protein expression and Abeta fragment generation. Curr. Alzheimer Res. 7, 566–577.
Davis, W. Jr. (2011). The ATP-binding cassette transporter-2 (ABCA2) regulates cholesterol homeostasis and low-density lipoprotein receptor metabolism in N2a neuroblastoma cells. Biochim. Biophys. Acta 1811, 1152–1164.
Davis, W. Jr., Boyd, J. T., Ile, K. E., and Tew, K. D. (2004). Human ATP-binding cassette transporter-2 (ABCA2) positively regulates low-density lipoprotein receptor expression and negatively regulates cholesterol esterification in Chinese hamster ovary cells. Biochim. Biophys. Acta 1683, 89–100.
Dwyer, S., Williams, H., Jones, I., Jones, L., Walters, J., Craddock, N., Owen, M. J., and O’Donovan, M. C. (2011). Investigation of rare non-synonymous variants at ABCA13 in schizophrenia and bipolar disorder. Mol. Psychiatry 16, 790–791.
Ehehalt, R., Keller, P., Haass, C., Thiele, C., and Simons, K. (2003). Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 160, 113–123.
ElAli, A., and Hermann, D. M. (2011). ATP-binding cassette transporters and their roles in protecting the brain. Neuroscientist 17, 423–436.
Fitzgerald, M. L., Xavier, R., Haley, K. J., Welti, R., Goss, J. L., Brown, C. E., Zhuang, D. Z., Bell, S. A., Lu, N., McKee, M., Seed, B., and Freeman, M. W. (2007). ABCA3 inactivation in mice causes respiratory failure, loss of pulmonary surfactant, and depletion of lung phosphatidylglycerol. J. Lipid Res. 48, 621–632.
Fluhrer, R., Capell, A., Westmeyer, G., Willem, M., Hartung, B., Condron, M. M., Teplow, D. B., Haass, C., and Walter, J. (2002). A non-amyloidogenic function of BACE-2 in the secretory pathway. J. Neurochem. 81, 1011–1020.
Fukumoto, H., Deng, A., Irizarry, M. C., Fitzgerald, M. L., and Rebeck, G. W. (2002). Induction of the cholesterol transporter ABCA1 in central nervous system cells by liver X receptor agonists increases secreted Abeta levels. J. Biol. Chem. 277, 48508–48513.
Gottesman, M. M., Fojo, T., and Bates, S. E. (2002). Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer 2, 48–58.
Hammel, M., Michel, G., Hoefer, C., Klaften, M., Muller-Hocker, J., de Angelis, M. H., and Holzinger, A. (2007). Targeted inactivation of the murine Abca3 gene leads to respiratory failure in newborns with defective lamellar bodies. Biochem. Biophys. Res. Commun. 359, 947–951.
Hartz, A. M., Miller, D. S., and Bauer, B. (2010). Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol. Pharmacol. 77, 715–723.
Hayashi, H. (2011). Lipid metabolism and glial lipoproteins in the central nervous system. Biol. Pharm. Bull. 34, 453–461.
Hayashi, M., be-Dohmae, S., Okazaki, M., Ueda, K., and Yokoyama, S. (2005). Heterogeneity of high density lipoprotein generated by ABCA1 and ABCA7. J. Lipid Res. 46, 1703–1711.
Heimerl, S., Bosserhoff, A. K., Langmann, T., Ecker, J., and Schmitz, G. (2007). Mapping ATP-binding cassette transporter gene expression profiles in melanocytes and melanoma cells. Melanoma Res. 17, 265–273.
Higgins, C. F. (1992). ABC transporters: from microorganisms to man. Annu. Rev. Cell Biol. 8, 67–113.
Higgins, C. F., Hiles, I. D., Salmond, G. P., Gill, D. R., Downie, J. A., Evans, I. J., Holland, I. B., Gray, L., Buckel, S. D., and Bell, A. W. (1986). A family of related ATP-binding subunits coupled to many distinct biological processes in bacteria. Nature 323, 448–450.
Hirsch-Reinshagen, V., Burgess, B. L., and Wellington, C. L. (2009). Why lipids are important for Alzheimer disease? Mol. Cell. Biochem. 326, 121–129.
Hirsch-Reinshagen, V., Chan, J. Y., Wilkinson, A., Tanaka, T., Fan, J., Ou, G., Maia, L. F., Singaraja, R. R., Hayden, M. R., and Wellington, C. L. (2007). Physiologically regulated transgenic ABCA1 does not reduce amyloid burden or amyloid-beta peptide levels in vivo. J. Lipid Res. 48, 914–923.
Hirsch-Reinshagen, V., Maia, L. F., Burgess, B. L., Blain, J. F., Naus, K. E., McIsaac, S. A., Parkinson, P. F., Chan, J. Y., Tansley, G. H., Hayden, M. R., Poirier, J., Van, N. W., and Wellington, C. L. (2005). The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J. Biol. Chem. 280, 43243–43256.
Hirsch-Reinshagen, V., Zhou, S., Burgess, B. L., Bernier, L., McIsaac, S. A., Chan, J. Y., Tansley, G. H., Cohn, J. S., Hayden, M. R., and Wellington, C. L. (2004). Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J. Biol. Chem. 279, 41197–41207.
Holland, I. B., Cole, S. P. C., Kuchler, K., and Higgins, C. F. (2002). ABC Proteins: From Bacteria to Man. London: Academic Press.
Hollingworth, P., Harold, D., Sims, R., Gerrish, A., Lambert, J. C., Carrasquillo, M. M., Abraham, R., Hamshere, M. L., Pahwa, J. S., Moskvina, V., Dowzell, K., Jones, N., Stretton, A., Thomas, C., Richards, A., Ivanov, D., Widdowson, C., Chapman, J., Lovestone, S., Powell, J., Proitsi, P., Lupton, M. K., Brayne, C., Rubinsztein, D. C., Gill, M., Lawlor, B., Lynch, A., Brown, K. S., Passmore, P. A., Craig, D., McGuinness, B., Todd, S., Holmes, C., Mann, D., Smith, A. D., Beaumont, H., Warden, D., Wilcock, G., Love, S., Kehoe, P. G., Hooper, N. M., Vardy, E. R., Hardy, J., Mead, S., Fox, N. C., Rossor, M., Collinge, J., Maier, W., Jessen, F., Ruther, E., Schurmann, B., Heun, R., Kolsch, H., van den Bussche, H., Heuser, I., Kornhuber, J., Wiltfang, J., Dichgans, M., Frolich, L., Hampel, H., Gallacher, J., Hull, M., Rujescu, D., Giegling, I., Goate, A. M., Kauwe, J. S., Cruchaga, C., Nowotny, P., Morris, J. C., Mayo, K., Sleegers, K., Bettens, K., Engelborghs, S., De Deyn, P. P., Van, B. C., Livingston, G., Bass, N. J., Gurling, H., McQuillin, A., Gwilliam, R., Deloukas, P., Al-Chalabi, A., Shaw, C. E., Tsolaki, M., Singleton, A. B., Guerreiro, R., Muhleisen, T. W., Nothen, M. M., Moebus, S., Jockel, K. H., Klopp, N., Wichmann, H. E., Pankratz, V. S., Sando, S. B., Aasly, J. O., Barcikowska, M., Wszolek, Z. K., Dickson, D. W., Graff-Radford, N. R., Petersen, R. C., van Duijn, C. M., Breteler, M. M., Ikram, M. A., DeStefano, A. L., Fitzpatrick, A. L., Lopez, O., Launer, L. J., Seshadri, S., Berr, C., Campion, D., Epelbaum, J., Dartigues, J. F., Tzourio, C., Alperovitch, A., Lathrop, M., Feulner, T. M., Friedrich, P., Riehle, C., Krawczak, M., Schreiber, S., Mayhaus, M., Nicolhaus, S., Wagenpfeil, S., Steinberg, S., Stefansson, H., Stefansson, K., Snaedal, J., Bjornsson, S., Jonsson, P. V., Chouraki, V., Genier-Boley, B., Hiltunen, M., Soininen, H., Combarros, O., Zelenika, D., Delepine, M., Bullido, M. J., Pasquier, F., Mateo, I., Frank-Garcia, A., Porcellini, E., Hanon, O., Coto, E., Alvarez, V., Bosco, P., Siciliano, G., Mancuso, M., Panza, F., Solfrizzi, V., Nacmias, B., Sorbi, S., Bossu, P., Piccardi, P., Arosio, B., Annoni, G., Seripa, D., Pilotto, A., Scarpini, E., Galimberti, D., Brice, A., Hannequin, D., Licastro, F., Jones, L., Holmans, P. A., Jonsson, T., Riemenschneider, M., Morgan, K., Younkin, S. G., Owen, M. J., O’Donovan, M., Amouyel, P., and Williams, J. (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 43, 429–435.
Hu, Y., Wang, M., Veverka, K., Garcia, F. U., and Stearns, M. E. (2007). The ABCA5 protein: a urine diagnostic marker for prostatic intraepithelial neoplasia. Clin. Cancer Res. 13, 929–938.
Huang, D., Gao, Q., Guo, L., Zhang, C., Jiang, W., Li, H., Wang, J., Han, X., Shi, Y., and Lu, S. H. (2009). Isolation and identification of cancer stem-like cells in esophageal carcinoma cell lines. Stem Cells Dev. 18, 465–473.
Ikeda, Y., be-Dohmae, S., Munehira, Y., Aoki, R., Kawamoto, S., Furuya, A., Shitara, K., Amachi, T., Kioka, N., Matsuo, M., Yokoyama, S., and Ueda, K. (2003). Posttranscriptional regulation of human ABCA7 and its function for the apoA-I-dependent lipid release. Biochem. Biophys. Res. Commun. 311, 313–318.
Ile, K. E., Davis, W. Jr., Boyd, J. T., Soulika, A. M., and Tew, K. D. (2004). Identification of a novel first exon of the human ABCA2 transporter gene encoding a unique N-terminus. Biochim. Biophys. Acta 1678, 22–32.
Iwamoto, N., be-Dohmae, S., Sato, R., and Yokoyama, S. (2006). ABCA7 expression is regulated by cellular cholesterol through the SREBP2 pathway and associated with phagocytosis. J. Lipid Res. 47, 1915–1927.
Jehle, A. W., Gardai, S. J., Li, S., Linsel-Nitschke, P., Morimoto, K., Janssen, W. J., Vandivier, R. W., Wang, N., Greenberg, S., Dale, B. M., Qin, C., Henson, P. M., and Tall, A. R. (2006). ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J. Cell Biol. 174, 547–556.
Jones, L., Holmans, P. A., Hamshere, M. L., Harold, D., Moskvina, V., Ivanov, D., Pocklington, A., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Pahwa, J. S., Jones, N., Stretton, A., Morgan, A. R., Lovestone, S., Powell, J., Proitsi, P., Lupton, M. K., Brayne, C., Rubinsztein, D. C., Gill, M., Lawlor, B., Lynch, A., Morgan, K., Brown, K. S., Passmore, P. A., Craig, D., McGuinness, B., Todd, S., Holmes, C., Mann, D., Smith, A. D., Love, S., Kehoe, P. G., Mead, S., Fox, N., Rossor, M., Collinge, J., Maier, W., Jessen, F., Schurmann, B., Heun, R., Kolsch, H., van den Bussche, H., Heuser, I., Peters, O., Kornhuber, J., Wiltfang, J., Dichgans, M., Frolich, L., Hampel, H., Hull, M., Rujescu, D., Goate, A. M., Kauwe, J. S., Cruchaga, C., Nowotny, P., Morris, J. C., Mayo, K., Livingston, G., Bass, N. J., Gurling, H., McQuillin, A., Gwilliam, R., Deloukas, P., Al-Chalabi, A., Shaw, C. E., Singleton, A. B., Guerreiro, R., Muhleisen, T. W., Nothen, M. M., Moebus, S., Jockel, K. H., Klopp, N., Wichmann, H. E., Ruther, E., Carrasquillo, M. M., Pankratz, V. S., Younkin, S. G., Hardy, J., O’Donovan, M. C., Owen, M. J., and Williams, J. (2010). Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS ONE 5, e13950. doi:10.1371/journal.pone.0013950
Kaminski, W. E., Orso, E., Diederich, W., Klucken, J., Drobnik, W., and Schmitz, G. (2000). Identification of a novel human sterol-sensitive ATP-binding cassette transporter (ABCA7). Biochem. Biophys. Res. Commun. 273, 532–538.
Kaminski, W. E., Piehler, A., Pullmann, K., Porsch-Ozcurumez, M., Duong, C., Bared, G. M., Buchler, C., and Schmitz, G. (2001a). Complete coding sequence, promoter region, and genomic structure of the human ABCA2 gene and evidence for sterol-dependent regulation in macrophages. Biochem. Biophys. Res. Commun. 281, 249–258.
Kaminski, W. E., Wenzel, J. J., Piehler, A., Langmann, T., and Schmitz, G. (2001b). ABCA6, a novel a subclass ABC transporter. Biochem. Biophys. Res. Commun. 285, 1295–1301.
Kaminski, W. E., Piehler, A., and Wenzel, J. J. (2006). ABC A-subfamily transporters: structure, function and disease. Biochim. Biophys. Acta 1762, 510–524.
Kang, M. H., Singaraja, R., and Hayden, M. R. (2010). Adenosine-triphosphate-binding cassette transporter-1 trafficking and function. Trends Cardiovasc. Med. 20, 41–49.
Karjalainen, M. K., Haataja, R., and Hallman, M. (2008). Haplotype analysis of ABCA3: association with respiratory distress in very premature infants. Ann. Med. 40, 56–65.
Katzov, H., Chalmers, K., Palmgren, J., Andreasen, N., Johansson, B., Cairns, N. J., Gatz, M., Wilcock, G. K., Love, S., Pedersen, N. L., Brookes, A. J., Blennow, K., Kehoe, P. G., and Prince, J. A. (2004). Genetic variants of ABCA1 modify Alzheimer disease risk and quantitative traits related to beta-amyloid metabolism. Hum. Mutat. 23, 358–367.
Kelsell, D. P., Norgett, E. E., Unsworth, H., Teh, M. T., Cullup, T., Mein, C. A., Dopping-Hepenstal, P. J., Dale, B. A., Tadini, G., Fleckman, P., Stephens, K. G., Sybert, V. P., Mallory, S. B., North, B. V., Witt, D. R., Sprecher, E., Taylor, A. E., Ilchyshyn, A., Kennedy, C. T., Goodyear, H., Moss, C., Paige, D., Harper, J. I., Young, B. D., Leigh, I. M., Eady, R. A., and O’Toole, E. A. (2005). Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am. J. Hum. Genet. 76, 794–803.
Kielar, D., Kaminski, W. E., Liebisch, G., Piehler, A., Wenzel, J. J., Mohle, C., Heimerl, S., Langmann, T., Friedrich, S. O., Bottcher, A., Barlage, S., Drobnik, W., and Schmitz, G. (2003). Adenosine triphosphate binding cassette (ABC) transporters are expressed and regulated during terminal keratinocyte differentiation: a potential role for ABCA7 in epidermal lipid reorganization. J. Invest. Dermatol. 121, 465–474.
Kim, W. S., Fitzgerald, M. L., Kang, K., Okuhira, K., Bell, S. A., Manning, J. J., Koehn, S. L., Lu, N., Moore, K. J., and Freeman, M. W. (2005). Abca7 null mice retain normal macrophage phosphatidylcholine and cholesterol efflux activity despite alterations in adipose mass and serum cholesterol levels. J. Biol. Chem. 280, 3989–3995.
Kim, W. S., Guillemin, G. J., Glaros, E. N., Lim, C. K., and Garner, B. (2006). Quantitation of ATP-binding cassette subfamily-A transporter gene expression in primary human brain cells. Neuroreport 17, 891–896.
Kim, W. S., Hill, A. F., Fitzgerald, M. L., Freeman, M. W., Evin, G., and Garner, B. (2011). Wild type and Tangier disease ABCA1 mutants modulate cellular amyloid-beta production independent of cholesterol efflux activity. J. Alzheimers Dis. 27, 441–452.
Kim, W. S., Rahmanto, A. S., Kamili, A., Rye, K. A., Guillemin, G. J., Gelissen, I. C., Jessup, W., Hill, A. F., and Garner, B. (2007). Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J. Biol. Chem. 282, 2851–2861.
Kim, W. S., Weickert, C. S., and Garner, B. (2008). Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J. Neurochem. 104, 1145–1166.
Knight, H. M., Pickard, B. S., Maclean, A., Malloy, M. P., Soares, D. C., McRae, A. F., Condie, A., White, A., Hawkins, W., McGhee, K., van, B. M., MacIntyre, D. J., Starr, J. M., Deary, I. J., Visscher, P. M., Porteous, D. J., Cannon, R. E., St, C. D., Muir, W. J., and Blackwood, D. H. (2009). A cytogenetic abnormality and rare coding variants identify ABCA13 as a candidate gene in schizophrenia, bipolar disorder, and depression. Am. J. Hum. Genet. 85, 833–846.
Koldamova, R., Fitz, N. F., and Lefterov, I. (2010). The role of ATP-binding cassette transporter A1 in Alzheimer’s disease and neurodegeneration. Biochim. Biophys. Acta 1801, 824–830.
Koldamova, R., Staufenbiel, M., and Lefterov, I. (2005). Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J. Biol. Chem. 280, 43224–43235.
Koldamova, R. P., Lefterov, I. M., Ikonomovic, M. D., Skoko, J., Lefterov, P. I., Isanski, B. A., DeKosky, S. T., and Lazo, J. S. (2003). 22R-hydroxycholesterol and 9-cis-retinoic acid induce ATP-binding cassette transporter A1 expression and cholesterol efflux in brain cells and decrease amyloid beta secretion. J. Biol. Chem. 278, 13244–13256.
Kolsch, H., Lutjohann, D., Jessen, F., Von, B. K., Schmitz, S., Urbach, H., Maier, W., and Heun, R. (2006). Polymorphism in ABCA1 influences CSF 24S-hydroxycholesterol levels but is not a major risk factor of Alzheimer’s disease. Int. J. Mol. Med. 17, 791–794.
Krohn, M., Lange, C., Hofrichter, J., Scheffler, K., Stenzel, J., Steffen, J., Schumacher, T., Bruning, T., Plath, A. S., Alfen, F., Schmidt, A., Winter, F., Rateitschak, K., Wree, A., Gsponer, J., Walker, L. C., and Pahnke, J. (2011). Cerebral amyloid-beta proteostasis is regulated by the membrane transport protein ABCC1 in mice. J. Clin. Invest. 121, 3924–3931.
Kubo, Y., Sekiya, S., Ohigashi, M., Takenaka, C., Tamura, K., Nada, S., Nishi, T., Yamamoto, A., and Yamaguchi, A. (2005). ABCA5 resides in lysosomes, and ABCA5 knockout mice develop lysosomal disease-like symptoms. Mol. Cell. Biol. 25, 4138–4149.
Kuhnke, D., Jedlitschky, G., Grube, M., Krohn, M., Jucker, M., Mosyagin, I., Cascorbi, I., Walker, L. C., Kroemer, H. K., Warzok, R. W., and Vogelgesang, S. (2007). MDR1-P-Glycoprotein (ABCB1) mediates transport of Alzheimer’s amyloid-beta peptides – implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol. 17, 347–353.
Kunig, A. M., Parker, T. A., Nogee, L. M., Abman, S. H., and Kinsella, J. P. (2007). ABCA3 deficiency presenting as persistent pulmonary hypertension of the newborn. J. Pediatr. 151, 322–324.
Lam, F. C., Liu, R., Lu, P., Shapiro, A. B., Renoir, J. M., Sharom, F. J., and Reiner, P. B. (2001). beta-Amyloid efflux mediated by p-glycoprotein. J. Neurochem. 76, 1121–1128.
Langmann, T., Klucken, J., Reil, M., Liebisch, G., Luciani, M. F., Chimini, G., Kaminski, W. E., and Schmitz, G. (1999). Molecular cloning of the human ATP-binding cassette transporter 1 (hABC1): evidence for sterol-dependent regulation in macrophages. Biochem. Biophys. Res. Commun. 257, 29–33.
Langmann, T., Mauerer, R., Zahn, A., Moehle, C., Probst, M., Stremmel, W., and Schmitz, G. (2003). Real-time reverse transcription-PCR expression profiling of the complete human ATP-binding cassette transporter superfamily in various tissues. Clin. Chem. 49, 230–238.
Lawn, R. M., Wade, D. P., Garvin, M. R., Wang, X., Schwartz, K., Porter, J. G., Seilhamer, J. J., Vaughan, A. M., and Oram, J. F. (1999). The Tangier disease gene product ABC1 controls the cellular apolipoprotein-mediated lipid removal pathway. J. Clin. Invest. 104, R25–R31.
Lefevre, C., Audebert, S., Jobard, F., Bouadjar, B., Lakhdar, H., Boughdene-Stambouli, O., Blanchet-Bardon, C., Heilig, R., Foglio, M., Weissenbach, J., Lathrop, M., Prud’Homme, J. F., and Fischer, J. (2003). Mutations in the transporter ABCA12 are associated with lamellar ichthyosis type 2. Hum. Mol. Genet. 12, 2369–2378.
Li, Y., Tacey, K., Doil, L., van, L. R., Garcia, V., Rowland, C., Schrodi, S., Leong, D., Lau, K., Catanese, J., Sninsky, J., Nowotny, P., Holmans, P., Hardy, J., Powell, J., Lovestone, S., Thal, L., Owen, M., Williams, J., Goate, A., and Grupe, A. (2004). Association of ABCA1 with late-onset Alzheimer’s disease is not observed in a case-control study. Neurosci. Lett. 366, 268–271.
Linsel-Nitschke, P., Jehle, A. W., Shan, J., Cao, G., Bacic, D., Lan, D., Wang, N., and Tall, A. R. (2005). Potential role of ABCA7 in cellular lipid efflux to apoA-I. J. Lipid Res. 46, 86–92.
Linton, K. J., and Higgins, C. F. (1998). The Escherichia coli ATP-binding cassette (ABC) proteins. Mol. Microbiol. 28, 5–13.
Liu, K., Doms, R. W., and Lee, V. M. (2002). Glu11 site cleavage and N-terminally truncated A beta production upon BACE overexpression. Biochemistry 41, 3128–3136.
Luciani, M. F., Denizot, F., Savary, S., Mattei, M. G., and Chimini, G. (1994). Cloning of two novel ABC transporters mapping on human chromosome 9. Genomics 21, 150–159.
Mace, S., Cousin, E., Ricard, S., Genin, E., Spanakis, E., Lafargue-Soubigou, C., Genin, B., Fournel, R., Roche, S., Haussy, G., Massey, F., Soubigou, S., Brefort, G., Benoit, P., Brice, A., Campion, D., Hollis, M., Pradier, L., Benavides, J., and Deleuze, J. F. (2005). ABCA2 is a strong genetic risk factor for early-onset Alzheimer’s disease. Neurobiol. Dis. 18, 119–125.
Mack, J. T., Beljanski, V., Soulika, A. M., Townsend, D. M., Brown, C. B., Davis, W., and Tew, K. D. (2007). “Skittish” Abca2 knockout mice display tremor, hyperactivity, and abnormal myelin ultrastructure in the central nervous system. Mol. Cell. Biol. 27, 44–53.
Mack, J. T., Brown, C. B., and Tew, K. D. (2008). ABCA2 as a therapeutic target in cancer and nervous system disorders. Expert Opin. Ther. Targets 12, 491–504.
Maden, M. (2007). Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 8, 755–765.
Martinez-Mir, A., Paloma, E., Allikmets, R., Ayuso, C., del, R. T., Dean, M., Vilageliu, L., Gonzalez-Duarte, R., and Balcells, S. (1998). Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat. Genet. 18, 11–12.
Mata, N. L., Weng, J., and Travis, G. H. (2000). Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 97, 7154–7159.
Maugeri, A., Klevering, B. J., Rohrschneider, K., Blankenagel, A., Brunner, H. G., Deutman, A. F., Hoyng, C. B., and Cremers, F. P. (2000). Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am. J. Hum. Genet. 67, 960–966.
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., Yarasheski, K. E., and Bateman, R. J. (2010). Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330, 1774.
Michaki, V., Guix, F. X., Vennekens, K., Munck, S., Dingwall, C., Davis, J. B., Townsend, D. M., Tew, K. D., Feguin, F., De, S. B., Dotti, C. G., and Wahle, T. (2011). Downregulation of the ATP-binding cassette transporter 2 (Abca2) reduces Amyloid-beta production by altering Nicastrin maturation and intracellular localization. J. Biol. Chem. 287, 1100–1111.
Minster, R. L., Dekosky, S. T., and Kamboh, M. I. (2008). No association of DAPK1 and ABCA2 SNPs on chromosome 9 with Alzheimer’s disease. Neurobiol. Aging 30, 1890–1891.
Molday, R. S. (2007). ATP-binding cassette transporter ABCA4: molecular properties and role in vision and macular degeneration. J. Bioenerg. Biomembr. 39, 507–517.
Nagao, K., Takahashi, K., Azuma, Y., Takada, M., Kimura, Y., Matsuo, M., Kioka, N., and Ueda, K. (2011a). ATP hydrolysis-dependent conformational changes in the extracellular domain of ABCA1 are associated with apoA-I binding. J. Lipid Res. 53, 126–136.
Nagao, K., Tomioka, M., and Ueda, K. (2011b). Function and regulation of ABCA1 – membrane meso-domain organization and reorganization. FEBS J. 278, 3190–3203.
Nagase, T., Ishikawa, K., Suyama, M., Kikuno, R., Hirosawa, M., Miyajima, N., Tanaka, A., Kotani, H., Nomura, N., and Ohara, O. (1998). Prediction of the coding sequences of unidentified human genes. XII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 5, 355–364.
Napoli, I., and Neumann, H. (2009). Microglial clearance function in health and disease. Neuroscience 158, 1030–1038.
Natsuga, K., Akiyama, M., Kato, N., Sakai, K., Sugiyama-Nakagiri, Y., Nishimura, M., Hata, H., Abe, M., Arita, K., Tsuji-Abe, Y., Onozuka, T., Aoyagi, S., Kodama, K., Ujiie, H., Tomita, Y., and Shimizu, H. (2007). Novel ABCA12 mutations identified in two cases of non-bullous congenital ichthyosiform erythroderma associated with multiple skin malignant neoplasia. J. Invest. Dermatol. 127, 2669–2673.
Neufeld, E. B., Remaley, A. T., Demosky, S. J., Stonik, J. A., Cooney, A. M., Comly, M., Dwyer, N. K., Zhang, M., Blanchette-Mackie, J., Santamarina-Fojo, S., and Brewer, H. B. Jr. (2001). Cellular localization and trafficking of the human ABCA1 transporter. J. Biol. Chem. 276, 27584–27590.
Ohtsuki, S., Kamoi, M., Watanabe, Y., Suzuki, H., Hori, S., and Terasaki, T. (2007). Correlation of induction of ATP binding cassette transporter A5 (ABCA5) and ABCB1 mRNAs with differentiation state of human colon tumor. Biol. Pharm. Bull. 30, 1144–1146.
Ohtsuki, S., Watanabe, Y., Hori, S., Suzuki, H., Bhongsatiern, J., Fujiyoshi, M., Kamoi, M., Kamiya, N., Takanaga, H., and Terasaki, T. (2004). mRNA expression of the ATP-binding cassette transporter subfamily A (ABCA) in rat and human brain capillary endothelial cells. Biol. Pharm. Bull. 27, 1437–1440.
Orso, E., Broccardo, C., Kaminski, W. E., Bottcher, A., Liebisch, G., Drobnik, W., Gotz, A., Chambenoit, O., Diederich, W., Langmann, T., Spruss, T., Luciani, M. F., Rothe, G., Lackner, K. J., Chimini, G., and Schmitz, G. (2000). Transport of lipids from Golgi to plasma membrane is defective in tangier disease patients and Abc1-deficient mice. Nat. Genet. 24, 192–196.
Panzenboeck, U., Balazs, Z., Sovic, A., Hrzenjak, A., Levak-Frank, S., Wintersperger, A., Malle, E., and Sattler, W. (2002). ABCA1 and scavenger receptor class B, type I, are modulators of reverse sterol transport at an in vitro blood-brain barrier constituted of porcine brain capillary endothelial cells. J. Biol. Chem. 277, 42781–42789.
Parmentier, L., Clepet, C., Boughdene-Stambouli, O., Lakhdar, H., Blanchet-Bardon, C., Dubertret, L., Wunderle, E., Pulcini, F., Fizames, C., and Weissenbach, J. (1999). Lamellar ichthyosis: further narrowing, physical and expression mapping of the chromosome 2 candidate locus. Eur. J. Hum. Genet. 7, 77–87.
Peelman, F., Labeur, C., Vanloo, B., Roosbeek, S., Devaud, C., Duverger, N., Denefle, P., Rosier, M., Vandekerckhove, J., and Rosseneu, M. (2003). Characterization of the ABCA transporter subfamily: identification of prokaryotic and eukaryotic members, phylogeny and topology. J. Mol. Biol. 325, 259–274.
Petry, F., Kotthaus, A., and Hirsch-Ernst, K. I. (2003). Cloning of human and rat ABCA5/Abca5 and detection of a human splice variant. Biochem. Biophys. Res. Commun. 300, 343–350.
Petry, F., Ritz, V., Meineke, C., Middel, P., Kietzmann, T., Schmitz-Salue, C., and Hirsch-Ernst, K. I. (2006). Subcellular localization of rat Abca5, a rat ATP-binding-cassette transporter expressed in Leydig cells, and characterization of its splice variant apparently encoding a half-transporter. Biochem. J. 393, 79–87.
Piehler, A., Kaminski, W. E., Wenzel, J. J., Langmann, T., and Schmitz, G. (2002). Molecular structure of a novel cholesterol-responsive A subclass ABC transporter, ABCA9. Biochem. Biophys. Res. Commun. 295, 408–416.
Piehler, A. P., Hellum, M., Wenzel, J. J., Kaminski, E., Haug, K. B., Kierulf, P., and Kaminski, W. E. (2008). The human ABC transporter pseudogene family: evidence for transcription and gene-pseudogene interference. BMC Genomics 9, 165. doi:10.1186/1471-2164-9-165
Piehler, A. P., Wenzel, J. J., Olstad, O. K., Haug, K. B., Kierulf, P., and Kaminski, W. E. (2006). The human ortholog of the rodent testis-specific ABC transporter Abca17 is a ubiquitously expressed pseudogene (ABCA17P) and shares a common 5′ end with ABCA3. BMC Mol. Biol. 7, 28. doi:10.1186/1471-2199-7-28
Prades, C., Arnould, I., Annilo, T., Shulenin, S., Chen, Z. Q., Orosco, L., Triunfol, M., Devaud, C., Maintoux-Larois, C., Lafargue, C., Lemoine, C., Denefle, P., Rosier, M., and Dean, M. (2002). The human ATP binding cassette gene ABCA13, located on chromosome 7p12.3, encodes a 5058 amino acid protein with an extracellular domain encoded in part by a 4.8-kb conserved exon. Cytogenet. Genome Res. 98, 160–168.
Puglielli, L., Tanzi, R. E., and Kovacs, D. M. (2003). Alzheimer’s disease: the cholesterol connection. Nat. Neurosci. 6, 345–351.
Quezada, C. A., Garrido, W. X., Gonzalez-Oyarzun, M. A., Rauch, M. C., Salas, M. R., San Martin, R. E., Claude, A. A., Yanez, A. J., Slebe, J. C., and Carcamo, J. G. (2008). Effect of tacrolimus on activity and expression of P-glycoprotein and ATP-binding cassette transporter A5 (ABCA5) proteins in hematoencephalic barrier cells. Biol. Pharm. Bull. 31, 1911–1916.
Reppe, S., Refvem, H., Gautvik, V. T., Olstad, O. K., Hovring, P. I., Reinholt, F. P., Holden, M., Frigessi, A., Jemtland, R., and Gautvik, K. M. (2010). Eight genes are highly associated with BMD variation in postmenopausal Caucasian women. Bone 46, 604–612.
Reynolds, C. A., Hong, M. G., Eriksson, U. K., Blennow, K., Bennet, A. M., Johansson, B., Malmberg, B., Berg, S., Wiklund, F., Gatz, M., Pedersen, N. L., and Prince, J. A. (2009). A survey of ABCA1 sequence variation confirms association with dementia. Hum. Mutat. 30, 1348–1354.
Rodriguez-Rodriguez, E., Mateo, I., Llorca, J., Sanchez-Quintana, C., Infante, J., Garcia-Gorostiaga, I., Sanchez-Juan, P., Berciano, J., and Combarros, O. (2007). Association of genetic variants of ABCA1 with Alzheimer’s disease risk. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 964–968.
Rust, S., Rosier, M., Funke, H., Real, J., Amoura, Z., Piette, J. C., Deleuze, J. F., Brewer, H. B., Duverger, N., Denefle, P., and Assmann, G. (1999). Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat. Genet. 22, 352–355.
Saito, T., Yamada, K., Wang, Y., Tanaka, Y., Ohtomo, K., Ishikawa, K., and Inagaki, N. (2007). Expression of ABCA2 protein in both non-myelin-forming and myelin-forming Schwann cells in the rodent peripheral nerve. Neurosci. Lett. 414, 35–40.
Sakai, H., Tanaka, Y., Tanaka, M., Ban, N., Yamada, K., Matsumura, Y., Watanabe, D., Sasaki, M., Kita, T., and Inagaki, N. (2007a). ABCA2 deficiency results in abnormal sphingolipid metabolism in mouse brain. J. Biol. Chem. 282, 19692–19699.
Sakai, K., Akiyama, M., Sugiyama-Nakagiri, Y., McMillan, J. R., Sawamura, D., and Shimizu, H. (2007b). Localization of ABCA12 from Golgi apparatus to lamellar granules in human upper epidermal keratinocytes. Exp. Dermatol. 16, 920–926.
Sarkadi, B., Homolya, L., Szakacs, G., and Varadi, A. (2006). Human multidrug resistance ABCB and ABCG transporters: participation in a chemoimmunity defense system. Physiol. Rev. 86, 1179–1236.
Sasaki, M., Shoji, A., Kubo, Y., Nada, S., and Yamaguchi, A. (2003). Cloning of rat ABCA7 and its preferential expression in platelets. Biochem. Biophys. Res. Commun. 304, 777–782.
Seetharaman, S., Barrand, M. A., Maskell, L., and Scheper, R. J. (1998). Multidrug resistance-related transport proteins in isolated human brain microvessels and in cells cultured from these isolates. J. Neurochem. 70, 1151–1159.
Shawahna, R., Uchida, Y., Decleves, X., Ohtsuki, S., Yousif, S., Dauchy, S., Jacob, A., Chassoux, F., Daumas-Duport, C., Couraud, P. O., Terasaki, T., and Scherrmann, J. M. (2011). Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol. Pharm. 8, 1332–1341.
Shibata, N., Kawarai, T., Lee, J. H., Lee, H. S., Shibata, E., Sato, C., Liang, Y., Duara, R., Mayeux, R. P., St George-Hyslop, P. H., and Rogaeva, E. (2006). Association studies of cholesterol metabolism genes (CH25H, ABCA1 and CH24H) in Alzheimer’s disease. Neurosci. Lett. 391, 142–146.
Shukla, A., Hillegass, J. M., MacPherson, M. B., Beuschel, S. L., Vacek, P. M., Pass, H. I., Carbone, M., Testa, J. R., and Mossman, B. T. (2010). Blocking of ERK1 and ERK2 sensitizes human mesothelioma cells to doxorubicin. Mol. Cancer 9, 314.
Shulenin, S., Nogee, L. M., Annilo, T., Wert, S. E., Whitsett, J. A., and Dean, M. (2004). ABCA3 gene mutations in newborns with fatal surfactant deficiency. N. Engl. J. Med. 350, 1296–1303.
Simons, M., Keller, P., De, S. B., Beyreuther, K., Dotti, C. G., and Simons, K. (1998). Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 95, 6460–6464.
Soichi, O., Masanori, N., Hideo, T., Kazunori, A., Nobuya, I., and Jun-ichi, K. (2007). Clinical significance of ABCA2′ a possible molecular marker for oligodendrogliomas. Neurosurgery 60, 707–714.
Stahlman, M. T., Besnard, V., Wert, S. E., Weaver, T. E., Dingle, S., Xu, Y., von, Z. K., Olson, S. J., and Whitsett, J. A. (2007). Expression of ABCA3 in developing lung and other tissues. J. Histochem. Cytochem. 55, 71–83.
Stolzing, A., and Grune, T. (2004). Neuronal apoptotic bodies: phagocytosis and degradation by primary microglial cells. FASEB J. 18, 743–745.
Su, A. I., Wiltshire, T., Batalov, S., Lapp, H., Ching, K. A., Block, D., Zhang, J., Soden, R., Hayakawa, M., Kreiman, G., Cooke, M. P., Walker, J. R., and Hogenesch, J. B. (2004). A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U.S.A. 101, 6062–6067.
Sun, H., Molday, R. S., and Nathans, J. (1999). Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor-specific ATP-binding cassette transporter responsible for Stargardt disease. J. Biol. Chem. 274, 8269–8281.
Sundar, P. D., Feingold, E., Minster, R. L., DeKosky, S. T., and Kamboh, M. I. (2007). Gender-specific association of ATP-binding cassette transporter 1 (ABCA1) polymorphisms with the risk of late-onset Alzheimer’s disease. Neurobiol. Aging 28, 856–862.
Tachikawa, M., Watanabe, M., Hori, S., Fukaya, M., Ohtsuki, S., Asashima, T., and Terasaki, T. (2005). Distinct spatio-temporal expression of ABCA and ABCG transporters in the developing and adult mouse brain. J. Neurochem. 95, 294–304.
Tanaka, A. R., be-Dohmae, S., Ohnishi, T., Aoki, R., Morinaga, G., Okuhira, K., Ikeda, Y., Kano, F., Matsuo, M., Kioka, N., Amachi, T., Murata, M., Yokoyama, S., and Ueda, K. (2003a). Effects of mutations of ABCA1 in the first extracellular domain on subcellular trafficking and ATP binding/hydrolysis. J. Biol. Chem. 278, 8815–8819.
Tanaka, Y., Yamada, K., Zhou, C. J., Ban, N., Shioda, S., and Inagaki, N. (2003b). Temporal and spatial profiles of ABCA2-expressing oligodendrocytes in the developing rat brain. J. Comp. Neurol. 455, 353–367.
Tanaka, N., Abe-Dohmae, S., Iwamoto, N., Fitzgerald, M. L., and Yokoyama, S. (2010). Helical apolipoproteins of high-density lipoprotein enhance phagocytosis by stabilizing ATP-binding cassette transporter A7. J. Lipid Res. 51, 2591–2599.
Tanaka, N., Abe-Dohmae, S., Iwamoto, N., Fitzgerald, M. L., and Yokoyama, S. (2011a). HMG-CoA reductase inhibitors enhance phagocytosis by upregulating ATP-binding cassette transporter A7. Atherosclerosis 217, 407–414.
Tanaka, N., Abe-Dohmae, S., Iwamoto, N., and Yokoyama, S. (2011b). Roles of ATP-binding cassette transporter A7 in cholesterol homeostasis and host defense system. J. Atheroscler. Thromb. 18, 274–281.
Tatsuta, T., Naito, M., Oh-hara, T., Sugawara, I., and Tsuruo, T. (1992). Functional involvement of P-glycoprotein in blood-brain barrier. J. Biol. Chem. 267, 20383–20391.
Timmins, J. M., Lee, J. Y., Boudyguina, E., Kluckman, K. D., Brunham, L. R., Mulya, A., Gebre, A. K., Coutinho, J. M., Colvin, P. L., Smith, T. L., Hayden, M. R., Maeda, N., and Parks, J. S. (2005). Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J. Clin. Invest. 115, 1333–1342.
Tishler, D. M., Weinberg, K. I., Hinton, D. R., Barbaro, N., Annett, G. M., and Raffel, C. (1995). MDR1 gene expression in brain of patients with medically intractable epilepsy. Epilepsia 36, 1–6.
Tsuruoka, S., Ishibashi, K., Yamamoto, H., Wakaumi, M., Suzuki, M., Schwartz, G. J., Imai, M., and Fujimura, A. (2002). Functional analysis of ABCA8, a new drug transporter. Biochem. Biophys. Res. Commun. 298, 41–45.
Tusnady, G. E., Sarkadi, B., Simon, I., and Varadi, A. (2006). Membrane topology of human ABC proteins. FEBS Lett. 580, 1017–1022.
Unamuno, P., Pierola, J. M., Fernandez, E., Roman, C., and Velasco, J. A. (1987). Harlequin foetus in four siblings. Br. J. Dermatol. 116, 569–572.
van Helvoort, A., Smith, A. J., Sprong, H., Fritzsche, I., Schinkel, A. H., Borst, P., and van, M. G. (1996). MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell 87, 507–517.
Vedhachalam, C., Chetty, P. S., Nickel, M., Dhanasekaran, P., Lund-Katz, S., Rothblat, G. H., and Phillips, M. C. (2010). Influence of apolipoprotein (Apo) A-I structure on nascent high density lipoprotein (HDL) particle size distribution. J. Biol. Chem. 285, 31965–31973.
Vedhachalam, C., Duong, P. T., Nickel, M., Nguyen, D., Dhanasekaran, P., Saito, H., Rothblat, G. H., Lund-Katz, S., and Phillips, M. C. (2007a). Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J. Biol. Chem. 282, 25123–25130.
Vedhachalam, C., Ghering, A. B., Davidson, W. S., Lund-Katz, S., Rothblat, G. H., and Phillips, M. C. (2007b). ABCA1-induced cell surface binding sites for ApoA-I. Arterioscler. Thromb. Vasc. Biol. 27, 1603–1609.
Vetrivel, K. S., Cheng, H., Lin, W., Sakurai, T., Li, T., Nukina, N., Wong, P. C., Xu, H., and Thinakaran, G. (2004). Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 279, 44945–44954.
Vogelgesang, S., Jedlitschky, G., Brenn, A., and Walker, L. C. (2011). The role of the ATP-binding cassette transporter P-Glycoprotein in the transport of beta-amyloid across the blood-brain barrier. Curr. Pharm. Des. 17, 2778–2786.
Vogelgesang, S., Warzok, R. W., Cascorbi, I., Kunert-Keil, C., Schroeder, E., Kroemer, H. K., Siegmund, W., Walker, L. C., and Pahnke, J. (2004). The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 1, 121–125.
Vulevic, B., Chen, Z., Boyd, J. T., Davis, W. Jr., Walsh, E. S., Belinsky, M. G., and Tew, K. D. (2001). Cloning and characterization of human adenosine 5′-triphosphate-binding cassette, sub-family A, transporter 2 (ABCA2). Cancer Res. 61, 3339–3347.
Wahrle, S., Das, P., Nyborg, A. C., McLendon, C., Shoji, M., Kawarabayashi, T., Younkin, L. H., Younkin, S. G., and Golde, T. E. (2002). Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 9, 11–23.
Wahrle, S. E., Jiang, H., Parsadanian, M., Hartman, R. E., Bales, K. R., Paul, S. M., and Holtzman, D. M. (2005). Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem. 280, 43236–43242.
Wahrle, S. E., Jiang, H., Parsadanian, M., Kim, J., Li, A., Knoten, A., Jain, S., Hirsch-Reinshagen, V., Wellington, C. L., Bales, K. R., Paul, S. M., and Holtzman, D. M. (2008). Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Invest. 118, 671–682.
Wahrle, S. E., Jiang, H., Parsadanian, M., Legleiter, J., Han, X., Fryer, J. D., Kowalewski, T., and Holtzman, D. M. (2004). ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem. 279, 40987–40993.
Wahrle, S. E., Shah, A. R., Fagan, A. M., Smemo, S., Kauwe, J. S., Grupe, A., Hinrichs, A., Mayo, K., Jiang, H., Thal, L. J., Goate, A. M., and Holtzman, D. M. (2007). Apolipoprotein E levels in cerebrospinal fluid and the effects of ABCA1 polymorphisms. Mol. Neurodegener. 2, 7.
Wakaumi, M., Ishibashi, K., Ando, H., Kasanuki, H., and Tsuruoka, S. (2005). Acute digoxin loading reduces ABCA8A mRNA expression in the mouse liver. Clin. Exp. Pharmacol. Physiol. 32, 1034–1041.
Wang, F., and Jia, J. (2007). Polymorphisms of cholesterol metabolism genes CYP46 and ABCA1 and the risk of sporadic Alzheimer’s disease in Chinese. Brain Res. 1147, 34–38.
Wang, N., Lan, D., Gerbod-Giannone, M., Linsel-Nitschke, P., Jehle, A. W., Chen, W., Martinez, L. O., and Tall, A. R. (2003). ATP-binding cassette transporter A7 (ABCA7) binds apolipoprotein A-I and mediates cellular phospholipid but not cholesterol efflux. J. Biol. Chem. 278, 42906–42912.
Wang, Y., Yamada, K., Tanaka, Y., Ishikawa, K., and Inagaki, N. (2005). Expression of ABCA2 protein in human vestibular schwannoma and peripheral nerve. J. Neurol. Sci. 232, 59–63.
Warren, M. S., Zerangue, N., Woodford, K., Roberts, L. M., Tate, E. H., Feng, B., Li, C., Feuerstein, T. J., Gibbs, J., Smith, B., de Morais, S. M., Dower, W. J., and Koller, K. J. (2009). Comparative gene expression profiles of ABC transporters in brain microvessel endothelial cells and brain in five species including human. Pharmacol. Res. 59, 404–413.
Wavrant-De, V. F., Compton, D., Womick, M., Arepalli, S., Adighibe, O., Li, L., Perez-Tur, J., and Hardy, J. (2007). ABCA1 polymorphisms and Alzheimer’s disease. Neurosci. Lett. 416, 180–183.
Wellington, C. L., Brunham, L. R., Zhou, S., Singaraja, R. R., Visscher, H., Gelfer, A., Ross, C., James, E., Liu, G., Huber, M. T., Yang, Y. Z., Parks, R. J., Groen, A., Fruchart-Najib, J., and Hayden, M. R. (2003). Alterations of plasma lipids in mice via adenoviral-mediated hepatic overexpression of human ABCA1. J. Lipid Res. 44, 1470–1480.
Weng, J., Mata, N. L., Azarian, S. M., Tzekov, R. T., Birch, D. G., and Travis, G. H. (1999). Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell 98, 13–23.
Wenzel, J. J., Kaminski, W. E., Piehler, A., Heimerl, S., Langmann, T., and Schmitz, G. (2003). ABCA10, a novel cholesterol-regulated ABCA6-like ABC transporter. Biochem. Biophys. Res. Commun. 306, 1089–1098.
Wenzel, J. J., Piehler, A., and Kaminski, W. E. (2007). ABC A-subclass proteins: gatekeepers of cellular phospho- and sphingolipid transport. Front. Biosci. 12, 3177–3193.
Wertz, P. W. (2000). Lipids and barrier function of the skin. Acta Derm. Venereol. Suppl. (Stockh.) 208, 7–11.
Wollmer, M. A., Kapaki, E., Hersberger, M., Muntwyler, J., Brunner, F., Tsolaki, M., Akatsu, H., Kosaka, K., Michikawa, M., Molyva, D., Paraskevas, G. P., Lutjohann, D., von, E. A., Hock, C., Nitsch, R. M., and Papassotiropoulos, A. (2006). Ethnicity-dependent genetic association of ABCA2 with sporadic Alzheimer’s disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141, 534–536.
Wollmer, M. A., Streffer, J. R., Lutjohann, D., Tsolaki, M., Iakovidou, V., Hegi, T., Pasch, T., Jung, H. H., Bergmann, K., Nitsch, R. M., Hock, C., and Papassotiropoulos, A. (2003). ABCA1 modulates CSF cholesterol levels and influences the age at onset of Alzheimer’s disease. Neurobiol. Aging 24, 421–426.
Xiong, H., Callaghan, D., Jones, A., Bai, J., Rasquinha, I., Smith, C., Pei, K., Walker, D., Lue, L. F., Stanimirovic, D., and Zhang, W. (2009). ABCG2 is upregulated in Alzheimer’s brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for Abeta(1-40) peptides. J. Neurosci. 29, 5463–5475.
Yamano, G., Funahashi, H., Kawanami, O., Zhao, L. X., Ban, N., Uchida, Y., Morohoshi, T., Ogawa, J., Shioda, S., and Inagaki, N. (2001). ABCA3 is a lamellar body membrane protein in human lung alveolar type II cells. FEBS Lett. 508, 221–225.
Yanagisawa, K. (2007). Role of gangliosides in Alzheimer’s disease. Biochim. Biophys. Acta 1768, 1943–1951.
Ye, D., Hoekstra, M., Out, R., Meurs, I., Kruijt, J. K., Hildebrand, R. B., Van Berkel, T. J., and Van, E. M. (2008). Hepatic cell-specific ATP-binding cassette (ABC) transporter profiling identifies putative novel candidates for lipid homeostasis in mice. Atherosclerosis 196, 650–658.
Ye, D., Meurs, I., Ohigashi, M., Calpe-Berdiel, L., Habets, K. L., Zhao, Y., Kubo, Y., Yamaguchi, A., Van Berkel, T. J., Nishi, T., and Van, E. M. (2010). Macrophage ABCA5 deficiency influences cellular cholesterol efflux and increases susceptibility to atherosclerosis in female LDLr knockout mice. Biochem. Biophys. Res. Commun. 395, 387–394.
Yokota, T., Matsumura, Y., Ban, N., Matsubayashi, T., and Inagaki, N. (2008). Heterozygous ABCA3 mutation associated with non-fatal evolution of respiratory distress. Eur. J. Pediatr. 167, 691–693.
Yvan-Charvet, L., Wang, N., and Tall, A. R. (2010). Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler. Thromb. Vasc. Biol. 30, 139–143.
Zhao, L. X., Zhou, C. J., Tanaka, A., Nakata, M., Hirabayashi, T., Amachi, T., Shioda, S., Ueda, K., and Inagaki, N. (2000). Cloning, characterization and tissue distribution of the rat ATP-binding cassette (ABC) transporter ABC2/ABCA2. Biochem. J. 350(Pt 3), 865–872.
Zhou, C., Zhao, L., Inagaki, N., Guan, J., Nakajo, S., Hirabayashi, T., Kikuyama, S., and Shioda, S. (2001). Atp-binding cassette transporter ABC2/ABCA2 in the rat brain: a novel mammalian lysosome-associated membrane protein and a specific marker for oligodendrocytes but not for myelin sheaths. J. Neurosci. 21, 849–857.
Keywords: ATP-binding cassette transporter, Alzheimer’s disease
Citation: Piehler AP, Özcürümez M and Kaminski WE (2012) A-subclass ATP-binding cassette proteins in brain lipid homeostasis and neurodegeneration. Front. Psychiatry 3:17. doi: 10.3389/fpsyt.2012.00017
Received: 29 December 2011; Paper pending published: 17 January 2012;
Accepted: 19 February 2012; Published online: 05 March 2012.
Edited by:
Silke Vogelgesang, University of Greifswald, GermanyReviewed by:
Michele Peters, Universitätsmedizin Greifswald, GermanyGabriele Jedlitschky, Universitätsmedizin Greifswald, Germany
Copyright: © 2012 Piehler, Özcürümez and Kaminski. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Wolfgang E. Kaminski, Institute for Clinical Chemistry, Medical Faculty Mannheim, University of Heidelberg, 68167 Mannheim, Germany. e-mail:d29sZmdhbmcua2FtaW5za2lAdW1tLmRl
†Armin P. Piehler and Mustafa Özcürümez have contributed equally to this work.