 Aaron Y. L. Cheung1,2
Aaron Y. L. Cheung1,2

- 1 Program in Developmental and Stem Cell Biology, The Hospital for Sick Children, Toronto, ON, Canada
- 2 Department of Molecular Genetics, University of Toronto, Toronto, ON, Canada
- 3 Department of Biochemistry and Molecular Biology, Pennsylvania State University, College of Medicine, Hershey, PA, USA
Rett syndrome (RTT) is a neurodevelopmental disorder that affects girls due primarily to heterozygous mutations in the X-linked gene encoding methyl-CpG binding protein 2 (MECP2). Random X-chromosome inactivation (XCI) results in cellular mosaicism in which some cells express wild-type (WT) MECP2 while other cells express mutant MECP2. The generation of patient-specific human induced pluripotent stem cells (hiPSCs) facilitates the production of RTT-hiPSC-derived neurons in vitro to investigate disease mechanisms and identify novel drug treatments. The generation of RTT-hiPSCs has been reported by many laboratories, however, the XCI status of RTT-hiPSCs has been inconsistent. Some report RTT-hiPSCs retain the inactive X-chromosome (post-XCI) of the founder somatic cell allowing isogenic RTT-hiPSCs that express only the WT or mutant MECP2 allele to be isolated from the same patient. Post-XCI RTT-hiPSCs-derived neurons retain this allele-specific expression pattern of WT or mutant MECP2. Conversely, others report RTT-hiPSCs in which the inactive X-chromosome of the founder somatic cell reactivates (pre-XCI) upon reprogramming into RTT-hiPSCs. Pre-XCI RTT-hiPSC-derived neurons exhibit random XCI resulting in cellular mosaicism with respect to WT and mutant MECP2 expression. Here we review and attempt to interpret the inconsistencies in XCI status of RTT-hiPSCs generated to date by comparison to other pluripotent systems in vitro and in vivo and the methods used to analyze XCI. Finally, we discuss the relative strengths and weaknesses of post- and pre-XCI hiPSCs in the context of RTT, and other X-linked and autosomal disorders for translational medicine.
Introduction
Rett syndrome [RTT (MIM 312750)] is a neurodevelopmental disorder that primarily affects young girls at an incidence of 1 in 10,000 live female births (Chahrour and Zoghbi, 2007). RTT girls develop normally until 6–18 months of age when they enter developmental arrest. Clinical features of RTT include microcephaly, stereotypic hand-washing movements, autistic features, loss of language, and mental retardation (Hagberg et al., 1983). Genetically, over 95% of RTT patients carry a heterozygous mutation in the X-linked gene encoding methyl-CpG binding protein 2 (MECP2; Amir et al., 1999). MECP2 functions as a transcriptional regulator by binding to the genome in a DNA methylation-dependent manner via its methyl-CpG binding domain and recruiting chromatin remodeling proteins via its transcriptional repression domain (Nan et al., 1993, 1997, 1998; Chahrour et al., 2008; Ben-Shachar et al., 2009; Skene et al., 2010). Other genes less commonly implicated in RTT include CDKL5 and FOXG1, which are located on chromosome X and 14, respectively (Scala et al., 2005; Ariani et al., 2008).
A complexity of the RTT story is that the MECP2 gene is located on the X-chromosome and is influenced by X-chromosome inactivation (XCI). XCI is the mammalian strategy to equalize X-linked gene dosage between XX females and XY males and involves transcriptionally silencing the majority of genes on one X-chromosome in females (Escamilla-Del-Arenal et al., 2011; Yang et al., 2011). This process initiates early in development; in the embryo proper this occurs at about the time of implantation. At its onset, XCI is random and either the maternally or paternally inherited X-chromosome is silenced in each cell. Subsequently, that X-chromosome remains the inactive X-chromosome (Xi) throughout all future cell divisions (Escamilla-Del-Arenal et al., 2011).
Regulation of XCI in both human and mouse requires the presence in cis of XIST, a 17 kb non-coding RNA (ncRNA; Brockdorff et al., 1991; Brown et al., 1991, 1992; Penny et al., 1996). XIST is only expressed from the Xi and its RNA product closely associates with or “coats” the chromosome (Brown et al., 1991, 1992; Clemson et al., 1996). Therefore, a key developmental event is to upregulate Xist from the future Xi. Both cis and trans-acting factors have been identified in mouse through the use of transgenes and targeted deletions (Donohoe et al., 2009; Barakat et al., 2011). In cis Xist is positively and negatively regulated by adjacent sequences and transcripts that include at least four ncRNAs (Debrand et al., 1999; Lee et al., 1999; Ogawa and Lee, 2003; Augui et al., 2007; Tian et al., 2010; Barakat et al., 2011). Perhaps the best characterized negative regulator is Tsix, an ncRNA antisense to Xist (Lee et al., 1999). While XIST is conserved between human and mouse, at least some events at the onset of XCI must differ between the species, as most ncRNAs including TSIX are poorly conserved (Chureau et al., 2002; Migeon et al., 2002).
Upon Xist upregulation, the Xi is heavily epigenetically remodeled, in many ways similar to other silenced genes throughout the genome. Epigenetic marks associated with the Xi include CpG island promoter DNA methylation (Hellman and Chess, 2007; Sharp et al., 2011), incorporation of histone variant MacroH2A (Costanzi and Pehrson, 1998), and modification of core histones (Keohane et al., 1996; de Napoles et al., 2004; Kohlmaier et al., 2004). An early event that follows XIST accumulation is the recruitment of the polycomb complex PRC2 that induces histone H3 trimethylation at lysine 27 (H3K27me3; Plath et al., 2003; Silva et al., 2003; Marks et al., 2009). Other epigenetic features, such as DNA methylation, accumulate later, and are important in the maintenance of XCI. Altogether these many alterations function with XIST to create a silenced nuclear compartment (Chow et al., 2010) that is spatially sequestered to the periphery of the nucleus and is cytologically recognizable as the darkly staining Barr body (Barr and Bertram, 1949).
Because of the random nature of XCI, RTT females are mosaic with both wild-type (WT) cells that inactivate the X-chromosome harboring the mutant allele and mutant cells that have inactivated the WT allele. Although XCI is random in most cases resulting in a 50:50 XCI ratio, it can deviate from this ratio. This results in a wide range of clinical presentations for RTT patients, even among related individuals carrying identical MECP2 mutant alleles, depending on the extent of favorable skewing (Archer et al., 2007).
One model to study RTT is to employ the technology of induced pluripotency. Human induced pluripotent stem cells (hiPSCs) are pluripotent stem cells generated from somatic cells by the introduction of a combination of pluripotency associated genes such as OCT4, SOX2, along with either KLF4 and c-MYC or NANOG and LIN28 via retroviral or lentiviral vectors (Takahashi et al., 2007; Yu et al., 2007; Park et al., 2008). Most importantly, hiPSCs are similar to human embryonic stem cells (hESCs) functionally as they are pluripotent and can potentially differentiate into any desired cell type when provided with the appropriate cues, but do not have the ethical issues surrounding hESCs (Murry and Keller, 2008). For these reasons, hiPSCs have huge potential in translational medicine such as disease modeling, drug screening, and cellular therapy. Indeed, patient-specific hiPSCs have been generated for a multitude of diseases, including many with a neurological basis, in which disease phenotypes have been recapitulated in vitro and proof-of-principle drug screening has been performed (Saha and Jaenisch, 2009; Ross and Ellis, 2010; Han et al., 2011; Marchetto et al., 2011; Zhu et al., 2011).
The generation of patient-specific hiPSCs from RTT girls has been an area of intense research as several groups have reported the generation of such cells (Hotta et al., 2009; Marchetto et al., 2010; Amenduni et al., 2011; Ananiev et al., 2011; Cheung et al., 2011; Kim et al., 2011; Pomp et al., 2011). RTT-hiPSCs generated by different groups have similar properties as they carry pathogenic mutations in MECP2 or CDKL5 and are pluripotent in vitro and in vivo. Most importantly, RTT-hiPSCs can be differentiated into affected neurons and exhibit RTT-associated phenotypes in vitro and can be rescued by transgene expression or drug treatments. However, with the generation of RTT-hiPSCs from multiple groups, the XCI status of RTT-hiPSCs, and more generally, female hiPSCs, has been variable. Some groups (Amenduni et al., 2011; Ananiev et al., 2011; Cheung et al., 2011; Pomp et al., 2011) reported the generation of RTT-hiPSCs that retain the Xi (post-XCI) from the founder somatic cell it was derived from, while others (Kim et al., 2009; Marchetto et al., 2010) reported the generation of some RTT-hiPSCs that reactivate the Xi of the founder somatic cell and hence carry two active X-chromosomes (pre-XCI).
Here we review the differences in XCI status reported in the RTT-hiPSC literature and attempt to discuss the differences and interpret the inconsistencies between post- and pre-XCI RTT-hiPSCs by considering the XCI status described in other pluripotent systems from mouse and human, both in vitro and in vivo. Given the variations in XCI status reported in the human pluripotent stem cell (hPSC) literature, we discuss different methods to accurately evaluate the XCI status. We discuss the advantages and disadvantages of post- and pre-XCI RTT-hiPSCs for translational medicine. Finally, we discuss the significant impact XCI has on hiPSCs generated from female individuals, especially those affected by X-linked disorders other than RTT, but also apparently unaffected females who may or may not be carriers of X-linked mutations, and perhaps also autosomal disorders.
XCI in the Mouse Pluripotent System
To place the relationship between XCI and RTT-hiPSCs into context, it is important to consider the XCI status of other pluripotent stem cell systems such as hiPSCs, hESCs, mouse induced pluripotent stem cells (miPSCs), and mouse embryonic stem cells (mESCs), and how they compare to their in vivo counterparts (Fan and Tran, 2011; van den Berg et al., 2011; Minkovsky et al., 2012). At embryonic day (e)3.5 of mouse embryogenesis, preimplantation epiblast cells carry two active X-chromosomes (Xa; Rastan, 1982; Takagi et al., 1982). Subsequently, random XCI ensues at gastrulation at around e5.5 (Rastan, 1982; Takagi et al., 1982). Xist RNA is expressed at low levels on both Xa until random XCI ensues in which the Xa represses Xist RNA while the Xi upregulates Xist RNA (Mak et al., 2004; Okamoto et al., 2004). Consistently, mESCs isolated from the preimplantation epiblast cells of the blastocysts at e3.5 carry two Xa, expressing Xist RNA in a biallelic manner (Evans and Kaufman, 1981; Martin, 1981; Lee et al., 1999). Similarly, the generation of miPSCs is accompanied by reactivation of the Xi in the founder somatic cell and hence carry two Xa followed by random XCI upon differentiation (Maherali et al., 2007; Stadtfeld et al., 2008a). This indicates that the generation of miPSCs involves the complete erasure of XCI. Subsequently, pluripotent stem cells known as mouse epiblast stem cells (mEpiSCs) have been isolated from the postimplantation epiblast at around e5.5 (Brons et al., 2007; Tesar et al., 2007). mEpiSCs carry an Xi similar to their in vivo counterpart in which random XCI has already ensued (Guo et al., 2009).
XCI in hESCs
HESCs are isolated from the preimplantation epiblast cells of the blastocyst (Thomson et al., 1998; Reubinoff et al., 2000). Studies of XCI in human embryos are much more limited than in mouse, but intriguingly suggest differences in XCI timing and XIST RNA expression (van den Berg et al., 2009; Okamoto et al., 2011). Unlike mouse, XIST RNA is upregulated in preimplantation blastocysts (van den Berg et al., 2009; Okamoto et al., 2011). The role of XIST RNA association in XCI at this early timepoint is not yet clear as two recent studies have shown different results; female preimplantation blastocysts had monoallelic XIST RNA upregulation and XCI hallmarks in one study (van den Berg et al., 2009), whereas another reported XIST RNA accumulation from all X-chromosomes, males and females, without gene silencing (Okamoto et al., 2011). Whether such heterogeneity exists between different human embryos or variability is introduced upon culturing, it appears that human XIST RNA coating and XCI are not strictly coupled.
Similarly, the XCI status of hESCs has been highly variable (Dhara and Benvenisty, 2004; Enver et al., 2005; Hoffman et al., 2005; Adewumi et al., 2007; Hall et al., 2008; Shen et al., 2008; Silva et al., 2008; Liu and Sun, 2009; Dvash et al., 2010; Diaz Perez et al., 2012). The XCI status of hESCs can be categorized into three classes as proposed by Lee and colleagues (Silva et al., 2008). Class I hESCs are in a pre-XCI state, express X-linked genes in a biallelic fashion, lack XCI marks such as XIST RNA and H3K27me3, and initiate XCI upon differentiation accompanied by upregulation of the same marks (Dhara and Benvenisty, 2004; Hall et al., 2008; Silva et al., 2008; Dvash et al., 2010; Diaz Perez et al., 2012). The most defining feature of Class I hESCs is initiation of random XCI upon differentiation, resulting in random monoallelic expression of X-linked genes (Dhara and Benvenisty, 2004). To date, few Class I hESCs have been isolated using conventional hESC conditions in atmospheric oxygen concentrations (20% O2) and basic FGF (bFGF)/knockout serum (KOSR; Dhara and Benvenisty, 2004; Hall et al., 2008; Silva et al., 2008; Dvash et al., 2010; Diaz Perez et al., 2012). Most hESCs have initiated XCI and thus are in a post-XCI state that can be subdivided according to the presence (Class II) or absence (Class III) of XCI marks such as XIST RNA and H3K27me3 (Hoffman et al., 2005; Hall et al., 2008; Shen et al., 2008; Silva et al., 2008; Liu and Sun, 2009; Dvash et al., 2010; Diaz Perez et al., 2012). Class II hESCs carry an Xi with a non-random skewing pattern resulting in non-random monoallelic expression of X-linked genes (Hoffman et al., 1987; Hall et al., 2008; Shen et al., 2008; Silva et al., 2008; Liu and Sun, 2009). This is likely due to clonal expansion of cells with one of the parental X-chromosomes inactivated (Liu and Sun, 2009). Class III hESCs can also retain an Xi despite the absence of such XCI marks (Shen et al., 2008; Silva et al., 2008; Diaz Perez et al., 2012). This indicates that XCI marks are not an accurate evaluation of XCI status in hESCs. Although most genes stay silenced in Class III hESCs, previously silenced X-linked genes may reactivate on a small scale accompanied by DNA hypomethylation of their promoters (Shen et al., 2008; Dvash et al., 2010). A recent analysis suggests that this reactivation may occur on a much larger scale with entire regions of the X-chromosome arms being reactivated (Bruck and Benvenisty, 2011). Overall, such sporadic X-linked gene reactivation appears higher than in somatic cells but is likely akin to levels in mouse cells that have lost Xist RNA (Wutz and Jaenisch, 2000; Csankovszki et al., 2001; Zhang et al., 2007). A defining feature of Class III hESCs is that they have already initiated XCI despite having lost the XCI marks. Therefore, they cannot initiate XCI again and do not upregulate XCI marks upon differentiation (Shen et al., 2008; Silva et al., 2008; Dvash et al., 2010; Diaz Perez et al., 2012).
HESCs with these three classes of XCI are hypothesized to be in a continuum and interrelated (Silva et al., 2008; Diaz Perez et al., 2012). It is thought that Class I hESCs represent the most pristine pluripotent stem cells present in the human blastocyst which contains two Xa (Okamoto et al., 2011). However, the culturing of Class I hESCs can result in a spontaneous transition into Class II hESCs in which XCI initiates and upregulates XCI marks (Silva et al., 2008). Class II hESCs, upon culture and/or cellular stresses such as freeze/thaw cycles, can also lose XCI marks such as XIST RNA and repressive chromatin marks, and thus transition into Class III hESCs (Hall et al., 2008; Shen et al., 2008; Silva et al., 2008; Dvash et al., 2010; Diaz Perez et al., 2012). Together, these data suggest that the XCI status of hESCs is subject to extensive epigenetic fluidity with respect to XCI.
Given the lack of correlation between XCI marks and XCI status in hESCs, Benvenisty and colleagues used an X-chromosome wide microarray analysis to investigate the expression level of a large number of X-linked genes as a functional read out of XCI in hPSCs, including hESCs and hiPSCs (Bruck and Benvenisty, 2011). Their microarray analysis revealed that hPSCs can be classified into No XCI, Full XCI, or Partial XCI categories. These three categories are defined by the proportion of X-linked genes that express twofold (i.e., two Xa) in comparison to male hPSCs. No XCI indicates most X-linked genes (>85%) express at twofold levels compared to male hPSCs. Full XCI indicates few X-linked genes (<15%) express at twofold levels compared to male hPSCs. Partial XCI indicates reactivation of a proportion (15–85%) of X-linked genes. Interestingly, it was observed that the location of partial inactivation tends to surround the XIST locus. XIST RNA expression, by quantitative reverse transcription polymerase chain reaction (qRT-PCR), was only partially correlated with these three states and would not have been able to distinguish between Full XCI and Partial XCI states. The term No XCI used in this study is fitting as it is not clear whether these hPSCs were generated directly with a pre-XCI phenotype, or from partial-XCI hPSCs that have reactivated X-linked genes on a chromosomal level resulting in an apparent pre-XCI phenotype. Thus, X-chromosome wide expression analysis via microarray could be used in determining the XCI status of the entire X-chromosome, but this method alone cannot identify pre-XCI hPSCs.
The two classifications above proposed by Lee and colleagues and Benvenisty and colleagues are both invaluable (Silva et al., 2008; Bruck and Benvenisty, 2011). XCI based solely on XCI marks such as XIST RNA and H3K27me3 is insufficient and suggests that candidate X-linked gene expression analysis in an allele-specific manner is crucial to functionally read out the XCI status of hESCs. In contrast, Benvenisty and colleagues categorized the hPSCs in their study based solely on expression of the entire X-chromosome. For the purpose of this review and to be consistent with the terminology used in most of the hESC conversion studies and the hiPSC literature discussed below, we prefer to categorize hPSCs as either pre-XCI or post-XCI. Pre-XCI hPSCs are identical to Class I hPSCs and carry two Xa without any signs of XCI initiation. Upon differentiation, pre-XCI hPSCs will initiate XCI and upregulate XCI marks in a random pattern resulting in random monoallelic expression of X-linked genes. Post-XCI hPSCs encompass both Class II and III hPSCs in that they have already initiated the process of XCI and carry an Xi regardless of the presence or absence of XCI marks. Differentiation of post-XCI hPSCs will yield a non-random XCI skewing pattern resulting in a non-random monoallelic expression of X-linked genes.
The largely post-XCI state of hESCs may be explained by the fact that they are thought to represent a cell type that is developmentally later than mESCs (Rossant, 2008; Silva and Smith, 2008; Nichols and Smith, 2009). It is thought that although hESCs are isolated from the preimplantation epiblast of the blastocyst, where there could be two Xa (Okamoto et al., 2011), they may in fact represent cells of the postimplantation epiblast where XCI has likely ensued as in the murine postimplantation epiblast (Rastan, 1982; Takagi et al., 1982). This is supported by the fact that hESCs are more similar to the murine in vitro counterpart of the postimplantation epiblast, mEpiSCs, than the murine in vitro counterpart of the preimplantation epiblast, mESCs. The pluripotent state of mESCs is maintained by cytokines including leukemia inhibitory factor (LIF) and bone morphogenic protein (BMP) 4 which activates the Jak/Stat pathway and induces inhibitor-of-differentiation proteins, respectively (Smith et al., 1988; Williams et al., 1988; Niwa et al., 1998; Matsuda et al., 1999; Ying et al., 2003). On the other hand, hESCs, similar to mEpiSCs, are maintained by fibroblast growth factor (FGF) which induces mitogen-activated protein kinases Erk1/2 and activin/nodal signaling (Thomson et al., 1998; Vallier et al., 2004; James et al., 2005; Brons et al., 2007; Kunath et al., 2007; Stavridis et al., 2007; Tesar et al., 2007). Furthermore, gene expression profiling reveals similarities between hESCs and mEpiSCs, but distinct from mESCs (Tesar et al., 2007). Therefore it is not surprising that hESCs are mostly in a post-XCI state similar to mEpiSCs (Guo et al., 2009).
Conversion of Mouse and Human Pluripotent Stem Cells to the Naïve State
To explain why hESCs isolated from the preimplantation epiblast would more closely resemble cells isolated from the postimplantation epiblast, it is important to consider that upon isolation, they are not frozen in developmental time in culture (Nichols and Smith, 2009). Indeed, it may be unnatural for hESCs (and mESCs) to expand in a pre-XCI state as this does not occur in development (Hall et al., 2008). Therefore, hESCs may preferentially undergo XCI during derivation and expansion. On the other hand, female mESCs appear to utilize a different strategy to accommodate for two Xa by frequently losing one X-chromosome (Zvetkova et al., 2005). Therefore, although hESCs may have a pre-XCI status during their isolation, they may continue to progress into the postimplantation epiblast stage (representative of mEpiSCs) and become post-XCI. To distinguish these two pluripotent stages, mESCs are classified to be in a naïve state, whereas mEpiSCs (and likely hESCs) are classified to be in a primed state (Silva and Smith, 2008; Nichols and Smith, 2009). Further evidence that these two stages are biologically relevant comes from experiments that show mESCs and mEpiSCs are distinct states that can be converted between one another. When cultured in conditions consisting of small molecules (known as the 2i cocktail) that inhibit FGF stimulation of mitogen-activated protein kinases Erk1/2 and constitutive activity of glycogen synthase kinase-3, mESCs are maintained in the most pristine naïve state of pluripotency (Ying et al., 2008). On the other hand, mESCs can be differentiated into mEpiSCs when placed in the culture conditions of the latter while introduction of a Klf4 transgene and growth in 2i conditions can convert mEpiSCs back to a naïve state reminiscent of mESCs (Guo et al., 2009). These studies suggest that the external milieu to which pluripotent stem cells are exposed has a significant impact on their pluripotent state and their XCI status.
Since the identification of optimal conditions for naïve pluripotency for mESCs and the hypothesis that hESCs may be more similar to mEpiSCs, attempts have been made to define conditions to isolate hESCs in a more naïve state of pluripotency in order to derive hESCs equivalent to mESCs. One approach that was examined was the isolation of hESCs under physiological oxygen concentrations (5% O2; Lengner et al., 2010). It was proposed that the atmospheric oxygen concentration (20% O2), in which conventional propagation of hESCs is performed is hyperoxic in comparison to the blastocyst in vivo, and may represent a suboptimal culture condition for hESCs. Indeed, when hESCs were established from human embryos in 5% O2, pre-XCI hESCs were derived that were capable of random XCI upon differentiation. On the other hand, hESCs derived under 20% O2 were all post-XCI with a non-random monoallelic expression pattern. Furthermore, when 5% O2 pre-XCI hESCs were subsequently exposed to 20% O2 or cellular stress (such as freeze/thaw), XCI ensued. This suggests that conventional hESC culture conditions (i.e., 20% O2) and other cellular stresses are detrimental for capturing and maintaining hESCs in a pre-XCI state. Conversely, post-XCI hESCs were not able to go through X-chromosome reactivation (XCR) when placed back in hypoxic conditions or after treatment with antioxidants. This suggests that XCI is irreversible (Lengner et al., 2010) or that other strategies are required for XCR (Hanna et al., 2010).
To that end, primed hESCs, with an Xi, have been converted to naïve hESCs, with two Xa, by continuous transgene expression of OCT4 and KLF4, or KLF4 and KLF2, combined with defined conditions including 2i and LIF (Hanna et al., 2010). The converted naïve hESCs revert back to a primed state when placed back in conventional hESC conditions (bFGF and KOSR) demonstrating that the naïve and primed states are interchangeable in hESCs when given the correct environmental cues along with exogenous transgenes. However, naïve hESCs were only maintained under continuous transgene expression demonstrating the need for further optimal culture conditions without genetic manipulation to capture this naïve pluripotency state. To this end, the authors identified Forskolin, a protein kinase A pathway agonist which induces the expression of KLF4 and KLF2. Forskolin can substitute for the continuous ectopic transgene expression although its effect was transient as naïve hESCs were only maintained for a limited number of passages. Similar methods in generating naïve hiPSCs can also be achieved by the exogenous transgene expression of OCT4, SOX2, KLF4, c-MYC, and NANOG in the presence of LIF although the XCI status of these cells was not examined (Buecker et al., 2010). Together, these data suggest that XCR is possible via the introduction of exogenous transgenes. However, if naïve hESCs were to be used clinically, non-integrating approaches will be preferred.
To that end, two small molecules, Sodium Butyrate (SB) and 3-deazaneplanocin A (DZNep), have been recently identified to promote the conversion and isolation of Class I hESCs (Diaz Perez et al., 2012). Note that here we employ the Class I, II, III, system to be consistent with the description by the authors (Diaz Perez et al., 2012). SB is a histone deacetylase inhibitor which can convert hESCs to a more naïve state similar to mESCs (Ware et al., 2009). DZNep depletes enhancer of zeste homolog2 (EZH2) resulting in a global decrease of H3K27me3 (Tan et al., 2007; Fiskus et al., 2009; Musch et al., 2010). When Class II hESCs were treated with SB or SB + DZNep, Class I hESCs emerged as indicated by the absence of H3K27me3 and lack of H3K4me3 exclusion on the X-chromosome, indicative of XCR (Diaz Perez et al., 2012). However, this effect was not 100% efficient, as only a proportion of cells within a culture of Class II hESCs can be converted. In contrast, Class III hESCs were not able to proceed through XCR by the same treatment. Conversely, treatment with SB or DZNep or both prevented Class I hESCs from transitioning into Class II, at least during the time frame (10 passages) analyzed by the authors. Altogether, these data suggest that although pre-XCI hESCs can be isolated, suboptimal culture conditions prevent facile maintenance of naïve hESCs in a pre-XCI state (Hanna et al., 2010; Lengner et al., 2010; Diaz Perez et al., 2012). Hence, hESCs continue to progress along the developmental timeline to stabilize in a primed state as post-XCI hESCs (Nichols and Smith, 2009).
XCI in hiPSCs
The XCI status of female hiPSCs has been extensively studied by Plath and colleagues. They found that most female hiPSCs in their study were post-XCI as indicated by XIST RNA and enrichment of the polycomb repressive complex EZH2 which mediates enrichment of H3K27me3, and depletion of active histone marks H3K18ac and H3K4me3 from the Xi (Tchieu et al., 2010). Furthermore, post-XCI hiPSCs expressed the X-linked genes XIST, ATRX, and PDHA1 in a non-random monoallelic fashion revealed by allele-specific SNP analysis. This suggests that during reprogramming, hiPSCs inherit the Xi from the founder somatic cell. Post-XCI hiPSCs were prone to losing XCI marks such as XIST RNA, EZH2, macroH2A1, and H4K20me1 upon extended passaging. However, these hiPSCs retained a transcriptionally silent Xi. This reiterates that evaluation of XCI marks such as XIST RNA and chromatin marks and their mediators is not sufficient to determine XCI status in hPSCs (Shen et al., 2008; Silva et al., 2008; Lengner et al., 2010; Diaz Perez et al., 2012). Finally, these data suggest that the non-random XCI nature of female hiPSCs can be exploited to generate isogenic control (expressing WT protein) and mutant (expressing mutant protein) hiPSCs from the same individual carrying heterozygous mutations in X-linked genes. Indeed, the authors were able to isolate isogenic control and mutant hiPSCs from Duchenne muscular dystrophy (DMD) carriers with a heterozygous mutation in the X-linked DYSTROPHIN gene (Tchieu et al., 2010).
Several groups have generated hiPSCs from RTT patients (Marchetto et al., 2010; Amenduni et al., 2011; Ananiev et al., 2011; Cheung et al., 2011; Kim et al., 2011; Pomp et al., 2011). The heterozygous mutation of the X-linked gene MECP2 in RTT-hiPSCs has prompted extensive analysis of their XCI status as it directly affects their functional read out (Figure 1). Post-XCI RTT-hiPSCs with non-random XCI will yield isogenic WT or mutant MECP2 expressing RTT-hiPSCs. Differentiation of post-XCI RTT-hiPSCs will yield cultures that maintain this non-random monoallelic expression pattern allowing the direct comparison of WT and mutant neurons (Figure 1A). Conversely, pre-XCI RTT-hiPSCs will carry two Xa and upon differentiation would yield a mosaic culture of WT or mutant MECP2 expressing neurons similar to RTT patients (Figure 1B).
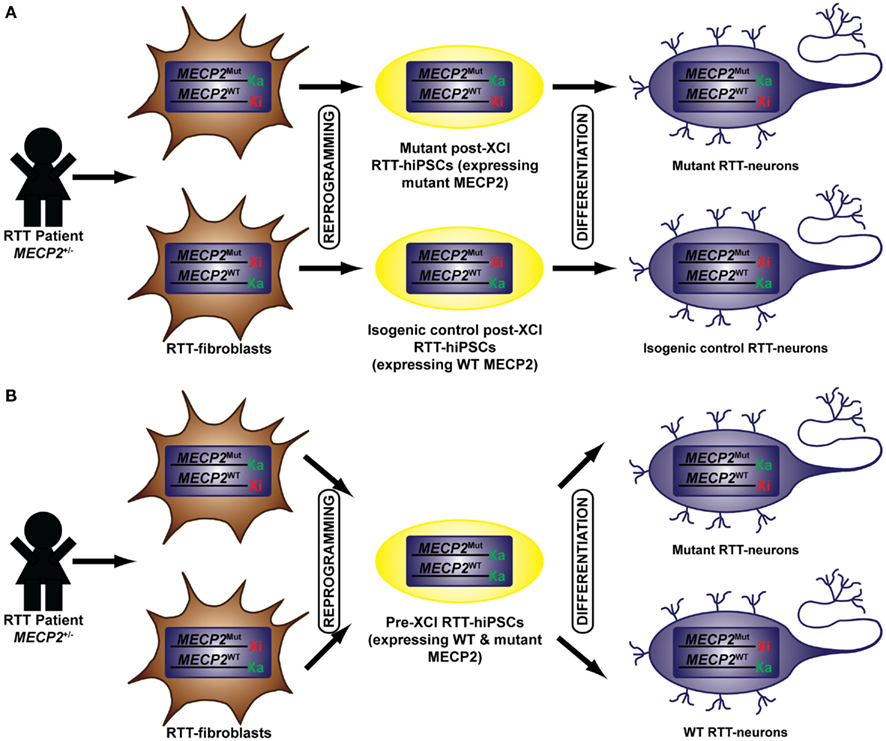
Figure 1. Generation of pre- and post-XCI RTT-hiPSCs. RTT-hiPSCs have been generated from RTT-fibroblasts isolated from RTT patients who carry a heterozygous mutation in MECP2. The reprogramming of RTT-fibroblasts have yielded RTT-hiPSCs that are in a post-XCI (A) or pre-XCI (B) state. Post-XCI RTT-hiPSCs retains an Xi from the founder somatic cell and express MECP2 in a non-random monoallelic manner. This results in the generation of mutant and isogenic control RTT-hiPSCs depending on whether the Xi harbors the WT or mutant MECP2. The differentiation of post-XCI RTT-hiPSCs retains this XCI pattern allowing homogeneous cultures of neurons that express WT or mutant MECP2. Pre-XCI RTT-hiPSCs carry two Xa and express the WT and mutant MECP2 in a biallelic fashion. The differentiation of RTT-hiPSCs into neurons initiate random XCI resulting in a mosaic culture of neurons that either express WT or mutant MECP2. Mut, mutant.
Post-XCI RTT-hiPSCs
Four studies report the generation of RTT-hiPSCs in a post-XCI state (Amenduni et al., 2011; Ananiev et al., 2011; Cheung et al., 2011; Pomp et al., 2011). In our study, X-chromosome activity and mosaicism were evaluated by XIST RNA-FISH and H3K27me3 immunofluorescence (IF; Cheung et al., 2011). We found RTT-hiPSCs that were positive or negative for these XCI marks within the same culture. The RTT-hiPSCs that are negative for XCI marks could be interpreted as: (1) loss of an X-chromosome, (2) two Xa, or (3) an Xi that has subsequently lost XCI marks but remains inactive. We rejected the first hypothesis as RTT-hiPSCs carried two X-chromosomes as assayed by X-centromere DNA-FISH and karyotyping. The second possibility, two Xa, was ruled out as RTT-hiPSC-derived neurons had a non-random XCI skewing pattern, as measured by the androgen receptor methylation (AR) assay (Allen et al., 1992), which is inconsistent with the presence of two Xa which would have otherwise resulted in random XCI upon differentiation. Therefore we favored the explanation that RTT-hiPSCs negative for XCI marks are post-XCI with an Xi that has subsequently lost those marks. Post-XCI RTT-hiPSCs had non-random XCI consistent with the fact that reprogramming retains the Xi in the founder somatic cell (Cheung et al., 2011). Finally, expression analysis, by direct sequencing of cDNA or qRT-PCR using WT MECP2-specific primers, confirmed that post-XCI RTT-hiPSCs and their neuronal derivatives expressed WT or mutant MECP2 in non-random monoallelic manner. This confirms the findings by Plath and colleagues that the non-random monoallelic post-XCI status of female hiPSCs can be exploited to generate isogenic control (expressing WT MECP2) and mutant (expressing mutant MECP2) hiPSCs from the same individual (Tchieu et al., 2010). Similarly, Renieri and colleagues and Chang and colleagues isolated isogenic control and mutant RTT-hiPSCs from some female patients carrying heterozygous mutations in the X-linked CDKL5 gene and MECP2, respectively (Amenduni et al., 2011; Ananiev et al., 2011). RTT-hiPSCs were post-XCI as they displayed non-random XCI skewing by AR assay and expressed CDKL5 or MECP2 in a non-random monoallelic manner allowing the isolation of isogenic control and mutant RTT-hiPSCs. Altogether, these studies demonstrate that post-XCI RTT-hiPSCs can be isolated, and become a particularly strong model system that allows direct comparison of mutant and isogenic control RTT-hiPSCs from the same individual (Amenduni et al., 2011; Ananiev et al., 2011; Cheung et al., 2011).
Colman and colleagues examined the dynamics of XCI during RTT-hiPSC establishment in order to implement strategies that were necessary to isolate pairs of mutant and isogenic control RTT-hiPSCs (Pomp et al., 2011). Similar to the RTT-hiPSCs described above (Amenduni et al., 2011; Ananiev et al., 2011; Cheung et al., 2011), RTT-hiPSCs displayed a non-random post-XCI pattern (Pomp et al., 2011). However, they were unable to isolate pairs of mutant and isogenic control post-XCI RTT-hiPSCs from the same individual. For example, RTT-hiPSCs generated from p.T158M MECP2 fibroblasts were all expressing WT MECP2, while RTT-hiPSCs generated from 1155del32 MECP2 fibroblasts were all expressing mutant MECP2. They referred to the expressing and silent X-chromosome as dominant (Xd) and unfavored (Xu), respectively. Fibroblasts carrying an active Xd had a higher reprogramming efficiency (50 ∼ 100 fold) than those carrying an active Xu. Interestingly, although early passage fibroblasts were mosaic for the two MECP2 alleles, serial passaging skewed the XCI toward the Xd in a predictable manner mirrored during reprogramming. This loss of mosaicism was due to a proliferative advantage of those fibroblasts carrying an active Xd, and an increase in DNA damage in fibroblasts carrying an active Xu. The authors hypothesized that this was a consequence of critically shortened telomeres resulting in telomere-associated DNA damage and senescence in fibroblasts expressing Xu.
To address the role of telomeres in reprogramming, Colman and colleagues introduced an exogenous telomerase reverse transcriptase (TERT) transgene into fibroblasts to prevent skewing (Pomp et al., 2011). Such TERT expression allowed the generation of isogenic control and mutant RTT-hiPSCs from the same individual. The reprogramming bias toward fibroblasts with an active Xd can also be overcome by increasing the number of hiPSC lines characterized, or by subcloning the fibroblasts expressing Xu prior to reprogramming. However, this remedy will not work on fibroblasts with a totally skewed population in which all the fibroblasts with an active Xu may have already been lost. To restore this lost population, they promoted naïve conversion of post-XCI RTT-hiPSCs to pre-XCI RTT-hiPSCs by culturing in 2i/LIF conditions as discussed above (Hanna et al., 2010). The resulting pre-XCI RTT-hiPSCs expressed both the WT and mutant MECP2 allele in a biallelic fashion and lost H3K27me3 suggesting XCR (Pomp et al., 2011). Upon differentiation, pre-XCI RTT-hiPSCs underwent random XCI and generated a mosaic culture of fibroblasts that expressed either the WT or mutant MECP2, thereby restoring the lost population expressing Xu. Although not demonstrated, the reprogramming of such fibroblasts expressing Xu should also yield isogenic control and mutant post-XCI RTT-hiPSCs. Together, these data suggest that there is an inherent difference in the two parental X-chromosomes, independent of the MECP2 mutation, which impacts the reprogramming efficiency of any given cell. This will ultimately impact one’s ability to generate isogenic control and mutant post-XCI RTT-hiPSCs from fibroblasts that have been extensively passaged. For example, we were only able to isolate isogenic control and mutant RTT-hiPSCs from primary fibroblasts carrying the Δ3–4 MECP2 mutation (Cheung et al., 2011). Similarly, Chang and colleagues and Renieri and colleagues isolated isogenic control RTT-hiPSCs from primary Q347X-fibroblasts (Amenduni et al., 2011), and primary R294X-fibroblasts (Ananiev et al., 2011), respectively. This is consistent with the findings by Colman and colleagues who suggest that the mosaic fibroblast culture is lost overtime. Therefore, the generation of isogenic control and mutant post-XCI RTT-hiPSCs should be more efficient from primary fibroblasts at earlier passages while the mosaic culture is still present (Pomp et al., 2011).
Pre-XCI RTT-hiPSCs
Two groups have isolated pre- and post-XCI RTT-hiPSCs (Marchetto et al., 2010; Kim et al., 2011). To evaluate the XCI status of RTT-hiPSCs, Muotri and colleagues performed XIST RNA-FISH and H3K27me3 IF (Marchetto et al., 2010). They reported both pre- and post-XCI RTT-hiPSCs based on the absence or presence of these XCI marks, respectively, in which the former was the focus for the rest of their study. Their pre-XCI RTT-hiPSCs initiated XCI upon differentiation into neurons as they induced the expression of XIST RNA and H3K27me3. To determine whether there was random XCI, the authors focused on RTT-hiPSCs from a patient carrying an 1155del32 mutation which results in a truncated MECP2 protein. By using a C-terminus MECP2 antibody, they could distinguish between the WT and mutant protein via IF. The pre-XCI RTT-hiPSCs were homogeneously MECP2-positive, suggesting biallelic expression of MECP2 as expected for two Xa. When pre-XCI RTT-hiPSCs were differentiated into neurons, there was a mosaic expression of MECP2-positive and -negative neurons. Furthermore, western blot analysis of pre-XCI RTT-hiPSC-derived neurons showed a reduction in MECP2 protein levels. Based on these findings, the authors conclude that pre-XCI RTT-hiPSCs initiate random XCI upon differentiation. However, the AR assay revealed that the pre-XCI RTT-hiPSC-derived neurons showed extreme skewing (96:4 to 98:2) inconsistent with random XCI. Thus, the neurons preferentially inactivated the AR gene on one parental X-chromosome but not randomly as would be expected. These results have been interpreted as suggesting that the neurons preferentially inactivated the parental X-chromosome carrying WT MECP2 (Walsh and Hochedlinger, 2010). However, it is also possible that during neuronal differentiation, dividing neuronal progenitors carrying a particular Xi may have a proliferative advantage causing the observed skewing in the resulting neurons (Pomp et al., 2011).
Park and colleagues were also able to isolate both pre- and post-XCI RTT-hiPSCs (Kim et al., 2011). Pre-XCI and post-XCI RTT-hiPSCs expressed MECP2 in a biallelic and monoallelic manner, respectively. Furthermore, the authors measured the total transcript levels of MECP2 via qRT-PCR and found that pre-XCI RTT-hiPSCs expressed MECP2 twofold in comparison to male hESCs while post-XCI RTT-hiPSCs expressed MECP2 at similar levels compared to male hESCs. With regards to XCI marks, pre-XCI and post-XCI RTT-hiPSCs were absent and present for H3K27me3, respectively. Based on these findings, the authors also concluded that they had isolated pre-XCI RTT-hiPSCs which carry two Xa and express MECP2 in a biallelic manner.
Inconsistencies between the XCI Status of RTT-hiPSCs
The generation of RTT-hiPSCs with different XCI status is worth reviewing (Marchetto et al., 2010; Amenduni et al., 2011; Ananiev et al., 2011; Cheung et al., 2011; Kim et al., 2011; Pomp et al., 2011). These differences are unlikely to be due to the starting fibroblasts as identical fibroblast cell lines were often used, as were similar culture conditions using bFGF/KOSR on mouse embryonic fibroblasts (MEFs; Table 1). One exception was that Muotri and colleagues used feeder-free conditions with mTeSR1 on matrigel (Marchetto et al., 2010). Whether this subtle difference can affect the XCI status of RTT-hiPSCs remains to be determined, although it is worth noting that the mTeSR1/matrigel conditions act as a substitute for the bFGF/KOSR conditions (Ludwig et al., 2006). Finally, reprogramming methods were similar between the different groups, as all used retroviral transduction of OCT4, SOX2, KLF4, and c-MYC, consistent with the lack of correlation between reprogramming methods and XCI status of female hiPSCs (Tchieu et al., 2010; Ananiev et al., 2011). Therefore, the starting cell type, culture conditions, and reprogramming methods were similar and are not strong candidates to explain the differences observed in the XCI status of RTT-hiPSCs.
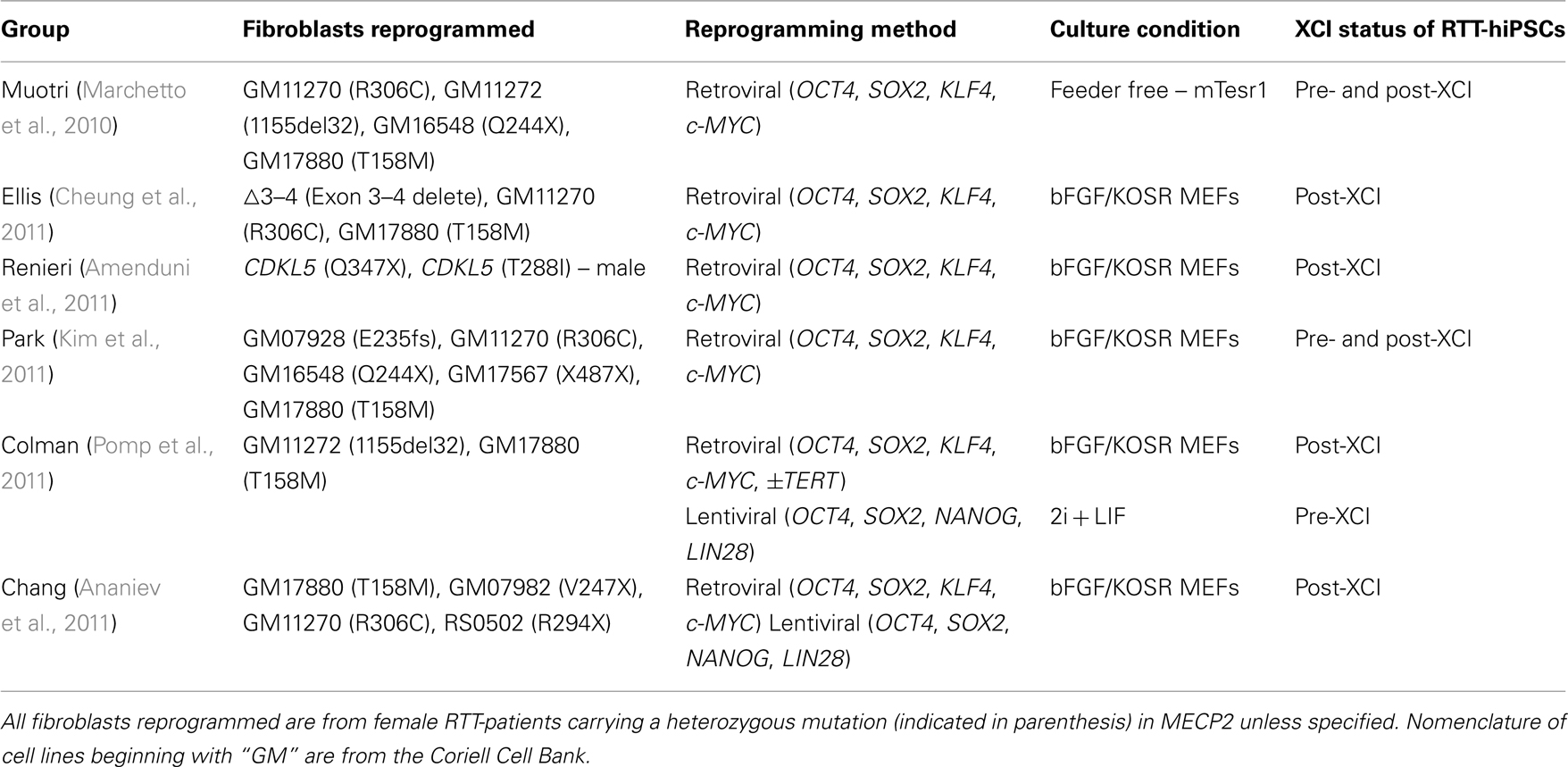
Table 1. Summary of RTT-fibroblasts reprogrammed, method of reprogramming, and the outcome of XCI status in RTT-hiPSCs.
Although we have opted to use the post- and pre-XCI nomenclature for this review, it is worth revisiting the classification system proposed by Lee and colleagues (Silva et al., 2008). Most RTT-hiPSCs reported are either Class II or III as they are post-XCI, exhibit non-random XCI skewing resulting in non-random monoallelic expression of MECP2 and maintain this expression pattern upon differentiation into neurons (Amenduni et al., 2011; Ananiev et al., 2011; Cheung et al., 2011; Pomp et al., 2011). XCI marker analyses have identified examples of RTT-hiPSCs that can be classified as Class II or III based on XIST RNA-FISH and/or H3K27me3 IF (Cheung et al., 2011; Kim et al., 2011; Pomp et al., 2011), while other studies cannot conclusively place their RTT-hiPSCs within Class II or III (Amenduni et al., 2011; Ananiev et al., 2011). On the other hand, the pre-XCI RTT-hiPSCs reported by Park and colleagues lacked H3K27me3 marks and expressed MECP2 twofold compared to male hESCs, consistent with a Class I assignment (Kim et al., 2011). However, this approach does not exclude the possibility of Class III hiPSCs that have lost XCI marks resulting in reactivation of X-linked genes (Silva et al., 2008). Likewise, the undifferentiated pre-XCI RTT-hiPSCs reported by Muotri and colleagues lacked XCI marks, such as XIST RNA and H3K27me3, and expressed MECP2 in a biallelic manner (Marchetto et al., 2010). Upon differentiation, the RTT-hiPSCs initiated XCI by upregulating XCI marks and exhibited a mosaic expression of WT and mutant MECP2 consistent with a Class I hiPSC. However, the apparent non-random skewing revealed by the AR assay in the RTT-hiPSC-derived neurons revealed a pattern characteristic of a Class II/III hiPSC. Overall, these data remain difficult to interpret. Further experiments to conclusively demonstrate the isolation of pre-XCI RTT-hiPSCs equivalent to Class I are required.
Evaluation of XCI in RTT-hiPSCs
Given the epigenetic fluidity that exists in hPSCs in the context of XCI (Hall et al., 2008; Shen et al., 2008; Silva et al., 2008; Dvash et al., 2010; Lengner et al., 2010; Tchieu et al., 2010; Pomp et al., 2011; Diaz Perez et al., 2012), it is critical to review how the XCI status was determined in RTT-hiPSCs and review the strengths and weaknesses of different technical approaches to accurately evaluate XCI (Tables 2 and 3). The most common method of determining the XCI status in RTT-hiPSCs was the evaluation of XCI markers such as XIST by RNA-FISH and repressive chromatin marks and their mediators such as H3K27me3 and EZH2 by IF (Table 2). However, the evaluation of XCI based solely on XIST RNA and/or chromatin marks and their mediators is insufficient to define the XCI status of hPSCs (Table 3). Another method that has been used for evaluation of XCI in RTT-hiPSCs is the AR assay, a method that measures the DNA methylation status of the X-linked AR gene (Table 2; Allen et al., 1992). This method has two advantages, (1) it measures DNA methylation, which is a likely epigenetic candidate that maintains the Xi in a transcriptionally repressed state in the absence of XISTRNA and other repressive chromatin marks, and (2) it measures the skewing of XCI (Table 3). Although not demonstrated, it may be possible to identify pre-XCI hPSCs with the AR assay, as they will display no signal indicative of two unmethylated Xa.
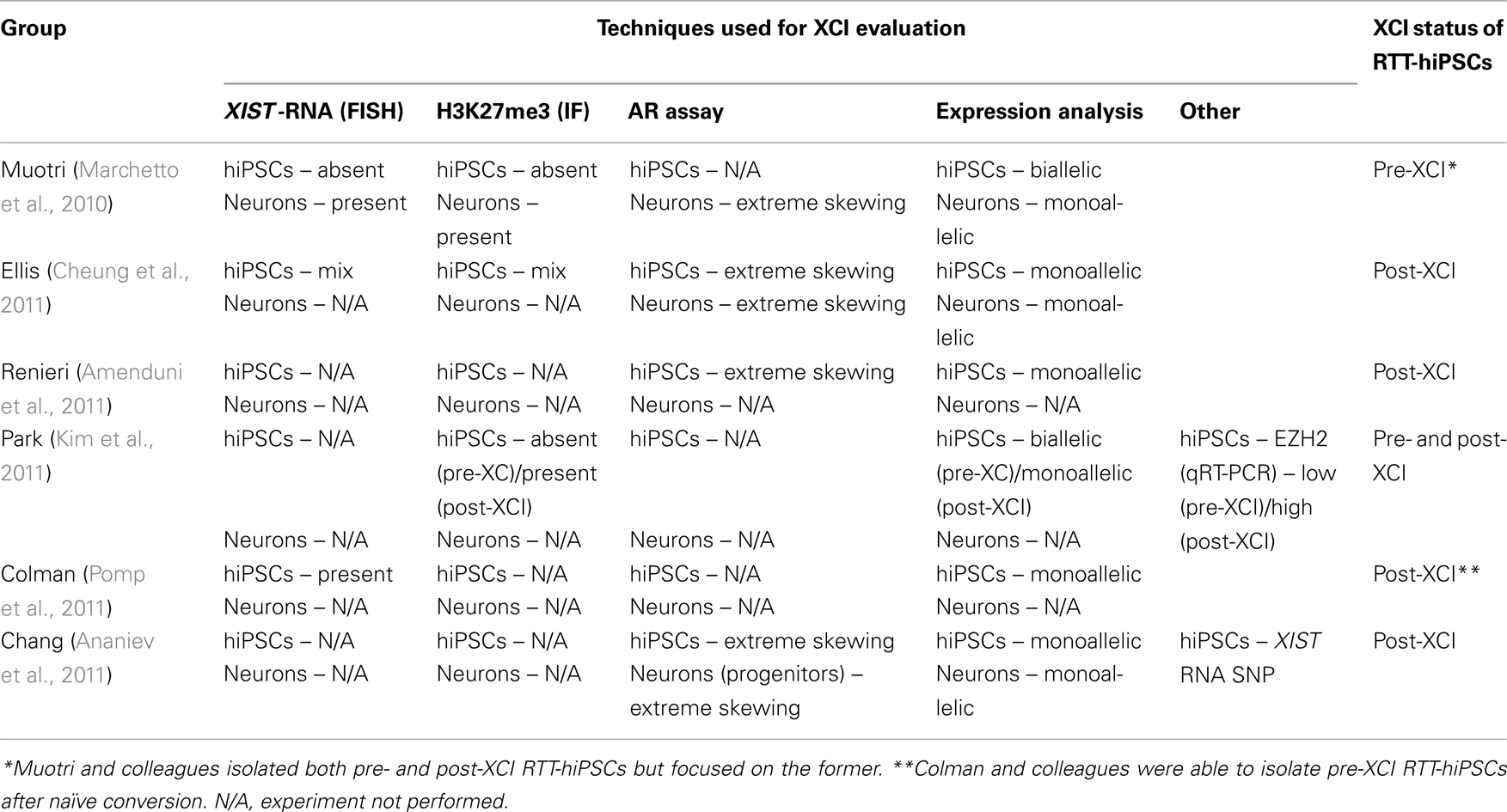
Table 2. Summary of RTT-hiPSCs generated to date and their XCI status as determined by different methods.

Table 3. Commonly used techniques to evaluate XCI status in hPSCs.
The importance of XCI is its functional outcome on the expression status of X-linked genes. For these reasons, expression analyses must be performed (Table 3). Preferably, expression analysis is done in a quantitative and allele-specific manner such as direct sequencing of cDNA or qRT-PCR using allele-specific primers at the population level. More stringently, allele-specific expression can be done at the single cell level by using antibodies (via IF) or probes (via RNA-FISH) that can distinguish between the WT or mutant allele. Whenever possible, it is preferable to measure the expression of both WT and mutant alleles although, in some cases, it may be difficult to design primers/antibodies/probes that can distinguish the two. Allele-specific expression analysis at the population level is important to distinguish non-random monoallelic expression of post-XCI RTT-hiPSCs from biallelic pre-XCI RTT-hiPSCs. Although biallelic expression may represent pre-XCI RTT-hiPSCs, the mix of two clones of post-XCI RTT-hiPSCs that express alternative parental X-chromosomes would also show the same expression profile. This mixture of two clones could be revealed by the presence of two different-sized peaks in the AR assay. The clonality of pre-XCI RTT-hiPSCs could also be confirmed by subcloning multiple sublines to examine whether they contain identical or distinct retroviral transgene integration sites by southern blot analysis.
Allele-specific expression analysis of MECP2 only measures the XCI status of that particular locus and may not represent the status of the entire the X-chromosome. Therefore, it may be worthwhile to use other approaches to measure the transcription level of the entire X-chromosome. This includes Cot1 RNA-FISH which highlight areas of actively transcribed heterogeneous nuclear RNA (hnRNA; van den Berg et al., 2009). The Xi is devoid of Cot1 RNA indicative of a transcriptionally silent compartment. This technique has been most widely used in the hESC literature (Hoffman et al., 1987; Hall et al., 2008; Silva et al., 2008). However, Cot1 RNA-FISH may not detect small scale reactivation of X-linked genes. Therefore, a more stringent method will be to perform X-chromosome wide expression analysis via microarray to measure the transcriptional activity of all X-linked genes (Table 3). It is important to note that the detection of twofold differences from the microarray inferred by male:female expression comparisons could be difficult and that allele-specific expression analysis (mentioned above) should be performed as independent validation. Given that these X-chromosome wide expression analyses have not been reported for RTT-hiPSC and their neuronal derivatives, we conclude that the methods employed to date on RTT-hiPSCs cannot unambiguously exclude the possibility of partial-XCI in either the post- or pre-XCI RTT-hiPSCs.
Finally, it is informative to investigate the XCI status of RTT-hiPSCs not only in their undifferentiated state but also upon differentiation. Pre-XCI RTT-hiPSCs should initiate XCI upon differentiation and exhibit a random mode of XCI revealed by the AR assay, accompanied by the upregulation of XCI marks such as XIST RNA and H3K27me3 (Dhara and Benvenisty, 2004; Hall et al., 2008; Silva et al., 2008; Dvash et al., 2010). Upon differentiation, X-linked genes should be expressed in a random monoallelic fashion (Dhara and Benvenisty, 2004) at the single cell level, which could be identified by RNA-FISH or IF. Differentiation of post-XCI RTT-hiPSCs, regardless of the presence or absence of XCI marks, will show a continued presence or absence of XCI marks, respectively, resulting in non-random XCI skewing and monoallelic expression pattern of X-linked genes (Hoffman et al., 1987; Hall et al., 2008; Shen et al., 2008; Silva et al., 2008; Liu and Sun, 2009; Dvash et al., 2010; Lengner et al., 2010; Tchieu et al., 2010; Cheung et al., 2011; Pomp et al., 2011; Diaz Perez et al., 2012). Altogether, the analysis of XCI upon differentiation of RTT-hiPSCs into neurons will be critical to conclusively demonstrate the isolation of pre-XCI RTT-hiPSCs in comparison to post-XCI RTT-hiPSCs.
Pros and Cons of Post- and Pre-XCI RTT-hiPSCs
The generation of both post- and pre-XCI RTT-hiPSCs offers investigators two different cell types to study RTT (Marchetto et al., 2010; Amenduni et al., 2011; Ananiev et al., 2011; Cheung et al., 2011; Kim et al., 2011; Pomp et al., 2011). Post-XCI RTT-hiPSCs are advantageous as they allow the generation of isogenic control and mutant RTT-hiPSCs from the same patient (Amenduni et al., 2011; Ananiev et al., 2011; Cheung et al., 2011; Pomp et al., 2011). For disease phenotyping, healthy control hiPSCs are essential and isogenic cells from the same patient eliminate genetic background effects that exist between individuals that may have an affect on subsequent phenotyping. Furthermore, isogenic RTT-hiPSCs may respond to directed differentiation cues in a more uniform manner compared to hPSCs generated from different individuals (Osafune et al., 2008; Hu et al., 2010). Directed differentiation of post-XCI RTT-hiPSCs will yield homogeneous cultures of neurons that express either WT or mutant MECP2 allowing simpler analysis of a population of cells without influence from the opposite allele being expressed as in pre-XCI RTT-hiPSCs (Cheung et al., 2011). Finally, the generation of isogenic control and mutant RTT-hiPSCs allow the mixing and matching of WT and mutant expressing cells in different proportions which provides an opportunity to study the effects of XCI skewing as observed in RTT patients (Archer et al., 2007). Furthermore, it will also allow the mixing and matching of different cell types such as neurons and glia to study the non-cell autonomous effects of non-neuronal cell types in RTT as has become apparent in the recent RTT literature (Ballas et al., 2009; Maezawa et al., 2009; Maezawa and Jin, 2010; Lioy et al., 2011).
However, there are also potential disadvantages to post-XCI RTT-hiPSCs. From a biological standpoint, one could question whether post-XCI RTT-hiPSCs are fully reprogrammed given that the Xi did not reactivate. This is likely in part due to suboptimal culture conditions and reprogramming methods required to capture and stabilize RTT-hiPSCs in a pre-XCI state (Nichols and Smith, 2009; Hanna et al., 2010; Lengner et al., 2010; Diaz Perez et al., 2012). Although post-XCI RTT-hiPSCs have been shown to exhibit non-random monoallelic expression of MECP2, the possibility that they have partial-XCI cannot be excluded (Amenduni et al., 2011; Ananiev et al., 2011; Cheung et al., 2011; Pomp et al., 2011). Additionally, it is important to note that although isogenic control and mutant post-XCI RTT-hiPSCs are described as isogenic, they are not truly isogenic by the strictest definition as illustrated by the following two points. First, all post-XCI RTT-hiPSCs generated to date were reprogrammed by integrating viral vectors (Amenduni et al., 2011; Ananiev et al., 2011; Cheung et al., 2011; Pomp et al., 2011). Given that each RTT-hiPSC line is expected to be an independent clone, they will harbor unique vector integration sites. Therefore, methods of reprogramming such as non-integrating viral vectors (Stadtfeld et al., 2008b; Seki et al., 2010), non-replicative episomal vectors (Yu et al., 2009), and/or mRNA- (Warren et al., 2010), miRNA- (Anokye-Danso et al., 2011; Miyoshi et al., 2011), or protein- (Kim et al., 2009) based reprogramming methods will be required to overcome this limitation. Secondly, the process of reprogramming itself leads to the accumulation of diverse abnormalities in the genome of hiPSCs (Mayshar et al., 2010; Gore et al., 2011; Hussein et al., 2011; Laurent et al., 2011; Lister et al., 2011). Therefore, each hiPSC line will carry unique abnormalities that may affect the subsequent phenotype of each hiPSC line. It is thus imperative to study more than one hiPSC line per individual, affected and/or unaffected, to ensure the validity of any phenotypes.
Pre-XCI RTT-hiPSCs are advantageous because their differentiation initiates random XCI allowing generation of a mosaic culture of neurons expressing WT or mutant MECP2 (Marchetto et al., 2010; Kim et al., 2011). This provides a clinically relevant system as RTT patients are mosaic with respect to WT and mutant MECP2 expression. However, such a mosaic culture may introduce a new variable as one needs to ensure that all differentiation experiments yield similar XCI skewing ratios. Furthermore, the mosaic culture creates difficulty when interpreting results, as any population analysis reads out both WT and mutant MECP2 expressing cells. One needs to perform single cell analysis and visualize WT and mutant cells separately, such as by using C-terminus specific antibodies of MECP2 that will not detect truncation mutations (i.e., 1155del32; Marchetto et al., 2010). However, this may be difficult on the more common point mutations of MECP2 due to the lack of mutation-specific antibodies (Percy et al., 2007; Marchetto et al., 2010; Cheung et al., 2011). Similar to the case of post-XCI RTT-hiPSCs, although pre-XCI RTT-hiPSCs have been shown to exhibit biallelic expression of MECP2, it cannot be excluded that pre-XCI hiPSCs may have partial-XCI (Marchetto et al., 2010; Kim et al., 2011). Nevertheless, Muotri and colleagues have extensively studied RTT phenotypes in pre-XCI RTT-hiPSC-derived neurons, and reported defects in glutamatergic synapse numbers, spine density, soma size, calcium signaling, and electrophysiological properties (Marchetto et al., 2010). Interestingly, Park and colleagues found a defect in neuronal differentiation from pre-XCI RTT-hiPSCs (Kim et al., 2011), which is novel in comparison to the RTT literature (Bauman et al., 1995; Chen et al., 2001; Kishi and Macklis, 2004; Marchetto et al., 2010; Ananiev et al., 2011) and warrants further investigation. Reassuringly, this phenotype was also observed in their mutant post-XCI RTT-hiPSCs (Kim et al., 2011). Both these studies demonstrate the potential of using pre-XCI RTT-hiPSCs for disease modeling (Marchetto et al., 2010; Kim et al., 2011).
Impact of XCI in Other Diseases
XCI in female hiPSCs has implications in other X-linked diseases including Fragile X Syndrome (FMR1), α-thalassemia (ATRX), Coffin-Lowry Syndrome (RSK2), DMD (DYSTROPHIN), Lesch-Nyhan Syndrome (HPRT), and Wiskott Aldrich Syndrome (WASP). In fact, many mental-disorder related genes are concentrated on the X-chromosome (Skuse, 2005; Gecz et al., 2009). DMD-hiPSCs have been generated and were shown to be post-XCI in a non-random pattern allowing the generation of isogenic control and mutant hiPSCs (Tchieu et al., 2010). HiPSCs have also been generated from Fragile X Syndrome patients although they were from male patients (Urbach et al., 2010). Perhaps more intriguing is the possibility of generating isogenic control and mutant post-XCI hiPSCs from carriers of X-linked diseases who otherwise may be apparently healthy. This will be particular advantageous in cases in which the proband is a male but has an unaffected mother and/or sisters who are carriers. Finally, the generation of post-XCI hiPSCs poses a unique opportunity for cellular therapy as a mosaic population of cells from a female patient can be reprogrammed into post-XCI hiPSCs in which hiPSCs can expressing the WT allele be used as a source for normal cells for cellular therapy purposes (Tchieu et al., 2010).
In the case of autosomal disorders, although XCI would not affect the primary gene of interest, female post-XCI hiPSCs generated from these patients may have a completely different X-chromosome transcriptome as each hiPSC line will carry either a paternal or maternal Xa. Therefore any heterozygous X-linked genes will be differentially expressed. It may be important to screen post-XCI hiPSCs, generated from female patients carrying an autosomal disorder, as well as unaffected females, by the AR assay to ensure that multiple post-XCI hiPSC lines used for downstream applications carry the same parental Xi.
Conclusion and Future Remarks
The generation of hiPSCs from RTT patients represents an inexhaustible source for in vitro derived patient-specific neurons, assuming that RTT-hiPSCs can be expanded indefinitely with a normal karyotype and stable genome. These neurons are useful for investigating the pathogenesis of RTT and have potential for use in drug screens and identification of novel compounds for therapy (Marchetto et al., 2010; Amenduni et al., 2011; Ananiev et al., 2011; Cheung et al., 2011; Kim et al., 2011; Pomp et al., 2011). For this potential to be realized, efficient protocols that direct differentiation into adult stage neurons of defined subtypes may be required (Hansen et al., 2011). The X-linked nature of MECP2 has prompted the extensive investigation of the XCI status of RTT-hiPSCs. Both post- and pre-XCI RTT-hiPSCs have been reported each with their own advantages and disadvantages for subsequent use. However, given the variation in XCI status seen in RTT-hiPSCs, it is imperative to accurately evaluate the XCI status in hiPSCs generated from RTT patients. Indeed, the XCI status of hiPSCs also has huge impact on other X-linked diseases, unaffected females who may or may not carry X-linked mutations, and perhaps also autosomal diseases.
To accurately evaluate the XCI status of RTT-hiPSCs, we propose the following workflow. RTT-hiPSCs should be screened first using the AR assay to distinguish potential pre-XCI RTT-hiPSCs that will show no peaks from potential post-XCI RTT-hiPSCs that retain an Xi as revealed by one peak. The AR assay is advantageous as a screening method given its relative technical ease to perform on a large number of samples. To confirm the candidate pre-XCI RTT-hiPSCs identified by the AR assay retain two Xa, allele-specific expression analysis of MECP2 by cDNA sequencing, qRT-PCR, RNA-FISH, or IF in an allele-specific manner will show a biallelic expression pattern. XCI marks will be absent as revealed by XIST RNA-FISH or H3K27me3 IF. It is important to differentiate candidate pre-XCI RTT-hiPSCs into neurons to demonstrate random XCI, as revealed by two equal peaks in the AR assay, accompanied by upregulation of XCI marks. Allele-specific expression analysis, preferably by RNA-FISH or IF at the single cell level, will yield a random monoallelic expression of MECP2. Only pre-XCI RTT-hiPSCs that meet these criteria will generate the desired unskewed mix of neurons expressing either the WT or mutant MECP2 allele. As these bona fide pre-XCI RTT-hiPSCs are passaged, it will be important to continue to assess their XCI status periodically, by performing AR assays for example, to ensure they are not transitioning into post- or partial-XCI RTT-hiPSCs.
The candidate post-XCI RTT-hiPSCs identified by presence of one peak in the initial AR assay could be divided into two groups with alternative parental X-chromosomes inactivated based on the different peak size of the AR assay. This will ensure that mutant and isogenic control RTT-hiPSCs can be isolated, and will facilitate identification of RTT-hiPSCs that express the Xu if the starting fibroblasts have been extensively passaged. Allele-specific MECP2 expression analysis of post-XCI RTT-hiPSCs will show non-random monoallelic expression and identify isogenic control and mutant RTT-hiPSCs. Post-XCI RTT-hiPSCs will be either positive or negative for XCI marks as revealed by XIST RNA-FISH or H3K27me3 IF. Differentiation of post-XCI RTT-hiPSCs will produce neurons that maintain the non-random XCI skewing pattern in AR assays and non-random monoallelic expression pattern of MECP2 by allele-specific expression analysis by RNA-FISH or IF at the single cell level. These post-XCI RTT-hiPSCs should also be subjected to XCI analyses periodically upon passaging, for example by AR assays, to ensure they do not transition into partial-XCI that have reactivated large regions of the X-chromosome.
The possibility that reported post- and pre-XCI RTT-hiPSCs may in fact exhibit partial-XCI has not yet been tested. Both post- and pre-XCI RTT-hiPSCs do however recapitulate the expected neuronal maturation and electrophysiological defects indicative of RTT. To fully classify XCI in the RTT-hiPSCs, it would be informative to perform X-chromosome wide expression analysis using microarrays. Nevertheless, the impact of a partial-XCI state on disease phenotyping in vitro remains to be determined.
Finally, to facilitate the isolation and stable maintenance of pre-XCI hPSCs that carry two Xa and are capable of initiating random XCI upon differentiation, it is important to continue to identify the optimal culture conditions that can capture this naïve state (Nichols and Smith, 2009). Efforts are underway to try to capture this pristine state of hPSCs by modifying oxygen concentrations, addition of exogenous transcription factors, small molecules, and/or using the 2i regimen, in which the latter has been successfully applied in the mouse system (Ying et al., 2008; Buecker et al., 2010; Hanna et al., 2010; Lengner et al., 2010; Diaz Perez et al., 2012). Generation of pre-XCI hPSCs will provide an invaluable in vitro system to study XCI in humans which will be essential as the mouse may be not be quite as similar as once thought (van den Berg et al., 2009, 2011; Fan and Tran, 2011; Okamoto et al., 2011; Minkovsky et al., 2012).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Canadian Institute for Health Research (MOP102649, IG194505, RMF92090 to James Ellis); the Ontario Ministry for Research and Innovation to James Ellis; the Beta Sigma Phi International Endowment Fund to James Ellis; the National Institutes of Health (HD056452 to Laura Carrel); Aaron Y. L. Cheung is supported by the Natural Sciences and Engineering Research Council of Canada, Ontario Student Opportunity Trust Funds (University of Toronto), and the Graduate Studies Endowment Funds (University of Toronto).
References
Adewumi, O., Aflatoonian, B., Ahrlund-Richter, L., Amit, M., Andrews, P. W., Beighton, G., Bello, P. A., Benvenisty, N., Berry, L. S., Bevan, S., Blum, B., Brooking, J., Chen, K. G., Choo, A. B., Churchill, G. A., Corbel, M., Damjanov, I., Draper, J. S., Dvorak, P., Emanuelsson, K., Fleck, R. A., Ford, A., Gertow, K., Gertsenstein, M., Gokhale, P. J., Hamilton, R. S., Hampl, A., Healy, L. E., Hovatta, O., Hyllner, J., Imreh, M. P., Itskovitz-Eldor, J., Jackson, J., Johnson, J. L., Jones, M., Kee, K., King, B. L., Knowles, B. B., Lako, M., Lebrin, F., Mallon, B. S., Manning, D., Mayshar, Y., McKay, R. D., Michalska, A. E., Mikkola, M., Mileikovsky, M., Minger, S. L., Moore, H. D., Mummery, C. L., Nagy, A., Nakatsuji, N., O’Brien, C. M., Oh, S. K., Olsson, C., Otonkoski, T., Park, K. Y., Passier, R., Patel, H., Patel, M., Pedersen, R., Pera, M. F., Piekarczyk, M. S., Pera, R. A., Reubinoff, B. E., Robins, A. J., Rossant, J., Rugg-Gunn, P., Schulz, T. C., Semb, H., Sherrer, E. S., Siemen, H., Stacey, G. N., Stojkovic, M., Suemori, H., Szatkiewicz, J., Turetsky, T., Tuuri, T., van den Brink, S., Vintersten, K., Vuoristo, S., Ward, D., Weaver, T. A., Young, L. A., and Zhang, W. (2007). Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat. Biotechnol. 25, 803–816.
Allen, R. C., Zoghbi, H. Y., Moseley, A. B., Rosenblatt, H. M., and Belmont, J. W. (1992). Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 51, 1229–1239.
Amenduni, M., De Filippis, R., Cheung, A. Y., Disciglio, V., Epistolato, M. C., Ariani, F., Mari, F., Mencarelli, M. A., Hayek, Y., Renieri, A., Ellis, J., and Meloni, I. (2011). iPS cells to model CDKL5-related disorders. Eur. J. Hum. Genet. 19, 1246–1255.
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188.
Ananiev, G., Williams, E. C., Li, H., and Chang, Q. (2011). Isogenic pairs of wild type and mutant induced pluripotent stem cell (iPSC) lines from Rett syndrome patients as in vitro disease model. PLoS ONE 6, e25255. doi:10.1371/journal.pone.0025255
Anokye-Danso, F., Trivedi, C. M., Juhr, D., Gupta, M., Cui, Z., Tian, Y., Zhang, Y., Yang, W., Gruber, P. J., Epstein, J. A., and Morrisey, E. E. (2011). Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 8, 376–388.
Archer, H., Evans, J., Leonard, H., Colvin, L., Ravine, D., Christodoulou, J., Williamson, S., Charman, T., Bailey, M. E., Sampson, J., de Klerk, N., and Clarke, A. (2007). Correlation between clinical severity in patients with Rett syndrome with a p.R168X or p.T158M MECP2 mutation, and the direction and degree of skewing of X-chromosome inactivation. J. Med. Genet. 44, 148–152.
Ariani, F., Hayek, G., Rondinella, D., Artuso, R., Mencarelli, M. A., Spanhol-Rosseto, A., Pollazzon, M., Buoni, S., Spiga, O., Ricciardi, S., Meloni, I., Longo, I., Mari, F., Broccoli, V., Zappella, M., and Renieri, A. (2008). FOXG1 is responsible for the congenital variant of Rett syndrome. Am. J. Hum. Genet. 83, 89–93.
Augui, S., Filion, G. J., Huart, S., Nora, E., Guggiari, M., Maresca, M., Stewart, A. F., and Heard, E. (2007). Sensing X chromosome pairs before X inactivation via a novel X-pairing region of the Xic. Science 318, 1632–1636.
Ballas, N., Lioy, D. T., Grunseich, C., and Mandel, G. (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 12, 311–317.
Barakat, T. S., Gunhanlar, N., Pardo, C. G., Achame, E. M., Ghazvini, M., Boers, R., Kenter, A., Rentmeester, E., Grootegoed, J. A., and Gribnau, J. (2011). RNF12 activates Xist and is essential for X chromosome inactivation. PLoS Genet. 7, e1002001. doi:10.1371/journal.pgen.1002001
Barr, M. L., and Bertram, E. G. (1949). A morphological distinction between neurones of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis. Nature 163, 676.
Bauman, M. L., Kemper, T. L., and Arin, D. M. (1995). Pervasive neuroanatomic abnormalities of the brain in three cases of Rett’s syndrome. Neurology 45, 1581–1586.
Ben-Shachar, S., Chahrour, M., Thaller, C., Shaw, C. A., and Zoghbi, H. Y. (2009). Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet. 18, 2431–2442.
Brockdorff, N., Ashworth, A., Kay, G. F., Cooper, P., Smith, S., McCabe, V. M., Norris, D. P., Penny, G. D., Patel, D., and Rastan, S. (1991). Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 351, 329–331.
Brons, I. G., Smithers, L. E., Trotter, M. W., Rugg-Gunn, P., Sun, B., Chuva de Sousa Lopes, S. M., Howlett, S. K., Clarkson, A., Ahrlund-Richter, L., Pedersen, R. A., and Vallier, L. (2007). Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 448, 191–195.
Brown, C. J., Ballabio, A., Rupert, J. L., Lafreniere, R. G., Grompe, M., Tonlorenzi, R., and Willard, H. F. (1991). A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349, 38–44.
Brown, C. J., Hendrich, B. D., Rupert, J. L., Lafreniere, R. G., Xing, Y., Lawrence, J., and Willard, H. F. (1992). The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 71, 527–542.
Bruck, T., and Benvenisty, N. (2011). Meta-analysis of the heterogeneity of X chromosome inactivation in human pluripotent stem cells. Stem Cell Res 6, 187–193.
Buecker, C., Chen, H. H., Polo, J. M., Daheron, L., Bu, L., Barakat, T. S., Okwieka, P., Porter, A., Gribnau, J., Hochedlinger, K., and Geijsen, N. (2010). A murine ESC-like state facilitates transgenesis and homologous recombination in human pluripotent stem cells. Cell Stem Cell 6, 535–546.
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., and Zoghbi, H. Y. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229.
Chahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437.
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331.
Cheung, A. Y., Horvath, L. M., Grafodatskaya, D., Pasceri, P., Weksberg, R., Hotta, A., Carrel, L., and Ellis, J. (2011). Isolation of MECP2-null Rett syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum. Mol. Genet. 20, 2103–2115.
Chow, J. C., Ciaudo, C., Fazzari, M. J., Mise, N., Servant, N., Glass, J. L., Attreed, M., Avner, P., Wutz, A., Barillot, E., Greally, J. M., Voinnet, O., and Heard, E. (2010). LINE-1 activity in facultative heterochromatin formation during X chromosome inactivation. Cell 141, 956–969.
Chureau, C., Prissette, M., Bourdet, A., Barbe, V., Cattolico, L., Jones, L., Eggen, A., Avner, P., and Duret, L. (2002). Comparative sequence analysis of the X-inactivation center region in mouse, human, and bovine. Genome Res. 12, 894–908.
Clemson, C. M., McNeil, J. A., Willard, H. F., and Lawrence, J. B. (1996). XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J. Cell Biol. 132, 259–275.
Costanzi, C., and Pehrson, J. R. (1998). Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 393, 599–601.
Csankovszki, G., Nagy, A., and Jaenisch, R. (2001). Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J. Cell Biol. 153, 773–784.
de Napoles, M., Mermoud, J. E., Wakao, R., Tang, Y. A., Endoh, M., Appanah, R., Nesterova, T. B., Silva, J., Otte, A. P., Vidal, M., Koseki, H., and Brockdorff, N. (2004). Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell 7, 663–676.
Debrand, E., Chureau, C., Arnaud, D., Avner, P., and Heard, E. (1999). Functional analysis of the DXPas34 locus, a 3′ regulator of Xist expression. Mol. Cell. Biol. 19, 8513–8525.
Dhara, S. K., and Benvenisty, N. (2004). Gene trap as a tool for genome annotation and analysis of X chromosome inactivation in human embryonic stem cells. Nucleic Acids Res. 32, 3995–4002.
Diaz Perez, S. V., Kim, R., Li, Z., Marquez, V. E., Patel, S., Plath, K., and Clark, A. T. (2012). Derivation of new human embryonic stem cell lines reveals rapid epigenetic progression in vitro that can be prevented by chemical modification of chromatin. Hum. Mol. Genet. 21, 751–764.
Donohoe, M. E., Silva, S. S., Pinter, S. F., Xu, N., and Lee, J. T. (2009). The pluripotency factor Oct4 interacts with Ctcf and also controls X-chromosome pairing and counting. Nature 460, 128–132.
Dvash, T., Lavon, N., and Fan, G. (2010). Variations of X chromosome inactivation occur in early passages of female human embryonic stem cells. PLoS ONE 5, e11330. doi:10.1371/journal.pone.0011330
Enver, T., Soneji, S., Joshi, C., Brown, J., Iborra, F., Orntoft, T., Thykjaer, T., Maltby, E., Smith, K., Abu Dawud, R., Jones, M., Matin, M., Gokhale, P., Draper, J., and Andrews, P. W. (2005). Cellular differentiation hierarchies in normal and culture-adapted human embryonic stem cells. Hum. Mol. Genet. 14, 3129–3140.
Escamilla-Del-Arenal, M., da Rocha, S. T., and Heard, E. (2011). Evolutionary diversity and developmental regulation of X-chromosome inactivation. Hum. Genet. 130, 307–327.
Evans, M. J., and Kaufman, M. H. (1981). Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156.
Fan, G., and Tran, J. (2011). X chromosome inactivation in human and mouse pluripotent stem cells. Hum. Genet. 130, 217–222.
Fiskus, W., Wang, Y., Sreekumar, A., Buckley, K. M., Shi, H., Jillella, A., Ustun, C., Rao, R., Fernandez, P., Chen, J., Balusu, R., Koul, S., Atadja, P., Marquez, V. E., and Bhalla, K. N. (2009). Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood 114, 2733–2743.
Gecz, J., Shoubridge, C., and Corbett, M. (2009). The genetic landscape of intellectual disability arising from chromosome X. Trends Genet. 25, 308–316.
Gore, A., Li, Z., Fung, H. L., Young, J. E., Agarwal, S., Antosiewicz-Bourget, J., Canto, I., Giorgetti, A., Israel, M. A., Kiskinis, E., Lee, J. H., Loh, Y. H., Manos, P. D., Montserrat, N., Panopoulos, A. D., Ruiz, S., Wilbert, M. L., Yu, J., Kirkness, E. F., Izpisua Belmonte, J. C., Rossi, D. J., Thomson, J. A., Eggan, K., Daley, G. Q., Goldstein, L. S., and Zhang, K. (2011). Somatic coding mutations in human induced pluripotent stem cells. Nature 471, 63–67.
Guo, G., Yang, J., Nichols, J., Hall, J. S., Eyres, I., Mansfield, W., and Smith, A. (2009). Klf4 reverts developmentally programmed restriction of ground state pluripotency. Development 136, 1063–1069.
Hagberg, B., Aicardi, J., Dias, K., and Ramos, O. (1983). A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann. Neurol. 14, 471–479.
Hall, L. L., Byron, M., Butler, J., Becker, K. A., Nelson, A., Amit, M., Itskovitz-Eldor, J., Stein, J., Stein, G., Ware, C., and Lawrence, J. B. (2008). X-inactivation reveals epigenetic anomalies in most hESC but identifies sublines that initiate as expected. J. Cell. Physiol. 216, 445–452.
Han, S. S., Williams, L. A., and Eggan, K. C. (2011). Constructing and deconstructing stem cell models of neurological disease. Neuron 70, 626–644.
Hanna, J., Cheng, A. W., Saha, K., Kim, J., Lengner, C. J., Soldner, F., Cassady, J. P., Muffat, J., Carey, B. W., and Jaenisch, R. (2010). Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc. Natl. Acad. Sci. U.S.A. 107, 9222–9227.
Hansen, D. V., Rubenstein, J. L., and Kriegstein, A. R. (2011). Deriving excitatory neurons of the neocortex from pluripotent stem cells. Neuron 70, 645–660.
Hellman, A., and Chess, A. (2007). Gene body-specific methylation on the active X chromosome. Science 315, 1141–1143.
Hoffman, E. P., Brown, R. H. Jr., and Kunkel, L. M. (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928.
Hoffman, L. M., Hall, L., Batten, J. L., Young, H., Pardasani, D., Baetge, E. E., Lawrence, J., and Carpenter, M. K. (2005). X-inactivation status varies in human embryonic stem cell lines. Stem Cells 23, 1468–1478.
Hotta, A., Cheung, A. Y., Farra, N., Vijayaragavan, K., Seguin, C. A., Draper, J. S., Pasceri, P., Maksakova, I. A., Mager, D. L., Rossant, J., Bhatia, M., and Ellis, J. (2009). Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat. Methods 6, 370–376.
Hu, B. Y., Weick, J. P., Yu, J., Ma, L. X., Zhang, X. Q., Thomson, J. A., and Zhang, S. C. (2010). Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. U.S.A. 107, 4335–4340.
Hussein, S. M., Batada, N. N., Vuoristo, S., Ching, R. W., Autio, R., Narva, E., Ng, S., Sourour, M., Hamalainen, R., Olsson, C., Lundin, K., Mikkola, M., Trokovic, R., Peitz, M., Brustle, O., Bazett-Jones, D. P., Alitalo, K., Lahesmaa, R., Nagy, A., and Otonkoski, T. (2011). Copy number variation and selection during reprogramming to pluripotency. Nature 471, 58–62.
James, D., Levine, A. J., Besser, D., and Hemmati-Brivanlou, A. (2005). TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development 132, 1273–1282.
Keohane, A. M., O’Neill L, P., Belyaev, N. D., Lavender, J. S., and Turner, B. M. (1996). X-inactivation and histone H4 acetylation in embryonic stem cells. Dev. Biol. 180, 618–630.
Kim, D., Kim, C. H., Moon, J. I., Chung, Y. G., Chang, M. Y., Han, B. S., Ko, S., Yang, E., Cha, K. Y., Lanza, R., and Kim, K. S. (2009). Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 4, 472–476.
Kim, K. Y., Hysolli, E., and Park, I. H. (2011). Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 108, 14169–14174.
Kishi, N., and Macklis, J. D. (2004). MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell. Neurosci. 27, 306–321.
Kohlmaier, A., Savarese, F., Lachner, M., Martens, J., Jenuwein, T., and Wutz, A. (2004). A chromosomal memory triggered by Xist regulates histone methylation in X inactivation. PLoS Biol. 2, E171. doi:10.1371/journal.pbio.0020171
Kunath, T., Saba-El-Leil, M. K., Almousailleakh, M., Wray, J., Meloche, S., and Smith, A. (2007). FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development 134, 2895–2902.
Laurent, L. C., Ulitsky, I., Slavin, I., Tran, H., Schork, A., Morey, R., Lynch, C., Harness, J. V., Lee, S., Barrero, M. J., Ku, S., Martynova, M., Semechkin, R., Galat, V., Gottesfeld, J., Izpisua Belmonte, J. C., Murry, C., Keirstead, H. S., Park, H. S., Schmidt, U., Laslett, A. L., Muller, F. J., Nievergelt, C. M., Shamir, R., and Loring, J. F. (2011). Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell 8, 106–118.
Lee, J. T., Davidow, L. S., and Warshawsky, D. (1999). Tsix, a gene antisense to Xist at the X-inactivation centre. Nat. Genet. 21, 400–404.
Lengner, C. J., Gimelbrant, A. A., Erwin, J. A., Cheng, A. W., Guenther, M. G., Welstead, G. G., Alagappan, R., Frampton, G. M., Xu, P., Muffat, J., Santagata, S., Powers, D., Barrett, C. B., Young, R. A., Lee, J. T., Jaenisch, R., and Mitalipova, M. (2010). Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell 141, 872–883.
Lioy, D. T., Garg, S. K., Monaghan, C. E., Raber, J., Foust, K. D., Kaspar, B. K., Hirrlinger, P. G., Kirchhoff, F., Bissonnette, J. M., Ballas, N., and Mandel, G. (2011). A role for glia in the progression of Rett’s syndrome. Nature 475, 497–500.
Lister, R., Pelizzola, M., Kida, Y. S., Hawkins, R. D., Nery, J. R., Hon, G., Antosiewicz-Bourget, J., O’Malley, R., Castanon, R., Klugman, S., Downes, M., Yu, R., Stewart, R., Ren, B., Thomson, J. A., Evans, R. M., and Ecker, J. R. (2011). Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 471, 68–73.
Liu, W., and Sun, X. (2009). Skewed X chromosome inactivation in diploid and triploid female human embryonic stem cells. Hum. Reprod. 24, 1834–1843.
Ludwig, T. E., Bergendahl, V., Levenstein, M. E., Yu, J., Probasco, M. D., and Thomson, J. A. (2006). Feeder-independent culture of human embryonic stem cells. Nat. Methods 3, 637–646.
Maezawa, I., and Jin, L. W. (2010). Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 30, 5346–5356.
Maezawa, I., Swanberg, S., Harvey, D., LaSalle, J. M., and Jin, L. W. (2009). Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J. Neurosci. 29, 5051–5061.
Maherali, N., Sridharan, R., Xie, W., Utikal, J., Eminli, S., Arnold, K., Stadtfeld, M., Yachechko, R., Tchieu, J., Jaenisch, R., Plath, K., and Hochedlinger, K. (2007). Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 1, 55–70.
Mak, W., Nesterova, T. B., de Napoles, M., Appanah, R., Yamanaka, S., Otte, A. P., and Brockdorff, N. (2004). Reactivation of the paternal X chromosome in early mouse embryos. Science 303, 666–669.
Marchetto, M. C., Brennand, K. J., Boyer, L., and Gage, F. H. (2011). Induced pluripotent stem cells (iPSC) and neurologic disease modeling: progress and promises. Hum. Mol. Genet. 20, R109–R115.
Marchetto, M. C., Carromeu, C., Acab, A., Yu, D., Yeo, G. W., Mu, Y., Chen, G., Gage, F. H., and Muotri, A. R. (2010). A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539.
Marks, H., Chow, J. C., Denissov, S., Francoijs, K. J., Brockdorff, N., Heard, E., and Stunnenberg, H. G. (2009). High-resolution analysis of epigenetic changes associated with X inactivation. Genome Res. 19, 1361–1373.
Martin, G. R. (1981). Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. U.S.A. 78, 7634–7638.
Matsuda, T., Nakamura, T., Nakao, K., Arai, T., Katsuki, M., Heike, T., and Yokota, T. (1999). STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J. 18, 4261–4269.
Mayshar, Y., Ben-David, U., Lavon, N., Biancotti, J. C., Yakir, B., Clark, A. T., Plath, K., Lowry, W. E., and Benvenisty, N. (2010). Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 7, 521–531.
Migeon, B. R., Lee, C. H., Chowdhury, A. K., and Carpenter, H. (2002). Species differences in TSIX/Tsix reveal the roles of these genes in X-chromosome inactivation. Am. J. Hum. Genet. 71, 286–293.
Minkovsky, A., Patel, S., and Plath, K. (2012). Pluripotency and the transcriptional inactivation of the female mammalian X chromosome. Stem Cells 30, 48–54.
Miyoshi, N., Ishii, H., Nagano, H., Haraguchi, N., Dewi, D. L., Kano, Y., Nishikawa, S., Tanemura, M., Mimori, K., Tanaka, F., Saito, T., Nishimura, J., Takemasa, I., Mizushima, T., Ikeda, M., Yamamoto, H., Sekimoto, M., Doki, Y., and Mori, M. (2011). Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 8, 633–638.
Murry, C. E., and Keller, G. (2008). Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell 132, 661–680.
Musch, T., Oz, Y., Lyko, F., and Breiling, A. (2010). Nucleoside drugs induce cellular differentiation by caspase-dependent degradation of stem cell factors. PLoS ONE 5, e10726. doi:10.1371/journal.pone.0010726
Nan, X., Campoy, F. J., and Bird, A. (1997). MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88, 471–481.
Nan, X., Meehan, R. R., and Bird, A. (1993). Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 21, 4886–4892.
Nan, X., Ng, H. H., Johnson, C. A., Laherty, C. D., Turner, B. M., Eisenman, R. N., and Bird, A. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389.
Niwa, H., Burdon, T., Chambers, I., and Smith, A. (1998). Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 12, 2048–2060.
Ogawa, Y., and Lee, J. T. (2003). Xite, X-inactivation intergenic transcription elements that regulate the probability of choice. Mol. Cell 11, 731–743.
Okamoto, I., Otte, A. P., Allis, C. D., Reinberg, D., and Heard, E. (2004). Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 303, 644–649.
Okamoto, I., Patrat, C., Thepot, D., Peynot, N., Fauque, P., Daniel, N., Diabangouaya, P., Wolf, J. P., Renard, J. P., Duranthon, V., and Heard, E. (2011). Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature 472, 370–374.
Osafune, K., Caron, L., Borowiak, M., Martinez, R. J., Fitz-Gerald, C. S., Sato, Y., Cowan, C. A., Chien, K. R., and Melton, D. A. (2008). Marked differences in differentiation propensity among human embryonic stem cell lines. Nat. Biotechnol. 26, 313–315.
Park, I. H., Zhao, R., West, J. A., Yabuuchi, A., Huo, H., Ince, T. A., Lerou, P. H., Lensch, M. W., and Daley, G. Q. (2008). Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451, 141–146.
Penny, G. D., Kay, G. F., Sheardown, S. A., Rastan, S., and Brockdorff, N. (1996). Requirement for Xist in X chromosome inactivation. Nature 379, 131–137.
Percy, A. K., Lane, J. B., Childers, J., Skinner, S., Annese, F., Barrish, J., Caeg, E., Glaze, D. G., and MacLeod, P. (2007). Rett syndrome: North American database. J. Child Neurol. 22, 1338–1341.
Plath, K., Fang, J., Mlynarczyk-Evans, S. K., Cao, R., Worringer, K. A., Wang, H., de la Cruz, C. C., Otte, A. P., Panning, B., and Zhang, Y. (2003). Role of histone H3 lysine 27 methylation in X inactivation. Science 300, 131–135.
Pomp, O., Dreesen, O., Leong, D. F., Meller-Pomp, O., Tan, T. T., Zhou, F., and Colman, A. (2011). Unexpected X chromosome skewing during culture and reprogramming of human somatic cells can be alleviated by exogenous telomerase. Cell Stem Cell 9, 156–165.
Rastan, S. (1982). Timing of X-chromosome inactivation in postimplantation mouse embryos. J. Embryol. Exp. Morphol. 71, 11–24.
Reubinoff, B. E., Pera, M. F., Fong, C. Y., Trounson, A., and Bongso, A. (2000). Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat. Biotechnol. 18, 399–404.
Ross, P. J., and Ellis, J. (2010). Modeling complex neuropsychiatric disease with induced pluripotent stem cells. F1000 Biol. Rep. 2, 84.
Saha, K., and Jaenisch, R. (2009). Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell 5, 584–595.
Scala, E., Ariani, F., Mari, F., Caselli, R., Pescucci, C., Longo, I., Meloni, I., Giachino, D., Bruttini, M., Hayek, G., Zappella, M., and Renieri, A. (2005). CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J. Med. Genet. 42, 103–107.
Seki, T., Yuasa, S., Oda, M., Egashira, T., Yae, K., Kusumoto, D., Nakata, H., Tohyama, S., Hashimoto, H., Kodaira, M., Okada, Y., Seimiya, H., Fusaki, N., Hasegawa, M., and Fukuda, K. (2010). Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 7, 11–14.
Sharp, A. J., Stathaki, E., Migliavacca, E., Brahmachary, M., Montgomery, S. B., Dupre, Y., and Antonarakis, S. E. (2011). DNA methylation profiles of human active and inactive X chromosomes. Genome Res. 21, 1592–1600.
Shen, Y., Matsuno, Y., Fouse, S. D., Rao, N., Root, S., Xu, R., Pellegrini, M., Riggs, A. D., and Fan, G. (2008). X-inactivation in female human embryonic stem cells is in a nonrandom pattern and prone to epigenetic alterations. Proc. Natl. Acad. Sci. U.S.A. 105, 4709–4714.
Silva, J., Mak, W., Zvetkova, I., Appanah, R., Nesterova, T. B., Webster, Z., Peters, A. H., Jenuwein, T., Otte, A. P., and Brockdorff, N. (2003). Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev. Cell 4, 481–495.
Silva, S. S., Rowntree, R. K., Mekhoubad, S., and Lee, J. T. (2008). X-chromosome inactivation and epigenetic fluidity in human embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 105, 4820–4825.
Skene, P. J., Illingworth, R. S., Webb, S., Kerr, A. R., James, K. D., Turner, D. J., Andrews, R., and Bird, A. P. (2010). Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 37, 457–468.
Smith, A. G., Heath, J. K., Donaldson, D. D., Wong, G. G., Moreau, J., Stahl, M., and Rogers, D. (1988). Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature 336, 688–690.
Stadtfeld, M., Maherali, N., Breault, D. T., and Hochedlinger, K. (2008a). Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell 2, 230–240.
Stadtfeld, M., Nagaya, M., Utikal, J., Weir, G., and Hochedlinger, K. (2008b). Induced pluripotent stem cells generated without viral integration. Science 322, 945–949.
Stavridis, M. P., Lunn, J. S., Collins, B. J., and Storey, K. G. (2007). A discrete period of FGF-induced Erk1/2 signalling is required for vertebrate neural specification. Development 134, 2889–2894.
Takagi, N., Sugawara, O., and Sasaki, M. (1982). Regional and temporal changes in the pattern of X-chromosome replication during the early post-implantation development of the female mouse. Chromosoma 85, 275–286.
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., and Yamanaka, S. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872.
Tan, J., Yang, X., Zhuang, L., Jiang, X., Chen, W., Lee, P. L., Karuturi, R. K., Tan, P. B., Liu, E. T., and Yu, Q. (2007). Pharmacologic disruption of polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 21, 1050–1063.
Tchieu, J., Kuoy, E., Chin, M. H., Trinh, H., Patterson, M., Sherman, S. P., Aimiuwu, O., Lindgren, A., Hakimian, S., Zack, J. A., Clark, A. T., Pyle, A. D., Lowry, W. E., and Plath, K. (2010). Female human iPSCs retain an inactive X chromosome. Cell Stem Cell 7, 329–342.
Tesar, P. J., Chenoweth, J. G., Brook, F. A., Davies, T. J., Evans, E. P., Mack, D. L., Gardner, R. L., and McKay, R. D. (2007). New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448, 196–199.
Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., and Jones, J. M. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147.
Tian, D., Sun, S., and Lee, J. T. (2010). The long noncoding RNA, Jpx, is a molecular switch for X chromosome inactivation. Cell 143, 390–403.
Urbach, A., Bar-Nur, O., Daley, G. Q., and Benvenisty, N. (2010). Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 6, 407–411.
Vallier, L., Reynolds, D., and Pedersen, R. A. (2004). Nodal inhibits differentiation of human embryonic stem cells along the neuroectodermal default pathway. Dev. Biol. 275, 403–421.
van den Berg, I. M., Galjaard, R. J., Laven, J. S., and van Doorninck, J. H. (2011). XCI in preimplantation mouse and human embryos: first there is remodelling. Hum. Genet. 130, 203–215.
van den Berg, I. M., Laven, J. S., Stevens, M., Jonkers, I., Galjaard, R. J., Gribnau, J., and van Doorninck, J. H. (2009). X chromosome inactivation is initiated in human preimplantation embryos. Am. J. Hum. Genet. 84, 771–779.
Walsh, R. M., and Hochedlinger, K. (2010). Modeling Rett syndrome with stem cells. Cell 143, 499–500.
Ware, C. B., Wang, L., Mecham, B. H., Shen, L., Nelson, A. M., Bar, M., Lamba, D. A., Dauphin, D. S., Buckingham, B., Askari, B., Lim, R., Tewari, M., Gartler, S. M., Issa, J. P., Pavlidis, P., Duan, Z., and Blau, C. A. (2009). Histone deacetylase inhibition elicits an evolutionarily conserved self-renewal program in embryonic stem cells. Cell Stem Cell 4, 359–369.
Warren, L., Manos, P. D., Ahfeldt, T., Loh, Y. H., Li, H., Lau, F., Ebina, W., Mandal, P. K., Smith, Z. D., Meissner, A., Daley, G. Q., Brack, A. S., Collins, J. J., Cowan, C., Schlaeger, T. M., and Rossi, D. J. (2010). Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7, 618–630.
Williams, R. L., Hilton, D. J., Pease, S., Willson, T. A., Stewart, C. L., Gearing, D. P., Wagner, E. F., Metcalf, D., Nicola, N. A., and Gough, N. M. (1988). Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 336, 684–687.
Wutz, A., and Jaenisch, R. (2000). A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Mol. Cell 5, 695–705.
Yang, C., Chapman, A. G., Kelsey, A. D., Minks, J., Cotton, A. M., and Brown, C. J. (2011). X-chromosome inactivation: molecular mechanisms from the human perspective. Hum. Genet. 130, 175–185.
Ying, Q. L., Nichols, J., Chambers, I., and Smith, A. (2003). BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 115, 281–292.
Ying, Q. L., Wray, J., Nichols, J., Batlle-Morera, L., Doble, B., Woodgett, J., Cohen, P., and Smith, A. (2008). The ground state of embryonic stem cell self-renewal. Nature 453, 519–523.
Yu, J., Hu, K., Smuga-Otto, K., Tian, S., Stewart, R., Slukvin, II and Thomson, J. A. (2009). Human induced pluripotent stem cells free of vector and transgene sequences. Science 324, 797–801.
Yu, J., Vodyanik, M. A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J. L., Tian, S., Nie, J., Jonsdottir, G. A., Ruotti, V., Stewart, R., Slukvin, II and Thomson, J. A. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920.
Zhang, L. F., Huynh, K. D., and Lee, J. T. (2007). Perinucleolar targeting of the inactive X during S phase: evidence for a role in the maintenance of silencing. Cell 129, 693–706.
Zhu, H., Lensch, M. W., Cahan, P., and Daley, G. Q. (2011). Investigating monogenic and complex diseases with pluripotent stem cells. Nat. Rev. Genet. 12, 266–275.
Keywords: Rett syndrome, human induced pluripotent stem cells, X-chromosome inactivation
Citation: Cheung AYL, Horvath LM, Carrel L and Ellis J (2012) X-chromosome inactivation in Rett syndrome human induced pluripotent stem cells. Front. Psychiatry 3:24. doi: 10.3389/fpsyt.2012.00024
Received: 01 December 2011; Accepted: 05 March 2012;
Published online: 23 March 2012.
Edited by:
Daniela Tropea, Trinity College Dublin, IrelandReviewed by:
Ju Wang, University of Virginia, USAAlan Colman, A*STAR Institute of Medical Biology, Singapore
Copyright: © 2012 Cheung, Horvath, Carrel and Ellis. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: James Ellis, Program in Developmental and Stem Cell Biology, The Hospital for Sick Children, Toronto Medical Discovery Tower, 13-310, 101 College Street, Toronto, ON, Canada M5G 1L7. e-mail:amVsbGlzQHNpY2traWRzLmNh