 Andrea Wolf
Andrea Wolf
- Department of Pharmaceutical Sciences, College of Pharmacy, University of Minnesota, Duluth, MN, USA
Alzheimer’s disease (AD) is considered the “disease of the twenty-first century.” With a 10-fold increase in global incidence over the past 100 years, AD is now reaching epidemic proportions and by all projections, AD patient numbers will continue to rise. Despite intense research efforts, AD remains a mystery and effective therapies are still unavailable. This represents an unmet need resulting in clinical, social, and economic problems. Over the last decade, a new AD research focus has emerged: ATP-binding cassette (ABC) transporters. In this article, we provide an overview of the ABC transporters ABCA1, ABCA2, P-glycoprotein (ABCB1), MRP1 (ABCC1), and BCRP (ABCG2), all of which are expressed in the brain and have been implicated in AD. We summarize recent findings on the role of these five transporters in AD, and discuss their potential to serve as therapeutic targets.
Introduction
Worldwide, more than 25 million patients suffer from Alzheimer’s disease (AD), and it is projected that by 2050 this number will have quadrupled to over 100 million patients (Abbott, 2011). This development will raise global AD health care costs to unprecedented dimensions (Brookmeyer et al., 2007; WHO, 2007). Besides the cost factor, AD is a devastating disease. AD patients decline mentally and physically and transform from functioning human beings into helpless dependents, while fading out of life (Mega et al., 1996).
Currently, few FDA-approved AD medications are on the market. Those that are available can cause severe side effects and, if effective, alleviate disease symptoms up to 6–12 months only in patients with mild to moderate AD (Ellis, 2005; Seltzer, 2007). Moreover, most patients respond poorly or not at all to therapy. Thus, there is clearly a great unmet need for new therapies that target the underlying cause(s) of AD, reverse symptoms, or prevent AD completely. However, despite decades of research efforts, AD is still a mystery and effective therapies remain unavailable for millions of patients.
At the molecular level, two proteins – amyloid-β (Aβ) and tau – are characteristic signatures of AD. Aβ is a small peptide that is generated through cleavage of amyloid precursor protein (APP); the most common Aβ isoforms in AD are the neurotoxic peptides Aβ40 and Aβ42. The physiological function of Aβ is still unclear, but in healthy individuals Aβ brain levels are low, whereas in AD patients Aβ levels can be increased up to 100-fold in the brain, where it aggregates to plaques (Gravina et al., 1995; Hardy and Selkoe, 2002). Tau proteins are microtubule-associated proteins that interact with tubulin and promote its insertion into microtubules, thereby modulating stability and flexibility of axonal microtubules. Phosphorylation of tau, on the other hand, results in disruption of microtubule organization and structure, and in AD hyperphosphorylation of tau proteins causes microtubular collapse and results in the formation of neurofibrillary tangles inside neurons. Both Aβ plaques and neurofibrillary tau tangles are hallmarks of AD pathology and are thought to contribute to neurodegeneration and dementia (Wolozin et al., 1986; Wood et al., 1986; Hardy and Selkoe, 2002).
Over the last 10 years a new AD research field emerged with a focus on ATP-binding cassette (ABC) transporters at the blood-brain barrier and in other cells of the CNS. ABC transporters utilize ATP to move their substrates across membranes of organelles, cells, and tissues. The substrates of ABC transporters involved with AD include cholesterol, sterols, lipids, peptides, metabolites, and xenobiotics including toxins and a large number of therapeutic drugs. Thus, ABC transporters are an integral part of many physiological processes and biochemical pathways, and play an important role in maintaining the body’s homeostasis by extruding metabolites and limiting uptake of xenobiotics. Only recently have researchers discovered that ABC transporters also play a critical role in diseases, where processes and pathways involving ABC transporters are altered. For some ABC transporters – ABCA1, ABCA2, P-gp, MRP1, and BCRP – such a role is emerging in AD and other CNS disorders associated with high Aβ brain levels.
If, and how exactly, ABC transporters contribute to AD pathophysiology is unknown. This lack of understanding provides opportunities for exciting new discoveries, and more importantly, holds the promise to unravel details of disease processes and identify targets that could help in the development of novel therapeutic strategies for AD. In this review, we summarize current knowledge about ABCA1, ABCA2, P-glycoprotein (ABCB1), MRP1 (ABCC1), and BCRP (ABCG2) in AD, and discuss their potential as therapeutic targets.
ABCA1 and ABCA2
Background
ABCA1 and ABCA2 are prototypic members of the ABCA subfamily that comprises 12 transporter proteins (Kaminski et al., 2006). Luciani et al. (1994) identified both the Abca1 and Abca2 genes in several mouse tissues including the brain. The ABCA1 protein shares approximately 50% sequence homology with ABCA2 and although the transporters are localized in different cell compartments, both proteins are involved in lipid transport and metabolism (Garewal et al., 1986; Davis et al., 2004; Mack et al., 2007).
ABCA1
ABCA1, also known as cholesterol efflux regulatory protein, is a plasma membrane protein that is highly expressed in the adrenal gland and uterus. ABCA1 has also been detected in the CNS in neurons, astrocytes, microglia, and in epithelial cells of the choroid plexus (Figure 1; Koldamova et al., 2003; Kim et al., 2006; Fujiyoshi et al., 2007). In porcine brain capillary endothelial cells, Abca1 protein has been localized to the basolateral plasma membrane (Panzenboeck et al., 2002). In the periphery, ABCA1 mediates cholesterol and phospholipid efflux from cells for transfer onto lipid-free or lipid-poor apoprotein A-1 (ApoA-1) particles, the main component of high density lipoproteins (HDL; Lawn et al., 1999). Fully loaded, mature HDL particles then transport ApoA-1-bound cholesterol and lipids from the tissues to the liver for excretion or recycling. In 1999, researchers found that mutations in the ABCA1 gene, resulting in a non-functional ABCA1 transporter protein are the cause of Tangier disease, a rare autosomal recessive disorder characterized by high cellular cholesterol and low plasma HDL levels (Bodzioch et al., 1999). In individuals carrying two copies of the mutant ABCA1 gene, these changes in lipid metabolism drastically increase the risk for cardiovascular disease.
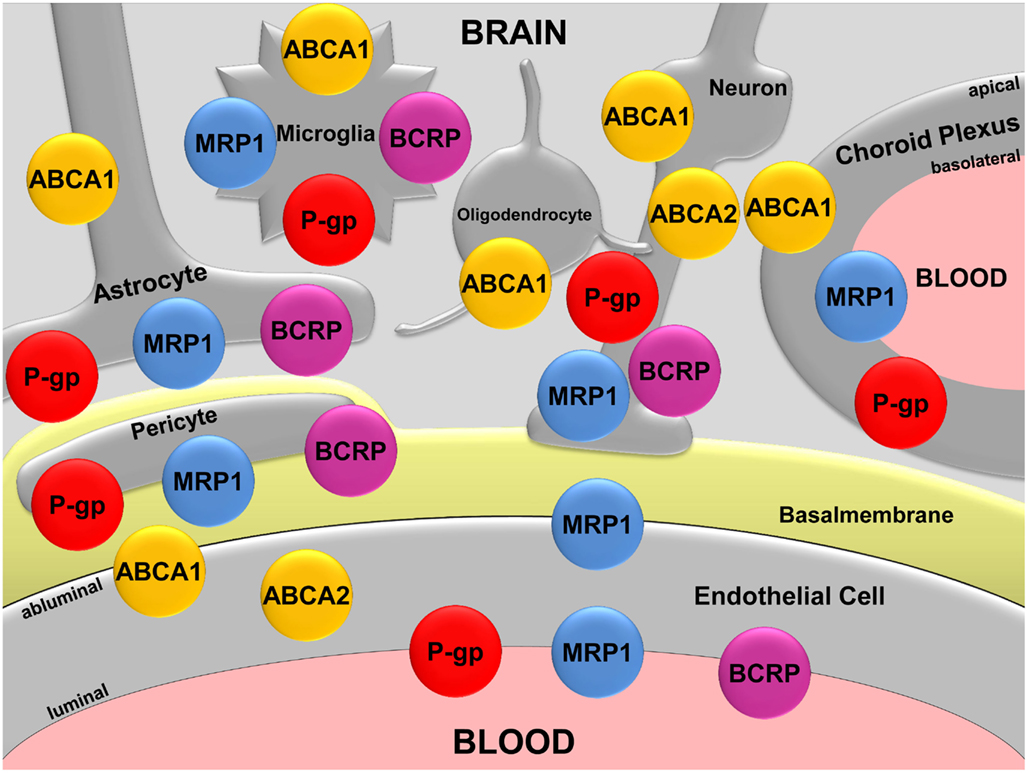
Figure 1. Map of the ABC transporters ABCA1, ABCA2, P-gp, MRP1, and BCRP in the CNS. Modified according to Hartz and Bauer (2011).
In the CNS, ABCA1 mediates cholesterol and phospholipid efflux from astrocytes and microglia thereby facilitating lipidation of ApoE, which is the major apoprotein in the brain (Panzenboeck et al., 2002; Hirsch-Reinshagen et al., 2004). Consequently, ABCA1-deficiency results in poorly lipidated ApoE particles, but also in a thus far unexplained decrease in ApoE brain levels (Hirsch-Reinshagen et al., 2004; Wahrle et al., 2004).
ABCA2
ABCA2 protein is predominantly expressed in the brain, where it has been detected at the protein level in neurons, oligodendrocytes, and brain capillary endothelial cells (Figure 1; Zhou et al., 2001; Ohtsuki et al., 2004; Broccardo et al., 2006; Shawahna et al., 2011). The primary location of ABCA2 is in the lysosomal membrane, where it transports waste products from the cytoplasm into the lysosomal compartment. In addition, studies suggest that ABCA2 plays a role in neural transmembrane lipid transport, macrophage lipid metabolism, and neural development (Kaminski et al., 2001; Zhou et al., 2001; Tachikawa et al., 2005; Broccardo et al., 2006). However, in contrast to ABCA1, the function of ABCA2 is not well-established.
ABCA1 and ABCA2 in AD
ABCA1
ABCA1-deficiency, ApoE, and Aβ brain levels. Changes in brain cholesterol metabolism are linked to an increased risk of AD. One important factor for brain cholesterol homeostasis is ApoE, which supplies brain cells with cholesterol and lipids for development, repair, and nerve growth. In 1993, Margaret Pericak-Vance and her team conducted linkage and association analyses and discovered that ApoE plays a critical role in AD (Corder et al., 1993). Of the three major ApoE isoforms, ApoE-ε4 bears the highest genetic risk factor for late-onset sporadic AD (Strittmatter et al., 1993), and individuals carrying two ApoE-ε4 alleles have a 10–30 times higher risk of developing AD compared to individuals not carrying E-ε4 alleles (Farrer et al., 1997). In the AD field, this increased risk is thought to be due to ApoE-ε4 directly interacting with Aβ, thereby favoring Aβ aggregation and plaque formation (Holtzman et al., 2000). Since ABCA1 promotes cholesterol and phospholipid efflux to ApoE particles and influences lipidation of apolipoproteins and ApoE brain levels, researchers postulated a potential role for ABCA1 in AD pathology.
In this regard, Hirsch-Reinshagen et al. (2004) found that Abca1-deficiency in mice leads to reduced lipid efflux to ApoE and, for an unknown reason, also in a dramatic decrease of ApoE brain levels. The same group also showed in two hAPP-overexpressing AD mouse models (Tg-SwDI/B; APP/PS1) that Abca1-deficiency reduces ApoE brain levels, but has no effect on Aβ brain levels (Hirsch-Reinshagen et al., 2005). In contrast, two other studies demonstrate that a lack of Abca1 in hAPP-overexpressing mice (APP23 and PDAPP models) decreases both ApoE lipidation and ApoE brain levels, and also increases brain levels of insoluble Aβ40 and Aβ42 (Koldamova et al., 2005a; Wahrle et al., 2005). From these observations, the authors concluded that poorly lipidated ApoE has a high affinity to Aβ and promotes Aβ fibrillogenesis.
ABCA1 upregulation, ApoE, and Aβ brain levels. Several groups studied the impact of inducing Abca1 protein in mouse on ApoE and Aβ brain levels. ABCA1 gene transcription is controlled by the ligand-activated nuclear receptors LXRα and β (liver X receptor), which are key regulators of cholesterol and lipid metabolism (Whitney et al., 2002). Fukumoto et al. (2002) conducted the first in vitro study on the effect of LXR activation on Abca1 protein expression. Using mouse Neuro2A cells, the authors showed that upregulation of Abca1 protein levels by various LXR activators increased secretion of both Aβ40 and Aβ42. This effect was reduced when Abca1 expression was blocked with RNAi (Fukumoto et al., 2002). However, these findings could not be reproduced in subsequent in vitro studies. In a variety of mouse and human cell lines transfected with Swedish or wild-type APP, LXR-mediated Abca1 and ABCA1 induction reduced Aβ production and secretion (Koldamova et al., 2003; Sun et al., 2003; Brown et al., 2004; Kim et al., 2007, 2009). This observation is consistent with findings from in vivo studies using LXR agonists in AD mouse models. Koldamova et al. (2005b) showed that treatment of 11-week-old APP23 mice with T0901317, a synthetic LXR ligand, increased Abca1 protein expression and significantly decreased brain levels of Aβ40 and Aβ42. Similar results were also published by other groups (Burns et al., 2006; Lefterov et al., 2007; Riddell et al., 2007; Jiang et al., 2008). Even in 40-week-old APP/PS1 mice, LXR-mediated upregulation of Abca1 and ApoE protein levels improved cognitive impairment in the novel object recognition test, while cognition remained unchanged in APP/PS1-Abca1-deficient mice. Although a reduction in Aβ burden was observed, guanidine-soluble or mature Aβ levels were not significantly reduced in mice with increased Abca1 expression compared to Abca1-deficient mice (Donkin et al., 2010). These observations are consistent with findings by Vanmierlo et al. (2009) who also demonstrated that LXR activation improved memory function in aged APP/PS1 mice without reducing Aβ deposition.
Zelcer et al. used a different approach and quantified Aβ plaques in the brains of APP/PS1 mice lacking either LXRα or LXRβ. LXR-deficiency resulted in reduced Abca1 and ApoE protein levels, and increased Aβ brain levels compared to wild-type mice (Zelcer et al., 2007).
To further clarify the role of Abca1 in AD, Wahrle et al. (2008) generated Abca1-overexpressing PDAPP mice. In these mice, Aβ load was significantly decreased at 12 months of age, and with sixfold Abca1 protein overexpression, Aβ deposition was almost absent. Furthermore, hippocampal ApoE levels were significantly decreased in Abca1-overexpressing PDAPP mice, the proportion of insoluble ApoE was increased, and ApoE showed increased lipidation (Wahrle et al., 2008).
Although Abca1 upregulation has been shown to decrease Aβ brain burden in AD mouse models and exert a beneficial effect, its role in AD is not fully understood. ABCA1 does not directly transport Aβ (Akanuma et al., 2008), rather it acts as a cholesterol and phospholipid transporter. Thus, ABCA1 affects ApoE lipidation and brain cholesterol homeostasis, factors that have been shown to influence APP processing and Aβ aggregation (Simons et al., 1998; Puglielli et al., 2003). Abca1-deficiency in mice results in poorly lipidated ApoE and in reduced brain levels of soluble ApoE (Koldamova et al., 2005a; Wahrle et al., 2005). Interestingly, low ApoE brain levels per se are associated with decreased Aβ deposition, while poorly lipidated ApoE has been discussed to have an amyloidogenic effect in vivo (Koldamova et al., 2005a; Wahrle et al., 2005). The underlying mechanism(s) of these observations is/are largely unknown and more studies are required to unravel the complex interrelation between ABCA1, brain lipid metabolism, and AD.
ABCA1 expression in AD. Recent studies also analyzed ABCA1 expression in post-mortem brain tissue samples from AD patients. Akram et al. found increased ABCA1 mRNA and protein expression in hippocampal brain tissue from AD patients compared to age-matched control subjects. ABCA1 mRNA expression correlated with the severity of dementia, Braak score (tau tangles), and Aβ plaque density, whereas ABCA1 protein expression correlated with dementia severity and Braak score, but not with plaque density (Akram et al., 2010). Kim et al. (2010) also found increased ABCA1 protein and mRNA levels in hippocampal tissue from AD patients compared to age-matched controls.
ABCA1 polymorphisms and AD. A total of 14 studies have been conducted to investigate the association of ABCA1 gene polymorphisms with the risk for AD (Wollmer et al., 2003; Katzov et al., 2004; Li et al., 2004; Kolsch et al., 2006; Shibata et al., 2006; Chu et al., 2007; Rodriguez-Rodriguez et al., 2007, 2009, 2010; Sundar et al., 2007; Wahrle et al., 2007; Wang and Jia, 2007; Wavrant-De Vrieze et al., 2007; Reynolds et al., 2009). Eight of these studies link single nucleotide polymorphisms (SNP) in the ABCA1 gene to an increased risk for AD (Katzov et al., 2004; Shibata et al., 2006; Chu et al., 2007; Rodriguez-Rodriguez et al., 2007; Sundar et al., 2007; Wang and Jia, 2007; Wavrant-De Vrieze et al., 2007; Reynolds et al., 2009). In summary, the SNP rs2230808 was associated with increased tau and Aβ brain levels, the C-alleles of the G-395C polymorphisms were associated with decreased cerebrospinal fluid (CSF) levels of 24-hydroxycholesterol, and the rs2230805 allele was associated with reduced Aβ42 CSF levels (Wollmer et al., 2003; Katzov et al., 2004; Kolsch et al., 2006; Reynolds et al., 2009). In addition, individuals carrying polymorphisms of the ABCA1 and the Niemann-Pick C1 genes, and individuals with polymorphisms of ABCA1 in conjunction with polymorphisms of hydroxy-methylglutaryl-coenzyme A reductase, have an increased risk for AD (Rodriguez-Rodriguez et al., 2009, 2010).
ABCA2
In contrast to ABCA1, little is known about ABCA2 in AD. Recently, researchers identified a synonymous SNP (rs908832) in exon 14 that is associated with a high risk for sporadic, early-onset AD, but not late-onset AD (Mace et al., 2005). These results were confirmed by Wollmer et al.; however, their data indicate that the genetic association of ABCA2 with sporadic AD depends on ethnicity. While these authors found a strong association of rs908832 with AD in a Western European population, no such association was found for Southern Europeans (Wollmer et al., 2006).
In addition, a number of independent results from different studies point to a possible role of ABCA2 in AD pathology: (1) ABCA2 protein has been found to co-localize with both APP and Aβ in vitro (Chen et al., 2004); (2) ABCA2 overexpression increases APP and Aβ-levels and upregulates a number of genes associated with resistance to oxidative stress (Chen et al., 2004); (3) β-secretase-1, which is involved in the generation of Aβ40 and Aβ42, is decreased in an Abca2-overexpressing mouse neuroblastoma N2a cell line (Davis, 2010); (4), ABCA2 protein expression is increased in temporal lobe samples from AD patients (Chen et al., 2004); and most recently (5) ABCA2 downregulation with siRNA reduces Aβ production in vitro, which is an interesting finding given that this is the opposite of what has been found for ABCA1 (Michaki et al., 2012).
ABCA1 and ABCA2 as Potential Therapeutic Target in AD
Although ABCA1 and ABCA2 are members of the same ABC transporter subfamily, they are different with regard to their physiological functions and potential involvement in AD pathophysiology. In the CNS, ABCA1 is important for proper lipidation of apoproteins. Reduced ABCA1 protein levels result in lipid-poor ApoE, which seems to increase Aβ aggregation. Several studies demonstrated that LXR-mediated Abca1 upregulation increases ApoE levels, decreases Aβ brain levels, and has beneficial effects on cognition in AD mouse models (Koldamova et al., 2005b; Burns et al., 2006; Lefterov et al., 2007; Riddell et al., 2007; Jiang et al., 2008; Donkin et al., 2010). Since numerous compounds are available to activate LXR, this approach might be a potential therapeutic strategy for AD. However, in addition to ABCA1 upregulation, activating LXR also increases expression levels of ABCG1, another lipid transporter of the ABC superfamily, and ApoE. This is of relevance, since both ABCG1 and ApoE have been implicated with AD as well (Corder et al., 1993; Wollmer et al., 2007). Consequently, the beneficial effects of LXR activation could be a result of upregulating one, two, or all of these proteins, and possibly include other proteins that control lipid transport and metabolism, such as Cyp7A1 or cholesterol ester transfer protein (Repa et al., 2000; Tall et al., 2000; Honzumi et al., 2010). Thus, due to the complexity of the LXR regulatory network, interpretation of study results is difficult and more research is needed to discern the roles of ABCA1, ABCG1, ApoE, and possibly other proteins in AD.
In contrast to ABCA1, overexpression of ABCA2 increases APP synthesis, alters APP processing, and elevates Aβ brain levels (Chen et al., 2004; Davis, 2010). This suggests that ABCA2 downregulation could potentially reduce Aβ brain levels and have a beneficial effect on cognition. To date, no studies exist that clarify ABCA2 function under physiological and pathophysiological conditions and properly test the hypothesis that ABCA2 downregulation could be a potential therapeutic strategy in AD.
P-Glycoprotein
Background
Juliano and Ling (1976) discovered a 170 kDa transmembrane protein in a Chinese hamster ovary cell line and named it P-glycoprotein (P-gp). Shortly thereafter, in 1983, P-gp was found in a human tumor cell line (Kartner et al., 1983), and in 1988 it was detected in normal liver tissue and small intestine mucosa (Hitchins et al., 1988). Since then, P-gp has become the most prominent transporter of the ABC family. In humans, P-gp is encoded by the ABCB1 gene (MDR1), and in rodents by the Abc1a (Mdr1a) and Abc1b (Mdr1b) genes (Ueda et al., 1986; Hsu et al., 1989).
A number of barrier and excretory tissues throughout the human body express P-gp, including intestine, liver, kidney, and the blood-placenta and blood-testis barriers. In the CNS, P-gp has relatively low expression levels in neurons, astrocytes, pericytes, microglia, and the epithelial cells of the choroid plexus (Figure 1; Lee and Bendayan, 2004). In contrast, the highest P-gp expression levels in the body are found in the brain capillary endothelium comprising the blood-brain barrier (Thiebaut et al., 1987; Cordon-Cardo et al., 1989).
P-gp is a potent and efficient efflux transporter that prevents a variety of structurally diverse compounds, primarily amphiphilic and slightly cationic organic chemicals, including therapeutic drugs and toxicants, from entering the brain and pumps metabolic waste products out of the brain. Thus, P-gp plays a crucial role in neuroprotection, brain detoxification, and overall CNS homeostasis (Schinkel, 1999; Schinkel and Jonker, 2003). Therefore, deletion or inhibition of P-gp can result in significantly increased brain uptake of its substrates. For example, Mdr1a knockout mice showed a 10- to 50-fold increase in brain concentrations of vinblastine and ivermectin, respectively (Schinkel et al., 1994). Likewise, direct inhibition of P-gp with valspodar (PSC833) increased brain levels of methadone, digoxin, paclitaxel, and colchicine by 5- to 10-fold (Desrayaud et al., 1997; Mayer et al., 1997; Fellner et al., 2002; Rodriguez et al., 2004).
Although it is well-established that P-gp can handle a large number of exogenous compounds (Schinkel, 1999), it is still unknown if P-gp has endogenous substrates. Recent reports show that P-gp effectively transports Aβ at the blood-brain barrier, indicating that amyloid peptides may be endogenous P-gp substrates (Cirrito et al., 2005; Hartz et al., 2010). Importantly, these findings link blood-brain barrier P-gp to AD and are described in the following section.
P-Glycoprotein in AD
Lam et al. (2001) were the first to suggest that P-gp is capable of transporting Aβ. In their study, the authors co-transfected HEK293 cells with human MDR1 and the gene coding for the human amyloid precursor protein hAPP, and observed increased Aβ secretion from APP/MDR1-co-transfected cells compared to cells that were only transfected with hAPP, but not with MDR1. Moreover, treatment with P-gp inhibitors significantly decreased Aβ secretion in MDR1-transfected cells. However, since cellular APP levels were increased in MDR1-transfected cells, it was unclear if increased Aβ secretion was due to P-gp-mediated transport or a result of higher intracellular Aβ levels. In additional experiments using inside-out membrane vesicles isolated from P-gp overexpressing CHRB30 cells, Lam et al. also showed that Aβ40 and Aβ42 transport is ATP-dependent. From this study Lam et al. (2001) concluded that P-gp facilitates Aβ transport and proposed that ABC transporters could, in general, act as Aβ efflux pumps.
Consistent with these findings, Kuhnke et al. (2007) demonstrated increased basal-to-apical transport of FITC-labeled Aβ40 and Aβ42 in MDR1-transfected LLC-PK1 cells compared to LLC-PK1 parental cells; this effect was blocked by the P-gp inhibitor cyclosporine A. The authors then reversed the experiment and showed that Aβ40 and Aβ42 inhibit transport of the fluorescent P-gp substrate rhodamine 123, suggesting Aβ transport is competitive. Lastly, using inside-out membrane vesicles from MDR1-transfected and parental LLC-PK1 cells, Kuhnke et al. (2007) demonstrated that transport of both rhodamine 123 and FITC-labeled Aβ in vesicles from MDR1-transfected LLC cells is ATP-dependent. Thus, Kuhnke et al. provided further evidence indicating that P-gp transports Aβ.
In a comprehensive in vivo study, Cirrito et al. (2005) provided first evidence for a direct link between blood-brain barrier P-gp and Aβ brain deposition. Using Mdrla/b−/− double-knockout mice, the authors first showed that brain clearance of Aβ40 and Aβ42 was significantly lower compared to that in control animals 30 min after intracerebral microinjection of the amyloid peptides. Then, the authors dosed transgenic hAPP-overexpressing mice, a well-established AD model (Tg2576 mice; Hsiao et al., 1996), with the selective P-gp inhibitor XR9576 (tariquidar) and measured Aβ brain concentrations by microdialysis. As early as 8 h after injecting hAPP mice with the P-gp inhibitor, Aβ levels in the brain interstitial fluid were significantly increased compared to untreated hAPP control mice (Cirrito et al., 2005). Consistent with this, 12-month-old P-gp-deficient hAPP mice had a higher Aβ brain load compared to hAPP mice expressing P-gp. Thus, Cirrito et al. (2005) demonstrated for the first time that reduced blood-brain barrier P-gp activity due to an inhibitor or a genetic lack of P-gp increases Aβ brain uptake and reduces Aβ brain clearance in an AD mouse model, which further substantiates that P-gp could be a contributing factor in AD pathology.
Several groups studied P-gp expression at the human blood-brain barrier and correlated the results with Aβ load in the surrounding brain tissue. Vogelgesang et al. (2002) analyzed vascular P-gp protein expression and cerebral Aβ load in post-mortem brain tissue samples from non-demented elderly patients and found that P-gp levels were low in the vasculature surrounding Aβ plaques. In another study, the same authors analyzed post-mortem brain tissue samples from non-demented elderly patients with cerebral amyloid angiopathy (CAA), a disease caused by Aβ deposition in the cerebrovasculature. In these samples, brain capillaries with Aβ deposition showed no P-gp immunoreactivity, whereas P-gp immunostaining was increased in capillaries with no Aβ deposition compared to control samples (Vogelgesang et al., 2004). In a recent study Wijesuriya et al. (2010) detected 25% lower P-gp protein expression levels in hippocampal blood vessels in post-mortem brain samples from AD patients than in samples from age-matched non-demented patients. Jeynes and Provias (2011) made similar observations in post-mortem cerebral cortex tissue samples from 10 AD patients: in areas with a high density of Aβ40 plaques and neurofibrillary tangles, respectively, P-gp protein expression levels were decreased compared to levels found in tissue samples from non-demented individuals. Thus, these studies indicate an inverse correlation between Aβ deposition and vascular P-gp expression at the human blood-brain barrier in AD, that is: high Aβ brain levels correlate with low vascular P-gp levels and vice versa.
In a recent study, van Assema et al. (2012) used positron emission tomography and the P-gp substrate (R)-[11C]verapamil to assess blood-brain barrier P-gp function in AD patients with that of age-matched healthy control subjects. The authors found an increase in the non-displaceable binding potential of (R)-[11C]verapamil in several cortical brain regions of AD patients compared to healthy control individuals. These are critical data since they provide first direct evidence for reduced P-gp function at the blood-brain barrier of AD patients.
Zlokovic and Frangione (2003) proposed the “transport-clearance hypothesis” of AD. This hypothesis states that increased Aβ brain accumulation in AD is due to reduced Aβ clearance from brain to blood. Based on Zlokovic’s Aβ transport-clearance hypothesis and the above-mentioned previous findings, we postulated that Aβ clearance from brain to blood occurs in two steps, and that each step must be facilitated by a transporter or receptor since Aβ is a peptide that does not easily cross the plasma membrane by itself. In the first step, Aβ must pass from the brain parenchyma through the abluminal plasma membrane of the capillary endothelium into brain capillary endothelial cells. Studies suggest that the low-density lipoprotein receptor-related protein 1 (LRP1) facilitates this step (Shibata et al., 2000; Deane et al., 2004; Fujiyoshi et al., 2011). In the second step, Aβ must pass from brain capillary endothelial cells across the luminal plasma membrane into the blood of the neurovasculature. We hypothesized that (1) P-glycoprotein mediates this critical, because rate-limiting, second step of the neurovascular Aβ clearance process, that (2) it is this P-gp-mediated step in the neurovascular Aβ clearance process that is impaired in AD, and consequently, that (3) restoring blood-brain barrier P-gp lowers Aβ brain levels in AD.
We addressed these hypotheses in a recent study (Hartz et al., 2010). First, we demonstrated P-gp-mediated transport of fluorescein-labeled Aβ42 in intact, living isolated mouse brain capillaries ex vivo. This was the first direct evidence that P-gp mediates Aβ transport at the blood-brain barrier. In contrast, our data showed that Bcrp and Mrp2 are not involved in Aβ42 transport in isolated brain capillaries from mouse (Hartz et al., 2010). Second, using the transgenic hAPP-overexpressing mouse model (Tg2576 mice) we showed that P-gp is compromised at the blood-brain barrier. We found a 70% decrease in P-gp transport activity and a 60% decrease in P-gp protein expression in isolated brain capillaries from hAPP mice compared to age-matched wild-type mice. These findings demonstrate that, similar to findings in humans, P-gp expression and function are significantly reduced at the blood-brain barrier in mice with high Aβ brain levels. Next, we showed that dosing hAPP mice with pregnenolone-16α-carbonitrile (PCN) once daily for 7 days to activate the pregnane X receptor (PXR) completely restored P-gp expression and transport activity in brain capillaries. As a consequence, Aβ40 and Aβ42 levels were reduced in capillaries from hAPP mice by approximately 30%. Furthermore, PCN-treatment lowered Aβ40 and Aβ42 brain levels by approximately 50–65% compared to untreated hAPP mice. Thus, our study confirms that blood-brain barrier P-gp plays a role in the Aβ brain clearance process, which suggests that restoring blood-brain barrier P-gp could potentially be used as a new therapeutic strategy to lower Aβ brain levels in AD (Hartz et al., 2010).
In addition to countless physiological and pharmacological studies on P-gp, the number of pharmacogenomic studies on its gene ABCB1 is increasing. Yet, the only published pharmacogenomic association study on ABCB1 and AD is by Kohen et al. (2011). These authors hypothesized the existence of a link between common ABCB1 SNPs such as G2677T/A and C3435T and the age of AD onset and/or Aβ42 CSF levels in AD patients. However, no significant association was found and it remains to be elucidated if ABCB1 polymorphisms that may alter transport function and/or expression play a role in AD.
In summary, research over the last 10 years indicates that P-gp at the blood-brain barrier is part of the process involved in clearing Aβ from brain to blood. These findings put P-gp in the spotlight as potential therapeutic target in AD, which will be discussed in the next section.
P-Glycoprotein as Potential Therapeutic Target in AD
Given its critical role in the neurovascular Aβ clearance system, P-gp could potentially be used as a therapeutic target in AD. As mentioned above, activation of the nuclear receptor PXR to restore blood-brain barrier P-gp, and thus, to reduce Aβ brain burden and slow cognitive decline has been proposed as a novel strategy (Hartz et al., 2010). In this regard, Loeb et al. (2004) showed in a randomized, triple-blind and controlled clinical trial that treating patients with mild to moderate AD with the antibiotics doxycycline and rifampicin daily for 3 months significantly slowed cognitive decline compared to patients in the control group. This effect lasted for up to 9 months beyond the treatment period, and the authors stated that the mechanism for this positive clinical benefit was unknown, but unlikely to be due to the antibiotic effect. Interestingly, both doxycycline and rifampicin activate PXR (Mealey et al., 2002; Yasuda et al., 2008). Thus, one possible explanation for slowed cognitive decline in these patients is that doxycycline and rifampicin activated PXR, which increased blood-brain barrier P-gp levels, and in turn could have increased Aβ brain clearance, lowered Aβ brain levels, and thus, reduced progressive decline of cognition. This hypothesis, however, remains to be verified.
Another strategy is to unravel the underlying mechanism/s that reduce/s blood-brain barrier P-gp expression levels and functional activity in AD. Knowledge of this/these mechanism/s could help identify targets to protect P-gp, maintain Aβ brain clearance, prevent or slow Aβ brain accumulation, and thereby prevent or delay AD. In a recent study, Brenn et al. investigated the effect of Aβ on blood-brain barrier P-gp and LRP expression in mice. Aβ40 and Aβ42 were administered through a subcutaneously implanted ALZET Mini-Osmotic pump for 24 h. The authors showed that only Aβ42 significantly decreased both Abcb1 and LRP1 mRNA expression levels in vivo, but no changes were observed at the protein level (Brenn et al., 2011). However, this could have been due to peripheral Aβ administration, rather than administering Aβ to the brain, which would have mimicked AD pathology more closely. Other studies addressing the mechanism/s that reduce blood-brain barrier P-gp in AD are not available.
Clearly, studies indicate that restoring P-gp expression and function could be effective in enhancing Aβ clearance from the brain and lower Aβ brain levels (Cirrito et al., 2005; Hartz et al., 2010). However, the concept that restoring P-gp at the blood-brain barrier could serve as a valid therapeutic strategy to lower Aβ brain load, reduce cognitive decline, delay onset and slow progression of AD now has to be critically evaluated.
MRP1
Background
MRP1 was discovered by Cole et al. (1992) in the H69AR cancer cell line. Unlike P-gp, which mainly handles amphiphilic and slightly cationic chemicals, MRP1 substrates are mainly organic anions. The MRP1 substrate spectrum includes endogenous compounds like leukotriene C4, glutathione, glucuronide, and sulfate conjugates, as well as a variety of exogenous compounds such as chemotherapeutics, HIV protease inhibitors, antibiotics, and toxins (Loe et al., 1996; Conseil et al., 2005). In the human body, MRP1 is expressed in most tissues, including kidney, lung, placenta, and liver. In the brain, MRP1 protein expression has been detected in astrocytes, microglia, pericytes, neurons, the basolateral membrane of choroid plexus cells, and the luminal and abluminal membranes of brain capillary endothelial cells (Figure 1; Schinkel, 2001; Soontornmalai et al., 2006; Roberts et al., 2008). Based on this expression pattern and substrate spectrum, the main function of MRP1 is extrusion of glutathione, glucuronide, and sulfate metabolites, as well as cell protection against potentially harmful metabolites, xenobiotics, and drugs. Since increasing evidence indicates involvement of ABC transporters in neurodegenerative diseases, researchers were prompted to investigate MRP1 in AD.
MRP1 in AD
Currently, two studies implicate MRP1 (ABCC1) with AD. The first study is concerned with the effect of 4-hydroxy-2-trans-non-enal (HNE) on the metabolizing enzyme glutathione-S-transferase-α (GST-α) and MRP1 in AD (Sultana and Butterfield, 2004). HNE is a neurotoxic product of membrane lipid peroxidation (Mark et al., 1997). Under physiological conditions, HNE is first inactivated by GST-α through conjugation with glutathione, and then, in a second step, the HNE-glutathione conjugate is transported out of the cell by MRP1 to limit toxicity (Loe et al., 1996).
In AD, however, due to increased oxidative stress, high levels of HNE are present in the brain (Lovell et al., 1997). In this regard, Sultana and Butterfield showed that HNE co-immunoprecipitates with both GST-α and MRP1 to a greater extent in post-mortem hippocampal tissue samples from AD patients compared to samples from aged-matched control individuals. The authors concluded that excess HNE in AD brain leads to covalent binding of HNE to GST-α and MRP1, and speculated that this could impair GST-α and MRP1 function, resulting in cellular accumulation of GST-α and MRP1 substrates, which could promote neurotoxicity. However, more studies are required to verify this conclusion and to demonstrate the proposed sequence of events.
The second study that implicates Mrp1 with AD was recently published by Krohn et al. (2011). The authors used double transgenic mice expressing both human APP and human presenilin-1 (APP/PS1), and, in addition, generated APP/PS1 mice lacking Abcb1 (P-gp), Abcg2 (Bcrp), or Abcc1 (Mrp1), respectively. Krohn et al. showed an age-dependent increase of Aβ40 and Aβ42 (guanidine-soluble fraction) and increased coverage and size of Aβ plaques in the cortex of APP/PS1 × Abcc1−/− mice (Mrp1-deficient) compared to control APP/PS1 mice. The number and size of Aβ plaques and Aβ levels in the brains of APP/PS1 × Abcb1−/− mice (P-gp-deficient) were also elevated compared to control animals; no difference was observed between APP/PS1 × Abcg2−/− mice (Bcrp-deficient) and control animals. Using a Transwell assay with primary cultured endothelial cell monolayers from standard Abcc1−/− and wild-type mice, Krohn et al. also showed that abluminal-to-luminal Aβ42 transport was significantly reduced in Mrp1-deficient cells.
In additional experiments, the authors tested if activation of Mrp1 reduces Aβ brain levels by dosing APP/PS1 and APP/PS1 × Abcc1−/− mice with thiethylperazine twice daily for 30 days. Thiethylperazine is a phenothiazine derivative that has been shown to increase Mrp1 activity in mature human red blood cells (Wesolowska et al., 2009). ELISA measurements showed that soluble Aβ42 brain levels were significantly reduced in thiethylperazine-treated APP/PS1 but not in untreated APP/PS1 × Abcc1−/− mice. Furthermore, both number and size of Aβ plaques were reduced, and brain levels of both aggregated and soluble Aβ42 were lowered, although the decrease in aggregated Aβ42 was not significant. A reduction in Aβ brain levels was also observed in APP/PS1 × Abcb1−/− mice, suggesting that activation of Mrp1 was the reason for reduced Aβ brain levels (Krohn et al., 2011).
MRP1 as Potential Therapeutic Target in AD
Currently, the report by Krohn et al. (2011) is the first and only evidence that suggests a potentially important role for Mrp1 in Aβ removal from the mouse brain in vivo. More importantly, the authors provide proof-of-principle of a potential new therapeutic strategy to treat AD. Three critical points remain to be addressed. First, the detailed molecular mechanism by which thiethylperazine affects Mrp1, and possibly other transporters in mouse (and eventually in humans), needs to be elucidated. Such knowledge is critical given that thiethylperazine seems to activate Mrp1 while inhibiting P-gp at same time. In addition, although thiethylperazine dosing decreased Aβ brain load, the direct effect on blood-brain barrier Mrp1 expression and/or functional activity in vivo remains unknown. Second, MRP1 localization at the blood-brain barrier in humans and rodents is controversial. While some studies show MRP1 to be localized in the luminal plasma membrane, other studies detected MRP1 expression in the abluminal plasma membrane (Nies et al., 2004; Zhang et al., 2004; Soontornmalai et al., 2006; Kilic et al., 2008). However, a recent study by Roberts et al. (2008) indicates that rat Mrp1 is expressed at both the luminal and abluminal membranes of the brain capillary endothelium. Based on this finding, MRP1-mediated Aβ transport could occur in two directions: (1) into the brain by abluminal MRP1, and (2) out of the brain into the capillary lumen by luminal MRP1. Third and last, direct evidence is available that P-gp transports Aβ at the blood-brain barrier, but such direct proof is not yet available for MRP1. Together, more studies are required to confirm existing data and unequivocally demonstrate that MRP1 is involved in Aβ brain clearance and a valid target for AD treatment.
BCRP
Background
ABCG2 is the gene that codes for the breast cancer resistance protein (BCRP), which was first discovered in 1998 in the human breast cancer cell line MCF-7/AdrVp (Allikmets and Dean, 1998; Doyle et al., 1998). BCRP is a half-transporter that functions as a homodimer or oligomer, and despite its name, is expressed throughout the human body (Nakanishi et al., 2003; Xu et al., 2004). BCRP has been found in placenta, testis, GI tract, kidney, liver canalicular membrane, and in hematopoietic stem cells (Maliepaard et al., 2001; Scharenberg et al., 2002; Bart et al., 2004; Huls et al., 2008). In the CNS, BCRP expression has been detected in pericytes, microglia, astrocytes, in neural progenitor, and stem cells (Figure 1; Islam et al., 2005; Mouthon et al., 2006; Lee et al., 2007; Shimizu et al., 2008), and it is highly expressed in the luminal membrane of brain capillary endothelial cells (Cooray et al., 2002; Eisenblatter and Galla, 2002; Zhang et al., 2003; Dauchy et al., 2008). BCRP tissue distribution and substrate spectrum greatly overlap with those of P-gp, and recent evidence indicates that at the blood-brain barrier, BCRP and P-gp work together in a “coordinated” fashion to transport chemotherapeutic drugs (Schinkel and Jonker, 2003; de Vries et al., 2007; Chen et al., 2009). In this regard, researchers currently study the involvement of BCRP in Aβ transport across the blood-brain barrier and its possible role in AD.
BCRP in AD
The recently described concept that BCRP and P-gp “team up and work together” in transporting chemotherapeutics at the blood-brain barrier may possibly be extended to their handling of Aβ. In a review article, Pahnke et al. (2008) stated that BCRP partially compensates for a loss in P-gp-mediated Aβ clearance in hAPP-overexpressing AD mouse models that are P-gp-deficient at the blood-brain barrier. However, these data appear to be unpublished, and thus, the details of this work are unclear. In contrast, the same group showed that there was no difference in Aβ load and plaque size in APP/PS1 × Abcg2−/− mice compared to control APP/PS1 mice, indicating that Bcrp is not involved in the process that clears Aβ from the brain (Krohn et al., 2011). Consistent with this, we showed that Bcrp is not involved in Aβ42 transport in isolated brain capillaries of wild-type and hAPP mice (Hartz et al., 2010).
In another study, Xiong et al. analyzed cerebral cortex samples from AD patients and patients with AD/CAA with qPCR, Western blotting, and immunohistochemistry. Expression of both BCRP mRNA and protein were significantly increased in samples from AD/CAA patients compared to control samples from age-matched non-demented individuals. This same trend was observed in brain tissue samples from AD patients (Xiong et al., 2009). In contrast, Wijesuriya et al. (2010) found no changes in BCRP protein expression in hippocampal blood vessels of samples from AD patients when compared to control samples from age-matched non-demented individuals. In further experiments, Xiong et al. used triple-transgenic 3XTg mice that overexpress human APP, presenilin-1, and tau, and that constitute the only AD model that exhibits both Aβ and tau pathology. The authors showed that capillary Bcrp protein expression was increased in brain sections from 10-month-old 3XTg mice that had high Aβ brain deposits in the parenchyma and spatially close to brain capillaries compared to wild-type mice. To test if Aβ affects BCRP transport function, Xiong et al. then exposed hABCG2-transfected HEK293 cells to Aβ40. Accumulation of the fluorescent BCRP substrate Hoechst 33342 was significantly higher in cells treated with Aβ40 compared to untreated and empty-vector transfected cells, suggesting that Aβ interacts with BCRP, thereby impairing BCRP-mediated efflux transport. The authors also dosed Abcg2 knockout mice intravenously with Cy5.5-labeled Aβ40 and, using in vivo imaging, found significantly higher Cy5.5 fluorescence in the brains of Bcrp knockout mice compared to brains from wild-type mice. This effect was not observed after injecting Cy5.5-labeled scrambled Aβ40 (Xiong et al., 2009). From these data the authors concluded that BCRP may act as a gatekeeper at the blood-brain barrier to prevent Aβ from entering the brain.
In another study, Tai et al. (2009) used the human brain endothelial cell line hCMEC/D3 and showed that P-gp and BCRP inhibitors reduced Aβ40 efflux from cells preloaded with 125I-Aβ40. Inhibition of P-gp or BCRP increased apical-to-basolateral, but not basolateral-to-apical permeation of 125I-Aβ40. The authors concluded that both P-gp and BCRP transport Aβ40 and that P-gp and BCRP prevent apical-to-basolateral transport, but may not be involved in basolateral-to-apical Aβ transport.
A potential protective role of BCRP against oxidative stress in AD was proposed by Shen et al. (2010). Using ABCG2-overexpressing HEK293 cells, the authors demonstrated that BCRP inhibits NFκB activation, which reduces oxidative stress. Consistent with this, Shen et al. found increased levels of phosphorylated IκB-α, which leads to NFκB activation, and higher Aβ40 levels in brain tissue from Abcg2−/− mice compared to wild-type mice. Based on these findings, the authors proposed that upregulation of blood-brain barrier BCRP in AD is an adaptive response to reduce oxidative stress through inhibition of the NFκB signaling pathway.
Finally, only one pharmacogenomic study is available where an association between BCRP polymorphisms and AD was investigated. In this genome-wide association study that involved screening the potential CSF biomarkers Aβ42, total tau, and p-tau181 across the genomes of patient samples, Kim et al. (2011) identified one SNP (rs11724427) in the ABCG2 gene that is possibly associated with a higher risk for AD. However, it remains to be seen if ABCG2 polymorphisms play a role in AD etiology and pathology.
BCRP as Potential Therapeutic Target in AD
Soon after the first studies on P-gp and AD were published, researchers hypothesized that BCRP also transports Aβ and contributes to Aβ brain clearance. However, the data that are available are conflicting and, at this point, the picture of BCRP in AD is inconsistent and incomplete. While some studies show that BCRP transports Aβ (Tai et al., 2009; Xiong et al., 2009), other studies demonstrate that Aβ is not transported by BCRP (Hartz et al., 2010; Krohn et al., 2011). Data from AD patient brain samples are also conflicting: BCRP protein levels at the blood-brain barrier have been reported to be unchanged (Wijesuriya et al., 2010), but another study shows that BCRP expression is increased (Xiong et al., 2009). The latter is contrary to what one would expect if BCRP contributed to Aβ clearance from brain to blood. One explanation that has been provided for increased BCRP in AD is that it may act as a gatekeeper at the blood-brain barrier to prevent Aβ from entering the brain (Xiong et al., 2009). However, this is inconsistent with current knowledge of AD etiology indicating that Aβ accumulation in the brain is not due to increased Aβ uptake from the blood, but rather due to reduced Aβ clearance from the brain.
Several issues could contribute to these conflicting findings. For example, changes in transporter expression and/or activity could depend on the AD model, patient or animal age, and/or inter-individual differences between patients or research animals. Differences could also exist between AD in humans and modeled AD in research animals. In addition, cell lines of non-brain origin may not accurately represent the in vivo blood-brain barrier and provide a misleading assumption of BCRP expression and/or activity in the brain. Clearly, future studies need to elucidate if BCRP is involved in Aβ brain clearance and pathology. Answering this question will determine if BCRP could be a potential therapeutic target in AD.
Future perspectives
An ideal AD medication corrects the pathophysiological cause of AD, reverses, or at least halts, cognitive decline, prevents disease onset, is effective in most patients with little side effects, and is affordable. However, currently available AD therapies have not met these criteria, and it is obvious that novel strategies are desperately needed. Based on current knowledge, we propose that, under normal physiological conditions, Aβ clearance from the brain is, at least in part, based on an interplay between ABCA1, LRP1, P-gp, and MRP1 (and possibly BCRP; Figure 2). We postulate that a decrease in function of ABCA1, LRP1, P-gp, or MRP1 (and possibly BCRP), or a decrease in function of some or all of these proteins reduces Aβ brain clearance, resulting in increased Aβ brain levels, which may pose a risk for AD. Thus, one potential new strategy that emerges from this picture is targeting ABC transporters at the blood-brain barrier and in other CNS cells. However, this approach is still in its infancy, and future research needs to address, amongst others, the following basic questions:
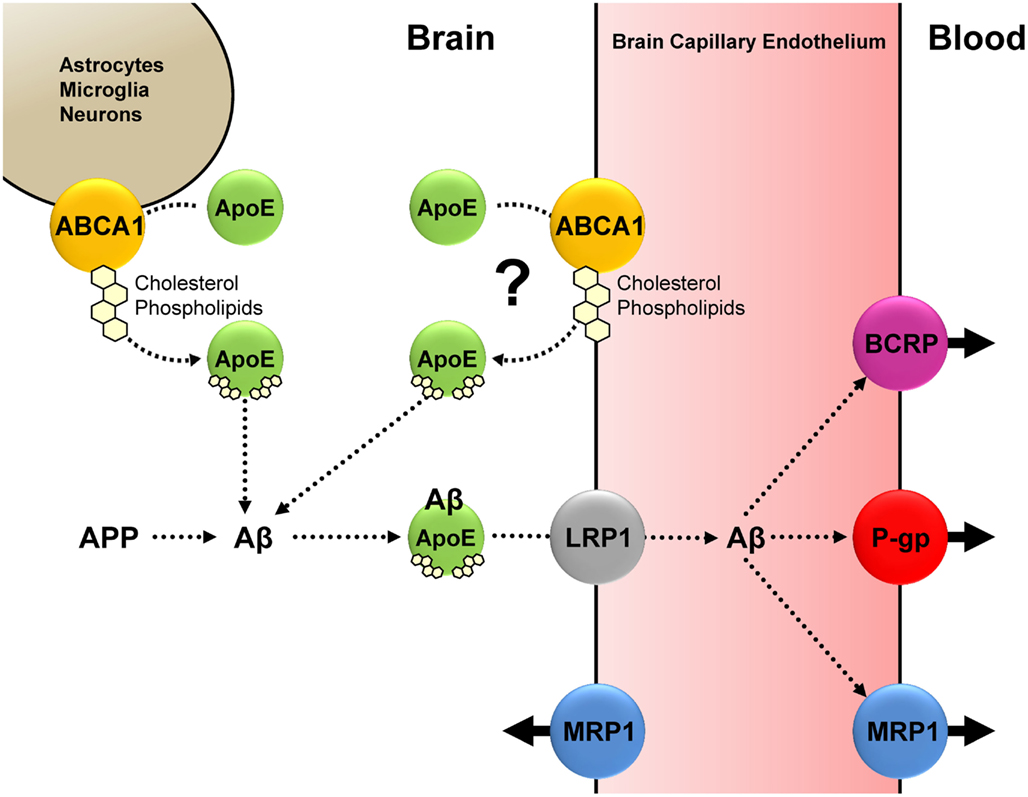
Figure 2. Based on current knowledge, we propose the following interplay between Aβ, ApoE, ABCA1, LRP1, P-gp, MRP1 (and possibly BCRP): Under normal physiological conditions, ABCA1-mediated cholesterol and phospholipid efflux from astrocytes, microglia, neurons (and possibly endothelial cells) facilitates ApoE lipidation. Lipidated ApoE associates with Aβ, and the ApoE-Aβ complex is taken up at the abluminal membrane of the brain capillary endothelium; this step is facilitated by LRP1. P-gp and MRP1 (and possibly BCRP) mediate Aβ efflux from the brain capillary endothelium into the blood. We postulate that a decrease in function of either ABCA1, LRP1, P-gp, or MRP1 (and possibly BCRP), or a decrease in function of some or all of these proteins reduces Aβ brain clearance, resulting in increased Aβ brain levels, which may pose a risk for AD.
(1) Do ABC transporters contribute to AD, and if so, what is the contribution of each transporter?
(2) How does AD affect ABC transporters in the CNS?
(3) Where do ABC transporters fit in the larger “map” of AD pathophysiology?
Answering these questions may still not bring us closer to a viable strategy that can be translated into the clinic. And targeting ABC transporters alone may also be an ineffective approach. Given the complexity of AD, it is unlikely to find a “magic bullet.” However, combining new strategies with existing ones that target AD from different angles may increase the chance of finding an effective therapy and may provide flexible therapy modules for individualized treatment tailored to patients’ needs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Britt Johnson and Emma Soldner for editorial assistance. The project described was supported by Grant Number 1R01AG039621 from the National Institute on Aging. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Aging or the National Institutes of Health.
References
Akanuma, S., Ohtsuki, S., Doi, Y., Tachikawa, M., Ito, S., Hori, S., Asashima, T., Hashimoto, T., Yamada, K., Ueda, K., Iwatsubo, T., and Terasaki, T. (2008). ATP-binding cassette transporter A1 (ABCA1) deficiency does not attenuate the brain-to-blood efflux transport of human amyloid-beta peptide (1-40) at the blood-brain barrier. Neurochem. Int. 52, 956–961.
Akram, A., Schmeidler, J., Katsel, P., Hof, P. R., and Haroutunian, V. (2010). Increased expression of cholesterol transporter ABCA1 is highly correlated with severity of dementia in AD hippocampus. Brain Res. 1318, 167–177.
Allikmets, R., and Dean, M. (1998). Cloning of novel ABC transporter genes. Meth. Enzymol. 292, 116–130.
Bart, J., Hollema, H., Groen, H. J., De Vries, E. G., Hendrikse, N. H., Sleijfer, D. T., Wegman, T. D., Vaalburg, W., and Van Der Graaf, W. T. (2004). The distribution of drug-efflux pumps, P-gp, BCRP, MRP1 and MRP2, in the normal blood-testis barrier and in primary testicular tumours. Eur. J. Cancer 40, 2064–2070.
Bodzioch, M., Orso, E., Klucken, J., Langmann, T., Bottcher, A., Diederich, W., Drobnik, W., Barlage, S., Buchler, C., Porsch-Ozcurumez, M., Kaminski, W. E., Hahmann, H. W., Oette, K., Rothe, G., Aslanidis, C., Lackner, K. J., and Schmitz, G. (1999). The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 22, 347–351.
Brenn, A., Grube, M., Peters, M., Fischer, A., Jedlitschky, G., Kroemer, H. K., Warzok, R. W., and Vogelgesang, S. (2011). Beta-amyloid downregulates MDR1-P-glycoprotein (Abcb1) expression at the blood-brain barrier in mice. Int. J. Alzheimers Dis. 2011, 690121.
Broccardo, C., Nieoullon, V., Amin, R., Masmejean, F., Carta, S., Tassi, S., Pophillat, M., Rubartelli, A., Pierres, M., Rougon, G., Nieoullon, A., Chazal, G., and Chimini, G. (2006). ABCA2 is a marker of neural progenitors and neuronal subsets in the adult rodent brain. J. Neurochem. 97, 345–355.
Brookmeyer, R., Johnson, E., Ziegler-Graham, K., and Arrighi, H. M. (2007). Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 3, 186–191.
Brown, J. III, Theisler, C., Silberman, S., Magnuson, D., Gottardi-Littell, N., Lee, J. M., Yager, D., Crowley, J., Sambamurti, K., Rahman, M. M., Reiss, A. B., Eckman, C. B., and Wolozin, B. (2004). Differential expression of cholesterol hydroxylases in Alzheimer’s disease. J. Biol. Chem. 279, 34674–34681.
Burns, M. P., Vardanian, L., Pajoohesh-Ganji, A., Wang, L., Cooper, M., Harris, D. C., Duff, K., and Rebeck, G. W. (2006). The effects of ABCA1 on cholesterol efflux and Abeta levels in vitro and in vivo. J. Neurochem. 98, 792–800.
Chen, Y., Agarwal, S., Shaik, N. M., Chen, C., Yang, Z., and Elmquist, W. F. (2009). P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J. Pharmacol. Exp. Ther. 330, 956–963.
Chen, Z. J., Vulevic, B., Ile, K. E., Soulika, A., Davis, W. Jr., Reiner, P. B., Connop, B. P., Nathwani, P., Trojanowski, J. Q., and Tew, K. D. (2004). Association of ABCA2 expression with determinants of Alzheimer’s disease. FASEB J. 18, 1129–1131.
Chu, L. W., Li, Y., Li, Z., Tang, A. Y., Cheung, B. M., Leung, R. Y., Yik, P. Y., Jin, D. Y., and Song, Y. Q. (2007). A novel intronic polymorphism of ABCA1 gene reveals risk for sporadic Alzheimer’s disease in Chinese. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 1007–1013.
Cirrito, J. R., Deane, R., Fagan, A. M., Spinner, M. L., Parsadanian, M., Finn, M. B., Jiang, H., Prior, J. L., Sagare, A., Bales, K. R., Paul, S. M., Zlokovic, B. V., Piwnica-Worms, D., and Holtzman, D. M. (2005). P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Invest. 115, 3285–3290.
Cole, S. P., Bhardwaj, G., Gerlach, J. H., Mackie, J. E., Grant, C. E., Almquist, K. C., Stewart, A. J., Kurz, E. U., Duncan, A. M., and Deeley, R. G. (1992). Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 258, 1650–1654.
Conseil, G., Deeley, R. G., and Cole, S. P. (2005). Polymorphisms of MRP1 (ABCC1) and related ATP-dependent drug transporters. Pharmacogenet. Genomics 15, 523–533.
Cooray, H. C., Blackmore, C. G., Maskell, L., and Barrand, M. A. (2002). Localisation of breast cancer resistance protein in microvessel endothelium of human brain. Neuroreport 13, 2059–2063.
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., Roses, A. D., Haines, J. L., and Pericak-Vance, M. A. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923.
Cordon-Cardo, C., O’Brien, J. P., Casals, D., Rittman-Grauer, L., Biedler, J. L., Melamed, M. R., and Bertino, J. R. (1989). Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. U.S.A. 86, 695–698.
Dauchy, S., Dutheil, F., Weaver, R. J., Chassoux, F., Daumas-Duport, C., Couraud, P. O., Scherrmann, J. M., De Waziers, I., and Decleves, X. (2008). ABC transporters, cytochromes P450 and their main transcription factors: expression at the human blood-brain barrier. J. Neurochem. 107, 1518–1528.
Davis, W. Jr. (2010). The ATP-binding cassette transporter-2 (ABCA2) increases endogenous amyloid precursor protein expression and Abeta fragment generation. Curr. Alzheimer Res. 7, 566–577.
Davis, W. Jr., Boyd, J. T., Ile, K. E., and Tew, K. D. (2004). Human ATP-binding cassette transporter-2 (ABCA2) positively regulates low-density lipoprotein receptor expression and negatively regulates cholesterol esterification in Chinese hamster ovary cells. Biochim. Biophys. Acta 1683, 89–100.
de Vries, N. A., Zhao, J., Kroon, E., Buckle, T., Beijnen, J. H., and Van Tellingen, O. (2007). P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin. Cancer Res. 13, 6440–6449.
Deane, R., Wu, Z., Sagare, A., Davis, J., Du Yan, S., Hamm, K., Xu, F., Parisi, M., Larue, B., Hu, H. W., Spijkers, P., Guo, H., Song, X., Lenting, P. J., Van Nostrand, W. E., and Zlokovic, B. V. (2004). LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43, 333–344.
Desrayaud, S., Guntz, P., Scherrmann, J. M., and Lemaire, M. (1997). Effect of the P-glycoprotein inhibitor, SDZ PSC 833, on the blood and brain pharmacokinetics of colchicine. Life Sci. 61, 153–163.
Donkin, J. J., Stukas, S., Hirsch-Reinshagen, V., Namjoshi, D., Wilkinson, A., May, S., Chan, J., Fan, J., Collins, J., and Wellington, C. L. (2010). ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J. Biol. Chem. 285, 34144–34154.
Doyle, L. A., Yang, W., Abruzzo, L. V., Krogmann, T., Gao, Y., Rishi, A. K., and Ross, D. D. (1998). A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 95, 15665–15670.
Eisenblatter, T., and Galla, H. J. (2002). A new multidrug resistance protein at the blood-brain barrier. Biochem. Biophys. Res. Commun. 293, 1273–1278.
Ellis, J. M. (2005). Cholinesterase inhibitors in the treatment of dementia. J. Am. Osteopath. Assoc. 105, 145–158.
Farrer, L. A., Cupples, L. A., Haines, J. L., Hyman, B., Kukull, W. A., Mayeux, R., Myers, R. H., Pericak-Vance, M. A., Risch, N., and Van Duijn, C. M. (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356.
Fellner, S., Bauer, B., Miller, D. S., Schaffrik, M., Fankhanel, M., Spruss, T., Bernhardt, G., Graeff, C., Farber, L., Gschaidmeier, H., Buschauer, A., and Fricker, G. (2002). Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J. Clin. Invest. 110, 1309–1318.
Fujiyoshi, M., Ohtsuki, S., Hori, S., Tachikawa, M., and Terasaki, T. (2007). 24S-hydroxycholesterol induces cholesterol release from choroid plexus epithelial cells in an apical- and apoE isoform-dependent manner concomitantly with the induction of ABCA1 and ABCG1 expression. J. Neurochem. 100, 968–978.
Fujiyoshi, M., Tachikawa, M., Ohtsuki, S., Ito, S., Uchida, Y., Akanuma, S., Kamiie, J., Hashimoto, T., Hosoya, K., Iwatsubo, T., and Terasaki, T. (2011). Amyloid-beta peptide(1-40) elimination from cerebrospinal fluid involves low-density lipoprotein receptor-related protein 1 at the blood-cerebrospinal fluid barrier. J. Neurochem. 118, 407–415.
Fukumoto, H., Deng, A., Irizarry, M. C., Fitzgerald, M. L., and Rebeck, G. W. (2002). Induction of the cholesterol transporter ABCA1 in central nervous system cells by liver X receptor agonists increases secreted Abeta levels. J. Biol. Chem. 277, 48508–48513.
Garewal, H. S., Ahmann, F. R., Schifman, R. B., and Celniker, A. (1986). ATP assay: ability to distinguish cytostatic from cytocidal anticancer drug effects. J. Natl. Cancer Inst. 77, 1039–1045.
Gravina, S. A., Ho, L., Eckman, C. B., Long, K. E., Otvos, L. Jr., Younkin, L. H., Suzuki, N., and Younkin, S. G. (1995). Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43). J. Biol. Chem. 270, 7013–7016.
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356.
Hartz, A. M., and Bauer, B. (2011). ABC transporters in the CNS – an inventory. Curr. Pharm. Biotechnol. 12, 656–673.
Hartz, A. M., Miller, D. S., and Bauer, B. (2010). Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol. Pharmacol. 77, 715–723.
Hirsch-Reinshagen, V., Maia, L. F., Burgess, B. L., Blain, J. F., Naus, K. E., Mcisaac, S. A., Parkinson, P. F., Chan, J. Y., Tansley, G. H., Hayden, M. R., Poirier, J., Van Nostrand, W., and Wellington, C. L. (2005). The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J. Biol. Chem. 280, 43243–43256.
Hirsch-Reinshagen, V., Zhou, S., Burgess, B. L., Bernier, L., Mcisaac, S. A., Chan, J. Y., Tansley, G. H., Cohn, J. S., Hayden, M. R., and Wellington, C. L. (2004). Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J. Biol. Chem. 279, 41197–41207.
Hitchins, R. N., Harman, D. H., Davey, R. A., and Bell, D. R. (1988). Identification of a multidrug resistance associated antigen (P-glycoprotein) in normal human tissues. Eur. J. Cancer Clin. Oncol. 24, 449–454.
Holtzman, D. M., Fagan, A. M., Mackey, B., Tenkova, T., Sartorius, L., Paul, S. M., Bales, K., Ashe, K. H., Irizarry, M. C., and Hyman, B. T. (2000). Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann. Neurol. 47, 739–747.
Honzumi, S., Shima, A., Hiroshima, A., Koieyama, T., Ubukata, N., and Terasaka, N. (2010). LXRalpha regulates human CETP expression in vitro and in transgenic mice. Atherosclerosis 212, 139–145.
Hsiao, K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S., Yang, F., and Cole, G. (1996). Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102.
Hsu, S. I., Lothstein, L., and Horwitz, S. B. (1989). Differential overexpression of three mdr gene family members in multidrug-resistant J774.2 mouse cells. Evidence that distinct P-glycoprotein precursors are encoded by unique mdr genes. J. Biol. Chem. 264, 12053–12062.
Huls, M., Brown, C. D., Windass, A. S., Sayer, R., Van Den Heuvel, J. J., Heemskerk, S., Russel, F. G., and Masereeuw, R. (2008). The breast cancer resistance protein transporter ABCG2 is expressed in the human kidney proximal tubule apical membrane. Kidney Int. 73, 220–225.
Islam, M. O., Kanemura, Y., Tajria, J., Mori, H., Kobayashi, S., Shofuda, T., Miyake, J., Hara, M., Yamasaki, M., and Okano, H. (2005). Characterization of ABC transporter ABCB1 expressed in human neural stem/progenitor cells. FEBS Lett. 579, 3473–3480.
Jeynes, B., and Provias, J. (2011). An investigation into the role of P-glycoprotein in Alzheimer’s disease lesion pathogenesis. Neurosci. Lett. 487, 389–393.
Jiang, Q., Lee, C. Y., Mandrekar, S., Wilkinson, B., Cramer, P., Zelcer, N., Mann, K., Lamb, B., Willson, T. M., Collins, J. L., Richardson, J. C., Smith, J. D., Comery, T. A., Riddell, D., Holtzman, D. M., Tontonoz, P., and Landreth, G. E. (2008). ApoE promotes the proteolytic degradation of Abeta. Neuron 58, 681–693.
Juliano, R. L., and Ling, V. (1976). A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 455, 152–162.
Kaminski, W. E., Piehler, A., Pullmann, K., Porsch-Ozcurumez, M., Duong, C., Bared, G. M., Buchler, C., and Schmitz, G. (2001). Complete coding sequence, promoter region, and genomic structure of the human ABCA2 gene and evidence for sterol-dependent regulation in macrophages. Biochem. Biophys. Res. Commun. 281, 249–258.
Kaminski, W. E., Piehler, A., and Wenzel, J. J. (2006). ABC A-subfamily transporters: structure, function and disease. Biochim. Biophys. Acta 1762, 510–524.
Kartner, N., Riordan, J. R., and Ling, V. (1983). Cell surface P-glycoprotein associated with multidrug resistance in mammalian cell lines. Science 221, 1285–1288.
Katzov, H., Chalmers, K., Palmgren, J., Andreasen, N., Johansson, B., Cairns, N. J., Gatz, M., Wilcock, G. K., Love, S., Pedersen, N. L., Brookes, A. J., Blennow, K., Kehoe, P. G., and Prince, J. A. (2004). Genetic variants of ABCA1 modify Alzheimer disease risk and quantitative traits related to beta-amyloid metabolism. Hum. Mutat. 23, 358–367.
Kilic, E., Spudich, A., Kilic, U., Rentsch, K. M., Vig, R., Matter, C. M., Wunderli-Allenspach, H., Fritschy, J. M., Bassetti, C. L., and Hermann, D. M. (2008). ABCC1: a gateway for pharmacological compounds to the ischaemic brain. Brain 131, 2679–2689.
Kim, S., Swaminathan, S., Shen, L., Risacher, S. L., Nho, K., Foroud, T., Shaw, L. M., Trojanowski, J. Q., Potkin, S. G., Huentelman, M. J., Craig, D. W., Dechairo, B. M., Aisen, P. S., Petersen, R. C., Weiner, M. W., and Saykin, A. J. (2011). Genome-wide association study of CSF biomarkers Abeta1-42, t-tau, and p-tau181p in the ADNI cohort. Neurology 76, 69–79.
Kim, W. S., Bhatia, S., Elliott, D. A., Agholme, L., Kagedal, K., Mccann, H., Halliday, G. M., Barnham, K. J., and Garner, B. (2010). Increased ATP-binding cassette transporter A1 expression in Alzheimer’s disease hippocampal neurons. J. Alzheimers Dis. 21, 193–205.
Kim, W. S., Chan, S. L., Hill, A. F., Guillemin, G. J., and Garner, B. (2009). Impact of 27-hydroxycholesterol on amyloid-beta peptide production and ATP-binding cassette transporter expression in primary human neurons. J. Alzheimers Dis. 16, 121–131.
Kim, W. S., Guillemin, G. J., Glaros, E. N., Lim, C. K., and Garner, B. (2006). Quantitation of ATP-binding cassette subfamily-A transporter gene expression in primary human brain cells. Neuroreport 17, 891–896.
Kim, W. S., Rahmanto, A. S., Kamili, A., Rye, K. A., Guillemin, G. J., Gelissen, I. C., Jessup, W., Hill, A. F., and Garner, B. (2007). Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J. Biol. Chem. 282, 2851–2861.
Kohen, R., Shofer, J. B., Korvatska, O., Petrie, E. C., Wang, L. Y., Schellenberg, G. D., Peskind, E. R., and Wilkinson, C. W. (2011). ABCB1 genotype and CSF beta-amyloid in Alzheimer disease. J. Geriatr. Psychiatry. Neurol. 24, 63–66.
Koldamova, R., Staufenbiel, M., and Lefterov, I. (2005a). Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J. Biol. Chem. 280, 43224–43235.
Koldamova, R. P., Lefterov, I. M., Staufenbiel, M., Wolfe, D., Huang, S., Glorioso, J. C., Walter, M., Roth, M. G., and Lazo, J. S. (2005b). The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer’s disease. J. Biol. Chem. 280, 4079–4088.
Koldamova, R. P., Lefterov, I. M., Ikonomovic, M. D., Skoko, J., Lefterov, P. I., Isanski, B. A., Dekosky, S. T., and Lazo, J. S. (2003). 22R-hydroxycholesterol and 9-cis-retinoic acid induce ATP-binding cassette transporter A1 expression and cholesterol efflux in brain cells and decrease amyloid beta secretion. J. Biol. Chem. 278, 13244–13256.
Kolsch, H., Lutjohann, D., Jessen, F., Von Bergmann, K., Schmitz, S., Urbach, H., Maier, W., and Heun, R. (2006). Polymorphism in ABCA1 influences CSF 24S-hydroxycholesterol levels but is not a major risk factor of Alzheimer’s disease. Int. J. Mol. Med. 17, 791–794.
Krohn, M., Lange, C., Hofrichter, J., Scheffler, K., Stenzel, J., Steffen, J., Schumacher, T., Bruning, T., Plath, A. S., Alfen, F., Schmidt, A., Winter, F., Rateitschak, K., Wree, A., Gsponer, J., Walker, L. C., and Pahnke, J. (2011). Cerebral amyloid-beta proteostasis is regulated by the membrane transport protein ABCC1 in mice. J. Clin. Invest. 121, 3924–3931.
Kuhnke, D., Jedlitschky, G., Grube, M., Krohn, M., Jucker, M., Mosyagin, I., Cascorbi, I., Walker, L. C., Kroemer, H. K., Warzok, R. W., and Vogelgesang, S. (2007). MDR1-P-Glycoprotein (ABCB1) mediates transport of Alzheimer’s amyloid-beta peptides – implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol. 17, 347–353.
Lam, F. C., Liu, R., Lu, P., Shapiro, A. B., Renoir, J. M., Sharom, F. J., and Reiner, P. B. (2001). beta-Amyloid efflux mediated by p-glycoprotein. J. Neurochem. 76, 1121–1128.
Lawn, R. M., Wade, D. P., Garvin, M. R., Wang, X., Schwartz, K., Porter, J. G., Seilhamer, J. J., Vaughan, A. M., and Oram, J. F. (1999). The Tangier disease gene product ABC1 controls the cellular apolipoprotein-mediated lipid removal pathway. J. Clin. Invest. 104, R25–R31.
Lee, G., Babakhanian, K., Ramaswamy, M., Prat, A., Wosik, K., and Bendayan, R. (2007). Expression of the ATP-binding cassette membrane transporter, ABCG2, in human and rodent brain microvessel endothelial and glial cell culture systems. Pharm. Res. 24, 1262–1274.
Lee, G., and Bendayan, R. (2004). Functional expression and localization of P-glycoprotein in the central nervous system: relevance to the pathogenesis and treatment of neurological disorders. Pharm. Res. 21, 1313–1330.
Lefterov, I., Bookout, A., Wang, Z., Staufenbiel, M., Mangelsdorf, D., and Koldamova, R. (2007). Expression profiling in APP23 mouse brain: inhibition of Abeta amyloidosis and inflammation in response to LXR agonist treatment. Mol. Neurodegener. 2, 20.
Li, Y., Tacey, K., Doil, L., Van Luchene, R., Garcia, V., Rowland, C., Schrodi, S., Leong, D., Lau, K., Catanese, J., Sninsky, J., Nowotny, P., Holmans, P., Hardy, J., Powell, J., Lovestone, S., Thal, L., Owen, M., Williams, J., Goate, A., and Grupe, A. (2004). Association of ABCA1 with late-onset Alzheimer’s disease is not observed in a case-control study. Neurosci. Lett. 366, 268–271.
Loe, D. W., Almquist, K. C., Deeley, R. G., and Cole, S. P. (1996). Multidrug resistance protein (MRP)-mediated transport of leukotriene C4 and chemotherapeutic agents in membrane vesicles. Demonstration of glutathione-dependent vincristine transport. J. Biol. Chem. 271, 9675–9682.
Loeb, M. B., Molloy, D. W., Smieja, M., Standish, T., Goldsmith, C. H., Mahony, J., Smith, S., Borrie, M., Decoteau, E., Davidson, W., Mcdougall, A., Gnarpe, J., O’D, O. M., and Chernesky, M. (2004). A randomized, controlled trial of doxycycline and rifampin for patients with Alzheimer’s disease. J. Am. Geriatr. Soc. 52, 381–387.
Lovell, M. A., Ehmann, W. D., Mattson, M. P., and Markesbery, W. R. (1997). Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer’s disease. Neurobiol. Aging 18, 457–461.
Luciani, M. F., Denizot, F., Savary, S., Mattei, M. G., and Chimini, G. (1994). Cloning of two novel ABC transporters mapping on human chromosome 9. Genomics 21, 150–159.
Mace, S., Cousin, E., Ricard, S., Genin, E., Spanakis, E., Lafargue-Soubigou, C., Genin, B., Fournel, R., Roche, S., Haussy, G., Massey, F., Soubigou, S., Brefort, G., Benoit, P., Brice, A., Campion, D., Hollis, M., Pradier, L., Benavides, J., and Deleuze, J. F. (2005). ABCA2 is a strong genetic risk factor for early-onset Alzheimer’s disease. Neurobiol. Dis. 18, 119–125.
Mack, J. T., Townsend, D. M., Beljanski, V., and Tew, K. D. (2007). The ABCA2 transporter: intracellular roles in trafficking and metabolism of LDL-derived cholesterol and sterol-related compounds. Curr. Drug Metab. 8, 47–57.
Maliepaard, M., Scheffer, G. L., Faneyte, I. F., Van Gastelen, M. A., Pijnenborg, A. C., Schinkel, A. H., Van De Vijver, M. J., Scheper, R. J., and Schellens, J. H. (2001). Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 61, 3458–3464.
Mark, R. J., Lovell, M. A., Markesbery, W. R., Uchida, K., and Mattson, M. P. (1997). A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J. Neurochem. 68, 255–264.
Mayer, U., Wagenaar, E., Dorobek, B., Beijnen, J. H., Borst, P., and Schinkel, A. H. (1997). Full blockade of intestinal P-glycoprotein and extensive inhibition of blood-brain barrier P-glycoprotein by oral treatment of mice with PSC833. J. Clin. Invest. 100, 2430–2436.
Mealey, K. L., Barhoumi, R., Burghardt, R. C., Safe, S., and Kochevar, D. T. (2002). Doxycycline induces expression of P glycoprotein in MCF-7 breast carcinoma cells. Antimicrob. Agents Chemother. 46, 755–761.
Mega, M. S., Cummings, J. L., Fiorello, T., and Gornbein, J. (1996). The spectrum of behavioral changes in Alzheimer’s disease. Neurology 46, 130–135.
Michaki, V., Guix, F. X., Vennekens, K., Munck, S., Dingwall, C., Davis, J. B., Townsend, D. M., Tew, K. D., Feiguin, F., De Strooper, B., Dotti, C. G., and Wahle, T. (2012). Down-regulation of the ATP-binding cassette transporter 2 (Abca2) reduces Amyloid-beta production by altering nicastrin maturation and intracellular localization. J. Biol. Chem. 287, 1100–1111.
Mouthon, M. A., Fouchet, P., Mathieu, C., Sii-Felice, K., Etienne, O., Lages, C. S., and Boussin, F. D. (2006). Neural stem cells from mouse forebrain are contained in a population distinct from the “side population.” J. Neurochem. 99, 807–817.
Nakanishi, T., Doyle, L. A., Hassel, B., Wei, Y., Bauer, K. S., Wu, S., Pumplin, D. W., Fang, H. B., and Ross, D. D. (2003). Functional characterization of human breast cancer resistance protein (BCRP, ABCG2) expressed in the oocytes of Xenopus laevis. Mol. Pharmacol. 64, 1452–1462.
Nies, A. T., Jedlitschky, G., Konig, J., Herold-Mende, C., Steiner, H. H., Schmitt, H. P., and Keppler, D. (2004). Expression and immunolocalization of the multidrug resistance proteins, MRP1-MRP6 (ABCC1-ABCC6), in human brain. Neuroscience 129, 349–360.
Ohtsuki, S., Watanabe, Y., Hori, S., Suzuki, H., Bhongsatiern, J., Fujiyoshi, M., Kamoi, M., Kamiya, N., Takanaga, H., and Terasaki, T. (2004). mRNA expression of the ATP-binding cassette transporter subfamily A (ABCA) in rat and human brain capillary endothelial cells. Biol. Pharm. Bull. 27, 1437–1440.
Pahnke, J., Wolkenhauer, O., Krohn, M., and Walker, L. C. (2008). Clinico-pathologic function of cerebral ABC transporters – implications for the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 5, 396–405.
Panzenboeck, U., Balazs, Z., Sovic, A., Hrzenjak, A., Levak-Frank, S., Wintersperger, A., Malle, E., and Sattler, W. (2002). ABCA1 and scavenger receptor class B, type I, are modulators of reverse sterol transport at an in vitro blood-brain barrier constituted of porcine brain capillary endothelial cells. J. Biol. Chem. 277, 42781–42789.
Puglielli, L., Tanzi, R. E., and Kovacs, D. M. (2003). Alzheimer’s disease: the cholesterol connection. Nat. Neurosci. 6, 345–351.
Repa, J. J., Turley, S. D., Lobaccaro, J. A., Medina, J., Li, L., Lustig, K., Shan, B., Heyman, R. A., Dietschy, J. M., and Mangelsdorf, D. J. (2000). Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 289, 1524–1529.
Reynolds, C. A., Hong, M. G., Eriksson, U. K., Blennow, K., Bennet, A. M., Johansson, B., Malmberg, B., Berg, S., Wiklund, F., Gatz, M., Pedersen, N. L., and Prince, J. A. (2009). A survey of ABCA1 sequence variation confirms association with dementia. Hum. Mutat. 30, 1348–1354.
Riddell, D. R., Zhou, H., Comery, T. A., Kouranova, E., Lo, C. F., Warwick, H. K., Ring, R. H., Kirksey, Y., Aschmies, S., Xu, J., Kubek, K., Hirst, W. D., Gonzales, C., Chen, Y., Murphy, E., Leonard, S., Vasylyev, D., Oganesian, A., Martone, R. L., Pangalos, M. N., Reinhart, P. H., and Jacobsen, J. S. (2007). The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol. Cell. Neurosci. 34, 621–628.
Roberts, L. M., Black, D. S., Raman, C., Woodford, K., Zhou, M., Haggerty, J. E., Yan, A. T., Cwirla, S. E., and Grindstaff, K. K. (2008). Subcellular localization of transporters along the rat blood-brain barrier and blood-cerebral-spinal fluid barrier by in vivo biotinylation. Neuroscience 155, 423–438.
Rodriguez, M., Ortega, I., Soengas, I., Suarez, E., Lukas, J. C., and Calvo, R. (2004). Effect of P-glycoprotein inhibition on methadone analgesia and brain distribution in the rat. J. Pharm. Pharmacol. 56, 367–374.
Rodriguez-Rodriguez, E., Mateo, I., Infante, J., Llorca, J., Garcia-Gorostiaga, I., Vazquez-Higuera, J. L., Sanchez-Juan, P., Berciano, J., and Combarros, O. (2009). Interaction between HMGCR and ABCA1 cholesterol-related genes modulates Alzheimer’s disease risk. Brain Res. 1280, 166–171.
Rodriguez-Rodriguez, E., Mateo, I., Llorca, J., Sanchez-Quintana, C., Infante, J., Garcia-Gorostiaga, I., Sanchez-Juan, P., Berciano, J., and Combarros, O. (2007). Association of genetic variants of ABCA1 with Alzheimer’s disease risk. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 964–968.
Rodriguez-Rodriguez, E., Vazquez-Higuera, J. L., Sanchez-Juan, P., Mateo, I., Pozueta, A., Martinez-Garcia, A., Frank, A., Valdivieso, F., Berciano, J., Bullido, M. J., and Combarros, O. (2010). Epistasis between intracellular cholesterol trafficking-related genes (NPC1 and ABCA1) and Alzheimer’s disease risk. J. Alzheimers Dis. 21, 619–625.
Scharenberg, C. W., Harkey, M. A., and Torok-Storb, B. (2002). The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood 99, 507–512.
Schinkel, A. H. (1999). P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv. Drug Deliv. Rev. 36, 179–194.
Schinkel, A. H. (2001). The roles of P-glycoprotein and MRP1 in the blood-brain and blood-cerebrospinal fluid barriers. Adv. Exp. Med. Biol. 500, 365–372.
Schinkel, A. H., and Jonker, J. W. (2003). Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv. Drug Deliv. Rev. 55, 3–29.
Schinkel, A. H., Smit, J. J., Van Tellingen, O., Beijnen, J. H., Wagenaar, E., Van Deemter, L., Mol, C. A., Van Der Valk, M. A., Robanus-Maandag, E. C., Te Riele, H. P., Berns, A. J. M., and Borst, P. (1994). Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 77, 491–502.
Seltzer, B. (2007). Is long-term treatment of Alzheimer’s disease with cholinesterase inhibitor therapy justified? Drugs Aging 24, 881–890.
Shawahna, R., Uchida, Y., Decleves, X., Ohtsuki, S., Yousif, S., Dauchy, S., Jacob, A., Chassoux, F., Daumas-Duport, C., Couraud, P. O., Terasaki, T., and Scherrmann, J. M. (2011). Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol. Pharm. 8, 1332–1341.
Shen, S., Callaghan, D., Juzwik, C., Xiong, H., Huang, P., and Zhang, W. (2010). ABCG2 reduces ROS-mediated toxicity and inflammation: a potential role in Alzheimer’s disease. J. Neurochem. 114, 1590–1604.
Shibata, M., Yamada, S., Kumar, S. R., Calero, M., Bading, J., Frangione, B., Holtzman, D. M., Miller, C. A., Strickland, D. K., Ghiso, J., and Zlokovic, B. V. (2000). Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 106, 1489–1499.
Shibata, N., Kawarai, T., Lee, J. H., Lee, H. S., Shibata, E., Sato, C., Liang, Y., Duara, R., Mayeux, R. P., St George-Hyslop, P. H., and Rogaeva, E. (2006). Association studies of cholesterol metabolism genes (CH25H, ABCA1 and CH24H) in Alzheimer’s disease. Neurosci. Lett. 391, 142–146.
Shimizu, F., Sano, Y., Maeda, T., Abe, M. A., Nakayama, H., Takahashi, R., Ueda, M., Ohtsuki, S., Terasaki, T., Obinata, M., and Kanda, T. (2008). Peripheral nerve pericytes originating from the blood-nerve barrier expresses tight junctional molecules and transporters as barrier-forming cells. J. Cell. Physiol. 217, 388–399.
Simons, M., Keller, P., De Strooper, B., Beyreuther, K., Dotti, C. G., and Simons, K. (1998). Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 95, 6460–6464.
Soontornmalai, A., Vlaming, M. L., and Fritschy, J. M. (2006). Differential, strain-specific cellular and subcellular distribution of multidrug transporters in murine choroid plexus and blood-brain barrier. Neuroscience 138, 159–169.
Strittmatter, W. J., Saunders, A. M., Schmechel, D., Pericak-Vance, M., Enghild, J., Salvesen, G. S., and Roses, A. D. (1993). Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 1977–1981.
Sultana, R., and Butterfield, D. A. (2004). Oxidatively modified GST and MRP1 in Alzheimer’s disease brain: implications for accumulation of reactive lipid peroxidation products. Neurochem. Res. 29, 2215–2220.
Sun, Y., Yao, J., Kim, T. W., and Tall, A. R. (2003). Expression of liver X receptor target genes decreases cellular amyloid beta peptide secretion. J. Biol. Chem. 278, 27688–27694.
Sundar, P. D., Feingold, E., Minster, R. L., Dekosky, S. T., and Kamboh, M. I. (2007). Gender-specific association of ATP-binding cassette transporter 1 (ABCA1) polymorphisms with the risk of late-onset Alzheimer’s disease. Neurobiol. Aging 28, 856–862.
Tachikawa, M., Watanabe, M., Hori, S., Fukaya, M., Ohtsuki, S., Asashima, T., and Terasaki, T. (2005). Distinct spatio-temporal expression of ABCA and ABCG transporters in the developing and adult mouse brain. J. Neurochem. 95, 294–304.
Tai, L. M., Loughlin, A. J., Male, D. K., and Romero, I. A. (2009). P-glycoprotein and breast cancer resistance protein restrict apical-to-basolateral permeability of human brain endothelium to amyloid-beta. J. Cereb. Blood Flow Metab. 29, 1079–1083.
Thiebaut, F., Tsuruo, T., Hamada, H., Gottesman, M. M., Pastan, I., and Willingham, M. C. (1987). Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. U.S.A. 84, 7735–7738.
Ueda, K., Cornwell, M. M., Gottesman, M. M., Pastan, I., Roninson, I. B., Ling, V., and Riordan, J. R. (1986). The mdr1 gene, responsible for multidrug-resistance, codes for P-glycoprotein. Biochem. Biophys. Res. Commun. 141, 956–962.
van Assema, D. M., Lubberink, M., Boellaard, R., Schuit, R. C., Windhorst, A. D., Scheltens, P., Lammertsma, A. A., and van Berckel, B. N. (2012). P-glycoprotein function at the blood-brain barrier: effects of age and gender. Mol. Imaging Biol. [Epub ahead of print].
Vanmierlo, T., Rutten, K., Dederen, J., Bloks, V. W., Van Vark-Van Der Zee, L. C., Kuipers, F., Kiliaan, A., Blokland, A., Sijbrands, E. J., Steinbusch, H., Prickaerts, J., Lutjohann, D., and Mulder, M. (2009). Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol. Aging 32, 1262–1272.
Vogelgesang, S., Cascorbi, I., Schroeder, E., Pahnke, J., Kroemer, H. K., Siegmund, W., Kunert-Keil, C., Walker, L. C., and Warzok, R. W. (2002). Deposition of Alzheimer’s beta-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics 12, 535–541.
Vogelgesang, S., Warzok, R. W., Cascorbi, I., Kunert-Keil, C., Schroeder, E., Kroemer, H. K., Siegmund, W., Walker, L. C., and Pahnke, J. (2004). The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 1, 121–125.
Wahrle, S. E., Jiang, H., Parsadanian, M., Hartman, R. E., Bales, K. R., Paul, S. M., and Holtzman, D. M. (2005). Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem. 280, 43236–43242.
Wahrle, S. E., Jiang, H., Parsadanian, M., Kim, J., Li, A., Knoten, A., Jain, S., Hirsch-Reinshagen, V., Wellington, C. L., Bales, K. R., Paul, S. M., and Holtzman, D. M. (2008). Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Invest. 118, 671–682.
Wahrle, S. E., Jiang, H., Parsadanian, M., Legleiter, J., Han, X., Fryer, J. D., Kowalewski, T., and Holtzman, D. M. (2004). ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem. 279, 40987–40993.
Wahrle, S. E., Shah, A. R., Fagan, A. M., Smemo, S., Kauwe, J. S., Grupe, A., Hinrichs, A., Mayo, K., Jiang, H., Thal, L. J., Goate, A. M., and Holtzman, D. M. (2007). Apolipoprotein E levels in cerebrospinal fluid and the effects of ABCA1 polymorphisms. Mol. Neurodegener. 2, 7.
Wang, F., and Jia, J. (2007). Polymorphisms of cholesterol metabolism genes CYP46 and ABCA1 and the risk of sporadic Alzheimer’s disease in Chinese. Brain Res. 1147, 34–38.
Wavrant-De Vrieze, F., Compton, D., Womick, M., Arepalli, S., Adighibe, O., Li, L., Perez-Tur, J., and Hardy, J. (2007). ABCA1 polymorphisms and Alzheimer’s disease. Neurosci. Lett. 416, 180–183.
Wesolowska, O., Molnar, J., Ocsovszki, I., and Michalak, K. (2009). Differential effect of phenothiazines on MRP1 and P-glycoprotein activity. In vivo 23, 943–947.
Whitney, K. D., Watson, M. A., Collins, J. L., Benson, W. G., Stone, T. M., Numerick, M. J., Tippin, T. K., Wilson, J. G., Winegar, D. A., and Kliewer, S. A. (2002). Regulation of cholesterol homeostasis by the liver X receptors in the central nervous system. Mol. Endocrinol. 16, 1378–1385.
WHO. (2007). Every 72 Seconds Someone in America Develops Alzheimer’s. Chicago: Alzheimer’s Association, 1–28.
Wijesuriya, H. C., Bullock, J. Y., Faull, R. L., Hladky, S. B., and Barrand, M. A. (2010). ABC efflux transporters in brain vasculature of Alzheimer’s subjects. Brain Res. 1358, 228–238.
Wollmer, M. A., Kapaki, E., Hersberger, M., Muntwyler, J., Brunner, F., Tsolaki, M., Akatsu, H., Kosaka, K., Michikawa, M., Molyva, D., Paraskevas, G. P., Lutjohann, D., Von Eckardstein, A., Hock, C., Nitsch, R. M., and Papassotiropoulos, A. (2006). Ethnicity-dependent genetic association of ABCA2 with sporadic Alzheimer’s disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141B, 534–536.
Wollmer, M. A., Sleegers, K., Ingelsson, M., Zekanowski, C., Brouwers, N., Maruszak, A., Brunner, F., Huynh, K. D., Kilander, L., Brundin, R. M., Hedlund, M., Giedraitis, V., Glaser, A., Engelborghs, S., De Deyn, P. P., Kapaki, E., Tsolaki, M., Daniilidou, M., Molyva, D., Paraskevas, G. P., Thal, D. R., Barcikowska, M., Kuznicki, J., Lannfelt, L., Van Broeckhoven, C., Nitsch, R. M., Hock, C., and Papassotiropoulos, A. (2007). Association study of cholesterol-related genes in Alzheimer’s disease. Neurogenetics 8, 179–188.
Wollmer, M. A., Streffer, J. R., Lutjohann, D., Tsolaki, M., Iakovidou, V., Hegi, T., Pasch, T., Jung, H. H., Bergmann, K., Nitsch, R. M., Hock, C., and Papassotiropoulos, A. (2003). ABCA1 modulates CSF cholesterol levels and influences the age at onset of Alzheimer’s disease. Neurobiol. Aging 24, 421–426.
Wolozin, B. L., Pruchnicki, A., Dickson, D. W., and Davies, P. (1986). A neuronal antigen in the brains of Alzheimer patients. Science 232, 648–650.
Wood, J. G., Mirra, S. S., Pollock, N. J., and Binder, L. I. (1986). Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc. Natl. Acad. Sci. U.S.A. 83, 4040–4043.
Xiong, H., Callaghan, D., Jones, A., Bai, J., Rasquinha, I., Smith, C., Pei, K., Walker, D., Lue, L. F., Stanimirovic, D., and Zhang, W. (2009). ABCG2 is upregulated in Alzheimer’s brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for Abeta(1-40) peptides. J. Neurosci. 29, 5463–5475.
Xu, J., Liu, Y., Yang, Y., Bates, S., and Zhang, J. T. (2004). Characterization of oligomeric human half-ABC transporter ATP-binding cassette G2. J. Biol. Chem. 279, 19781–19789.
Yasuda, K., Ranade, A., Venkataramanan, R., Strom, S., Chupka, J., Ekins, S., Schuetz, E., and Bachmann, K. (2008). A comprehensive in vitro and in silico analysis of antibiotics that activate pregnane X receptor and induce CYP3A4 in liver and intestine. Drug Metab. Dispos. 36, 1689–1697.
Zelcer, N., Khanlou, N., Clare, R., Jiang, Q., Reed-Geaghan, E. G., Landreth, G. E., Vinters, H. V., and Tontonoz, P. (2007). Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 10601–10606.
Zhang, W., Mojsilovic-Petrovic, J., Andrade, M. F., Zhang, H., Ball, M., and Stanimirovic, D. B. (2003). The expression and functional characterization of ABCG2 in brain endothelial cells and vessels. FASEB J. 17, 2085–2087.
Zhang, Y., Schuetz, J. D., Elmquist, W. F., and Miller, D. W. (2004). Plasma membrane localization of multidrug resistance-associated protein homologs in brain capillary endothelial cells. J. Pharmacol. Exp. Ther. 311, 449–455.
Zhou, C., Zhao, L., Inagaki, N., Guan, J., Nakajo, S., Hirabayashi, T., Kikuyama, S., and Shioda, S. (2001). Atp-binding cassette transporter ABC2/ABCA2 in the rat brain: a novel mammalian lysosome-associated membrane protein and a specific marker for oligodendrocytes but not for myelin sheaths. J. Neurosci. 21, 849–857.
Keywords: ABC transporter, Alzheimer’s disease, blood-brain barrier, ABCA1, ABCA2, P-glycoprotein, MRP1, BCRP
Citation: Wolf A, Bauer B and Hartz AMS (2012) ABC transporters and the Alzheimer’s disease enigma. Front. Psychiatry 3:54. doi: 10.3389/fpsyt.2012.00054
Received: 01 February 2012; Paper pending published: 19 February 2012;
Accepted: 15 May 2012; Published online: 04 June 2012.
Edited by:
Silke Vogelgesang, University of Greifswald, GermanyReviewed by:
Silke Vogelgesang, University of Greifswald, GermanyGabriele Jedlitschky, Universitätsmedizin Greifswald, Germany
Copyright: © 2012 Wolf, Bauer and Hartz. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Anika M. S. Hartz, Department of Pharmaceutical Sciences, College of Pharmacy, University of Minnesota, 1110 Kirby Dr, 232 Life Science, Duluth, MN 55812, USA. e-mail:YW1oYXJ0ekBkLnVtbi5lZHU=