 Julia M. Gohlke1
Julia M. Gohlke1 Emily J. Dhurandhar2 Christoph U. Correll3 Elaine H. Morrato4 John W. Newcomer5
Emily J. Dhurandhar2 Christoph U. Correll3 Elaine H. Morrato4 John W. Newcomer5 Gary Remington6 Henry A. Nasrallah7
Gary Remington6 Henry A. Nasrallah7 Stephen Crystal8 Ginger Nicol9 Adipogenic and Metabolic Effects of APDs Conference Speakers†
Stephen Crystal8 Ginger Nicol9 Adipogenic and Metabolic Effects of APDs Conference Speakers† David B. Allison2*
David B. Allison2*- 1 Department of Environmental Health Sciences, School of Public Health, University of Alabama at Birmingham, Birmingham, AL, USA
- 2 Department of Biostatisticxss, School of Public Health, University of Alabama at Birmingham, Birmingham, AL, USA
- 3 Department of Psychiatry, The Zucker Hillside Hospital, Glen Oaks, NY, USA
- 4 Department of Health Systems, Management and Policy, Colorado School of Public Health, University of Colorado Anschutz Medical Campus, Aurora, CO, USA
- 5 Department of Psychiatry and Behavioral Sciences, Miller School of Medicine, University of Miami, Miami, FL, USA
- 6 University of Toronto and Centre for Addiction and Mental Health, Toronto, ON, Canada
- 7 University of Cincinnati College of Medicine, Cincinnati, OH, USA
- 8 Institute for Health, Health Care Policy and Aging Research, Rutgers University, New Brunswick, NJ, USA
- 9 Department of Psychiatry, Washington University, St. Louis, MO, USA
Although offering many benefits for several psychiatric disorders, antipsychotic drugs (APDs) as a class have a major liability in their tendency to promote adiposity, obesity, and metabolic dysregulation in an already metabolically vulnerable population. The past decade has witnessed substantial research aimed at investigating the mechanisms of these adverse effects and mitigating them. On July 11 and 12, 2011, with support from 2 NIH institutes, leading experts convened to discuss current research findings and to consider future research strategies. Five areas where significant advances are being made emerged from the conference: (1) methodological issues in the study of APD effects; (2) unique characteristics and needs of pediatric patients; (3) genetic components underlying susceptibility to APD-induced metabolic effects; (4) APD effects on weight gain and adiposity in relation to their acute effects on glucose regulation and diabetes risk; and (5) the utility of behavioral, dietary, and pharmacological interventions in mitigating APD-induced metabolic side effects. This paper summarizes the major conclusions and important supporting data from the meeting.
Introduction
It is clear that weight gain is a ubiquitous side effect of antipsychotic drugs (APDs; Allison et al., 1999) and that APDs are also associated with an increased risk of complications related to obesity, including insulin resistance, hyperlipidemia, type 2 diabetes mellitus, and cardiovascular disease (Lieberman et al., 2005; Newcomer, 2005; Daumit et al., 2008; De Hert et al., 2011a). However, the specific expression of APD-induced metabolic side effects varies widely across APDs and across individuals. Although patients with mental disorders in general have increased morbidity and mortality, it is unclear how APDs specifically affect both psychiatric illness and also metabolic function in order to yield the observed clinical outcomes.
It is widely accepted that most of the APDs induce significant weight gain, with median weight changes of +0.2, −0.1, +0.7, and +0.3 kg/month are observed for haloperidol, ziprasidone (ZIP), olanzapine (OLZ), and risperidone (RISP), respectively, following 1 year of treatment (Parsons et al., 2009). Studies conducted within a first-episode schizophrenia sample, such as in the Comparison of Atypicals for First-Episode Psychosis (CAFÉ) study (McEvoy et al., 2007) or the European First-Episode Schizophrenia Trial (EUFEST) study (Kahn et al., 2008), and in antipsychotic-naïve youth (Correll et al., 2009) have shown that all antipsychotics are associated with varying degrees of significant weight gain (Table 1). Moreover, when switching from an antipsychotic with a higher weight gain risk to one with a lower risk, patients tend to lose weight (Newcomer et al., 2008). Results of the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study suggest that female patients may be at a higher risk for weight gain; in fact 36.6% of male and 54.2% of female patients (and an overall rate of 42.7%) met the metabolic syndrome criteria (McEvoy et al., 2005). However, findings on predictors of APD related weight gain have been mixed and seem to be influenced by prior treatment effects (Correll et al., 2011).
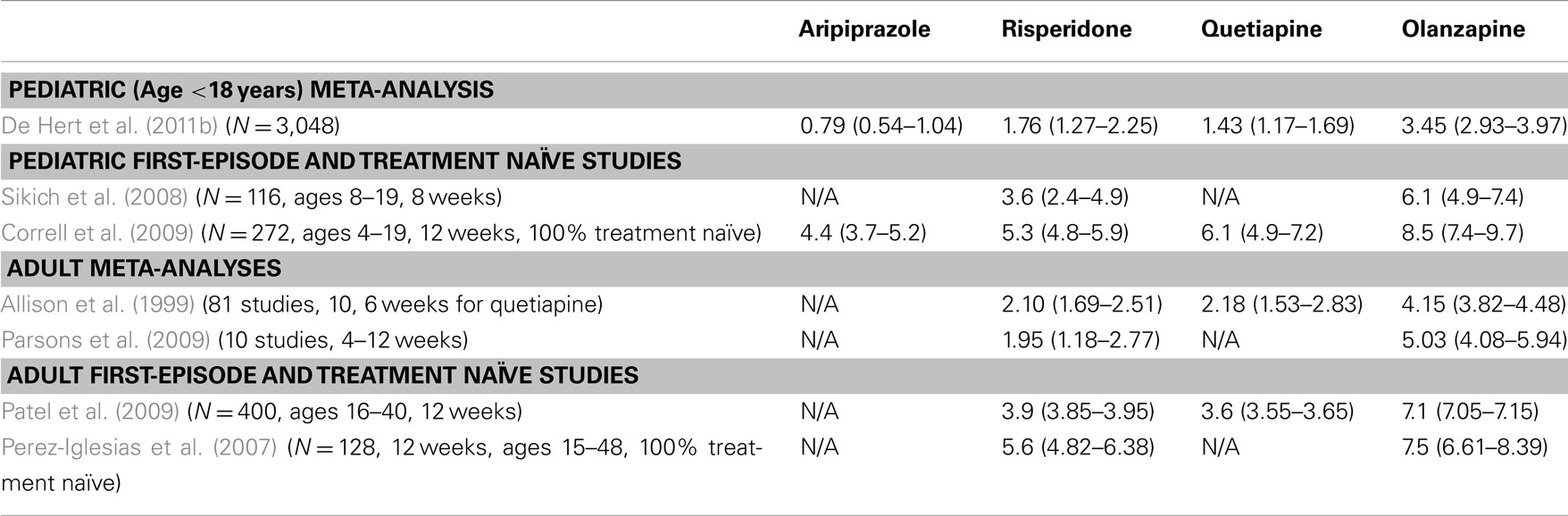
Table 1. Mean (95% CI) antipsychotic-induced weight gain in pediatric and adult patients (kg).
The outcomes of a 2005 meeting underscored the significant contribution of APDs to obesity-related conditions in psychiatric patients and the need for further research on weight-management programs tailored to this patient population (Allison et al., 2009b). Since then, significant research efforts have focused on APD-induced weight gain and related metabolic consequences. In July of 2011, a follow-up conference to examine the current state of the field was funded by the National Institute of Mental Health, with additional funds from the National Institute of Diabetes and Digestive and Kidney Diseases. The conference consisted of expert presentations addressing topics ranging from clinical findings to molecular mechanisms of APD-induced weight gain. The discussion below reflects a distillation of the conference proceedings, highlighting progress, key pharmacoepidemiological observations, and future needs in five key research areas. These areas include: (1) methodological issues in the study of APD effects; (2) unique characteristics and needs of pediatric patients; (3) genetic and life-style risk factors underlying susceptibility to APD-induced metabolic effects; (4) APD effects on weight gain and adiposity in relation to their acute effects on glucose regulation and diabetes risk; and (5) the utility of behavioral, dietary, and pharmacological interventions in mitigating APD-induced metabolic side effects.
Methodological Issues in the Study of APD Effects
Two major categories of methodological issues were identified during the conference: first, statistical challenges, and second, the detection and proper reporting of adverse effects of APDs. In the category of statistical issues, two in particular were highlighted: 1. Dealing with missing data; and, 2. analysis of heterogeneity in response.
Missing data often arise when patients drop out of a study, discontinue antipsychotic treatment, or switch to a different antipsychotic drug, which produces a sample that is no longer a population in which treatment assignment is guaranteed to be independent of all pre-assignment covariates. This is a particular challenge for studies of antipsychotic drug metabolic effects, because treatment discontinuation and switches are common, often take place relatively soon after treatment initiation, and do not occur randomly, being likely to be associated with clinical characteristics that may in turn be associated with treatment outcomes (informative censoring) in ways that may not be fully adjusted for by available covariates. Most commonly, the last observation carried forward (LOCF) technique is implemented; however, this method is not likely to capture what would have occurred if the patients who dropped out had remained in the study. Alternatively, several mixed models and multiple imputation can be used to test treatment effects (Elobeid et al., 2009), although caution is warranted when using mixed models approaches with small sample sizes and a large number of measurements, because Type 1 error rates can become inflated (Ahn et al., 2000). Of course, it must be kept in mind that the use of mixed models or multiple imputation to conduct intention to treat (ITT) analyses, allows one to obtain unbiased estimates of treatment assignment only under certain assumptions, the critical one being that the data are missing at random (MAR), meaning that the missing data are MAR conditional on other variables in the model. This is a less restrictive assumption than missing completely at random (MCAR) which is required for analysis of completers only to be valid, but not an easily testable assumption (Little, 1988). Moreover, these methods test the effect of treatment assignment, not treatment per se, and there is no easy way nor universally accepted procedure that solves this problem at the analytic level. Only designs which ensure perfect conformity of treatment to treatment assignment and eliminate drop-outs entirely can unequivocally solve such problems (Little and Rubin, 2002). Empirical data can be used to inform expected dropout rates for weight loss trial design (Elobeid et al., 2009), to ensure a large enough sample size for adequate power. Additionally, it should be noted that the type of analysis used affects power, and that mixed models and multiple imputation have comparable power, and are preferred to LOCF methods (Elobeid et al., 2009).
A second statistical and methodological challenge is the analysis of heterogeneity in response to a given treatment, such as inter-individual variation in APD-induced weight gain, and in analyzing the contribution of genetic factors to this heterogeneity. It is clear that genetic contributions to inter-individual variability in treatment response can be profound (Bronikowski et al., 2006). However, detecting treatment response heterogeneity in randomized controlled trials requires a specific study design, because variation in outcome within the treatment group does not necessarily indicate treatment response heterogeneity. Conceptually, true treatment response for an individual is the difference between the change in the outcome variable after a pre-determined treatment period and the change in the outcome variable during the same time period had treatment not occurred (Gadbury et al., 2001). This difference can be discussed theoretically, but cannot be observed in reality. Commonly, studies seek to account for heterogeneity in weight gain response to APD treatment by correlating with baseline BMI or other baseline covariates. Such analyses may suffer from flaws, however, because any correlation may be a result of regression to the mean, such that obese persons gain relatively less weight and thin persons gain relatively more weight after an obesogenic treatment (Allison et al., 2009a).
Several alternative methods can be used to identify treatment response heterogeneity and covariates that may predict response. Instead of a parallel group design, which can detect only a treatment effect, a repeated-period crossover design in which each patient receives both treatments at least twice is useful for detection of treatment-patient interactions and treatment response heterogeneity (Senn, 2001). In addition, a simple method to calculate the upper and lower bounds of treatment response heterogeneity in a parallel groups has been proposed by Gadbury et al. (2001). Lastly, Whole Genome Prediction can be used to predict which individuals may respond better or worse to a particular treatment (de los Campos et al., 2010), and although the method does not necessarily explain why or which genes may be involved, it is a strong prediction tool that uses all genetic markers available instead of just a few.
Finally, there are methodological challenges in detecting adverse effects of APDs. Postmarketing pharmacovigilance for adverse effects of drugs has improved with the FDA Amendment Act of 2007, which allowed for the creation of an active surveillance system using electronic data from health care providers via the Sentinel Initiative (FDA, 2011). Nonetheless, observational pharmacoepidemiology studies examining APD-induced weight gain suffer from several confounders (Ramaswamy et al., 2006). For example, patients taking antipsychotics often have higher background rates of metabolic disorders, longitudinal follow-up to detect metabolic abnormalities from chronic APD use is limited, the validity of using diagnosis codes may be questionable (particularly for ascertaining obesity), there may be differential disease surveillance rates, and use of over-the-counter weight loss products may not be tracked. Data on family history, BMI status, dietary intake, and level of physical activity (known risk factors for metabolic abnormalities) are lacking in electronic claims data and represent sources of unmeasured confounding. Electronic medical records offer promise for studies of outcomes such as BMI, glucose, and lipid profiles, but careful exploration of the completeness of such data sources and the potential for confounding through informative absence of data (e.g., a higher likelihood of values being recorded when overweight or other metabolic problems exist) is a necessary initial step in such studies. Linking specific antipsychotic exposure to weight gain is also challenging because of frequent medication switching to optimize clinical response, as well as drug selection bias, e.g., drugs with a greater propensity for weight gain may be used less often in patients who are already overweight. Other psychotropic medications prescribed to patients with psychosis, including antidepressants and mood stabilizers, can also contribute to weight gain. These methodological challenges lead to delays in drug causality determinations and result in delayed changes in clinical practice and policy decisions. However, despite these challenges, sufficient clinical and pharmacoepidemiologic evidence has been generated over time to result in changes in antipsychotic risk mitigation, including FDA labeling and ADA guidelines that recommends baseline assessment and monitoring of weight, hyperglycemia, and hyperlipidemia. A review of Medicaid datasets for 3-states suggests metabolic monitoring is not routinely practiced and did not increase substantially in the year following publication of ADA guidelines (Figure 1). Probably, the most notable risk mitigation response has been a decline in OLZ prescribing practices (Morrato et al., 2010a). Some states, such as New York, have implemented quality improvement initiatives aimed at reducing the use of antipsychotic medications associated with higher risk of cardiometabolic disorders, such as OLZ, among patients who have risk factors such as diabetes or pre-diabetes, hypertension, hyperlipidemia, or cardiovascular disease (NYDH, 2010).
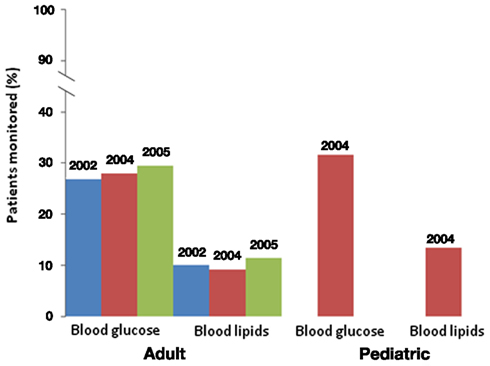
Figure 1. Blood glucose and lipid monitoring in adult and pediatric patients. In adult patients, monitoring did not significantly change after the Dec. 2003 FDA warning. Monitoring is also low in pediatric patients after the warning. Data from Morrato et al. (2010a,b).
Pediatric Patients and APDs
APD use in children (as young as 2 years of age) has increased substantially over the past two decades (Olfson et al., 2006); in particular, the off-label use of atypical antipsychotics in youth has increased in the past decade (Zito et al., 2007; Crystal et al., 2009). This increasing prescription of antipsychotic agents in children occurs against the concerning backdrop of childhood obesity in the general population; childhood onset of obesity is known to be associated with increased obesity-related mortality risk in early adulthood (Morrison et al., 2007). Most recent estimates indicate that approximately 17% of the US population of youth ages 2–19 are considered obese, with an additional 15% of the pediatric population at risk for developing obesity (Ogden et al., 2010).
APDs are commonly used to treat bipolar disorder and early onset schizophrenia in pediatric patients. In addition, APDs are increasingly used to treat non-psychotic disorders, such as autism, spectrum disorders, oppositional personality disorder, Tourette Syndrome, attention deficit hyperactivity disorder (ADHD), and pervasive developmental disorder. APDs are also increasingly used to treat oppositional, irritable, and aggressive behaviors across diagnoses (Connor et al., 2006; Cooper et al., 2006), with limited evidence to support such off-label use. Children who are publicly insured (e.g., Medicaid) are more commonly prescribed psychotropic medications than privately insured children (Olfson et al., 2002), and children in foster care who are publicly insured represent the population of publicly insured children with the greatest likelihood of being prescribed psychotropic medications (Zito et al., 2008; Dosreis et al., 2011). Four percent of children aged 6–17 years of age enrolled in Medicaid were using antipsychotics as of 2005, which is a 40–50% increase since 2001, and most of these prescriptions (approximately 50%) were for the treatment of ADHD or disruptive behavior disorders (Crystal et al., 2009). The majority of children taking antipsychotics in this Medicaid cohort were concomitantly taking other psychotropic drugs, whereas a minority were treated with psychotherapy (Crystal et al., 2009). Similar trends are seen in privately insured youth, although the annual percentage of enrollees using APDs in this group was only 0.93% as of 2007 (Olfson et al., 2010). Several issues for concern arise in this context, such as the low monitoring of metabolic risk in both public- and privately insured populations (Haupt et al., 2009; Morrato et al., 2010b), and additional metabolic risk conferred by polypharmacy (McIntyre and Jerrell, 2008), contributed to by poor access to high quality medical and mental health care related to socio-economic disparity. This concurrent rise in APD use in children along with the paucity of information on the safety of such medications in children has led to a concerted effort among researchers, clinicians, and policymakers to better understand this issue.
Data from commercially insured (Haupt et al., 2009) and Medicaid beneficiaries (Morrato et al., 2010b) indicate that most persons using APDs do not receive glucose and dyslipidemia monitoring (Figure 1), and that monitoring rates are lowest in the youngest age groups. In a restrospective new-APD user cohort study of 3-state Medicaid claims data from 2004 to 2006, rates of fasting glucose and lipid testing were higher in non-diabetic youth treated with APDs than in a control population of non-diabetic youth (Morrato et al., 2010b). However, rates of incident diabetes and hyperlipidemia during the study period, defined as either a new diagnosis code or new prescription for anti-diabetes or hyperlipidemia treatment, were on average higher in the APD-treated group, with lower rates of anti-diabetes or hyperlipidemia treatment noted in the APD-treated group.
In large observational databases like the multistate Medicaid analysis, youth taking antipsychotics compared to albuterol-treated controls have higher rates of treatment-incident diagnosis of diabetes and hyperlipidemia, without higher anti-diabetes or hyperlipidemia treatment in the APD-treated population. This is associated with the adverse medical outcomes that could be predicted; unfortunately at this stage and indication that it is not properly treated. However, these observational studies are to a large extent hypothesis-generating; clinical trials are needed to test hypotheses and evaluate treatment-related metabolic changes over time.
Similar or greater weight gain is seen across APDs in children as in adults, although relative weight gain tends to be higher in children (Shin et al., 2008), with some indication that perhaps initial antipsychotic exposure accounts for the higher weight gain observed in younger patients (Table 1). The non-randomized Second-Generation Antipsychotic Treatment Indications, Effectiveness, and Tolerability in Youth (SATIETY) cohort study was one of the first studies to specifically include antipsychotic-naïve patients (Correll et al., 2009). The SATIETY study enrolled antipsychotic-naïve patients aged 4–19 years (N = 272), with non-randomized assignment to either aripiprazole (ARI), quetiapine (QUET), risperidone (RISP), or olanzapine (OLZ). Although there was potential for channeling bias at baseline in that many children who were overweight or obese at baseline were assigned to treatment with aripiprazole, weight gain was apparent in all treatment groups after 12 weeks, including in the ARI treatment group, which in previously exposed youth showed only minimal weight gain (De Hert et al., 2011b). Olanzapine treatment was associated with the highest weight gain (avg. 8.5 kg), with QUET, RISP, and ARI producing somewhat less, but still substantial weight gain (avg. 4.4 kg; Table 1). Cholesterol and triglyceride levels also increased significantly in the OLZ and QUET groups, triglycerides increased significantly with RISP, but ARI did not cause significant lipid increases despite significant weight gain. By contrast, glucose, insulin, and insulin resistance increased significantly only with OLZ, at least during the first 12 weeks of antipsychotic exposure. The researchers suggested that the increased weight gain seen in pediatric populations can be fully explained by a lack of prior exposure to antipsychotics, because the degree of metabolic effects was consistent with those seen in adult treatment-naïve patients (Correll et al., 2011).
The recently completed NIH-funded MEAC (Metabolic Effects of Antipsychotics in Children) study (MH72912, PI: Newcomer) was the first to specifically evaluate gold-standard metabolic outcomes in children and adolescents during an initial 12 weeks of exposure to antipsychotics. Antipsychotic-naive participants aged 6–18 with clinically significant aggression and irritability (score of >18 on Aberrant Child Behavior Checklist Irritability Subscale) in the setting of one or more DSM-IV diagnosis indicating a disruptive behavior disorder were enrolled. Participants (N = 144) were randomized to specific antipsychotic treatments (RISP, OLZ, ARI) following baseline assessments. Baseline and 12 week measures include body composition analysis with Dual Energy X-ray Absorptiometry (DEXA) and abdominal MRI, as well as metabolic testing including hyperinsulinemic euglycemic glucose clamps with stable isotopomer tracing. Primary endpoints were change in whole-body and abdominal adiposity, and whole-body and tissue-specific insulin sensitivity. In the pooled group, combining all treatments, irritability scores were significantly improved. However, during the 12 week trial, a mean DEXA-measured 2.4 (±3.1)% gain in total percent body fat was noted, corresponding to a 10 (±7) lb weight gain, and an associated mean decrease in clamp-measured whole-body insulin sensitivity were observed. Concerningly, greater weight gain was observed in the youngest age group. Half of the participants had a primary diagnosis of ADHD and were concurrently treated with a psychostimulant during study participation. There was no clear relationship between stimulant treatment and weight/adiposity change compared to non-stimulant treated participants, reflecting results of a recent study also suggested that stimulant medication may not mitigate the metabolic effects of APDs in children (Penzner et al., 2009). These results also support what was seen in large observational database studies; the weight gain observed during antipsychotic treatment is largely attributable to AP drug effect. Results from long-term follow-up studies such as the Treatment of Early Onset Schizophrenia Spectrum (TEOSS) study suggest that similar rates of weight gain continue during extended AP treatment, with a leveling off at 1 year after initial exposure (Findling et al., 2010), and that molindone and RISP may exhibit more delayed-onset weight gain than does OLZ. Overall, these studies highlight the need for more frequent cardiometabolic monitoring in pediatric patients after the initial 3 months of treatment and for additional research addressing the long-term cardiometabolic effects of pediatric exposures.
Genetic Components Underlying Susceptibility to APD-Induced Metabolic Effects
Given that the risk of metabolic syndrome risk associated with APD use differs across ethnic groups, it can be inferred that genetic factors may be playing a mediating role. In the general population, rates of metabolic syndrome are lower in black and Hispanic patients than in white patients, whereas rates of diabetes and CVD are higher in black and Hispanic patients when compared to white patients (Park et al., 2003). Ethnic differences in lipid profiles have also been noted, and may lead to under-diagnosis of metabolic syndrome in blacks (Gaillard et al., 2009; Sumner, 2009). In addition, the linear relationship between waist circumference and insulin resistance seen in white patients does not hold true for black or Hispanic patients (Nelson et al., 2008; Sumner et al., 2008). The rates of death from CVD in Hispanic and black patients receiving clozapine (CLOZ) are 4.3 and 11.5 times the rates in whites, respectively (Henderson et al., 2005).
Several genes have been identified that may mediate the effect of APD-induced weight gain. A genome-wide association study (GWAS) was conducted with the SATIETY cohort to determine whether differential metabolic responses to antipsychotics are associated with genetic markers. The analysis was conducted on those exposed to QUET, RISP, and ARI and approximately 20 single nucleotide polymorphisms (SNPs) on Chr 18, near the melanocortin four receptor gene (MC4R), were associated with weight gain after antipsychotic exposure (Malhotra et al., 2012). These results are consistent with other studies implicating rare MC4R mutations in early onset obesity (Loos et al., 2008) and more common variants downstream of MC4R that are associated with obesity (Loos, 2011), and suggest that variants in this region may confer susceptibility to weight gain while taking APDs. As there was no randomized control group in this study to allow for testing of an APD by genotype interaction, it is unclear if these SNPs moderated the effects of APDs on weight gain or were merely associated with weight gain among people taking APDs. In addition, SNPs in the serotonin 2c receptor (5HT2C) promoter region (Reynolds et al., 2002) and Prohormone Convertase 1 (PCSK1) gene were associated with APD related weight gain in the SATIETY cohort and other obesity studies (Kilpelainen et al., 2009). PCSK1 metabolizes an MC4R ligand, suggesting that the genetic variants found to date may converge on the proopiomelanocortin (POMC) pathway (Lett et al., 2012). Ultimately, a clearer understanding of the role of specific genes in treatment effect heterogeneity holds much promise for the development of genetically informed, personalized treatment approaches, but further research is needed before such approaches are practical on a routine basis (Lett et al., 2012).
Several genetic factors may also mediate risk of developing CVD when taking APDs. Associations have been detected between metabolic syndrome risk (Ellingrod et al., 2008; van Winkel et al., 2010) and variants in the methylenetetrahydrofolate reductase (MTHFR) and catechol-o-methyl transferase (COMT) genes was detected in schizophrenia patients, which suggests that perturbations in the Aldo Met cycle may be related to schizophrenia and APD-induced metabolic effects. The Aldo Met cycle regulates homocysteine levels, a marker associated with CVD risk; therefore these gene variants may particularly affect CVD risk.
Other evidence suggests genes may interact with certain nutrients to modify CVD risk when taking APDs. For example, n−3 fatty acid administration may offer a protective effect and interact with gene variants (Amminger et al., 2010). When endothelial functioning in schizophrenia patients was evaluated in relation to n−3 fatty acid intake, endothelial function was lower in patients than in healthy subjects, and the relationship between n−3 fatty acid intake and endothelial function was modified by antipsychotic exposure (Ellingrod et al., 2011). In addition, the COMT Val variant showed a significant association with physical activity in these patients (Lott et al., 2012). Finally, preliminary evidence suggests that folate supplementation, which may restore balance of the Aldo Met cycle, improves endothelial function in schizophrenia patients with metabolic syndrome (Ellingrod et al., 2012).
APD Effects on Weight Gain and Adiposity in Relation to Their Acute Effects on Glucose Regulation and Diabetes Risk
Not surprisingly, increases in treatment-related adiposity predict insulin resistance in adults and children (Haupt et al., 2007; Nicol and Newcomer, 2008; Newcomer et al., 2009). Consistent with relative weight gain across the APDs, results from a recent post-marketing survey across three U.S. insurer databases suggest significant increased risk of diabetes for users of OLZ and CLOZ but not ARI, ZIP, RISP, or QUET, when compared to users of haloperidol (Yood et al., 2009). An important aspect of this study was the inclusion of other non-psychiatric medications as covariates (alpha and beta blockers, corticosteroids, oral contraceptives, statins, etc.). Recent mechanistic studies described below suggest that APD-associated insulin resistance can occur without weight gain.
While the specific mechanism or mechanisms of APD-induced weight gain are not fully understood, studies have shown that affinity for the type 1 Histamine receptor explains the majority of the variance in weight gain between APDs (Kroeze et al., 2003), with the proposed orexigenic mechanism being related to selective activation of hypothalamic AMP-kinase (Kim et al., 2007). However, the promiscuity of the various APD drugs for interacting with multiple receptor systems, many of which have direct ties to regulating energy balance, further complicates understanding the mechanism(s) by which APDs induce weight gain. APDs are almost exclusively dopamine receptor 2 (DRD2) antagonists, with the exception of partial agonists like ARI. All the second-generation APDs also antagonize serotonin 2A and 2C receptors, with variable antagonist activity at histamine, muscarinic and α-adrenergic receptors and some agents are agonists or partial agonists at the serotonin receptor 1A (e.g., ARI). Because of the differential weight gain associated with different APDs, previous studies have suggested that APD binding to the serotonin 2c receptor (5HT2c), histamine receptor (H1), and the dopamine D2 receptor (DRD2) plays an important role in APD-induced weight gain (Nasrallah, 2008; Reynolds and Kirk, 2010).
Olanzapine and clozapine, which exhibit a particularly high affinity for histamine and muscarinic receptors, are associated with the most weight gain in patients and also cause increases in fasting glucose, insulin, and triglycerides. The role of the neural control of food intake and glucose homeostasis in this effect is currently being explored. The role of the vagal nerve in insulin release during and just after meals suggests that meal ingestion may uncover effects that cannot be assessed in an intravenous glucose intervention study. For example, a 12-day inpatient study of healthy individuals given OLZ and ARI showed no increase in body weight or food intake, but increased hunger (Teff, unpublished data). Furthermore, no change in glucose production, small decreases in glucose disposal, and a doubling of postprandial insulin response were noted with OLZ, but not ARI. These results suggest that a compensatory postprandial response of OLZ may explain the insulin resistance seen in longer term studies. This study raises the question of whether a sublingual route that avoids first-pass hepatic metabolism may alleviate this effect, although the largest randomized trial to date does not seem to support this assertion (Karagianis et al., 2009). However, as noted above, the weight gain potential of individual agents and not adiposity-independent effects, is the primary factor associated with variation in insulin sensitivity during treatment. It can be noted that there is some evidence in animals for adiposity-independent effects on insulin sensitivity (Houseknecht et al., 2007), but adiposity-independent effects in humans have not been demonstrated. Moreover, these results also raise the question of whether decreases in insulin sensitivity precede or follow weight gain.
Results from animal models suggest OLZ may alter the adipose tissue profile in addition to affecting insulin secretion. A canine model showed no significant changes in body weight or caloric intake, but a doubling of body fat, after 6 weeks of treatment with OLZ but not RISP (Ader et al., 2005). This model suggests OLZ promotes fat deposition associated with PPARγ expression, hepatic insulin resistance, and impairment of the pancreatic beta cell compensatory response. However, no impairment of glucose tolerance was observed, and insulin levels were increased to a greater extent than C-peptide, which is co-secreted with insulin. These data suggest that OLZ treatment may alter both insulin secretion and metabolic clearance of the hormone by the liver. In a canine model of preexisting obesity via high-fat diet, OLZ was associated with additional weight gain and adiposity compared with ARI and placebo. The volume of visceral fat cells was dramatically increased in the OLZ-treated animals. Other studies have suggested increased adipocyte inflammation in OLZ-treated animals (Victoriano et al., 2010) and activation of the TGFβ pathway (Cohen et al., 2012). Importantly, while OLZ treatment impaired insulin secretory function in vivo, perfusion of these pancreatic islets in vitro demonstrated no significant defect of insulin release. These data indicate that OLZ disturb pancreatic function not by direct damage to pancreatic β-cells, but rather via interference with signals to the pancreas, possibly secondary to an effect on the central nervous system.
While development of a rodent model that reflects the magnitude of weight gain seen in humans has been difficult due to rapid metabolism in rodents (Kapur et al., 2000; Remington, 2010), research looking at acute effects have shown that hepatic glucose production increases with CLOZ and OLZ, and insulin and C-peptide responses to glucose load are reduced (Chintoh et al., 2009). Subsequent studies using specific receptor blockers suggest that D2/D3, M3, and 5HT2A antagonism may play a role in the APD-induced acute glucose dysregulation (Hahn et al., 2011).
Although APDs increase body fat, evidence suggests they may result in a paradoxical reduction of free fatty acid levels and upregulation of fatty acid utilization. Metabolomic and metabolic phenotyping after OLZ administration in rodents revealed increased adiposity without a concomitant change in body weight, decreased physical activity, an acute decrease in free fatty acids, decreased lipolysis, and increased whole-body lipid oxidation (Albaugh et al., 2011a, 2012). These studies are consistent with three human studies that also showed acute decreases in free fatty acids with APD treatment (Kaddurah-Daouk et al., 2007; Vidarsdottir et al., 2010; Albaugh et al., 2011b). In addition, OLZ administration is associated with a shift in relative fuel consumption to fat oxidation, as measured by a rapid decline in the respiratory exchange ratio (RER) during the dark cycle without the normal rise in RER following meal ingestion (Albaugh et al., 2012). A decline in RER would indicate greater consumption of fat as fuel in comparison to carbohydrate or protein. It is unclear how these metabolic effects may or may not contribute to adiposity and insulin resistance associated with APD use, but suggests that high free fatty acids and lipotoxicity are unlikely to play a role.
The hypothesis that APDs may affect appetite regulation has also been investigated. The Effect of Antipsychotics on Appetite Regulation (ADAPT) study is an ongoing trial evaluating APD-induced changes in appetite-regulating hormones in a treatment-naïve population over 4 months (Brownley, 2011). Preliminary data indicate no changes in pre- or post-prandial insulin or glucose after an overnight fast resulting from APD use and APD reduced caloric intake on average, despite some, though not all, individuals gaining a significant amount of weight (Brownley, unpublished data). An increase in hunger was reported in those who gained weight, but a decrease was reported in those who lost weight. ADP-induced weight gain was associated with decreased ghrelin levels and increased peptide tyrosine-tyrosine (PYY), and resting energy expenditure was decreased. The increases in PYY suggest compensation in peripheral release in response to down-regulated central PYY receptors and implicate perturbation of the reward system in the antipsychotic-induced metabolic changes. Such implications are supported by the results of Wang and Huang who demonstrated decreases in central PYY binding densities in rat brain after chronic administration of OLZ, but not haloperidol (Wang and Huang, 2008).
Similar to the differences seen in human populations, OLZ and CLOZ cause increased lipid accumulation in the nematode C. elegans, whereas haloperidol and fluphenazine did not (Dwyer et al., 2005). Consistent with the hypothesis of APD-induced appetite regulation disturbances, foraging behavior in C. elegans is increased following CLOZ and OLZ exposure and is associated with insulin receptor-controlled activation of Akt and subsequent FOXO (DAF-16) nuclear localization (Weeks et al., 2011).
Behavioral, Dietary, and Pharmacological Interventions to Mitigate APD-Induced Metabolic Side Effects
The differences in incidence of obesity and metabolic complications in patients taking APDs compared to the general population may be in part due to lack of preventative care for metabolic diseases in the psychiatric patient population (Newcomer and Hennekens, 2007). Particularly low rates of treatment for hypertension, dyslipidemia, and diabetes were evident in the patients enrolled in the CATIE trial when compared to the general population (Nasrallah et al., 2006). Furthermore, the Food and Drug Administration (FDA) warnings for APDs have not produced substantial changes in practice, such as increased lipid and glucose monitoring, in both publicly and privately insured populations of patients (Haupt et al., 2009; Morrato et al., 2009, 2010a). The gap in monitoring is particularly evident in pediatric patients (Haupt et al., 2009). In one Medicaid study, glucose and lipid testing among APD users varied significantly between states and counties; patients with diagnosed cardiometabolic co-morbidity, serious mental illness, persistent use of APDs, and with greater frequency of non-psychiatric medical office visits were more likely to be screened (Morrato et al., 2011). More effort is needed to understand reasons for screening disparities in order to inform risk management quality improvement interventions. For example, recent studies have shown that rates of metabolic monitoring have increased in some Medicaid populations (Moeller et al., 2011) and significant improvements can be made in Community Mental Health Centers by applying principles of diffusion of innovation theory (Morrato et al., 2010b; Nicol et al., 2011).
The attitudes of psychiatric patients toward weight gain and their desire to lose weight are consistent with those of the general population (Strassnig et al., 2005; Ganguli and Strassnig, 2011). The WAIST study introduced weight loss behavioral therapy in a group setting in a schizophrenia patient population (Brar et al., 2005). The therapy included self-monitoring actions such as daily weighing, using a pedometer, using a food diary, and the additional incentive of a monetary reward (Brar et al., 2005). Therapy also included making eating a specific activity, slowing the rate of eating, and getting over the inhibition of wasting food. Physical activity was increased mainly by using a walking in place video program. As an active control, social skills training was implemented as an alternative behavioral therapy. After 14 weeks, weight loss was significantly greater in the behavioral therapy group than in the social skills training and usual care groups, with one-quarter of the patients losing more than 5% of their body weight at the endpoint (Brar et al., 2005). Currently, research is focused on determining whether behavioral interventions are associated with increased maintenance of weight loss after 2 years in those patients who did lose weight.
Several pharmacologic interventions have been tested in patients taking APDs to counteract the weight gain and metabolic complications associated with these drugs. Metformin, and topiramate have demonstrated efficacy in weight loss trials in patients receiving APD treatment with an average weight loss of approximately 2–3 kg after 8–12 weeks of treatment; fenfluramine, sibutramine, and reboxetine, show similar effects but have been removed from the market due to side effect concerns (Maayan et al., 2010). Metformin not only reduces weight, but also improves fasting insulin and insulin resistance in first-episode patients more efficiently than does a life-style intervention alone, but is even more effective when combined with a life-style intervention (Wu et al., 2008). Slow-release metformin induces weight loss even after long-term CLOZ treatment (Carrizo et al., 2009). Interestingly, metformin may act through improving leptin sensitivity, and certain mutations in leptin may prevent metformin action (Fernandez et al., 2010). Topiramate improves triglycerides, glucose, leptin, cholesterol, and blood pressure in OLZ-treated patients in addition to promoting weight loss (Narula et al., 2010). Notably, however, all drug and behavioral interventions produced modest weight loss. Improved interventions are needed, and the appropriateness of bariatric surgery in this population could be explored.
Some evidence suggests that using pharmacologic agents for weight loss may interfere with the therapeutic effects of antipsychotic treatment (Baptista et al., 2008). In one study, the treatment algorithm producing the most weight loss via metformin administration suggested reduced improvement in depression (Hoffmann et al., 2011). In addition, topiramate is associated with a worsening of cognitive functions, and amantadine may worsen psychosis (Baptista et al., 2008). Thus, patients taking amantadine and topiramate to reduce weight gain should be monitored carefully.
Switching of antipsychotic medications has been investigated for effects on weight gain and metabolic complications. Overall, switching from RISP or OLZ (medium and high weight gain drugs) to ZIP or ARI (lower weight gain drugs) does produce weight loss and modestly improves triglycerides and cholesterol (Mukundan et al., 2010; Stroup et al., 2011). Unfortunately, at least for a subgroup of patients, drugs that cause less weight gain may not be as effective in treating psychosis as are those that cause more weight gain, although the CATIE data shows a very modest correlation between clinical improvement and increase in BMI (Hermes et al., 2011). In the CATIE trial, patients who stayed on the same medication and avoided switching were more likely to stay in treatment, and those taking an effective medication were more likely to regress if they switched (Essock et al., 2006). In another trial, switching from the higher risk OLZ to lower risk RISP or QUET did not result in increased psychiatric risk, and patients who switched gained less weight than did those who stayed on OLZ (Rosenheck et al., 2009). Regarding recommendations for clinicians, a meta-analysis of APD clinical trials suggests that CLOZ, OLZ, and RISP appear to most consistently show superior efficacy (Leucht et al., 2009a,b; Glick et al., 2011); for first-episode schizophrenic patients, however, who are both most sensitive to APD adverse effects and have higher therapeutic response rates, antipsychotics showing less weight gain liability should be prescribed first and only those patients who do not respond should be switched to potentially more effective, higher weight gain treatments (Buchanan et al., 2010; Agid et al., 2011).
Several pretreatment or early treatment characteristics have been associated with APD outcomes and may be useful for predicting APD response. Although the initial analysis of the CATIE trial dataset suggested that patients with metabolic syndrome did not have lower cognitive performance than did patients without metabolic syndrome (Meyer et al., 2005), a recent re-analysis suggests a significant, although small and clinically irrelevant, negative association between BMI and the Positive and Negative Symptom Scale (PANSS) total score, and this association did not differ across antipsychotic medications (Hermes et al., 2011). However, findings regarding associations of weight gain with improvement in symptoms after antipsychotic treatment are varied and controversial (Correll et al., 2011; De Hert et al., 2011a) and Kelly, unpublished data).
Concluding Remarks
In conclusion, the conference presentations highlighted progress in many novel areas of APD-induced metabolic dysfunction research. Recent results from pharmacogenetic studies are suggestive of mechanistic pathways, yet methodological considerations will be important in designing future studies to tease apart APD-gene relationships. More problematic weight gain trajectories are evident in the growing number of pediatric patients being prescribed APDs, although long-term studies are needed to determine if metabolic consequences in pediatric patients lead to greater or earlier disease risk in later life since we have much less information on long-term effects than short-term effects, and long-term effects are very difficult to track with existing treatment designs. This dilemma arguably demonstrates the importance of a conservative, cautious approach in dosing (start low, go slow) and careful metabolic monitoring of patients. Trials of discontinuation after long-term use might also be useful. Novel studies investigating acute effects of APD exposure on metabolic dysfunction in animal and human studies suggest immediate effects on glucose regulation may play a role in APD-induced metabolic dysfunction. Lastly, combined behavioral, dietary, and pharmacological interventions are showing some promise in mitigating APD-induced metabolic side effects, but novel agents are needed that can more completely prevent or reverse ADP related weight gain and metabolic abnormalities.
Future research is needed to determine mechanistically how APDs lead to metabolic disturbances and weight gain in order to facilitate development of effective combination therapies to negate these effects or to develop new drugs that do not induce weight gain. In addition, future work should also determine what factors predict weight gain so that clinical guidelines for prescribing APDs and managing weight gain and its consequences can be improved.
Conflict of Interest Statement
David B. Allison has, anticipates, or has had financial interests with the Frontiers Foundation; Vivus, Inc.; Kraft Foods; University of Wisconsin; University of Arizona; Paul, Weiss, Wharton and Garrison LLP; and Sage. In the last 36months, Christoph U. Correll has been a consultant and/or advisor to or has received honoraria from: Actelion, Alexza; AstraZeneca, Biotis, Boehringer-Ingelheim, Bristol-Myers Squibb, Cephalon, Desitin, Eli Lilly, GSK, IntraCellular Therapies, Lundbeck, Medavante, Medicure, Medscape, Merck, National Institute of Mental Health, Novartis, Ortho-McNeill/Janssen/J&J, Otsuka, Pfizer, ProPhase, Schering-Plough, Sepracor/Sunovion, Supernus, Takeda, Teva, and Vanda. He has received research support from BMS, Feinstein Institute for Medical Research, Janssen/J&J, National Institute of Mental Health (NIMH), National Alliance for Research in Schizophrenia and Depression (NARSAD), and Otsuka. Stephen Crystal, Emily J. Dhurandhar, and Julia M. Gohlke declare no commercial or financial relationships that could be construed as a potential conflict of interest. In the past 3 years, Elaine H. Morrato has received research support from Janssen Pharmaceuticals, Inc and Pfizer, Inc. In the last 12months, Henry A. Nasrallah received research support from Forest, Lilly Otsuka, Roche and Shire and served as a consultant for Genentech, Janssen, Merck, Novartis, Sunovion, Gruenthal, Boehringer-Ingelheim, and Lundbeck and was on the speakers bureau of Janssen, Merck, Novartis, and Sunovion. In the last 36months, JWN has received research support from NIMH, Bristol-Myers Squibb, and Pfizer, Inc. He has received consultant fees and/or been on data safety monitoring committees for AstraZeneca Pharmaceuticals, Bristol-Myers Squibb, BioVail, H. Lundbeck, Janssen Pharmaceutica, Obecure, Otsuka Pharmaceuticals, Sepracor, Inc., Solvay Pharma, Inc., Teva Pharmaceutical, Dainippon Sumitomo Pharma America, Inc., Organon Pharmaceuticals USA Inc., Schering-Plough/Merck, Vivus, Inc., Consultant to Litigation and Boehringer-Ingelheim. He has received royalties/patents/other income from Compact Clinicals/Jones and Bartlett Publishing. Ginger Nicol is Instructor of Child Psychiatry at Washington University in St. Louis, MO, USA and has no significant financial conflict of interest in compliance with the Washington University School of Medicine Conflict of Interest Policy. Ginger Nicol has received research funding from the NIMH, NARSAD, the Dana Brown Charitable Trust Foundation, the Sidney R. Baer, Jr. Foundation, and the CHADS Coalition for Mental Health. She also receives grant support from Pfizer, Inc. for an investigator-initiated clinical trial. She receives royalties from Jones & Barlett Learning for development of a pediatric metabolic monitoring form and has consulted to MedScape. In the last 3 years, Gary Remington has received research support from the following external funding agencies: Canadian Institutes of Health Research (CIHR), Schizophrenia Society of Ontario (SSO), and the Canadian Diabetes Association (CDA). He has also received support from Novartis Canada, Medicure Inc., and Neurocrine Bioscience. He has received consultant fees from Laboratorios Farmacéuticos ROVI, Novartis, and Roche, as well as speaker’s fees from Novartis.
Acknowledgments
Funding for the conference was made possible (in part) by (R13MH086166) from the National Institute of Mental Health (NIMH) and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK).
Footnote
- †Additional speakers at the Conference on Adipogenic and Metabolic Effects of Antipsychotic Drugs include: Marilyn Ader-Cedars-Sinai Medical Center, Trino Baptista-Los Andes University Medical School, Kimberly A. Brownley-University of North Carolina at Chapel Hill, Tony Cohn-University of Toronto, Donard S. Dwyer-Louisiana State University, Rohan Ganguli-University of Pittsburgh, Margaret Hahn-University of Toronto, David C. Henderson-Harvard University, Deanna Kelly University of Maryland, Christopher J. Lynch-Penn State University, Anil K. Malhotra-Albert Einstein College of Medicine, Vicki L. Ellingrod Ringold-University of Michigan, and Karen Teff-University of Pennsylvania
References
Ader, M., Kim, S. P., Catalano, K. J., Ionut, V., Hucking, K., Richey, J. M., Kabir, M., and Bergman, R. N. (2005). Metabolic dysregulation with atypical antipsychotics occurs in the absence of underlying disease: a placebo-controlled study of olanzapine and risperidone in dogs. Diabetes 54, 862–871.
Agid, O., Arenovich, T., Sajeev, G., Zipursky, R. B., Kapur, S., Foussias, G., and Remington, G. (2011). An algorithm-based approach to first-episode schizophrenia: response rates over 3 prospective antipsychotic trials with a retrospective data analysis. J. Clin. Psychiatry 72, 1439–1444.
Ahn, C., Tonidandel, S., and Overall, J. E. (2000). Issues in use of SAS PROC.MIXED to test the significance of treatment effects in controlled clinical trials. J. Biopharm. Stat. 10, 265–286.
Albaugh, V. L., Judson, J. G., She, P., Lang, C. H., Maresca, K. P., Joyal, J. L., and Lynch, C. J. (2011a). Olanzapine promotes fat accumulation in male rats by decreasing physical activity, repartitioning energy and increasing adipose tissue lipogenesis while impairing lipolysis. Mol. Psychiatry 16, 569–581.
Albaugh, V. L., Singareddy, R., Mauger, D., and Lynch, C. J. (2011b). A double blind, placebo-controlled, randomized crossover study of the acute metabolic effects of olanzapine in healthy volunteers. PLoS ONE 6, e22662. doi:10.1371/journal.pone.0022662
Albaugh, V. L., Vary, T. C., Ilkayeva, O., Wenner, B. R., Maresca, K. P., Joyal, J. L., Breazeale, S., Elich, T. D., Lang, C. H., and Lynch, C. J. (2012). Atypical antipsychotics rapidly and inappropriately switch peripheral fuel utilization to lipids, impairing metabolic flexibility in rodents. Schizophr. Bull. 38, 153–166.
Allison, D. B., Loebel, A. D., Lombardo, I., Romano, S. J., and Siu, C. O. (2009a). Understanding the relationship between baseline BMI and subsequent weight change in antipsychotic trials: effect modification or regression to the mean? Psychiatry Res. 170, 172–176.
Allison, D. B., Newcomer, J. W., Dunn, A. L., Blumenthal, J. A., Fabricatore, A. N., Daumit, G. L., Cope, M. B., Riley, W. T., Vreeland, B., Hibbeln, J. R., and Alpert, J. E. (2009b). Obesity among those with mental disorders: a National Institute of Mental Health meeting report. Am. J. Prev. Med. 36, 341–350.
Allison, D. B., Mentore, J. L., Heo, M., Chandler, L. P., Cappelleri, J. C., Infante, M. C., and Weiden, P. J. (1999). Antipsychotic-induced weight gain: a comprehensive research synthesis. Am. J. Psychiatry 156, 1686–1696.
Amminger, G. P., Schafer, M. R., Papageorgiou, K., Klier, C. M., Cotton, S. M., Harrigan, S. M., MacKinnon, A., McGorry, P. D., and Berger, G. E. (2010). Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Arch. Gen. Psychiatry 67, 146–154.
Baptista, T., Elfakih, Y., Uzcategui, E., Sandia, I., Talamo, E., Araujo De Baptista, E., and Beaulieu, S. (2008). Pharmacological management of atypical antipsychotic-induced weight gain. CNS Drugs 22, 477–495.
Brar, J. S., Ganguli, R., Pandina, G., Turkoz, I., Berry, S., and Mahmoud, R. (2005). Effects of behavioral therapy on weight loss in overweight and obese patients with schizophrenia or schizoaffective disorder. J. Clin. Psychiatry 66, 205–212.
Bronikowski, A. M., Morgan, T. J., Garland, T. Jr., and Carter, P. A. (2006). The evolution of aging and age-related physical decline in mice selectively bred for high voluntary exercise. Evolution 60, 1494–1508.
Brownley, K. A. (2011). Effect of Antipsychotics on Appetite Regulation (ADAPT). Available at: http://clinicaltrials.gov/ct2/show/study/NCT00903916?view=results [Accessed].
Buchanan, R. W., Kreyenbuhl, J., Kelly, D. L., Noel, J. M., Boggs, D. L., Fischer, B. A., Himelhoch, S., Fang, B., Peterson, E., Aquino, P. R., and Keller, W. (2010). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr. Bull. 36, 71–93.
Carrizo, E., Fernandez, V., Connell, L., Sandia, I., Prieto, D., Mogollon, J., Valbuena, D., Fernandez, I., De Baptista, E. A., and Baptista, T. (2009). Extended release metformin for metabolic control assistance during prolonged clozapine administration: a 14 week, double-blind, parallel group, placebo-controlled study. Schizophr. Res. 113, 19–26.
Chintoh, A. F., Mann, S. W., Lam, L., Giacca, A., Fletcher, P., Nobrega, J., and Remington, G. (2009). Insulin resistance and secretion in vivo: effects of different antipsychotics in an animal model. Schizophr. Res. 108, 127–133.
Cohen, T., Sundaresh, S., and Levine, F. (2012). Antipsychotics activate the TGFbeta pathway effector SMAD3. Mol. Psychiatry. doi:10.1038/mp.2011.186
Connor, D. F., Carlson, G. A., Chang, K. D., Daniolos, P. T., Ferziger, R., Findling, R. L., Hutchinson, J. G., Malone, R. P., Halperin, J. M., Plattner, B., Post, R. M., Reynolds, D. L., Rogers, K. M., Saxena, K., and Steiner, H. (2006). Juvenile maladaptive aggression: a review of prevention, treatment, and service configuration and a proposed research agenda. J. Clin. Psychiatry 67, 808–820.
Cooper, W. O., Arbogast, P. G., Ding, H., Hickson, G. B., Fuchs, D. C., and Ray, W. A. (2006). Trends in prescribing of antipsychotic medications for US children. Ambul. Pediatr. 6, 79–83.
Correll, C. U., Lencz, T., and Malhotra, A. K. (2011). Antipsychotic drugs and obesity. Trends. Mol. Med. 17, 97–107.
Correll, C. U., Manu, P., Olshanskiy, V., Napolitano, B., Kane, J. M., and Malhotra, A. K. (2009). Cardiometabolic risk of second-generation antipsychotic medications during first-time use in children and adolescents. JAMA 302, 1765–1773.
Crystal, S., Olfson, M., Huang, C., Pincus, H., and Gerhard, T. (2009). Broadened use of atypical antipsychotics: safety, effectiveness, and policy challenges. Health Aff. 28, w770–w781.
Daumit, G. L., Goff, D. C., Meyer, J. M., Davis, V. G., Nasrallah, H. A., McEvoy, J. P., Rosenheck, R., Davis, S. M., Hsiao, J. K., Stroup, T. S., and Lieberman, J. A. (2008). Antipsychotic effects on estimated 10-year coronary heart disease risk in the CATIE schizophrenia study. Schizophr. Res. 105, 175–187.
De Hert, M., Detraux, J., Van Winkel, R., Yu, W., and Correll, C. U. (2011a). Metabolic and cardiovascular adverse effects associated with antipsychotic drugs. Nat. Rev. Endocrinol. 8, 114–126.
De Hert, M., Dobbelaere, M., Sheridan, E. M., Cohen, D., and Correll, C. U. (2011b). Metabolic and endocrine adverse effects of second-generation antipsychotics in children and adolescents: a systematic review of randomized, placebo controlled trials and guidelines for clinical practice. Eur. Psychiatry 26, 144–158.
de los Campos, G., Gianola, D., and Allison, D. B. (2010). Predicting genetic predisposition in humans: the promise of whole-genome markers. Nat. Rev. Genet. 11, 880–886.
Dosreis, S., Yoon, Y., Rubin, D. M., Riddle, M. A., Noll, E., and Rothbard, A. (2011). Antipsychotic treatment among youth in foster care. Pediatrics 128, e1459–e1466.
Dwyer, D. S., Donohoe, D., Lu, X. H., and Aamodt, E. J. (2005). Mechanistic connections between glucose/lipid disturbances and weight gain induced by antipsychotic drugs. Int. Rev. Neurobiol. 65, 211–247.
Ellingrod, V. L., Miller, D. D., Taylor, S. F., Moline, J., Holman, T., and Kerr, J. (2008). Metabolic syndrome and insulin resistance in schizophrenia patients receiving antipsychotics genotyped for the methylenetetrahydrofolate reductase (MTHFR) 677C/T and 1298A/C variants. Schizophr. Res. 98, 47–54.
Ellingrod, V. L., Taylor, S. F., Brook, R. D., Evans, S. J., Zollner, S. K., Grove, T. B., Gardner, K. M., Bly, M. J., Pop-Busui, R., and Dalack, G. (2011). Dietary, lifestyle and pharmacogenetic factors associated with arteriole endothelial-dependent vasodilatation in schizophrenia patients treated with atypical antipsychotics (AAPs). Schizophr. Res. 130, 20–26.
Ellingrod, V. L., Taylor, S. F., Dalack, G., Grove, T. B., Bly, M. J., Brook, R. D., Zollner, S. K., and Pop-Busui, R. (2012). Risk factors associated with metabolic syndrome in bipolar and schizophrenia subjects treated with antipsychotics: the role of folate pharmacogenetics. J. Clin. Psychopharmacol. 32, 261–265.
Elobeid, M. A., Padilla, M. A., McVie, T., Thomas, O., Brock, D. W., Musser, B., Lu, K., Coffey, C. S., Desmond, R. A., St-Onge, M. P., Gadde, K. M., Heymsfield, S. B., and Allison, D. B. (2009). Missing data in randomized clinical trials for weight loss: scope of the problem, state of the field, and performance of statistical methods. PLoS ONE 4, e6624. doi:10.1371/journal.pone.0006624
Essock, S. M., Covell, N. H., Davis, S. M., Stroup, T. S., Rosenheck, R. A., and Lieberman, J. A. (2006). Effectiveness of switching antipsychotic medications. Am. J. Psychiatry 163, 2090–2095.
FDA. (2011). FDA’s Sentinel Initiative: Transforming How We Monitor the Safety of FDA-Regulated Products. Available at: http://www.fda.gov/Safety/FDAsSentinelInitiative/ucm2007250.htm [Accessed].
Fernandez, E., Carrizo, E., Fernandez, V., Connell, L., Sandia, I., Prieto, D., Mogollon, J., Valbuena, D., Fernandez, I., De Baptista, E. A., and Baptista, T. (2010). Polymorphisms of the LEP- and LEPR genes, metabolic profile after prolonged clozapine administration and response to the antidiabetic metformin. Schizophr. Res. 121, 213–217.
Findling, R. L., Johnson, J. L., McClellan, J., Frazier, J. A., Vitiello, B., Hamer, R. M., Lieberman, J. A., Ritz, L., McNamara, N. K., Lingler, J., Hlastala, S., Pierson, L., Puglia, M., Maloney, A. E., Kaufman, E. M., Noyes, N., and Sikich, L. (2010). Double-blind maintenance safety and effectiveness findings from the Treatment of Early Onset Schizophrenia Spectrum (TEOSS) study. J. Am. Acad. Child Adolesc. Psychiatry 49, 583–594.
Gadbury, G. L., Iyer, H. K., and Allison, D. B. (2001). Evaluating subject-treatment interaction when comparing two treatments. J. Biopharm. Stat. 11, 313–333.
Gaillard, T., Schuster, D., and Osei, K. (2009). Metabolic syndrome in Black people of the African diaspora: the paradox of current classification, definition and criteria. Ethn. Dis. 19, S2-1–S2-7.
Ganguli, R., and Strassnig, M. (2011). Prevention of metabolic syndrome in serious mental illness. Psychiatr. Clin. North Am. 34, 109–125.
Glick, I. D., Correll, C. U., Altamura, A. C., Marder, S. R., Csernansky, J. G., Weiden, P. J., Leucht, S., and Davis, J. M. (2011). Mid-term and long-term efficacy and effectiveness of antipsychotic medications for schizophrenia: a data-driven, personalized clinical approach. J. Clin. Psychiatry 72, 1616–1627.
Hahn, M., Chintoh, A., Giacca, A., Xu, L., Lam, L., Mann, S., Fletcher, P., Guenette, M., Cohn, T., Wolever, T., Arenovich, T., and Remington, G. (2011). Atypical antipsychotics and effects of muscarinic, serotonergic, dopaminergic and histaminergic receptor binding on insulin secretion in vivo: an animal model. Schizophr. Res. 131, 90–95.
Haupt, D. W., Fahnestock, P. A., Flavin, K. A., Schweiger, J. A., Stevens, A., Hessler, M. J., Maeda, J., Yingling, M., and Newcomer, J. W. (2007). Adiposity and insulin sensitivity derived from intravenous glucose tolerance tests in antipsychotic-treated patients. Neuropsychopharmacology 32, 2561–2569.
Haupt, D. W., Rosenblatt, L. C., Kim, E., Baker, R. A., Whitehead, R., and Newcomer, J. W. (2009). Prevalence and predictors of lipid and glucose monitoring in commercially insured patients treated with second-generation antipsychotic agents. Am. J. Psychiatry 166, 345–353.
Henderson, D. C., Nguyen, D. D., Copeland, P. M., Hayden, D. L., Borba, C. P., Louie, P. M., Freudenreich, O., Evins, A. E., Cather, C., and Goff, D. C. (2005). Clozapine, diabetes mellitus, hyperlipidemia, and cardiovascular risks and mortality: results of a 10-year naturalistic study. J. Clin. Psychiatry 66, 1116–1121.
Hermes, E., Nasrallah, H., Davis, V., Meyer, J., McEvoy, J., Goff, D., Davis, S., Stroup, T. S., Swartz, M., Lieberman, J., and Rosenheck, R. (2011). The association between weight change and symptom reduction in the CATIE schizophrenia trial. Schizophr. Res. 128, 166–170.
Hoffmann, V. P., Case, M., and Jacobson, J. G. (2011). Assessment of treatment algorithms including amantadine, metformin, and zonisamide for the prevention of weight gain with olanzapine: a randomized controlled open-label study. J. Clin. Psychiatry 73, 216–223.
Houseknecht, K. L., Robertson, A. S., Zavadoski, W., Gibbs, E. M., Johnson, D. E., and Rollema, H. (2007). Acute effects of atypical antipsychotics on whole-body insulin resistance in rats: implications for adverse metabolic effects. Neuropsychopharmacology 32, 289–297.
Kaddurah-Daouk, R., McEvoy, J., Baillie, R. A., Lee, D., Yao, J. K., Doraiswamy, P. M., and Krishnan, K. R. (2007). Metabolomic mapping of atypical antipsychotic effects in schizophrenia. Mol. Psychiatry 12, 934–945.
Kahn, R. S., Fleischhacker, W. W., Boter, H., Davidson, M., Vergouwe, Y., Keet, I. P., Gheorghe, M. D., Rybakowski, J. K., Galderisi, S., Libiger, J., Hummer, M., Dollfus, S., Lopez-Ibor, J. J., Hranov, L. G., Gaebel, W., Peuskens, J., Lindefors, N., Riecher-Rossler, A., and Grobbee, D. E. (2008). Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: an open randomised clinical trial. Lancet 371, 1085–1097.
Kapur, S., Wadenberg, M. L., and Remington, G. (2000). Are animal studies of antipsychotics appropriately dosed? Lessons from the bedside to the bench. Can. J. Psychiatry 45, 241–246.
Karagianis, J., Grossman, L., Landry, J., Reed, V. A., De Haan, L., Maguire, G. A., Hoffmann, V. P., and Milev, R. (2009). A randomized controlled trial of the effect of sublingual orally disintegrating olanzapine versus oral olanzapine on body mass index: the PLATYPUS Study. Schizophr. Res. 113, 41–48.
Kilpelainen, T. O., Bingham, S. A., Khaw, K. T., Wareham, N. J., and Loos, R. J. (2009). Association of variants in the PCSK1 gene with obesity in the EPIC-Norfolk study. Hum. Mol. Genet. 18, 3496–3501.
Kim, S. F., Huang, A. S., Snowman, A. M., Teuscher, C., and Snyder, S. H. (2007). From the Cover: Antipsychotic drug-induced weight gain mediated by histamine H1 receptor-linked activation of hypothalamic AMP-kinase. Proc. Natl. Acad. Sci. U.S.A. 104, 3456–3459.
Kroeze, W. K., Hufeisen, S. J., Popadak, B. A., Renock, S. M., Steinberg, S., Ernsberger, P., Jayathilake, K., Meltzer, H. Y., and Roth, B. L. (2003). H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology 28, 519–526.
Lett, T. A., Wallace, T. J., Chowdhury, N. I., Tiwari, A. K., Kennedy, J. L., and Muller, D. J. (2012). Pharmacogenetics of antipsychotic-induced weight gain: review and clinical implications. Mol. Psychiatry 17, 242–266.
Leucht, S., Corves, C., Arbter, D., Engel, R. R., Li, C., and Davis, J. M. (2009a). Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet 373, 31–41.
Leucht, S., Komossa, K., Rummel-Kluge, C., Corves, C., Hunger, H., Schmid, F., Asenjo Lobos, C., Schwarz, S., and Davis, J. M. (2009b). A meta-analysis of head-to-head comparisons of second-generation antipsychotics in the treatment of schizophrenia. Am. J. Psychiatry 166, 152–163.
Lieberman, J. A., Stroup, T. S., McEvoy, J. P., Swartz, M. S., Rosenheck, R. A., Perkins, D. O., Keefe, R. S., Davis, S. M., Davis, C. E., Lebowitz, B. D., Severe, J., and Hsiao, J. K. (2005). Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N. Engl. J. Med. 353, 1209–1223.
Little, R. J. A. (1988). A test of missing completely at random for multivariate data with missing values. J. Am. Stat. Assoc. 83, 1198–1202.
Little, R. J. A., and Rubin, D. B. (2002). Statistical Analysis with Missing Data, 2nd Edn. Hoboken, NJ: John Wiley and Sons, Inc.
Loos, R. J. (2011). The genetic epidemiology of melanocortin 4 receptor variants. Eur. J. Pharmacol. 660, 156–164.
Loos, R. J., Lindgren, C. M., Li, S., Wheeler, E., Zhao, J. H., Prokopenko, I., Inouye, M., Freathy, R. M., Attwood, A. P., Beckmann, J. S., Berndt, S. I., Jacobs, K. B., Chanock, S. J., Hayes, R. B., Bergmann, S., Bennett, A. J., Bingham, S. A., Bochud, M., Brown, M., Cauchi, S., Connell, J. M., Cooper, C., Smith, G. D., Day, I., Dina, C., De, S., Dermitzakis, E. T., Doney, A. S., Elliott, K. S., Elliott, P., Evans, D. M., Sadaf Farooqi, I., Froguel, P., Ghori, J., Groves, C. J., Gwilliam, R., Hadley, D., Hall, A. S., Hattersley, A. T., Hebebrand, J., Heid, I. M., Lamina, C., Gieger, C., Illig, T., Meitinger, T., Wichmann, H. E., Herrera, B., Hinney, A., Hunt, S. E., Jarvelin, M. R., Johnson, T., Jolley, J. D., Karpe, F., Keniry, A., Khaw, K. T., Luben, R. N., Mangino, M., Marchini, J., McArdle, W. L., McGinnis, R., Meyre, D., Munroe, P. B., Morris, A. D., Ness, A. R., Neville, M. J., Nica, A. C., Ong, K. K., O’Rahilly, S., Owen, K. R., Palmer, C. N., Papadakis, K., Potter, S., Pouta, A., Qi, L., Randall, J. C., Rayner, N. W., Ring, S. M., Sandhu, M. S., Scherag, A., Sims, M. A., Song, K., Soranzo, N., Speliotes, E. K., Syddall, H. E., Teichmann, S. A., Timpson, N. J., Tobias, J. H., Uda, M., Vogel, C. I., Wallace, C., Waterworth, D. M., Weedon, M. N., Willer, C. J., Wraight, V. L., Yuan, X., Zeggini, E., Hirschhorn, J. N., Strachan, D. P., Ouwehand, W. H., Caulfield, M. J., Samani, N. J., Frayling, T. M., Vollenweider, P., Waeber, G., Mooser, V., Deloukas, P., McCarthy, M. I., Wareham, N. J., Barroso, I., Jacobs, K. B., Chanock, S. J., Hayes, R. B., Lamina, C., Gieger, C., Illig, T., Meitinger, T., Wichmann, H. E., Kraft, P., Hankinson, S. E., Hunter, D. J., Hu, F. B., Lyon, H. N., Voight, B. F., Ridderstrale, M., Groop, L., Scheet, P., Sanna, S., Abecasis, G. R., Albai, G., Nagaraja, R., Schlessinger, D., Jackson, A. U., Tuomilehto, J., Collins, F. S., Boehnke, M., and Mohlke, K. L. (2008). Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat. Genet. 40, 768–775.
Lott, S. A., Burghardt, P. R., Burghardt, K. J., Bly, M. J., Grove, T. B., and Ellingrod, V. L. (2012). The influence of metabolic syndrome, physical activity and genotype on catechol-O-methyl transferase promoter-region methylation in schizophrenia. Pharmacogenomics J. doi: 10.1038/tpj.2012.6
Maayan, L., Vakhrusheva, J., and Correll, C. U. (2010). Effectiveness of medications used to attenuate antipsychotic-related weight gain and metabolic abnormalities: a systematic review and meta-analysis. Neuropsychopharmacology 35, 1520–1530.
Malhotra, A. K., Correll, C. U., Chowdhury, N. I., Müller, D. J., Gregersen, P. K., Lee, A. T., Tiwari, A. K., Kane, J. M., Fleischhacker, W. W., Kahn, R. S., Ophoff, R. A., Lieberman, J. A., Meltzer, H. Y., Lencz, T., and Kennedy, J. L. (2012). Common variants near the melanocortin 4 receptor gene are associated with severe antipsychotic drug-induced weight gain. Arch. Gen. Psychiatry. doi: 10.1001/archgenpsychiatry.2012.191. [Epub ahead of print].
McEvoy, J. P., Lieberman, J. A., Perkins, D. O., Hamer, R. M., Gu, H., Lazarus, A., Sweitzer, D., Olexy, C., Weiden, P., and Strakowski, S. D. (2007). Efficacy and tolerability of olanzapine, quetiapine, and risperidone in the treatment of early psychosis: a randomized, double-blind 52-week comparison. Am. J. Psychiatry 164, 1050–1060.
McEvoy, J. P., Meyer, J. M., Goff, D. C., Nasrallah, H. A., Davis, S. M., Sullivan, L., Meltzer, H. Y., Hsiao, J., Scott Stroup, T., and Lieberman, J. A. (2005). Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the clinical antipsychotic trials of intervention effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr. Res. 80, 19–32.
McIntyre, R. S., and Jerrell, J. M. (2008). Metabolic and cardiovascular adverse events associated with antipsychotic treatment in children and adolescents. Arch. Pediatr. Adolesc. Med. 162, 929–935.
Meyer, J. M., Nasrallah, H. A., McEvoy, J. P., Goff, D. C., Davis, S. M., Chakos, M., Patel, J. K., Keefe, R. S., Stroup, T. S., and Lieberman, J. A. (2005). The clinical antipsychotic trials of intervention effectiveness (CATIE) schizophrenia trial: clinical comparison of subgroups with and without the metabolic syndrome. Schizophr. Res. 80, 9–18.
Moeller, K. E., Rigler, S. K., Mayorga, A., Nazir, N., and Shireman, T. I. (2011). Quality of monitoring for metabolic effects associated with second generation antipsychotics in patients with schizophrenia on public insurance. Schizophr. Res. 126, 117–123.
Morrato, E. H., Druss, B., Hartung, D. M., Valuck, R. J., Allen, R., Campagna, E., and Newcomer, J. W. (2010a). Metabolic testing rates in 3 state medicaid programs after FDA warnings and ADA/APA recommendations for second-generation antipsychotic drugs. Arch. Gen. Psychiatry 67, 17–24.
Morrato, E. H., Nicol, G. E., Maahs, D., Druss, B. G., Hartung, D. M., Valuck, R. J., Campagna, E., and Newcomer, J. W. (2010b). Metabolic screening in children receiving antipsychotic drug treatment. Arch. Pediatr. Adolesc. Med. 164, 344–351.
Morrato, E. H., Druss, B. G., Hartung, D. M., Valuck, R. J., Thomas, D., Allen, R., Campagna, E., and Newcomer, J. W. (2011). Small area variation and geographic and patient-specific determinants of metabolic testing in antipsychotic users. Pharmacoepidemiol. Drug. Saf. 20, 66–75.
Morrato, E. H., Newcomer, J. W., Kamat, S., Baser, O., Harnett, J., and Cuffel, B. (2009). Metabolic screening after the American Diabetes Association’s consensus statement on antipsychotic drugs and diabetes. Diabetes Care 32, 1037–1042.
Morrison, J. A., Friedman, L. A., and Gray-McGuire, C. (2007). Metabolic syndrome in childhood predicts adult cardiovascular disease 25 years later: the princeton lipid research clinics follow-up study. Pediatrics 120, 340–345.
Mukundan, A., Faulkner, G., Cohn, T., and Remington, G. (2010). Antipsychotic switching for people with schizophrenia who have neuroleptic-induced weight or metabolic problems. Cochrane Database Syst. Rev. 12, CD006629.
Narula, P. K., Rehan, H. S., Unni, K. E., and Gupta, N. (2010). Topiramate for prevention of olanzapine associated weight gain and metabolic dysfunction in schizophrenia: a double-blind, placebo-controlled trial. Schizophr. Res. 118, 218–223.
Nasrallah, H. A. (2008). Atypical antipsychotic-induced metabolic side effects: insights from receptor-binding profiles. Mol. Psychiatry 13, 27–35.
Nasrallah, H. A., Meyer, J. M., Goff, D. C., McEvoy, J. P., Davis, S. M., Stroup, T. S., and Lieberman, J. A. (2006). Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr. Res. 86, 15–22.
Nelson, T. L., Bessesen, D. H., and Marshall, J. A. (2008). Relationship of abdominal obesity measured by DXA and waist circumference with insulin sensitivity in hispanic and non-hispanic white individuals: The San Luis Valley Diabetes Study. Diabetes Metab. Res. Rev. 24, 33–40.
Newcomer, J. W. (2005). Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs 19(Suppl. 1), 1–93.
Newcomer, J. W., Campos, J. A., Marcus, R. N., Breder, C., Berman, R. M., Kerselaers, W., L’Italien G, J., Nys, M., Carson, W. H., and McQuade, R. D. (2008). A multicenter, randomized, double-blind study of the effects of aripiprazole in overweight subjects with schizophrenia or schizoaffective disorder switched from olanzapine. J. Clin. Psychiatry 69, 1046–1056.
Newcomer, J. W., and Hennekens, C. H. (2007). Severe mental illness and risk of cardiovascular disease. JAMA 298, 1794–1796.
Newcomer, J. W., Ratner, R. E., Eriksson, J. W., Emsley, R., Meulien, D., Miller, F., Leonova-Edlund, J., Leong, R. W., and Brecher, M. (2009). A 24-week, multicenter, open-label, randomized study to compare changes in glucose metabolism in patients with schizophrenia receiving treatment with olanzapine, quetiapine, or risperidone. J. Clin. Psychiatry 70, 487–499.
Nicol, G., and Newcomer, J. (2008). Review: children and adolescents with schizophrenia spectrum disorders respond to antipsychotics, but are susceptible to adverse events. Evid. Based Ment. Health 11, 81.
Nicol, G. E., Morrato, E. H., Johnson, M. C., Campagna, E., Yingling, M. D., Pham, V., and Newcomer, J. W. (2011). Best practices: implementation of a glucose screening program based on diffusion of innovation theory methods. Psychiatr. serv. 62, 12–14.
NYDH. (2010). “The PSYCKES-CQI initiative: improving the quality of psychotropic prescribing practices in New York State,” in 2010 Handbook, eds T. Cheney, V. Hackethal, E. Kealey, J. Kutok, T. Lawinski, A. McCollom, J. Masterson, M. Perkins, S. Ricketts, K. Sherman, and A. Whited (New York State Office of Mental Health). Available at: http://www.psyckes.com/PSContent/CQI_Handbook.chapter_3.quality_concerns.cardiometabolic_risk.pdf
Ogden, C. L., Carroll, M. D., Curtin, L. R., Lamb, M. M., and Flegal, K. M. (2010). Prevalence of high body mass index in US children and adolescents, 2007–2008. JAMA 303, 242–249.
Olfson, M., Blanco, C., Liu, L., Moreno, C., and Laje, G. (2006). National trends in the outpatient treatment of children and adolescents with antipsychotic drugs. Arch. Gen. Psychiatry 63, 679–685.
Olfson, M., Crystal, S., Huang, C., and Gerhard, T. (2010). Trends in antipsychotic drug use by very young, privately insured children. J. Am. Acad. Child Adolesc. Psychiatry 49, 13–23.
Olfson, M., Marcus, S. C., Weissman, M. M., and Jensen, P. S. (2002). National trends in the use of psychotropic medications by children. J. Am. Acad. Child Adolesc. Psychiatry 41, 514–521.
Park, Y. W., Zhu, S., Palaniappan, L., Heshka, S., Carnethon, M. R., and Heymsfield, S. B. (2003). The metabolic syndrome: prevalence and associated risk factor findings in the US population from the third national health and nutrition examination survey, 1988–1994. Arch. Intern. Med. 163, 427–436.
Parsons, B., Allison, D. B., Loebel, A., Williams, K., Giller, E., Romano, S., and Siu, C. (2009). Weight effects associated with antipsychotics: a comprehensive database analysis. Schizophr. Res. 110, 103–110.
Patel, J. K., Buckley, P. F., Woolson, S., Hamer, R. M., Mcevoy, J. P., Perkins, D. O., and Lieberman, J. A. (2009). Metabolic profiles of second-generation antipsychotics in early psychosis: findings from the CAFE study. Schizophr. Res. 111, 9–16.
Penzner, J. B., Dudas, M., Saito, E., Olshanskiy, V., Parikh, U. H., Kapoor, S., Chekuri, R., Gadaleta, D., Avedon, J., Sheridan, E. M., Randell, J., Malhotra, A. K., Kane, J. M., and Correll, C. U. (2009). Lack of effect of stimulant combination with second-generation antipsychotics on weight gain, metabolic changes, prolactin levels, and sedation in youth with clinically relevant aggression or oppositionality. J. Child Adolesc. Psychopharmacol. 19, 563–573.
Perez-Iglesias, R., Crespo-Facorro, B., Amado, J. A., Garcia-Unzueta, M. T., Ramirez-Bonilla, M. L., Gonzalez-Blanch, C., Martinez-Garcia, O., and Vazquez-Barquero, J. L. (2007). A 12-week randomized clinical trial to evaluate metabolic changes in drug-naive, first-episode psychosis patients treated with haloperidol, olanzapine, or risperidone. J. Clin. Psychiatry 68, 1733–1740.
Ramaswamy, K., Masand, P. S., and Nasrallah, H. A. (2006). Do certain atypical antipsychotics increase the risk of diabetes? A critical review of 17 pharmacoepidemiologic studies. Ann. clin. psychiatry 18, 183–194.
Remington, G. (2010). Antipsychotic dosing: still a work in progress. Am. J. Psychiatry 167, 623–625.
Reynolds, G. P., and Kirk, S. L. (2010). Metabolic side effects of antipsychotic drug treatment – pharmacological mechanisms. Pharmacol. Ther. 125, 169–179.
Reynolds, G. P., Zhang, Z. J., and Zhang, X. B. (2002). Association of antipsychotic drug-induced weight gain with a 5-HT2C receptor gene polymorphism. Lancet 359, 2086–2087.
Rosenheck, R. A., Davis, S., Covell, N., Essock, S., Swartz, M., Stroup, S., McEvoy, J., and Lieberman, J. (2009). Does switching to a new antipsychotic improve outcomes? Data from the CATIE trial. Schizophr. Res. 107, 22–29.
Shin, L., Bregman, H., Frazier, J., and Noyes, N. (2008). An overview of obesity in children with psychiatric disorders taking atypical antipsychotics. Harv. Rev. Psychiatry 16, 69–79.
Sikich, L., Frazier, J. A., Mcclellan, J., Findling, R. L., Vitiello, B., Ritz, L., Ambler, D., Puglia, M., Maloney, A. E., Michael, E., De Jong, S., Slifka, K., Noyes, N., Hlastala, S., Pierson, L., Mcnamara, N. K., Delporto-Bedoya, D., Anderson, R., Hamer, R. M., and Lieberman, J. A. (2008). Double-blind comparison of first- and second-generation antipsychotics in early-onset schizophrenia and schizo-affective disorder: findings from the treatment of early-onset schizophrenia spectrum disorders (TEOSS) study. Am. J. Psychiatry 165, 1420–1431.
Strassnig, M., Brar, J. S., and Ganguli, R. (2005). Self-reported body weight perception and dieting practices in community-dwelling patients with schizophrenia. Schizophr. Res. 75, 425–432.
Stroup, T. S., McEvoy, J. P., Ring, K. D., Hamer, R. H., Lavange, L. M., Swartz, M. S., Rosenheck, R. A., Perkins, D. O., Nussbaum, A. M., and Lieberman, J. A. (2011). A randomized trial examining the effectiveness of switching from olanzapine, quetiapine, or risperidone to aripiprazole to reduce metabolic risk: comparison of antipsychotics for metabolic problems (CAMP). Am. J. Psychiatry 168, 947–956.
Sumner, A. E. (2009). Ethnic differences in triglyceride levels and high-density lipoprotein lead to under diagnosis of the metabolic syndrome in black children and adults. J. Pediatr. 155 (Suppl. 7), e7–e11.
Sumner, A. E., Sen, S., Ricks, M., Frempong, B. A., Sebring, N. G., and Kushner, H. (2008). Determining the waist circumference in african americans which best predicts insulin resistance. Obesity (Silver Spring) 16, 841–846.
van Winkel, R., Rutten, B. P., Peerbooms, O., Peuskens, J., Van Os, J., and De Hert, M. (2010). MTHFR and risk of metabolic syndrome in patients with schizophrenia. Schizophr. Res. 121, 193–198.
Victoriano, M., De Beaurepaire, R., Naour, N., Guerre-Millo, M., Quignard-Boulange, A., Huneau, J. F., Mathe, V., Tome, D., and Hermier, D. (2010). Olanzapine-induced accumulation of adipose tissue is associated with an inflammatory state. Brain Res. 1350, 167–175.
Vidarsdottir, S., De Leeuw Van Weenen, J. E., Frolich, M., Roelfsema, F., Romijn, J. A., and Pijl, H. (2010). Effects of olanzapine and haloperidol on the metabolic status of healthy men. J. Clin. Endocrinol. Metab. 95, 118–125.
Wang, Q., and Huang, X. F. (2008). Effects of chronic treatment of olanzapine and haloperidol on peptide YY binding densities in the rat brain. Exp. Neurol. 209, 261–267.
Weeks, K. R., Dwyer, D. S., and Aamodt, E. J. (2011). Clozapine and lithium require Caenorhabditis elegans beta-arrestin and serum- and glucocorticoid-inducible kinase to affect Daf-16 (Foxo) localization. J. Neurosci. Res. 89, 1658–1665.
Wu, R. R., Zhao, J. P., Jin, H., Shao, P., Fang, M. S., Guo, X. F., He, Y. Q., Liu, Y. J., Chen, J. D., and Li, L. H. (2008). Lifestyle intervention and metformin for treatment of antipsychotic-induced weight gain: a randomized controlled trial. JAMA 299, 185–193.
Yood, M. U., Delorenze, G., Quesenberry, C. P. Jr., Oliveria, S. A., Tsai, A. L., Willey, V. J., McQuade, R., Newcomer, J., and L’Italien, G. (2009). The incidence of diabetes in atypical antipsychotic users differs according to agent–results from a multisite epidemiologic study. Pharmacoepidemiol. Drug. Saf. 18, 791–799.
Zito, J. M., Safer, D. J., Sai, D., Gardner, J. F., Thomas, D., Coombes, P., Dubowski, M., and Mendez-Lewis, M. (2008). Psychotropic medication patterns among youth in foster care. Pediatrics 121, e157–e163.
Keywords: antipsychotic drugs, obesity, diabetes, schizophrenia, adiposity, pediatric populations, pharmacologic interventions, behavioral interventions
Citation: Gohlke JM, Dhurandhar EJ, Correll CU, Morrato EH, Newcomer JW, Remington G, Nasrallah HA, Crystal S, Nicol G, Adipogenic and Metabolic Effects of APDs Conference Speakers and Allison DB (2012) Recent advances in understanding and mitigating adipogenic and metabolic effects of antipsychotic drugs. Front. Psychiatry 3:62. doi: 10.3389/fpsyt.2012.00062
Received: 10 April 2012; Accepted: 06 June 2012;
Published online: 28 June 2012.
Edited by:
Andrew C. McCreary, Brains On-Line, NetherlandsCopyright: © 2012 Gohlke, Dhurandhar, Correll, Morrato, Newcomer, Remington, Nasrallah, Crystal, Nicol, Adipogenic and Metabolic Effects of APDs Conference Speakers and Allison. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: David B. Allison, Department of Biostatistics, School of Public Health, University of Alabama at Birmingham, 1665 University Blvd, RPHB 420C, Birmingham, AL 35294-0022, USA. e-mail:ZGFsbGlzb25AdWFiLmVkdQ==