

- Department of Neurobiology, Evelyn F. McKnight Brain Institute, University of Alabama at Birmingham, Birmingham, AL, USA
Post-traumatic stress disorder (PTSD) is a psychiatric condition characterized by intrusive and persistent memories of a psychologically traumatic event that leads to significant functional and social impairment in affected individuals. The molecular bases underlying persistent outcomes of a transient traumatic event have remained elusive for many years, but recent studies in rodents have implicated epigenetic modifications of chromatin structure and DNA methylation as fundamental mechanisms for the induction and stabilization of fear memory. In addition to mediating adaptations to traumatic events that ultimately cause PTSD, epigenetic mechanisms are also involved in establishing individual differences in PTSD risk and resilience by mediating long-lasting effects of genes and early environment on adult function and behavior. In this review, we discuss the current evidence for epigenetic regulation of PTSD in human studies and in animal models and comment on ways in which these models can be expanded. In addition, we identify key outstanding questions in the study of epigenetic mechanisms of PTSD in the context of rapidly evolving technologies that are constantly updating and adjusting our understanding of epigenetic modifications and their functional roles. Finally, we discuss the potential application of epigenetic approaches in identifying markers of risk and resilience that can be utilized to promote early intervention and develop therapeutic strategies to combat PTSD after symptom onset.
Post-traumatic stress disorder (PTSD) is a devastating psychiatric condition that develops in the aftermath of a traumatic life event. It is characterized by persistent and intrusive memories that interfere with daily functioning, often to the point of physical and emotional disability (American Psychiatric Association, 2000). In recent years, epigenetic mechanisms in the central nervous system emerged as the long sought-after link between transient environmental stimuli, including trauma, and persistent changes in gene expression and behavior (Zovkic et al., 2013). Epigenetic mechanisms were initially implicated in behavior through their ability to shape stress- and mood-associated neural and behavioral outcomes caused by early life experiences (Champagne and Meaney, 2001; Meaney, 2001; Fish et al., 2004; Weaver et al., 2004a,b). Shortly after, the epigenetic mechanisms once thought to be stable across the lifespan were found to be dynamically regulated during learning and memory in adult rodents (Levenson and Sweatt, 2005, 2006; Chwang et al., 2006; Levenson et al., 2006; Lubin and Sweatt, 2007; Miller and Sweatt, 2007). This temporal duality implicated epigenetic mechanisms in shaping individual variation in risk and resilience in response to early life experiences and in mediating environmental triggers of psychiatric disorders in adulthood (McGowan et al., 2009; Labonte et al., 2012; Russo et al., 2012; Zovkic and Sweatt, 2013).
Vast individual differences in PTSD prevalence among people exposed to traumatic events (Kessler et al., 1995), the contribution of early life events to adult risk for psychopathology (Yehuda and Bierer, 2009; Kremen et al., 2012; Russo et al., 2012), and the ability of stressors to regulate gene expression (Cullinan et al., 1995) pointed to epigenetic involvement in PTSD before any direct evidence for a link was available (e.g., Yehuda and Bierer, 2009). Soon, human studies provided evidence linking epigenetics with PTSD, either as a consequence of trauma or as a mediator of gene-environment interactions in PTSD risk and resilience (e.g., Ressler et al., 2011; Skelton et al., 2012; Klengel et al., 2013; Norrholm et al., 2013). While comparatively few animal studies have directly investigated epigenetic involvement in PTSD, the related literature on the epigenetics of stress and fear conditioning provides important insights into the role of these mechanisms in establishing persistent fear in response to stressful stimuli (Zovkic and Sweatt, 2013). Thus, one of our goals is to review the relatively small literature that directly addresses the involvement of epigenetics in PTSD and put it into the broader context of epigenetics in stress and fear learning. Given that approximately one in eight individuals who experience traumatic life events develop PTSD (Kessler et al., 1995), we will place particular emphasis on individual differences in PTSD and the need for animal models to consider this variation in studies of risk and resilience. Finally, to encourage direct investigation of epigenetic mechanisms in animal models of PTSD, we will provide a brief technical overview of the rapidly evolving approaches for measuring DNA methylation that can serve as a launching pad for researchers interested in applying these tools to their own work. Overall, we will explore the idea that trauma induces long-lasting epigenetic changes that mediate PTSD-associated outcomes, and that the exact changes elicited are influenced by individual-specific epigenetic landscape on which the traumatic event occurs.
Overview of Epigenetics in PTSD
Post-traumatic stress disorder is a multifaceted disorder that involves the deregulation of stress-responsive endocrine systems and of neurotransmitters and neuromodulators involved in arousal, depression, and anxiety (Baker et al., 2012). Although studies focused on identifying the relative contributions of these various systems have shaped the current understanding of the biological basis of PTSD, further progress will be driven by identifying common factors that drive and maintain their deregulation. Epigenetic modifications are emerging as one such factor because of their role in mediating gene expression for various components of the endocrine system, signaling molecules, and transcription factors (Champagne et al., 2006; Weaver et al., 2007; Chang et al., 2012; Russo et al., 2012; Klengel et al., 2013). In turn, epigenetic modifications are themselves regulated by upstream signaling cascades that converge to mediate epigenetic modifications in the nucleus, whereas existing epigenetic states alter the efficacy of the upstream signaling cascades on gene expression and behavior (Levenson et al., 2004; Fischer et al., 2007; Roozendaal et al., 2010; Graff and Tsai, 2011; Graff et al., 2011). In addition to providing an integrated perspective on psychiatric disorders, this bidirectionality offers promise for epigenetically based pharmacotherapies, which can reduce the threshold for, and amplify the response to, upstream signals (Graff and Tsai, 2011).
Epigenetic Mechanisms
DNA methylation initially gained notoriety for its role in the maintenance of cellular identity and heritable changes in gene expression throughout the cell cycle. DNA is initially methylated at the 5′ position of the cytosine–pyrimidine (5mC) ring by de novo DNA methyltransferase enzymes DNMT3a and 3b (Chiang et al., 1996; Turker, 1999; Bird, 2002; Cheng et al., 2010). This pattern of methylation is maintained throughout cell division by the maintenance DNMT1, which methylates cytosines opposite a methylated strand to allow for self-perpetuation of DNA methylation across cell divisions (Santos et al., 2005; Cheng et al., 2010). Given the energetic cost of removing a covalent 5mC bond and its role in the maintenance and propagation of cellular identity, 5mC was classically considered to be an irreversible mark. This notion has held largely true for the maintenance of cell-specific gene expression, but the classical view was challenged by evidence of active DNA demethylation in response to environmental stimuli in the brain, estrogen stimulation in cultured cells, and in muscle after exercise (Levenson et al., 2006; Miller and Sweatt, 2007; Kangaspeska et al., 2008; Metivier et al., 2008; Barres et al., 2012).
Indeed, 1.4% of measured CpGs (cytosines adjacent to guanines) in the granule cells in the dentate gyrus of the hippocampus exhibited altered DNA methylation in response to neuronal activation (Guo et al., 2011a), indicating that active DNA methylation and demethylation are a normal outcome of neuronal activity. Mechanisms of active DNA demethylation have been elusive, with limited success of early efforts to identify enzymes that remove 5mC directly. However, recent studies have provided a flurry of evidence suggesting that active demethylation is a stepwise process in which modifications of 5mC create a base mismatch that is replaced by an unmethylated cytosine through base excision repair (BER) (Wu and Zhang, 2010). Oxidation of 5mC to 5-hydroxymethylcytosine (5hmC) has emerged as a key step in active DNA demethylation in the brain and is accomplished by the ten-eleven translocation (TET) family of methyl-cytosine-dioxygenases (Tahiliani et al., 2009; Guo et al., 2011b; Ito et al., 2011). The resulting 5hmC is further modified by AID/APOBEC deaminases to yield 5 hydroxymethyluridine (5hmU) that is recognized by TDG and MBD4 to generate apurinic/apyrimidinic (AP) sites, which are cleaved by AP endonucleases and subjected to BER for replacement with an unmodified cytosine (5mC) (Ma et al., 2009a,b; Fritz and Papavasiliou, 2010; Bhutani et al., 2011; Guo et al., 2011b; Niehrs and Schafer, 2012; Grayson and Guidotti, 2013). Gadd45 proteins have also been implicated in this process, presumably by linking the AID/APOBEC deaminases with glycosylases to promote BER, which is particularly relevant for activity induced demethylation given the high sensitivity of Gadd45 to neural activity (Ma et al., 2009a,b). The much higher abundance (10-fold) of 5hmC in the rodent and human brain relative to other tissues (Kriaucionis and Heintz, 2009; Munzel et al., 2010; Jin et al., 2011; Szulwach et al., 2011), its increased levels over development (Munzel et al., 2010; Song et al., 2011b), and its regulation by neuronal activity (Guo et al., 2011b), suggest that this modification may be particularly relevant for cognitive function.
DNA methylation was initially identified as a mechanism for persistent repression of gene activity by the recruitment of co-repressor complexes (Karymov et al., 2001; Drewell et al., 2002) and the direct interference of 5mC with binding of transcriptional machinery (Iguchi-Ariga and Schaffner, 1989) to gene promoters. However, recent studies have shown that this is not always the case and that 5mC may at times be associated with gene activation. For example, although DNMTs are traditionally implicated in gene silencing (Chen et al., 2002), DNMT3a2 was recently associated with gene activation and improved cognitive function (Kotini et al., 2011) and de novo DNMTs may even be involved in DNA demethylation (Chen et al., 2012a). New evidence also suggests that the effect of DNA methylation on transcription may depend on the specific site on which the modification occurs, with 5mC at gene promoters and regulatory elements promoting gene repression and 5mC in gene bodies promoting gene activity (Hellman and Chess, 2007; Zilberman and Henikoff, 2007; Maunakea et al., 2010; Shenker and Flanagan, 2012). Although gene repression has been associated with the methylation of CpG islands in gene promoters, recent studies suggest that only 6.8% of CpGs are found in CpG islands (Rollins et al., 2006) and CpG islands are not ubiquitous to all human promoters (Deaton and Bird, 2011). In fact, newly identified CpG-island flanking regions termed CpG shores exhibit comparatively low CpG density and higher variability in methylation in cancer compared to CpG islands (Irizarry et al., 2009), implicating these sites in gene regulation. It is not clear to what extent these site-dependent functional differences are attributable to 5mC compared to 5hmC, since recent studies have demonstrated that 5hmC is depleted from promoters and intergenic regions and is enriched in gene bodies of actively transcribed cerebellar genes (Mellen et al., 2012). In contrast, a 5hmC peak that appeared 900 bp 5′ of the transcription start site was not correlated with transcription (Mellen et al., 2012), perhaps suggesting an alternative function. Of note, 5mC in the latter study was depleted from the bodies of actively transcribed genes and there was a tendency for a negative association between intragenic 5mC and gene expression in some cell types more than others (Mellen et al., 2012). Overall, however, intragenic 5hmC:5mC ratio was a better predictor of gene expression than 5mC or 5hmC alone (Mellen et al., 2012), pointing to the importance of considering both modifications in relation to transcription in different cell types. More broadly, these data indicate that the role for DNA methylation is more heterogeneous than initially suspected and that further studies will be required to fully understand the relationship between DNA methylation and transcription.
Many effects of DNA methylation are exerted through interactions with DNA-binding factors that recruit co-activators or co-repressors to mediate gene transcription and post-translational modifications (PTMs) of histones (Strahl and Allis, 2000; Ooi et al., 2007; Borrelli et al., 2008). DNA is packaged into nucleosomes, which are the building blocks of chromatin and are composed of ∼147 bp of DNA wrapped around an octamer of 2 each of histones H2A, H2B, H3, and H4 (Quina et al., 2006). Histones are critical regulators of DNA accessibility and chromatin compression or openness is determined in part by PTMs of histones (Strahl and Allis, 2000). Histones contain protruding tails that can be modified in a number of ways, most notably through acetylation, methylation, and phosphorylation (Strahl and Allis, 2000). As with DNA methylation, the pattern of histone modifications appears to differ based on the genomic region of interest, notably between introns and exons and around the transription start site (TSS) (Lieb and Clarke, 2005; Zilberman and Henikoff, 2007; Choi et al., 2010; Huff et al., 2010).
Of these various chromatin modifications, histone acetylation has received the most attention and is typically involved in the activation of gene expression (Mujtaba et al., 2007), whereas histone methylation can be involved in either activation or repression (Nakayama et al., 2001; Peters and Schubeler, 2005). Histone acetylation is catalyzed by histone acetyltransferases (HATs) and the reverse deacetylation reaction is performed by a large number of histone deacetylases (HDACs) (Hebbes et al., 1988; Wade, 2001). The binding of these enzymes is influenced by DNA methylation and associated methyl-CpG binding proteins, particularly MeCP2, which recruits HDACs to gene promoters to facilitate histone deacetylation (Lorincz et al., 2004; Liu et al., 2005). Recent evidence from the model plant Arabidopsis thaliana and from treatment with the HDAC inhibitor (HDACi) valproic acid (VPA) suggests that the reverse reaction may also be true, with HATs participating in DNA demethylation (Tremolizzo et al., 2002, 2005a,b; Detich et al., 2003; Qian et al., 2012). Indeed, the methyl-CpG binding protein MeCP2 interacts with CREB to promote histone acetylation (Chahrour et al., 2008), although it is not clear whether this relationship occurs through MeCP2 interactions with 5hmC and 5mC (Mellen et al., 2012).
MeCP2 binds to both 5mC and 5hmC in the cerebellum, but binding at 5hmC was associated with accessible chromatin and gene activation, whereas binding at 5mC was associated with closed chromatin and gene repression (Mellen et al., 2012). These effects were mediated by different residues on MeCP2, with a mutation in R133C preferentially altering binding to 5hmC compared to 5mC (Mellen et al., 2012). Other have also reported chromatin-regulating effects of neurally expressed MeCP2, with a weak association of MeCP2 with open chromatin and a strong association with closed chromatin (Thambirajah et al., 2012). Overall, there is some overlap in DNA-binding factors that interact with 5mC and 5hmC, but each modification also has unique interacting partners and different affinities for certain MBDs and other DNA-binding proteins (Hashimoto et al., 2012; Spruijt et al., 2013). These data suggest that 5hmC may be a transcriptional regulator in its own right rather than having a sole function as an in intermediary in active DNA demethylation.
The rapid advancements in techniques for detecting 5mC and 5hmC (see Box 1) and the application of whole-genome sequencing approaches have led to an unprecedented rate of growth in our understanding of epigenetic mechanisms in the regulation of gene expression. The wealth of new information has challenged many of the classical views of epigenetics in general and DNA methylation in particular, with many important implications for interpreting past and future data in the field. The most direct lesson from recent studies is that epigenetic modifications have diverse roles in gene regulation and that their effect on gene expression cannot be directly inferred from examining epigenetic modifications alone.
Box 1. Review of tools for measurement of hydroxymethylation.
Global 5hmC analysis
Several options exist for the global quantification of nucleotide variants implicated in epigenetic regulation, which include 5-methylcytosine (5mC), 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC), respectively. Currently, various antibodies raised against these epigenetic markers are available (Inoue et al., 2011; Ito et al., 2011) and have been employed in combination with either an enzyme-linked immunosorbent assay (ELISA) or dot-blot analysis for global detection (Koh et al., 2011; Li and Liu, 2011). In addition, some have taken advantage of the enzyme T4 phage β-glucosyltransferase (T4-GβT) and its ability to specifically add glucose to 5hmC to generate β-glucosyl-5-hmC (5gmC) (Terragni et al., 2012). Using this process, a radio-labeled glucose can then be transferred to 5hmC for sensitive and accurate detection of its global levels (Lian et al., 2012).
Another strategy to identify nucleotide modifications involves the use of thin layer chromatography (TLC) coupled with radio labeling. This approach provides a high level of sensitivity and was implemented by several labs leading to the initial discoveries of both 5hmC (Kriaucionis and Heintz, 2009; Tahiliani et al., 2009) and 5caC (He et al., 2011; Ito et al., 2011), respectively. However, of all the methods mentioned above, the gold standard remains the use of liquid chromatography (LC) followed by MS to resolve and precisely measure nucleosides containing these epigenetic marks (Globisch et al., 2010; Munzel et al., 2010; Ito et al., 2011).
Loci specific analysis and 5hmC enrichment
hMeDIP: A number of methods are now available for the measurement of 5hmC levels at the resolution of individual genomic loci. The first, hydroxymethyl-DNA immunoprecipitation (hMeDIP) requires the enrichment of 5hmC-containing DNA, typically with an antibody raised against 5hmC or its sodium bisulfite converted derivative, 5-methylsulfonate (CMS) (Pastor et al., 2011). Alternatively, enrichment can be achieved through T4-GβT-mediated conversion of 5hmC to 5gmC, followed by incubation with J-binding protein-1 (JBP-1); a protein shown to have a strong affinity for the modified base (Robertson et al., 2011). Likewise, some groups have gone on to process 5gmC even further through oxidation and biotinlyation steps (Pastor et al., 2011) (GLIB) or with selective chemical labeling (Song et al., 2011a) (hMe-Seal), allowing for the enrichment of biotinylated 5hmC residues using streptavidin beads. Using either the GLIB or hMe-Seal strategies allow for improved hMeDIP fidelity as well as the enrichment of genomic regions containing low amounts of 5hmC.
Restriction enzyme digestion: Recently, a number of restriction enzymes whose activity is blocked either by 5hmC itself or after 5hmC glycosylation have been characterized (Zheng et al., 2010; Cohen-Karni et al., 2011; Davis and Vaisvila, 2011; Kinney et al., 2011; Song et al., 2011b; Szwagierczak et al., 2011; Wang et al., 2011; Xu et al., 2011). The latter of these enzymes have been termed glucosyl-5-hydroxymethylcytosine sensitive restriction endonucleases (GSREs) and several companies now offer commercial kits to investigators. Due to their unique properties, these enzymes can be used to identify specific 5hmc-containing loci provided the region of interest contains the enzymes recognition site. Difficulties arising due to lack of compatible restriction endonuclease recognition sites in genomic regions of interest may be overcome using several different GRSEs. This technique has been further extended using several GSREs in combination, which enables the determination of both 5hmC and 5mC content at the same genomic location (Davis and Vaisvila, 2011).
Both techniques outlined here are amenable for analysis using quantitative PCR analysis for 5hmC detection at individual loci or for whole-genome profiling, microarray analysis, and next generation sequencing.
Single-base resolution sequencing
Although hMeDIP and GRSE analysis can both be useful tools for the analysis of 5hmC distribution on a genome-wide scale, they are limited in their resolution due to DNA fragmentation size and capture technology. To overcome these limitations, several groups have developed techniques to achieve the sequencing of 5hmC residues at a single-base resolution. The first involves the use of third-generation sequencing technology called single-molecule, real-time (SMRT) sequencing which monitors the incorporation of individual nucleotides by DNA polymerase in real time (Eid et al., 2009) and can detect DNA base modifications like 5mC and 5hmC compared to unmodified cytosine due to changes in DNA polymerase kinetics (Flusberg et al., 2010). This technique has now been combined with hMe-Seal to chemically label 5hmC in an effort to distinguish it further from 5hmC and C during DNA polymerization (Song et al., 2012). A second method termed oxidative bisulfite sequencing (oxBS-Seq) involves comparison of a traditional bisulfite sequenced sample (BS-seq) to that which has undergone oxidative conversion of 5hmC to 5fC, prior to bisulfite conversion (Booth et al., 2012). While BS-seq can convert both 5mC and 5hmC to cytosines (Cs), oxBS-Seq only allows for conversion of 5mC sites to Cs. Thus, the amount of 5hmC present at a particular nucleotide position can then be determined by subtraction of the oxBS-Seq sample from that of the BS-Seq sample. Finally, Tet-assisted bisulfite sequencing (TAB-seq) is a technique which combines the protection of 5hmC by T4-GBT glycosylation with Tet-mediated oxidation of 5mCs to 5caCs (Yu et al., 2012). After bisulfite conversion all 5caCs are converted to Ts, leaving only protected 5hmC residues as Cs, resulting in a straight-forward interpretation of 5hmC data at a base-by-base resolution.
Although we have focused heavily on DNA methylation, even the typically activating histone acetylation mark has been found on bivalent promoters that can be either activated or repressed (Lin et al., 2007; Vastenhouw and Schier, 2012) and HATs and HDACs tend to colocalize on the same genomic sites (Peserico and Simone, 2011). Similarly, in addition to their role in oxidation of 5mC to 5hmC, TET proteins may have additional regulatory roles, as exemplified by TET1 interaction with a repressive complex that might have a role in gene repression independent of its role in DNA demethylation (Bhutani et al., 2011; Ito et al., 2011; Wu and Zhang, 2011; Williams et al., 2012).
Another critical point is that many of the earlier studies did not distinguish between 5mC and 5hmC, making it difficult to draw inferences regarding the relative contribution of each. Although many approaches have been developed to differentiate the two, many techniques still encompass both modifications (see Box 1). Finally, growing evidence for distinct functions of DNA methylation at different genomic sites indicates that the common approach of using a single primer per gene may not be sufficient to detect changes in DNA methylation, or that the direction of the change may depend on the region studied (i.e., intragenic vs. promoter; CpG island vs. CpG-island shore). One solution to this approach is to do whole-genome sequencing, although a cost effective alternative is to utilize multiple primers to represent different regions relevant for detecting functional effects. An additional confound in neurobiology is the difficulty in distinguishing between epigenetic modifications occurring in neurons and glia, which will require more wide-spread use of cell sorting approaches and neuron-specific labels to improve our understanding of epigenetic changes in these distinct cell types.
Animal Models
Well-designed animal models are critical for uncovering epigenetic mechanisms of PTSD by allowing for the control of the temporal parameters needed to understand predisposition, onset, maintenance, and treatment of the disorder. An appropriate animal model must ensure face and etiological validity by mimicking the conditions of the disorder seen in human patients, which primarily involves exposure to a traumatic event in the case of PTSD (van der Staay et al., 2009). The traumatic event has been modeled in a number of ways, including exposure to an aversive shock in fear conditioning, exposure to a predator, social defeat, restraint, or underwater holding (Adamec and Shallow, 1993; Richter-Levin, 1998; Cohen et al., 2000a,b; Siegmund and Wotjak, 2007; Zoladz et al., 2008; Zovkic and Sweatt, 2013). Given the association between trauma and symptom severity in human patients (Koenen and Uddin, 2010; Uddin et al., 2010), stimuli that can deliver graded levels of trauma are preferred in an animal model (Siegmund and Wotjak, 2007). Shock delivery in fear conditioning is particularly amenable to manipulating gradations in stimulus intensity and duration (Siegmund and Wotjak, 2007), although different degrees of trauma intensity can also be achieved in predator exposure models by exposing rodents to cat odor alone, a caged cat, or to a direct encounter with a cat (Zoladz et al., 2008; Goswami et al., 2012). The etiological validity of these models can be further improved by combining the traumatic experience with additional risk factors that further enhance vulnerability to PTSD, such as the lack of social support and persistent exposure to chronic mild stress (Zoladz et al., 2008). In one example, the number of rodents expressing PTSD symptoms was increased by combining predator odor exposure with 31 days of social instability stress in an effort to mimic the lack of social support seen in individuals vulnerable to PTSD (Zoladz et al., 2008).
As an extension of the face validity criterion, animal models must include dependent variables that parallel the diverse symptoms of PTSD in order to generate testable predictions for research and treatment in human patients (van der Staay et al., 2009). PTSD patients exhibit a wide range of symptoms, including hyperarousal, increased startle, emotional blunting, and social withdrawal, which can be included as outcome variables in response to a variety of traumatic stimuli (Siegmund and Wotjak, 2007; Uddin et al., 2010; Skelton et al., 2012). These symptoms are induced by fear conditioning and predator exposure models of PTSD and are typically assessed after a prolonged stress-free period (Zovkic and Sweatt, 2013). For example, fear conditioning can be used to assess intensity-dependent freezing upon re-exposure to the training context to test for the expression of associative fear and upon exposure to a novel, neutral tone to provide an index of non-associative fear sensitization, hyperarousal, and startle (Siegmund and Wotjak, 2007). Importantly, the expression of sensitized fear to a novel auditory cue was found to increase with time after shock exposure (Siegmund and Wotjak, 2007), which is particularly important in light of the proposed revisions to DSM-V that emphasize delayed symptom onset over the initial fear response (Resick and Miller, 2009; Friedman et al., 2011a,b). Indeed, many patients who are diagnosed with PTSD show little to no emotional response upon initial exposure to trauma, with symptoms developing only with the passage of time (Shalev et al., 2000; Griffin, 2008; Friedman et al., 2011a,b). Associative fear is particularly amenable to delayed behavioral assessment (Miller et al., 2010) and has the advantage of modeling the re-experiencing of fear in PTSD patients by presenting animals with an aversive cue or context without the need for re-exposure to the traumatic stimulus (i.e., footshock) (Zovkic and Sweatt, 2013).
Models of Predisposition to PTSD
A difficulty with PTSD studies in humans is the poor ability to distinguish between pre-existing risk factors and trauma-induced outcomes. Co-twin studies, in which only one twin has experienced trauma, found that certain parameters that were thought to be caused by trauma may actually be pre-existing risk factors for PTSD, including impaired cognitive function and reduced hippocampal volume (Kremen et al., 2012). It can also be difficult to separate out epigenetic outcomes from epigenetic risk factors in non-controlled studies of human patients, although cumulative effects of trauma on DNA methylation of immune system genes suggest that traumatic experiences are key drivers of epigenetic outcomes in this scenario (Uddin et al., 2010).
Animal models can be particularly valuable in this regard by evaluating temporal parameters of variables identified in human studies to determine their relative role in conferring risk for PTSD or outcomes of trauma. An evaluation of individual differences is a key component for the success of such models (Yehuda and Bierer, 2009). The emphasis of animal models of PTSD tends to be on the stress exposure (induction) and the associated depressive and anxious phenotypes (read out), which are valuable tools for defining the cognitive, molecular, and neuroanatomical outcomes of stress exposure, but are less productive in explaining the relatively low incidence of persistent psychopathology in response to trauma (Yehuda et al., 2006). Individual differences in rodents can be investigated by classifying animals according to natural variation in behavior, by utilizing genetic predictors of risk and selective breeding strategies based on traits associated with risk and resilience (Scharf and Schmidt, 2012). Cohen and Zohar (2004) developed a model of classifying rodents according to natural variation in responses to predator exposure, in which ∼22% of the rats meet the PTSD criteria based on behavioral, endocrine, and sympathetic markers. The ad hoc classification system used in these studies is extremely useful for understanding long-term adaptations in behavioral and molecular systems that distinguish vulnerable and resilient individuals after trauma, but such models do not provide any information on the source of vulnerability or resilience before trauma (Siegmund and Wotjak, 2007; Zovkic and Sweatt, 2013). Selective breeding of vulnerable and resilient mice can help circumvent this problem by allowing for the assessment of pre-existing differences prior to trauma (Siegmund and Wotjak, 2007). For example, strain differences have been noted between PTSD vulnerable B6N and the resilient B6JOla mice, with B6N exhibiting blunted affect and reduced sociability in the absence of any exposure to trauma (Siegmund and Wotjak, 2007). Genetic mouse models can also be used to evaluate the role of risk genes identified in human studies and these models can be combined with developmental stressors to probe for gene-environment interactions at distinct stages of development.
History of stress exposure, particularly in early life, is another essential factor in predisposing individuals to PTSD in adulthood (Scharf and Schmidt, 2012). Although most rodent studies of early environment do not relate behavioral and biochemical outcomes to PTSD directly, their relevance to PTSD has been widely recognized because of the emphasis on the stress-responsive neural circuitry and the related behavioral outcomes (Yehuda and Bierer, 2009).
A widely studied model utilizes variation in maternal licking and grooming (LG) and arched back nursing (ABN) of pups as an index of early life experience. In this model, the offspring of high LG-ABN mothers are less anxious and responsive to stressors compared to the offspring of low LG-ABN dams (Weaver, 2009). However, the extent to which one maternal style is “better” than the other is not clear, as the offspring of high and low LG mothers may each be better adapted to different environmental circumstances (Bagot et al., 2009). However, the offspring of low LG mothers are phenotypically similar to the pups that underwent maternal separation stress (≥3 h separation) and high LG offspring are similar to those who underwent a milder brief separation (15 min), often referred to as handling, in the first 7–14 days of life (Anisman et al., 1998; Francis et al., 2002; Wilber and Wellman, 2009), indicating that low LG-ABN is akin to a more stressful upbringing. Utilization of maternal maltreatment models with an unambiguous stress exposure in early life (Roth and Sweatt, 2011) will be particularly useful in clarifying the association between stressful upbringing and behavior. Moreover, combining these early life models with adult models of PTSD (Cohen and Zohar, 2004) can provide critical information regarding the interaction between early life experience and the later risk and resilience for PTSD.
PTSD and the HPA Axis
The HPA Axis
Many studies of individual differences in risk and resilience have focused on the hypothalamic-pituitary-adrenal (HPA) axis because of its central role in adaptation to stress. The diagnosis of PTSD requires that symptom onset be preceded by a traumatic event and PTSD was recently reclassified as a traumatic and stress-related disorder instead of an anxiety disorder in the newly released DSM-V (Friedman et al., 2011a,b). Stressful encounters induce the release of corticotropin releasing hormone (CRH) from the hypothalamus into the pituitary gland to stimulate the release of adrenocorticotropin hormone (ACTH) and activate the release of glucocorticoids (GCs; cortisol in people, corticosterone in rodents) from the adrenals (Novak et al., 2013). The acute GC response allows individuals to deal with stressors by mobilizing resources, adjusting heart rate, and regulating immune function in the short term, whereas deregulation of GC release over prolonged time periods is detrimental to health, mood, and cognition (Sorrells and Sapolsky, 2007).
GC receptors (GR) are found throughout the brain and are particularly abundant in the hippocampus, where they influence memory formation, cognitive function, and initiate negative feedback to reinstate baseline GC levels after stress (Novak et al., 2013). The HPA axis is a critical regulator of the response to trauma in adults and the deregulation of this response has been implicated in the development of PTSD (Yehuda et al., 2004, 2009). PTSD patients generally exhibit reduced levels of GCs, increased negative feedback, abnormal GR expression, and higher levels of CRH compared to non-traumatized controls (Yehuda et al., 2002; Rohleder et al., 2004; de Kloet et al., 2008), indicating that a blunted GC response to stress may be a risk factor for PTSD (Yehuda and McFarlane, 1995; Yehuda and LeDoux, 2007).
Exposure to GCs stimulates a range of epigenetic modifications that are though to be mediated by the GR (Roozendaal et al., 2010; Yang et al., 2012). The GR is a transcriptional regulator that translocates to the nucleus upon ligand binding, where it activates or represses transcription through actions on the glucocorticoid response element (GRE) (Cairns et al., 1991). According to some estimates, GCs regulate between 1000 and 2000 genes in cortical neurons (Kino, 2007; Sun et al., 2008) and chronic stressors regulate gene expression and histone modifications in neural regions associated with cognitive function, affect, and motivation (Tsankova et al., 2004, 2006; Covington et al., 2011a,b). Effects of GR on transcription are closely tied to epigenetic regulation, as exposure to stress or treatment with glucocorticoids results in the persistent and gene-specific demethylation of GREs in peripherally derived human cells and the rodent brain (Unternaehrer et al., 2012; Yang et al., 2012; Klengel et al., 2013). In addition, exposure to stress in early life alters the expression of epigenetic regulators themselves, as evidenced by increased levels of DNMT3a in the placenta and the brain of pups of stressed mothers (Jensen Pena et al., 2012). Moreover, maternal maltreatment and natural variation in maternal care induce modifications of DNA methylation and expression of target genes that persist into adulthood in the rodent brain (Weaver et al., 2002; Zhang et al., 2006; Roth and Sweatt, 2011). In addition to the direct effects of GR in the nucleus, the GR mediates histone acetylation at target genes indirectly through the activation of the PKA-pCREB/CBP pathway via its membrane-bound receptor (Roozendaal et al., 2010). Overall, these studies demonstrate the ability of stressors to regulate epigenetic modifications, presumably through the actions of GCs on the GR receptor, although other potential pathways, such as sympathetic and noradrenergic responses, cannot be ruled out (Geracioti et al., 2001; Bierer et al., 2006; Videlock et al., 2008).
In addition to regulating epigenetic modifications, the HPA axis is itself epigenetically regulated, particularly during development, when the GR, CRH, and the GC inactivating enzyme HSD11B2 exhibit persistent alterations in DNA methylation and gene expression in response to maternal stress and natural variation in maternal behavior (Weaver, 2009; Roth and Sweatt, 2011; Jensen Pena et al., 2012). It has been shown, for example, that maternal deprivation between postnatal days 2 and 13 leads to hypomethylation of the cyclic AMP-response element (CRE) in the CRH promoter in the hypothalamus (Chen et al., 2012b), which is particularly relevant given the higher levels of CRH in patients with PTSD (de Kloet et al., 2008). Similarly, offspring of low LG mothers exhibit higher GR methylation, reduced GR expression, and higher reactivity to stress and anxiety in adulthood compared to the offspring of high LG mothers (Weaver, 2009). This effect is established in the first 7 days of life, when high levels of LG induce increased NGFI-1 expression, an upregulation of histone acetylation, and a decrease in DNA methylation at the NGFI-1 binding site of the GR promoter (Weaver et al., 2002, 2004a; Weaver, 2009). Extensive individual differences in DNA methylation of the GR promoter have been reported in people and include increased methylation of the NGFI-1 binding site on the GR promoter and decreased GR expression in the hippocampus of suicide victims with a history of childhood abuse compared to non-abused suicide victims or controls (McGowan et al., 2009).
It thus appears that elevated activity of DNA-binding molecules, such as GR and NGFI-I, may regulate binding to their respective response elements by reducing DNA methylation at the appropriate sites. This relatively well characterized system of maternal regulation of GR expression illustrates the close bidirectional relationship between epigenetic mechanisms and upstream signaling cascades. Moreover, the observation that persistent epigenetic changes are established in the first 7 days of life (Szyf et al., 2007) suggests that long-lasting effects of stress, maternal behavior, and GC exposure are amplified during sensitive periods of development, although the GR promoter is also subject to epigenetic regulation in adulthood (Witzmann et al., 2012).
Depending on the type of maternal care, epigenetic programing of GC release and GR expression may promote vulnerable or resilient phenotypes in adulthood (Dudley et al., 2011). On the surface, some parallels can be drawn between high LG mothers and the children of Holocaust survivors who had lower levels of GCs and rated their mothers as overprotective (Yehuda and Bierer, 2009). Based on such evidence, Yehuda and Bierer (2008) argued that reduced GC levels in high LG offspring parallel reduced cortisol, increased lymphocyte GR expression, and increased GR sensitivity seen in human PTSD patients (Yehuda et al., 1998, 2002; Yehuda, 2002a,b), although others interpret the reduced cortisol response to stress as protective (Barha et al., 2007). However, there are also reports of reduced levels of GR in leukocytes of PTSD patients (de Kloet et al., 2007), which parallels the reduced levels of GR density seen in low LG offspring. Furthermore, prenatal maternal depression/anxiety leads to increased methylation within the GR promoter at a predicted NGFI-A binding site in infants (Oberlander et al., 2008), likely leading to an overall reduction in GR expression. It is difficult to draw comparisons based on circulating GC and peripheral GR levels alone and studies of GR density in post-mortem brains of PTSD patients are needed to draw clearer parallels. The contention that the offspring of high LG mothers exhibit parallel HPA axis activity to PTSD patients (Yehuda and Bierer, 2009) is consistent with the view that some stress exposure at distinct stages of development confers resilience to PTSD in adults through “stress inoculation” (Ricon et al., 2012). Given the phenotypic similarities between outcomes associated with maternal separation stress and low LG offspring (Anisman et al., 1998; Francis et al., 2002; Wilber and Wellman, 2009), the concept of protective effects of “stress inoculation” (Ricon et al., 2012), and the adaptive function of an appropriate GC response to stress (Yehuda and McEwen, 2004), some have suggested that low LG offspring are actually better adapted to deal with stressful environments than high LG offspring (Bagot et al., 2009).
Gene × Environment × Epigenome Interactions
Individual differences in adult risk and resilience to PTSD are shaped by a combination of genetic and environmental factors. Although genes and environment can produce shifts in risk and resilience on their own, the strongest phenotypic effects are associated with gene-environment interactions that are mediated by epigenetic modifications (Caspi and Moffitt, 2006; Chertkow-Deutsher et al., 2010; Dudley et al., 2011; Russo et al., 2012; Klengel et al., 2013; Zovkic and Sweatt, 2013). Environmental factors are particularly influential during early life, when they interact with epigenetic mechanisms to lastingly alter the expression of a broad range of genes (Sundermann et al., 2012, 2013).
Studies of stress exposure in rodent strains with differential susceptibility to risk and resilience have reported distinct epigenetic and behavioral outcomes, although the specific genes involved in mediating these effects have not been identified. For example, vulnerable BALB/c (BALB) mice exhibited differences in H3 acetylation and DNA methylation of the glial cell-derived neurotrophic factor (GDNF) in the nucleus accumbens compared to the resilient C57BL/6 (B6) mice (Uchida et al., 2011). In addition, BALB mice showed a specific increase in MeCP2-HDAC2 association and B6 showed an increase in MeCP2-CREB association at the GDNF promoter, which were associated with increased and decreased GDNF expression in B6 and BALB mice, respectively. Vulnerability and resilience were also associated with opposite changes in DNA methylation of the same genes (Elliott et al., 2010), and with different levels of intronic DNA methylation and expression of dlgap2, a gene coding for PSD95 (Chertkow-Deutsher et al., 2010). In contrast, rodents classified as vulnerable or resilient based on behavioral responses to acute unavoidable stress exhibited differential regulation of distinct genes, with upregulation of neuroendocrine-related genes, growth hormone, and prolactin genes in vulnerable rats, and the downregulation of interferon-β and leukemia inhibitory factor in the frontal cortex of resilient rats (Benatti et al., 2012). Furthermore, Oh et al. (2013) found that an adverse maternal environment in rodents was associated with differentially methylated sites that clustered in the bodies of genes associated with cell adhesion and neurotransmitter receptors in the hippocampus. Finally, a study investigating the effects of environmental stress in non-human primates found an association between increased stress reactivity and higher overall methylation (Kinnally et al., 2011), which led them to speculate that the overall increase in methylation constrained plasticity through gene repression. However, the degree to which this hypothesis will hold true in light of evidence for an activating role of DNA methylation remains to be determined. Overall, these studies demonstrate that similar environmental experiences can produce extremely different outcomes through epigenetic interactions with individual-specific factors that mediate vulnerability and resilience.
The HPA Axis
Studies of human patients have found genotype-specific effects of early environment on adult risk for PTSD to be mediated by epigenetic modifications of genes associated with the HPA axis (Koenen and Uddin, 2010; Ressler et al., 2011; Klengel et al., 2013). Fkbp5 is a negative regulator of GR activity that has received much attention in this regard, in part because of reduced Fkbp5 and cortisol levels in the patient population (Yehuda and Bierer, 2009; Yehuda et al., 2009; Koenen and Uddin, 2010; Klengel et al., 2013). Single nucleotide polymorphisms (SNPs) in the Fkbp5 gene are associated with predisposition to PTSD, wherein the same allele confers either risk or resilience in the presence or absence of childhood adversity, respectively (Xie et al., 2010). Specifically, SNPs around the GRE in the human Fkbp5 gene increased risk for PTSD only in individuals who experienced childhood trauma (Binder et al., 2008; Yehuda et al., 2009). This effect was epigenetically mediated, in that GC exposure was associated with greater Fkbp5 GRE demethylation and higher Fkbp5 expression in carriers of the risk compared with the protective allele (Klengel et al., 2013). These data are in contrast to reports of reduced Fkbp5 expression in PTSD patients (Yehuda et al., 2009), but the discrepancy may in part be explained by variability across peripherally derived cell types and the sorting of data according to genetic variation.
PAC1R, a receptor for PACAP (pituitary adenylate cyclase-activating peptide), is another gene involved in stress responsiveness that has been implicated in PTSD (Dias and Ressler, 2013). PACAP is elevated in the blood of female, but not male, patients with PTSD and an SNP in the estrogen response element of the PAC1R gene interacts with trauma to mediate risk for PTSD in affected individuals (Ressler et al., 2011). Moreover, DNA methylation of the PAC1R gene is positively associated with PTSD (Ressler et al., 2011), again emphasizing the role of epigenetic factors as mediators of gene-environment associations. These data provide critical insights into the factors involved in establishing vulnerable phenotypes during development, but comparisons of epigenetic responses in vulnerable and resilient adult rodents are required to understand how traumatic experiences produce different outcomes in those individuals.
Synaptic Plasticity and Neurotransmitter Genes
In addition to the HPA axis, gene-environment-epigenome interactions have been reported for neuromodulators, neurotransmitters, and other molecules relevant for synaptic function and plasticity. For example, the serotonergic system is of clear relevance to PTSD given its well documented, albeit complex role in the regulation of emotional circuitry and fear learning (Rainnie, 1999; Koponen et al., 2005; Christianson et al., 2010), which has prompted an interest in polymorphisms of the serotonin transporter gene SLC6A4. A recent meta-analysis found that a short allele with an SNP in the 5-HTT promoter was associated with reduced transcriptional efficiency and increased sensitivity to stress (Karg et al., 2011). As with Fkbp5, association of the risk allele with PTSD occurs primarily through interactions with environmental experiences (Kilpatrick et al., 2007; Koenen et al., 2009; Xie et al., 2009), although some discrepancies have been reported (Mellman et al., 2009; Koenen et al., 2011). Epigenetic mediation of gene-environment interactions in PTSD has also been reported for other genes related to synaptic function, such as genes coding for Neuropeptide Y (NPY), BDNF, and molecules involved in the noradrenergic and dopaminergic systems (Boulle et al., 2012; Russo et al., 2012; Skelton et al., 2012; Wu et al., 2013).
In contrast to Fkpb5 and SLC6A4, carriers of the 9R allele of the dopamine transporter gene SLC6A3 were at greater risk of developing PTSD compared to 10R/10R allele carriers irrespective of environmental factors (Chang et al., 2012), highlighting the independent contribution of certain genes and the need to consider environmental interactions on a gene-by-gene basis. Furthermore, the risk for PTSD was highest in 9R allele carriers who also had high promoter methylation (Chang et al., 2012), indicating that epigenetic factors may mediate the contribution of genetic risk factors, irrespective of environmental factors.
It is important to note that the studies discussed thus far have primarily utilized a candidate-gene approach that allows for a thorough assessment of polymorphisms in a single gene, but is not suited for identifying novel genetic candidates involved in mediating risk and resilience. A recent genome-wide association study (GWAS) of trauma-exposed individuals identified a single polymorphism in the gene encoding the retinoic acid orphan receptor A (RORA), to be associated with PTSD after applying stringent genome-wide significance criteria (Logue et al., 2012). The high-risk SNP was associated with increased risk for PTSD in response to lower levels of trauma compared to individuals with the protective allele. RORA is a member of the NR1 subfamily of nuclear hormone receptors with neuroprotective functions (Jolly et al., 2011), which led the authors to hypothesize that genetically mediated differences in RORA levels are associated with an impaired stress response (Logue et al., 2012). Studies such as this will be critical for identifying novel candidates for PTSD risk and resilience in distinct populations.
Epigenetic Mediation of Risk and Resilience
Although genetic factors are essential mediators of risk and resilience, epigenetic mechanisms also mediate the effects of severe trauma independently of predisposing factors (Chang et al., 2012). Indeed, the extent of epigenetic modifications observed in patients is amplified by the severity and the number of traumatic events experienced (Uddin et al., 2010; Koenen et al., 2011) and distinct methylation profiles of the serotonin transporter gene are differentially associated with risk and resilience even after controlling for genotype (Koenen et al., 2011). Furthermore, observations of altered DNA methylation in response to childhood abuse and bullying (Ouellet-Morin et al., 2012) indicate that epigenetic modifications can predispose individuals to risk or resilience over and above the variability induced by genetic factors, as found with maternal behavior in rodents. In fact, the sensitive period for establishing lasting effects of stress appears to extend beyond early life into juvenile and adolescent periods of development (McCormick and Mathews, 2010; Dudley et al., 2011), as evidenced by site-specific demethylation of fkbp5 and reduced expression of DNMT1 in the mouse dentate gyrus of juvenile pups that were given 4 weeks of corticosterone treatment (Yang et al., 2012).
Although individual differences in PTSD risk are well established, relatively few studies have evaluated the specific mechanisms that underlie resilience. It is not yet clear whether resilience reflects the addition of a protective factor or the absence/reduction of a vulnerability factor (Russo et al., 2012). Studies of gene expression have found higher blood levels of NPY in resilient Special Forces soldiers (Morgan et al., 2000, 2002) and higher levels of c-Fos, FosB, and ΔFosB in resilient mice (Covington et al., 2010; Lehmann and Herkenham, 2011; Adamec et al., 2012), although genome-wide studies have reported altered expression in a comparable number of genes in resilient and vulnerable populations (Benatti et al., 2012). Some evidence suggests that risk and resilience are associated with opposite epigenetic modifications on similar genes, wherein resilience is associated with increased methylation and decreased expression of CRH and vulnerability is associated with demethylation and increased expression of CRH and fkbp5 in response to chronic stress (Elliott et al., 2010).
Epigenetic writers themselves are also candidates for mediating risk and resilience, in part through managing an appropriate balance between epigenetic repressors and epigenetic activators (Zovkic et al., 2013). Under baseline conditions, epigenetic repressors, such as HDACs, keep gene expression in check, whereas neuronal signaling increases gene expression in part by relieving such inhibition (Lattal et al., 2007). This repressive action of HDACs and other molecules has been termed the molecular break pad, reflecting the involvement of these molecules in regulating the magnitude and the duration of gene expression (Lattal and Wood, 2013). For example, reduction of HDAC3 expression prolongs the expression of immediate early genes involved in learning and memory (McQuown and Wood, 2011; McQuown et al., 2011) and HDAC inhibition or knockdown enhances fear learning in rodents (Levenson et al., 2004; Guan et al., 2009; Graff and Tsai, 2011; Lesburgueres et al., 2011). An implication of these studies is that a disrupted balance favoring activators over repressors may promote PTSD vulnerability by increasing the intensity of the traumatic memory, which is consistent with reports of reduced HDAC2 expression in the post-mortem brains of human patients suffering from depression (Sun et al., 2013).
However, not all HDAC subtypes are memory repressors. For example, sirtuins are class III HDACs that have been implicated in improved cognition (Kim et al., 2007) and hippocampal overexpression of HDAC1 was recently associated with improved extinction of fear memory (Bahari-Javan et al., 2012). Although sirtuins tend to be regulated by a distinct class of compounds, many pharmacological HDACi influence a broad range of HDACs, making it difficult to associate behavioral outcomes with specific HDAC subtypes. For example, although chronic social defeat was found to selectively increase the expression of the memory repressor HDAC2, treatment with the HDACi mass spectrometry (MS)-275 had an anti-depressant effect in these rodents (Guan et al., 2009; Covington et al., 2011b). This discrepancy may in part be associated with the relative specificity of MS-275 for HDAC1 over other HDACs, including HDAC2 (reviewed in Grayson and Guidotti, 2013). Similarly, TSA is an HDACi that actually increases HDAC1 expression in cell culture (Ajamian et al., 2004) and given the positive role of this HDAC on fear extinction (Bahari-Javan et al., 2012), TSA-induced enhancement of cognitive function may at least partly be a reflection of enhanced expression of HDAC1.
In addition to HDACs, DNMTs are emerging as another class of multifunctional epigenetic regulators with distinct modifications in PTSD, as evidenced by hypomethylation of DNMT3B gene and hypermethylation of the DNMT3L gene (Uddin et al., 2010). HDACs and DNMTs are only few of many epigenetic regulators involved in mediating the balance between activators and repressors and a thorough understanding of the potential shifts in their balance will require the consideration of these enzymes in the context of other epigenetic mediators, including histone acetylases, methylases, and demethylases, TET proteins, and other enzymes that directly mediate epigenetic modifications. Complementing these studies with genome-wide changes in gene expression will provide valuable information regarding the effects of specific modifications on genes associated with risk and resilience.
Cognitive Aspects of PTSD: Cause and Outcome
In addition to deregulation of the stress response, PTSD involves the development of strong cognitive associations between the stressful/fear-inducing event and the context or cue in which the event took place. This aspect is strongly tied into PTSD symptom expression, in that the stimuli or cues that are similar to those associated with trauma can on their own trigger re-experiencing of traumatic memories long after the event has passed (Ehlers et al., 2004). Epigenetic mechanisms are particularly relevant in this regard because of their role in the persistent stabilization of memory for transient fear-inducing stimuli (Zovkic et al., 2013). Existing literature on the epigenetics of fear learning has been conducted primarily in the context of cognition, but the use of fear conditioning models in PTSD makes these studies directly relevant to the disorder. Epigenetic modifications in fear learning have been extensively reviewed in a recent paper (Zovkic and Sweatt, 2013) and will be discussed here only briefly to illustrate the current state of understanding and the future directions required to advance the application of these models to mechanistic and clinical studies of PTSD.
A major advantage of animal models of fear learning is the ability to assess fear expression at defined time points after the fear-inducing stimulus to track the molecular mechanisms involved in the establishment and perpetuation of fear memory. Assessment of fear learning shortly after shock (2 h) best reflects the initial response to trauma, but this is only an index of short term memory and is not well suited for assessing persistent symptoms of PTSD, particularly given the lack of an initial emotional response in many patients with PTSD (Friedman et al., 2011a,b). However, this is a critical time point for evaluating epigenetic mechanisms of fear memory consolidation that are required for stabilizing the memory over time (Zovkic et al., 2013). Molecular assessment at this time point may reveal different patterns or intensities of gene expression and associated epigenetic modifications in vulnerable and resilient individuals that can inform efforts to identify mechanisms for pathological fear formation. Consolidation of cued and contextual fear conditioning is mediated by epigenetic modifications in the amygdala, whereas epigenetic modifications in the hippocampus are specifically involved in mediating contextual fear learning (Levenson et al., 2004; Lubin and Sweatt, 2007; Miller et al., 2008; Monsey et al., 2011). Consolidation of hippocampus-dependent fear memory is typically evaluated 24 h after training, whereas consolidation of remote memory becomes evident at least 7 days after training, when the memory from the hippocampus has been “downloaded” to the cortex for maintenance (Frankland et al., 2004; Ding et al., 2008). A focus on the remote time point is especially relevant because it provides the best approximation of the persistent and intrusive memories seen in patients with PTSD. Given the well-defined nature of the brain regions involved in fear consolidation and maintenance, the straight-forward behavioral read out of fear learning (i.e., freezing), and the ease with which stimulus (i.e., shock) intensity can be modified, this model provides an excellent platform on which to build our understanding of epigenetic mechanisms of PTSD. For a full review of epigenetic mechanisms of fear learning and their implications for PTSD, please refer to a recent review from our lab (Zovkic and Sweatt, 2013).
Memory Consolidation
It is now well established that epigenetic mechanisms are essential mediators of fear memory consolidation. Histone acetylation, phosphorylation, and methylation play a critical role in hippocampus- and amygdala-dependent consolidation across a range of conditioning paradigms (Chwang et al., 2006; Levenson et al., 2006; Lattal et al., 2007; Lubin and Sweatt, 2007; Bredy and Barad, 2008; Lubin et al., 2008; Miller et al., 2008; Maddox and Schafe, 2011; Monsey et al., 2011; Stafford et al., 2012) and interact with DNA methylation to mediate memory formation (Miller et al., 2008; Maddox and Schafe, 2011). Pharmacological or genetic interference with DNA methylation impairs fear memory (Miller and Sweatt, 2007; Miller et al., 2008, 2010; Feng et al., 2010), whereas enhancement of histone acetylation through HDAC inhibition enhances memory formation through reduced threshold for memory formation (Levenson et al., 2004; Bredy and Barad, 2008; Miller et al., 2008; Graff and Tsai, 2011; Maddox and Schafe, 2011; Monsey et al., 2011). Similarly, knocking out gadd45, which is involved in DNA demethylation, improves memory performance (Sultan et al., 2012), indicating that memory may be enhanced through a paradoxical upregulation of both DNA methylation and histone acetylation. Although a number of explanations have been offered for this paradox (Zovkic et al., 2013), recent advances implicating DNA methylation in gene activation may also contribute to a resolution. Nevertheless, the observation of improved memory with HDACi indicates that individuals with lower levels of HDACs and higher levels of enzymes involved in DNA demethylation may be more sensitive to developing fear responses that are disproportional to the traumatic event. This proposition is particularly relevant in the context of the molecular breakpad hypothesis, in which synaptic activity overrides the normally repressive effects of HDACs on transcription (Lattal and Wood, 2013), in that lower levels of HDACs may allow subthreshold levels of synaptic activity to produce a strong memory. Thus, an essential aim of PTSD research must be to fully characterize the relationship between different epigenetic enzymes and PTSD, as they relate to the consolidation of traumatic experiences. Advances have already been made in studies of group differences under baseline conditions in studies of human patients, which found reduced levels of HDAC2 and altered methylation of DNMT3L genes in PTSD (Uddin et al., 2010; Sun et al., 2013).
Memory Maintenance
Hippocampus-dependent memory consolidation occurs within hours of fear conditioning, when the memory begins a gradual process of downloading to the cortex for maintenance (Frankland and Bontempi, 2005). Epigenetic mechanisms have been implicated in the initial memory transfer process in which histone modifications in the cortex serve as a memory transfer tag (Lesburgueres et al., 2011) and as a persistent mark for memory maintenance (Graff et al., 2012). In contrast to the transient changes in DNA methylation observed in the hippocampus, fear conditioning is associated with a persistent increase in DNA methylation and decreased expression of the gene coding for the memory suppressor calcineurin up to 30 days after fear conditioning (Miller et al., 2010). DNA methylation is critical for memory maintenance at this time point, such that blocking DNA methylation immediately before a 30 day memory test impairs recall (Miller et al., 2010). The role of DNA methylation in memory persistence is of particular relevance to PTSD given the persistent re-experiencing of the traumatic event, indicating that manipulation of the epigenome at appropriate time points may allow for interference with previously established traumatic memories (see Zovkic and Sweatt, 2013 for an expanded discussion of therapeutic implications).
Memory Reconsolidation and Extinction
Persistent and intrusive memory recall is the foremost feature of PTSD in humans (Orr et al., 1993). Studies of molecular events associated with recall are critical for understanding the molecular mechanisms that underlie the strength of the pathological memory and for identifying potential treatments. Memory recall is an active process that renders the memory labile and employs molecular mechanisms to re-stabilize the original memory (Bredy and Barad, 2008; McKenzie and Eichenbaum, 2011; Pitman et al., 2011; Lattal and Wood, 2013). From a treatment perspective, interference with reconsolidation provides an opportunity to destabilize and even erase the original traumatic memory and this avenue is being actively pursued in patients with PTSD (Pitman, 2011). A recalled memory also becomes subject to a second active process termed extinction. Extinction occurs when a previously learned association is altered by new information, such as the safety of a conditioned stimulus in which a shock had previously occurred (Bouton, 2004). In contrast to disrupted reconsolidation, which effectively promotes forgetting, enhancing extinction allows for new learning in which the fear-inducing component of the memory can be selectively disrupted while leaving the initial memory relatively intact (Lattal and Wood, 2013). PTSD is often considered to be a disorder of extinction (Koenigs and Grafman, 2009) and many studies with rodents have found impaired extinction in at-risk individuals. For example, low LG and maternally separated offspring exhibit enhanced fear conditioning and impaired extinction (Champagne et al., 2008; Bagot et al., 2009; Callaghan and Richardson, 2011), whereas others associated brief maternal separation (15 min) with impaired fear extinction in spite of similar rates of initial conditioning (Wilber et al., 2007, 2009; Stevenson et al., 2009). Deficits in extinction have also been associated with the serotonin transporter in rodents (Wellman et al., 2007; Narayanan et al., 2011) and in people (Hartley et al., 2012), indicating that genetic risk for PTSD may be partly mediated by impaired fear extinction.
A major challenge in applying extinction and reconsolidation as a long-term treatment is the relative transience of extinction relative to the original learning event and the requirement for pharmacological intervention for robust interference with reconsolidation (Pitman, 2011). However, recent studies in rodents suggest that extinction may be an effective treatment strategy when combined with manipulations of the epigenome that can stabilize extinction learning over time (Lattal et al., 2007; Bredy and Barad, 2008; Lattal and Wood, 2013). Specifically, administration of HDACi promotes extinction of fear learning and robust extinction of cocaine preference that is resistant to subsequent reinstatement by cocaine (Malvaez et al., 2010), indicating that HDAC inhibition combined with extinction may promote adaptive learning that can combat deleterious adaptations. The effects of HDACi on extinction are dependent primarily on the HDAC1 subtype (Bahari-Javan et al., 2012) and are most effective in the infralimbic cortex (Stafford et al., 2012), which is consistent with evidence that HATs mediate extinction specifically in this brain region (Marek et al., 2011; Wei et al., 2012). This regional specificity is of functional relevance because it selectively alters extinction without impacting initial learning (Wei et al., 2012) and of translational relevance because lesions of the ventromedial prefrontal cortex (vmPFC) impair long-term extinction in people (Quirk et al., 2000) and reduced vmPFC activity has been reported in patients with PTSD (Koenigs and Grafman, 2009). These studies point to a dissociation between fear learning and fear extinction, wherein deficits in extinction are a critical characteristic of PTSD (Schnurr and Friedman, 2008) and may be more relevant to treatment because of the temporal gap between trauma and treatment onset. This temporal and regional difference also has implications for HDAC treatment, in that HDAC inhibition around the time of trauma exposure increases fear memory, whereas HDAC inhibition in association with fear extinction enhances the extinction of fear memory. This distinction is important to keep in mind when developing therapies for PTSD.
Extinction, in the form of exposure therapy, is heavily utilized treating PTSD (Schnurr and Friedman, 2008) and some effort has been made toward implementing reconsolidation based approaches in treatment (Pitman, 2011). However, more work is needed to identify the best conditions under which behavioral therapies are most effective when combined with pharmacological therapies. Clinical studies suggest that contextual parameters utilized in extinction and reconsolidation based therapies are critical for treatment efficacy, in that similarity between treatment conditions and those associated with the initial trauma has a major impact on treatment outcomes (Pitman, 2011). In humans and in animal models, this concept is typically investigated under the umbrella of fear generalization, whereby neutral cues can elicit fear responses based on their degree of similarity to cues or contexts associated with traumatic stimuli (Dunsmoor et al., 2009; Iordanova and Honey, 2012), but we are not aware of studies that have directly investigated epigenetic mechanisms in relation to fear generalization. In humans, there is increased use of virtual reality-based therapies to better reconstruct traumatic scenarios (Davis et al., 2006a,b) and similar approaches can be used to predict traumatic scenarios for soldiers before going into combat. Nevertheless, it is still necessary for animal studies to combine a thorough behavioral assessment with epigenetically based pharmacological interventions to identify the appropriate conditions for subsequent testing in human patients.
Epigenetics in Diagnosis and Intervention
Epigenetic modifications may be particularly strong candidates for use in assessing at-risk individuals for early intervention, as well as for PTSD treatment through epigenetically based therapies. An important first step toward such use of epigenetics involves the establishment of “epigenetic signatures” that serve as diagnostic markers and help identify targets for directed treatment. The idea of an epigenetic signature is driven by the assumption that PTSD is associated with consistent alterations in the epigenome across individuals and that these modifications can be used as markers of risk, resilience, and symptom severity. We have discussed much of the substantial progress that has been made toward identifying common epigenetic modifications in at-risk and resilient populations (see discussion of gene-environment-epigenome interactions above) and systematic investigations of these modifications can be of tremendous importance for directing early intervention (Uddin et al., 2010). Indeed, studies in animal models have exhibited encouraging results regarding the reversal of risk factors and cognitive deficits through enrichment and epigenetic intervention in juvenile and adult rodents (e.g., Francis et al., 2002; Fischer et al., 2007), indicating that early detection of risk may be key for PTSD prevention. Epigenetic markers will also be useful in PTSD diagnosis and in tracking therapeutic efficacy after trauma.
Although it is unlikely that any two individuals will have identical epigenetic profiles in response to trauma, it is conceivable that a substantial amount of overlap in essential genes will be found across patients, as evidenced by reports of altered DNMT, FKBP5, SLC6A4, and GR methylation reviewed above. Moreover, recent studies have demonstrated similar levels of DNA methylation of SLC6A4 and FKBP5 genes in the blood and the brain (Suomi, 2011; Klengel et al., 2013), indicating that the epigenetic status of certain genes in the brain may be indicated by changes in the blood. However, differences in function between immune cells and neurons must be taken into account, as early life stressors are known to modify immune cell function (Lam et al., 2012) and immune system alterations are commonly observed in PTSD (Smith et al., 2011). The degree to which concordance between brain and blood is replicable and applicable to other genes remains to be determined, but such concordance is less relevant for many clinical purposes than the ability of peripheral markers to reliably predict PTSD symptoms.
Nevertheless, tracking changes in pre- and post-treatment epigenetic profiles would be particularly useful for identifying genes that are modified by behavioral and epigenetic interventions as a first step in developing targeted therapies. In addition, it may be possible to identify the changes that are associated with persistent positive outcomes and to use this information to tailor more effective therapies. Indeed, an extinction-specific role of HDAC1 has already been identified for fear learning and a library of such markers would provide a valuable tool for designing epigenetic therapies to specifically enhance the efficacy of corresponding behavioral therapies. The efficacy of epigenetic intervention is evidenced by the fact that anti-depressant drugs produce epigenetic modifications (Tsankova et al., 2004, 2006) and that epigenetic drugs have anti-depressant effects (Covington et al., 2011b).
Epigenetic therapies, including HDACi, may be particularly strong candidates for PTSD treatment because they do not target a single neurotransmitter system and often exhibit therapeutic or behavioral effects only under appropriate signaling conditions (Roozendaal et al., 2010; Graff and Tsai, 2011). These pharmacological features are relevant to many psychiatric disorders, including PTSD, which exhibit deregulation of multiple neurotransmitter and signaling molecules (e.g., serotonin, NPY, GR, Fkpb5, BDNF, and CRH) and are typically treated by drugs such as fluoxetine that are targeted against specific neurotransmitter systems (Steckler and Risbrough, 2012). It is now evident that up- and down-regulation of the various deregulated molecules is at least partly mediated by altered DNA methylation and histone acetylation (Costa et al., 2003; Dong et al., 2007; Kinnally et al., 2011; Koenen et al., 2011; Klengel et al., 2013), indicating that epigenetic drugs may stabilize function across a range of disrupted molecules. For example, upregulation of FKBP5 in high-risk individuals is associated with reduced methylation of the GRE, which is demethylated by exposure to glucocorticoids during early development (Klengel et al., 2013). Similarly, reduced levels of reelin and 67-kDA glutamate decarboxylase (GAD67) have been attributed to DNA hypermethylation in mice (Costa et al., 2003; Dong et al., 2007).
On the flip side, many of the molecules we are discussing also alter gene expression through downstream effects on the epigenome and some evidence suggests that the bidirectional relationship across levels of regulation is a key driver or appropriate and stimulus-specific effects on behavior. For example, PKA-pCREB/CBP signaling and GR receptor activation drive changes in histone acetylation and hippocampus-dependent learning (Levenson et al., 2004; Roozendaal et al., 2010; Maddox and Schafe, 2011; Monsey et al., 2011), but HDACi could only enhance learning in the presence of GR activation (Roozendaal et al., 2010), indicating that increasing histone acetylation exerts behavioral effects only in the presence of appropriate signaling.
In addition to driving gene expression in mental disorders, epigenetic drugs may also normalize deregulated gene expression. For example, reduced gene expression and increased DNA methylation in schizophrenia were reversed by treatment with HDACi and this effect was associated with an improvement in psychotic symptoms (Costa et al., 2003; Tremolizzo et al., 2005a; Dong et al., 2007). Similarly, positive outcomes were reported for the HDACi valproic acid when given in conjunction with antipsychotics (Wassef et al., 2000, 2001; Grayson et al., 2010) or anti-depressants (Schroeder et al., 2007). Additionally, studies in rodents reported enhanced extinction of fear (Lattal et al., 2007; Bredy and Barad, 2008) and reduced depression (Covington et al., 2011b) in response to HDACi treatment, indicating that these drugs may be efficacious through effects on cognitive and affective processes. Even treatments that do not directly manipulate the epigenome produce epigenetic changes, with behavioral enrichment improving cognitive deficits through similar effects on chromatin regulation as HDACi (Fischer et al., 2007). Thus, a combination of environmental and pharmacological treatments may provide strongest candidates for early intervention or treatment. However, it is important to caution that outcomes associated with HDAC treatment in humans have been mixed (Narayan and Dragunow, 2010) and may be partly attributed to administration of adjunct therapies and the stage of the disease at the time of treatment (Tsai review). The development of more specific drugs and under appropriate conditions will be essential for improving the clinical efficacy of epigenetic therapies.
A number of issues must be addressed in designing epigenetic drugs with greater efficacy, including improved blood-brain-barrier (BBB) permeability and increased specificity for brain regions and HDAC subtypes (Grayson et al., 2010). HDACs are differentially distributed throughout the brain (Broide et al., 2007) and a growing number of studies point to distinct effects of specific HDAC subtypes in distinct brain regions and behavioral phenomena (Kim et al., 2007; Bahari-Javan et al., 2012; Sun et al., 2013). There is already a range of drugs that have different levels of regional and subtype specificity, with one study reporting that systemic injections of the BBB-permeable MS-275 (an HDACi selective for HDAC1 over HDAC3 and 8) selectively enhanced H3 acetylation in the frontal cortex and the hippocampus, but not the striatum (Simonini et al., 2006). In that study, MS-275 was more potent than valproic acid, which induced similar changes in acetylation across brain regions (Simonini et al., 2006). With more studies demonstrating HDAC-subtype specific effects on neural function and behavior (Kim et al., 2007; Bahari-Javan et al., 2012; Hanson et al., 2013), drugs that can be systemically administered and produce selective effects will be critical for targeted treatments. Interestingly, studies of human T23 bladder and MDA breast carcinoma, as well as HL60 cells, found a similar proportion of genes to be up- and down-regulated in response to treatment with HDACi (Glaser et al., 2003; Halsall et al., 2012), indicating that the effects of HDACi are not exclusively activating. Indeed, a high level of complexity and balance between memory activators and repressors necessitates that some genes are turned off in order for others to be effectively turned on (Zovkic et al., 2013).
Discussions of epigenetic treatments rightfully caution about the potential for wide-spread and non-specific effects on gene expression. However, microarray analysis of changes in hippocampal gene expression in response to l-methionine treatment only affected 1% of all the genes (Weaver et al., 2006), indicating that manipulations of epigenetic precursors and enzymes may produce surprisingly specific outcomes. Similarly, treatment of HL60 cells with three commonly used HDACi (VPA, TSA, SAHA) altered the expression of approximately 9% of genes, with only minor increases in histone acetylation observed at gene promoters (Halsall et al., 2012).
Moreover, manipulations of HDACs result in at least partly discriminant changes in acetylation, with HDAC1 having no effect on AcH3K14 and AcH4K5 (Bahari-Javan et al., 2012) and MS-275 altering AcH3K9 without affecting H4K12 (Simonini et al., 2006; Peleg et al., 2010). In addition to gene- and residue-specific effects, HDAC2 selectively enhances inhibitory synaptic function in the CA1 (Hanson et al., 2013), indicating that there is potential for specificity among epigenetic therapies. Much work is still needed to improve target specificity and the mechanisms of HDACi actions are not always clear, given the potential for the disruption of repressive complexes and effects on non-histone targets, which can be difficult to tease apart from effects of these drugs on histone acetylation (Zovkic et al., 2013). Thus, the development of increasingly specific therapies and a full characterization of the effects of existing therapies are important for developing drugs that can influence specific types of neural activity in selected brain regions.
Future Directions
In the present review, we focused heavily on developmental and genetic factors that predispose or protect individuals from PTSD. In many of these studies, the degree to which a manipulation is adaptive in adulthood is determined by reactivity to acute stressors outside of the context of PTSD. While these studies provide valuable insights into variable responses to trauma, a thorough assessment of persistent symptoms over prolonged periods of time is required to better model the lasting and intrusive nature of PTSD in people. Fear conditioning is one example of a model that is amenable to testing at different time points that can be used to characterize epigenetic changes that extend beyond the initial response to trauma and include trauma maintenance (see Tables 1 and 2 for summary of epigenetic modifications in fear conditioning and PTSD models). It will also be important to combine developmental models with models of PTSD that sort animals according to symptom development, severity, and persistence. This will be especially useful in distinguishing between early life experiences acting as stress inoculators vs. risk inducers in relation to adult risk and resilience. Ideally, early life manipulations will be combined with genetic models to reflect the gene-environment interactions identified in human risk populations. It is important to note that the rapid pace at which novel techniques are developed poses an opportunity and a challenge for conducting cutting edge research that can shed new light on our understanding of PTSD and psychiatric disorders. Most rapid advancements are particularly evident for techniques differentiating between DNA methylation and hydroxymethylation. Given the role of 5hmC in DNA demethylation and the distinct association of each modification with gene regulation, we have included a summary of new approaches to facilitate their application in studies of PTSD (see Box 1).
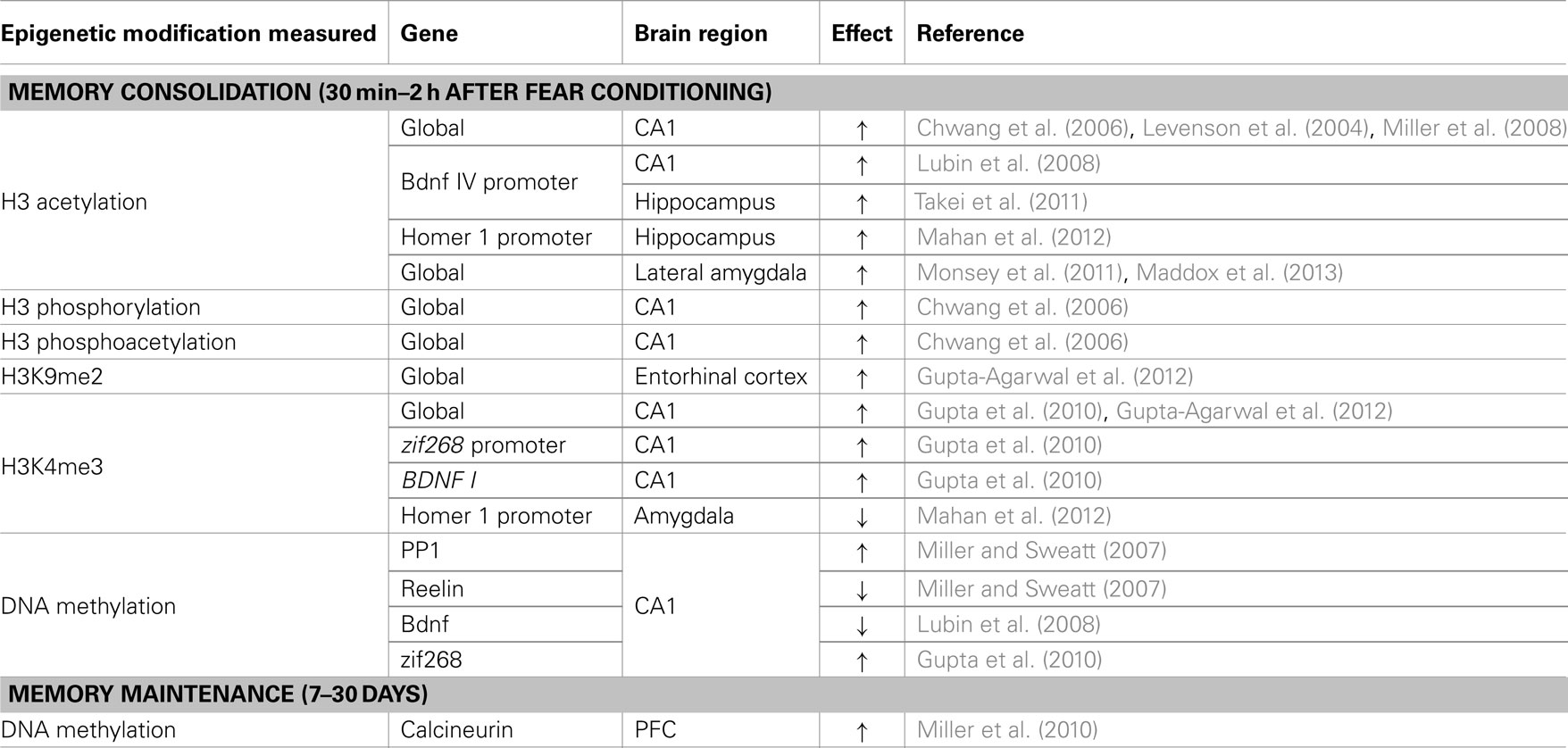
Table 1. A summary. of epigenetic modifications reported in rodent models of fear conditioning.
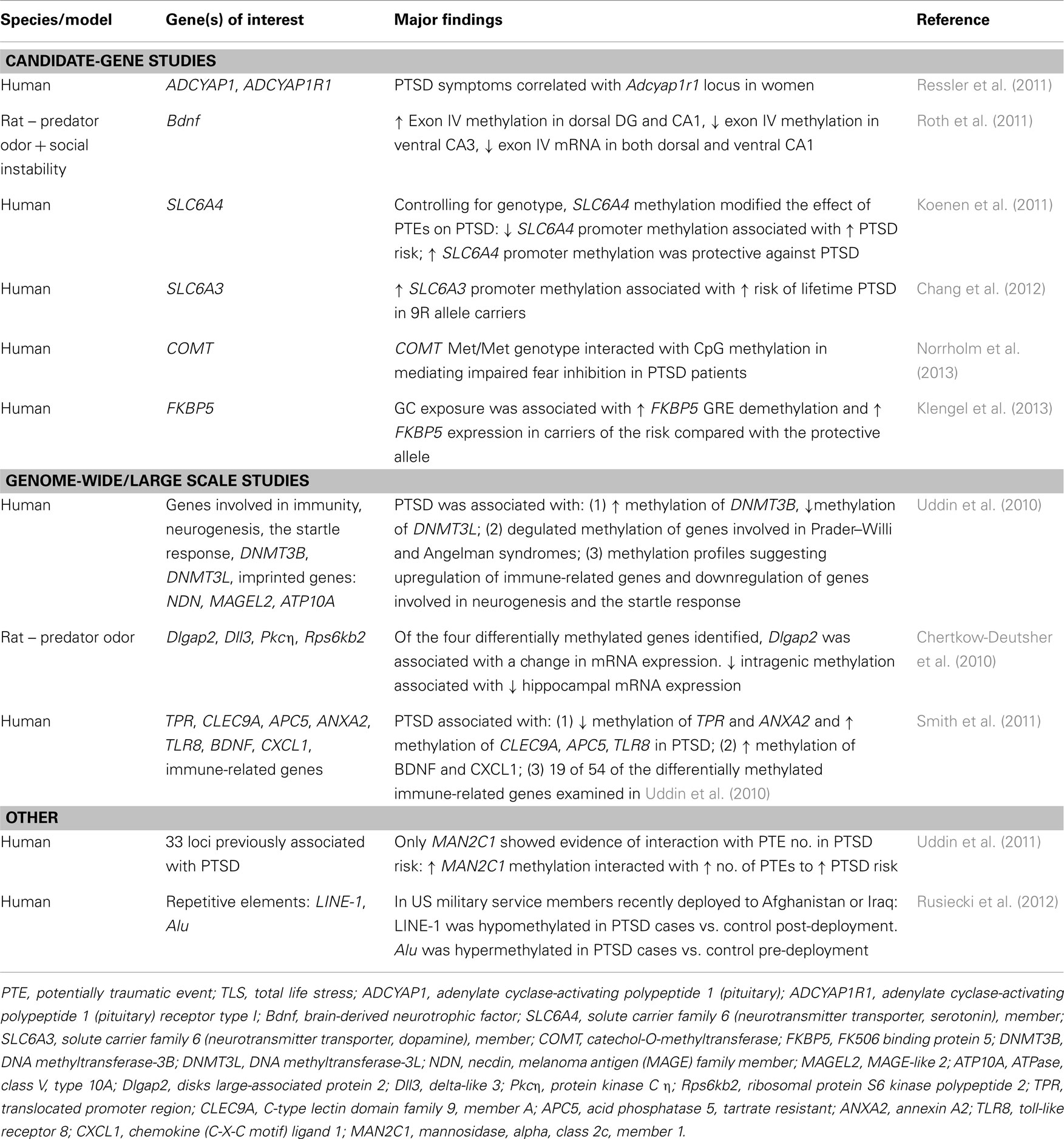
Table 2. A summary of epigenetic modifications in human and animal models of PTSD.
In addition to affecting gene expression through the direct interaction with transcriptional machinery, DNA methylation is increasingly being recognized as an incredibly complex regulator of neuronal function and metaplasticity (Baker-Andresen et al., 2013). For example, de novo DNA methylation was shown to prevent RNA polymerase stalling, thus providing a mechanism by which DNA methylation can regulate alternative splicing (Shukla et al., 2011). The long-term regulation of alternative splicing by DNA methylation may affect the responsivity of neurons to future stimuli (Baker-Andresen et al., 2013); thus investigations into the epigenetic regulation of alternative splicing are warranted in delineating factors that convey resilience to PTSD. Additionally, DNA methylation has been implicated in the regulation of retrotransposons, such as the positioning of long interspersed nuclear elements-1 (LINE-1) (Muotri et al., 2010). This is important, as the insertion of LINE-1 into various genomic regions may increase gene length, which is associated with decreased efficiency of expression (Castillo-Davis et al., 2002). Interestingly, a recent report demonstrated that a post-deployment diagnosis of PTSD was associated with hypomethylation of LINE-1, suggesting that methylation of these elements may play a role in mediated risk or resilience (Rusiecki et al., 2012). The existence of 5hmc and of distinct cofactors that interact with 5mc and 5hmc (Spruijt et al., 2013) further complicates the interpretation and calls for the use of techniques that distinguish between these modifications (see Box 1). Such approaches will be critical for identifying epigenetic signatures that differentiate between risk and resilience.
Finally, although women are more likely to develop PTSD than men, mot rodent studies have been conducted only in males (Mulchahey et al., 2001; Pope et al., 2003). Thus, a greater focus on females is warranted and may help us understand the basis for relative resilience in men, with testosterone providing one possible candidate for resilience (Russo et al., 2012). Some existing evidence suggests that sex differences are will be relevant for understanding all aspects of PTSD, with prenatal stress producing altered levels of DNMT1 expression, hippocampal GR methylation, and hypothalamic CRH methylation in adult male, but not female, offspring (Mueller and Bale, 2008). In addition, evidence from rodent studies suggests that sex differences vary for distinct components of PTSD, with females exhibiting vulnerability in tests of motivation and affect, while exhibiting resilience in non-stressful cognitive tasks (Luine, 2002).
Conclusion
In this review, we have demonstrated that epigenetic mechanisms are fundamental regulators of predisposition and resilience to PTSD, primarily through mediating gene-environment interactions during sensitive periods of development. Epigenetic mechanisms are also essential mediators of proximal causes of PTSD because of their role in stabilizing persistent outcomes of transient traumatic events. Although much progress has been made toward identifying epigenetic contributions to PTSD, ongoing shifts in our fundamental understanding of epigenetic modifications and their function call for the use of new technologies to clarify the role of these modifications in psychiatric disorders. As our appreciation of the complex roles of epigenetic mechanisms increases, so will our ability to create new and effective interventions for this incredibly complex and devastating human disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors’ work is supported by NIH Grants MH091122, MH57014, NS 057098, AG031722, and NR012686 (J. David Sweatt) and NSERC-PDF (Iva B. Zovkic).
References
Adamec, R. E., and Shallow, T. (1993). Lasting effects on rodent anxiety of a single exposure to a cat. Physiol. Behav. 54, 101–109. doi:10.1016/0031-9384(93)90050-P
Adamec, R., Toth, M., Haller, J., Halasz, J., and Blundell, J. (2012). A comparison of activation patterns of cells in selected prefrontal cortical and amygdala areas of rats which are more or less anxious in response to predator exposure or submersion stress. Physiol. Behav. 105, 628–638. doi:10.1016/j.physbeh.2011.09.016
Ajamian, F., Salminen, A., and Reeben, M. (2004). Selective regulation of class I and class II histone deacetylases expression by inhibitors of histone deacetylases in cultured mouse neural cells. Neurosci. Lett. 365, 64–68. doi:10.1016/j.neulet.2004.04.087
American Psychiatric Association. (2000). Diagnostic and Statistical Manual of Mental Disorders. Washington: American Psychiatric Publishing.
Anisman, H., Zaharia, M. D., Meaney, M. J., and Merali, Z. (1998). Do early-life events permanently alter behavioral and hormonal responses to stressors? Int. J. Dev. Neurosci. 16, 149–164. doi:10.1016/S0736-5748(98)00025-2
Bagot, R. C., Van Hasselt, F. N., Champagne, D. L., Meaney, M. J., Krugers, H. J., and Joels, M. (2009). Maternal care determines rapid effects of stress mediators on synaptic plasticity in adult rat hippocampal dentate gyrus. Neurobiol. Learn. Mem. 92, 292–300. doi:10.1016/j.nlm.2009.03.004
Bahari-Javan, S., Maddalena, A., Kerimoglu, C., Wittnam, J., Held, T., Bahr, M., et al. (2012). HDAC1 regulates fear extinction in mice. J. Neurosci. 32, 5062–5073. doi:10.1523/JNEUROSCI.0079-12.2012
Baker, D. G., Nievergelt, C. M., and O’Connor, D. T. (2012). Biomarkers of PTSD: neuropeptides and immune signaling. Neuropharmacology 62, 663–673. doi:10.1016/j.neuropharm.2011.02.027
Baker-Andresen, D., Ratnu, V. S., and Bredy, T. W. (2013). Dynamic DNA methylation: a prime candidate for genomic metaplasticity and behavioral adaptation. Trends Neurosci. 36, 3–13. doi:10.1016/j.tins.2012.09.003
Barha, C. K., Pawluski, J. L., and Galea, L. A. (2007). Maternal care affects male and female offspring working memory and stress reactivity. Physiol. Behav. 92, 939–950. doi:10.1016/j.physbeh.2007.06.022
Barres, R., Yan, J., Egan, B., Treebak, J. T., Rasmussen, M., Fritz, T., et al. (2012). Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 15, 405–411. doi:10.1016/j.cmet.2012.01.001
Benatti, C., Valensisi, C., Blom, J. M., Alboni, S., Montanari, C., Ferrari, F., et al. (2012). Transcriptional profiles underlying vulnerability and resilience in rats exposed to an acute unavoidable stress. J. Neurosci. Res. 90, 2103–2115. doi:10.1002/jnr.23100
Bhutani, N., Burns, D. M., and Blau, H. M. (2011). DNA demethylation dynamics. Cell 146, 866–872. doi:10.1016/j.cell.2011.08.042
Bierer, L. M., Tischler, L., Labinsky, E., Cahill, S., Foa, E., and Yehuda, R. (2006). Clinical correlates of 24-h cortisol and norepinephrine excretion among subjects seeking treatment following the world trade center attacks on 9/11. Ann. N. Y. Acad. Sci. 1071, 514–520. doi:10.1196/annals.1364.055
Binder, E. B., Bradley, R. G., Liu, W., Epstein, M. P., Deveau, T. C., Mercer, K. B., et al. (2008). Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. 299, 1291–1305. doi:10.1001/jama.299.11.1291
Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21. doi:10.1101/gad.947102
Booth, M. J., Branco, M. R., Ficz, G., Oxley, D., Krueger, F., Reik, W., et al. (2012). Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 336, 934–937. doi:10.1126/science.1220671
Borrelli, E., Nestler, E. J., Allis, C. D., and Sassone-Corsi, P. (2008). Decoding the epigenetic language of neuronal plasticity. Neuron 60, 961–974. doi:10.1016/j.neuron.2008.10.012
Boulle, F., Van Den Hove, D. L., Jakob, S. B., Rutten, B. P., Hamon, M., Van Os, J., et al. (2012). Epigenetic regulation of the BDNF gene: implications for psychiatric disorders. Mol. Psychiatry 17, 584–596. doi:10.1038/mp.2011.107
Bouton, M. E. (2004). Context and behavioral processes in extinction. Learn. Mem. 11, 485–494. doi:10.1101/lm.78804
Bredy, T. W., and Barad, M. (2008). The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn. Mem. 15, 39–45. doi:10.1101/lm.801108
Broide, R. S., Redwine, J. M., Aftahi, N., Young, W., Bloom, F. E., and Winrow, C. J. (2007). Distribution of histone deacetylases 1-11 in the rat brain. J. Mol. Neurosci. 31, 47–58. doi:10.1007/BF02686117
Cairns, W., Cairns, C., Pongratz, I., Poellinger, L., and Okret, S. (1991). Assembly of a glucocorticoid receptor complex prior to DNA binding enhances its specific interaction with a glucocorticoid response element. J. Biol. Chem. 266, 11221–11226.
Callaghan, B. L., and Richardson, R. (2011). Maternal separation results in early emergence of adult-like fear and extinction learning in infant rats. Behav. Neurosci. 125, 20–28. doi:10.1037/a0022008
Caspi, A., and Moffitt, T. E. (2006). Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat. Rev. Neurosci. 7, 583–590. doi:10.1038/nrn1925
Castillo-Davis, C. I., Mekhedov, S. L., Hartl, D. L., Koonin, E. V., and Kondrashov, F. A. (2002). Selection for short introns in highly expressed genes. Nat. Genet. 31, 415–418.
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi:10.1126/science.1153252
Champagne, D. L., Bagot, R. C., Van Hasselt, F., Ramakers, G., Meaney, M. J., De Kloet, E. R., et al. (2008). Maternal care and hippocampal plasticity: evidence for experience-dependent structural plasticity, altered synaptic functioning, and differential responsiveness to glucocorticoids and stress. J. Neurosci. 28, 6037–6045. doi:10.1523/JNEUROSCI.0526-08.2008
Champagne, F., and Meaney, M. J. (2001). Like mother, like daughter: evidence for non-genomic transmission of parental behavior and stress responsivity. Prog. Brain Res. 133, 287–302. doi:10.1016/S0079-6123(01)33022-4
Champagne, F. A., Weaver, I. C., Diorio, J., Dymov, S., Szyf, M., and Meaney, M. J. (2006). Maternal care associated with methylation of the estrogen receptor-alpha1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology 147, 2909–2915. doi:10.1210/en.2005-1119
Chang, S. C., Koenen, K. C., Galea, S., Aiello, A. E., Soliven, R., Wildman, D. E., et al. (2012). Molecular variation at the SLC6A3 locus predicts lifetime risk of PTSD in the Detroit Neighborhood Health Study. PLoS ONE 7:e39184. doi:10.1371/journal.pone.0039184
Chen, C. C., Wang, K. Y., and Shen, C. K. (2012a). The mammalian de novo DNA methyltransferases DNMT3A and DNMT3B are also DNA 5-hydroxymethylcytosine dehydroxymethylases. J. Biol. Chem. 287, 33116–33121. doi:10.1074/jbc.C112.406975
Chen, D. Y., Bambah-Mukku, D., Pollonini, G., and Alberini, C. M. (2012b). Glucocorticoid receptors recruit the CaMKIIalpha-BDNF-CREB pathways to mediate memory consolidation. Nat. Neurosci. 15, 1707–1714. doi:10.1038/nn.3266
Chen, T., Ueda, Y., Xie, S., and Li, E. (2002). A novel Dnmt3a isoform produced from an alternative promoter localizes to euchromatin and its expression correlates with active de novo methylation. J. Biol. Chem. 277, 38746–38754. doi:10.1074/jbc.M205312200
Cheng, X., Hashimoto, H., Horton, J. R., and Zhang, X. (2010). “Mechanisms of DNA methylation, methyl-CpG recognition, and demethylation in mammals,” in Handbook of Epigenetics, ed. T. O. Tollefsbol (Oxford: Academic Press), 9–24.
Chertkow-Deutsher, Y., Cohen, H., Klein, E., and Ben-Shachar, D. (2010). DNA methylation in vulnerability to post-traumatic stress in rats: evidence for the role of the post-synaptic density protein Dlgap2. Int. J. Neuropsychopharmacol. 13, 347–359. doi:10.1017/S146114570999071X
Chiang, P. K., Gordon, R. K., Tal, J., Zeng, G. C., Doctor, B. P., Pardhasaradhi, K., et al. (1996). S-adenosylmethionine and methylation. FASEB J. 10, 471–480.
Choi, J., Jang, H., Kim, H., Kim, S. T., Cho, E. J., and Youn, H. D. (2010). Histone demethylase LSD1 is required to induce skeletal muscle differentiation by regulating myogenic factors. Biochem. Biophys. Res. Commun. 401, 327–332. doi:10.1016/j.bbrc.2010.09.014
Christianson, J. P., Ragole, T., Amat, J., Greenwood, B.N., Strong, P. V., Paul, E. D., et al. (2010). 5-hydroxytryptamine 2C receptors in the basolateral amygdala are involved in the expression of anxiety after uncontrollable traumatic stress. Biol. Psychiatry 67, 339–345. doi:10.1016/j.biopsych.2009.09.011
Chwang, W. B., O’Riordan, K. J., Levenson, J. M., and Sweatt, J. D. (2006). ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn. Mem. 13, 322–328. doi:10.1101/lm.152906
Cohen, H., Benjamin, J., Geva, A. B., Matar, M. A., Kaplan, Z., and Kotler, M. (2000a). Autonomic dysregulation in panic disorder and in post-traumatic stress disorder: application of power spectrum analysis of heart rate variability at rest and in response to recollection of trauma or panic attacks. Psychiatry Res. 96, 1–13. doi:10.1016/S0165-1781(00)00195-5
Cohen, H., Benjamin, J., Kaplan, Z., and Kotler, M. (2000b). Administration of high-dose ketoconazole, an inhibitor of steroid synthesis, prevents posttraumatic anxiety in an animal model. Eur. Neuropsychopharmacol. 10, 429–435. doi:10.1016/S0924-977X(00)00105-X
Cohen, H., and Zohar, J. (2004). An animal model of posttraumatic stress disorder: the use of cut-off behavioral criteria. Ann. N. Y. Acad. Sci. 1032, 167–178. doi:10.1196/annals.1314.014
Cohen-Karni, D., Xu, D., Apone, L., Fomenkov, A., Sun, Z., Davis, P. J., et al. (2011). The MspJI family of modification-dependent restriction endonucleases for epigenetic studies. Proc. Natl. Acad. Sci. U.S.A. 108, 11040–11045. doi:10.1073/pnas.1018448108
Costa, E., Grayson, D. R., and Guidotti, A. (2003). Epigenetic downregulation of GABAergic function in schizophrenia: potential for pharmacological intervention? Mol. Interv. 3, 220–229. doi:10.1124/mi.3.4.220
Covington, H. E., III, Lobo, M. K., Maze, I., Vialou, V., Hyman, J. M., Zaman, S., et al. (2010). Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J. Neurosci. 30, 16082–16090. doi:10.1523/JNEUROSCI.1731-10.2010
Covington, H. E. III, Maze, I., Sun, H., Bomze, H. M., Demaio, K. D., Wu, E. Y., et al. (2011a). A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron 71, 656–670. doi:10.1016/j.neuron.2011.06.007
Covington, H. E. III, Vialou, V. F., Laplant, Q., Ohnishi, Y. N., and Nestler, E. J. (2011b). Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neurosci. Lett. 493, 122–126. doi:10.1016/j.neulet.2011.02.022
Cullinan, W. E., Herman, J. P., Battaglia, D. F., Akil, H., and Watson, S. J. (1995). Pattern and time course of immediate early gene expression in rat brain following acute stress. Neuroscience 64, 477–505. doi:10.1016/0306-4522(94)00355-9
Davis, M., Myers, K. M., Chhatwal, J., and Ressler, K. J. (2006a). Pharmacological treatments that facilitate extinction of fear: relevance to psychotherapy. NeuroRx 3, 82–96. doi:10.1016/j.nurx.2005.12.008
Davis, M., Ressler, K., Rothbaum, B. O., and Richardson, R. (2006b). Effects of D-cycloserine on extinction: translation from preclinical to clinical work. Biol. Psychiatry 60, 369–375. doi:10.1016/j.biopsych.2006.03.084
Davis, T., and Vaisvila, R. (2011). High sensitivity 5-hydroxymethylcytosine detection in Balb/C brain tissue. J. Vis. Exp. 48, e2661–e2663. doi:10.3791/2661
de Kloet, C. S., Vermetten, E., Bikker, A., Meulman, E., Geuze, E., Kavelaars, A., et al. (2007). Leukocyte glucocorticoid receptor expression and immunoregulation in veterans with and without post-traumatic stress disorder. Mol. Psychiatry 12, 443–453.
de Kloet, C. S., Vermetten, E., Geuze, E., Lentjes, E. G., Heijnen, C. J., Stalla, G. K., et al. (2008). Elevated plasma corticotrophin-releasing hormone levels in veterans with posttraumatic stress disorder. Prog. Brain Res. 167, 287–291. doi:10.1016/S0079-6123(07)67025-3
Deaton, A. M., and Bird, A. (2011). CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022. doi:10.1101/gad.2037511
Detich, N., Bovenzi, V., and Szyf, M. (2003). Valproate induces replication-independent active DNA demethylation. J. Biol. Chem. 278, 27586–27592. doi:10.1074/jbc.M303740200
Dias, B. G., and Ressler, K. J. (2013). PACAP and the PAC1 receptor in post-traumatic stress disorder. Neuropsychopharmacology 38, 245–246. doi:10.1038/npp.2012.147
Ding, H. K., Teixeira, C. M., and Frankland, P. W. (2008). Inactivation of the anterior cingulate cortex blocks expression of remote, but not recent, conditioned taste aversion memory. Learn. Mem. 15, 290–293. doi:10.1101/lm.905008
Dong, E., Guidotti, A., Grayson, D. R., and Costa, E. (2007). Histone hyperacetylation induces demethylation of reelin and 67-kDa glutamic acid decarboxylase promoters. Proc. Natl. Acad. Sci. U.S.A. 104, 4676–4681. doi:10.1073/pnas.0700529104
Drewell, R. A., Goddard, C. J., Thomas, J. O., and Surani, M. A. (2002). Methylation-dependent silencing at the H19 imprinting control region by MeCP2. Nucleic Acids Res. 30, 1139–1144. doi:10.1093/nar/30.5.1139
Dudley, K. J., Li, X., Kobor, M. S., Kippin, T. E., and Bredy, T. W. (2011). Epigenetic mechanisms mediating vulnerability and resilience to psychiatric disorders. Neurosci. Biobehav. Rev. 35, 1544–1551. doi:10.1016/j.neubiorev.2010.12.016
Dunsmoor, J. E., Mitroff, S. R., and Labar, K. S. (2009). Generalization of conditioned fear along a dimension of increasing fear intensity. Learn. Mem. 16, 460–469. doi:10.1101/lm.1431609
Ehlers, A., Hackmann, A., and Michael, T. (2004). Intrusive re-experiencing in post-traumatic stress disorder: phenomenology, theory, and therapy. Memory 12, 403–415. doi:10.1080/09658210444000025
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138. doi:10.1126/science.1162986
Elliott, E., Ezra-Nevo, G., Regev, L., Neufeld-Cohen, A., and Chen, A. (2010). Resilience to social stress coincides with functional DNA methylation of the Crf gene in adult mice. Nat. Neurosci. 13, 1351–1353. doi:10.1038/nn.2642
Feng, J., Zhou, Y., Campbell, S. L., Le, T., Li, E., Sweatt, J. D., et al. (2010). Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 13, 423–430. doi:10.1038/nn.2514
Fischer, A., Sananbenesi, F., Wang, X., Dobbin, M., and Tsai, L. H. (2007). Recovery of learning and memory is associated with chromatin remodelling. Nature 447, 178–182. doi:10.1038/nature05772
Fish, E. W., Shahrokh, D., Bagot, R., Caldji, C., Bredy, T., Szyf, M., et al. (2004). Epigenetic programming of stress responses through variations in maternal care. Ann. N. Y. Acad. Sci. 1036, 167–180. doi:10.1196/annals.1330.011
Flusberg, B. A., Webster, D. R., Lee, J. H., Travers, K. J., Olivares, E. C., Clark, T. A., et al. (2010). Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–465. doi:10.1038/nmeth.1459
Francis, D. D., Diorio, J., Plotsky, P. M., and Meaney, M. J. (2002). Environmental enrichment reverses the effects of maternal separation on stress reactivity. J. Neurosci. 22, 7840–7843.
Frankland, P. W., and Bontempi, B. (2005). The organization of recent and remote memories. Nat. Rev. Neurosci. 6, 119–130. doi:10.1038/nrn1607
Frankland, P. W., Bontempi, B., Talton, L. E., Kaczmarek, L., and Silva, A. J. (2004). The involvement of the anterior cingulate cortex in remote contextual fear memory. Science 304, 881–883. doi:10.1126/science.1094804
Friedman, M. J., Resick, P. A., Bryant, R. A., and Brewin, C. R. (2011a). Considering PTSD for DSM-5. Depress. Anxiety 28, 750–769. doi:10.1002/da.20767
Friedman, M. J., Resick, P. A., Bryant, R. A., Strain, J., Horowitz, M., and Spiegel, D. (2011b). Classification of trauma and stressor-related disorders in DSM-5. Depress. Anxiety 28, 737–749. doi:10.1002/da.20845
Fritz, E. L., and Papavasiliou, F. N. (2010). Cytidine deaminases: AIDing DNA demethylation? Genes Dev. 24, 2107–2114. doi:10.1101/gad.1963010
Geracioti, T. D. Jr., Baker, D. G., Ekhator, N. N., West, S. A., Hill, K. K., Bruce, A. B., et al. (2001). CSF norepinephrine concentrations in posttraumatic stress disorder. Am. J. Psychiatry 158, 1227–1230. doi:10.1176/appi.ajp.158.8.1227
Glaser, K. B., Staver, M. J., Waring, J. F., Stender, J., Ulrich, R. G., and Davidsen, S. K. (2003). Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol. Cancer Ther. 2, 151–163.
Globisch, D., Munzel, M., Muller, M., Michalakis, S., Wagner, M., Koch, S., et al. (2010). Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 5:e15367. doi:10.1371/journal.pone.0015367
Goswami, S., Samuel, S., Sierra, O. R., Cascardi, M., and Pare, D. (2012). A rat model of post-traumatic stress disorder reproduces the hippocampal deficits seen in the human syndrome. Front. Behav. Neurosci. 6:26. doi:10.3389/fnbeh.2012.00026
Graff, J., Kim, D., Dobbin, M. M., and Tsai, L. H. (2011). Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol. Rev. 91, 603–649. doi:10.1152/physrev.00012.2010
Graff, J., and Tsai, L. H. (2011). Cognitive enhancement: a molecular memory booster. Nature 469, 474–475. doi:10.1038/469474a
Graff, J., Woldemichael, B. T., Berchtold, D., Dewarrat, G., and Mansuy, I. M. (2012). Dynamic histone marks in the hippocampus and cortex facilitate memory consolidation. Nat. Commun. 3, 991. doi:10.1038/ncomms1997
Grayson, D. R., and Guidotti, A. (2013). The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology 38, 138–166. doi:10.1038/npp.2012.125
Grayson, D. R., Kundakovic, M., and Sharma, R. P. (2010). Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol. Pharmacol. 77, 126–135. doi:10.1124/mol.109.061333
Griffin, M. G. (2008). A prospective assessment of auditory startle alterations in rape and physical assault survivors. J. Trauma. Stress 21, 91–99. doi:10.1002/jts.20300
Guan, J. S., Haggarty, S. J., Giacometti, E., Dannenberg, J. H., Joseph, N., Gao, J., et al. (2009). HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60. doi:10.1038/nature07925
Guo, J. U., Ma, D. K., Mo, H., Ball, M. P., Jang, M. H., Bonaguidi, M. A., et al. (2011a). Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 14, 1345–1351. doi:10.1038/nn.2900
Guo, J. U., Su, Y., Zhong, C., Ming, G. L., and Song, H. (2011b). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434. doi:10.1016/j.cell.2011.03.022
Gupta, S., Kim, S. Y., Artis, S., Molfese, D. L., Schumacher, A., Sweatt, J. D., et al. (2010). Histone methylation regulates memory formation. J. Neurosci. 30, 3589–3599. doi:10.1523/JNEUROSCI.3732-09.2010
Gupta-Agarwal, S., Franklin, A. V., Deramus, T., Wheelock, M., Davis, R. L., McMahon, L. L., et al. (2012). G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. J. Neurosci. 32, 5440–5453. doi:10.1523/JNEUROSCI.0147-12.2012
Halsall, J., Gupta, V., O’Neill, L. P., Turner, B. M., and Nightingale, K. P. (2012). Genes are often sheltered from the global histone hyperacetylation induced by HDAC inhibitors. PLoS ONE 7:e33453. doi:10.1371/journal.pone.0033453
Hanson, J. E., Deng, L., Hackos, D. H., Lo, S. C., Lauffer, B. E., Steiner, P., et al. (2013). Histone deacetylase 2 cell autonomously suppresses excitatory and enhances inhibitory synaptic function in CA1 pyramidal neurons. J. Neurosci. 33, 5924–5929. doi:10.1523/JNEUROSCI.3162-12.2013
Hartley, C. A., McKenna, M. C., Salman, R., Holmes, A., Casey, B. J., Phelps, E. A., et al. (2012). Serotonin transporter polyadenylation polymorphism modulates the retention of fear extinction memory. Proc. Natl. Acad. Sci. U.S.A. 109, 5493–5498. doi:10.1073/pnas.1202044109
Hashimoto, H., Liu, Y., Upadhyay, A. K., Chang, Y., Howerton, S. B., Vertino, P. M., et al. (2012). Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 40, 4841–4849. doi:10.1093/nar/gks155
He, Y. F., Li, B. Z., Li, Z., Liu, P., Wang, Y., Tang, Q., et al. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. doi:10.1126/science.1210944
Hebbes, T. R., Thorne, A. W., and Crane-Robinson, C. (1988). A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. 7, 1395–1402.
Hellman, A., and Chess, A. (2007). Gene body-specific methylation on the active X chromosome. Science 315, 1141–1143. doi:10.1126/science.1136352
Huff, J. T., Plocik, A. M., Guthrie, C., and Yamamoto, K. R. (2010). Reciprocal intronic and exonic histone modification regions in humans. Nat. Struct. Mol. Biol. 17, 1495–1499. doi:10.1038/nsmb.1924
Iguchi-Ariga, S. M., and Schaffner, W. (1989). CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev. 3, 612–619. doi:10.1101/gad.3.5.612
Inoue, A., Shen, L., Dai, Q., He, C., and Zhang, Y. (2011). Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 21, 1670–1676. doi:10.1038/cr.2011.189
Iordanova, M. D., and Honey, R. C. (2012). Generalization of contextual fear as a function of familiarity: the role of within- and between-context associations. J. Exp. Psychol. Anim. Behav. Process. 38, 315–321. doi:10.1037/a0028689
Irizarry, R. A., Ladd-Acosta, C., Wen, B., Wu, Z., Montano, C., Onyango, P., et al. (2009). The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 41, 178–186. doi:10.1038/ng.298
Ito, S., Shen, L., Dai, Q., Wu, S. C., Collins, L. B., Swenberg, J. A., et al. (2011). Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303. doi:10.1126/science.1210597
Jensen Pena, C., Monk, C., and Champagne, F. A. (2012). Epigenetic effects of prenatal stress on 11beta-hydroxysteroid dehydrogenase-2 in the placenta and fetal brain. PLoS ONE 7:e39791. doi:10.1371/journal.pone.0039791
Jin, S. G., Wu, X., Li, A. X., and Pfeifer, G. P. (2011). Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 39, 5015–5024. doi:10.1093/nar/gkr120
Jolly, S., Journiac, N., Naudet, F., Gautheron, V., Mariani, J., and Vernet-Der Garabedian, B. (2011). Cell-autonomous and non-cell-autonomous neuroprotective functions of RORalpha in neurons and astrocytes during hypoxia. J. Neurosci. 31, 14314–14323. doi:10.1523/JNEUROSCI.1443-11.2011
Kangaspeska, S., Stride, B., Metivier, R., Polycarpou-Schwarz, M., Ibberson, D., Carmouche, R. P., et al. (2008). Transient cyclical methylation of promoter DNA. Nature 452, 112–115. doi:10.1038/nature06640
Karg, K., Burmeister, M., Shedden, K., and Sen, S. (2011). The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch. Gen. Psychiatry 68, 444–454. doi:10.1001/archgenpsychiatry.2010.189
Karymov, M. A., Tomschik, M., Leuba, S. H., Caiafa, P., and Zlatanova, J. (2001). DNA methylation-dependent chromatin fiber compaction in vivo and in vitro: requirement for linker histone. FASEB J. 15, 2631–2641. doi:10.1096/fj.01-0345com
Kessler, R. C., Sonnega, A., Bromet, E., Hughes, M., and Nelson, C. B. (1995). Posttraumatic stress disorder in the National Comorbidity Survey. Arch. Gen. Psychiatry 52, 1048–1060. doi:10.1001/archpsyc.1995.03950240066012
Kilpatrick, D. G., Koenen, K. C., Ruggiero, K. J., Acierno, R., Galea, S., Resnick, H. S., et al. (2007). The serotonin transporter genotype and social support and moderation of posttraumatic stress disorder and depression in hurricane-exposed adults. Am. J. Psychiatry 164, 1693–1699. doi:10.1176/appi.ajp.2007.06122007
Kim, D., Nguyen, M. D., Dobbin, M. M., Fischer, A., Sananbenesi, F., Rodgers, J. T., et al. (2007). SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 26, 3169–3179. doi:10.1038/sj.emboj.7601758
Kinnally, E. L., Feinberg, C., Kim, D., Ferguson, K., Leibel, R., Coplan, J. D., et al. (2011). DNA methylation as a risk factor in the effects of early life stress. Brain Behav. Immun. 25, 1548–1553. doi:10.1016/j.bbi.2011.05.001
Kinney, S. M., Chin, H. G., Vaisvila, R., Bitinaite, J., Zheng, Y., Esteve, P. O., et al. (2011). Tissue-specific distribution and dynamic changes of 5-hydroxymethylcytosine in mammalian genomes. J. Biol. Chem. 286, 24685–24693. doi:10.1074/jbc.M110.217083
Kino, T. (2007). Tissue glucocorticoid sensitivity: beyond stochastic regulation on the diverse actions of glucocorticoids. Horm. Metab. Res. 39, 420–424. doi:10.1055/s-2007-980193
Klengel, T., Mehta, D., Anacker, C., Rex-Haffner, M., Pruessner, J. C., Pariante, C. M., et al. (2013). Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat. Neurosci. 16, 33–41. doi:10.1038/nn.3275
Koenen, K. C., Aiello, A. E., Bakshis, E., Amstadter, A. B., Ruggiero, K. J., Acierno, R., et al. (2009). Modification of the association between serotonin transporter genotype and risk of posttraumatic stress disorder in adults by county-level social environment. Am. J. Epidemiol. 169, 704–711. doi:10.1093/aje/kwn397
Koenen, K. C., and Uddin, M. (2010). FKBP5 polymorphisms modify the effects of childhood trauma. Neuropsychopharmacology 35, 1623–1624. doi:10.1038/npp.2010.60
Koenen, K. C., Uddin, M., Chang, S. C., Aiello, A. E., Wildman, D. E., Goldmann, E., et al. (2011). SLC6A4 methylation modifies the effect of the number of traumatic events on risk for posttraumatic stress disorder. Depress. Anxiety 28, 639–647. doi:10.1002/da.20825
Koenigs, M., and Grafman, J. (2009). Posttraumatic stress disorder: the role of medial prefrontal cortex and amygdala. Neuroscientist 15, 540–548. doi:10.1177/1073858409333072
Koh, K. P., Yabuuchi, A., Rao, S., Huang, Y., Cunniff, K., Nardone, J., et al. (2011). Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 8, 200–213. doi:10.1016/j.stem.2011.01.008
Koponen, E., Rantamaki, T., Voikar, V., Saarelainen, T., Macdonald, E., and Castren, E. (2005). Enhanced BDNF signaling is associated with an antidepressant-like behavioral response and changes in brain monoamines. Cell Mol. Neurobiol. 25, 973–980. doi:10.1007/s10571-005-8468-z
Kotini, A. G., Mpakali, A., and Agalioti, T. (2011). Dnmt3a1 upregulates transcription of distinct genes and targets chromosomal gene clusters for epigenetic silencing in mouse embryonic stem cells. Mol. Cell. Biol. 31, 1577–1592. doi:10.1128/MCB.01093-10
Kremen, W. S., Koenen, K. C., Afari, N., and Lyons, M. J. (2012). Twin studies of posttraumatic stress disorder: differentiating vulnerability factors from sequelae. Neuropharmacology 62, 647–653. doi:10.1016/j.neuropharm.2011.03.012
Kriaucionis, S., and Heintz, N. (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930. doi:10.1126/science.1169786
Labonte, B., Suderman, M., Maussion, G., Navaro, L., Yerko, V., Mahar, I., et al. (2012). Genome-wide epigenetic regulation by early-life trauma. Arch. Gen. Psychiatry 69, 722–731. doi:10.1001/archgenpsychiatry.2011.2287
Lam, L. L., Emberly, E., Fraser, H. B., Neumann, S. M., Chen, E., Miller, G. E., et al. (2012). Factors underlying variable DNA methylation in a human community cohort. Proc. Natl. Acad. Sci. U.S.A. 109(Suppl. 2), 17253–17260. doi:10.1073/pnas.1121249109
Lattal, K. M., Barrett, R. M., and Wood, M. A. (2007). Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav. Neurosci. 121, 1125–1131. doi:10.1037/0735-7044.121.5.1125
Lattal, K. M., and Wood, M. A. (2013). Epigenetics and persistent memory: implications for reconsolidation and silent extinction beyond the zero. Nat. Neurosci. 16, 124–129. doi:10.1038/nn.3302
Lehmann, M. L., and Herkenham, M. (2011). Environmental enrichment confers stress resiliency to social defeat through an infralimbic cortex-dependent neuroanatomical pathway. J. Neurosci. 31, 6159–6173. doi:10.1523/JNEUROSCI.0577-11.2011
Lesburgueres, E., Gobbo, O. L., Alaux-Cantin, S., Hambucken, A., Trifilieff, P., and Bontempi, B. (2011). Early tagging of cortical networks is required for the formation of enduring associative memory. Science 331, 924–928. doi:10.1126/science.1196164
Levenson, J. M., O’Riordan, K. J., Brown, K. D., Trinh, M. A., Molfese, D. L., and Sweatt, J. D. (2004). Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 279, 40545–40559. doi:10.1074/jbc.M402229200
Levenson, J. M., Roth, T. L., Lubin, F. D., Miller, C. A., Huang, I. C., Desai, P., et al. (2006). Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 281, 15763–15773. doi:10.1074/jbc.M511767200
Levenson, J. M., and Sweatt, J. D. (2005). Epigenetic mechanisms in memory formation. Nat. Rev. Neurosci. 6, 108–118. doi:10.1038/nrn1604
Levenson, J. M., and Sweatt, J. D. (2006). Epigenetic mechanisms: a common theme in vertebrate and invertebrate memory formation. Cell. Mol. Life Sci. 63, 1009–1016. doi:10.1007/s00018-006-6026-6
Li, W., and Liu, M. (2011). Distribution of 5-hydroxymethylcytosine in different human tissues. J. Nucleic Acids 2011, 870726. doi:10.4061/2011/870726
Lian, C. G., Xu, Y., Ceol, C., Wu, F., Larson, A., Dresser, K., et al. (2012). Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 150, 1135–1146. doi:10.1016/j.cell.2012.07.033
Lieb, J. D., and Clarke, N. D. (2005). Control of transcription through intragenic patterns of nucleosome composition. Cell 123, 1187–1190. doi:10.1016/j.cell.2005.12.010
Lin, J. M., Collins, P. J., Trinklein, N. D., Fu, Y., Xi, H., Myers, R. M., et al. (2007). Transcription factor binding and modified histones in human bidirectional promoters. Genome Res. 17, 818–827. doi:10.1101/gr.5623407
Liu, X., Noll, D. M., Lieb, J. D., and Clarke, N. D. (2005). DIP-chip: rapid and accurate determination of DNA-binding specificity. Genome Res. 15, 421–427. doi:10.1101/gr.3256505
Logue, M. W., Baldwin, C., Guffanti, G., Melista, E., Wolf, E. J., Reardon, A. F., et al. (2012). A genome-wide association study of post-traumatic stress disorder identifies the retinoid-related orphan receptor alpha (RORA) gene as a significant risk locus. Mol. Psychiatry 17, 1–6. doi:10.1038/mp.2012.113
Lorincz, M. C., Dickerson, D. R., Schmitt, M., and Groudine, M. (2004). Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Mol. Biol. 11, 1068–1075. doi:10.1038/nsmb840
Lubin, F. D., Roth, T. L., and Sweatt, J. D. (2008). Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 28, 10576–10586. doi:10.1523/JNEUROSCI.1786-08.2008
Lubin, F. D., and Sweatt, J. D. (2007). The IkappaB kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron 55, 942–957. doi:10.1016/j.neuron.2007.07.039
Luine, V. (2002). Sex differences in chronic stress effects on memory in rats. Stress 5, 205–216. doi:10.1080/1025389021000010549
Ma, D. K., Guo, J. U., Ming, G. L., and Song, H. (2009a). DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation. Cell Cycle 8, 1526–1531. doi:10.4161/cc.8.10.8500
Ma, D. K., Jang, M. H., Guo, J. U., Kitabatake, Y., Chang, M. L., Pow-Anpongkul, N., et al. (2009b). Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 323, 1074–1077. doi:10.1126/science.1166859
Maddox, S. A., and Schafe, G. E. (2011). Epigenetic alterations in the lateral amygdala are required for reconsolidation of a Pavlovian fear memory. Learn. Mem. 18, 579–593. doi:10.1101/lm.2243411
Maddox, S. A., Watts, C. S., and Schafe, G. E. (2013). p300/CBP histone acetyltransferase activity is required for newly acquired and reactivated fear memories in the lateral amygdala. Learn. Mem. 20, 109–119. doi:10.1101/lm.029157.112
Mahan, A. L., Mou, L., Shah, N., Hu, J. H., Worley, P. F., and Ressler, K. J. (2012). Epigenetic modulation of Homer1a transcription regulation in amygdala and hippocampus with pavlovian fear conditioning. J. Neurosci. 32, 4651–4659. doi:10.1523/JNEUROSCI.3308-11.2012
Malvaez, M., Sanchis-Segura, C., Vo, D., Lattal, K. M., and Wood, M. A. (2010). Modulation of chromatin modification facilitates extinction of cocaine-induced conditioned place preference. Biol. Psychiatry 67, 36–43. doi:10.1016/j.biopsych.2009.07.032
Marek, R., Coelho, C. M., Sullivan, R. K., Baker-Andresen, D., Li, X., Ratnu, V., et al. (2011). Paradoxical enhancement of fear extinction memory and synaptic plasticity by inhibition of the histone acetyltransferase p300. J. Neurosci. 31, 7486–7491. doi:10.1523/JNEUROSCI.0133-11.2011
Maunakea, A. K., Nagarajan, R. P., Bilenky, M., Ballinger, T. J., D’souza, C., Fouse, S. D., et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257. doi:10.1038/nature09165
McCormick, C. M., and Mathews, I. Z. (2010). Adolescent development, hypothalamic-pituitary-adrenal function, and programming of adult learning and memory. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 756–765. doi:10.1016/j.pnpbp.2009.09.019
McGowan, P. O., Sasaki, A., D’Alessio, A. C., Dymov, S., Labonte, B., Szyf, M., et al. (2009). Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 12, 342–348. doi:10.1038/nn.2270
McKenzie, S., and Eichenbaum, H. (2011). Consolidation and reconsolidation: two lives of memories? Neuron 71, 224–233. doi:10.1016/j.neuron.2011.06.037
McQuown, S. C., Barrett, R. M., Matheos, D. P., Post, R. J., Rogge, G. A., Alenghat, T., et al. (2011). HDAC3 is a critical negative regulator of long-term memory formation. J. Neurosci. 31, 764–774. doi:10.1523/JNEUROSCI.5052-10.2011
McQuown, S. C., and Wood, M. A. (2011). HDAC3 and the molecular brake pad hypothesis. Neurobiol. Learn. Mem. 96, 27–34. doi:10.1016/j.nlm.2011.04.002
Meaney, M. J. (2001). Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu. Rev. Neurosci. 24, 1161–1192. doi:10.1146/annurev.neuro.24.1.1161
Mellen, M., Ayata, P., Dewell, S., Kriaucionis, S., and Heintz, N. (2012). MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151, 1417–1430. doi:10.1016/j.cell.2012.11.022
Mellman, T. A., Alim, T., Brown, D. D., Gorodetsky, E., Buzas, B., Lawson, W. B., et al. (2009). Serotonin polymorphisms and posttraumatic stress disorder in a trauma exposed African American population. Depress. Anxiety 26, 993–997. doi:10.1002/da.20627
Metivier, R., Gallais, R., Tiffoche, C., Le Peron, C., Jurkowska, R. Z., Carmouche, R. P., et al. (2008). Cyclical DNA methylation of a transcriptionally active promoter. Nature 452, 45–50. doi:10.1038/nature06544
Miller, C. A., Campbell, S. L., and Sweatt, J. D. (2008). DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol. Learn. Mem. 89, 599–603. doi:10.1016/j.nlm.2007.07.016
Miller, C. A., Gavin, C. F., White, J. A., Parrish, R. R., Honasoge, A., Yancey, C. R., et al. (2010). Cortical DNA methylation maintains remote memory. Nat. Neurosci. 13, 664–666. doi:10.1038/nn.2560
Miller, C. A., and Sweatt, J. D. (2007). Covalent modification of DNA regulates memory formation. Neuron 53, 857–869. doi:10.1016/j.neuron.2007.02.022
Monsey, M. S., Ota, K. T., Akingbade, I. F., Hong, E. S., and Schafe, G. E. (2011). Epigenetic alterations are critical for fear memory consolidation and synaptic plasticity in the lateral amygdala. PLoS ONE 6:e19958. doi:10.1371/journal.pone.0019958
Morgan, C. A. III, Rasmusson, A. M., Wang, S., Hoyt, G., Hauger, R. L., and Hazlett, G. (2002). Neuropeptide-Y, cortisol, and subjective distress in humans exposed to acute stress: replication and extension of previous report. Biol. Psychiatry 52, 136–142. doi:10.1016/S0006-3223(02)01319-7
Morgan, C. A. III, Wang, S., Southwick, S. M., Rasmusson, A., Hazlett, G., Hauger, R. L., et al. (2000). Plasma neuropeptide-Y concentrations in humans exposed to military survival training. Biol. Psychiatry 47, 902–909. doi:10.1016/S0006-3223(99)00239-5
Mueller, B. R., and Bale, T. L. (2008). Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci. 28, 9055–9065. doi:10.1523/JNEUROSCI.1424-08.2008
Mujtaba, S., Zeng, L., and Zhou, M. M. (2007). Structure and acetyl-lysine recognition of the bromodomain. Oncogene 26, 5521–5527. doi:10.1038/sj.onc.1210618
Mulchahey, J. J., Ekhator, N. N., Zhang, H., Kasckow, J. W., Baker, D. G., and Geracioti, T. D. Jr. (2001). Cerebrospinal fluid and plasma testosterone levels in post-traumatic stress disorder and tobacco dependence. Psychoneuroendocrinology 26, 273–285. doi:10.1016/S0306-4530(00)00052-4
Munzel, M., Globisch, D., Bruckl, T., Wagner, M., Welzmiller, V., Michalakis, S., et al. (2010). Quantification of the sixth DNA base hydroxymethylcytosine in the brain. Angew. Chem. Int. Ed. Engl. 49, 5375–5377. doi:10.1002/anie.201002033
Muotri, A. R., Marchetto, M. C., Coufal, N. G., Oefner, R., Yeo, G., Nakashima, K., et al. (2010). L1 retrotransposition in neurons is modulated by MeCP2. Nature 468, 443–446. doi:10.1038/nature09544
Nakayama, J., Rice, J. C., Strahl, B. D., Allis, C. D., and Grewal, S. I. (2001). Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292, 110–113. doi:10.1126/science.1060118
Narayan, P., and Dragunow, M. (2010). Pharmacology of epigenetics in brain disorders. Br. J. Pharmacol. 159, 285–303. doi:10.1111/j.1476-5381.2009.00526.x
Narayanan, V., Heiming, R. S., Jansen, F., Lesting, J., Sachser, N., Pape, H. C., et al. (2011). Social defeat: impact on fear extinction and amygdala-prefrontal cortical theta synchrony in 5-HTT deficient mice. PLoS ONE 6:e22600. doi:10.1371/journal.pone.0022600
Niehrs, C., and Schafer, A. (2012). Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol. 22, 220–227. doi:10.1016/j.tcb.2012.01.002
Norrholm, S. D., Jovanovic, T., Smith, A. K., Binder, E., Klengel, T., Conneely, K., et al. (2013). Differential genetic and epigenetic regulation of catechol-O-methyltransferase is associated with impaired fear inhibition in posttraumatic stress disorder. Front. Behav. Neurosci. 7:30. doi:10.3389/fnbeh.2013.00030
Novak, M. A., Hamel, A. F., Kelly, B. J., Dettmer, A. M., and Meyer, J. S. (2013). Stress, the HPA axis, and nonhuman primate well-being: a review. Appl. Anim. Behav. Sci. 143, 135–149. doi:10.1016/j.applanim.2012.10.012
Oberlander, T. F., Weinberg, J., Papsdorf, M., Grunau, R., Misri, S., and Devlin, A. M. (2008). Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 3, 97–106. doi:10.4161/epi.3.2.6034
Oh, J. E., Chambwe, N., Klein, S., Gal, J., Andrews, S., Gleason, G., et al. (2013). Differential gene body methylation and reduced expression of cell adhesion and neurotransmitter receptor genes in adverse maternal environment. Transl. Psychiatry 3, e218. doi:10.1038/tp.2012.130
Ooi, S. K., Qiu, C., Bernstein, E., Li, K., Jia, D., Yang, Z., et al. (2007). DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717. doi:10.1038/nature05987
Orr, S. P., Pitman, R. K., Lasko, N. B., and Herz, L. R. (1993). Psychophysiological assessment of posttraumatic stress disorder imagery in World War II and Korean combat veterans. J. Abnorm. Psychol. 102, 152–159. doi:10.1037/0021-843X.102.1.152
Ouellet-Morin, I., Wong, C. C., Danese, A., Pariante, C. M., Papadopoulos, A. S., Mill, J., et al. (2012). Increased serotonin transporter gene (SERT) DNA methylation is associated with bullying victimization and blunted cortisol response to stress in childhood: a longitudinal study of discordant monozygotic twins. Psychol. Med. 10, 1–11. doi:10.1017/S0033291712002784
Pastor, W. A., Pape, U. J., Huang, Y., Henderson, H. R., Lister, R., Ko, M., et al. (2011). Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 473, 394–397. doi:10.1038/nature10102
Peleg, S., Sananbenesi, F., Zovoilis, A., Burkhardt, S., Bahari-Javan, S., Agis-Balboa, R. C., et al. (2010). Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 328, 753–756. doi:10.1126/science.1186088
Peserico, A., and Simone, C. (2011). Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011, 371832. doi:10.1155/2011/371832
Peters, A. H., and Schubeler, D. (2005). Methylation of histones: playing memory with DNA. Curr. Opin. Cell Biol. 17, 230–238. doi:10.1016/j.ceb.2005.04.013
Pitman, J. L., Huetteroth, W., Burke, C. J., Krashes, M. J., Lai, S. L., Lee, T., et al. (2011). A pair of inhibitory neurons are required to sustain labile memory in the Drosophila mushroom body. Curr. Biol. 21, 855–861. doi:10.1016/j.cub.2011.03.069
Pitman, R. K. (2011). Will reconsolidation blockade offer a novel treatment for posttraumatic stress disorder? Front. Behav. Neurosci. 5:11. doi:10.3389/fnbeh.2011.00011
Pope, H. G. Jr., Cohane, G. H., Kanayama, G., Siegel, A. J., and Hudson, J. I. (2003). Testosterone gel supplementation for men with refractory depression: a randomized, placebo-controlled trial. Am. J. Psychiatry 160, 105–111. doi:10.1176/appi.ajp.160.1.105
Qian, W., Miki, D., Zhang, H., Liu, Y., Zhang, X., Tang, K., et al. (2012). A histone acetyltransferase regulates active DNA demethylation in Arabidopsis. Science 336, 1445–1448. doi:10.1126/science.1219416
Quina, A. S., Buschbeck, M., and Di Croce, L. (2006). Chromatin structure and epigenetics. Biochem. Pharmacol. 72, 1563–1569. doi:10.1016/j.bcp.2006.06.016
Quirk, G. J., Russo, G. K., Barron, J. L., and Lebron, K. (2000). The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J. Neurosci. 20, 6225–6231.
Rainnie, D. G. (1999). Serotonergic modulation of neurotransmission in the rat basolateral amygdala. J. Neurophysiol. 82, 69–85.
Resick, P. A., and Miller, M. W. (2009). Posttraumatic stress disorder: anxiety or traumatic stress disorder? J. Trauma Stress 22, 384–390. doi:10.1002/jts.20437
Ressler, K. J., Mercer, K. B., Bradley, B., Jovanovic, T., Mahan, A., Kerley, K., et al. (2011). Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature 470, 492–497. doi:10.1038/nature09856
Richter-Levin, G. (1998). Acute and long-term behavioral correlates of underwater trauma – potential relevance to stress and post-stress syndromes. Psychiatry Res. 79, 73–83. doi:10.1016/S0165-1781(98)00030-4
Ricon, T., Toth, E., Leshem, M., Braun, K., and Richter-Levin, G. (2012). Unpredictable chronic stress in juvenile or adult rats has opposite effects, respectively, promoting and impairing resilience. Stress 15, 11–20. doi:10.3109/10253890.2011.572207
Robertson, A. B., Dahl, J. A., Vagbo, C. B., Tripathi, P., Krokan, H. E., and Klungland, A. (2011). A novel method for the efficient and selective identification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 39, e55. doi:10.1093/nar/gkr051
Rohleder, N., Joksimovic, L., Wolf, J. M., and Kirschbaum, C. (2004). Hypocortisolism and increased glucocorticoid sensitivity of pro-Inflammatory cytokine production in Bosnian war refugees with posttraumatic stress disorder. Biol. Psychiatry 55, 745–751. doi:10.1016/j.biopsych.2003.11.018
Rollins, R. A., Haghighi, F., Edwards, J. R., Das, R., Zhang, M. Q., Ju, J., et al. (2006). Large-scale structure of genomic methylation patterns. Genome Res. 16, 157–163. doi:10.1101/gr.4362006
Roozendaal, B., Hernandez, A., Cabrera, S. M., Hagewoud, R., Malvaez, M., Stefanko, D. P., et al. (2010). Membrane-associated glucocorticoid activity is necessary for modulation of long-term memory via chromatin modification. J. Neurosci. 30, 5037–5046. doi:10.1523/JNEUROSCI.5717-09.2010
Roth, T. L., and Sweatt, J. D. (2011). Epigenetic marking of the BDNF gene by early-life adverse experiences. Horm. Behav. 59, 315–320. doi:10.1016/j.yhbeh.2010.05.005
Roth, T. L., Zoladz, P. R., Sweatt, J. D., and Diamond, D. M. (2011). Epigenetic modification of hippocampal Bdnf DNA in adult rats in an animal model of post-traumatic stress disorder. J. Psychiatr. Res. 45, 919–926. doi:10.1016/j.jpsychires.2011.01.013
Rusiecki, J. A., Chen, L., Srikantan, V., Zhang, L., Yan, L., Polin, M. L., et al. (2012). DNA methylation in repetitive elements and post-traumatic stress disorder: a case-control study of US military service members. Epigenomics 4, 29–40. doi:10.2217/epi.11.116
Russo, S. J., Murrough, J. W., Han, M. H., Charney, D. S., and Nestler, E. J. (2012). Neurobiology of resilience. Nat. Neurosci. 15, 1475–1484. doi:10.1038/nn.3234
Santos, K. F., Mazzola, T. N., and Carvalho, H. F. (2005). The prima donna of epigenetics: the regulation of gene expression by DNA methylation. Braz. J. Med. Biol. Res. 38, 1531–1541. doi:10.1590/S0100-879X2005001000010
Scharf, S. H., and Schmidt, M. V. (2012). Animal models of stress vulnerability and resilience in translational research. Curr. Psychiatry Rep. 14, 159–165. doi:10.1007/s11920-012-0256-0
Schnurr, P. P., and Friedman, M. J. (2008). Treatments for PTSD: understanding the evidence. PTSD Res. Q. 19, 1–6.
Schroeder, F. A., Lin, C. L., Crusio, W. E., and Akbarian, S. (2007). Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol. Psychiatry 62, 55–64. doi:10.1016/j.biopsych.2006.06.036
Shalev, A. Y., Peri, T., Brandes, D., Freedman, S., Orr, S. P., and Pitman, R. K. (2000). Auditory startle response in trauma survivors with posttraumatic stress disorder: a prospective study. Am. J. Psychiatry 157, 255–261. doi:10.1176/appi.ajp.157.2.255
Shenker, N., and Flanagan, J. M. (2012). Intragenic DNA methylation: implications of this epigenetic mechanism for cancer research. Br. J. Cancer 106, 248–253. doi:10.1038/bjc.2011.550
Shukla, S., Kavak, E., Gregory, M., Imashimizu, M., Shutinoski, B., Kashlev, M., et al. (2011). CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479, 74–79. doi:10.1038/nature10442
Siegmund, A., and Wotjak, C. T. (2007). A mouse model of posttraumatic stress disorder that distinguishes between conditioned and sensitised fear. J. Psychiatr. Res. 41, 848–860. doi:10.1016/j.jpsychires.2006.07.017
Simonini, M. V., Camargo, L. M., Dong, E., Maloku, E., Veldic, M., Costa, E., et al. (2006). The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 103, 1587–1592. doi:10.1073/pnas.0510341103
Skelton, K., Ressler, K. J., Norrholm, S. D., Jovanovic, T., and Bradley-Davino, B. (2012). PTSD and gene variants: new pathways and new thinking. Neuropharmacology 62, 628–637. doi:10.1016/j.neuropharm.2011.02.013
Smith, A. K., Conneely, K. N., Kilaru, V., Mercer, K. B., Weiss, T. E., Bradley, B., et al. (2011). Differential immune system DNA methylation and cytokine regulation in post-traumatic stress disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156B, 700–708. doi:10.1002/ajmg.b.31212
Song, C.-X., Clark, T. A., Lu, X.-Y., Kislyuk, A., Dai, Q., Turner, S. W., et al. (2012). Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nat. Methods 9, 75–77. doi:10.1038/nmeth.1779
Song, C. X., Szulwach, K. E., Fu, Y., Dai, Q., Yi, C., Li, X., et al. (2011a). Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 29, 68–72. doi:10.1038/nbt.1732
Song, C. X., Yu, M., Dai, Q., and He, C. (2011b). Detection of 5-hydroxymethylcytosine in a combined glycosylation restriction analysis (CGRA) using restriction enzyme Taq(alpha)I. Bioorg. Med. Chem. Lett. 21, 5075–5077. doi:10.1016/j.bmcl.2011.03.118
Sorrells, S. F., and Sapolsky, R. M. (2007). An inflammatory review of glucocorticoid actions in the CNS. Brain Behav. Immun. 21, 259–272. doi:10.1016/j.bbi.2006.11.006
Spruijt, C. G., Gnerlich, F., Smits, A. H., Pfaffeneder, T., Jansen, P. W., Bauer, C., et al. (2013). Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 152, 1146–1159. doi:10.1016/j.cell.2013.02.004
Stafford, J. M., Raybuck, J. D., Ryabinin, A. E., and Lattal, K. M. (2012). Increasing histone acetylation in the hippocampus-infralimbic network enhances fear extinction. Biol. Psychiatry 72, 25–33. doi:10.1016/j.biopsych.2011.12.012
Steckler, T., and Risbrough, V. (2012). Pharmacological treatment of PTSD – established and new approaches. Neuropharmacology 62, 617–627. doi:10.1016/j.neuropharm.2011.06.012
Stevenson, C. W., Spicer, C. H., Mason, R., and Marsden, C. A. (2009). Early life programming of fear conditioning and extinction in adult male rats. Behav. Brain Res. 205, 505–510. doi:10.1016/j.bbr.2009.08.005
Strahl, B. D., and Allis, C. D. (2000). The language of covalent histone modifications. Nature 403, 41–45. doi:10.1038/47412
Sultan, F. A., Wang, J., Tront, J., Liebermann, D. A., and Sweatt, J. D. (2012). Genetic deletion of Gadd45b, a regulator of active DNA demethylation, enhances long-term memory and synaptic plasticity. J. Neurosci. 32, 17059–17066. doi:10.1523/JNEUROSCI.1747-12.2012
Sun, H., Kennedy, P. J., and Nestler, E. J. (2013). Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology 38, 124–137. doi:10.1038/npp.2012.73
Sun, Y., Tao, Y. G., Kagan, B. L., He, Y., and Simons, S. S. Jr. (2008). Modulation of transcription parameters in glucocorticoid receptor-mediated repression. Mol. Cell Endocrinol. 295, 59–69. doi:10.1016/j.mce.2008.05.008
Sundermann, O., Hauschildt, M., and Ehlers, A. (2013). Perceptual processing during trauma, priming and the development of intrusive memories. J. Behav. Ther. Exp. Psychiatry. 44, 213–220. doi:10.1016/j.jbtep.2012.10.001
Sundermann, O., Onwumere, J., Bebbington, P., and Kuipers, E. (2012). Social networks and support in early psychosis: potential mechanisms. Epidemiol. Psychiatr. Sci. 22, 147–150. doi:10.1017/S2045796012000601
Suomi, S. J. (2011). Risk, resilience, and gene-environment interplay in primates. J. Can. Acad. Child Adolesc. Psychiatry 20, 289–297.
Szulwach, K. E., Li, X., Li, Y., Song, C. X., Wu, H., Dai, Q., et al. (2011). 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 14, 1607–1616. doi:10.1038/nn.2959
Szwagierczak, A., Brachmann, A., Schmidt, C. S., Bultmann, S., Leonhardt, H., and Spada, F. (2011). Characterization of PvuRts1I endonuclease as a tool to investigate genomic 5-hydroxymethylcytosine. Nucleic Acids Res. 39, 5149–5156. doi:10.1093/nar/gkr118
Szyf, M., Weaver, I., and Meaney, M. (2007). Maternal care, the epigenome and phenotypic differences in behavior. Reprod. Toxicol. 24, 9–19. doi:10.1016/j.reprotox.2007.05.001
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. doi:10.1126/science.1170116
Takei, S., Morinobu, S., Yamamoto, S., Fuchikami, M., Matsumoto, T., and Yamawaki, S. (2011). Enhanced hippocampal BDNF/TrkB signaling in response to fear conditioning in an animal model of posttraumatic stress disorder. J. Psychiatr. Res. 45, 460–468. doi:10.1016/j.jpsychires.2010.08.009
Terragni, J., Bitinaite, J., Zheng, Y., and Pradhan, S. (2012). Biochemical characterization of recombinant beta-glucosyltransferase and analysis of global 5-hydroxymethylcytosine in unique genomes. Biochemistry 51, 1009–1019. doi:10.1021/bi3008643
Thambirajah, A. A., Ng, M. K., Frehlick, L. J., Li, A., Serpa, J. J., Petrotchenko, E. V., et al. (2012). MeCP2 binds to nucleosome free (linker DNA) regions and to H3K9/H3K27 methylated nucleosomes in the brain. Nucleic Acids Res. 40, 2884–2897. doi:10.1093/nar/gkr1066
Tremolizzo, L., Carboni, G., Ruzicka, W. B., Mitchell, C. P., Sugaya, I., Tueting, P., et al. (2002). An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc. Natl. Acad. Sci. U.S.A. 99, 17095–17100. doi:10.1073/pnas.262658999
Tremolizzo, L., Doueiri, M. S., Dong, E., Grayson, D. R., Davis, J., Pinna, G., et al. (2005a). Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biol. Psychiatry 57, 500–509. doi:10.1016/j.biopsych.2004.11.046
Tremolizzo, L., Rodriguez-Menendez, V., Sala, G., Di Francesco, J. C., and Ferrarese, C. (2005b). Valproate and HDAC inhibition: a new epigenetic strategy to mitigate phenotypic severity in ALS? Amyotroph. Lateral Scler. Other Motor Neuron Disord. 6, 185–186. doi:10.1080/14660820510033614
Tsankova, N. M., Berton, O., Renthal, W., Kumar, A., Neve, R. L., and Nestler, E. J. (2006). Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 9, 519–525. doi:10.1038/nn1659
Tsankova, N. M., Kumar, A., and Nestler, E. J. (2004). Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J. Neurosci. 24, 5603–5610. doi:10.1523/JNEUROSCI.0589-04.2004
Turker, M. S. (1999). The establishment and maintenance of DNA methylation patterns in mouse somatic cells. Semin. Cancer Biol. 9, 329–337. doi:10.1006/scbi.1999.0133
Uchida, S., Hara, K., Kobayashi, A., Otsuki, K., Yamagata, H., Hobara, T., et al. (2011). Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron 69, 359–372. doi:10.1016/j.neuron.2010.12.023
Uddin, M., Aiello, A. E., Wildman, D. E., Koenen, K. C., Pawelec, G., De Los Santos, R., et al. (2010). Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc. Natl. Acad. Sci. U.S.A. 107, 9470–9475. doi:10.1073/pnas.0910794107
Uddin, M., Galea, S., Chang, S. C., Aiello, A. E., Wildman, D. E., De Los Santos, R., et al. (2011). Gene expression and methylation signatures of MAN2C1 are associated with PTSD. Dis. Markers 30, 111–121. doi:10.3233/DMA-2011-0750
Unternaehrer, E., Luers, P., Mill, J., Dempster, E., Meyer, A. H., Staehli, S., et al. (2012). Dynamic changes in DNA methylation of stress-associated genes (OXTR, BDNF) after acute psychosocial stress. Transl. Psychiatry 2, e150. doi:10.1038/tp.2012.77
van der Staay, F. J., Arndt, S. S., and Nordquist, R. E. (2009). Evaluation of animal models of neurobehavioral disorders. Behav. Brain Funct. 5, 11. doi:10.1186/1744-9081-5-11
Vastenhouw, N. L., and Schier, A. F. (2012). Bivalent histone modifications in early embryogenesis. Curr. Opin. Cell Biol. 24, 374–386. doi:10.1016/j.ceb.2012.03.009
Videlock, E. J., Peleg, T., Segman, R., Yehuda, R., Pitman, R. K., and Shalev, A. Y. (2008). Stress hormones and post-traumatic stress disorder in civilian trauma victims: a longitudinal study. Part II: the adrenergic response. Int. J. Neuropsychopharmacol. 11, 373–380. doi:10.1017/S1461145707008139
Wade, P. A. (2001). Methyl CpG binding proteins: coupling chromatin architecture to gene regulation. Oncogene 20, 3166–3173. doi:10.1038/sj.onc.1204340
Wang, H., Guan, S., Quimby, A., Cohen-Karni, D., Pradhan, S., Wilson, G., et al. (2011). Comparative characterization of the PvuRts1I family of restriction enzymes and their application in mapping genomic 5-hydroxymethylcytosine. Nucleic Acids Res. 39, 9294–9305. doi:10.1093/nar/gkr607
Wassef, A. A., Dott, S. G., Harris, A., Brown, A., O’boyle, M., Meyer, W. J. III, et al. (2000). Randomized, placebo-controlled pilot study of divalproex sodium in the treatment of acute exacerbations of chronic schizophrenia. J. Clin. Psychopharmacol. 20, 357–361. doi:10.1097/00004714-200006000-00011
Wassef, A. A., Hafiz, N. G., Hampton, D., and Molloy, M. (2001). Divalproex sodium augmentation of haloperidol in hospitalized patients with schizophrenia: clinical and economic implications. J. Clin. Psychopharmacol. 21, 21–26. doi:10.1097/00004714-200102000-00006
Weaver, I. C. (2009). Shaping adult phenotypes through early life environments. Birth Defects Res. C Embryo Today 87, 314–326. doi:10.1002/bdrc.20164
Weaver, I. C., Cervoni, N., Champagne, F. A., D’Alessio, A. C., Sharma, S., Seckl, J. R., et al. (2004a). Epigenetic programming by maternal behavior. Nat. Neurosci. 7, 847–854. doi:10.1038/nn1276
Weaver, I. C., Diorio, J., Seckl, J. R., Szyf, M., and Meaney, M. J. (2004b). Early environmental regulation of hippocampal glucocorticoid receptor gene expression: characterization of intracellular mediators and potential genomic target sites. Ann. N. Y. Acad. Sci. 1024, 182–212. doi:10.1196/annals.1321.099
Weaver, I. C., D’Alessio, A. C., Brown, S. E., Hellstrom, I. C., Dymov, S., Sharma, S., et al. (2007). The transcription factor nerve growth factor-inducible protein a mediates epigenetic programming: altering epigenetic marks by immediate-early genes. J. Neurosci. 27, 1756–1768. doi:10.1523/JNEUROSCI.4164-06.2007
Weaver, I. C., Meaney, M. J., and Szyf, M. (2006). Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc. Natl. Acad. Sci. U.S.A. 103, 3480–3485. doi:10.1073/pnas.0507526103
Weaver, I. C., Szyf, M., and Meaney, M. J. (2002). From maternal care to gene expression: DNA methylation and the maternal programming of stress responses. Endocr. Res. 28, 699. doi:10.1081/ERC-120016989
Wei, W., Coelho, C. M., Li, X., Marek, R., Yan, S., Anderson, S., et al. (2012). p300/CBP-associated factor selectively regulates the extinction of conditioned fear. J. Neurosci. 32, 11930–11941. doi:10.1523/JNEUROSCI.0178-12.2012
Wellman, C. L., Izquierdo, A., Garrett, J. E., Martin, K. P., Carroll, J., Millstein, R., et al. (2007). Impaired stress-coping and fear extinction and abnormal corticolimbic morphology in serotonin transporter knock-out mice. J. Neurosci. 27, 684–691. doi:10.1523/JNEUROSCI.4595-06.2007
Wilber, A. A., Southwood, C. J., Sokoloff, G., Steinmetz, J. E., and Wellman, C. L. (2007). Neonatal maternal separation alters adult eyeblink conditioning and glucocorticoid receptor expression in the interpositus nucleus of the cerebellum. Dev. Neurobiol. 67, 1751–1764. doi:10.1002/dneu.20549
Wilber, A. A., Southwood, C. J., and Wellman, C. L. (2009). Brief neonatal maternal separation alters extinction of conditioned fear and corticolimbic glucocorticoid and NMDA receptor expression in adult rats. Dev. Neurobiol. 69, 73–87. doi:10.1002/dneu.20691
Wilber, A. A., and Wellman, C. L. (2009). Neonatal maternal separation-induced changes in glucocorticoid receptor expression in posterior interpositus interneurons but not projection neurons predict deficits in adult eyeblink conditioning. Neurosci. Lett. 460, 214–218. doi:10.1016/j.neulet
Williams, K., Christensen, J., and Helin, K. (2012). DNA methylation: TET proteins-guardians of CpG islands? EMBO Rep. 13, 28–35. doi:10.1038/embor.2011.233
Witzmann, S. R., Turner, J. D., Meriaux, S. B., Meijer, O. C., and Muller, C. P. (2012). Epigenetic regulation of the glucocorticoid receptor promoter 1(7) in adult rats. Epigenetics 7, 1290–1301. doi:10.4161/epi.22363
Wu, G., Feder, A., Cohen, H., Kim, J. J., Calderon, S., Charney, D. S., et al. (2013). Understanding resilience. Front. Behav. Neurosci. 7:10. doi:10.3389/fnbeh.2013.00010
Wu, H., and Zhang, Y. (2011). Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 25, 2436–2452. doi:10.1101/gad.179184.111
Wu, S. C., and Zhang, Y. (2010). Active DNA demethylation: many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 11, 607–620. doi:10.1038/nrm2975
Xie, P., Kranzler, H. R., Poling, J., Stein, M. B., Anton, R. F., Brady, K., et al. (2009). Interactive effect of stressful life events and the serotonin transporter 5-HTTLPR genotype on posttraumatic stress disorder diagnosis in 2 independent populations. Arch. Gen. Psychiatry 66, 1201–1209. doi:10.1001/archgenpsychiatry.2009.153
Xie, P., Kranzler, H. R., Poling, J., Stein, M. B., Anton, R. F., Farrer, L. A., et al. (2010). Interaction of FKBP5 with childhood adversity on risk for post-traumatic stress disorder. Neuropsychopharmacology 35, 1684–1692. doi:10.1038/npp.2010.37
Xu, S. Y., Corvaglia, A. R., Chan, S. H., Zheng, Y., and Linder, P. (2011). A type IV modification-dependent restriction enzyme SauUSI from Staphylococcus aureus subsp. aureus USA300. Nucleic Acids Res. 39, 5597–5610. doi:10.1093/nar/gkr098
Yang, X., Ewald, E. R., Huo, Y., Tamashiro, K. L., Salvatori, R., Sawa, A., et al. (2012). Glucocorticoid-induced loss of DNA methylation in non-neuronal cells and potential involvement of DNMT1 in epigenetic regulation of Fkbp5. Biochem. Biophys. Res. Commun. 420, 570–575. doi:10.1016/j.bbrc.2012.03.035
Yehuda, R. (2002a). Clinical relevance of biologic findings in PTSD. Psychiatr. Q. 73, 123–133. doi:10.1023/A:1015055711424
Yehuda, R. (2002b). Current status of cortisol findings in post-traumatic stress disorder. Psychiatr. Clin. North Am. 25, 341–368. doi:10.1016/S0193-953X(02)00027-8
Yehuda, R., and Bierer, L. M. (2008). Transgenerational transmission of cortisol and PTSD risk. Prog. Brain Res. 167, 121–135. doi:10.1016/S0079-6123(07)67009-5
Yehuda, R., and Bierer, L. M. (2009). The relevance of epigenetics to PTSD: implications for the DSM-V. J. Trauma Stress 22, 427–434. doi:10.1002/jts.20448
Yehuda, R., Bierer, L. M., Sarapas, C., Makotkine, I., Andrew, R., and Seckl, J. R. (2009). Cortisol metabolic predictors of response to psychotherapy for symptoms of PTSD in survivors of the World Trade Center attacks on September 11, 2001. Psychoneuroendocrinology 34, 1304–1313. doi:10.1016/j.psyneuen.2009.03.018
Yehuda, R., Flory, J. D., Southwick, S., and Charney, D. S. (2006). Developing an agenda for translational studies of resilience and vulnerability following trauma exposure. Ann. N. Y. Acad. Sci. 1071, 379–396. doi:10.1196/annals.1364.028
Yehuda, R., Golier, J. A., Halligan, S. L., Meaney, M., and Bierer, L. M. (2004). The ACTH response to dexamethasone in PTSD. Am. J. Psychiatry 161, 1397–1403. doi:10.1176/appi.ajp.161.8.1397
Yehuda, R., Halligan, S. L., and Bierer, L. M. (2002). Cortisol levels in adult offspring of Holocaust survivors: relation to PTSD symptom severity in the parent and child. Psychoneuroendocrinology 27, 171–180. doi:10.1016/S0306-4530(01)00043-9
Yehuda, R., and LeDoux, J. (2007). Response variation following trauma: a translational neuroscience approach to understanding PTSD. Neuron 56, 19–32. doi:10.1016/j.neuron.2007.09.006
Yehuda, R., and McEwen, B. S. (2004). Protective and damaging effects of the biobehavioral stress response: cognitive, systemic and clinical aspects: ISPNE XXXIV meeting summary. Psychoneuroendocrinology 29, 1212–1222. doi:10.1016/j.psyneuen.2004.01.006
Yehuda, R., and McFarlane, A. C. (1995). Conflict between current knowledge about posttraumatic stress disorder and its original conceptual basis. Am. J. Psychiatry 152, 1705–1713.
Yehuda, R., McFarlane, A. C., and Shalev, A. Y. (1998). Predicting the development of posttraumatic stress disorder from the acute response to a traumatic event. Biol. Psychiatry 44, 1305–1313. doi:10.1016/S0006-3223(98)00276-5
Yu, M., Hon, G. C., Szulwach, K. E., Song, C. X., Zhang, L., Kim, A., et al. (2012). Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 149, 1368–1380. doi:10.1016/j.cell.2012.04.027
Zhang, T. Y., Bagot, R., Parent, C., Nesbitt, C., Bredy, T. W., Caldji, C., et al. (2006). Maternal programming of defensive responses through sustained effects on gene expression. Biol. Psychol. 73, 72–89. doi:10.1016/j.biopsycho.2006.01.009
Zheng, Y., Cohen-Karni, D., Xu, D., Chin, H. G., Wilson, G., Pradhan, S., et al. (2010). A unique family of Mrr-like modification-dependent restriction endonucleases. Nucleic Acids Res. 38, 5527–5534. doi:10.1093/nar/gkq327
Zilberman, D., and Henikoff, S. (2007). Genome-wide analysis of DNA methylation patterns. Development 134, 3959–3965. doi:10.1242/dev.001131
Zoladz, P. R., Conrad, C. D., Fleshner, M., and Diamond, D. M. (2008). Acute episodes of predator exposure in conjunction with chronic social instability as an animal model of post-traumatic stress disorder. Stress 11, 259–281. doi:10.1080/10253890701768613
Zovkic, I. B., Guzman-Karlsson, M. C., and Sweatt, J. D. (2013). Epigenetic regulation of memory formation and maintenance. Learn. Mem. 20, 61–74. doi:10.1101/lm.026575.112
Keywords: PTSD, epigenetics, stress, DNA methylation, chromatin, individual differences
Citation: Zovkic IB, Meadows JP, Kaas GA and Sweatt JD (2013) Interindividual variability in stress susceptibility: a role for epigenetic mechanisms in PTSD. Front. Psychiatry 4:60. doi: 10.3389/fpsyt.2013.00060
Received: 30 April 2013; Accepted: 11 June 2013;
Published online: 26 June 2013.
Edited by:
Tania L. Roth, University of Delaware, USAReviewed by:
Timothy Bredy, The University of Queensland, AustraliaHerb Lachman, Yeshiva University, USA
Copyright: © 2013 Zovkic, Meadows, Kaas and Sweatt. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: J. David Sweatt, Department of Neurobiology, Evelyn F. McKnight Brain Institute, University of Alabama at Birmingham, 1010 Shelby Building, 1825 University Boulevard, Birmingham, AL 35294-2182, USA e-mail:ZHN3ZWF0dEB1YWIuZWR1