 Kate Merritt
Kate Merritt Philip McGuire
Philip McGuire Alice Egerton
Alice Egerton- Department of Psychosis Studies, Institute of Psychiatry, King’s College London, London, UK
The glutamate hypothesis of schizophrenia, proposed over two decades ago, originated following the observation that administration of drugs that block NMDA glutamate receptors, such as ketamine, could induce schizophrenia-like symptoms. Since then, this hypothesis has been extended to describe how glutamate abnormalities may disturb brain function and underpin psychotic symptoms and cognitive impairments. The glutamatergic system is now a major focus for the development of new compounds in schizophrenia. Relationships between regional brain glutamate function and symptom severity can be investigated using proton magnetic resonance spectroscopy (1H-MRS) to estimate levels of glutamatergic metabolites in vivo. Here we briefly review the 1H-MRS studies that have explored relationships between glutamatergic metabolites, symptoms, and cognitive function in clinical samples. While some of these studies suggest that more severe symptoms may be associated with elevated glutamatergic function in the anterior cingulate, studies in larger patient samples selected on the basis of symptom severity are required.
Introduction
Accumulating evidence suggests that glutamatergic dysfunction may contribute to the pathogenesis of schizophrenia, and the symptoms and cognitive deficits associated with the disorder (1). The glutamatergic system presents an attractive therapeutic target, as dopaminergic antipsychotics have little effect on negative symptoms or cognitive impairment, yet these features are better predictors of social and functional outcome than positive symptoms (2, 3). Here we briefly review the existing evidence linking abnormal glutamatergic transmission to cognitive, negative, and positive symptoms of schizophrenia.
The observation that administration of antagonists at the N-methyl-d-aspartate glutamate receptor complex (NMDAR) such as phencyclidine (PCP) or ketamine to healthy volunteers induces effects which resemble aspects of schizophrenia symptomatology forms a cornerstone of the glutamate hypothesis of schizophrenia (4–6). NMDAR antagonists also worsen positive, negative, and cognitive symptoms in patients with schizophrenia (7, 8). While dopamine-stimulating drugs such as amphetamine also produce positive “psychotic”-like effects, negative-type symptoms, and cognitive deficits are far more prominently elicited by ketamine than amphetamine administration (9). Neuroimaging studies indicate that these effects of ketamine are mediated by changes in activity in the frontal and cingulate cortices and the thalamus (10–13).
While pharmacological studies have provided evidence linking NMDAR dysfunction to these symptom domains, direct associations between glutamatergic function and symptom severity may be provided by neuroimaging studies. In a single photon emission tomography study using the NMDAR radiotracer 123I-CNS-1261 in schizophrenia, the availability of NMDAR in the hippocampus was negatively associated with the severity of symptoms, especially negative symptoms (14). A ketamine challenge study in healthy volunteers using the same radiotracer linked regional NMDAR binding to the induction of negative (but not positive) symptoms, particularly in the thalamus (15). These approaches are currently limited by a lack of availability of suitable radiotracers (1).
An alternative is to use proton magnetic resonance spectroscopy (1H-MRS) to estimate the concentration of glutamatergic metabolites. MRI scanners with field strengths of 3 T or above can resolve glutamate, at least for the most part, from its metabolite glutamine (16). At lower field strengths glutamate and glutamine are reported in combination, as Glx. A limitation of 1H-MRS is that the glutamate concentration estimates are not specific to neuronal glutamate, and that changes in glutamate levels cannot be specifically attributed to altered neurotransmission over other metabolic processes (17). However the majority (∼80%) of glutamine synthesis reflects cycling of neurotransmitter glutamate (17), and clinical studies at 4 T in minimally treated patients with schizophrenia have reported higher Gln/Glu ratios (18) or higher glutamine levels (19) in the anterior cingulate cortex (ACC). This is consistent with increased glutamatergic neurotransmission, but could also result from a deficiency in the conversion of glutamine to glutamate. However the majority of 1H-MRS studies in psychosis have used field strengths <4 T and thereby were unable to accurately quantify glutamine concentrations. Thus, the results from these studies at lower field strengths cannot be specifically attributed to changes in glutamate neurotransmission. Nonetheless, increases in frontal glutamatergic neurotransmission in schizophrenia are broadly consistent with the NMDA receptor hypofunction hypothesis, as administration of NMDAR antagonists to rats increases glutamate release as detected by microdialysis (20), and 1H-MRS studies report elevated Gln/Glu ratios (21), or increased glutamate levels with no changes in glutamine (22). 1H-MRS studies with ketamine in man at 4 T reveal increased ACC glutamine (23), and at 3 T reveal increased ACC glutamate (24). One mechanism through which increases in glutamate release may occur is via NMDAR dysfunction on GABA-ergic interneurons, leading to the disinhibition of glutamatergic pyramidal cells, as described by the NMDAR hypofunction hypothesis (25).
Individual 1H-MRS glutamate studies in schizophrenia have produced some inconsistent findings. In general, studies at higher field strengths suggest that frontal glutamine function is elevated in the early stages of psychosis (18, 19), whereas the findings in chronic schizophrenia are more variable (26–30). This could reflect effects of antipsychotic medication. For example, one cross-sectional and one longitudinal study reported higher Glx levels in the PFC of unmedicated, but not medicated schizophrenia, in comparison to controls (31, 32), although longitudinal studies have reported no effect of medication on anterior cingulate glutamate or glutamine levels (18, 33, 34). It could be that symptom severity contributes to variability in glutamatergic metabolites between patients early or late in the illness, as samples involving patients in the early phase of psychosis usually comprise non- or minimally medicated patients who are relatively symptomatic, whereas studies in chronic schizophrenia often involve patients who have been treated for long periods and have less severe or more stable symptoms (1). In relation to this, a recent meta-analysis of 1H-MRS glutamatergic studies in major depressive disorder found lower levels of Glx in the ACC which were only significant when remitted patients were excluded from the analysis (35).
In order to better understand the possible relationships between 1H-MRS glutamate, glutamine, and Glx levels and symptom severity, we identified publications that have reported associations with severity along symptom domains. These included studies in individuals at risk of psychosis, and patients with first-episode psychosis or established schizophrenia (Table 1).
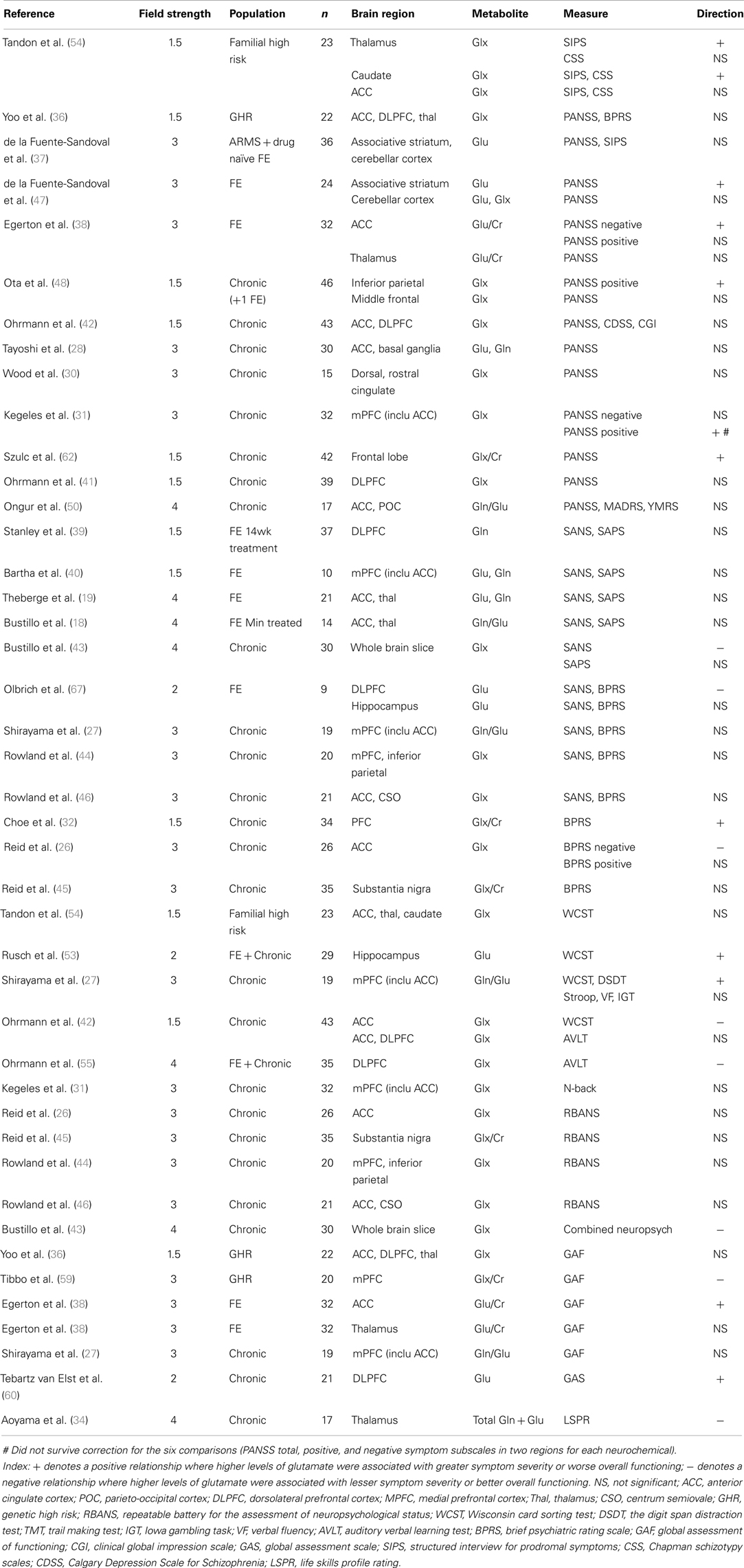
Table 1. Summary of articles reporting high levels of glutamate metabolites associated with greater or lesser severity of symptoms.
Relationships between Glutamate Function and Positive Symptoms
Several studies have investigated associations between regional glutamatergic metabolite levels and the severity of positive psychotic symptoms (Table 1), and most found no association between regional Glx, glutamate or glutamine levels and positive symptom severity, in either high genetic or clinical risk populations (36, 37), first-episode psychosis (18, 19, 37–40) or chronic schizophrenia (26–28, 30, 41–46).
However, many of these studies involved small patient samples, relied on post hoc correlational analyses, and patients in whom the severity and/or the variance in severity of symptoms was low, due to either being sub-clinical threshold in the at risk studies or due to the presence of antipsychotic medication in established schizophrenia. A recent study pooling both medicated and unmedicated patients, where unmedicated patients possessed elevated Glx in medial prefrontal cortex (mPFC), detected an association between positive symptom severity and mPFC Glx, although this did not survive correction for multiple comparisons (31). Another study in the PFC found that treatment reduced the level of Glx/Cr in chronic patients, and associated the change in Glx with improvement in total BPRS score (32). Moreover a recent study found that 4 weeks of antipsychotic treatment in first-episode psychosis patients reduced Glx in the striatum, and this was associated with improvement in PANSS score (47).
A notable exception is the study of Ota et al. (48), which directly compared patients experiencing exacerbated psychotic symptoms to healthy controls and stable patients, and found that increases in Glx in inferior parietal white matter were specific to the group currently experiencing exacerbated psychotic symptoms (48). In line with this, our own studies which have compared glutamate levels in patients according to symptom severity have found higher ACC glutamate levels in first-episode psychosis patients who are still symptomatic following treatment compared to those in remission (38) and in patients with treatment-resistant schizophrenia compared to those who respond to medication (49). These differences may be independent of medication effects, as groups either did not differ according to medication (38, 49) or the symptomatic group were actually receiving higher medication doses (48), and as longitudinal studies have not reported changes in cortical glutamate in relation to changes in positive symptoms following antipsychotic treatment (18, 33, 34). This suggests that glutamate and Glx levels may be selectively elevated in patients whose positive symptoms are not well controlled by conventional antipsychotic medication.
Relationships between Glutamate Function and Negative Symptoms
We recently reported that greater severity of PANSS negative symptoms was associated with higher levels of glutamate in the ACC in first-episode psychosis (38). Although there were several methodological differences, this contrasts with the study of Reid et al. (26), which reported that negative symptoms were associated with lower levels of ACC Glx in chronic schizophrenia. Other studies investigating correlations between ACC glutamatergic metabolites and negative symptoms have found no significant relationship (18, 19, 28, 42, 46, 50). We are not aware of any studies which have specifically compared regional levels of glutamatergic metabolites in patient groups selected according to differences in negative symptom severity.
As listed in Table 1, in brain regions other than the ACC many studies have failed to detect significant relationships between negative symptoms and regional glutamate, glutamine, or Glx levels in high genetic or clinical risk groups (36, 37), first-episode psychosis (18, 37, 39, 40), or chronic schizophrenia (27, 30, 31, 41, 44, 45, 48).
A general consideration of studies investigating relationships between glutamate markers and positive and negative symptoms is the scale used to score symptom severity. As detailed in Table 1, scores on a number of scales have been used and while these scales are highly correlated there are also some important differences in the clinical items included (51). As most studies examining relationships between brain glutamate measures and symptoms have relied on post hoc correlational analysis, it is of note that additional items are included on the SAPS/SANS compared to the PANSS, which in turn has additional items compared to the BPRS. Therefore use of the SAPS/SANS may be preferable as they provide a more detailed assessment of symptoms.
Relationships between Glutamate Measures and Cognitive Dysfunction
Relatively few studies have investigated relationships between glutamate measures and cognitive dysfunction in schizophrenia. The most commonly investigated task has been the Wisconsin card sort test (WCST): in schizophrenia, deficits on this task are associated with abnormal activation in the ACC and DLPFC (52). In a study of 19 patients with chronic schizophrenia at 3 T, poor performance on the WCST was associated with higher Gln/Glu ratios in the mPFC (including the ACC) (27). In contrast, a larger study of 43 patients with chronic schizophrenia at 1.5 T found that ACC, but not DLPFC, Glx levels were positively associated with WCST learning potential (42). In first-episode psychosis, an association between hippocampal, but not DLPFC, glutamate, and WCST errors was reported (53), and in a small sample of 16 genetic high risk individuals, no correlations between WCST performance and Glx levels in the caudate, ACC, or thalamus were detected (54).
Other tasks investigated include the Stroop, digit span distractibility test, auditory verbal learning test (AVLT), N-back task, Iowa gambling task and verbal fluency test (Table 1). Of these, there are reports of a positive association between mPFC Gln/Glu ratio and impairments on the digit span distraction test, which probes short-term memory and selective attention (27), and of DLPFC Glx and verbal learning and memory on the AVLT (55). Overall, the findings have been inconsistent and further studies with larger sample sizes are required. While the above studies used a voxel of interest method, using whole brain slice proton echo planar spectroscopy at 4 T in 30 patients Bustillo et al. (43) detected a positive correlation between general cognitive performance in schizophrenia and Glx. Furthermore, subsequent path analyses suggested that the relationship between glutamate and cognitive performance may be associated with negative symptoms and unemployment (43).
The relationship between glutamate and cognition in schizophrenia has been further investigated by combining 1H-MRS with functional magnetic resonance imaging (fMRI) of the blood oxygen level dependent (BOLD) response to measure changes in regional brain activation as participants perform cognitive tasks. In subjects at clinical high risk of developing psychosis, reductions in thalamic glutamate were correlated with an elevated BOLD response in the prefrontal cortex during a verbal fluency task, whereas the converse association was observed in controls (56). In a study by Valli et al. (57), medial temporal lobe glutamate levels were correlated with hippocampal BOLD response during an episodic memory task in controls, whereas this coupling was absent in clinical high risk subjects (57). Finally one study showed that hippocampal Glx was related to inferior frontal gyrus activation during episodic memory in controls, and suggested that the absence of this positive coupling in medicated schizophrenia patients may underlie episodic memory deficits, reflecting the results seen in Valli et al. (57, 58). Further investigation of the relationships between regional levels of glutamatergic metabolites and abnormalities in regional brain activation during cognitive tasks may provide a more sensitive means to characterize the relationship between glutamate and cognition in schizophrenia.
Relationships between Glutamate Measures and Social and Occupational Functioning
Several studies have also reported correlations between brain glutamate levels and overall level of social and occupational functioning (Table 1). In genetically high risk subjects, lower levels of Glx/Cr in the mPFC were associated with lower levels of overall functioning (59). A longitudinal study at 4 T showed that loss of glutamate and glutamine in the thalamus, but not the ACC, over 7 years since first presentation correlated with impaired social functioning (34). In contrast, in first-episode psychosis higher levels of glutamate in the ACC, but not the thalamus, were associated with worse overall functioning (38), and in chronic schizophrenia higher DLPFC glutamate levels were also associated with worse overall functioning (60). Other studies have found no association between glutamate measures and overall functioning in chronic schizophrenia (27) or genetic high risk subjects (36).
Summary and Future Directions
1H-MRS studies relating regional glutamate measures to symptoms in schizophrenia have produced inconsistent findings. There may be several methodological reasons for this, including differences in the brain region investigated, and between samples such as medication, illness stage, and symptom severity. The use of sub-optimal technical approaches such as low field strengths together with small sample sizes may also underlie the conflicting findings. A recent study at 4 T indicates that elevations in the ratio of Gln/Glu are present in schizophrenia patients, consistent with elevated glutamatergic neurotransmission (18). However the majority of 1H-MRS studies in schizophrenia use field strengths <4 T which cannot reliably measure glutamine, and glutamate and Glx measures cannot be specifically attributed to glutamate neurotransmission (17). The increasing availability of higher field strength scanners may resolve some of the apparent inconsistencies in the literature.
The majority of studies that have investigated relationships between glutamate levels and symptom severity have applied correlational analysis, usually post hoc to the main study findings. A few studies have directly compared glutamatergic metabolite levels in patient groups on the basis of symptom severity, specifically those who were or were not currently experiencing symptom exacerbation (48), were or were not in symptomatic remission following initial treatment (38) or did or did not have treatment-resistant illness (49). All found higher levels of glutamate or Glx in the more symptomatic patient group.
The field would benefit from further studies pre-selecting groups of patients who differ in severity of negative symptoms or cognitive impairment, or longitudinal studies comparing within-subjects glutamate levels during periods of illness stability compared to relapse. Studies of the relationships between cognitive dysfunction and glutamate in schizophrenia may benefit from combination with fMRI to determine the efficiency of glutamate in supporting networks that subserve cognitive function. To date there are only a handful of published studies of this type. Finally, relationships between glutamate levels and symptom severity may be indirect; for example Stone et al. showed that hippocampal glutamate may interact with striatal dopamine to determine risk of psychosis (61), and a path analysis has suggested that negative symptoms may be secondary to poor cognition associated with low brain Glx (43).
One question is whether baseline levels of glutamate, glutamine, or Glx are predictive of subsequent outcome or response to treatment in schizophrenia. A recent study showed that elevated frontal Glx at baseline was associated with poor response after 4 weeks of antipsychotic treatment (62). This is consistent with the above findings of glutamatergic elevations in patients with schizophrenia whose symptoms have not responded well to treatment (38, 49).
The suggestion that symptoms that do not respond well to conventional antipsychotic treatment may have a glutamatergic basis (38, 49) warrants further investigation, as compounds that target the glutamatergic system may have particular efficacy in these patients. Meta-analyses conclude that of the agents which may improve NMDA receptor-mediated neurotransmission, the NMDAR co-agonist d-serine and the glycine transporter type 1 inhibitor sarcosine reduce total and negative symptoms as an adjuvant to antipsychotic medication (63, 64). Lamotrigine, which may inhibit glutamate release, blocks the psychomimetic effects of ketamine in healthy volunteers (65) and small trials of lamotrigine in chronic, often treatment-resistant or clozapine-treated patients found that it is beneficial in reducing symptoms (63, 66). Further work is required to determine the relationship between regional glutamate concentrations and the expression of symptoms at different stages of psychotic illness. This area may benefit from meta-analyses of the previously published findings and from new studies selecting patient samples a priori on the basis of symptom severity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Egerton A, Stone JM. The glutamate hypothesis of schizophrenia: neuroimaging and drug development. Curr Pharm Biotechnol (2012) 13(8):1500–12. doi: 10.2174/138920112800784961
2. Ventura J, Hellemann GS, Thames AD, Koellner V, Nuechterlein KH. Symptoms as mediators of the relationship between neurocognition and functional outcome in schizophrenia: a meta-analysis. Schizophr Res (2009) 113(2-3):189–99. doi:10.1016/j.schres.2009.03.035
3. Javitt DC. Treatment of negative and cognitive symptoms. Curr Psychiatry Rep (1999) 1(1):25–30. doi:10.1007/s11920-999-0007-z
4. Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans – psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry (1994) 51(3):199–214. doi:10.1001/archpsyc.1994.03950030035004
5. Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, Hershey T, et al. Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology (1999) 20(2):106–18. doi:10.1016/s0893-133x(98)00067-0
6. Malhotra AK, Pinals DA, Weingartner H, Sirocco K, Missar CD, Pickar D, et al. NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology (1996) 14(5):301–7. doi:10.1016/0893-133x(95)00137-3
7. Malhotra AK, Pinals DA, Adler CM, Elman I, Clifton A, Pickar D, et al. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology (1997) 17(3):141–50. doi:10.1016/S0893-133X(97)00036-5
8. Lahti AC, Koffel B, Laporte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology (1995) 13(1):9–19. doi:10.1016/0893-133X(94)00131-I
9. Krystal JH, Perry EB, Gueorguieva R, Belger A, Madonich SH, Abi-Dargham A, et al. Comparative and interactive human psychopharmacologic effects of ketamine and amphetamine – implications for glutamatergic and dopaminergic model psychoses and cognitive function. Arch Gen Psychiatry (2005) 62(9):985–95. doi:10.1001/archpsyc.62.9.985
10. Holcomb HH, Lahti AC, Medoff DR, Cullen T, Tamminga CA. Effects of noncompetitive NMDA receptor blockade on anterior cingulate cerebral blood flow in volunteers with schizophrenia. Neuropsychopharmacology (2005) 30(12):2275–82. doi:10.1038/sj.npp.1300824
11. Deakin JFW, Lees J, McKie S, Hallak JEC, Williams SR, Dursun SM. Glutamate and the neural basis of the subjective effects of ketamine. Arch Gen Psychiatry (2008) 65(2):154–64. doi:10.1001/archgenpsychiatry.2007.37
12. De Simoni S, Schwarz AJ, O’Daly OG, Marquand AF, Brittain C, Gonzales C, et al. Test-retest reliability of the BOLD pharmacological MRI response to ketamine in healthy volunteers. Neuroimage (2013) 64:75–90. doi:10.1016/j.neuroimage.2012.09.037
13. Holcomb HH, Lahti AC, Medoff DR, Weiler M, Tamminga CA. Sequential regional cerebral blood flow brain scans using PET with (H2O)-O-15 demonstrate ketamine actions in CNS dynamically. Neuropsychopharmacology (2001) 25(2):165–72. doi:10.1016/s0893-133x(01)00229-9
14. Pilowsky LS, Bressan RA, Stone JM, Erlandsson K, Mulligan RS, Krystal JH, et al. First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol Psychiatry (2006) 11(2):118–9. doi:10.1038/sj.mp.4001751
15. Stone JM, Erlandsson K, Arstad E, Squassante L, Teneggi V, Bressan RA, et al. Relationship between ketamine-induced psychotic symptoms and NMDA receptor occupancy – a I-123 CNS-1261 SPET study. Psychopharmacology (2008) 197(3):401–8. doi:10.1007/s00213-007-1047-x
16. Snyder J, Wilman A. Field strength dependence of PRESS timings for simultaneous detection of glutamate and glutamine from 1.5 to 7 T. J Magn Reson (2010) 203(1):66–72. doi:10.1016/j.jmr.2009.12.002
17. Rothman DL, De Feyter HM, de Graaf RA, Mason GF, Behar KL. C-13 MRS studies of neuroenergetics and neurotransmitter cycling in humans. NMR Biomed (2011) 24(8):943–57. doi:10.1002/nbm.1772
18. Bustillo JR, Rowland LM, Mullins P, Jung R, Chen H, Qualls C, et al. H-1-MRS at 4 Tesla in minimally treated early schizophrenia. Mol Psychiatry (2010) 15(6):629–36. doi:10.1038/mp.2009.121
19. Theberge J, Bartha R, Drost DJ, Menon RS, Malla A, Takhar J, et al. Glutamate and glutamine measured with 4.0 T proton MRS in never-treated patients with schizophrenia and healthy volunteers. Am J Psychiatry (2002) 159(11):1944–6. doi:10.1176/appi.ajp.159.11.1944
20. Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci (1997) 17(8):2921–7.
21. Iltis I, Koski DM, Eberly LE, Nelson CD, Deelchand DK, Valette J, et al. Neurochemical changes in the rat prefrontal cortex following acute phencyclidine treatment: an in vivo localized H-1 MRS study. NMR Biomed (2009) 22(7):737–44. doi:10.1002/nbm.1385
22. Kim SY, Lee H, Kim HJ, Bang E, Lee SH, Lee DW, et al. In vivo and ex vivo evidence for ketamine-induced hyperglutamatergic activity in the cerebral cortex of the rat: potential relevance to schizophrenia. NMR Biomed (2011) 24(10):1235–42. doi:10.1002/nbm.1681
23. Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, et al. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry (2005) 162(2):394–6. doi:10.1176/appi.ajp.162.2.394
24. Stone JM, Dietrich C, Edden R, Mehta MA, De Simoni S, Reed LJ, et al. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry (2012) 17(7):664–5. doi:10.1038/mp.2011.171
25. Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex Interneurons and pyramidal neurons. J Neurosci (2007) 27(43):11496–500. doi:10.1523/jneurosci.2213-07.2007
26. Reid MA, Stoeckel LE, White DM, Avsar KB, Bolding MS, Akella NS, et al. Assessments of function and biochemistry of the anterior cingulate cortex in schizophrenia. Biol Psychiatry (2010) 68(7):625–33. doi:10.1016/j.biopsych.2010.04.013
27. Shirayama Y, Obata T, Matsuzawa D, Nonaka H, Kanazawa Y, Yoshitome E, et al. Specific metabolites in the medial prefrontal cortex are associated with the neurocognitive deficits in schizophrenia: a preliminary study. Neuroimage (2010) 49(3):2783–90. doi:10.1016/j.neuroimage.2009.10.031
28. Tayoshi S, Sumitani S, Taniguchi K, Shibuya-Tayoshi S, Numata S, Iga J, et al. Metabolite changes and gender differences in schizophrenia using 3-Tesla proton magnetic resonance spectroscopy (H-1-MRS). Schizophr Res (2009) 108(1-3):69–77. doi:10.1016/j.schres.2008.11.014
29. Theberge J, Al-Semaan Y, Williamson PC, Menon RS, Neufeld RWJ, Rajakumar N, et al. Glutamate and glutamine in the anterior cingulate and thalamus of medicated patients with chronic schizophrenia and healthy comparison subjects measured with 4.0-T proton MRS. Am J Psychiatry (2003) 160(12):2231–3. doi:10.1176/appi.ajp.160.12.2231
30. Wood SJ, Yucel M, Wellard RM, Harrison BJ, Clarke K, Fornito A, et al. Evidence for neuronal dysfunction in the anterior cingulate of patients with schizophrenia: a proton magnetic resonance spectroscopy study at 3 T. Schizophr Res (2007) 94(1-3):328–31. doi:10.1016/j.schres.2007.05.008
31. Kegeles LS, Mao XL, Stanford AD, Girgis R, Ojeil N, Xu XY, et al. Elevated prefrontal cortex gamma-aminobutyric acid and glutamate-glutamine levels in schizophrenia measured in vivo with proton magnetic resonance spectroscopy. Arch Gen Psychiatry (2012) 69(5):449–59. doi:10.1001/archgenpsychiatry.2011.1519
32. Choe BY, Kim KT, Suh TS, Lee C, Paik IH, Bahk YW, et al. 1H magnetic resonance spectroscopy characterization of neuronal dysfunction in drug-naive, chronic schizophrenia. Acad Radiol (1994) 1(3):211–6. doi:10.1016/s1076-6332(05)80716-0
33. Theberge J, Williamson KE, Aoyama N, Drost DJ, Manchanda R, Malla AK, et al. Longitudinal grey-matter and glutamatergic losses in first-episode schizophrenia. Br J Psychiatry (2007) 191:325–34. doi:10.1192/bjp.bp.106.033670
34. Aoyama N, Theberge J, Drost DJ, Manchanda R, Northcott S, Neufeld RWJ, et al. Grey matter and social functioning correlates of glutamatergic metabolite loss in schizophrenia. Br J Psychiatry (2011) 198(6):448–56. doi:10.1192/bjp.bp.110.079608
35. Luykx JJ, Laban KG, van den Heuvel MP, Boks MPM, Mandl RCW, Kahn RS, et al. Region and state specific glutamate downregulation in major depressive disorder: a meta-analysis of H-1-MRS findings. Neurosci Biobehav Rev (2012) 36(1):198–205. doi:10.1016/j.neubiorev.2011.05.014
36. Yoo SY, Yeon S, Choi CH, Kang DH, Lee JM, Shin NY, et al. Proton magnetic resonance spectroscopy in subjects with high genetic risk of schizophrenia: investigation of anterior cingulate, dorsolateral prefrontal cortex and thalamus. Schizophr Res (2009) 111(1-3):86–93. doi:10.1016/j.schres.2009.03.036
37. de la Fuente-Sandoval C, Leon-Ortiz P, Favila R, Stephano S, Mamo D, Ramirez-Bermudez J, et al. Higher levels of glutamate in the associative-striatum of subjects with prodromal symptoms of schizophrenia and patients with first-episode psychosis. Neuropsychopharmacology (2011) 36(9):1781–91. doi:10.1038/npp.2011.65
38. Egerton A, Brugger S, Raffin M, Barker GJ, Lythgoe DJ, McGuire PK, et al. Anterior cingulate glutamate levels related to clinical status following treatment in first-episode schizophrenia. Neuropsychopharmacology (2012) 37(11):2515–21. doi:10.1038/npp.2012.113
39. Stanley JA, Williamson PC, Drost DJ, Rylett RJ, Carr TJ, Malla A, et al. An in vivo proton magnetic resonance spectroscopy study of schizophrenia patients. Schizophr Bull (1996) 22(4):597–609. doi:10.1093/schbul/22.4.597
40. Bartha R, Williamson PC, Drost DJ, Malla A, Carr TJ, Cortese L, et al. Measurement of glutamate and glutamine in the medial prefrontal cortex of never-treated schizophrenic patients and healthy controls by proton magnetic resonance spectroscopy. Arch Gen Psychiatry (1997) 54(10):959–65. doi:10.1001/archpsyc.1997.01830220085012
41. Ohrmann P, Siegmund A, Suslow T, Spitzberg K, Kersting A, Arolt V, et al. Evidence for glutamatergic neuronal dysfunction in the prefrontal cortex in chronic but not in first-episode patients with schizophrenia: a proton magnetic resonance spectroscopy study. Schizophr Res (2005) 73(2-3):153–7. doi:10.1016/j.schres.2004.08.021
42. Ohrmann P, Kugel H, Bauer J, Siegmund A, Kolkebeck K, Suslow T, et al. Learning potential on the WCST in schizophrenia is related to the neuronal integrity of the anterior cingulate cortex as measured by proton magnetic resonance spectroscopy. Schizophr Res (2008) 106(2-3):156–63. doi:10.1016/j.schres.2008.08.005
43. Bustillo JR, Chen HJ, Gasparovic C, Mullins P, Caprihan A, Qualls C, et al. Glutamate as a marker of cognitive function in schizophrenia: a proton spectroscopic imaging study at 4 Tesla. Biol Psychiatry (2011) 69(1):19–27. doi:10.1016/j.biopsych.2010.08.024
44. Rowland LM, Spieker EA, Francis A, Barker PB, Carpenter WT, Buchanan RW. White matter alterations in deficit schizophrenia. Neuropsychopharmacology (2009) 34(6):1514–22. doi:10.1038/npp.2008.207
45. Reid MA, Kraguljac NV, Avsar KB, White DM, den Hollander JA, Lahti AC. Proton magnetic resonance spectroscopy of the substantia nigra in schizophrenia. Schizophr Res (2013) 147(2-3):348–54. doi:10.1016/j.schres.2013.04.036
46. Rowland L, Kontson K, West J, Edden R, Zhu H, Wijtenburg S, et al. In vivo measurements of glutamate, GABA, and NAAG in schizophrenia. Schizophr Bull (2012) 39(5):1096–104. doi:10.1093/schbul/sbs092
47. de la Fuente-Sandoval C, León-Ortiz P, Azcárraga M, Stephano S, Favila R, Díaz-Galvis L, et al. Glutamate levels in the associative striatum before and after 4 weeks of antipsychotic treatment in first-episode psychosis: a longitudinal proton magnetic resonance spectroscopy study. JAMA Psychiatry (2013) 70(10):1057–66. doi:10.1001/jamapsychiatry.2013.289
48. Ota M, Ishikawa M, Sato N, Hori H, Sasayama D, Hattori K, et al. Glutamatergic changes in the cerebral white matter associated with schizophrenic exacerbation. Acta Psychiatr Scand (2012) 126(1):72–8. doi:10.1111/j.1600-0447.2012.01853.x
49. Demjaha A, Egerton A, Murray R, Kapur S, Howes O, Stone J, et al. Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biol Psychiatry (2013). [Epub ahead of print]. doi:10.1016/j.biopsych.2013.06.011
50. Ongur D, Jensen JE, Prescot AP, Stork C, Lundy M, Cohen BM, et al. Abnormal glutamatergic neurotransmission and neuronal-glial interactions in acute mania. Biol Psychiatry (2008) 64(8):718–26. doi:10.1016/j.biopsych.2008.05.014
51. Lyne JP, Kinsella A, O’Donoghue B. Can we combine symptom scales for collaborative research projects? J Psychiatr Res (2012) 46(2):233–8. doi:10.1016/j.jpsychires.2011.10.002
52. Wilmsmeier A, Ohrmann P, Suslow T, Siegmund A, Koelkebeck K, Rothermundt M, et al. Neural correlates of set-shifting: decomposing executive functions in schizophrenia. J Psychiatry Neurosci (2010) 35(5):321–9. doi:10.1503/jpn.090181
53. Rusch N, van Elst LT, Valerius G, Buechert M, Thiel T, Ebert D, et al. Neurochemical and structural correlates of executive dysfunction in schizophrenia. Schizophr Res (2008) 99(1-3):155–63. doi:10.1016/j.schres.2007.05.024
54. Tandon N, Bolo NR, Sanghavi K, Mathew IT, Francis AN, Stanley JA, et al. Brain metabolite alterations in young adults at familial high risk for schizophrenia using proton magnetic resonance spectroscopy. Schizophr Res (2013) 148(1-3):59–66. doi:10.1016/j.schres.2013.05.024
55. Ohrmann P, Siegmund A, Suslow T, Pedersen A, Spitzberg K, Kersting A, et al. Cognitive impairment and in vivo metabolites in first-episode neuroleptic-naive and chronic medicated schizophrenic patients: a proton magnetic resonance spectroscopy study. J Psychiatr Res (2007) 41(8):625–34. doi:10.1016/j.jpsychires.2006.07.002
56. Fusar-Poli P, Stone JM, Broome MR, Valli I, Mechelli A, McLean MA, et al. Thalamic glutamate levels as a predictor of cortical response during executive functioning in subjects at high risk for psychosis. Arch Gen Psychiatry (2011) 68(9):881–90. doi:10.1001/archgenpsychiatry.2011.46
57. Valli I, Stone J, Mechelli A, Bhattacharyya S, Raffin M, Allen P, et al. Altered medial temporal activation related to local glutamate levels in subjects with prodromal signs of psychosis. Biol Psychiatry (2011) 69(1):97–9. doi:10.1016/j.biopsych.2010.08.033
58. Hutcheson NL, Reid MA, White DM, Kraguljac NV, Avsar KB, Bolding MS, et al. Multimodal analysis of the hippocampus in schizophrenia using proton magnetic resonance spectroscopy and functional magnetic resonance imaging. Schizophr Res (2012) 140(1-3):136–42. doi:10.1016/j.schres.2012.06.039
59. Tibbo P, Hanstock C, Valiakalayil A, Allen P. 3-T proton MRS investigation of glutamate and glutamine in adolescents at high genetic risk for schizophrenia. Am J Psychiatry (2004) 161(6):1116–8. doi:10.1176/appi.ajp.161.6.1116
60. Tebartz van Elst L, Valerius G, Buchert M, Thiel T, Rusch N, Bubl E, et al. Increased prefrontal and hippocampal glutamate concentration in schizophrenia: evidence from a magnetic resonance spectroscopy study. Biol Psychiatry (2005) 58(9):724–30. doi:10.1016/j.biopsych.2005.04.041
61. Stone JM, Howes OD, Egerton A, Kambeitz J, Allen P, Lythgoe DJ, et al. Altered relationship between hippocampal glutamate levels and striatal dopamine function in subjects at ultra high risk of psychosis. Biol Psychiatry (2010) 68(7):599–602. doi:10.1016/j.biopsych.2010.05.034
62. Szulc A, Konarzewskaa B, Galinska-Skoka B, Lazarczyka J, Waszkiewicza N, Tarasowb E, et al. Proton magnetic resonance spectroscopy measures related to short-term symptomatic outcome in chronic schizophrenia. Neurosci Lett (2013) 547:37–41. doi:10.1016/j.neulet.2013.04.051
63. Tiihonen J, Wahlbeck K. Glutamatergic drugs for schizophrenia. Cochrane Database Syst Rev (2006). doi:10.1002/14651858.CD003730.pub2
64. Singh SP, Singh V. Meta-analysis of the efficacy of adjunctive NMDA receptor modulators in chronic schizophrenia. CNS Drugs (2011) 25(10):859–85. doi:10.2165/11586650-000000000-00000
65. Anand A, Charney DS, Oren DA, Berman RM, Hu XS, Cappiello A, et al. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine – support for hyperglutamatergic effects of N-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry (2000) 57(3):270–6. doi:10.1001/archpsyc.57.3.270
66. Chatterton JE, Awobuluyi M, Premkumar LS, Takahashi H, Talantova M, Shin Y, et al. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature (2002) 415(6873):793–8. doi:10.1038/nature715
Keywords: schizophrenia, psychosis, glutamate, NMDA, MRS, spectroscopy, imaging
Citation: Merritt K, McGuire P and Egerton A (2013) Relationship between glutamate dysfunction and symptoms and cognitive function in psychosis. Front. Psychiatry 4:151. doi: 10.3389/fpsyt.2013.00151
Received: 01 September 2013; Paper pending published: 30 September 2013;
Accepted: 07 November 2013; Published online: 26 November 2013.
Edited by:
André Schmidt, University of Basel, SwitzerlandReviewed by:
John H. Krystal, Yale School of Medicine, USAAnouk Marsman, Johns Hopkins University, USA
Copyright: © 2013 Merritt, McGuire and Egerton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kate Merritt, Department of Psychosis Studies, Institute of Psychiatry, King’s College London, De Crespigny Park, Denmark Hill, London SE5 8AF, UK e-mail:a2F0ZS5tZXJyaXR0QGtjbC5hYy51aw==