 Daniel H. Mathalon1,2*
Daniel H. Mathalon1,2* Kyung-Heup Ahn3,4,5Edward B. Perry Jr.3,4,5Hyun-Sang Cho3,6
Kyung-Heup Ahn3,4,5Edward B. Perry Jr.3,4,5Hyun-Sang Cho3,6 Brian J. Roach2Rebecca K. Blais3
Brian J. Roach2Rebecca K. Blais3 Savita Bhakta3Mohini Ranganathan3
Savita Bhakta3Mohini Ranganathan3 Judith M. Ford1,2
Judith M. Ford1,2 Deepak Cyril D’Souza3,4,5
Deepak Cyril D’Souza3,4,5
- 1Department of Psychiatry, University of California San Francisco, San Francisco, CA, USA
- 2Mental Health Service (116D), San Francisco VA Medical Center, San Francisco, CA, USA
- 3Department of Psychiatry, Yale University School of Medicine, New Haven, CT, USA
- 4Schizophrenia Biological Research Center (116A), VA Connecticut Healthcare System, West Haven, CT, USA
- 5Abraham Ribicoff Research Facilities, Connecticut Mental Health Center, New Haven, CT, USA
- 6Department of Psychiatry, Yonsei University College of Medicine, Seoul, South Korea
Background: N-methyl-d-aspartate (NMDA) receptor hypofunction has been implicated in the pathophysiology of schizophrenia and its associated neurocognitive impairments. The high rate of cigarette smoking in schizophrenia raises questions about how nicotine modulates putative NMDA receptor hypofunction in the illness. Accordingly, we examined the modulatory effects of brain nicotinic acetylcholine receptor (nAChR) stimulation on NMDA receptor hypofunction by examining the interactive effects of nicotine, a nAChR agonist, and ketamine, a non-competitive NMDA receptor antagonist, on behavioral and neurophysiological measures in healthy human volunteers.
Methods: From an initial sample of 17 subjects (age range 18–55 years), 8 subjects successfully completed 4 test sessions, each separated by at least 3 days, during which they received ketamine or placebo and two injections of nicotine or placebo in a double-blind, counterbalanced manner. Schizophrenia-like effects Positive and Negative Syndrome Scale, perceptual alterations Clinician Administered Dissociative Symptoms Scale, subjective effects Visual Analog Scale and auditory event-related brain potentials (mismatch negativity, MMN; P300) were assessed during each test session.
Results: Consistent with existing studies, ketamine induced transient schizophrenia-like behavioral effects. P300 was reduced and delayed by ketamine regardless of whether it was elicited by a target (P3b) or novel (P3a) stimulus, while nicotine only reduced the amplitude of P3a. Nicotine did not rescue P300 from the effects of ketamine; the interactions of ketamine and nicotine were not significant. While nicotine significantly reduced MMN amplitude, ketamine did not.
Conclusion: Nicotine failed to modulate ketamine-induced neurophysiological and behavioral effects in this preliminary study. Interestingly, ketamine reduced P3b amplitude and nicotine reduced P3a amplitude, suggesting independent roles of NMDA receptor and nAChR in the generation of P3b and P3a, respectively.
Introduction
Several lines of evidence support a glutamatergic hypothesis of schizophrenia involving N-methyl-d-aspartate (NMDA) receptor hypofunction (1–7). Studies with the NMDA receptor antagonist, ketamine, in healthy human subjects have been a cornerstone of the glutamatergic hypothesis of schizophrenia, producing clinical symptoms and cognitive impairments similar to those observed in schizophrenia (4, 5, 7–13). Nicotine has been shown to have cognitive enhancing effects in some (14–16), although not all (17, 18), studies. It has been suggested that the high rate of cigarette smoking in schizophrenia patients may reflect their efforts to use nicotine to “self-medicate” (19–21). In the present, exploratory study, we examined the interactive effects of ketamine and nicotine in healthy volunteers in order to determine whether impairments in brain function mediated by NMDA receptor hypofunction can be ameliorated by nicotine.
Parallel Effects of Ketamine and Schizophrenia on Clinical, Cognitive, and Neurophysiological Function
When administered at subanesthetic doses, ketamine produces an array of transient effects in healthy humans that resemble the positive and negative symptoms and cognitive deficits of schizophrenia (4, 7–13, 22–26). Ketamine has also been shown to produce some of the event-related brain potential (ERP) abnormalities observed in schizophrenia including reductions of two ERP components associated with controlled and automatic processing of deviant stimuli, P300 and mismatch negativity (MMN), as described below.
P300 is a positive voltage ERP component occurring about 300 ms following infrequent deviant stimuli interspersed among frequent “standard” stimuli, typically elicited in auditory or visual “oddball” tasks (27). P300 has been posited to reflect allocation of attentional resources (28–30), stimulus categorization (31), and contextual updating of working memory (32). Multiple brain regions have been implicated as neural generators of the P300, including the temporo-parietal junction and prefrontal cortex (33, 34). P300 comprises two subcomponents, the P3b and P3a that are differentially present depending on task conditions (27, 35–38). The P3b is primarily elicited by infrequent target stimuli, reflects top-down allocation of attention, and has a parietal scalp maximum (27, 36, 37). The P3a is primarily elicited by an infrequent non-target distractor or novel stimuli in an oddball sequence, reflects bottom-up orienting of attention, and has a fronto-central scalp maximum (27, 38–41).
P300 amplitude reduction, particularly auditory P3b, is one of the most widely replicated brain abnormalities observed in patients with schizophrenia (42, 43), although it is also reduced in a number of other psychiatric and neurological disorders (44, 45). Furthermore, several studies have shown that ketamine reduces P300 amplitude in healthy volunteers (46–51), consistent with the possibility that NMDA receptor hypofunction contributes to P300 amplitude reduction in schizophrenia. Interestingly, in the Knott et al. (47) study, the reduction of P300 amplitude by ketamine was only evident in the subgroup of non-smokers, consistent with a possible protective effect of nicotine.
Mismatch negativity is a negative ERP component elicited automatically by infrequent deviant auditory stimuli randomly interspersed among frequent “standard” stimuli (52, 53). MMN has been widely interpreted to reflect auditory sensory echoic memory because the detection of deviance requires an online representation of what has recently been “standard” in the auditory stream (52). More recently, interpretations of the MMN have emphasized its reflection of both short-term (seconds) and long-term (minutes to hours) synaptic plasticity in the service of auditory sensory/perceptual learning since the amplitude of the MMN to a deviant stimulus increases as a function of the number of repetitions of the preceding standard stimulus (54, 55). From this perspective, memory traces of the recent auditory past code predictions of future auditory events, with the MMN signaling a prediction error, when the auditory expectancy is violated by a deviant stimulus (54–56). Auditory deviance along a number of dimensions elicits MMN, including pitch, duration, intensity, and location, among others (52, 57). MMN generators have been localized to the auditory cortex and to the frontal lobes (52). MMN is considered to be largely pre-attentive (52), and it is typically elicited while subjects perform a distractor task.
Mismatch negativity has been shown to be reduced in schizophrenia, particularly the duration-deviant MMN (58–60). Moreover, MMN abnormalities, relative to P300 abnormalities, appear to be more specific to schizophrenia (61, 62). Non-competitive NMDA receptor antagonism by phencyclidine (PCP) and ketamine has been shown to reduce MMN in non-human primates (63, 64) and healthy volunteers (26, 46, 65–68), respectively. However, some studies failed to find an effect of ketamine on MMN (50) or showed the effect to depend on the type of auditory deviance or the underlying cortical source examined (65, 67). Recently, Knott and colleagues (66) reported that reduction of MMN with ketamine was only seen in people with a predisposition to experience auditory hallucinations and delusions. Moreover, this effect was blocked when these subjects were chewing nicotine gum, consistent with a possible protective effect of nicotine (66).
Effects of Nicotine on Neurocognitive and Neurophysiological Function
Nicotine has been shown to enhance cognitive functions including attention, episodic memory, and working memory in humans in some (14–16, 69, 70), but not all (17, 18), studies. The effects of nicotine administration or smoking on neurophysiological measures related to processing of deviant stimuli have been examined in a number of studies (71, 72). In experienced tobacco users, cigarette smoking or nicotine administration has been shown to increase P300 amplitude (73–75), especially in non-smokers (47), and/or to reduce P300 latency (74, 76, 77). However, some studies have failed to show such effects (78–81) or have found effects on visual, but not auditory, P300 (77). In addition, some studies (75, 82, 83), but not others (84), have shown reduced P300 amplitudes in chronic smokers, suggesting a distinction between the effects of acute and chronic exposure to nicotine. Indeed, Knott and colleagues reported that the enhancement of P300 produced by nicotine administration was only evident in non-smokers (47).
Regarding MMN, some studies have reported that nicotine or nicotinic agonists increase MMN amplitude (85–88) or shorten MMN latency (86, 89, 90), whereas others have failed to show such amplitude increases (89–91) or latency reductions (66, 88, 91) with nicotine administration.
Nicotine and Schizophrenia
There is a high prevalence of cigarette smoking among patients with schizophrenia (92–95). This may reflect an effort by schizophrenia patients to “self-medicate” clinical symptoms and a number of neurocognitive impairments including deficits in attention and memory (19) and deficient sensory gating (96). Thus, there is significant interest in developing nicotinic acetylcholine receptor (nAChR) agonists to target the neurocognitive symptoms of schizophrenia. Indeed, an alpha7 nicotinic agonist, 3-[(2,4-dimethoxy) benzylidene] Anabaseine (DMXAB) has been shown to produce significant improvements in cognitive function and P50 sensory gating in patients with schizophrenia (97).
Several studies have examined the effects of nicotine administration on MMN in patients with schizophrenia. Regarding MMN amplitude, a study of schizophrenic smokers showed that nicotine increases the amplitude and the latency of duration-deviant MMN, but shows no effect on frequency-deviant MMN (98). An earlier study from the same group examined the effects of nicotine on a mixed sample of smoking and non-smoking schizophrenia patients with high levels of auditory hallucinations and found no effects on MMN amplitudes in response to duration, frequency, and intensity deviants, although the latency of the intensity-deviant MMN was shortened by nicotine (99). Another study of non-smoking schizophrenia patients failed to show any effect of nicotine on a frequency-deviant MMN (90).
Nicotinic Receptor Modulation of Glutamatergic Neurotransmission
One possible mechanism by which nAChR agonists might enhance neurocognitive and neurophysiological function is facilitating glutamatergic neurotransmission via presynaptic nAChR (100) or via GABA interneurons (101, 102). Nicotine or nAChR agonists have been shown to facilitate glutamatergic transmission in rat prefrontal cortex (103, 104) and hippocampus (105). Specifically relevant to this study, nicotine attenuates or reverses memory and attentional deficits induced by the NMDA receptor antagonist MK-801 (dizocilpine) in rats (106, 107). Moreover, dizocilpine blocked nicotinic enhancement of memory consolidation in mice (108). Knott et al. (47) examined the effects of nicotine and a sub-perceptual dose of ketamine on P300 in men and women, smokers and non-smokers. In non-smokers, ketamine reduced P300, an effect that did not interact with nicotine. However, in the third assessment block, following the drug infusion, nicotine increased P300 amplitude on its own but further reduced P300 when combined with ketamine. In a subsequent study, Knott and colleagues (66) found that ketamine reduces the amplitude of MMN in healthy individuals with a high propensity toward hallucinatory experiences and/or delusional thinking, an effect that was blocked by nicotine. Similar effects were not evident in individuals with a low propensity toward these psychotic symptoms.
The Rationale and Hypotheses for Current Study
It is difficult to isolate and study the NMDA receptor hypofunction and its consequences in schizophrenia. The ketamine paradigm in healthy subjects offers a pharmacological model for investigating nicotine’s effect on putative NMDA receptor hypofunction in schizophrenia. Specifically, this study examined the effects of nAChR activation on NMDA receptor hypofunction by investigating the interactions of ketamine, a non-competitive NMDA receptor antagonist, and nicotine, a nAChR agonist, when administered to healthy volunteers in a placebo-controlled study over four test days. The neurophysiological outcome measures, chosen based on their established sensitivity to schizophrenia, consisted of: (1) two variants of the auditory P300, the P3b elicited by target stimuli and the P3a elicited by novel distractor stimuli, and (2) the MMN elicited automatically by a duration-deviant auditory stimulus. The primary and secondary hypotheses were that nicotine would attenuate the neurophysiological abnormalities and schizophrenia-like clinical symptoms induced by ketamine, respectively.
Materials and Methods
Research Participants
The study was approved by the Institutional Review Boards of VA Connecticut Healthcare System (West Haven, CT, USA) and Yale University School of Medicine (New Haven, CT, USA). Subjects were recruited via public advertisements and were paid for their study participation. Written informed consent was obtained from all subjects. Smokers who were not interested in quitting and lifetime non-smokers who had tried nicotine in the past were both invited to participate. Subjects were medically healthy by physical examination, history, electrocardiography, and laboratory testing. They had no history of a DSM-IV Axis-I disorder (other than nicotine dependence), major current or recent (<6 weeks) life stressors, and first-degree relative with a history of psychosis. Screening procedures included the Structured Clinical Interview for DSM-IV, Non-Patient Edition (109), selected sub-tests from the Wechsler Adult Intelligence Scale (Information, Vocabulary, Block Design, Picture Completion) to provide an estimate of general level of cognitive ability (110), and the Fagerström Test for Nicotine Dependence (111) to measure the severity of nicotine dependence in smokers. Subjects were instructed to refrain from consuming psychoactive substances from 1 week prior to testing through completion of the study. An outside informant identified by the subject was interviewed to corroborate information provided by potential subjects. Urine toxicology testing was performed at screening and on the morning of each test day to rule out recent illicit substance use. Subjects were instructed to fast overnight and abstain from smoking after 11:00 p.m. prior to arrival for each test day. They were excluded if breath carbon monoxide levels were higher than 10 ppm.
Seventeen subjects participated in at least one test day. Eight of the 17 subjects scheduled for four test days did not complete testing: 3 of 5 (60%) non-smokers and 5 of 12 (42%) smokers. The reasons for discontinuation were mostly related to adverse effects of ketamine or nicotine (n = 6). Adverse events and study discontinuations were reported to the VA Connecticut Human Studies Subcommittee. As with all of our prior ketamine studies, clinical follow-ups indicated that all adverse events associated with acute ketamine administration resolved spontaneously without any late appearing or persistent adverse effects. There were no significant differences in age, sex, education, smoking status, or Fagerström Nicotine Dependence scores between study completers and non-completers. Only nine subjects completed all four test days. Demographic data for these nine completers are presented in Table 1. Of the nine completers (five men, four women), one woman was excluded from the ERP analyses because she was too somnolent and impaired to perform the oddball task or the MMN distractor task during the Ketamine Alone test day. In addition, one man was dropped from the ERP MMN analysis because of technical problems running the MMN paradigm during his Nicotine Alone test day.

Table 1. Demographic data for study completers (N = 9).
Test Days
Across the four test days, subjects received ketamine and nicotine in a double-blind, randomized, 2 × 2 crossover design. On each test day, subjects received ketamine (a bolus of 0.26 mg/kg over 1 min, followed by maintenance infusion at 0.65 mg/kg/h × 2 h) or placebo (normal saline). Fifteen minutes after the ketamine bolus, subjects received an intravenous infusion of nicotine (1.0 μg/kg/min × 10 min) or placebo followed by another nicotine or placebo injection 70 min later. The reason for two spaced nicotine injections was to attempt to minimize the possibility of desensitization that is known to occur with nicotine exposure. The dose of nicotine administered with each infusion (1.0 μg/kg/min × 10 min) = 0.7 mg in a 70-kg individual. A regular cigarette contains about 1.2–1.4 mg nicotine and an average of 0.88 mg of nicotine is delivered to a smoker from each cigarette. The timing of procedures is detailed in Table 2. Behavioral ratings were obtained at baseline and repeated periodically after the administration of ketamine and nicotine, but ERP data were collected only once per test day. Plasma ketamine and nicotine levels were measured after each infusion to rule out any pharmacokinetic interactions.

Table 2. Study procedures.
Behavioral Measures
The schizophrenia-like clinical symptoms induced by ketamine were assessed using the Positive and Negative Syndrome Scale (PANSS) (112). Perceptual alterations were assessed using the Clinician Administered Dissociative Symptoms Scale (CADSS) (113). The following subjective states were rated by participants using 100-mm Visual Analog Scales (VASs) (10): Talkative, Happy, Drowsy, Tense, Dad, Calm, Depressed, Anxious, Energetic, Fearful, Mellow, High, Angry, Mania, Irritable, Tired, Hungry, and Craving.
Analysis of Nicotine and Ketamine Levels
Plasma ketamine and norketamine were assayed using the identical method as described in detail elsewhere (114). Plasma nicotine concentrations were assayed using reversed-phase high-performance liquid chromatography (HPLC) based on a modification of a previously described method (115, 116). The nicotine assay involved HPLC/MS operated in the APCI/SIM mode using deuterated nicotine as an internal standard. After addition of the internal standard the plasma is deproteinized with sulfosalicylic acid and the supernatant made alkaline and extracted with heptane methylene chloride 85:15. This solvent is then dried down via vacuum centrifuge. The residue is redissolved in ethanol and an aliquot is injected into the HPLC. The HPLC column (Nova Pak C18 30 cm × 3.9 mm, 4 μm) is run in the isocratic mode using methanol acetonitrile ammonium formate (pH 4.0) 32.5:42.5:35.0 as mobile phase. The standard curve encompassing a range of 1–200 ng/ml was linear with negligible intercept. Plasma controls containing 4, 40, and 80 ng/ml nicotine in six consecutive runs demonstrated an inter-assay relative standard deviation RSD of 8.6, 7.4, and 8.3%, respectively.
ERP Measures
Electroencephalography (EEG) data were recorded using a 23-channel Physiometrix electrode cap. The cap included one ground electrode placed on the forehead (FPz), and the mean of freely placed bilateral earlobe electrodes served as the reference channel. Vertical and horizontal electro-oculograms (VEOGs and HEOGs) were recorded and used to correct EEG data for eye blink and eye-movement artifacts. Electrode impedances were maintained at <5 kΩ. The data were recorded using Neuroscan Synamps amplifiers, which were calibrated prior to each session. Data were acquired using a 0.05–100-Hz band pass filter, and the sampling rate was 1000 Hz.
P300 was elicited during an auditory oddball target detection task. Three types of stimuli were delivered through Etymotic ER-3A insert earphones: (1) standard tones: 500 Hz pure tones (rise/fall 5 ms; 50 ms duration), (2) target tones: 1000 Hz pure tones (rise/fall 5 ms; 50 ms duration), and (3) novel distractor sounds, selected from a corpus of novel sounds (average duration of 250 ms) developed by Friedman et al. (117). All auditory stimuli were presented at an identical sound pressure level (~80 dB SPL C scale). The task was presented in three blocks. Each block comprised of 150 pseudo-randomized stimuli (80% standards, 10% targets, 10% novel distractors) presented with a stimulus onset asynchrony (SOA) of 1250 ms. Subjects were instructed to press a response button with the index finger of their dominant hand each time a target tone occurred, giving equal emphasis to speed and accuracy.
For the MMN paradigm, subjects were presented with a pseudorandom sequence of standard tones (90% probability; 633 Hz; 5 ms rise/fall time; 50 ms duration) and duration-deviant tones (10% probability; 633 Hz, 5 ms rise/fall time, 100 ms duration) presented at 80 dB SPL, with a 510-ms SOA. A long duration-deviant MMN paradigm was chosen because of some evidence that it may more sensitive to the effect of schizophrenia than other types of MMN (58, 60, 61). The MMN paradigm was presented in two blocks, with each block comprising 783 standard tones and 87 deviant tones. These tones were presented binaurally through earphone inserts, while subjects performed a visual AX-Continuous Performance Task (AX-CPT) (118). Because several of the behavioral performance files from this task were irretrievably corrupted resulting in many subjects with missing behavioral performance data, the AX-CPT performance data were not analyzed in the current study. Thus, the AX-CPT essentially served as the distractor task during MMN recording.
ERP Data Processing
As the MMN and P300 measures generally achieve their maximum amplitudes along the midline and do not typically show hemispheric lateralization, EEG data from the midline fronto-central sites (Fz, Cz) and fronto-central-parietal sites (Fz, Cz, Pz) were analyzed for the MMN and P300 components, respectively. The processing pipeline for the P300 elicited during the three-stimulus auditory oddball task involved the following steps: continuous data were separated into 1000 ms epochs time-locked to stimulus onset, with a 100-ms pre-stimulus baseline. VEOG and HEOG data were used to correct EEG for eye-movements and blinks with a regression-based algorithm (119). After baseline correction, epochs containing artifacts (voltages exceeding ±100 μV) were rejected. P300 was identified as the most positive peak in a 235- to 400-ms time window following stimulus onset; however, because target P3b and novelty P3a have different topographies, different rules were used for identifying their peaks. The target P3b peak was first identified at Pz, then a 50-ms window (±25 ms) surrounding this peak’s latency was used to identify target P3b peaks at other sites. Novelty P3a showed more scalp variability in peak latency than target P3b, particularly at frontal sites, leading us to adopt a more flexible peak identification approach. Novelty P3a peaks were first identified at all central and parietal sites. From the range of peak latencies obtained at central sites (C3, Cz, C4), minimum and maximum latencies were identified. By subtracting 50 ms from the minimum and adding 50 ms to the maximum, the search window for identification of P3a peaks at frontal sites was defined. Somewhat early latency cut-off (400 ms) for auditory P300s was chosen to avoid picking the second late positive component, which peaked around 550 ms (see Figure 1). Peak amplitudes and latencies for target P3b and novelty P3a were extracted from electrodes Fz, Cz, and Pz for statistical analyses.
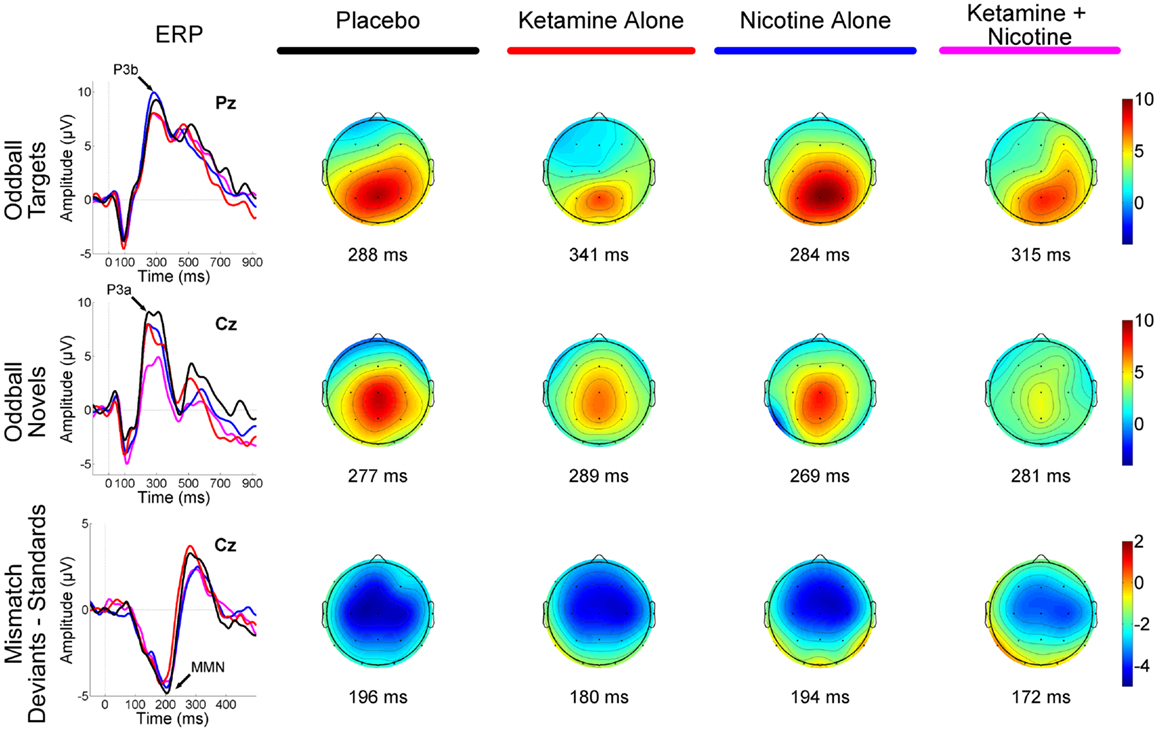
Figure 1. Event-related brain potential grand average waveforms (left) and corresponding topographic maps (right) are shown for placebo (black), ketamine alone (red), nicotine alone (blue), and ketamine + nicotine (magenta) days. ERPs, overlaid for each test day, are shown to oddball targets at Pz (top row), to oddball novels at Cz (middle row), and to difference waveforms (deviants-standards) at Cz. The oddball target elicited a P3b, the oddball novel elicited a P3a, and the deviant elicited a MMN, with each peak denoted by an arrow on the ERP waveforms. Amplitude in microvolts is on the y-axis, and latency in milliseconds is on the x-axis. Stimulus onset is at 0 ms. Negativity is plotted down. Scalp topography maps are shown for each test day for each stimulus, at the peak latency for P3b (top), P3a (middle), and MMN (bottom). Hot colors indicate positive voltage and cool colors, negative voltage.
The same eye-movement correction and artifact rejection criteria used in the P300 data processing pipeline were applied to the MMN standard and deviant trials, but these data were segmented into 550 ms epochs and baseline corrected using the 50-ms preceding tone onset. Standard and deviant trials remaining after artifact rejection were averaged separately, and the resulting ERP for the standard was subtracted from the deviant to create a difference wave. MMN was identified as the most negative peak between 90 and 270 ms post-tone onset in the difference wave at electrodes Fz and Cz. Peak amplitudes and latencies were extracted for statistical analyses.
Statistical Analyses
Behavioral data
Initially, behavioral data were examined descriptively using means, standard deviations, and graphs. Each measure was tested for normality using Kolmogorov-Smirnov test statistics and normal probability plots. All PANSS, CADSS, and VAS measures were highly skewed. Thus, these non-normal behavioral data were first ranked and then fitted using a mixed-effects model with an unstructured variance-covariance matrix and p-values adjusted for Analysis of Variance (ANOVA)-type statistics (ATS). In these models, Ketamine (active vs. placebo), Nicotine (active vs. placebo), and Time (−110, −19, +35, +110, and +180 min) were included as within-subjects explanatory factors, while Subject was the clustering factor. Time reflected the time point, in minutes, relative to Time 0 when the first intravenous infusion of active-nicotine or placebo-nicotine was initiated (see Table 2). All two- and three-way interactions were modeled. Significant interactions were followed by appropriate post hoc tests and graphical displays to interpret the effects. All results were considered statistically significant at p < 0.05. Bonferroni correction was applied within but not across domains. Thus, for example, a cut-off alpha level of 0.05/2 = 0.025 was used to declare effects significant for the two CADSS ratings (Subject- and Clinician-Rated).
ERP data
For the ERP data, which were collected once per test day, repeated-measures ANOVAs were conducted with Ketamine (active vs. placebo) and Nicotine (active vs. placebo) as within-subjects factors. The ANOVA models assessing P300 amplitudes and latencies included two additional within-subjects factors: Deviant Type (target vs. novel) and Lead (Fz vs. Cz vs. Pz). The ANOVA model for MMN amplitude and latency included one additional within-subjects factor: Lead (Fz vs. Cz). Analyses proceeded in a hierarchical fashion, with higher order interactions being parsed by examining lower order simple main effects and interactions. Ultimately, condition comparisons were tested with single degree of freedom contrasts.
Results
Behavioral Data
Ketamine produced significant increases in PANSS Total, CADSS Subject-Rated and Clinician-Rated Perceptual Alterations, and VAS subjective “High” ratings (see Figure 2). All Ketamine main effects and Ketamine × Time interactions were significant at p < 0.0001 (Table 3). Post hoc analyses showed significant effects of Ketamine at time points −19, +35, and +110 (all p < 0.05) for each of these measures. There were no significant main effects of Nicotine. Nor were there any significant Ketamine × Nicotine or Ketamine × Nicotine × Time interactions for any of these outcome measures.
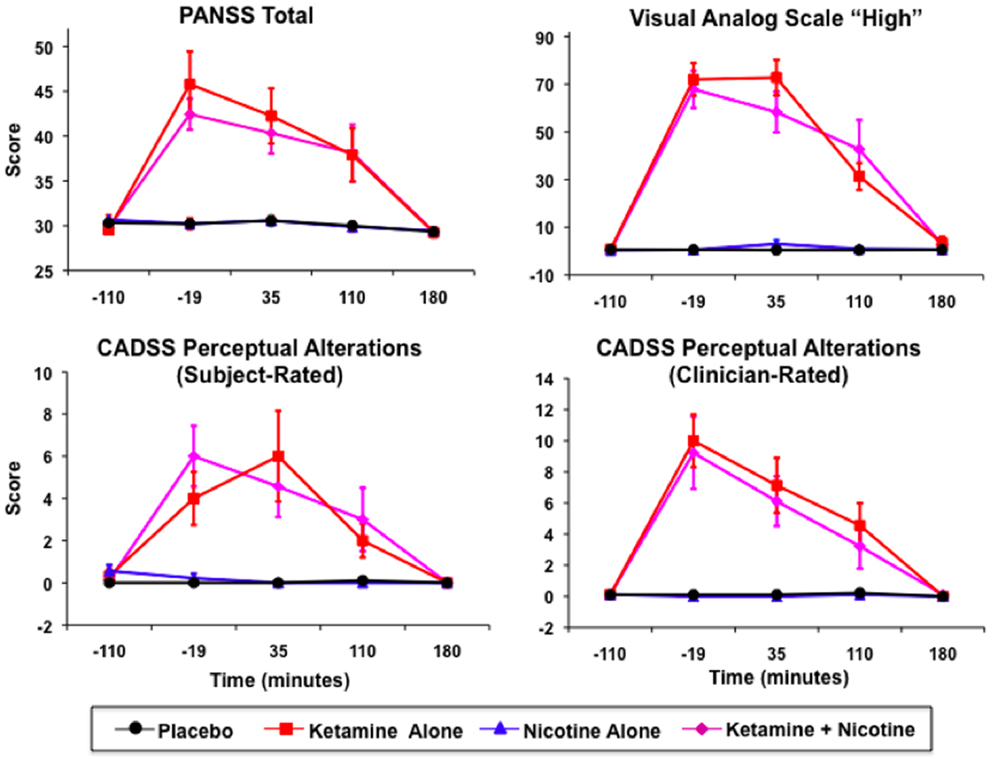
Figure 2. Mean and standard errors are plotted for Positive and Negative Syndrome Scale (PANSS) total scores (upper left), Visual Analog Scale of subjective states (upper right), subject-rate (lower-left) and clinician-rated (lower-right) perceptual alterations using the Clinician Administered Dissociative Symptoms Scale (CADSS). For each plot, values for each of the four test days are overlaid, for Placebo (black), Ketamine Alone (red), Nicotine Alone (blue), and Ketamine + Nicotine (magenta) days.
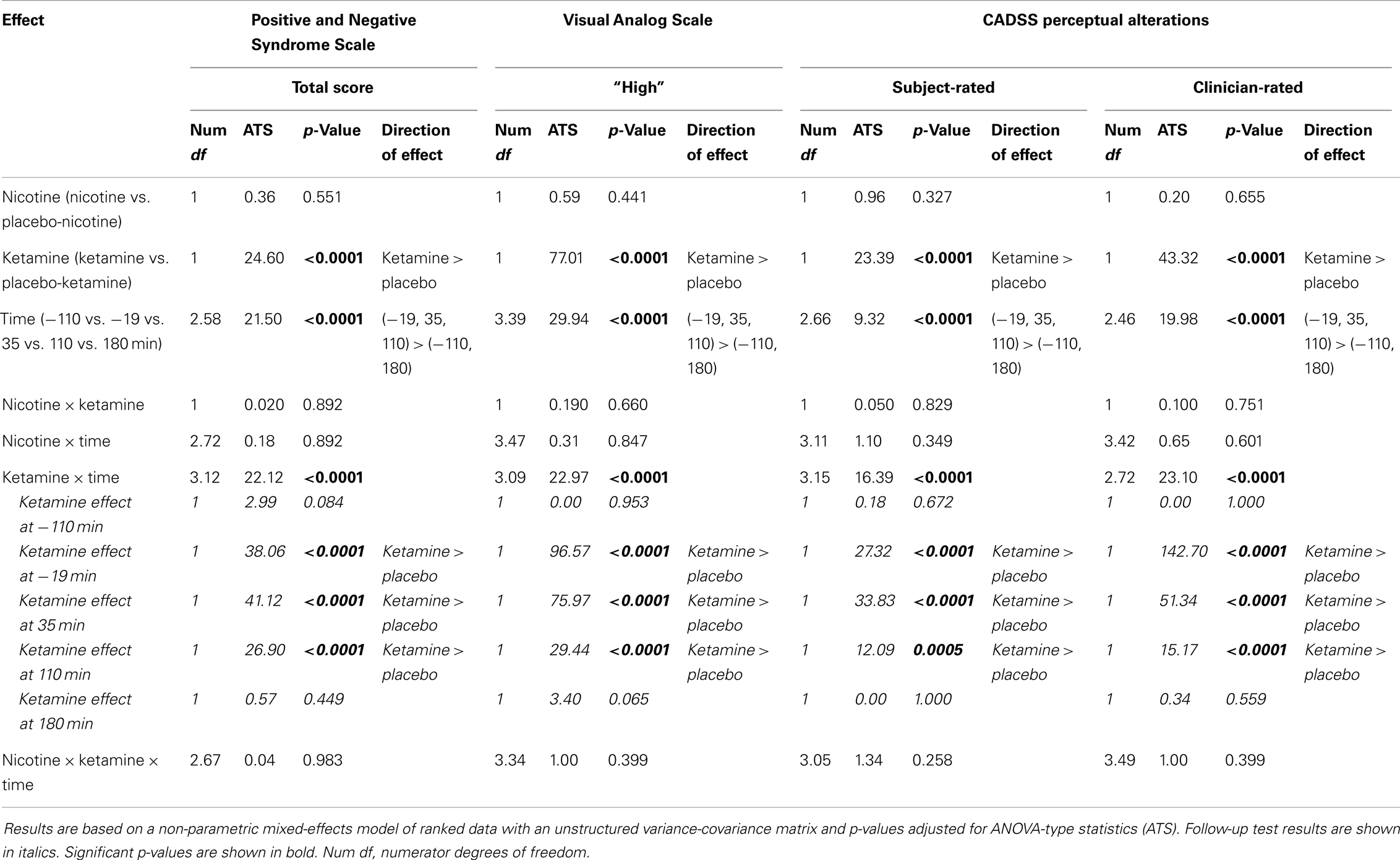
Table 3. Analysis of clinical scales.
P300 Amplitude
Event-related brain potential overlays showing P300 waves and topographic maps are shown in Figure 1, P300 peak amplitude and latency means are presented in Table 4 and histograms showing the effects of the drug conditions on P3b and P3a are shown in Figure 3. Results of the Ketamine × Nicotine × Deviant Type × Lead repeated-measures ANOVA for P300 amplitude and latency are presented in Table 5.
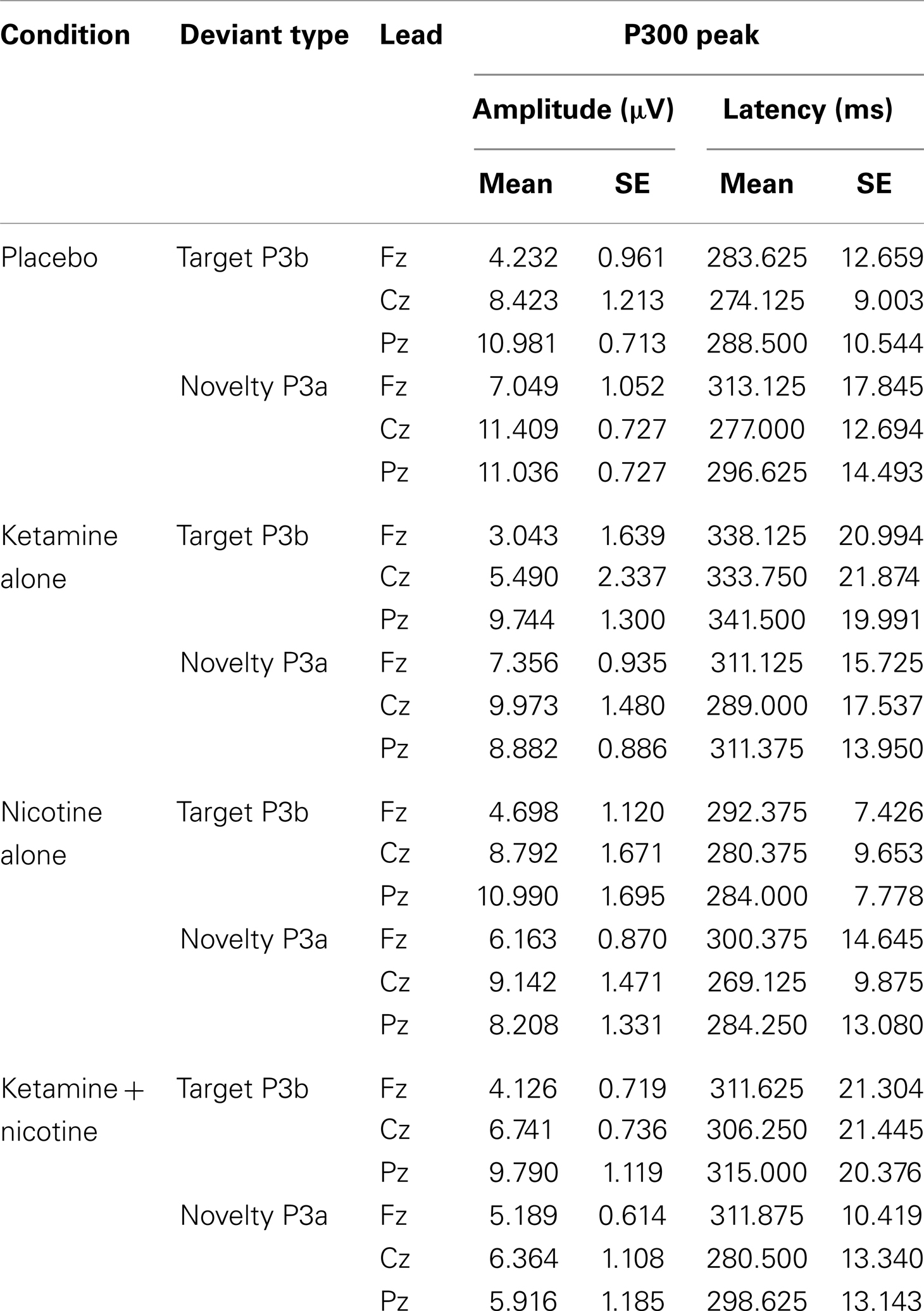
Table 4. Means and standard errors for auditory oddball P300 amplitude and latency across the four test sessions.
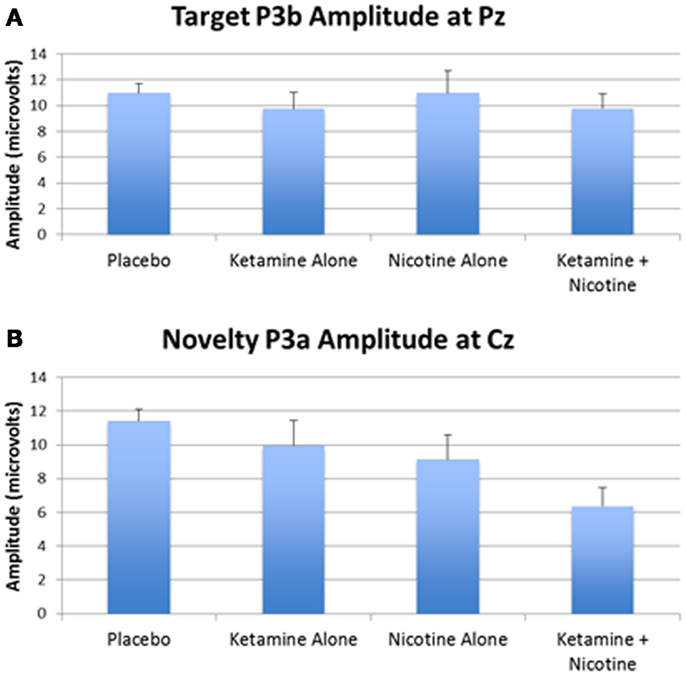
Figure 3. Mean P300 amplitude across four test days. (A) Mean target P3b amplitude for each test day. (B) Mean novelty P3a amplitude for each test day. Error bars indicate standard errors.
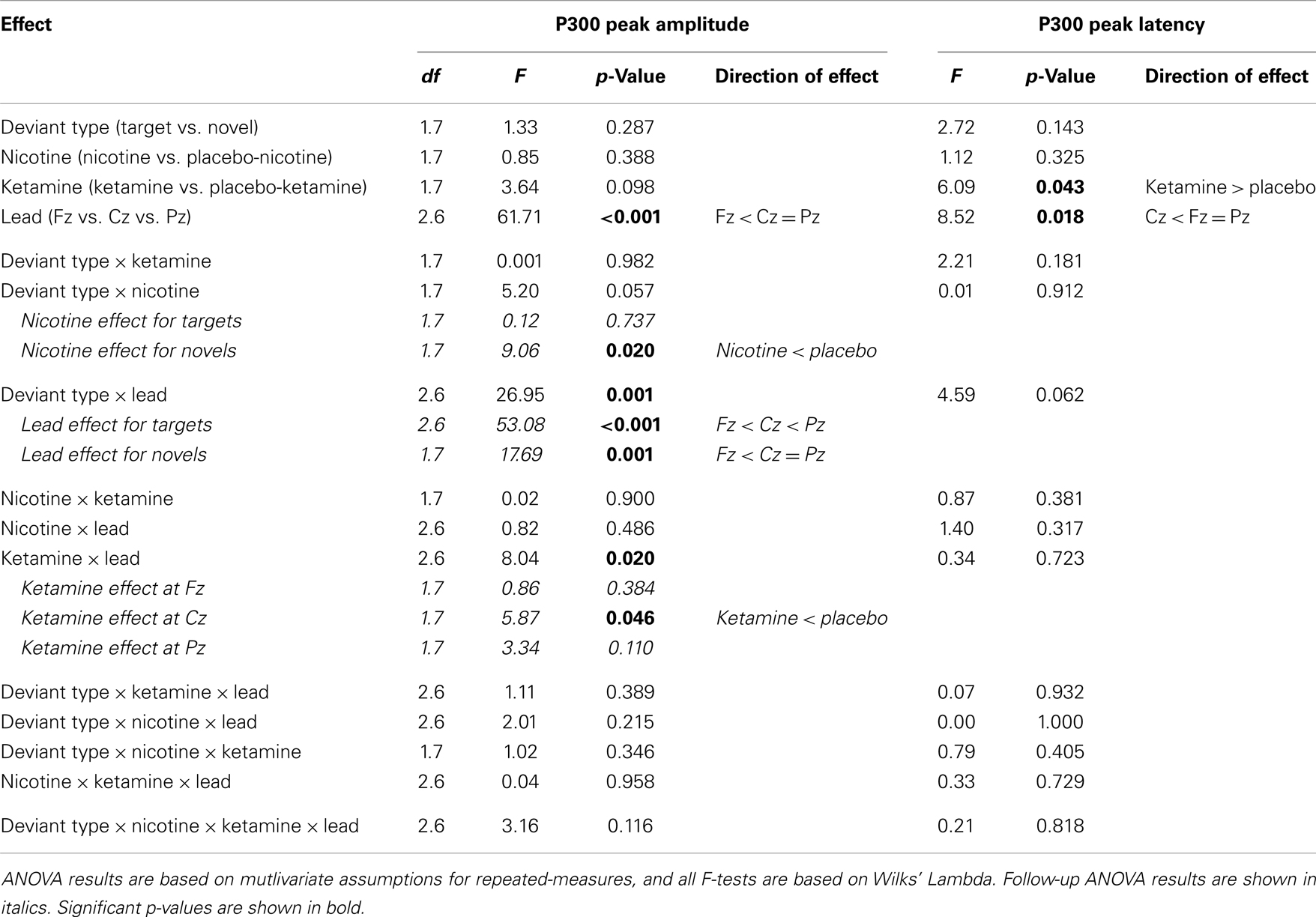
Table 5. Analysis of variance (ANOVA) of P300 peak amplitudes and latencies.
In terms of main effects, only the effect of Lead was significant, with contrasts indicating equivalent P300 amplitudes at Pz and Cz that were both larger than P300 amplitude at Fz. In terms of two-way interactions, there were significant Ketamine × Lead (p = 0.02) and Deviant Type × Lead (p = 0.001) effects, with a trend (p = 0.057) toward a Deviant Type × Nicotine effect. The Ketamine × Lead effect was parsed by examining the main effects of Ketamine separately for each of the three midline leads, with results showing ketamine to significantly reduce P300 amplitude at electrode Cz (p = 0.046), but not at Fz (p = 0.384) or Pz (p = 0.11). This ketamine-induced reduction of midline vertex P300 amplitude did not significantly depend on Deviant Type (p = 0.389). The Deviant Type × Lead effect was parsed by examining lead effects separately for targets and novels, both of which were significant. These Lead effects reflected the typical midline scalp topographies of target P3b amplitude (i.e., Fz < Cz < Pz) and novelty P3a amplitude (i.e., Fz < Cz = Pz). The Deviant Type × Nicotine trend was parsed by examining the main effect of Nicotine for each Deviant Type separately. Nicotine significantly reduced the amplitude of novelty P3a (p = 0.02) but not target P3b (p = 0.737). No other main effects or interactions were significant.
P300 Latency
There was a significant main effect of Ketamine (p = 0.043) indicating that ketamine delayed P300 latency by 25.44 ms relative to placebo (Tables 4 and 5). There was also a significant main effect of Lead (p = 0.018) indicating that P300’s peak latency was significantly shorter at Cz than at Pz and Fz. No other main effects or interactions were significant (see Table 5).
Mismatch Negativity Amplitude
Event-related brain potential overlays showing MMN difference waves and topographic maps are shown in Figure 1, MMN peak amplitude and latency means are presented in Table 6, and histograms showing the effects of the drug conditions on MMN are shown in Figure 4. The ANOVA results for MMN amplitude are presented in Table 7. None of the main effects were significant, but there were significant Nicotine × Ketamine and Nicotine × Ketamine × Lead interactions. The Nicotine × Ketamine × Lead three-way interaction was parsed by examining the Nicotine × Ketamine effect separately for Fz and Cz. The Nicotine × Ketamine effect was significant at Cz (p = 0.015) but not at Fz (p = 0.347). The Nicotine × Ketamine effect at Cz and the overall Nicotine × Ketamine two-way interaction (averaged over leads) were parsed by examining the main effect of each drug condition separately for the active and placebo days of the other drug condition. Nicotine Alone produced a trend level reduction of MMN amplitude relative to Placebo (p = 0.058 for Cz; p = 0.084 for average of Fz and Cz), but this Nicotine effect was not evident when Nicotine + Ketamine was compared to Ketamine Alone. In contrast, Ketamine did not significantly affect MMN amplitude when administered alone or along with Nicotine. No other interaction effects were significant.
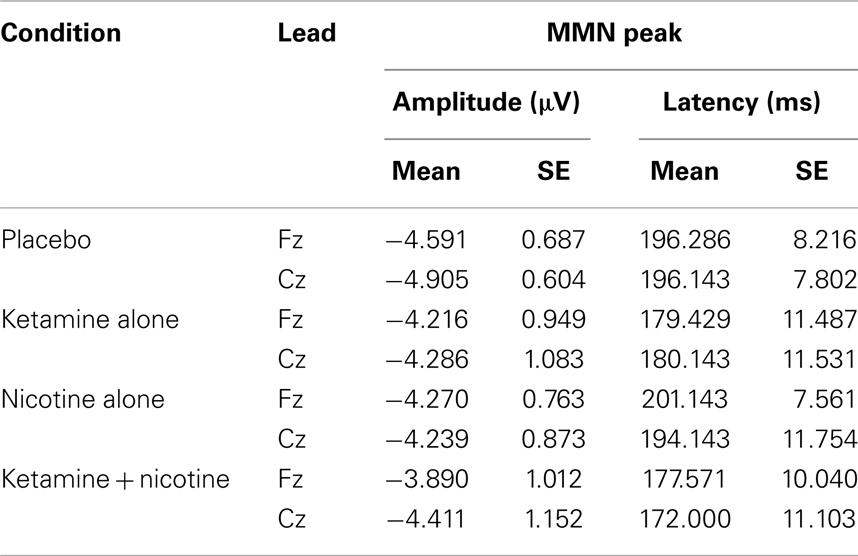
Table 6. Means and standard errors for mismatch negativity amplitude and latency across the four test sessions.
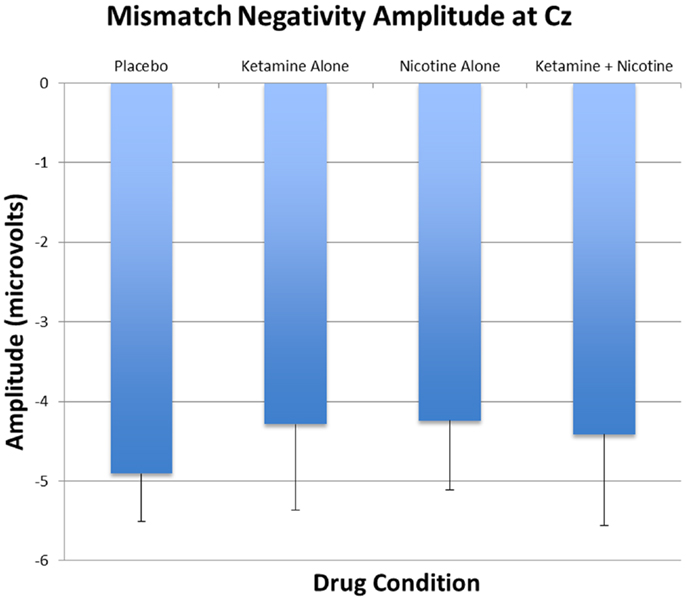
Figure 4. Mean and standard errors for MMN amplitudes recorded at Cz are plotted for the four test days.
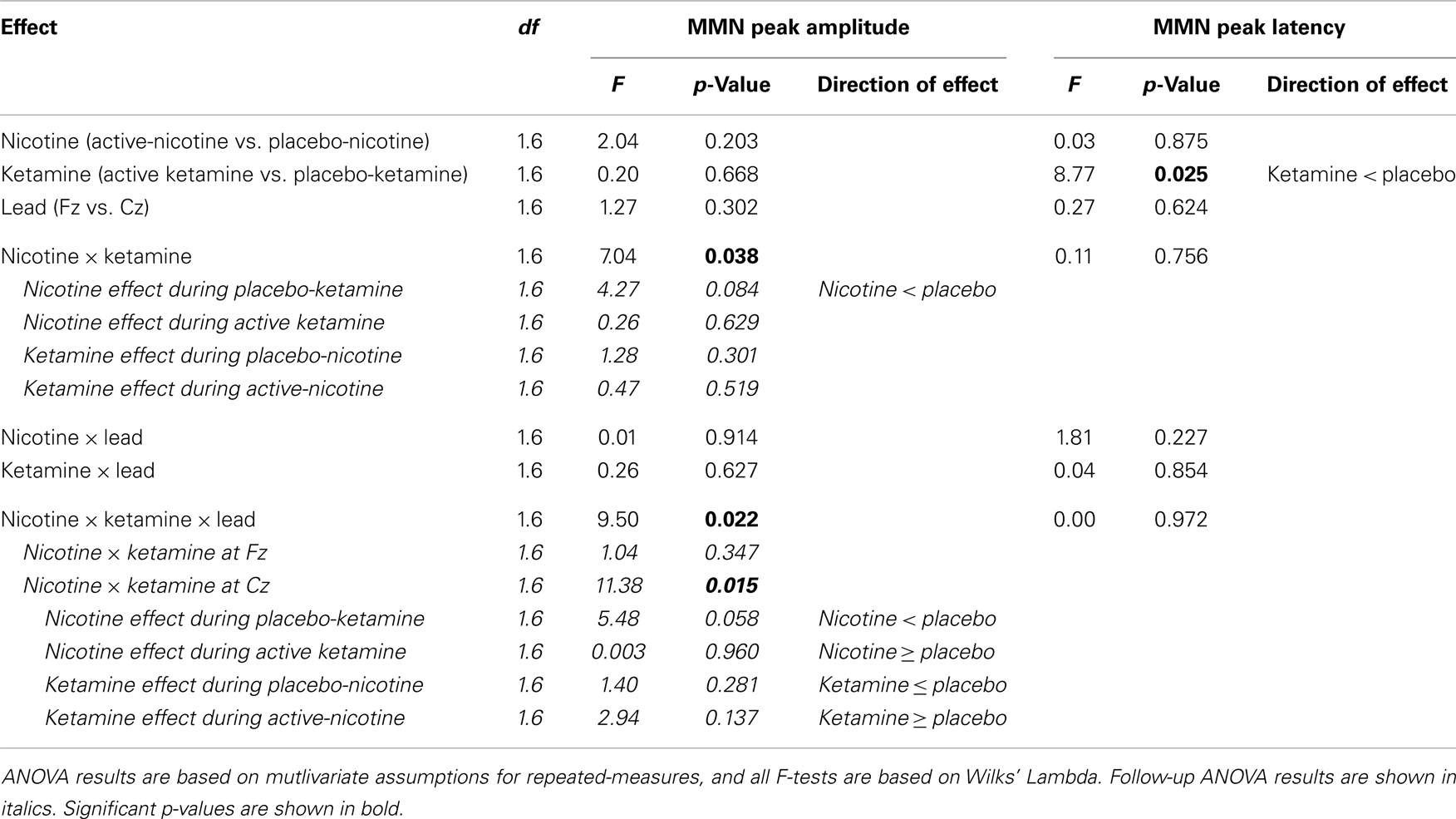
Table 7. Analysis of variance (ANOVA) of MMN peak amplitudes and latencies.
Mismatch Negativity Latency
Analysis of variance results for MMN latency are presented in Table 7. There was a significant main effect of ketamine (p = 0.025) indicating that ketamine shortened MMN latency by 19.64 ms relative to placebo. No other main effects or interactions were significant.
Plasma Drug Levels
Mean plasma levels for ketamine, norketamine, dehydroketamine, and nicotine are presented in Table 8. There were no significant differences in levels of plasma ketamine, norketamine, or dehydroketamine levels between the ketamine alone condition and the ketamine–nicotine condition. In addition, there were no significant differences in plasma nicotine levels between the nicotine alone condition and the ketamine–nicotine condition.
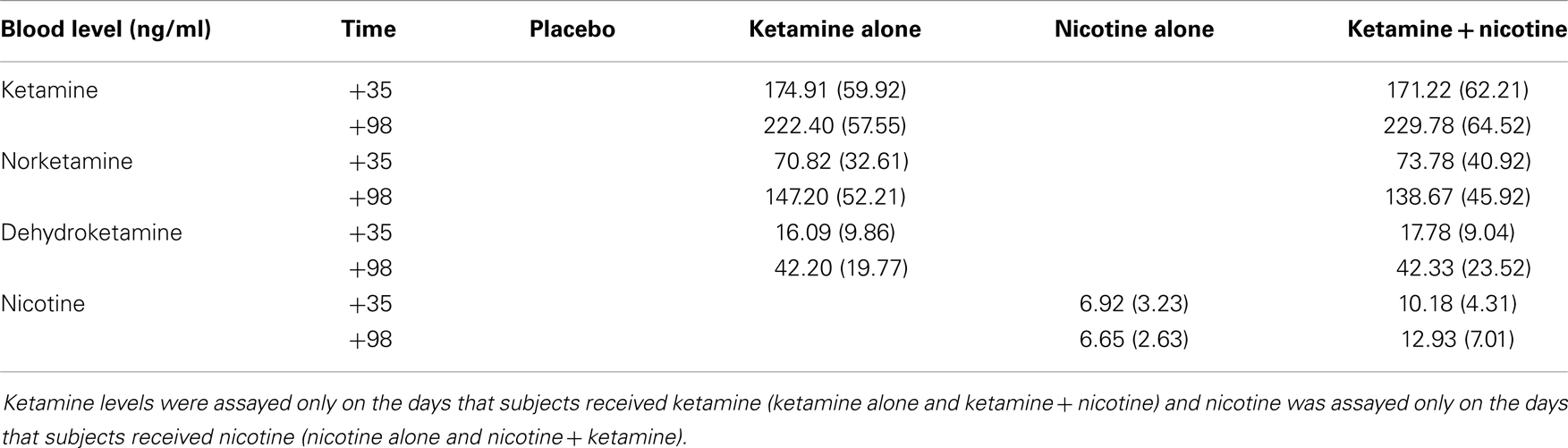
Table 8. Plasma ketamine, norketamine, dehydroketamine and nicotine levels.
Discussion
The principal findings of the current study are the differential effects of ketamine and nicotine on MMN and P300, and their interactive effects on MMN.
Effects of Ketamine
Consistent with previous studies (4, 5, 7–13, 120), ketamine induced transient schizophrenia-like behavioral effects in healthy subjects. In terms of the electrophysiological measures, ketamine decreased the amplitude and delayed the latency of P300, regardless of whether P300 was elicited by a target or novel stimulus. The decrease in amplitude is consistent with the other ERP studies showing ketamine to reduce P300 amplitude at parietal leads (46–51), although we did not observe the previously reported increase in novelty P3a at frontal leads with ketamine (51). Our results are also consistent with a prior study showing ketamine to decrease the amplitude of the late positive potential in a working memory task (9). These results provide evidence that glutamatergic neurotransmission at NMDA receptors contribute to P300 generation, both in response to infrequent target stimuli (P3b) and infrequent novel stimuli (P3a). Moreover, inasmuch as P300 amplitude reduction and latency delay are well established in schizophrenia (42, 43, 121, 122), our findings are consistent with the NMDA receptor hypofunction model of schizophrenia (1–3, 5, 6, 123, 124) and its possible role in mediating P300 deficits.
The current study did not find ketamine to significantly reduce MMN amplitude. This conflicts with a number of previous studies (26, 46, 65–68), but is consistent with some studies that either failed to show a ketamine effect on MMN (50) or showed the ketamine-induced MMN reduction to be limited to a subset of task conditions or cortical source locations (26, 65, 67), or to a subgroup of subjects (66). The discrepant results across these studies may be due to differences in the dosage and dosing schedule of ketamine. For example, the study with the most robust effects (26) used a high dose of ketamine (0.9 mg/kg), while the study with a non-significant result (50) used a relatively low dose (0.3 mg/kg). Heekeren and colleagues (65) also demonstrated dose-dependent changes in MMN amplitude using two different doses of ketamine (0.1–0.15 and 0.15–0.20 mg/kg). The absence of a significant ketamine effect on MMN in our study may also have been related to the relatively small size of our subject sample, resulting in limited power to detect an effect. For the ketamine vs. placebo effect on MMN amplitude during the placebo-nicotine day, the effect size (Cohen’s d) was estimated to be −0.43. This effect size, which appears to be smaller than the effects reported in prior studies showing ketamine to reduce MMN, would reach significance (p < 0.05) with a sample of about 25 subjects. This underscores the limited power in our current study, and points to an effect of ketamine on MMN that may emerge with moderate sample sizes.
However, it is noteworthy that our sample size was sufficiently large to detect robust psychotomimetic effects of ketamine, as well as significant ketamine-induced reductions and delays in the P300 ERP component. Thus, consistent with the report of Oranje and colleagues (50), the P300 ERP component appears to be more sensitive to the effects of NMDA receptor blockade than the MMN component. In terms of MMN latency, ketamine reduced MMN latency by about 19 ms. This effect has not been previously reported to our knowledge, and therefore should be regarded as preliminary pending replication in future studies.
Effects of Nicotine
To our knowledge, this is the first study to show nicotine to reduce the novelty P300 (P3a) in humans. This unexpected finding appears to conflict with the plethora of evidence showing nicotine to enhance cognitive functions, including attention (14–16). It is possible to construe the novelty P3a reduction by nicotine as a reflection of enhanced focus on the target detection task and reduced susceptibility to distraction by non-target distractors. However, such an interpretation is not consistent with other evidence showing P3a reduction in patients with schizophrenia (43, 121, 125–128) and patients with frontal lobe lesions (129–131), two conditions known to be associated with attentional impairments and increased distractibility. Thus, nicotine’s reduction of the P3a response to novel distractors is unlikely to be a reflection of its cognitive enhancing effects.
We did not observe significant effects of nicotine on target P300 (P3b), inconsistent with some prior reports showing nicotine to increase P300 amplitude (47, 73, 74) and decrease P300 latency (75, 77) in smokers. However, our results are consistent with other studies reporting no effects of nicotine on P300 (76, 78–80). Our study was relatively unique in its use of the intravenous route for nicotine administration, which may partially account for inconsistencies between our results and some prior studies. More generally, inconsistencies among studies may also be related to differences in the type of nicotinic agonist and dosage used, differences in sensory modality of the oddball task used to elicit the P300, and different representations of smokers and non-smokers in the study samples.
The differential effects of nicotine on P3a and P3b in our study is consistent with other evidence that the neuroanatomical (33) and neurochemical (71) underpinnings of P3a and P3b are at least partially dissociable. Polich and Criado (71) proposed dopaminergic/frontal processes for P3a generation and locus coeruleus-mediated noradrenergic/parietal processes for P3b generation. Evidence for this includes demonstrations that chronic abuse of different street drugs are associated with differential effects on P3a and P3b amplitudes (71). Nonetheless, roles for nicotinic cholinergic neurotransmission, as well as glutamatergic neurotransmission, have not figured prominently in prior neurochemical models of P300 generation.
Nicotine alone produced a trend level reduction of MMN amplitude, but this effect was not evident when comparing the Nicotine + Ketamine condition to Ketamine alone. These results conflict with some prior studies showing nicotine or nicotinic agonists to enhance MMN amplitude in response to duration (85), frequency (86, 87), or inter-stimulus interval (88) deviants in healthy volunteers, and similarly failed to corroborate studies showing nicotine to enhance duration-deviant MMN amplitude in schizophrenia patients (98). One possible reason for the discrepancy between our findings and those reported previously is that our study used an intravenous route of nicotine administration whereas prior studies used either gum (85, 88, 98) or a transdermal patch (87). While differences in the pharmacokinetics and pharmacodynamics between intravenous vs. gum and transdermal routes of administration have not been systematically studied’, it is likely that time to onset of action and peak levels achieved would differ between these modes of nicotine delivery, and such differences could account for variability in the effects of nicotine on MMN. At the same time, it should be noted that a number of studies using gum (47, 99) or transdermal patches (89, 90) failed to demonstrate an enhancement of MMN amplitude by nicotine. Moreover, the fact that our study focused on duration-deviant MMN, in part because of evidence that it is more sensitive to schizophrenia than other types of MMN (58, 60, 61) cannot be the reason we failed to observe enhancement by nicotine, since at least two prior studies have shown the amplitude of the duration-deviant MMN to be increased by nicotine [Ref. (85, 98); but, see Ref. (99)].
Combined Effect of Ketamine and Nicotine
Discordant with the study hypothesis, nicotine did not improve either the behavioral or neurophysiological abnormalities induced by ketamine. Of the many drugs tested in the ketamine model, few have been shown to reduce the schizophrenia-like behavioral and cognitive effects of ketamine in healthy human subjects. Lamotrigine, but not haloperidol or lorazepam, has been shown to reduce some of the behavioral and cognitive symptoms induced by ketamine in healthy volunteers (132–134). With previous findings from animal and human studies documenting cognitive enhancing effects of nicotine in humans (135–138) and animals (139, 140), including animal data showing nicotine to ameliorate NMDA-antagonist induced cognitive deficits (106, 107) or NMDA-antagonists to block cognitive enhancing effects of nicotine (108), it was surprising that nicotine did not show any tendency to reverse ketamine’s psychotomimetic or cognitive ERP effects. Inconsistencies among studies may be due to differences in nicotine dose, rate, and route of nicotine administration, and the smoking status of the subjects tested. Importantly, our results are consistent with other studies showing that nicotine did not block ketamine’s deleterious effects on P300 (47) or on neurocognitive test performance (141) in humans, suggesting that any pro-cognitive effects of nicotine may not be able to overcome the impairments produced by NMDA receptor blockade. However, our results were not consistent with a prior study that showed nicotine to prevent ketamine’s reduction on MMN amplitude, an effect that was only observed in the subgroup of healthy volunteers with a high propensity to have hallucinatory experiences (66). However, this prior study used a substantially lower dose of ketamine than used in the current study, and nicotine was administered with chewing gum rather than the intravenous route employed here.
Limitations
The main limitations of the current study include the small sample size, the high dropout rate, the heterogeneous smoking status of our sample, and the use of only one dose of nicotine. Future studies aimed at elucidating the effects of nicotine on patients with schizophrenia by using pharmacological models of psychosis in healthy volunteers must consider the fact that the large majority of schizophrenia patients are significantly dependent on nicotine. Accordingly, for studies about nAChR function to be relevant to schizophrenia, heavy smokers need to be included in the subject sample. However, the inclusion of nicotine-dependent heavy smokers in such studies raises the question of when to schedule the nicotine challenge relative to the timing of their last cigarette. Studying smokers who have been asked to abstain from smoking for several hours or more prior to study onset would mean studying them in a nicotine-withdrawal state. On the other hand, studying smokers who have smoked recently and are in a nicotine-satiated state may obscure the effects of intravenous nicotine. Further complicating this issue, studying non-smokers would result in high dropout routes because nicotine is generally unpleasant to non-smokers. Moreover, data from non-smokers may not generalize to schizophrenia patients, most of whom are heavy smokers.
In conclusion, the results of this study suggest that activation of nACH receptors does not influence ketamine’s psychotomimetic effects or physiological effects on MMN and P300 in healthy human volunteers. However, ketamine and nicotine appear to have independent effects on P3a, P3b, and MMN suggesting differential effects of nACH and NMDA receptor systems on these ERP components. Moreover, this is the first study to report a significant reduction in P3a amplitude by nicotine.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors also thank Angelina Genovese, R.N.C., M.B.A.; Elizabeth O’Donnell, R.N.; Brenda Breault, R.N., B.S.N.; Sonah Yoo, R.Ph.; Robert Sturwold, R.Ph.; and Willie Ford of the Neurobiological Studies Unit at the VA Connecticut Healthcare System, West Haven Campus for their central contributions to the success of this project. The authors also acknowledge the contributions of John Krystal, MD for his role in providing the impetus in conducting the study. The Department of Veterans Affairs Schizophrenia Biological Research Center and a NARSAD Young Investigator Award to Hyun-Sang Cho supported this study.
References
1. Coyle JT, Tsai G, Goff D. Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann N Y Acad Sci (2003) 1003:318–27. doi: 10.1196/annals.1300.020
2. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry (1991) 148:1301–8.
3. Javitt DC, Zukin SR, Heresco-Levy U, Umbricht D. Has an angel shown the way? Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Schizophr Bull (2012) 38:958–66. doi:10.1093/schbul/sbs069
4. Krystal JH, Anand A, Moghaddam B. Effects of NMDA receptor antagonists: implications for the pathophysiology of schizophrenia. Arch Gen Psychiatry (2002) 59:663–4. doi:10.1001/archpsyc.59.7.663
5. Krystal JH, D’Souza DC, Mathalon D, Perry E, Belger A, Hoffman R. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl) (2003) 169:215–33. doi:10.1007/s00213-003-1582-z
6. Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology (2012) 37:4–15. doi:10.1038/npp.2011.181
7. Moghaddam B, Krystal JH. Capturing the angel in “angel dust”: twenty years of translational neuroscience studies of NMDA receptor antagonists in animals and humans. Schizophr Bull (2012) 38:942–9. doi:10.1093/schbul/sbs075
8. Adler CM, Goldberg TE, Malhotra AK, Pickar D, Breier A. Effects of ketamine on thought disorder, working memory, and semantic memory in healthy volunteers. Biol Psychiatry (1998) 43:811–6. doi:10.1016/S0006-3223(97)00556-8
9. Ahn KH, Youn T, Cho SS, Ha TH, Ha KS, Kim MS, et al. N-methyl-D-aspartate receptor in working memory impairments in schizophrenia: event-related potential study of late stage of working memory process. Prog Neuropsychopharmacol Biol Psychiatry (2003) 27:993–9. doi:10.1016/S0278-5846(03)00159-3
10. Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry (1994) 51:199–214. doi:10.1001/archpsyc.1994.03950030035004
11. Lahti AC, Weiler MA, Tamara Michaelidis BA, Parwani A, Tamminga CA. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology (2001) 25:455–67. doi:10.1016/S0893-133X(01)00243-3
12. Malhotra AK, Pinals DA, Weingartner H, Sirocco K, Missar CD, Pickar D, et al. NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology (1996) 14:301–7. doi:10.1016/0893-133X(95)00137-3
13. Morgan CJ, Mofeez A, Brandner B, Bromley L, Curran HV. Acute effects of ketamine on memory systems and psychotic symptoms in healthy volunteers. Neuropsychopharmacology (2004) 29:208–18. doi:10.1038/sj.npp.1300342
14. Newhouse PA, Potter A, Singh A. Effects of nicotinic stimulation on cognitive performance. Curr Opin Pharmacol (2004) 4:36–46. doi:10.1016/j.coph.2003.11.001
15. Rezvani AH, Levin ED. Cognitive effects of nicotine. Biol Psychiatry (2001) 49:258–67. doi:10.1016/S0006-3223(00)01094-5
16. Swan GE, Lessov-Schlaggar CN. The effects of tobacco smoke and nicotine on cognition and the brain. Neuropsychol Rev (2007) 17:259–73. doi:10.1007/s11065-007-9035-9
17. Park S, Knopick C, McGurk S, Meltzer HY. Nicotine impairs spatial working memory while leaving spatial attention intact. Neuropsychopharmacology (2000) 22:200–9. doi:10.1016/S0893-133X(99)00098-6
18. Sakurai Y, Kanazawa I. Acute effects of cigarettes in non-deprived smokers on memory, calculation and executive functions. Hum Psychopharmacol (2002) 17:369–73. doi:10.1002/hup.424
19. George TP, Vessicchio JC, Termine A, Sahady DM, Head CA, Pepper WT, et al. Effects of smoking abstinence on visuospatial working memory function in schizophrenia. Neuropsychopharmacology (2002) 26:75–85. doi:10.1016/S0893-133X(01)00296-2
20. Kumari V, Postma P. Nicotine use in schizophrenia: the self medication hypotheses. Neurosci Biobehav Rev (2005) 29:1021–34. doi:10.1016/j.neubiorev.2005.02.006
21. Sacco KA, Bannon KL, George TP. Nicotinic receptor mechanisms and cognition in normal states and neuropsychiatric disorders. J Psychopharmacol (2004) 18:457–74. doi:10.1177/0269881104047273
22. Hetem LA, Danion JM, Diemunsch P, Brandt C. Effect of a subanesthetic dose of ketamine on memory and conscious awareness in healthy volunteers. Psychopharmacology (Berl) (2000) 152:283–8. doi:10.1007/s002130000511
23. Honey RA, Turner DC, Honey GD, Sharar SR, Kumaran D, Pomarol-Clotet E, et al. Subdissociative dose ketamine produces a deficit in manipulation but not maintenance of the contents of working memory. Neuropsychopharmacology (2003) 28:2037–44. doi:10.1038/sj.npp.1300272
24. Krystal JH, Bennett A, Abi-Saab D, Belger A, Karper LP, D’Souza DC, et al. Dissociation of ketamine effects on rule acquisition and rule implementation: possible relevance to NMDA receptor contributions to executive cognitive functions. Biol Psychiatry (2000) 47:137–43. doi:10.1016/S0006-3223(99)00097-9
25. Rowland LM, Astur RS, Jung RE, Bustillo JR, Lauriello J, Yeo RA. Selective cognitive impairments associated with NMDA receptor blockade in humans. Neuropsychopharmacology (2005) 30:633–9. doi:10.1038/sj.npp.1300642
26. Umbricht D, Schmid L, Koller R, Vollenweider FX, Hell D, Javitt DC. Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: implications for models of cognitive deficits in schizophrenia. Arch Gen Psychiatry (2000) 57:1139–47. doi:10.1001/archpsyc.57.12.1139
27. Polich J. Updating P300: an integrative theory of P3a and P3b. Clin Neurophysiol (2007) 118:2128–48. doi:10.1016/j.clinph.2007.04.019
28. Isreal JB, Chesney GL, Wickens CD, Donchin E. P300 and tracking difficulty: evidence for multiple resources in dual-task performance. Psychophysiology (1980) 17:259–73. doi:10.1111/j.1469-8986.1980.tb00146.x
29. Kramer AF, Strayer DL. Assessing the development of automatic processing: an application of dual-task and event-related brain potential methodologies. Biol Psychol (1988) 26:231–67. doi:10.1016/0301-0511(88)90022-1
30. Ravden D, Polich J. Habituation of P300 from visual stimuli. Int J Psychophysiol (1998) 30:359–65. doi:10.1016/S0167-8760(98)00039-7
31. Johnson R Jr, Donchin E. P300 and stimulus categorization: two plus one is not so different from one plus one. Psychophysiology (1980) 17:167–78. doi:10.1111/j.1469-8986.1980.tb00131.x
32. Donchin E, Coles M. Is the P300 component a manifestation of context updating? (Commentary on Verleger’s critique of the context updating model). Behav Brain Sci (1988) 11:357–74. doi:10.1017/S0140525X00058027
33. Halgren E, Marinkovic K, Chauvel P. Generators of the late cognitive potentials in auditory and visual oddball tasks. Electroencephalogr Clin Neurophysiol (1998) 106:156–64. doi:10.1016/S0013-4694(97)00119-3
34. Smith ME, Halgren E, Sokolik M, Baudena P, Musolino A, Liegeois-Chauvel C, et al. The intracranial topography of the P3 event-related potential elicited during auditory oddball. Electroencephalogr Clin Neurophysiol (1990) 76:235–48. doi:10.1016/0013-4694(90)90018-F
35. Dien J, Spencer KM, Donchin E. Parsing the late positive complex: mental chronometry and the ERP components that inhabit the neighborhood of the P300. Psychophysiology (2004) 41:665–78. doi:10.1111/j.1469-8986.2004.00193.x
36. Polich J, Kok A. Cognitive and biological determinants of P300: an integrative review. Biol Psychol (1995) 41:103–46. doi:10.1016/0301-0511(95)05130-9
37. Spencer KM, Dien J, Donchin E. Spatiotemporal analysis of the late ERP responses to deviant stimuli. Psychophysiology (2001) 38:343–58. doi:10.1111/1469-8986.3820343
38. Squires NK, Squires KC, Hillyard SA. Two varieties of long-latency positive waves evoked by unpredictable auditory stimuli in man. Electroencephalogr Clin Neuropysiol (1975) 38:387–401. doi:10.1016/0013-4694(75)90263-1
39. Comerchero MD, Polich J. P3a, perceptual distinctiveness, and stimulus modality. Brain Res Cogn Brain Res (1998) 7:41–8. doi:10.1016/S0926-6410(98)00009-3
40. Courchesne E, Hillyard S, Galambos R. Stimulus novelty, task relevance and the visual evoked potential in man. Electroencephalogr Clin Neurophysiol (1975) 39:131–43. doi:10.1016/0013-4694(75)90003-6
41. Goldstein A, Spencer KM, Donchin E. The influence of stimulus deviance and novelty on the P300 and novelty P3. Psychophysiology (2002) 39:781–90. doi:10.1111/1469-8986.3960781
42. Bramon E, Rabe-Hesketh S, Sham P, Murray RM, Frangou S. Meta-analysis of the P300 and P50 waveforms in schizophrenia. Schizophr Res (2004) 70:315–29. doi:10.1016/j.schres.2004.01.004
43. Jeon YW, Polich J. Meta-analysis of P300 and schizophrenia: patients, paradigms, and practical implications. Psychophysiology (2003) 40:684–701. doi:10.1111/1469-8986.00070
44. Charles G, Hansenne M. P 300 slow potential. Clinical interest in 3 mental diseases and neurobiology: a review. Encephale (1992) 18:225–236.
45. Hansenne M. The p300 cognitive event-related potential. II. Individual variability and clinical application in psychopathology. Neurophysiol Clin (2000) 30:211–31. doi:10.1016/S0987-7053(00)00224-0
46. Gunduz-Bruce H, Reinhart RM, Roach BJ, Gueorguieva R, Oliver S, D’Souza DC, et al. Glutamatergic modulation of auditory information processing in the human brain. Biol Psychiatry (2012) 71:969–77. doi:10.1016/j.biopsych.2011.09.031
47. Knott V, Millar AM, McIntosh JF, Shah DK, Fisher DJ, Blais CM, et al. Separate and combined effects of low dose ketamine and nicotine on behavioural and neural correlates of sustained attention. Biol Psychol (2011) 88:83–93. doi:10.1016/j.biopsycho.2011.06.012
48. Musso F, Brinkmeyer J, Ecker D, London MK, Thieme G, Warbrick T, et al. Ketamine effects on brain function – simultaneous fMRI/EEG during a visual oddball task. Neuroimage (2011) 58:508–25. doi:10.1016/j.neuroimage.2011.06.045
49. Oranje B, Gispen-de Wied CC, Westenberg HG, Kemner C, Verbaten MN, Kahn RS. Haloperidol counteracts the ketamine-induced disruption of processing negativity, but not that of the P300 amplitude. Int J Neuropsychopharmacol (2009) 12:823–32. doi:10.1017/S1461145708009814
50. Oranje B, van Berckel BN, Kemner C, van Ree JM, Kahn RS, Verbaten MN. The effects of a sub-anaesthetic dose of ketamine on human selective attention. Neuropsychopharmacology (2000) 22:293–302. doi:10.1016/S0893-133X(99)00118-9
51. Watson TD, Petrakis IL, Edgecombe J, Perrino A, Krystal JH, Mathalon DH. Modulation of the cortical processing of novel and target stimuli by drugs affecting glutamate and GABA neurotransmission. Int J Neuropsychopharmacol (2009) 12:357–70. doi:10.1017/S1461145708009334
52. Naatanen R, Alho K. Mismatch negativity – a unique measure of sensory processing in audition. Int J Neurosci (1995) 80:317–37. doi:10.3109/00207459508986107
53. Naatanen R, Gaillard AW, Mantysalo S. Early selective-attention effect on evoked potential reinterpreted. Acta Psychol (Amst) (1978) 42:313–29. doi:10.1016/0001-6918(78)90006-9
54. Stephan KE, Baldeweg T, Friston KJ. Synaptic plasticity and dysconnection in schizophrenia. Biol Psychiatry (2006) 59:929–39. doi:10.1016/j.biopsych.2005.10.005
55. Stephan KE, Friston KJ, Frith CD. Dysconnection in schizophrenia: from abnormal synaptic plasticity to failures of self-monitoring. Schizophr Bull (2009) 35:509–27. doi:10.1093/schbul/sbn176
56. Garrido MI, Kilner JM, Stephan KE, Friston KJ. The mismatch negativity: a review of underlying mechanisms. Neurophysiol Clin (2009) 120:453–63. doi:10.1016/j.clinph.2008.11.029
57. Naatanen R, Pakarinen S, Rinne T, Takegata R. The mismatch negativity (MMN): towards the optimal paradigm. Clin Neurophysiol (2004) 115:140–4. doi:10.1016/j.clinph.2003.04.001
58. Michie PT. What has MMN revealed about the auditory system in schizophrenia? Int J Psychophysiol (2001) 42:177–94. doi:10.1016/S0167-8760(01)00166-0
59. Todd J, Michie PT, Schall U, Karayanidis F, Yabe H, Naatanen R. Deviant matters: duration, frequency, and intensity deviants reveal different patterns of mismatch negativity reduction in early and late schizophrenia. Biol Psychiatry (2008) 63:58–64. doi:10.1016/j.biopsych.2007.02.016
60. Umbricht D, Krljes S. Mismatch negativity in schizophrenia: a meta-analysis. Schizophr Res (2005) 76:1–23. doi:10.1016/j.schres.2004.12.002
61. Naatanen R, Kahkonen S. Central auditory dysfunction in schizophrenia as revealed by the mismatch negativity (MMN) and its magnetic equivalent MMNm: a review. Int J Neuropsychopharmacol (2009) 12:125–35. doi:10.1017/S1461145708009322
62. Umbricht D, Koller R, Schmid L, Skrabo A, Grubel C, Huber T, et al. How specific are deficits in mismatch negativity generation to schizophrenia? Biol Psychiatry (2003) 53:1120–31. doi:10.1016/S0006-3223(02)01642-6
63. Javitt DC, Steinschneider M, Schroeder CE, Arezzo JC. Role of cortical N-methyl-D-aspartate receptors in auditory sensory memory and mismatch negativity generation: implications for schizophrenia. Proc Natl Acad Sci U S A (1996) 93:11962–7. doi:10.1073/pnas.93.21.11962
64. Javitt DC, Steinschneider M, Schroeder CE, Vaughan HG Jr, Arezzo JC. Detection of stimulus deviance within primate primary auditory cortex: intracortical mechanisms of mismatch negativity (MMN) generation. Brain Res (1994) 667:192–200. doi:10.1016/0006-8993(94)91496-6
65. Heekeren K, Daumann J, Neukirch A, Stock C, Kawohl W, Norra C, et al. Mismatch negativity generation in the human 5HT2A agonist and NMDA antagonist model of psychosis. Psychopharmacology (Berl) (2008) 199:77–88. doi:10.1007/s00213-008-1129-4
66. Knott V, Shah D, Millar A, McIntosh J, Fisher D, Blais C, et al. Nicotine, auditory sensory memory, and sustained attention in a human ketamine model of schizophrenia: moderating influence of a hallucinatory trait. Front Pharmacol (2012) 3:172. doi:10.3389/fphar.2012.00172
67. Kreitschmann-Andermahr I, Rosburg T, Demme U, Gaser E, Nowak H, Sauer H. Effect of ketamine on the neuromagnetic mismatch field in healthy humans. Brain Res Cogn Brain Res (2001) 12:109–16. doi:10.1016/S0926-6410(01)00043-X
68. Schmidt A, Bachmann R, Kometer M, Csomor PA, Stephan KE, Seifritz E, et al. Mismatch negativity encoding of prediction errors predicts S-ketamine-induced cognitive impairments. Neuropsychopharmacology (2012) 37:865–75. doi:10.1038/npp.2011.261
69. Froeliger B, Gilbert DG, McClernon FJ. Effects of nicotine on novelty detection and memory recognition performance: double-blind, placebo-controlled studies of smokers and nonsmokers. Psychopharmacology (Berl) (2009) 205:625–33. doi:10.1007/s00213-009-1571-y
70. Pineda JA, Herrera C, Kang C, Sandler A. Effects of cigarette smoking and 12-h abstention on working memory during a serial-probe recognition task. Psychopharmacology (Berl) (1998) 139:311–21. doi:10.1007/s002130050722
71. Polich J, Criado JR. Neuropsychology and neuropharmacology of P3a and P3b. Int J Psychophysiol (2006) 60:172–85. doi:10.1016/j.ijpsycho.2005.12.012
72. Pritchard W, Sokhadze E, Houlihan M. Effects of nicotine and smoking on event-related potentials: a review. Nicotine Tob Res (2004) 6:961–84. doi:10.1080/14622200412331324848
73. Houlihan ME, Pritchard WS, Robinson JH. Effects of smoking/nicotine on performance and event-related potentials during a short-term memory scanning task. Psychopharmacology (Berl) (2001) 156:388–96. doi:10.1007/s002130100751
74. Knott V, Bosman M, Mahoney C, Ilivitsky V, Quirt K. Transdermal nicotine: single dose effects on mood, EEG, performance, and event-related potentials. Pharmacol Biochem Behav (1999) 63:253–61. doi:10.1016/S0091-3057(99)00006-4
75. Kodama E, Morita K, Kinoshita S, Miyahira A, Nakamura J, Maeda H. Inhibitory effect of smoking on event-related potentials in schizophrenic patients: comparison with healthy subjects. Prog Neuropsychopharmacol Biol Psychiatry (1998) 22:1275–88. doi:10.1016/S0278-5846(98)00082-7
76. Edwards JA, Wesnes K, Warburton DM, Gale A. Evidence of more rapid stimulus evaluation following cigarette smoking. Addict Behav (1985) 10:113–26. doi:10.1016/0306-4603(85)90017-6
77. Houlihan ME, Pritchard WS, Robinson JH. Faster P300 latency after smoking in visual but not auditory oddball tasks. Psychopharmacology (Berl) (1996) 123:231–8. doi:10.1007/BF02246577
78. Knott V, McIntosh J, Millar A, Fisher D, Villeneuve C, Ilivitsky V, et al. Nicotine and smoker status moderate brain electric and mood activation induced by ketamine, an N-methyl-D-aspartate (NMDA) receptor antagonist. Pharmacol Biochem Behav (2006) 85:228–42. doi:10.1016/j.pbb.2006.08.005
79. Lindgren M, Molander L, Verbaan C, Lunell E, Rosen I. Electroencephalographic effects of intravenous nicotine – a dose-response study. Psychopharmacology (Berl) (1999) 145:342–50. doi:10.1007/s002130051067
80. Lindgren M, Stenberg G, Rosen I. Effects of nicotine in a bimodal attention task. Neuropsychobiology (1998) 38:42–9. doi:10.1159/000026515
81. Michel C, Hasenfratz M, Nil R, Battig K. Cardiovascular, electrocortical, and behavioral effects of nicotine chewing gum. Klin Wochenschr (1988) 66(Suppl 11):72–9.
82. Anokhin AP, Vedeniapin AB, Sirevaag EJ, Bauer LO, O’Connor SJ, Kuperman S, et al. The P300 brain potential is reduced in smokers. Psychopharmacology (Berl) (2000) 149:409–13. doi:10.1007/s002130000387
83. Neuhaus A, Bajbouj M, Kienast T, Kalus P, von Haebler D, Winterer G, et al. Persistent dysfunctional frontal lobe activation in former smokers. Psychopharmacology (Berl) (2006) 186:191–200. doi:10.1007/s00213-006-0366-7
84. Ascioglu M, Dolu N, Golgeli A, Suer C, Ozesmi C. Effects of cigarette smoking on cognitive processing. Int J Neurosci (2004) 114:381–90. doi:10.1080/00207450490270668
85. Baldeweg T, Wong D, Stephan KE. Nicotinic modulation of human auditory sensory memory: evidence from mismatch negativity potentials. Int J Psychophysiol (2006) 59:49–58. doi:10.1016/j.ijpsycho.2005.07.014
86. Dunbar G, Boeijinga PH, Demazieres A, Cisterni C, Kuchibhatla R, Wesnes K, et al. Effects of TC-1734 (AZD3480), a selective neuronal nicotinic receptor agonist, on cognitive performance and the EEG of young healthy male volunteers. Psychopharmacology (2007) 191:919–29. doi:10.1007/s00213-006-0675-x
87. Harkrider AW, Hedrick MS. Acute effect of nicotine on auditory gating in smokers and non-smokers. Hear Res (2005) 202:114–28. doi:10.1016/j.heares.2004.11.009
88. Martin LF, Davalos DB, Kisley MA. Nicotine enhances automatic temporal processing as measured by the mismatch negativity waveform. Nicotine Tob Res (2009) 11:698–706. doi:10.1093/ntr/ntp052
89. Inami R, Kirino E, Inoue R, Arai H. Transdermal nicotine administration enhances automatic auditory processing reflected by mismatch negativity. Pharmacol Biochem Behav (2005) 80:453–61. doi:10.1016/j.pbb.2005.01.001
90. Inami R, Kirino E, Inoue R, Suzuki T, Arai H. Nicotine effects on mismatch negativity in nonsmoking schizophrenic patients. Neuropsychobiology (2007) 56:64–72. doi:10.1159/000111536
91. Knott V, Heenan A, Shah D, Bolton K, Fisher D, Villeneuve C. Electrophysiological evidence of nicotine’s distracter-filtering properties in non-smokers. J Psychopharmacol (2011) 25:239–48. doi:10.1177/0269881109348158
92. Dalack GW, Healy DJ, Meador-Woodruff JH. Nicotine dependence in schizophrenia: clinical phenomena and laboratory findings. Am J Psychiatry (1998) 155:1490–501.
93. de Leon J, Diaz FJ. A meta-analysis of worldwide studies demonstrates an association between schizophrenia and tobacco smoking behaviors. Schizophr Res (2005) 76:135–57. doi:10.1016/j.schres.2005.02.010
94. Hughes JR, Hatsukami DK, Mitchell JE, Dahlgren LA. Prevalence of smoking among psychiatric outpatients. Am J Psychiatry (1986) 143:993–7.
95. Ziedonis DM, George TP. Schizophrenia and nicotine use: report of a pilot smoking cessation program and review of neurobiological and clinical issues. Schizophr Bull (1997) 23:247–54. doi:10.1093/schbul/23.2.247
96. Adler LE, Hoffer LD, Wiser A, Freedman R. Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am J Psychiatry (1993) 150:1856–61.
97. Olincy A, Harris JG, Johnson LL, Pender V, Kongs S, Allensworth D, et al. Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch Gen Psychiatry (2006) 63:630–8. doi:10.1001/archpsyc.63.6.630
98. Dulude L, Labelle A, Knott VJ. Acute nicotine alteration of sensory memory impairment in smokers with schizophrenia. J Clin Psychopharmacol (2010) 30:541–8. doi:10.1097/JCP.0b013e3181f0c9c6
99. Fisher DJ, Grant B, Smith DM, Borracci G, Labelle A, Knott VJ. Nicotine and the hallucinating brain: effects on mismatch negativity (MMN) in schizophrenia. Psychiatry Res (2012) 196:181–7. doi:10.1016/j.psychres.2012.01.026
100. McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science (1995) 269:1692–6. doi:10.1126/science.7569895
101. Alkondon M, Pereira EF, Eisenberg HM, Albuquerque EX. Nicotinic receptor activation in human cerebral cortical interneurons: a mechanism for inhibition and disinhibition of neuronal networks. J Neurosci (2000) 20:66–75.
102. Ji D, Dani JA. Inhibition and disinhibition of pyramidal neurons by activation of nicotinic receptors on hippocampal interneurons. J Neurophysiol (2000) 83:2682–90.
103. Gioanni Y, Rougeot C, Clarke PB, Lepouse C, Thierry AM, Vidal C. Nicotinic receptors in the rat prefrontal cortex: increase in glutamate release and facilitation of mediodorsal thalamo-cortical transmission. Eur J Neurosci (1999) 11:18–30. doi:10.1046/j.1460-9568.1999.00403.x
104. Lambe EK, Picciotto MR, Aghajanian GK. Nicotine induces glutamate release from thalamocortical terminals in prefrontal cortex. Neuropsychopharmacology (2003) 28:216–25. doi:10.1038/sj.npp.1300032
105. Radcliffe KA, Fisher JL, Gray R, Dani JA. Nicotinic modulation of glutamate and GABA synaptic transmission of hippocampal neurons. Ann N Y Acad Sci (1999) 868:591–610. doi:10.1111/j.1749-6632.1999.tb11332.x
106. Levin ED, Bettegowda C, Weaver T, Christopher NC. Nicotine-dizocilpine interactions and working and reference memory performance of rats in the radial-arm maze. Pharmacol Biochem Behav (1998) 61:335–40. doi:10.1016/S0091-3057(98)00109-9
107. Rezvani AH, Levin ED. Nicotinic-glutamatergic interactions and attentional performance on an operant visual signal detection task in female rats. Eur J Pharmacol (2003) 465:83–90. doi:10.1016/S0014-2999(03)01439-0
108. Ciamei A, Aversano M, Cestari V, Castellano C. Effects of MK-801 and nicotine combinations on memory consolidation in CD1 mice. Psychopharmacology (Berl) (2001) 154:126–30. doi:10.1007/s002130000584
109. Spitzer RL, Williams JB, Gibbon M, First MB. Structured Clinical Interview for DSM-III-R – Non-Patient Edition (SCID-NP, Version 1.0 with Supplement for DSM-IV). Washington, DC: American Psychiatric Press (1990).
110. Wechsler D. Manual for the Wechsler Adult Intelligence Scale. New York: Psychological Corporation (1955). 110 p.
111. Heatherton T, Kozlowski L, Frecker R, Fagerstrom K. The Fagerstrom test for nicotine dependence: a revision of the Fagerstrom tolerance questionnaire. Br J Addict (1991) 86:1119–27. doi:10.1111/j.1360-0443.1991.tb01879.x
112. Kay S, Fiszbein A, Opler L. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull (1987) 13:261–76. doi:10.1093/schbul/13.2.261
113. Bremner JD, Krystal JH, Putnam FW, Southwick SM, Marmar C, Charney DS, et al. Measurement of dissociative states with the Clinician-Administered Dissociative States Scale (CADSS). J Trauma Stress (1998) 11:125–36. doi:10.1023/A:1024465317902
114. Krystal JH, Perry EB Jr, Gueorguieva R, Belger A, Madonick SH, Abi-Dargham A, et al. Comparative and interactive human psychopharmacologic effects of ketamine and amphetamine: implications for glutamatergic and dopaminergic model psychoses and cognitive function. Arch Gen Psychiatry (2005) 62:985–94. doi:10.1001/archpsyc.62.9.985
115. Hariharan M, VanNoord T, Greden JF. A high-performance liquid-chromatographic method for routine simultaneous determination of nicotine and cotinine in plasma. Clin Chem (1988) 34:724–9.
116. Staley JK, Krishnan-Sarin S, Cosgrove KP, Krantzler E, Frohlich E, Perry E, et al. Human tobacco smokers in early abstinence have higher levels of beta2* nicotinic acetylcholine receptors than nonsmokers. J Neurosci (2006) 26:8707–14. doi:10.1523/JNEUROSCI.0546-06.2006
117. Friedman D, Simpson G, Hamberger M. Age-related changes in scalp topography to novel and target stimuli. Psychophysiology (1993) 30:383–96. doi:10.1111/j.1469-8986.1993.tb02060.x
118. Cohen JD, Barch DM, Carter C, Servan-Schreiber D. Context-processing deficits in schizophrenia: converging evidence from three theoretically motivated cognitive tasks. J Abnorm Psychol (1999) 108:120–33. doi:10.1037/0021-843X.108.1.120
119. Gratton G, Coles MG, Donchin E. A new method for off-line removal of ocular artifact. Electroencephalogr Clin Neurophysiol (1983) 55:468–84. doi:10.1016/0013-4694(83)90135-9
120. Cho HS, Perry EB, Gunduz-Bruce H, D’Souza DC. The ketamine paradigm in humans: relevance to psychotic disorders. In: Heresco-Levy U, Daniel C, Javitt DC, editors. Glutamate in Neuropsychiatric Disorders Trivandrum. Kerala, India: Research Signpost (2008). p. 99–131.
121. Mathalon DH, Ford JM, Pfefferbaum A. Trait and state aspects of P300 amplitude reduction in schizophrenia: a retrospective longitudinal study. Biol Psychiatry (2000) 47:434–49. doi:10.1016/S0006-3223(99)00277-2
122. Mathalon DH, Ford JM, Rosenbloom M, Pfefferbaum A. P300 reduction and prolongation with illness duration in schizophrenia. Biol Psychiatry (2000) 47:413–27. doi:10.1016/S0006-3223(99)00151-1
123. Abi-Saab WM, D’Souza DC, Moghaddam B, Krystal JH. The NMDA antagonist model for schizophrenia: promise and pitfalls. Pharmacopsychiatry (1998) 31(Suppl 2):104–9. doi:10.1055/s-2007-979354
124. Coyle JT. NMDA receptor and schizophrenia: a brief history. Schizophr Bull (2012) 38:920–6. doi:10.1093/schbul/sbs076
125. Alain C, Bernstein LJ, Cortese F, Yu H, Zipursky RB. Deficits in automatically detecting changes in conjunction of auditory features in patients with schizophrenia. Psychophysiology (2002) 39:599–606. doi:10.1111/1469-8986.3950599
126. Devrim-Ucok M, Keskin-Ergen HY, Ucok A. Novelty P3 and P3b in first-episode schizophrenia and chronic schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2006) 30:1426–34. doi:10.1016/j.pnpbp.2006.05.019
127. Grillon C, Courchesne E, Ameli R, Geyer MA, Braff DL. Increased distractibility in schizophrenic patients. Electrophysiologic and behavioral evidence. Arch Gen Psychiatry (1990) 47:171–9. doi:10.1001/archpsyc.1990.01810140071010
128. Merrin EL, Floyd TC. P300 responses to novel auditory stimuli in hospitalized schizophrenic patients. Biol Psychiatry (1994) 36:527–42. doi:10.1016/0006-3223(94)90617-3
129. Daffner KR, Mesulam MM, Holcomb PJ, Calvo V, Acar D, Chabrerie A, et al. Disruption of attention to novel events after frontal lobe injury in humans. J Neurol Neurosurg Psychiatry (2000) 68:18–24. doi:10.1136/jnnp.68.1.18
130. Daffner KR, Mesulam MM, Scinto LF, Acar D, Calvo V, Faust R, et al. The central role of the prefrontal cortex in directing attention to novel events. Brain (2000) 123(Pt 5):927–39. doi:10.1093/brain/123.5.927
131. Woods DL, Knight RT. Electrophysiologic evidence of increased distractibility after dorsolateral prefrontal lesions. Neurology (1986) 36:212–6. doi:10.1212/WNL.36.2.212
132. Anand A, Charney DS, Oren DA, Berman RM, Hu XS, Cappiello A, et al. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of N-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry (2000) 57:270–6. doi:10.1001/archpsyc.57.3.270
133. Krystal JH, D’Souza DC, Karper LP, Bennett A, Abi-Dargham A, Abi-Saab D, et al. Interactive effects of subanesthetic ketamine and haloperidol in healthy humans. Psychopharmacology (Berl) (1999) 145:193–204. doi:10.1007/s002130051049
134. Krystal JH, Karper LP, Bennett A, D’Souza DC, Abi-Dargham A, Morrissey K, et al. Interactive effects of subanesthetic ketamine and subhypnotic lorazepam in humans. Psychopharmacology (Berl) (1998) 135:213–29. doi:10.1007/s002130050503
135. Ernst M, Heishman SJ, Spurgeon L, London ED. Smoking history and nicotine effects on cognitive performance. Neuropsychopharmacology (2001) 25:313–9. doi:10.1016/S0893-133X(01)00257-3
136. Lawrence NS, Ross TJ, Stein EA. Cognitive mechanisms of nicotine on visual attention. Neuron (2002) 36:539–48. doi:10.1016/S0896-6273(02)01004-8
137. Mancuso G, Andres P, Ansseau M, Tirelli E. Effects of nicotine administered via a transdermal delivery system on vigilance: a repeated measure study. Psychopharmacology (Berl) (1999) 142:18–23. doi:10.1007/s002130050857
138. McClernon FJ, Gilbert DG, Radtke R. Effects of transdermal nicotine on lateralized identification and memory interference. Hum Psychopharmacol (2003) 18:339–43. doi:10.1002/hup.488
139. Arendash GW, Sanberg PR, Sengstock GJ. Nicotine enhances the learning and memory of aged rats. Pharmacol Biochem Behav (1995) 52:517–23. doi:10.1016/0091-3057(95)00119-H
140. Levin ED, Simon BB. Nicotinic acetylcholine involvement in cognitive function in animals. Psychopharmacology (Berl) (1998) 138:217–30. doi:10.1007/s002130050667
Keywords: schizophrenia, nicotine, ketamine, N-methyl-d-aspartate receptor, nicotinic acetylcholine receptor, event-related potential, mismatch negativity, P300
Citation: Mathalon DH, Ahn K-H, Perry EB Jr, Cho H-S, Roach BJ, Blais RK, Bhakta S, Ranganathan M, Ford JM and D’Souza DC (2014) Effects of nicotine on the neurophysiological and behavioral effects of ketamine in humans. Front. Psychiatry 5:3. doi: 10.3389/fpsyt.2014.00003
Received: 25 October 2013; Paper pending published: 19 November 2013;
Accepted: 07 January 2014; Published online: 24 January 2014.
Edited by:
André Schmidt, University of Basel, SwitzerlandReviewed by:
Kevin M. Spencer, Harvard Medical School, USAVerner Knott, University of Ottawa Institute of Mental Health Research, Canada
Copyright: © 2014 Mathalon, Ahn, Perry Jr, Cho, Roach, Blais, Bhakta, Ranganathan, Ford and D’Souza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel H. Mathalon, Mental Health Service (116D), San Francisco VA Medical Center, 4150 Clement Street, San Francisco, CA 94121, USA e-mail:ZGFuaWVsLm1hdGhhbG9uQHVjc2YuZWR1