 Adam J. Walker1,2
Adam J. Walker1,2 Yesul Kim1,2
Yesul Kim1,2 J. Blair Price1
J. Blair Price1 Rajas P. Kale1,3
Rajas P. Kale1,3 Jane A. McGillivray2
Jane A. McGillivray2 Michael Berk4,5,6,7
Michael Berk4,5,6,7 Susannah J. Tye1,2,8*
Susannah J. Tye1,2,8*- 1Department of Psychiatry and Psychology, Mayo Clinic, Rochester, MN, USA
- 2School of Psychology, Deakin University, Melbourne, VIC, Australia
- 3School of Engineering, Deakin University, Geelong, VIC, Australia
- 4School of Medicine, Deakin University, Geelong, VIC, Australia
- 5Department of Psychiatry, University of Melbourne, Melbourne, VIC, Australia
- 6Orygen Youth Health Research Centre, Melbourne, VIC, Australia
- 7The Florey Institute of Neuroscience and Mental Health, Melbourne, VIC, Australia
- 8Department of Psychiatry, University of Minnesota, Minneapolis, MN, USA
The mood disorder prodrome is conceptualized as a symptomatic, but not yet clinically diagnosable stage of an affective disorder. Although a growing area, more focused research is needed in the pediatric population to better characterize psychopathological symptoms and biological markers that can reliably identify this very early stage in the evolution of mood disorder pathology. Such information will facilitate early prevention and intervention, which has the potential to affect a person’s disease course. This review focuses on the prodromal characteristics, risk factors, and neurobiological mechanisms of mood disorders. In particular, we consider the influence of early-life stress, inflammation, and allostatic load in mediating neural mechanisms of neuroprogression. These inherently modifiable factors have known neuroadaptive and neurodegenerative implications, and consequently may provide useful biomarker targets. Identification of these factors early in the course of the disease will accordingly allow for the introduction of early interventions which augment an individual’s capacity for psychological resilience through maintenance of synaptic integrity and cellular resilience. A targeted and complementary approach to boosting both psychological and physiological resilience simultaneously during the prodromal stage of mood disorder pathology has the greatest promise for optimizing the neurodevelopmental potential of those individuals at risk of disabling mood disorders.
Introduction
There is increasing appreciation for the need to both identify and treat mood disorders during their earliest stages (1). Although some dispute remains, maladaptive changes in mood and behavior first become evident during the prodromal period (2). However, the low specificity of these changes makes the prodromal stage difficult to definitively characterize prior to disease onset (3). Observable changes in mood and general physiologic functioning can include increases in sadness, anhedonia, irritability, anger, and anxiety, together with alterations in sleep and energy (4). Correlating these symptoms with prodromal biomarkers offers an exciting juncture whereby targeted interventions could be opportunistically employed to prevent neurodegenerative changes from accruing as the disease progresses (5). The potential to intervene during the prodromal stage of psychiatric illness through the detection and remediation of novel biomarkers has perhaps been best studied in schizophrenia, wherein most individuals experience a lengthy prodromal period prior to the full emergence of diagnosable psychotic symptoms (6). As an exemplar, low levels of nervonic acid appear to be a risk factor for conversion from high-risk to frank psychosis (7), and this risk of conversion may be reduced by targeted omega-3 fatty acid supplementation (8). Encouraging results from this work have renewed interest in the early detection of affective disorders, particularly bipolar disorder, with the hope that earlier and more targeted interventions might slow disease progression (3, 9–12). This can significantly impact neuroprogression and subsequent disease course for the individual (13). This concept of “neuroprogression” refers to the cumulative restructuring of the central nervous system which in turn mediates the development and persistence of psychiatric illness (14, 15). This process results from disturbances in inflammatory mediators, neurotrophins, oxidative stress, and energy regulation (14, 15).
Biomarker Strategies for Prodromal Mood Disorders
Stress and Allostatic Load
Stress sensitization and early detection
Stress is one of the best-studied mediators by which genetic vulnerabilities are translated into mood disorder pathology through the process of neuroprogression (16–18). Numerous studies have demonstrated that both depression and bipolar disorder are more prevalent in individuals who have experienced adverse early-life events. This is partly because such experiences prime future physiologic and neural responses to stress, elicit a state of chronic inflammation (19), alter cellular mediators of plasticity and energy metabolism, and increase cellular “wear and tear” (20–22). Early-life stress (2) can be particularly deleterious because of its potential to influence the programing of the hypothalamic–pituitary–adrenal (HPA) axis (23) to induce persistent sensitization of neuroendocrine, autonomic, oxidative, and immune responses to stress. Over time these sensitized systems cumulatively contribute to the cellular and synaptic alterations underlying neuroprogression (21, 24–26). Specific examples include changes in reactivity of inflammatory cytokines [e.g., interleukin 6 (IL-6)] (25), alterations in markers for lipid peroxidation [e.g., 8-iso-prostaglandin F (2α)], oxidative damage to DNA (8-hydroxy-2′-deoxyguanosine) and RNA (8-hydroxyguanosine) (24), as well as altered cortisol, adrenocorticotropic hormone, and corticotrophin releasing factor responses (26). Identification of the state of physiologic and cellular resilience or sensitivity to stress may provide an important indicator of the level of neuroprogression and stress-mediated disease pathology for affective disorders, potentially prior to the initial manifestation of the mood episode (22).
One mechanism whereby HPA axis sensitization is likely to occur is through epigenetic regulation of stress response processes (21, 27). Evidence shows that exposure to various forms of stress result in multiple epigenetic changes in limbic regions as well as the HPA axis (21, 27). Interestingly, a recent study by Klendel and colleagues (18) found that only individuals who exhibited allele-specific DNA demethylation in functional glucocorticoid response elements of FK506 binding protein 5 (FKBP5), were prone to developing persistent cortisol dysregulation (18, 21). Further, this association was found to be dependent on an interaction effect with trauma in early life, suggesting that key developmental stages are directly related to stability of the observed effects across time (18). In another study, significant interactions between peripheral FKBP5 mRNA expression and disease progression were reported, suggesting that polymorphisms in the gene directly impact the extent of neuroendocrine dysregulation, and corresponding neuroprogression (28). The FKBP5 risk allele and corresponding levels of mRNA expression may represent useful biomarkers. These markers could be employed to identify individuals in the prodromal stages of stress-sensitive psychiatric disorders, such as major depression or bipolar disorder. Such detection would facilitate early intervention and could improve resilience and alleviate allostatic load in the prodromal individual.
Early-life stress and accumulation of allostatic load
Accumulation of allostatic load is a key mechanism through which early-life stress is thought to result in psychopathology (29). This is mediated via a series of enduring adaptive changes across a range of systems primed both to respond rapidly to challenge, as well as to restore homeostatic equilibrium (30). Adaptive allostatic mechanisms may fail when chronically challenged or when regulatory systems falter. This leads to a state of allostatic overload, which is thought to considerably impact the clinical course of mood disorders (31–33). Without sufficient opportunity for recovery, the brain and body are repeatedly exposed to molecular mediators of stress that can increase the level of cellular “wear and tear” (33). These mediators, which include metabolic factors, inflammatory cytokines, neurotrophins, and oxidative species, collectively impact an individual’s mental and physical resilience as outlined below [for more detailed reviews see Ref. (6, 34, 35)]. Both physiological (i.e., immune and/or metabolic) and psychological (i.e., bullying) stressors contribute significantly to allostatic load, and thus need to be considered together when assessing both risk and relative staging of mood disorder pathology (6, 34).
Enhancing an individual’s capacity to buffer the physiologic toll that accumulates through allostatic overload should be considered an important early intervention strategy. As allostatic load accumulates and attempts to maintain cellular homeostasis fail, cell danger signals are propagated and pro-apoptotic cell signaling pathways become increasingly engaged (36–39). This may play a role in medical comorbidities such as heart disease (40), as well as interfere with the therapeutic mechanisms of antidepressants and mood stabilizers to impair treatment efficacy (41–43). Internal stressors that activate the HPA axis and associated allostatic systems can limit an individual’s capacity for allostasis even prior to the onset of external stressors (36). For example, an endogenous load can build through the expression of homocysteine or inflammatory cytokines, limiting the capacity of adaptive responses in the face of subsequent stressors. Interventions that counter this load and reduce levels of proinflammatory mediators or interfere with their neuromodulatory actions could limit neuroprogression in both bipolar and unipolar depression, as well as enhance capacity for antidepressant efficacy (44–46).
Inflammatory Profile
Stress during earlier life is not only associated with disruption of the HPA axis, but may also serve to sensitize proinflammatory responses to future insults (47–49). Inflammatory mechanisms are increasingly appreciated for their critical role in mood disorder pathophysiology, in particular via their regulation of neuronal excitability, synaptic transmission, synaptic plasticity and neuronal survival (41, 50, 51). Of specific interest are proinflammatory mediators, such as cytokines [i.e., interleukin 1, IL-6, and tumor necrosis factor alpha (TNF-α)] and C-reactive protein (CRP). CRP is often used as a biomarker for inflammation in studies due to its relationship with proinflammatory cytokines and role in the immune response. As demonstrated by Slopen and colleagues (49), individuals at ages 10 and 15 who reported adverse life events at critical stages between the ages of 1.5 and 8 years were found to have significantly increased levels of CRP and IL-6. These heightened concentrations were correlated with immune activation and depressive-like symptoms. Notably, increased CRP levels have been used previously to predict depression severity and recurrence rates in males (48, 52).
There is a growing literature supporting the use of inflammatory biomarkers as predictors of ensuing mood disorder pathology (22). Research to date has been focused on investigating the relationship between inflammatory cytokines and affective disorders in adults; however, their specific role in early onset/adolescent psychopathology is less well explored (53). Cytokines are thought to influence neurodevelopment during key stages, such as adolescence, interacting with biological systems including those of stress hormones and gonadal hormones (53). As such, perturbation of inflammatory balance in adolescents may significantly contribute to neuroprogression and development of psychiatric illness (19, 53, 54). For example, elevated serum levels of TNF-α, IL-6, and interleukin-10 (IL-10) have been reported during the early stages of bipolar disorder (55), and CRP appears to be a biomarker of de novo depression risk (56).
As the mood disorder pathology progresses, an increasing number of proinflammatory cytokines are observed, including elevated levels of interferon gamma (IFN-γ) (22, 54, 55). Notably, increases in IFN-γ are associated with dysregulation of the tryptophan metabolite pathway via direct role in indoleamine 2,3-dioxygenase (IDO) activation. Activation of IDO is commonly found in later stages of mood disorders, and is a biomarker of depression-like behavior mediated by neural inflammation in animal models (48). Proinflammatory cytokines activate IDO, resulting in depletion of serotonin and augmentation of quinolinic acid (QUIN) metabolism over kynurenic acid (KYNA). Tryptophan metabolites (kynurenine, KYNA, 3-hydroxykynurenine, and QUIN) act as neuromodulators to influence behavioral, neuroendocrine, and neurochemical aspects of depression (57–60). Consequently, this accumulation of QUIN facilitates neurodegeneration over neuroprotection, impacting mood disorder neuroprogression and resultant disability (61).
It is noteworthy to mention several other findings regarding altered inflammation in youth with psychiatric pathology. Increased mRNA and protein expression levels of IL-1β, IL-6, and TNF-α were reported in the anterior prefrontal cortex of adolescent suicide victims compared with normal control subjects (62). Elevated levels of inflammatory cytokines (among others: TNF-α, IL-1β, IL-6, and IFN-γ) were also observed in the serum of pediatric patients who experienced first-episode psychosis, in addition to increased leukocyte counts and evidence of blood–brain barrier damage (63). Quantification of inflammatory biomarkers (e.g., TNF-α, IL-6, IL-10, or CRP) may thus prove useful for detecting individuals at risk for developing a mood disorder. A recent study by Byrne and colleagues (64) suggests that levels of peripheral cytokines (e.g., IFN-γ) and CRP in salivary samples may correlate with serum samples in young people. Salivary assay may prove to be a simpler, less invasive method of estimating peripheral levels of inflammatory markers in adolescents (64). This provides one avenue whereby prodromal individuals could potentially be identified and their disease onset delayed.
Diminished Synaptic Integrity
Homeostatic control of synaptic connections within key mood-related circuits plays a critical role in the etiology of mood disorders (65). Stress and inflammation as discussed in previous sections are implicated in disruption of synaptic signaling and integrity during the early stages of mood disorder pathogenesis. This is mediated in part through the inhibition of neurotrophin function, of which brain derived neurotrophic factor (BDNF) is the most thoroughly characterized. BDNF plays an important role in neuronal development, survival, and function, including activity-dependent synaptic plasticity (66). Synaptic plasticity is characterized by various processes, including synaptic remodeling, synaptogenesis, long-term potentiation, and long-term depression, all of which critically mediate the flow of electrochemical information throughout the central nervous system (67, 68). Stress, allostatic load, inflammation, antidepressants, and mood stabilizers exert major effects on signaling pathways that regulate cellular plasticity, suggesting these are critical neurobiological mediators of mood dysfunction and therapeutic intervention (69–72).
Glycogen synthase kinase-3 (GSK-3), part of the signaling cascade regulated by BDNF, plays an important role in synaptic homeostasis through regulation of synaptic deconsolidation (pruning) and glutamate receptor cycling (73). Increased GSK-3-mediated synaptic deconsolidation has been suggested to be an important factor contributing to reduced spine density in mood disorders (74). Additionally, levels of activated GSK-3 are increased in postmortem brain tissue from individuals with unipolar and bipolar depression (74). In addition to BDNF, GSK-3 is deactivated by signals originating from numerous signaling pathways demonstrated to be dysregulated in mood disorders (e.g., Wnt and PI3K pathways), and is either the direct or downstream target of many mood stabilizer and antidepressant medications (75). GSK-3 activity is modulated by serotonin and dopamine, and is a critical node at the intersection of multiple neurotransmitter and cell signaling cascades (68). As a result, GSK-3 modulates not only synaptic plasticity but also apoptotic mechanisms and, in turn, plays a critical role in mediating cellular resilience (75). For this reason, GSK-3 has received much attention for its potential to be targeted as an early intervention strategy during the prodrome period.
Identifying Impaired Cellular Resilience
Stress, allostatic overload, and neuroinflammation function together to impair synaptic plasticity and cellular resilience. Disrupted plasticity along with increased cellular vulnerability contributes significantly to the pathophysiology of mood disorders and directly to the neuroprogressive nature of the disease course (3, 76). Some of the key mechanisms of disease progression affecting cellular resilience include: oxidative stress, decreased neurotrophic factor expression, reduced neurogenesis, impaired regulation of calcium, altered endoplasmic reticulum and mitochondrial function, together with dysregulated energy metabolism and insulin signaling. Each of these mechanisms are mediated by allostatic overload and neuroinflammation [for detailed reviews see Ref. (3, 36, 76–78)]. Together, these processes demonstrate that in addition to synaptic integrity, maintenance of cellular homeostasis is critical for facilitating cellular resilience and attenuating mood disorder pathogenesis (79), which is also likely to enhance the capacity for treatment response during later stages of the disorder (80).
Cellular vulnerability and resilience are mediated by apoptotic and anti-apoptotic intracellular signaling cascades, respectively. Apoptosis is important for the regulation of developmental processes and prevention of cancerous growths. Excessive apoptosis in neuronal systems, however, leads to neurodegeneration and certain cell populations are at increased risk of stress-mediated apoptotic cell death (80). Apoptosis is a tightly regulated and energy-dependent process, which coordinates programed cell death in response to different stimuli (81). This can occur through stimulation of death receptor proteins [i.e., tumor necrosis factor (TNF) receptor] by cytokines of the TNF superfamily or in response to mitochondrial degradation. These stimuli result in activation of executioner caspases that function to coordinate cellular process necessary for apoptosis, including cessation of cell repair processes and cell cycle progression, cytoskeletal and nuclear disassembly, and flagging the cell for phagocytosis (82). Distinct classes of antidepressants and mood stabilizers have been demonstrated to facilitate cellular resilience to prevent progression of pro-apoptotic processes, and novel treatments are currently being developed to target these specific mechanisms (83). Biomarkers that characterize the level of neuronal vulnerability relative to resilience may prove useful as biomarkers of prodromal mood disorder pathology. This has been demonstrated for later stages of bipolar disorder (84), however more studies are needed to determine the utility of such cell danger biomarkers during the mood disorder prodrome (22).
Opportunities for Prevention and Intervention
Identifying Vulnerabilities and Building Resilience at the Cellular Level
Identification of individuals at risk of developing a mood disorder, or those in the prodromal stage, provides a potential opportunity to target these mechanisms for neuroprotective interventions that enhance cellular resilience, maintain synaptic plasticity and boost psychological resilience (Figure 1) (85). One of the longest held notions of brain plasticity is that certain critical periods or windows exist in development, during which circuitry is consolidated for lifetime functionality. Recently, there is a rising consensus that developmentally induced plasticity can, to an extent, be reversed by “re-opening” those windows of plasticity (86). Hyman and Nestler (87) have underscored the importance of shifting the brain into an “adaptive state” to necessitate the antidepressant response. Their theory of “initiation and adaption” is exemplified by psychotropic drugs wherein primary molecular targets that initiate alterations in brain function activate homeostatic mechanisms that return the system to an adaptive and treatment responsive state (87). Plasticity and cellular resilience are thus necessary for the efficacy of antidepressants and mood stabilizing treatments. McGorry and colleagues (6, 88) and others (89) have demonstrated this concept with pre-psychotic interventions, and repeatedly emphasized the need to take advantage of the “windows of opportunity” present within the prodromal stages of psychiatric disease (6, 88, 89). During this stage, the course of the disease remains theoretically plastic and amenable to intervention (90). Previous literature indicates that once risk or prodromal symptoms of mood disorders are identified, there is some (91), but not unequivocal (92) evidence that early intervention in adolescents can significantly reduce mood-related symptoms and incidence of fully diagnosable psychiatric disorders such as depression (93–95). Neuroprotective pharmacotherapies together with appropriate psychotherapy may reduce the risk of neuropsychiatric disease progression in young people which, together with allostatic load reducing behavioral interventions, may significantly slow the trajectory of the disease course into adulthood (6, 36, 96). Such interventions may include reducing lifestyle mediators of allostatic load (19, 97).
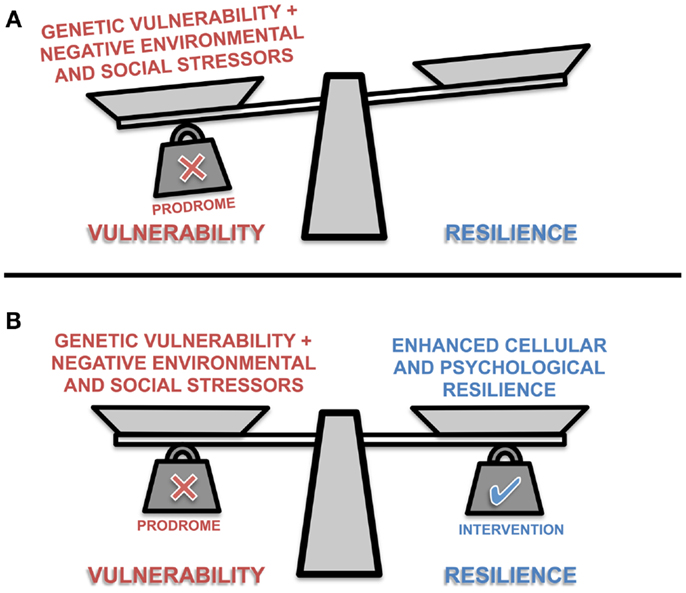
Figure 1. A representation of the conceptual balance between vulnerability and resilience in prodromal individuals. The scale’s balance beam teeters between vulnerability and resilience as scale pans are loaded with different positive and negative biological, psychological and social factors. The presence or absence of these factors influence the ability of the individual to cope with stressors, and maintain allostasis. (A) Prodromal individuals are somewhat predisposed to vulnerability; but with intervention (B) an individual may adopt more adaptive environmental coping strategies, support mechanisms, general healthy lifestyle choices, and/or receive pharmacological interventions that collectively enhance physiological and psychological resilience.
Cognitive and Behavioral Interventions to Buffer Stress and Build Resilience
Individuals provided with effective social and emotional support to help cope with stressors that are adverse and potentially taxing will be much better placed to limit associated biological costs and maintain allostasis (98). The absence of emotional or social support and the implementation of maladaptive coping strategies can enhance the toxic effects of stress and contribute to allostatic overload (98). Exposure to regular and controllable stressors over the course of childhood and adolescence is essential for the development of effective coping strategies. Through such exposure, an individual can develop a repertoire of these coping strategies. Mathew and Nanoo (99) found that adaptive coping strategies (e.g., employing self-control, accepting responsibilities, problem solving, seeking social support, or positive re-appraisal) are protective for suicide risk in adolescents. Conversely, maladaptive coping strategies, such as confrontation, distancing, and escape-avoidance were reported to be significant risk factors associated with adolescent suicide attempts (99). These findings provide evidence to support the notion that coping strategies can act as protective factors against both the development and progression of mood disorders. Importantly, educating children and adolescents in protective coping skills may be a promising intervention that could be implemented as early as elementary school. In recent years, patterns of threat perception such as optimism have attracted much attention in relation to later mood, coping, and immune change in response to stress (100, 101). Moreover, it has been found to be protective against the development of depressive symptoms in later life (102). Its potential role in buffering against the negative emotional consequence of adverse events has led to a view of optimism as an index of resilience (103). Optimists may also choose lifestyles that promote physical as well as mental health, thereby reducing other aspects of allostatic load.
Healthy lifestyle, similar to optimism, provides a solid foundation for adaptation, and increases available resources for buffering the neurodegenerative effects of stress. Specifically, previous literature highlights the importance of healthy diet, adequate sleep, avoidance of smoking, and sufficient exercise (104). A population-based study reported higher emotional well-being among physically active youths, independent of social class and health status (105). Across a 2-year period, Motl and colleagues (106) found changes in physical activity were inversely related to a change in depressive symptoms. Levels of physical activity in childhood can modulate the risk of adult depression (107). Exercise modulates many of the core biomarkers of neuroprogression, including inflammation, oxidative stress, and neurotrophins (108). Poor eating habits and sleep have been linked to the manifestation of toxic stress and unhealthy growth in pediatrics by disrupting the architecture of the plastic, adaptive brain (109). There is now extensive evidence that poor diet quality is a risk for adolescent depression (110), and new data suggests that maternal diet influences the mental health of offspring (111). Similarly, smoking increases the risk of mood and anxiety disorders, and appears to influence similar biological pathways (112, 113). Parents and care givers of younger children need to be informed of the potential impact that a healthy lifestyle can have in mitigating mood-related symptoms and problematic behaviors. Low-risk interventions such as those aforementioned are critical for enhancing both psychological and biological resilience to stress. When such perspectives and lifestyle health behaviors are consolidated early in childhood and adolescence, the cumulative effect may be meaningful (103).
Conclusion
Early intervention offers the possibility of altering the trajectory of mood disorder pathology. In so doing, we may curtail the progressive nature of the illness, both through neuroprotection and maintenance of peripheral health. Prevention and intervention treatments should go beyond stabilizing mood to include various and complementary strategies for reducing allostatic load, perhaps through psychoeducation and lifestyle-related interventions, including effective stress management. The combination of these techniques with specific pharmacotherapies may significantly improve functional outcomes by both reducing cellular insults and enhancing resilience. In so doing, this optimizes the capacity for maintenance of synaptic integrity and cellular resilience, which must be aggressively targeted as a therapeutic strategy during the prodromal stage of mood disorder pathology (90). This neuroprotective approach not only slows neuroprogression associated with the disease, but lays a foundation for more treatment-responsive outcomes during later stages.
Author Contributions
Adam J. Walker, Yesul Kim, J. Blair Price, Rajas P. Kale, Jane A. McGillivray, Michael Berk, and Susannah J. Tye each made contributions to the writing of this manuscript.
Conflict of Interest Statement
Michael Berk has received grant/research support from the NIH, Cooperative Research Centre, Simons Autism Foundation, Cancer Council of Victoria, Stanley Medical Research Foundation, MBF, NHMRC, Beyond Blue, Rotary Health, Geelong Medical Research Foundation, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, Meat and Livestock Board, Organon, Novartis, Mayne Pharma, Servier and Woolworths, has been a speaker for Astra Zeneca, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, Janssen Cilag, Lundbeck, Merck, Pfizer, Sanofi Synthelabo, Servier, Solvay, and Wyeth, and served as a consultant to Astra Zeneca, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, Janssen Cilag, Lundbeck, Merck, and Servier. The other authors have no conflicts to report.
Acknowledgments
This work was supported by a Mayo Minnesota Partnership grant to Susannah J. Tye. Adam J. Walker is supported by an Australian Postgraduate Award. Yesul Kim is supported by a Deakin University Award. Michael Berk is supported by a NHMRC Senior Principal Research Fellowship 1059660.
References
1. Madhusoodanan S. Preventive psychiatry: we are getting closer to fulfilling the promise of reducing mental illness. Ann Clin Psychiatry (2010) 22(4):217–8.
2. Skjelstad DV, Malt UF, Holte A. Symptoms and signs of the initial prodrome of bipolar disorder: a systematic review. J Affect Disord (2010) 126(1–2):1–13. doi: 10.1016/j.jad.2009.10.003
3. Berk M, Hallam K, Lucas N, Hasty M, McNeil CA, Conus P, et al. Early intervention in bipolar disorders: opportunities and pitfalls. Med J Aust (2007) 187(7 Suppl):S11–4.
4. Egeland JA, Hostetter AM, Pauls DL, Sussex JN. Prodromal symptoms before onset of manic-depressive disorder suggested by first hospital admission histories. J Am Acad Child Adolesc Psychiatry (2000) 39(10):1245–52. doi:10.1097/00004583-200010000-00011
5. Strakowski SM, DelBello MP, Zimmerman ME, Getz GE, Mills NP, Ret J, et al. Ventricular and periventricular structural volumes in first- versus multiple-episode bipolar disorder. Am J Psychiatry (2002) 159(11):1841–7. doi:10.1176/appi.ajp.159.11.1841
6. McGorry PD, Yung AR, Phillips LJ, Yuen HP, Francey S, Cosgrave EM, et al. Randomized controlled trial of interventions designed to reduce the risk of progression to first-episode psychosis in a clinical sample with subthreshold symptoms. Arch Gen Psychiatry (2002) 59(10):921–8. doi:10.1001/archpsyc.59.10.921
7. Amminger GP, Schafer MR, Klier CM, Slavik JM, Holzer I, Holub M, et al. Decreased nervonic acid levels in erythrocyte membranes predict psychosis in help-seeking ultra-high-risk individuals. Mol Psychiatry (2012) 17(12):1150–2. doi:10.1038/mp.2011.167
8. Amminger GP, Schafer MR, Papageorgiou K, Klier CM, Cotton SM, Harrigan SM, et al. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Arch Gen Psychiatry (2010) 67(2):146–54. doi:10.1001/archgenpsychiatry.2009.192
9. Bauer ME, Wieck A, Lopes RP, Teixeira AL, Grassi-Oliveira R. Interplay between neuroimmunoendocrine systems during post-traumatic stress disorder: a mini review. Neuroimmunomodulation (2010) 17(3):192–5. doi:10.1159/000258721
10. Leopold K, Ritter P, Correll CU, Marx C, Ozgurdal S, Juckel G, et al. Risk constellations prior to the development of bipolar disorders: rationale of a new risk assessment tool. J Affect Disord (2012) 136(3):1000–10. doi:10.1016/j.jad.2011.06.043
11. Luby JL, Navsaria N. Pediatric bipolar disorder: evidence for prodromal states and early markers. J Child Psychol Psychiatry (2010) 51(4):459–71. doi:10.1111/j.1469-7610.2010.02210.x
12. Schultze-Lutter F, Schimmelmann BG, Klosterkotter J, Ruhrmann S. Comparing the prodrome of schizophrenia-spectrum psychoses and affective disorders with and without psychotic features. Schizophr Res (2012) 138(2–3):218–22. doi:10.1016/j.schres.2012.04.001
13. Weissman MM, Wolk S, Goldstein RB, Moreau D, Adams P, Greenwald S, et al. Depressed adolescents grown up. JAMA (1999) 281(18):1707–13. doi:10.1001/jama.281.18.1707
14. Berk M, Kapczinski F, Andreazza AC, Dean OM, Giorlando F, Maes M, et al. Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci Biobehav Rev (2011) 35(3):804–17. doi:10.1016/j.neubiorev.2010.10.001
15. Gama CS, Kunz M, Magalhaes PV, Kapczinski F. Staging and neuroprogression in bipolar disorder: a systematic review of the literature. Rev Bras Psiquiatr (2013) 35(1):70–4. doi:10.1016/j.rbp.2012.09.001
16. Eley TC, Liang H, Plomin R, Sham P, Sterne A, Williamson R, et al. Parental familial vulnerability, family environment, and their interactions as predictors of depressive symptoms in adolescents. J Am Acad Child Adolesc Psychiatry (2004) 43(3):298–306. doi:10.1097/00004583-200403000-00011
17. Klengel T, Binder EB. Gene x environment interactions in the prediction of response to antidepressant treatment. Int J Neuropsychopharmacol (2013) 16(3):701–11. doi:10.1017/S1461145712001459
18. Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci (2013) 16(1):33–41. doi:10.1038/nn.3275
19. Berk M, Williams LJ, Jacka FN, O’Neil A, Pasco JA, Moylan S, et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med (2013) 11:200. doi:10.1186/1741-7015-11-200
20. Anda RF, Butchart A, Felitti VJ, Brown DW. Building a framework for global surveillance of the public health implications of adverse childhood experiences. Am J Prev Med (2010) 39(1):93–8. doi:10.1016/j.amepre.2010.03.015
21. Ehlert U. Understanding the trans-generational consequences of prenatal stress. J Psychosom Res (2013) 75(4):297–8. doi:10.1016/j.jpsychores.2013.09.002
22. Fries GR, Pfaffenseller B, Stertz L, Paz AV, Dargel AA, Kunz M, et al. Staging and neuroprogression in bipolar disorder. Curr Psychiatry Rep (2012) 14(6):667–75. doi:10.1007/s11920-012-0319-2
23. Lai MC, Huang LT. Effects of early life stress on neuroendocrine and neurobehavior: mechanisms and implications. Pediatr Neonatol (2011) 52(3):122–9. doi:10.1016/j.pedneo.2011.03.008
24. Aschbacher K, O’Donovan A, Wolkowitz OM, Dhabhar FS, Su Y, Epel E. Good stress, bad stress and oxidative stress: insights from anticipatory cortisol reactivity. Psychoneuroendocrinology (2013) 38(9):1698–708. doi:10.1016/j.psyneuen.2013.02.004
25. Carpenter LL, Gawuga CE, Tyrka AR, Lee JK, Anderson GM, Price LH. Association between plasma IL-6 response to acute stress and early-life adversity in healthy adults. Neuropsychopharmacology (2010) 35(13):2617–23. doi:10.1038/npp.2010.159
26. Heim C, Newport DJ, Heit S, Graham YP, Wilcox M, Bonsall R, et al. Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA (2000) 284(5):592–7. doi:10.1001/jama.284.5.592
27. Stankiewicz AM, Swiergiel AH, Lisowski P. Epigenetics of stress adaptations in the brain. Brain Res Bull (2013) 98(0):76–92. doi:10.1016/j.brainresbull.2013.07.003
28. Menke A, Klengel T, Rubel J, Bruckl T, Pfister H, Lucae S, et al. Genetic variation in FKBP5 associated with the extent of stress hormone dysregulation in major depression. Genes Brain Behav (2013) 12(3):289–96. doi:10.1111/gbb.12026
29. Howell BR, Sanchez MM. Understanding behavioral effects of early life stress using the reactive scope and allostatic load models. Dev Psychopathol (2011) 23(4):1001–16. doi:10.1017/S0954579411000460
30. McEwen BS, Stellar E. Stress and the individual. Mechanisms leading to disease. Arch Intern Med (1993) 153(18):2093–101. doi:10.1001/archinte.153.18.2093
31. Kapczinski F, Dias VV, Kauer-Sant’Anna M, Frey BN, Grassi-Oliveira R, Colom F, et al. Clinical implications of a staging model for bipolar disorders. Expert Rev Neurother (2009) 9(7):957–66. doi:10.1586/ern.09.31
32. Kapczinski F, Vieta E, Andreazza AC, Frey BN, Gomes FA, Tramontina J, et al. Allostatic load in bipolar disorder: implications for pathophysiology and treatment. Neurosci Biobehav Rev (2008) 32(4):675–92. doi:10.1016/j.neubiorev.2007.10.005
33. McEwen BS. Mood disorders and allostatic load. Biol Psychiatry (2003) 54(3):200–7. doi:10.1016/S0006-3223(03)00177-X
34. Moylan S, Maes M, Wray NR, Berk M. The neuroprogressive nature of major depressive disorder: pathways to disease evolution and resistance, and therapeutic implications. Mol Psychiatry (2013) 18(5):595–606. doi:10.1038/mp.2012.33
35. Pasco JA, Jacka FN, Williams LJ, Henry MJ, Nicholson GC, Kotowicz MA, et al. Leptin in depressed women: cross-sectional and longitudinal data from an epidemiologic study. J Affect Disord (2008) 107(1–3):221–5. doi:10.1016/j.jad.2007.07.024
36. Brietzke E, Kapczinski F, Grassi-Oliveira R, Grande I, Vieta E, McIntyre RS. Insulin dysfunction and allostatic load in bipolar disorder. Expert Rev Neurother (2011) 11(7):1017–28. doi:10.1586/ern.10.185
37. Gallo LC, Shivpuri S, Gonzalez P, Fortmann AL, de los Monteros KE, Roesch SC, et al. Socioeconomic status and stress in Mexican-American women: a multi-method perspective. J Behav Med (2013) 36(4):379–88. doi:10.1007/s10865-012-9432-2
38. Grande I, Magalhaes PV, Kunz M, Vieta E, Kapczinski F. Mediators of allostasis and systemic toxicity in bipolar disorder. Physiol Behav (2012) 106(1):46–50. doi:10.1016/j.physbeh.2011.10.029
39. Naviaux RK. Metabolic features of the cell danger response. Mitochondrion (2013). doi:10.1016/j.mito.2013.08.006
40. Wang YM, Liu XD, Zhang DF, Chen JH, Liu SZ, Berk M. The effects of apoptosis vulnerability markers on the myocardium in depression after myocardial infarction. BMC Med (2013) 11:32. doi:10.1186/1741-7015-11-32
41. Brietzke E, Kapczinski F. TNF-alpha as a molecular target in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry (2008) 32(6):1355–61. doi:10.1016/j.pnpbp.2008.01.006
42. Tye SJ. Allostatic overload: transcriptomic insights into the molecular basis of antidepressant resistance. Bipolar Disord (2013) 15(s1):1–163.
43. Walker AJ, Burnett SA, Hasebe K, McGillivray JA, Gray LJ, McGee SL, et al. Chronic adrenocorticotropic hormone treatment alters tricyclic antidepressant efficacy and prefrontal monoamine tissue levels. Behav Brain Res (2013) 242:76–83. doi:10.1016/j.bbr.2012.12.033
44. Almeida OP, Flicker L, Yeap BB, Alfonso H, McCaul K, Hankey GJ. Aspirin decreases the risk of depression in older men with high plasma homocysteine. Transl Psychiatry (2012):2. doi:10.1038/tp.2012.79
45. Goldstein BI, Kemp DE, Soczynska JK, McIntyre RS. Inflammation and the phenomenology, pathophysiology, comorbidity, and treatment of bipolar disorder: a systematic review of the literature. J Clin Psychiatry (2009) 70(8):1078–90. doi:10.4088/JCP.08r04505
46. Padmos RC, Van Baal GC, Vonk R, Wijkhuijs AJ, Kahn RS, Nolen WA, et al. Genetic and environmental influences on pro-inflammatory monocytes in bipolar disorder: a twin study. Arch Gen Psychiatry (2009) 66(9):957–65. doi:10.1001/archgenpsychiatry.2009.116
47. Danese A, Pariante CM, Caspi A, Taylor A, Poulton R. Childhood maltreatment predicts adult inflammation in a life-course study. Proc Natl Acad Sci U S A (2007) 104(4):1319–24. doi:10.1073/pnas.0610362104
48. Maes M, Berk M, Goehler L, Song C, Anderson G, Galecki P, et al. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med (2012) 10:66. doi:10.1186/1741-7015-10-66
49. Slopen N, Kubzansky LD, McLaughlin KA, Koenen KC. Childhood adversity and inflammatory processes in youth: a prospective study. Psychoneuroendocrinology (2013) 38(2):188–200. doi:10.1016/j.psyneuen.2012.05.013
50. Dantzer R, Kelley KW. Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun (2007) 21(2):153–60. doi:10.1016/j.bbi.2006.09.006
51. Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun (2011) 25(2):181–213. doi:10.1016/j.bbi.2010.10.015
52. Liukkonen T, Silvennoinen-Kassinen S, Jokelainen J, Rasanen P, Leinonen M, Meyer-Rochow VB, et al. The association between C-reactive protein levels and depression: results from the northern Finland 1966 birth Cohort study. Biol Psychiatry (2006) 60(8):825–30. doi:10.1016/j.biopsych.2006.02.016
53. Mills NT, Scott JG, Wray NR, Cohen-Woods S, Baune BT. Research review: the role of cytokines in depression in adolescents: a systematic review. J Child Psychol Psychiatry (2013) 54(8):816–35. doi:10.1111/jcpp.12080
54. Gabbay V, Klein RG, Alonso CM, Babb JS, Nishawala M, De Jesus G, et al. Immune system dysregulation in adolescent major depressive disorder. J Affect Disord (2009) 115(1–2):177–82. doi:10.1016/j.jad.2008.07.022
55. Kauer-Sant’Anna M, Kapczinski F, Andreazza AC, Bond DJ, Lam RW, Young LT, et al. Brain-derived neurotrophic factor and inflammatory markers in patients with early- vs. late-stage bipolar disorder. Int J Neuropsychopharmacol (2009) 12(4):447–58. doi:10.1017/S1461145708009310
56. Pasco JA, Nicholson GC, Williams LJ, Jacka FN, Henry MJ, Kotowicz MA, et al. Association of high-sensitivity C-reactive protein with de novo major depression. Br J Psychiatry (2010) 197(5):372–7. doi:10.1192/bjp.bp.109.076430
57. Biesmans S, Meert TF, Bouwknecht JA, Acton PD, Davoodi N, De Haes P, et al. Systemic immune activation leads to neuroinflammation and sickness behavior in mice. Mediators Inflamm (2013) 2013:271359. doi:10.1155/2013/271359
58. Erhardt S, Olsson SK, Engberg G. Pharmacological manipulation of kynurenic acid: potential in the treatment of psychiatric disorders. CNS Drugs (2009) 23(2):91–101. doi:10.2165/00023210-200923020-00001
59. Schiepers OJ, Wichers MC, Maes M. Cytokines and major depression. Prog Neuropsychopharmacol Biol Psychiatry (2005) 29(2):201–17. doi:10.1016/j.pnpbp.2004.11.003
60. Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci (2012) 13(7):465–77. doi:10.1038/nrn3257
61. Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflammation (2011) 8:94. doi:10.1186/1742-2094-8-94
62. Pandey GN, Rizavi HS, Ren X, Fareed J, Hoppensteadt DA, Roberts RC, et al. Proinflammatory cytokines in the prefrontal cortex of teenage suicide victims. J Psychiatr Res (2012) 46(1):57–63. doi:10.1016/j.jpsychires.2011.08.006
63. Falcone T, Carlton E, Lee C, Janigro M, Fazio V, Forcen FE, et al. Does systemic inflammation play a role in pediatric psychosis? Clin Schizophr Relat Psychoses (2013) 1–43. doi:10.3371/CSRP.FACA.030813
64. Byrne ML, O’Brien-Simpson NM, Reynolds EC, Walsh KA, Laughton K, Waloszek JM, et al. Acute phase protein and cytokine levels in serum and saliva: a comparison of detectable levels and correlations in a depressed and healthy adolescent sample. Brain Behav Immun (2013) 34:164–75. doi:10.1016/j.bbi.2013.08.010
65. Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science (2012) 338(6103):68–72. doi:10.1126/science.1222939
66. Duman RS, Voleti B. Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci (2012) 35(1):47–56. doi:10.1016/j.tins.2011.11.004
67. Mesulam MM. Neuroplasticity failure in Alzheimer’s disease: bridging the gap between plaques and tangles. Neuron (1999) 24(3):521–9. doi:10.1016/S0896-6273(00)81109-5
68. Zarate CA Jr, Singh J, Manji HK. Cellular plasticity cascades: targets for the development of novel therapeutics for bipolar disorder. Biol Psychiatry (2006) 59(11):1006–20. doi:10.1016/j.biopsych.2005.10.021
69. Drevets WC. Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Curr Opin Neurobiol (2001) 11(2):240–9. doi:10.1016/S0959-4388(00)00203-8
70. D’Sa C, Duman RS. Antidepressants and neuroplasticity. Bipolar Disord (2002) 4(3):183–94. doi:10.1034/j.1399-5618.2002.01203.x
71. Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron (2002) 34(1):13–25. doi:10.1016/S0896-6273(02)00653-0
72. Young LT. Neuroprotective effects of antidepressant and mood stabilizing drugs. J Psychiatry Neurosci (2002) 27(1):8–9.
73. Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci (2010) 11(7):459–73. doi:10.1038/nrn2867
74. Li X, Jope RS. Is glycogen synthase kinase-3 a central modulator in mood regulation? Neuropsychopharmacology (2010) 35(11):2143–54. doi:10.1038/npp.2010.105
75. Gould TD, Manji HK. Glycogen synthase kinase-3: a putative molecular target for lithium mimetic drugs. Neuropsychopharmacology (2005) 30(7):1223–37. doi:10.1038/sj.npp.1300731
76. Machado-Vieira R, Soeiro-De-Souza MG, Richards EM, Teixeira AL, Zarate CA Jr. Multiple levels of impaired neural plasticity and cellular resilience in bipolar disorder: developing treatments using an integrated translational approach. World J Biol Psychiatry (2013). doi:10.3109/15622975.2013.830775
77. Baek JH, Bernstein EE, Nierenberg AA. One-carbon metabolism and bipolar disorder. Aust N Z J Psychiatry (2013) 47(11):1013–8. doi:10.1177/0004867413502091
78. Nierenberg AA, Kansky C, Brennan BP, Shelton RC, Perlis R, Iosifescu DV. Mitochondrial modulators for bipolar disorder: a pathophysiologically informed paradigm for new drug development. Aust N Z J Psychiatry (2013) 47(1):26–42. doi:10.1177/0004867412449303
79. Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature (2008) 455(7215):894–902. doi:10.1038/nature07455
80. McKernan DP, Dinan TG, Cryan JF. “Killing the blues”: a role for cellular suicide (apoptosis) in depression and the antidepressant response? Prog Neurobiol (2009) 88(4):246–63. doi:10.1016/j.pneurobio.2009.04.006
81. Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol (2000) 1(2):120–9. doi:10.1038/35040009
82. Earnshaw WC. Apoptosis. A cellular poison cupboard. Nature (1999) 397(6718):387–9. doi:10.1038/17015
83. Dodd S, Maes M, Anderson G, Dean OM, Moylan S, Berk M. Putative neuroprotective agents in neuropsychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry (2013) 42:135–45. doi:10.1016/j.pnpbp.2012.11.007
84. Herberth M, Koethe D, Levin Y, Schwarz E, Krzyszton ND, Schoeffmann S, et al. Peripheral profiling analysis for bipolar disorder reveals markers associated with reduced cell survival. Proteomics (2011) 11(1):94–105. doi:10.1002/pmic.201000291
85. Maes M, Fisar Z, Medina M, Scapagnini G, Nowak G, Berk M. New drug targets in depression: inflammatory, cell-mediated immune, oxidative and nitrosative stress, mitochondrial, antioxidant, and neuroprogressive pathways. And new drug candidates – Nrf2 activators and GSK-3 inhibitors. Inflammopharmacology (2012) 20(3):127–50. doi:10.1007/s10787-011-0111-7
86. Davidson RJ, McEwen BS. Social influences on neuroplasticity: stress and interventions to promote well-being. Nat Neurosci (2012) 15(5):689–95. doi:10.1038/nn.3093
87. Hyman SE, Nestler EJ. Initiation and adaptation: a paradigm for understanding psychotropic drug action. Am J Psychiatry (1996) 153(2):151–62.
88. McGorry PD. Truth and reality in early intervention. Aust N Z J Psychiatry (2012) 46(4):313–6. doi:10.1177/0004867412442172
89. van der Gaag M, Smit F, Bechdolf A, French P, Linszen DH, Yung AR, et al. Preventing a first episode of psychosis: meta-analysis of randomized controlled prevention trials of 12 month and longer-term follow-ups. Schizophr Res (2013) 149(1–3):56–62. doi:10.1016/j.schres.2013.07.004
90. Berk M, Conus P, Kapczinski F, Andreazza AC, Yucel M, Wood SJ, et al. From neuroprogression to neuroprotection: implications for clinical care. Med J Aust (2010) 193(4 Suppl):S36–40.
91. Kessing LV, Hansen HV, Christensen EM, Dam H, Gluud C, Wetterslev J. Do young adults with bipolar disorder benefit from early intervention? J Affect Disord. doi:10.1016/j.jad.2013.10.001
92. Hansen HV, Christensen EM, Dam H, Gluud C, Wetterslev J, Kessing LV. The effects of centralised and specialised intervention in the early course of severe unipolar depressive disorder: a randomised clinical trial. PLoS One (2012) 7(3):e32950. doi:10.1371/journal.pone.0032950
93. Clarke GN, Hornbrook M, Lynch F, Polen M, Gale J, Beardslee W, et al. A randomized trial of a group cognitive intervention for preventing depression in adolescent offspring of depressed parents. Arch Gen Psychiatry (2001) 58(12):1127–34. doi:10.1001/archpsyc.58.12.1127
94. Harrington R, Rutter M, Fombonne E. Developmental pathways in depression: multiple meanings, antecedents, and endpoints. Dev Psychopathol (1996) 8:610–6. doi:10.1017/S095457940000732X
95. Kuo ES, Vander Stoep A, Herting JR, Grupp K, McCauley E. How to identify students for school-based depression intervention: can school record review be substituted for universal depression screening? J Child Adolesc Psychiatr Nurs (2013) 26(1):42–52. doi:10.1111/jcap.12010
96. Conus P, Ward J, Hallam KT, Lucas N, Macneil C, McGorry PD, et al. The proximal prodrome to first episode mania – a new target for early intervention. Bipolar Disord (2008) 10(5):555–65. doi:10.1111/j.1399-5618.2008.00610.x
97. Berk M, Sarris J, Coulson CE, Jacka FN. Lifestyle management of unipolar depression. Acta Psychiatr Scand Suppl (2013) 127(Suppl 443):38–54. doi:10.1111/acps.12124
98. Karatsoreos IN, McEwen BS. Resilience and vulnerability: a neurobiological perspective. F1000Prime Rep (2013) 5:13.
99. Mathew A, Nanoo S. Psychosocial stressors and patterns of coping in adolescent suicide attempters. Indian J Psychol Med (2013) 35(1):39–46. doi:10.4103/0253-7176.112200
100. Segerstrom SC, Taylor SE, Kemeny ME, Fahey JL. Optimism is associated with mood, coping, and immune change in response to stress. J Pers Soc Psychol (1998) 74(6):1646–55. doi:10.1037/0022-3514.74.6.1646
101. Wadee AA, Kuschke RH, Kometz S, Berk M. Personality factors, stress and immunity. Stress Health (2001) 17(1):25–40. doi:10.1002/1532-2998(200101)17:1<25::AID-SMI873>3.0.CO;2-N
102. Giltay EJ, Zitman FG, Kromhout D. Dispositional optimism and the risk of depressive symptoms during 15 years of follow-up: the Zutphen Elderly Study. J Affect Disord (2006) 91(1):45–52. doi:10.1016/j.jad.2005.12.027
103. Adler NE. Health disparities: what’s optimism got to do with it? J Adolesc Health (2007) 40(2):106–7. doi:10.1016/j.jadohealth.2006.12.003
104. Jorm AF. Mental health literacy: empowering the community to take action for better mental health. Am Psychol (2012) 67(3):231–43. doi:10.1037/a0025957
105. Steptoe A, Wardle J, Pollard TM, Canaan L, Davies GJ. Stress, social support and health-related behavior: a study of smoking, alcohol consumption and physical exercise. J Psychosom Res (1996) 41(2):171–80. doi:10.1016/0022-3999(96)00095-5
106. Motl RW, Birnbaum AS, Kubik MY, Dishman RK. Naturally occurring changes in physical activity are inversely related to depressive symptoms during early adolescence. Psychosom Med (2004) 66(3):336–42. doi:10.1097/01.psy.0000126205.35683.0a
107. Jacka FN, Pasco JA, Williams LJ, Leslie ER, Dodd S, Nicholson GC, et al. Lower levels of physical activity in childhood associated with adult depression. J Sci Med Sport (2011) 14(3):222–6. doi:10.1016/j.jsams.2010.10.458
108. Moylan S, Eyre HA, Maes M, Baune BT, Jacka FN, Berk M. Exercising the worry away: how inflammation, oxidative and nitrogen stress mediates the beneficial effect of physical activity on anxiety disorder symptoms and behaviours. Neurosci Biobehav Rev (2013) 37(4):573–84. doi:10.1016/j.neubiorev.2013.02.003
109. Garner AS, Shonkoff JP. Early childhood adversity, toxic stress, and the role of the pediatrician: translating developmental science into lifelong health. Pediatrics (2012) 129(1):e224–31. doi:10.1542/peds.2011-2662
110. Jacka FN, Kremer PJ, Berk M, de Silva-Sanigorski AM, Moodie M, Leslie ER, et al. A prospective study of diet quality and mental health in adolescents. PLoS One (2011) 6(9):e24805. doi:10.1371/journal.pone.0024805
111. Jacka FN, Ystrom E, Brantsaeter AL, Karevold E, Roth C, Haugen M, et al. Maternal and early postnatal nutrition and mental health of offspring by age 5 years: a prospective cohort study. J Am Acad Child Adolesc Psychiatry (2013) 52(10):1038–47. doi:10.1016/j.jaac.2013.07.002
112. Moylan S, Gustavson K, Karevold E, Overland S, Jacka FN, Pasco JA, et al. The impact of smoking in adolescence on early adult anxiety symptoms and the relationship between infant vulnerability factors for anxiety and early adult anxiety symptoms: the TOPP Study. PLoS One (2013) 8(5):e63252. doi:10.1371/journal.pone.0063252
Keywords: prodrome, depression, bipolar, biomarker, stress, inflammation, cellular resilience, plasticity
Citation: Walker AJ, Kim Y, Price JB, Kale RP, McGillivray JA, Berk M and Tye SJ (2014) Stress, inflammation, and cellular vulnerability during early stages of affective disorders: biomarker strategies and opportunities for prevention and intervention. Front. Psychiatry 5:34. doi: 10.3389/fpsyt.2014.00034
Received: 29 October 2013; Accepted: 17 March 2014;
Published online: 09 April 2014.
Edited by:
Stephanie Ameis, University of Toronto, CanadaReviewed by:
Vilma Gabbay, Mount Sinai School of Medicine, USAEllen Grishman, University of Texas Southwestern Medical Center, USA
Copyright: © 2014 Walker, Kim, Price, Kale, McGillivray, Berk and Tye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susannah J. Tye, Department of Psychiatry and Psychology, Mayo Clinic, 200 First Street SW, Rochester, MN 55905, USA e-mail:dHllLnN1c2FubmFoQG1heW8uZWR1