 Vaibhav A. Diwadkar
Vaibhav A. Diwadkar Angela Bustamante
Angela Bustamante Harinder Rai
Harinder Rai Monica Uddin
Monica Uddin- 1Department of Psychiatry and Behavioral Neurosciences, Wayne State University School of Medicine, Detroit, MI, USA
- 2Center for Molecular Medicine and Genetics, Wayne State University School of Medicine, Detroit, MI, USA
The recent sociodevelopmental cognitive model of schizophrenia/psychosis is a highly influential and compelling compendium of research findings. Here, we present logical extensions to this model incorporating ideas drawn from epigenetic mediation of psychiatric disease, and the plausible effects of epigenetics on the emergence of brain network function and dysfunction in adolescence. We discuss how gene–environment interactions, effected by epigenetic mechanisms, might in particular mediate the stress response (itself heavily implicated in the emergence of schizophrenia). Next, we discuss the plausible relevance of this framework for adolescent genetic risk populations, a risk group characterized by vexing and difficult-to-explain heterogeneity. We then discuss how exploring relationships between epigenetics and brain network dysfunction (a strongly validated finding in risk populations) can enhance understanding of the relationship between stress, epigenetics, and functional neurobiology, and the relevance of this relationship for the eventual emergence of schizophrenia/psychosis. We suggest that these considerations can expand the impact of models such as the sociodevelopmental cognitive model, increasing their explanatory reach. Ultimately, integration of these lines of research may enhance efforts of early identification, intervention, and treatment in adolescents at-risk for schizophrenia.
Introduction
Schizophrenia remains the most profoundly debilitating of psychiatric conditions (1, 2). General theories have struggled to capture the complexity of the disorder: genetic polymorphisms (3), neurodevelopment (4), and altered neurotransmission [dopamine (DA) and glutamate] (5, 6) have all being proposed as mediating factors in its emergence. A recently proposed “sociodevelopmental cognitive model” (7) has made compelling additions to the discourse on schizophrenia, with a specific emphasis on psychosis. A factorial combination of genetic and neurodevelopmental effects sensitize the DA system in early life. The disordered sensitivity subsequently leads to a disordered stress response that is further amplified by misattributed salience and paranoia. This cascading and recursive series of events eventually leads to the entrenchment of psychosis (and schizophrenia), explaining the life-long nature of the illness. This model is uniquely important because it integrates environmental, genetic, developmental, and molecular mechanisms (all converging on dysregulated DA release), providing a synthesis for several multi-disciplinary research agendas. Here, we attempt an incremental contribution to this synthesis suggesting that an expansion of this model may help elucidate the following:
(a) How do gene–environment interactions, effected by epigenetic mechanisms, mediate the stress response? The role of epigenetic mechanisms may be crucial in understanding why certain individuals at genetic risk eventually convert to schizophrenia but others with similar genetic vulnerability do not.
(b) In this context, the vexing problem of specific genetic at-risk populations is considered. Specifically, adolescents with one or both of whose parents have a diagnosis of schizophrenia form a “perfect storm” of genetic and neurodevelopmental contributors to risk for schizophrenia. These individuals present with extensive pre-morbid cognitive deficits (8) and sub-threshold clinical symptoms (9), yet a majority of them do not appear to develop the disorder. Whereas unexplained neurodevelopmental variation and resilience may explain this (10), we suggest that epigenetic mediation, particularly of genes mediating the stress response in adolescence, may explain some of this uncharacterized variance.
(c) Finally, we note the vast evidence of functioning brain network disruptions in schizophrenia, and the fact that these disruptions are now being characterized in at-risk populations, including children of patients, and suggest that epigenetic effects may mediate the shaping of functioning brain networks in the adolescent risk state, resulting in a highly variable and (currently) unpredictable pattern of conversion to psychosis (hence explaining the difficulty in estimating incidence rates of schizophrenia in at-risk groups).
In short, the proposed addendum motivates the role of epigenetics in the schizophrenia diathesis, the (potentially crucial) role of epigenetics in setting gene-expression levels that mediate the stress response, and ultimate causal (though presently unproven) effect on developing brain networks that sub-serve many of the cognitive functions impaired in schizophrenia. We note at the outset, that the proposed extensions remain speculative, yet seek to account for the relative under-representation of epigenetic considerations in schizophrenia-related research to date. In fact, epigenetics may provide a more proximate mediator of neuronal and behavioral effects than changes in the DNA sequence, and in turn these neuronal alterations may predispose individuals to schizophrenia, a question that has received comprehensive coverage in a recent canonical review (11). Moreover, the proposed additions also provide a prospective research impetus for studying particular sub-groups such as children of schizophrenia patients, a group that provides a particularly unique intersection of genetic risk, altered neurodevelopment, and environmental contributions (12–14). Finally, the notion of stress reactivity impacting brain network function is a particular extension of the seminal concept of “allostatic load” (15, 16), morphologic degeneration as a response to repeated adaptive responses to stress.
Genetics, Development, Environment: An Array of Interactions
Schizophrenia is an “epigenetic puzzle” (17). Apart from the rare variant of the illness that is childhood onset schizophrenia (18), the typical manifestations of schizophrenia occur in late adolescence and early adulthood (1). This relatively late onset suggests that a seemingly intractable array of interactions between genetically endowed vulnerability, and environmental effects may amplify genetic predisposition, leading to post-natal effects on brain plasticity and development in the critical adolescent period (2, 19). The role of genes in mediating the emergence of the disorder is likely to be extremely complex. After all, genes do not code for complex psychiatric disorders but for biological processes (20). Thus, dysfunctional genetic expression is likely to lead to dysfunctional biological processes, with psychiatric disorders an emergent phenomenon in this causal pathway (20, 21). Moreover, the lack of complete concordance even in monozygotic twins (22, 23), suggests that genes primarily confer vulnerability to the illness and that other factors that mediate gene-expression during pre- and post-natal developmental, life span, and environmental effects play a significant role in the transition to the illness.
Several proximate environmental factors may be highly relevant as noted in the sociodevelopmental cognitive model. Stress – narrowly defined as a real or employed threat to homeostasis (24) – assumes particular importance, primarily because adolescence is a period of dynamic stress both in terms of substantive neurodevelopmental turnover (25), and environmental influence (26). Repeated stress exposure in particular during critical developmental periods exerts untenable biophysical costs. These costs typically referred to as allostatic load, increase vulnerability for somatic disease (27), and notably exert tangible biological effects. For example, glucocorticoid elevations that result from chronic stress have been associated with medial temporal lobe atrophy across multiple disorders including mood disorders, post-traumatic stress disorder, and schizophrenia (28–30). Beyond medial temporal lobe regional atrophy, the documented molecular effects in the prefrontal cortex are suspected to ultimately impact frontal–striatal brain networks (31, 32). Elevated DA release during acute stress (33) adversely affects prefrontal pyramidal cells leading to a series of degenerative molecular events. The resultant dendritic spine loss in the infra-granular prefrontal cortex results in reductions in prefrontal-based network connectivity, particularly on prefrontal efferent pathways (34). These molecular effects are likely to have mesoscopic expressions; among them disordered prefrontal cortex related brain network function and organization that are hallmarks of schizophrenia (3, 35–37).
Stress and the Risk State for Schizophrenia
The risk state for schizophrenia offers a powerful framework for synthesizing multiple theoretical constructs of the disease (38), and disordered stress reactivity may play a key role in amplifying disposition for psychosis in the risk state (39). A critical challenge for high-risk research is navigating the relationship between multiple (and potentially non- or partially overlapping) risk groups each with different etiologies and defined based on different criteria (40). Here we consider prodromal subjects (41–46) in whom the role of stress has been heavily assessed, separately from adolescents with a genetic history of schizophrenia (including twins discordant for the illness and offspring of patients). The role of stress in the latter groups is relatively understudied. We note that the distinction does not imply exclusivity but rather criteria used to identify risk. Prodromal or clinical high-risk subjects (also on occasion referred to as “ultra high-risk”) are classified as such because they show non-specific yet considerably advanced clinical symptoms (47). Rates of conversion to psychosis within a short period after the emergence of clinical symptoms are high (estimates at 35%) (48). Genetic high-risk groups are identified typically on account of a family history of the illness itself; that is, not using clinical criteria. However, genetic high-risk groups may present with prodromal symptoms, hence these groups are not exclusive.
We will ultimately seek to drive our ideas in the direction of genetic risk in adolescence, largely because the prodromal question is heavily addressed in the sociodevelopmental model, whereas adolescent genetic risk is not. The adolescent genetic risk state presents a particularly vexing challenge, with substantial heterogeneity, and relative low rates of conversion to psychosis (9). The early identification of individuals who are likely to convert from the genetic risk state to actual schizophrenia (or psychosis?) thus remains a key issue to be addressed by future research efforts, as we propose here.
Prodromal subjects (sometimes referred to as “clinical high-risk”) present with a variety of symptoms that do not specifically warrant a diagnosis of schizophrenia, but include paranoia and impairment in social function. In general, prodromal patients have high rates of conversion to schizophrenia itself (48). For instance, multiple studies suggest that the average 12-month conversion rate in ultra high-risk samples not receiving any special anti-psychotic treatment is between 35 and 38% (48, 49). That a significant percentage of these individuals convert to psychosis is unsurprising because as noted, the prodromal state consists of highly advanced stage of clinical symptoms. Thus, these relatively non-specific symptoms that lead, and predict the presentation of the illness itself (38, 48, 50, 51) are considered the best clinical predictor of schizophrenia itself. Impaired neurobiology of the prodromal state is also relatively well understood: subjects are characterized by profound deficits in brain structure that are typically intermediate between healthy controls, and those observed in patients. Recent fMRI studies indicate substantive deficits in regional and brain network interactions (52–54) including frontal–striatal and frontal–limbic; cognitive and social neuroscience has established a crucial role for these networks in sub-serving basic mechanisms of memory, attention, and emotion. Heightened stress reactivity itself may be exacerbated by the presence of sub-threshold symptoms. For instance, prodromal subjects indicate heightened sensitivity to inter-personal interaction, an indirect measure of heightened stress (55), and a significant percent of prodromal subjects who have experienced trauma in their lives convert to psychosis (41). As noted, DA synthesis is increased in prodromal subjects, and the degree of synthesis is positively associated with the severity of sub-threshold clinical symptoms (56). Moreover, impaired stress sensitivity is also associated with a wide range of prodromal symptoms (44). The role of stress sensitivity, the hypothalamic–pituitary–adrenal (HPA) axis, and its impact on brain structures, has been heavily treated in the empirical and theoretical literature (43, 45, 57–59).
In contrast to the prodromal state, which includes individuals with a degree of existing symptoms, the genetic high-risk state encompasses individuals who are defined by having one (or more) parent(s) with schizophrenia, and who themselves may or may not evince symptoms of the disorder. The genetic high-risk state constitutes a partial complement of the clinical high-risk or prodromal state (these samples are often “enriched” by subjects with a family history of schizophrenia or psychosis providing overlap) (60). Genetic distance from a schizophrenia patient is a strong predictor of risk for the disease, and of the degree of biological impairments including brain structure, function, and behavior (61, 62). For example, children of schizophrenia patients being reared by the ill parent constitute a very particular and enigmatic high-risk sub-group (9, 13). These individuals have a genetic loading for the disease, but are also likely exposed to increased environmental stressors by virtue of being raised by their ill parent. Unlike with prodromal patients, conversion to psychosis in genetic high-risk groups is variable and lower.
Three principle longitudinal genetic high-risk studies are informative regarding lifetime incidence of schizophrenia in these groups. Between them, the New York (63), the Copenhagen high-risk projects (64), and a notable Israeli study (65) have provided evidence of lifetime incidences of narrowly defined schizophrenia at between 8 and 21%. While low, these rates constitute significantly elevated incidence rates relative to the sporadic incidence in the population (~1–2%). However, these rates are still notably lower than conversion rates in prodromal populations, a discrepancy that is somewhat surprising because the developmental psychopathology that characterizes prodromal patients is the very same one that is in play in adolescent high-risk subjects (45, 46). Subjects at genetic risk also show increased HPA axis sensitivity (59, 66), similar to what is observed in prodromal subjects, though the relationship to regional measures of brain integrity (e.g., pituitary size), is highly variable, and perhaps not informative as a biomarker (67). Heterogeneity is a cardinal characteristic of genetic risk groups (68, 69). Significant percentages of these subjects show attention deficits, working memory impairment, emotion dysregulation, and sub-threshold symptoms including negative symptoms (9, 70–75). Notably each of these cognitive, emotional, and clinical domains is highly impacted by stress sensitivity in adolescence (76, 77). Adolescent risk subjects also present with increased frequency of sub-threshold clinical symptoms including schizotypy and both positive and negative symptoms such as anhedonia (78–80), some of which have been associated with perceived stress (81, 82).
Understanding of altered DA synthesis in genetic risk groups is limited. A recent study in twins discordant for schizophrenia showed no increase in the elevation of striatal DA synthesis in the healthy twin (83) though the age range was well past the typical age of onset of the illness, and the healthy twin must retrospectively be classified as “low risk.” It is plausible the elevated striatal DA is not a marker of genetic risk per se, but might distinguish between adolescent sub-groups. Given that animal models and human studies have been highly informative in elucidating the impact of stress on neurobiology (32, 84), it is plausible that these effects might be quantifiable in neuroimaging data derived from such models in the context of risk for schizophrenia.
Brain Network Dysfunction in the Adolescent Risk State for Schizophrenia
The origins of psychiatric disorders lie in adolescence (85, 86), a developmental stage characterized by a unique set of vulnerabilities, where highly dynamic neurodevelopmental processes intersect with increasing environmental stressors (26, 87). The idea of “three-hits” in schizophrenia, which includes pre-natal insults (e.g., obstetric complications, exposure to infections in utero), neurodevelopmental processes and disease-related degeneration, predicts the emergence of reliable and identifiable abnormalities through the life span (10, 88, 89). Notably, the period from birth to early adulthood is characterized by significant potential for epigenetic dysfunction that can increase symptom severity, beginning with the emergence of sub-threshold symptoms in adolescence, and culminating (in some individuals) in psychotic symptoms in young adulthood (11). Moreover, brain network development remains highly tumultuous in this period and disordered brain network dynamics are likely to be a cardinal biological characteristic in adolescents at genetic risk for the illness (13).
Disordered frontal–striatal and frontal–limbic brain network interactions, a defining characteristic of schizophrenia (90, 91), are increasingly established in the adolescent genetic risk state. These interactions are well-understood for working memory and sustained attention, both domains particularly associated with these regions (92), with risk for schizophrenia (70), and with DA (93, 94). During working memory, adolescents at genetic risk for schizophrenia show inefficient regional responses as well as network interactions in frontal and striatal regions. During working memory-related recall, at-risk subjects hyper-activate frontal–striatal regions, specifically for correctly recalled items (95), an effect highly consistent with what has been documented in schizophrenia itself (96, 97) and with large studies assessing the relationship between genetic risk and prefrontal efficiency (98).
More impressively, network interactions are also inefficient. For instance, the degree of modulation by the dorsal anterior cingulate, the brain’s principle “cognitive control” structure (99), during working memory is significantly increased in at-risk subjects (100). Thus, when performing the task at levels comparable to typical control subjects, control-related “afferent signaling” from the dorsal anterior cingulate cortex is aberrantly increased in adolescents at genetic risk. This evidence of inefficient pair-wise network interactions is highly revealing of “dysconnection” in the adolescent risk state. Similar results have been observed in the domain of sustained attention, where again, frontal–striatal interactions are impaired in the risk state (80, 101). Genetic high-risk subjects are also characterized by disordered “effective connectivity” estimated from fMRI signals. Effective connectivity is noted as the most parsimonious “circuit diagram” replicating the observed dynamic relationships between acquired biological signals (102). Recent evidence suggests reduced effective thalamocortical (54) and frontal–limbic (103) effective connectivity in genetic risk groups. These and other studies establish a pattern of general brain network dysfunction in adolescents at genetic risk for schizophrenia, suggesting that dysfunction in cortical networks is a plausible “end-point” in a cascade of genetic and neurodevelopmental events.
However, this story on brain networks is incomplete, because these high-risk groups present with considerable heterogeneity in sub-clinical symptoms, and recent evidence suggests that this heterogeneity predicts fMRI responses. For example, high-risk subjects with sub-threshold negative symptoms show attenuated responses to rewarding social stimuli, particularly in regions of the limbic system, including the amygdala and the ventral prefrontal cortex (75). This pattern of responses is in fact similar to those seen in patients with frank depression, and suggests additional compelling evidence in support of stress mediating the emergence of negative symptoms that in turn affect functioning brain networks (44, 104–107).
Pathways and Epigenetic Mediation
Psychological stress is a major mediator of externally experienced (i.e., environmental) events, with relevance to both the central and peripheral nervous systems (108). Stress induces the release of corticotrophin releasing factor that activates the HPA axis to produce cortisol, and the sympathetic nervous system to produce norepinephrine and epinephrine. In some individuals, the initiation of an acute, adaptive “fight-or-flight” response in the face of threatening events becomes persistent and pathological. How this failure to return to homeostasis occurs in only a subset of individuals, resulting in a psychopathological state, remains to be fully elucidated. Stress is a clear risk factor for schizophrenia (109), and the biologic mechanisms linking stress, schizophrenia, and risk for schizophrenia are still being comprehensively characterized.
One candidate factor that may be a mediator in this causal chain is epigenetics, a field of increasing interest in mental illness, including risk for schizophrenia (110–112). Epigenetics, a term proposed nearly 70 years ago by Conrad Waddington, was born out of the terms “genetics” and “epigenesist,” narrowly referring to the study of causal relationships between genes and their phenotypic effects (113), but more recently associated with changes in gene activity independent of the DNA sequence, that may or may not be heritable, and that may also be modified through the life span. Epigenetic factors include DNA methylation which in vertebrates typically involves the addition of a methyl group to cytosine where cytosine and guanine occur on the same DNA strand; histone modifications, involving the addition (or removal) of chemical groups to the core proteins around which DNA is wound; and non-coding RNAs such as microRNAs (miRNAs), which bind to mRNAs to suppress gene-expression posttranscriptionally. Among these several mechanisms, DNA methylation is the most stable and the best studied within the context of psychiatric disorders, including schizophrenia, although emerging work suggests that miRNAs, which target multiple mRNA transcripts, serve as master regulators of developmental gene-expression patterns, and are responsive to stress (114), play an etiologic role in SCZ (115).
As mounting evidence fails to conclusively link individual genes to specific mental illnesses (116), epigenetic effects during critical developmental periods assumes increasing significance (11). In such a model, genetic etiology may be expressed in differentiated psychiatric phenotypes because epigenetic factors changing in response to external experiences vary across these phenotypes. Indeed, as potential regulators of DNA accessibility and activity, epigenetic factors through influences on gene-expression, offer a mechanism by which the environment – and, in particular, one’s response to the environment – can moderate the effects of genes (117). In the context of schizophrenia, models suggest that epigenetic deregulation of gene-expression at specific loci is highly unlikely, again given the highly polygenic nature of the illness. Rather, epigenetic effects may progressively impact gene-expression in salient neurodevelopmental gene networks during critical developmental periods, in response to environmental inputs (11). For example, the loss of synchronal activity of GABAergic interneurons in the prefrontal cortex might result from environmental stressors such as cannabis (118), which interact with the expression of vulnerability genes such as GAD1 that control GABA synthesis (119).
Previous work has shown that glucocorticoids (GC) such as cortisol induce epigenetic, DNA methylation changes in HPA axis genes (e.g., FK506 binding protein 5, FKBP5), both in neuronal [i.e., hippocampal (120, 121)] and peripheral [i.e., blood (121–123)] tissues, as well as in additional cells relevant to the HPA axis [i.e., pituitary cells (120)]. Moreover, GC-induced DNA methylation changes persist long after cessation of GC exposure (121–123), suggesting that stress-induced GC cascades have long lasting consequences for HPA axis function that may be accompanied by behavioral (mal)adaptations (121, 124).
These epigenetic mechanisms are of relevance to the previously noted role of stress as a major contributor in the emergence of cognitive impairments in first episode psychosis, in particular resulting from high stress sensitivity in this group (125). Stress sensitivity, a tendency to experience negative affect in response to negative environmental events (126), is a well-established risk factor for psychopathology (127), including schizophrenia (44, 128). This role has been clarified in recent work using experience sampling methods (ESM), where participants in prospective studies note their life experiences in real time. Using a twin-study design in a large longitudinal cohort of mono- and dizygotic twins, participants recorded multiple mood and daily life events with stress sensitivity defined as an increase in recorded negative affect to event unpleasantness. Notably, stress sensitivity showed relatively little genetic mediation and was almost exclusively environmentally determined (126). Whereas non-ESM investigations and some animal studies in models of schizophrenia (129) suggest a genetic, heritable component, the majority of variance still appears to be environmentally determined (130, 131). Thus, stress sensitivity is a labile characteristic that can change in response to environmental experiences to alter risk for psychopathology. Tracking epigenetic changes in stress-sensitive genes of the HPA axis, as well as additional stress-sensitive genes that interact with the HPA axis, might enable identification of a biologic mechanism that mediates risk for, and the emergence, of schizophrenia. Indeed, strong signatures of gene-expression differences in stress-related genes have been recently identified in post-mortem brain tissue in a manner that distinguishes schizophrenia patients from controls and from individuals with other psychiatric disorders (132). Many of these are likely accompanied by DNA methylation differences, as has been reported by studies performed on related genes in animal models (133).
Emerging evidence suggests that brain endophenotypes, as well as psychiatric outcomes, can be predicted by peripheral DNA methylation measurements. Notably, genes belonging to the HPA axis, as well as DA- and serotonin (5HT)-related genes, whose products interact those of the HPA axis, shape the stress response (109, 134, 135) and are known to show psychopathology-associated differences in blood (136–138). For example, recent work has shown that leukocyte DNA methylation in the serotonin transporter locus (SLC6A4) was higher among adult males who had experienced high childhood-limited physical aggression; moreover SLC6A4 DNA methylation was negatively correlated with serotonin synthesis in the orbitofrontal cortex, as measured by positron emission tomography (PET) (139). Similarly, leukocyte DNA methylation in the promoter region of the MAOA gene – whose product metabolizes monoamines such as serotonin and DA, is negatively associated with brain MAOA levels as measured by PET in healthy male adults (140). Structural imaging data analyses in relation to the FKBP5 locus discussed above have identified a negative association between DNA methylation in peripheral blood and volume of the right (but not left) hippocampal head (121). This observation is particularly noteworthy, as it suggests that lower FKBP5 DNA methylation in peripheral blood is associated not only with altered stress sensitivity (as indexed by a glucocorticoid receptor sensitivity assay within the same study), but also with structural brain differences in a brain region known to mediate stress reactivity (121). Finally, investigation of the COMT locus, a gene encoding an enzyme critical for degradation of DA and other catecholamines, has shown that, among Val/Val genotypes, subjects (all healthy adult males) with higher stress scores have reduced DNA methylation at a CpG site located in the promoter region of the gene (141). Moreover, DNA methylation at this site was positively correlated with working memory accuracy, with greater methylation predicting a greater percentage of correct responses (with results again limited to analysis of the Val/Val subjects); furthermore, fMRI demonstrated a negative correlation between DNA methylation at this site and bilateral PFC activity during the working memory task (141). Additional analyses showed an interaction between methylation and stress scores on bilateral prefrontal activity during working memory, indicating that greater stress, when combined with lower methylation, are associated with greater activity (141).
This last finding is especially noteworthy, because whereas stress–DNA methylation interactions have been reported for other stress-sensitive loci (142), the referenced study represents a direct demonstration of a heterogeneity in stress load that, when moderated by DNA methylation, impacts working memory. Clearly, greater stress and lower COMT DNA methylation correlate with reduced efficiency of prefrontal activity (141). This mechanism may be explained by the fact that disordered stress responses following prolonged stress exposure induces hyper-stimulation of prefrontal DA receptors (143, 144) that may be mediated by prefrontal glutamate neurotransmission (145). This hyper-stimulation in turn appears to affect the receptive field properties of prefrontal neurons during working memory (94). Patterns of network dysfunction in the genetic risk state may reflect brain network sensitivity to stress in the “pre-morbid” risk state that may be under as yet undiscovered epigenetic control. Thus, much of the unaccounted variance in schizophrenia previously construed as genetic, may likely be epigenetic (11, 146). Is it possible to assess epigenetic factors mediating the stress response in risk for schizophrenia, and the effects on brain network function?
The influence of stress on DNA methylation on HPA axis genes in blood is well established (121–123). Indeed, blood disperses GC hormones produced by the HPA axis throughout the body, which then regulates gene-expression in virtually all cell types (108). Thus, the broad reach of HPA axis activity, together with evidence that blood-derived DNA methylation in HPA axis genes is altered through stress (121, 147), provides ample biologic and clinical plausibility to our proposed hypothesis that stress sensitivity, measured in the periphery, can serve as an important – perhaps even predictive – index of transition from the genetic risk state into actual schizophrenia. Importantly, although GCs also influence DNA methylation and gene-expression in the CNS and neuronal cells (120, 121), our model does not suppose that this epigenetic measure in CNS tissues will match those in the periphery; rather, it proposes that DNA methylation in stress-sensitive, HPA-axis genes in the periphery will index the known dysregulation in brain function and connectivity in stress-sensitive regions of the brain among adolescents at genetic risk. Figure 1 provides an overview of an integrative approach and builds on previous considerations of epigenetic mechanisms in developmental psychopathology (11).
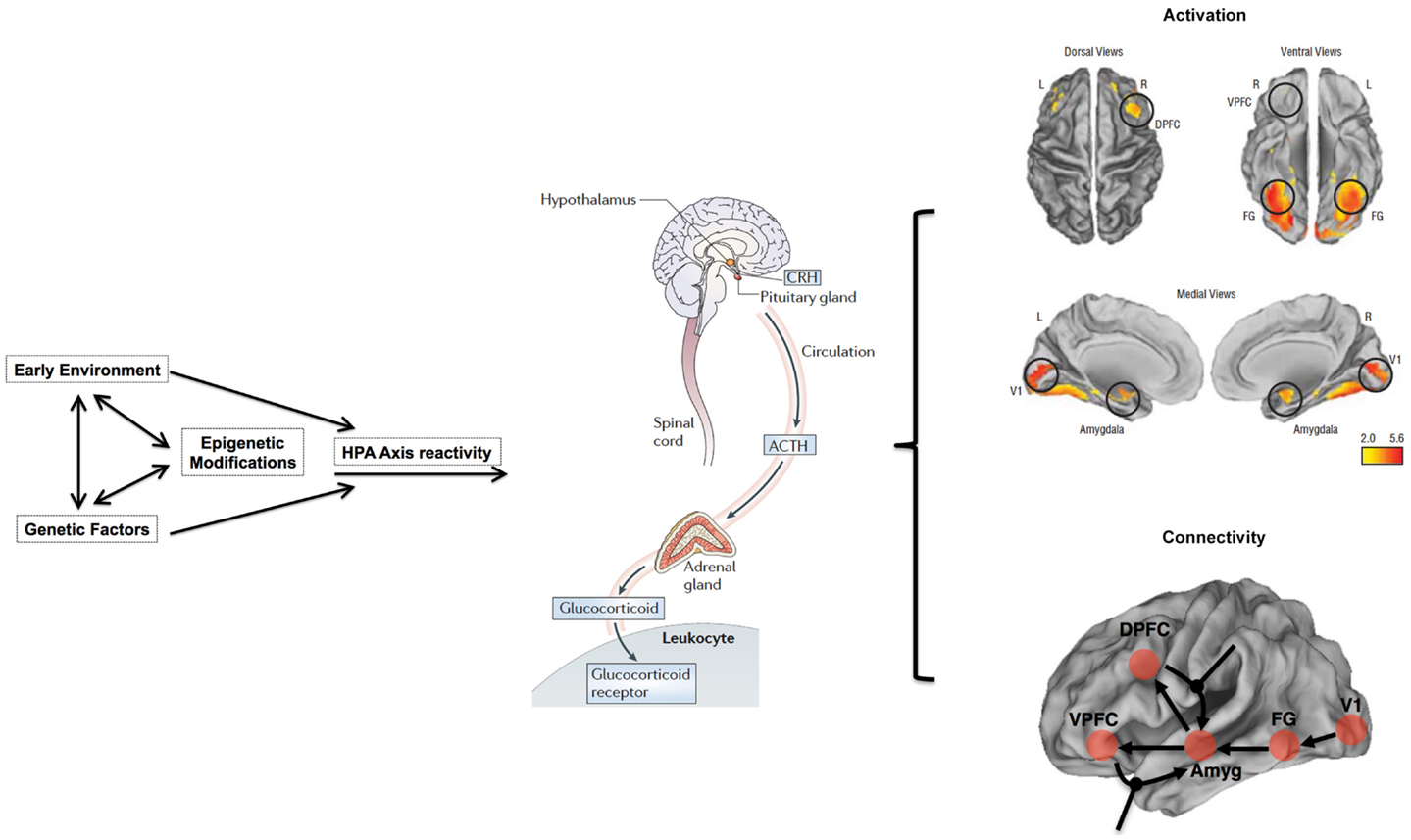
Figure 1. Overview of working model. HPA axis reactivity is determined both by intrinsic genetic factors and stressful environmental (including pre-natal) experiences. Stressful exposures induce a glucocorticoid (i.e., cortisol) cascade that then induces DNAm changes in HPA axis genes in the blood. These changes are expected to be more pronounced in at-risk adolescents, particularly those who may already exhibit sub-clinical psychopathology, such as negative symptoms. Risk-associated, blood-derived DNAm differences in HPA axis and related stress sensitivity genes are hypothesized to index metrics of brain function including activation patterns and effective connectivity in stress-sensitive brain regions. The activation patterns are reproduced from Diwadkar (13) and reflect engagement of an extended face-processing network in controls and high-risk subjects during a continuous emotion-processing task. These activations are most likely generated by complex dynamic interactions between brain networks that are represented in the figure below. The figure presents a putative combination of intrinsic connections between brain regions activated during such a task, and the contextual modulation of specific intrinsic connections by dynamic task elements. The role of effective connectivity analyses is to recover and estimate parameter values for intrinsic and modulatory connections that a) may be different in the diseased or risk state and b) may plausibly be under epigenetic mediation. The figure is adapted and reprinted from: Mehta and Binder (124), with permission from Elsevier; adapted by permission from Macmillan Publishers Ltd.: Frontiers in Neuropsychiatric Imaging and Stimulation (108). Reproduced with permission, Copyright © (2012) American Medical Association. All rights reserved.
Existing data support the hypothesis that schizophrenia-associated DNA methylation differences exist in stress-sensitive genes. Table 1 summarizes results from existing genome-scale studies that have been conducted in blood and brain in relation to schizophrenia, focusing specifically on the HPA axis genes involved in the glucocorticoid receptor complex (148), as well as representative DA- and serotonin-related genes, and genes that produce DNA methylation and have been shown to be responsive to glucocorticoid induction in both the brain and periphery [i.e., DNA methyltransferase 1, DNMT1; (120)]. As can be seen from the table, all of the genes show SCZ-related DNA methylation differences in brain derived tissue (149), and the majority (four of five) of GC-receptor chaperone complex genes show DNA methylation differences in the blood as well. Although we have limited our analysis to genome-wide studies of DNA methylation, additional candidate gene studies have linked stress-sensitive mental disorders to methylation differences in blood (142, 150, 151), suggesting that similar findings may be forthcoming for schizophrenia as additional studies are completed. Importantly, among these genes, some (but not all) have shown that DNA methylation levels can vary depending on local [e.g., Ref. (141)] or distal [e.g., Ref. (121)] DNA sequence variation – so-called “methQTLs” (methylation quantitative trait loci). Thus, as evidence accumulates regarding the existence of methQTLs, we note that analyses based on these proposed genes should take these into consideration.
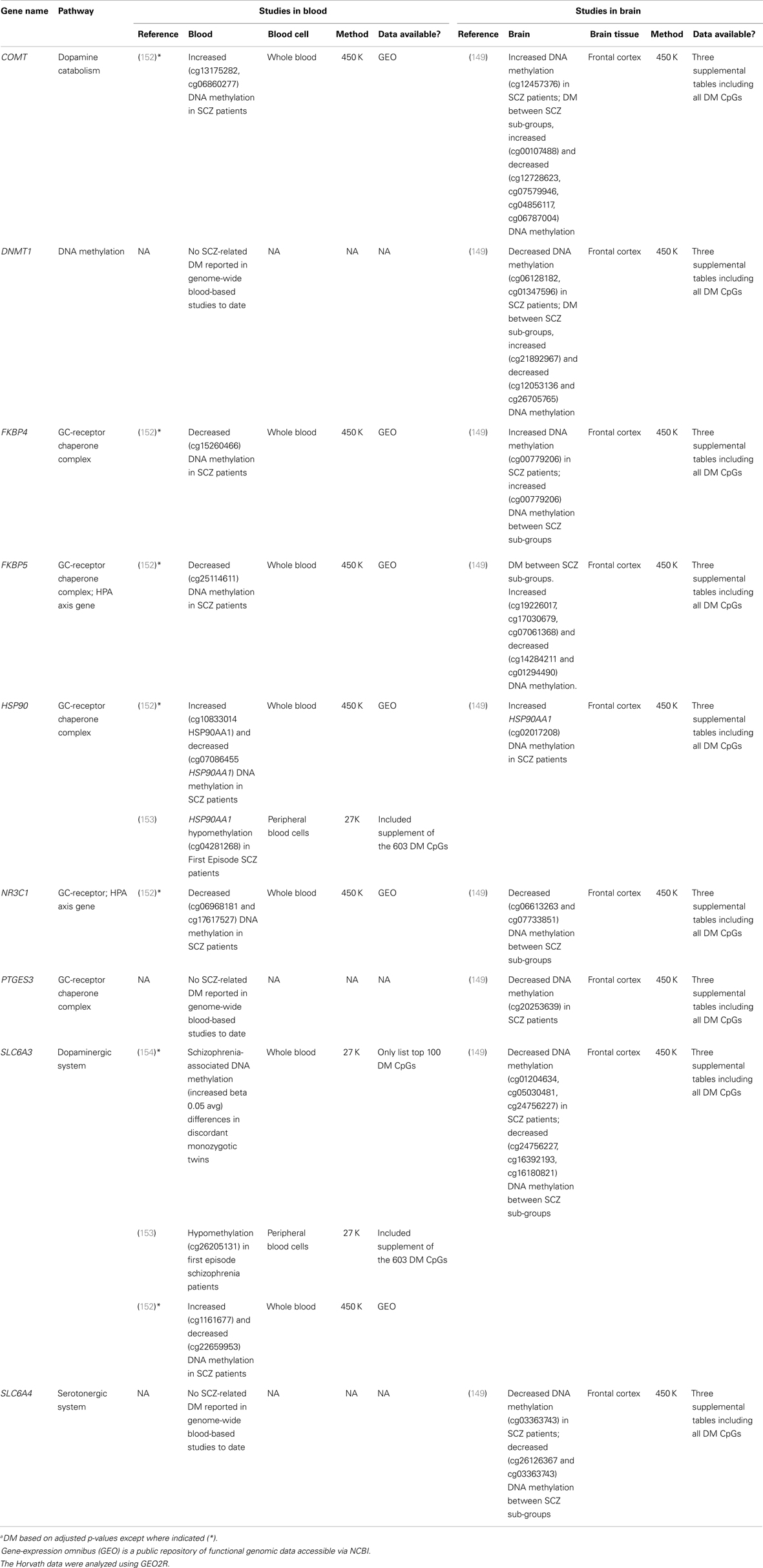
Table 1. Summary of genome-wide studies reporting differential DNA methylationa (DM) within stress-sensitive genes in blood or brain.
Conclusion
Incorporating epigenetic considerations into the sociodevelopmental model might provide a particular powerful explanatory framework for understanding genetic risk in adolescence. Regressive pressures from a combination of fixed genetic vulnerability for schizophrenia and epigenetic effects during adolescence are most likely to impact the development of neuronal network profiles (155, 156). As we noted earlier, advances in the analyses of fMRI signals now permit the estimate of effective connectivity and dysconnectivity between healthy, clinical, and at-risk populations, providing a significant framework for exploring brain dysfunction using a priori hypothesis (157). A focus on frontal–striatal and frontal–limbic dysconnectivity may be particularly warranted. A disordered stress response may cleave apart frontal–striatal and frontal–limbic neuronal network profiles in high-risk adolescents, providing a convergence of biological markers across multiple levels (genetic, epigenetic, and brain networks). Here, we have proposed that increased stress sensitivity (which can be indexed in the periphery) can help to unpack the heterogeneity among individuals at genetic high-risk of SCZ when linked to a strongly validated finding in genetic risk populations, namely brain network dysfunction. This framework may help to identify, among individuals at high genetic risk for SCZ, a subset who are likely to go on to develop the disorder. Our focus on stress-relevant genes does not exclude the possibility that genes in other pathways (e.g., dopaminergic, serotonergic, glutamatergic) may also be important; indeed, this focus may be considered a limitation of the proposed hypothesis. However, we believe that our proposed framework is a logical starting point for merging central and peripheral indicators of the potential for SCZ among HRS individuals. This framework may help extend the sociodevelopmental cognitive model into the realm of high-risk research. The presence of non-specific, sub-threshold symptoms continues to remain a significant clinical challenge for disorders such as schizophrenia and bipolar disorder (38, 158). Early intervention strategies will be boosted if biological markers can be interlinked to identify ultra high-risk adolescents. Our intent is to motivate this search for biological convergence hoping that this may lead to psychosis prediction and, ultimately, prevention.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Association for Research on Schizophrenia and Depression (NARSAD, now Brain Behavior Research Fund; Vaibhav A. Diwadkar), the Prechter World Bipolar Foundation (Vaibhav A. Diwadkar), the Lyckaki Young Fund from the State of Michigan, and the Children’s Research Center of Michigan (Monica Uddin). The agencies played no role in the shaping of the ideas presented herein.
References
1. Schultz SK, Andreasen NC. Schizophrenia. Lancet (1999) 353:1425–30. doi: 10.1016/S0140-6736(98)07549-7
2. Rapoport JL, Addington AM, Frangou S, Psych MR. The neurodevelopmental model of schizophrenia: update 2005. Mol Psychiatry (2005) 10:434–49.
3. Tan HY, Callicott JH, Weinberger DR. Prefrontal cognitive systems in schizophrenia: towards human genetic brain mechanisms. Cogn Neuropsychiatry (2009) 14:277–98. doi:10.1080/13546800903091665
4. Gogtay N. Cortical brain development in schizophrenia: insights from neuroimaging studies in childhood-onset schizophrenia. Schizophr Bull (2008) 34:30–6. doi:10.1093/schbul/sbm103
5. Castner SA, Williams GV. Tuning the engine of cognition: a focus on NMDA/D1 receptor interactions in prefrontal cortex. Brain Cogn (2007) 63:94–122. doi:10.1016/j.bandc.2006.11.002
6. Di Forti M, Lappin JM, Murray RM. Risk factors for schizophrenia – all roads lead to dopamine. Eur Neuropsychopharmacol (2007) 17(Suppl 2):S101–7. doi:10.1016/j.euroneuro.2007.02.005
7. Howes OD, Murray RM. Schizophrenia: an integrated sociodevelopmental-cognitive model. Lancet (2013) 383(9929):1677–87. doi:10.1016/S0140-6736(13)62036-X
8. Cornblatt B, Obuchowski M, Roberts S, Pollack S, Erlenmeyer-Kimling L. Cognitive and behavioral precursors of schizophrenia. Dev Psychopathol (1999) 11:487–508. doi:10.1017/S0954579499002175
9. Keshavan M, Montrose DM, Rajarethinam R, Diwadkar V, Prasad K, Sweeney JA. Psychopathology among offspring of parents with schizophrenia: relationship to premorbid impairments. Schizophr Res (2008) 103:114–20. doi:10.1016/j.schres.2008.03.006
10. Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci (2002) 25:409–32. doi:10.1146/annurev.neuro.25.112701.142754
11. Millan MJ. An epigenetic framework for neurodevelopmental disorders: from pathogenesis to potential therapy. Neuropharmacology (2013) 68:2–82. doi:10.1016/j.neuropharm.2012.11.015
12. Keshavan MS, Diwadkar VA, Montrose DM, Rajarathinam R, Sweeney JA. Premorbid indicators of risk for schizophrenia: a selective review and update. Schizophr Res (2005) 79:45–57. doi:10.1016/j.schres.2005.07.004
13. Diwadkar VA. Adolescent risk pathways toward schizophrenia: sustained attention and the brain. Curr Top Med Chem (2012) 12:2339–47. doi:10.2174/156802612805289962
14. Diwadkar VA, Ofen N. Disordered brain network function in adolescence: impact on thought, language and vulnerability for schizophrenia. In: Brambilla P, Marini A, editors. Brain Evolution, Language and Psychopathology in Schizophrenia. Oxford: Taylor and Francis Group (2014). p. 73–95.
15. McEwen BS. Stress, adaptation, and disease. Allostasis and allostatic load. Ann N Y Acad Sci (1998) 840:33–44. doi:10.1111/j.1749-6632.1998.tb09546.x
16. McEwen BS. Effects of adverse experiences for brain structure and function. Biol Psychiatry (2000) 48:721–31. doi:10.1016/S0006-3223(00)00964-1
17. Petronis A, Paterson AD, Kennedy JL. Schizophrenia: an epigenetic puzzle? Schizophr Bull (1999) 25:639–55. doi:10.1093/oxfordjournals.schbul.a033408
18. Gogtay N, Rapoport JL. Childhood-onset schizophrenia: insights from neuroimaging studies. J Am Acad Child Adolesc Psychiatry (2008) 47:1120–4. doi:10.1097/CHI.0b013e31817eed7a
19. Rapoport JL, Gogtay N. Brain neuroplasticity in healthy, hyperactive and psychotic children: insights from neuroimaging. Neuropsychopharmacology (2008) 33:181–97. doi:10.1038/sj.npp.1301553
20. Weinberger DR, Egan MF, Bertolino A, Callicott JH, Mattay VS, Lipska BK, et al. Prefrontal neurons and the genetics of schizophrenia. Biol Psychiatry (2001) 50:825–44. doi:10.1016/S0006-3223(01)01252-5
21. Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry (2005) 10:40–68.
22. Gottesman I, Shields J. A critical review of recent adoption, twin, and family studies of schizophrenia: behavioral genetics perspectives. Schizophr Bull (1976) 2:360–401. doi:10.1093/schbul/2.3.360
23. Gottesman II, Shields J. Schizophrenia: The Epigenetic Puzzle. New York: Cambridge University Press (1982).
24. Lupien SJ, Ouellet-Morin I, Hupbach A, Tu MT, Buss C, Walker D, et al. Beyond the stress concept: allostatic load – a developmental biological and cognitive perspective. In: Cicchetiti DJCD, editor. Developmental Psychopathology. Hoboken, NJ: John Wiley & Sons (2006). p. 578–628.
25. Feinberg I. Cortical pruning and the development of schizophrenia. Schizophr Bull (1990) 16:567–70. doi:10.1093/schbul/16.4.567
26. Dahl RE. Adolescent brain development: a period of vulnerabilities and opportunities. Keynote address. Ann N Y Acad Sci (2004) 1021:1–22. doi:10.1196/annals.1308.001
27. McEwen BS, Stellar E. Stress and the individual. Mechanisms leading to disease. Arch Intern Med (1993) 153:2093–101.
28. Bremner JD, Narayan M. The effects of stress on memory and the hippocampus throughout the life cycle: implications for childhood development and aging. Dev Psychopathol (1998) 10:871–85. doi:10.1017/S0954579498001916
29. Heckers S. Neuroimaging studies of the hippocampus in schizophrenia. Hippocampus (2001) 11:520–8. doi:10.1002/hipo.1068
30. Vythilingam M, Vermetten E, Anderson GM, Luckenbaugh D, Anderson ER, Snow J, et al. Hippocampal volume, memory, and cortisol status in major depressive disorder: effects of treatment. Biol Psychiatry (2004) 56:101–12. doi:10.1016/j.biopsych.2004.04.002
31. Bremner JD. Effects of traumatic stress on brain structure and function: relevance to early responses to trauma. J Trauma Dissociation (2005) 6:51–68. doi:10.1300/J229v06n02_06
32. Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci (2009) 10:410–22. doi:10.1038/nrn2648
33. Pruessner JC, Champagne F, Meaney MJ, Dagher A. Dopamine release in response to a psychological stress in humans and its relationship to early life maternal care: a positron emission tomography study using [11C]raclopride. J Neurosci (2004) 24:2825–31. doi:10.1523/JNEUROSCI.3422-03.2004
34. Arnsten AF. Prefrontal cortical network connections: key site of vulnerability in stress and schizophrenia. Int J Dev Neurosci (2011) 29:215–23. doi:10.1016/j.ijdevneu.2011.02.006
35. Bassett DS, Bullmore E, Verchinski BA, Mattay VS, Weinberger DR, Meyer-Lindenberg A. Hierarchical organization of human cortical networks in health and schizophrenia. J Neurosci (2008) 28:9239–48. doi:10.1523/JNEUROSCI.1929-08.2008
36. Arnsten AF, Paspalas CD, Gamo NJ, Yang Y, Wang M. Dynamic network connectivity: a new form of neuroplasticity. Trends Cogn Sci (2010) 14:365–75. doi:10.1016/j.tics.2010.05.003
37. Banyai M, Diwadkar VA, Erdi P. Model-based dynamical analysis of functional disconnection in schizophrenia. Neuroimage (2011) 58:870–7. doi:10.1016/j.neuroimage.2011.06.046
38. Fusar-Poli P, Borgwardt S, Bechdolf A, Addington J, Riecher-Rossler A, Schultze-Lutter F, et al. The psychosis high-risk state: a comprehensive state-of-the-art review. JAMA Psychiatry (2013) 70:107–20. doi:10.1001/jamapsychiatry.2013.269
39. Walker EF, Diforio D, Baum K. Developmental neuropathology and the precursors of schizophrenia. Acta Psychiatr Scand Suppl (1999) 395:12–9. doi:10.1111/j.1600-0447.1999.tb05978.x
40. Wood SJ, Reniers RL, Heinze K. Neuroimaging findings in the at-risk mental state: a review of recent literature. Can J Psychiatry (2013) 58:13–8.
41. Bechdolf A, Thompson A, Nelson B, Cotton S, Simmons MB, Amminger GP, et al. Experience of trauma and conversion to psychosis in an ultra-high-risk (prodromal) group. Acta Psychiatr Scand (2010) 121:377–84. doi:10.1111/j.1600-0447.2010.01542.x
42. Ruhrmann S, Schultze-Lutter F, Salokangas RK, Heinimaa M, Linszen D, Dingemans P, et al. Prediction of psychosis in adolescents and young adults at high risk: results from the prospective European prediction of psychosis study. Arch Gen Psychiatry (2010) 67:241–51. doi:10.1001/archgenpsychiatry.2009.206
43. Holtzman CW, Shapiro DI, Trotman HD, Walker EF. Stress and the prodromal phase of psychosis. Curr Pharm Des (2012) 18:527–33. doi:10.2174/138161212799316280
44. Devylder JE, Ben-David S, Schobel SA, Kimhy D, Malaspina D, Corcoran CM. Temporal association of stress sensitivity and symptoms in individuals at clinical high risk for psychosis. Psychol Med (2013) 43:259–68. doi:10.1017/S0033291712001262
45. Holtzman CW, Trotman HD, Goulding SM, Ryan AT, Macdonald AN, Shapiro DI, et al. Stress and neurodevelopmental processes in the emergence of psychosis. Neuroscience (2013) 249:172–91. doi:10.1016/j.neuroscience.2012.12.017
46. Walker EF, Trotman HD, Goulding SM, Holtzman CW, Ryan AT, McDonald A, et al. Developmental mechanisms in the prodrome to psychosis. Dev Psychopathol (2013) 25:1585–600. doi:10.1017/S0954579413000783
47. Woods SW, Miller TJ, McGlashan TH. The “prodromal” patient: both symptomatic and at-risk. CNS Spectr (2001) 6:223–32.
48. Cannon TD, Cadenhead K, Cornblatt B, Woods SW, Addington J, Walker E, et al. Prediction of psychosis in youth at high clinical risk: a multisite longitudinal study in North America. Arch Gen Psychiatry (2008) 65:28–37. doi:10.1001/archgenpsychiatry.2007.3
49. Klosterkotter J, Schultze-Lutter F, Ruhrmann S. Kraepelin and psychotic prodromal conditions. Eur Arch Psychiatry Clin Neurosci (2008) 258(Suppl 2):74–84. doi:10.1007/s00406-008-2010-5
50. Yung AR, Phillips LJ, Yuen HP, Francey SM, McFarlane CA, Hallgren M, et al. Psychosis prediction: 12-month follow up of a high-risk (“prodromal”) group. Schizophr Res (2003) 60:21–32. doi:10.1016/S0920-9964(02)00167-6
51. Fusar-Poli P, Howes OD, Allen P, Broome M, Valli I, Asselin MC, et al. Abnormal frontostriatal interactions in people with prodromal signs of psychosis: a multimodal imaging study. Arch Gen Psychiatry (2010) 67:683–91. doi:10.1001/archgenpsychiatry.2010.77
52. Benetti S, Mechelli A, Picchioni M, Broome M, Williams S, McGuire P. Functional integration between the posterior hippocampus and prefrontal cortex is impaired in both first episode schizophrenia and the at risk mental state. Brain (2009) 132:2426–36. doi:10.1093/brain/awp098
53. Crossley NA, Mechelli A, Fusar-Poli P, Broome MR, Matthiasson P, Johns LC, et al. Superior temporal lobe dysfunction and frontotemporal dysconnectivity in subjects at risk of psychosis and in first-episode psychosis. Hum Brain Mapp (2009) 30:4129–37. doi:10.1002/hbm.20834
54. Dauvermann MR, Whalley HC, Romaniuk L, Valton V, Owens DG, Johnstone EC, et al. The application of nonlinear dynamic causal modelling for fMRI in subjects at high genetic risk of schizophrenia. Neuroimage (2013) 73:16–29. doi:10.1016/j.neuroimage.2013.01.063
55. Masillo A, Day F, Laing J, Howes O, Fusar-Poli P, Byrne M, et al. Interpersonal sensitivity in the at-risk mental state for psychosis. Psychol Med (2012) 42:1835–45. doi:10.1017/S0033291711002996
56. Howes OD, Bose SK, Turkheimer F, Valli I, Egerton A, Valmaggia LR, et al. Dopamine synthesis capacity before onset of psychosis: a prospective [18F]-DOPA PET imaging study. Am J Psychiatry (2011) 168:1311–7. doi:10.1176/appi.ajp.2011.11010160
57. Corcoran C, Walker E, Huot R, Mittal V, Tessner K, Kestler L, et al. The stress cascade and schizophrenia: etiology and onset. Schizophr Bull (2003) 29:671–92. doi:10.1093/oxfordjournals.schbul.a007038
58. Howes OD, Montgomery AJ, Asselin MC, Murray RM, Grasby PM, McGuire PK. Molecular imaging studies of the striatal dopaminergic system in psychosis and predictions for the prodromal phase of psychosis. Br J Psychiatry Suppl (2007) 51:s13–8. doi:10.1192/bjp.191.51.s13
59. Thompson KN, Phillips LJ, Komesaroff P, Yuen HP, Wood SJ, Pantelis C, et al. Stress and HPA-axis functioning in young people at ultra high risk for psychosis. J Psychiatr Res (2007) 41:561–9. doi:10.1016/j.jpsychires.2006.05.010
60. Tandon N, Montrose D, Shah J, Rajarethinam RP, Diwadkar VA, Keshavan MS. Early prodromal symptoms can predict future psychosis in familial high-risk youth. J Psychiatr Res (2012) 46:105–10. doi:10.1016/j.jpsychires.2011.09.019
61. Cannon TD, Gasperoni TL, Van Erp TG, Rosso IM. Quantitative neural indicators of liability to schizophrenia: implications for molecular genetic studies. Am J Med Genet (2001) 105:16–9. doi:10.1002/1096-8628(20010108)105:1<16::AID-AJMG1046>3.3.CO;2-Q
62. Cannon TD, Thompson PM, Van Erp TG, Toga AW, Poutanen VP, Huttunen M, et al. Cortex mapping reveals regionally specific patterns of genetic and disease-specific gray-matter deficits in twins discordant for schizophrenia. Proc Natl Acad Sci U S A (2002) 99:3228–33. doi:10.1073/pnas.052023499
63. Erlenmeyer-Kimling L, Squires-Wheeler E, Adamo UH, Bassett AS, Cornblatt BA, Kestenbaugh CJ, et al. The New York high-risk project. Arch Gen Psychiatry (1995) 10:857–65. doi:10.1001/archpsyc.1995.03950220067013
64. Mednick SA, Parnas J, Schulsinger F. The Copenhagen High-Risk Project, 1962-1986. Schizophr Bull (1987) 13:485–95. doi:10.1093/schbul/13.3.485
65. Mirsky AF, Kugelmass S, Ingraham LJ, Frenkel E, Nathan M. Overview and summary: twenty-five year follow-up of high-risk children. Schizophr Bull (1995) 21:227–39. doi:10.1093/schbul/21.2.227
66. Collip D, Nicolson NA, Lardinois M, Lataster T, Van Os J, Myin-Germeys I, et al. Daily cortisol, stress reactivity and psychotic experiences in individuals at above average genetic risk for psychosis. Psychol Med (2011) 41:2305–15. doi:10.1017/S0033291711000602
67. Habets P, Collip D, Myin-Germeys I, Gronenschild E, Van Bronswijk S, Hofman P, et al. Pituitary volume, stress reactivity and genetic risk for psychotic disorder. Psychol Med (2012) 42:1523–33. doi:10.1017/S0033291711002728
68. Diwadkar VA, Montrose DM, Dworakowski D, Sweeney JA, Keshavan MS. Genetically predisposed offspring with schizotypal features: an ultra high-risk group for schizophrenia? Prog Neuropsychopharmacol Biol Psychiatry (2006) 30:230–8. doi:10.1016/j.pnpbp.2005.10.019
69. Keshavan MS, Gilbert AR, Diwadkar VA. Developmental hypotheses of schizophrenia. In: Lieberman JA, Stroup TS, Perkins DO, editors. Textbook of Schizophrenia. Arlington, VA: American Psychiatric Publishing (2006). p. 69–84.
70. Erlenmeyer-Kimling L, Cornblatt BA. A summary of attentional findings in the New York high-risk project. J Psychiatr Res (1992) 26:405–26. doi:10.1016/0022-3956(92)90043-N
71. Diwadkar V, Sweeney J, Boarts D, Montrose D, Keshavan M. Oculomotor delayed response abnormalities in young offspring and siblings at risk for schizophrenia. CNS Spectr (2001) 6(11):899–903.
72. Cornblatt BA. The New York high risk project to the hillside recognition and prevention (RAP) program. Am J Med Genet (2002) 114:956–66. doi:10.1002/ajmg.b.10520
73. Johnstone EC, Russell KD, Harrison LK, Lawrie SMJ. The Edinburgh high risk study: current status and future prospects. World Psychiatry (2003) 2:45–9.
74. Phillips LK, Seidman LJ. Emotion processing in persons at risk for schizophrenia. Schizophr Bull (2008) 34:888–903. doi:10.1093/schbul/sbn085
75. Barbour T, Pruitt P, Diwadkar VA. fMRI responses to emotional faces in children and adolescents at genetic risk for psychiatric illness share some of the features of depression. J Affect Disord (2012) 136:276–85. doi:10.1016/j.jad.2011.11.036
76. Charmandari E, Kino T, Souvatzoglou E, Chrousos GP. Pediatric stress: hormonal mediators and human development. Horm Res (2003) 59:161–79. doi:10.1159/000069325
77. Brown GR, Spencer KA. Steroid hormones, stress and the adolescent brain: a comparative perspective. Neuroscience (2013) 249:115–28. doi:10.1016/j.neuroscience.2012.12.016
78. Bassett AS, Collins EJ, Nuttall SE, Honer WG. Positive and negative symptoms in families with schizophrenia. Schizophr Res (1993) 11:9–19. doi:10.1016/0920-9964(93)90033-F
79. Kwapil TR. Social anhedonia as a predictor of the development of schizophrenia-spectrum disorders. J Abnorm Psychol (1998) 107:558–65. doi:10.1037/0021-843X.107.4.558
80. Diwadkar VA, Segel J, Pruitt P, Murphy ER, Keshavan MS, Radwan J, et al. Hypo-activation in the executive core of the sustained attention network in adolescent offspring of schizophrenia patients mediated by pre-morbid functional deficits. Psychiatry Res (2011) 192:91–9. doi:10.1016/j.pscychresns.2010.12.005
81. Brekke JS, Raine A, Thomson C. Cognitive and psychophysiological correlates of positive, negative, and disorganized symptoms in the schizophrenia spectrum. Psychiatry Res (1995) 57:241–50. doi:10.1016/0165-1781(95)02668-M
82. Horan WP, Brown SA, Blanchard JJ. Social anhedonia and schizotypy: the contribution of individual differences in affective traits, stress, and coping. Psychiatry Res (2007) 149:147–56. doi:10.1016/j.psychres.2006.06.002
83. Shotbolt P, Stokes PR, Owens SF, Toulopoulou T, Picchioni MM, Bose SK, et al. Striatal dopamine synthesis capacity in twins discordant for schizophrenia. Psychol Med (2011) 41:2331–8. doi:10.1017/S0033291711000341
84. Bremne JD, Vermetten E. Stress and development: behavioral and biological consequences. Dev Psychopathol (2001) 13:473–89. doi:10.1017/S0954579401003042
85. Keshavan MS, Diwadkar V, Rosenberg DR. Developmental biomarkers in schizophrenia and other psychiatric disorders: common origins, different trajectories? Epidemiol Psichiatr Soc (2005) 14:188–93. doi:10.1017/S1121189X00007934
86. Paus T, Keshavan M, Giedd JN. Why do many psychiatric disorders emerge during adolescence? Nat Rev Neurosci (2008) 9:947–57. doi:10.1038/nrn2513
87. Ballon JS, Kaur T, Marks I, Cadenhead KS. Social functioning in young people at risk for schizophrenia. Psychiatry Res (2007) 151:29–35. doi:10.1016/j.psychres.2006.10.012
88. Keshavan MS. Development, disease and degeneration in schizophrenia: a unitary pathophysiological model. J Psychiatr Res (1999) 33:513–21. doi:10.1016/S0022-3956(99)00033-3
89. Cannon TD, Van Erp TG, Bearden CE, Loewy R, Thompson P, Toga AW, et al. Early and late neurodevelopmental influences in the prodrome to schizophrenia: contributions of genes, environment, and their interactions. Schizophr Bull (2003) 29:653–69. doi:10.1093/oxfordjournals.schbul.a007037
90. Tan HY, Callicott JH, Weinberger DR. Dysfunctional and compensatory prefrontal cortical systems, genes and the pathogenesis of schizophrenia. Cereb Cortex (2007) 17(Suppl 1):i171–81. doi:10.1093/cercor/bhm069
91. Gore CD, Banyai M, Gray PJ, Diwadkar V, Erdi P. Pathological effects of cortical architecture on working memory in schizophrenia. Pharmacopsychiatry (2010) 43(Suppl 1):S92–7. doi:10.1055/s-0030-1251979
92. Posner MI. Imaging attention networks. Neuroimage (2012) 61:450–6. doi:10.1016/j.neuroimage.2011.12.040
93. Goldman-Rakic PS, Castner SA, Svensson TH, Siever LJ, Williams GV. Targeting the dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction. Psychopharmacology (Berl) (2004) 174:3–16. doi:10.1007/s00213-004-1793-y
94. Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AF. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci (2007) 10:376–84. doi:10.1038/nn1846
95. Diwadkar VA, Pruitt P, Zhang A, Radwan J, Keshavan MS, Murphy E, et al. The neural correlates of performance in adolescents at risk for schizophrenia: inefficiently increased cortico-striatal responses measured with fMRI. J Psychiatr Res (2012) 46:12–21. doi:10.1016/j.jpsychires.2011.09.016
96. Callicott JH, Bertolino A, Mattay VS, Langheim FJ, Duyn J, Coppola R, et al. Physiological dysfunction of the dorsolateral prefrontal cortex in schizophrenia revisited. Cereb Cortex (2000) 10:1078–92. doi:10.1093/cercor/10.11.1078
97. Jansma JM, Ramsey NF, Van Der Wee NJ, Kahn RS. Working memory capacity in schizophrenia: a parametric fMRI study. Schizophr Res (2004) 68:159–71. doi:10.1016/S0920-9964(03)00127-0
98. Walton E, Geisler D, Lee PH, Hass J, Turner JA, Liu J, et al. Prefrontal inefficiency is associated with polygenic risk for schizophrenia. Schizophr Bull (2013). doi:10.1093/schbul/sbt174
99. Paus T. Primate anterior cingulate cortex: where motor control, drive and cognition interface. Nat Rev Neurosci (2001) 2:417–24. doi:10.1038/35077500
100. Bakshi N, Pruitt P, Radwan J, Keshavan MS, Rajan U, Zajac-Benitez C, et al. Inefficiently increased anterior cingulate modulation of cortical systems during working memory in young offspring of schizophrenia patients. J Psychiatr Res (2011) 45:1067–76. doi:10.1016/j.jpsychires.2011.01.002
101. Diwadkar VA, Bakshi N, Gupta G, Pruitt P, White R, Eickhoff SB, et al. Dysfunction and disconnection in cortical–striatal networks during sustained attention: genetic risk for schizophrenia or bipolar disorder and its impact on brain network function. Front Psychiatry (2014) 5:50. doi:10.3389/fpsyt.2014.00050
102. Friston KJ. Models of brain function in neuroimaging. Annu Rev Psychol (2005) 56:57–87. doi:10.1146/annurev.psych.56.091103.070311
103. Diwadkar VA, Wadehra S, Pruitt P, Keshavan MS, Rajan U, Zajac-Benitez C, et al. Disordered cortico-limbic interactions during affective processing in children and adolescents at risk for schizophrenia revealed by fMRI and dynamic causal modeling. Arch Gen Psychiatry (2012) 69:231–42. doi:10.1001/archgenpsychiatry.2011.1349
104. Cohen AS, Docherty NM, Nienow T, Dinzeo T. Self-reported stress and the deficit syndrome of schizophrenia. Psychiatry (2003) 66:308–16.
105. Pizzagalli DA, Oakes TR, Fox AS, Chung MK, Larson CL, Abercrombie HC, et al. Functional but not structural subgenual prefrontal cortex abnormalities in melancholia. Mol Psychiatry (2004) 9(325):393–405. doi:10.1038/sj.mp.40014694001469
106. Soliman A, O’Driscoll GA, Pruessner J, Joober R, Ditto B, Streicker E, et al. Limbic response to psychosocial stress in schizotypy: a functional magnetic resonance imaging study. Schizophr Res (2011) 131:184–91. doi:10.1016/j.schres.2011.05.016
107. Strauss GP, Gold JM. A new perspective on anhedonia in schizophrenia. Am J Psychiatry (2012) 169:364–73. doi:10.1176/appi.ajp.2011.11030447
108. Irwin MR, Cole SW. Reciprocal regulation of the neural and innate immune systems. Nat Rev Immunol (2011) 11:625–32. doi:10.1038/nri3042
109. Walker E, Mittal V, Tessner K. Stress and the hypothalamic pituitary adrenal axis in the developmental course of schizophrenia. Annu Rev Clin Psychol (2008) 4:189–216. doi:10.1146/annurev.clinpsy.4.022007.141248
110. Grayson DR, Chen Y, Dong E, Kundakovic M, Guidotti A. From trans-methylation to cytosine methylation: evolution of the methylation hypothesis of schizophrenia. Epigenetics (2009) 4:144–9. doi:10.4161/epi.4.3.8534
111. Grayson DR. Schizophrenia and the epigenetic hypothesis. Epigenomics (2010) 2:341–4. doi:10.2217/epi.10.22
112. Grayson DR, Guidotti A. The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology (2013) 38:138–66. doi:10.1038/npp.2012.125
113. Van Speybroeck L. From epigenesis to epigenetics: the case of C. H. Waddington. Ann N Y Acad Sci (2002) 981:61–81. doi:10.1111/j.1749-6632.2002.tb04912.x
114. Serafini G, Pompili M, Innamorati M, Giordano G, Montebovi F, Sher L, et al. The role of microRNAs in synaptic plasticity, major affective disorders and suicidal behavior. Neurosci Res (2012) 73:179–90. doi:10.1016/j.neures.2012.04.001
115. Lett TA, Chakavarty MM, Felsky D, Brandl EJ, Tiwari AK, Goncalves VF, et al. The genome-wide supported microRNA-137 variant predicts phenotypic heterogeneity within schizophrenia. Mol Psychiatry (2013) 18:443–50. doi:10.1038/mp.2013.17
116. Cross-Disorder Group of the Psychiatric Genomics Lee C, Ripke SH, Neale S, Faraone BM, Purcell SV, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet (2013) 45:984–94. doi:10.1038/ng.2711
117. Rutter M, Moffit TE, Caspi A. Gene-environment interplay and psychopathology: multiple varieties but real effects. J Child Psychol Psychiatry (2006) 47:226–61. doi:10.1111/j.1469-7610.2005.01557.x
118. D’Souza DC, Sewell RA, Ranganathan M. Cannabis and psychosis/schizophrenia: human studies. Eur Arch Psychiatry Clin Neurosci (2009) 259:413–31. doi:10.1007/s00406-009-0024-2
119. Gonzalez-Burgos G, Hashimoto T, Lewis DA. Alterations of cortical GABA neurons and network oscillations in schizophrenia. Curr Psychiatry Rep (2010) 12:335–44. doi:10.1007/s11920-010-0124-8
120. Yang X, Ewald ER, Huo Y, Tamashiro KL, Salvatori R, Sawa A, et al. Glucocorticoid-induced loss of DNA methylation in non-neuronal cells and potential involvement of DNMT1 in epigenetic regulation of Fkbp5. Biochem Biophys Res Commun (2012) 420:570–5. doi:10.1016/j.bbrc.2012.03.035
121. Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci (2013) 16:33–41. doi:10.1038/nn.3275
122. Lee RS, Tamashiro KL, Yang X, Purcell RH, Harvey A, Willour VL, et al. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology (2010) 151:4332–43. doi:10.1210/en.2010-0225
123. Lee RS, Tamashiro KL, Yang X, Purcell RH, Huo Y, Rongione M, et al. A measure of glucocorticoid load provided by DNA methylation of Fkbp5 in mice. Psychopharmacology (Berl) (2011) 218:303–12. doi:10.1007/s00213-011-2307-3
124. Mehta D, Binder EB. Gene x environment vulnerability factors for PTSD: the HPA-axis. Neuropharmacology (2012) 62:654–62. doi:10.1016/j.neuropharm.2011.03.009
125. Aas M, Dazzan P, Mondelli V, Melle I, Murray RM, Pariante CM. A systematic review of cognitive function in first-episode psychosis, including a discussion on childhood trauma, stress, and inflammation. Front Psychiatry (2014) 4:182. doi:10.3389/fpsyt.2013.00182
126. Menne-Lothmann C, Jacobs N, Derom C, Thiery E, Van Os J, Wichers M. Genetic and environmental causes of individual differences in daily life positive affect and reward experience and its overlap with stress-sensitivity. Behav Genet (2012) 42:778–86. doi:10.1007/s10519-012-9553-y
127. Morris MC, Ciesla JA, Garber J. A prospective study of stress autonomy versus stress sensitization in adolescents at varied risk for depression. J Abnorm Psychol (2010) 119:341–54. doi:10.1037/a0019036
128. Collip D, Habets P, Marcelis M, Gronenschild E, Lataster T, Lardinois M, et al. Hippocampal volume as marker of daily life stress sensitivity in psychosis. Psychol Med (2012) 43(7):1377–87. doi:10.1017/S003329171200219X
129. Zimmerman EC, Bellaire M, Ewing SG, Grace AA. Abnormal stress responsivity in a rodent developmental disruption model of schizophrenia. Neuropsychopharmacology (2013) 38:2131–9. doi:10.1038/npp.2013.110
130. Federenko IS, Schlotz W, Kirschbaum C, Bartels M, Hellhammer DH, Wust S. The heritability of perceived stress. Psychol Med (2006) 36:375–85. doi:10.1017/S0033291705006616
131. Bogdan R, Pizzagalli DA. The heritability of hedonic capacity and perceived stress: a twin study evaluation of candidate depressive phenotypes. Psychol Med (2009) 39:211–8. doi:10.1017/S0033291708003619
132. Fillman SG, Sinclair D, Fung SJ, Webster MJ, Shannon Weickert C. Markers of inflammation and stress distinguish subsets of individuals with schizophrenia and bipolar disorder. Transl Psychiatry (2014) 4:e365. doi:10.1038/tp.2014.8
133. Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci (2004) 7:847–54. doi:10.1038/nn1276nn1276
134. Piazza PV, Barrot M, Rouge-Pont F, Marinelli M, Maccari S, Abrous DN, et al. Suppression of glucocorticoid secretion and antipsychotic drugs have similar effects on the mesolimbic dopaminergic transmission. Proc Natl Acad Sci U S A (1996) 93:15445–50. doi:10.1073/pnas.93.26.15445
135. Goel N, Bale TL. Sex differences in the serotonergic influence on the hypothalamic-pituitary-adrenal stress axis. Endocrinology (2010) 151:1784–94. doi:10.1210/en.2009-1180
136. Hanna GL, Yuwiler A, Cantwell DP. Whole blood serotonin in juvenile obsessive-compulsive disorder. Biol Psychiatry (1991) 29:738–44. doi:10.1016/0006-3223(91)90193-P
137. Sallee FR, Richman H, Beach K, Sethuraman G, Nesbitt L. Platelet serotonin transporter in children and adolescents with obsessive-compulsive disorder or Tourette’s syndrome. J Am Acad Child Adolesc Psychiatry (1996) 35:1647–56. doi:10.1097/00004583-199612000-00017
138. Delorme R, Betancur C, Callebert J, Chabane N, Laplanche JL, Mouren-Simeoni MC, et al. Platelet serotonergic markers as endophenotypes for obsessive-compulsive disorder. Neuropsychopharmacology (2005) 30:1539–47. doi:10.1038/sj.npp.1300752
139. Wang D, Szyf M, Benkelfat C, Provencal N, Turecki G, Caramaschi D, et al. Peripheral SLC6A4 DNA methylation is associated with in vivo measures of human brain serotonin synthesis and childhood physical aggression. PLoS One (2012) 7:e39501. doi:10.1371/journal.pone.0039501
140. Shumay E, Logan J, Volkow ND, Fowler JS. Evidence that the methylation state of the monoamine oxidase A (MAOA) gene predicts brain activity of MAO A enzyme in healthy men. Epigenetics (2012) 7:1151–60. doi:10.4161/epi.21976
141. Ursini G, Bollati V, Fazio L, Porcelli A, Iacovelli L, Catalani A, et al. Stress-related methylation of the catechol-O-methyltransferase Val 158 allele predicts human prefrontal cognition and activity. J Neurosci (2011) 31:6692–8. doi:10.1523/JNEUROSCI.6631-10.2011
142. Koenen KC, Uddin M, Chang SC, Aiello AE, Wildman DE, Goldmann E, et al. SLC6A4 methylation modifies the effect of the number of traumatic events on risk for posttraumatic stress disorder. Depress Anxiety (2011) 28(8):639–47. doi:10.1002/da.20825
143. Jay TM, Rocher C, Hotte M, Naudon L, Gurden H, Spedding M. Plasticity at hippocampal to prefrontal cortex synapses is impaired by loss of dopamine and stress: importance for psychiatric diseases. Neurotox Res (2004) 6:233–44. doi:10.1007/BF03033225
144. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III – the final common pathway. Schizophr Bull (2009) 35:549–62. doi:10.1093/schbul/sbp006
145. Moghaddam B. Stress activation of glutamate neurotransmission in the prefrontal cortex: implications for dopamine-associated psychiatric disorders. Biol Psychiatry (2002) 51:775–87. doi:10.1016/S0006-3223(01)01362-2
146. Hirvonen J, Hietala J. Dysfunctional brain networks and genetic risk for schizophrenia: specific neurotransmitter systems. CNS Neurosci Ther (2011) 17:89–96. doi:10.1111/j.1755-5949.2010.00223.x
147. Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics (2008) 3:97–106. doi:10.4161/epi.3.2.6034
148. Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology (2009) 34(Suppl 1):S186–95. doi:10.1016/j.psyneuen.2009.05.021
149. Wockner LF, Noble EP, Lawford BR, Young RM, Morris CP, Whitehall VL, et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl Psychiatry (2014) 4:e339. doi:10.1038/tp.2013.111
150. Zhao J, Goldberg J, Bremner JD, Vaccarino V. Association between promoter methylation of serotonin transporter gene and depressive symptoms: a monozygotic twin study. Psychosom Med (2013) 75:523–9. doi:10.1097/PSY.0b013e3182924cf4
151. Sipahi L, Wildman DE, Aiello AE, Koenen KC, Galea S, Abbas A, et al. Longitudinal epigenetic variation of DNA methyltransferase genes is associated with vulnerability to post-traumatic stress disorder. Psychol Med (2014). doi:10.1017/S0033291714000968
152. de Jong S, Boks MPM, Fuller TF, Strengman E, Janson E, de Kovel CG, et al. A gene co-expression network in whole blood of schizophrenia patients is independent of antipsychotic-use and enriched for brain-expressed genes. PLoS One (2012) 7:e39498. doi:10.1371/journal.pone.0039498
153. Nishioka M, Bundo M, Koike S, Takizawa R, Kakiuchi C, Araki T, et al. Comprehensive DNA methylation analysis of peripheral blood cells derived from patients with first-episode schizophrenia. J Hum Genet (2013) 58:91–7. doi:10.1038/jhg.2012.140
154. Dempster EL, Pidsley R, Schalkwyk LC, Owens S, Georgiades A, Kane F, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet (2011) 20:4786–96. doi:10.1093/hmg/ddr416
155. Casey BJ, Giedd JN, Thomas KM. Structural and functional brain development and its relation to cognitive development. Biol Psychol (2000) 54:241–57.
156. Casey BJ, Galvan A, Hare TA. Changes in cerebral functional organization during cognitive development. Curr Opin Neurobiol (2005) 15:239–44. doi:10.1016/j.conb.2005.03.012
157. Stephan KE, Roebroeck A. A short history of causal modeling of fMRI data. Neuroimage (2012) 62:856–63. doi:10.1016/j.neuroimage.2012.01.034
Keywords: schizophrenia, adolescence, risk, epigenetics, brain networks
Citation: Diwadkar VA, Bustamante A, Rai H and Uddin M (2014) Epigenetics, stress, and their potential impact on brain network function: a focus on the schizophrenia diatheses. Front. Psychiatry 5:71. doi: 10.3389/fpsyt.2014.00071
Received: 26 March 2014; Accepted: 04 June 2014;
Published online: 23 June 2014.
Edited by:
André Schmidt, University of Basel, SwitzerlandReviewed by:
Kwang-Hyuk Lee, University of Sheffield, UKGianluca Serafini, Sapienza University of Rome, Italy
Copyright: © 2014 Diwadkar, Bustamante, Rai and Uddin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vaibhav A. Diwadkar, Department of Psychiatry and Behavioral Neurosciences, Brain Imaging Research Division, Tolan Park Medical Building, Suite 5B, 3901 Chrysler Service Drive, Detroit, MI 48201, USA e-mail:dmRpd2Fka2FAbWVkLndheW5lLmVkdQ==