 Jennifer M. Wenzel
Jennifer M. Wenzel Joseph F. Cheer
Joseph F. Cheer- 1Department of Anatomy and Neurobiology, University of Maryland School of Medicine, Baltimore, MD, USA
- 2Department of Psychiatry, University of Maryland School of Medicine, Baltimore, MD, USA
The mesolimbic dopamine (DA) system plays an integral role in incentive motivation and reward seeking and a growing body of evidence identifies signal transduction at cannabinoid receptors as a critical modulator of this system. Indeed, administration of exogenous cannabinoids results in burst firing of DA neurons of the ventral tegmental area and increases extracellular DA in the nucleus accumbens (NAcc). Implementation of fast-scan cyclic voltammetry (FSCV) confirms the ability of cannabinoids to augment DA within the NAcc on a subsecond timescale. The use of FSCV along with newly developed highly selective pharmacological compounds advances our understanding of how cannabinoids influence DA transmission and highlights a role for endocannabinoid-modulated subsecond DAergic activation in the incentive motivational properties of not only external, but also internal reward-predictive cues. For example, our laboratory has recently demonstrated that in mice responding under a fixed-interval (FI) schedule for food reinforcement, fluctuations in NAcc DA signal the principal cue predictive of reinforcer availability – time. That is, as the interval progresses, NAcc DA levels decline leading to accelerated food seeking and the resulting characteristic FI scallop pattern of responding. Importantly, administration of WIN 55,212-2, a synthetic cannabinoid agonist, or JZL184, an indirect cannabinoid agonist, increases DA levels during the interval and disrupts this pattern of responding. Along with a wealth of other reports, these results illustrate the role of cannabinoid receptor activation in the regulation of DA transmission and the control of temporally guided reward seeking. The current review will explore the striatal beat frequency model of interval timing as it pertains to cannabinoid signaling and propose a neurocircuitry through which this system modulates interoceptive time cues.
Introduction
A wealth of psychology research has documented an integral role for environmental stimuli in guiding reward seeking behavior. Stimuli that repeatedly occur in conjunction with the presentation of reinforcers (for the sake of simplicity, “rewards”), themselves gain incentive value as reward predictors and work to motivate behavior (1). For example, the neutral auditory stimulus of a can of cat food being opened, through its repeated pairing with food delivery, serves as an incentive stimulus to the cat that is predictive of food-related stimuli (e.g., taste) and energizes the cat’s approach toward the food bowl. Of course, external motivational cues (e.g., sound of the can opener) frequently interact with internal cues (e.g., hunger) to guide behavior – i.e., if the cat is not hungry the likelihood that it will approach the food bowl is greatly reduced (2, 3). Further, through repeated pairing external cues may gain the ability to elicit internal cues, such as hunger (4) or the initiation of an internal clock that works to predict reward availability (5). While conditioned environmental cues guide advantageous instrumental behaviors, they also may support the development of detrimental behaviors such as drug abuse. Indeed, cues frequently paired with drug use (e.g., drug paraphernalia) develop incentive value that in turn promotes drug seeking and contributes to the relapse [see review in Ref. (6)]. Given the formidable influence of incentive cues on both positive and negative behaviors, it is important to better understand the neurobiological mechanisms subserving cue-driven behaviors. One system repeatedly implicated in incentive motivational processes is the mesolimbic dopamine (DA) system (7).
DAergic cell bodies resident to the ventral tegmental area (VTA) send their diffuse projections to various cortical and limbic regions such as the prefrontal cortex, amygdala, hippocampus, and the ventral striatum (nucleus accumbens, NAcc) (8). Together, this network comprises the mesolimbic DA system, a system that is highly implicated in the development and maintenance of reward seeking behaviors. A wide body of evidence supports a role for mesolimbic DA in reinforcement learning and motivation for incentive stimuli (9–12). For instance, natural reinforcers such as food and water, as well as drugs of abuse and brain stimulation reward (BSR), support operant behaviors through their ability to activate the mesolimbic system (13–17). Evidencing this view, DAergic lesions or antagonism attenuate approaches toward, or responding for, these reinforcers (18–21).
Single-unit recording data show that VTA DAergic neurons fire in two distinct modes: low-frequency (1–5 Hz) tonic activity and high-frequency (≥20 Hz) phasic activity, the latter of which is characterized by transient rapid bursts (<1 s) of cell firing (22). In the absence of salient stimuli midbrain DA neurons exhibit tonic pacemaker activity producing a steady DAergic “tone” on high-affinity inhibitory D2-like (D2, D3, D4) DA receptors of the mesolimbic system (23–25). This baseline DAergic tone is believed to facilitate long-term depression (LTD) at cortico-striatal synapses and suppress activity of the basal ganglia indirect pathway (26, 27). The presentation of motivationally relevant stimuli such as primary rewards, however, results in rapid burst firing of midbrain DA cells, which increases terminal DA sufficiently to occupy low-affinity excitatory D1-like (D1 and D5) receptors (24). D1 receptor activation following reward-related stimuli is proposed to result in enhanced long-term potentiation (LTP) at excitatory synapses and activation of the basal ganglia direct pathway, thereby motivating behavior (28). Therefore distinct patterns of DAergic cell activity provide a mechanism through which high- and low-affinity DA receptor populations may be differentially activated resulting in the conveyance of distinct reward-related information.
Interestingly, DA neurons adapt to reward presentations by signaling the difference in value of “expected” versus “received” rewards, or a reward prediction error (29, 30). In support of this theory, the presentation of an unexpected reward, such as reinforcer delivery in the initial stages of a conditioning paradigm, results in burst firing of midbrain DA neurons. Following repeated presentations, however, this phasic DA signal previously coupled to reward receipt now results from presentation of reward-predictive stimuli that precede reward delivery (29, 31). The magnitude of burst activity is greater to the reward-predictive cues when the probability of reward is high, but when the probability of reward is low DAergic cell activity is greater during reward receipt. Conversely, midbrain DA neurons cease their firing when no reward or an aversive stimulus is delivered, communicating a negative reward prediction error (30). It should be mentioned that non-appetitive auditory, tactile, or visual sensory stimuli can result in burst firing of midbrain DA cells (32–35), however, these cells appear to fire preferentially to reward-related stimuli (36). Thus, phasic activation of midbrain DA neurons transmits information about previous (expectancy) and current reward situations, making this form of signaling particularly important to the development of conditioned reward associations (37, 38). Indeed, Zweifel et al. (39) reported that genetic inactivation of N-methyl-d-aspartate (NMDA) glutamate receptors on DA neurons, a treatment that blocks the ability of DA neurons to burst fire, attenuates stimulus-response learning.
Measurement of Extracellular Dopamine Concentrations within the NAcc
While electrophysiological recordings provide valuable information about DAergic cell activity patterns, the relationship between DA cell firing and neurotransmitter release at terminal regions is not linear (40). Therefore, techniques that allow for the measurement of extracellular DA concentrations within discrete structures are critical to evaluate functional roles of regional DA release.
Tonic DA levels can be measured using in vivo microdialysis techniques, allowing for neurochemical analysis of brain dialysate with a temporal resolution of minutes [(41), for review see Ref. (42)]. A wealth of microdialysis data correlate reward-related phenomena with enhanced DA levels at mesolimbic terminal regions, such as the NAcc. For example, DA levels are elevated in target regions of the mesolimbic system following self-administration of either food (14, 15, 43), water (17), or drugs of abuse (44–50). However, a sample rate of minutes is insufficient to disentangle DA release related to reward receipt versus cue-evoked DA. Direct assessment of subsecond fluctuations in DA concentration due to phasic firing requires the use of techniques with greater temporal resolution, such as fast-scan cyclic voltammetry (FSCV). FSCV has consistently been utilized to measure subsecond transient changes in DA concentration within distinct brain areas [for review see Ref. (51)] of both anesthetized (52) and behaving animals (53, 54). However, FSCV cannot readily differentiate between norepinephrine and DA signals. Thus, voltammetric assessment of phasic DA activity is best suited for regions with low noradrenergic input, i.e., the NAcc.
Research employing FSCV demonstrates that stimuli promoting burst activity of DA neurons also produce transient increases in extracellular DA concentration (termed “transients”) at terminal fields of the mesolimbic system. For example, several studies show enhanced DA transient activity within the NAcc coincident with the presentation of a food reward or related reward-predictive cues (55–59) – stimuli known to result in phasic burst firing of midbrain DA neurons (30, 60, 61). Importantly, a wide body of FSCV data support a role for reward-evoked striatal DA as a reward prediction error signal. Indeed, enhanced phasic DA transmission is reliably observed following unexpected reward delivery or, after conditioning, in response to cues that predict reward (40, 55, 57, 59, 62–64). Further, in congruence with electrophysiological data, reward omission or administration of an aversive stimulus results in decreased extracellular DA within the ventral striatum (65–67).
Shifts in midbrain DA neuron activation from tonic low-frequency activity to phasic high-frequency burst firing likely result from changes in synaptic input from glutamate and gamma-aminobutyric (GABA) afferents to VTA DA cells. The VTA receives excitatory afferents from both sensory and cognitive regions, including glutamatergic afferents from the prefrontal cortex, the extended amygdala, and the laterodorsal and pedunculopontine tegmental nuclei (68–70) and inhibitory GABAergic input from the basal ganglia and rostromedial tegmental nucleus. DAergic neurons in brain slice preparations (i.e., without afferent input) exhibit pacemaker-like tonic activation but do not fire in bursts, thus DA cells are “conditional” rather than “intrinsic” bursters (71, 72). Indeed, burst firing of DA neurons requires glutamatergic input and the activation of DAergic cell NMDA glutamate receptors (73, 74). Conversely, GABAergic input to midbrain DA neurons dampens burst firing and returns the cell to baseline pacemaker-like activity (75). Thus, the maintenance of midbrain DAergic firing patterns requires a balance between excitatory and inhibitory VTA afferent signals. A key signaling network implicated in the maintenance of this balance is the endocannabinoid system.
A Brief Overview of the Endocannabinoid System
The endocannabinoid system, composed of endogenous cannabinoids (i.e., endocannabinoids), cannabinoid receptors, and the enzymes responsible for endocannabinoid synthesis and degradation, is a neuromodulatory network known to play a role in a number of neural processes, including learning and memory, motivation, reward, operant behavior, and neuroplasticity (76–79). Cannabinoid receptors were the first components of this system to be discovered. In the early 1990s, Δ9-tetrahydrocannabinol (THC), the primary psychoactive constituent of the cannabis plant, was found to produce its characteristic effects as a partial agonist of a G-protein-coupled receptor (GPCR) (Gi/o) isolated from neural cell lines and later named cannabinoid receptor type 1 (CB1) (80, 81). A few years after this discovery, a second cannabinoid receptor (cannabinoid receptor type 2; CB2) was identified (82). CB1 and CB2 receptors differ in anatomical distribution as well as function. Autoradiography studies show that CB1 is the primary cannabinoid receptor of the central nervous system (CNS) with a wide distribution throughout the brain and periphery and the highest concentrations of CB1 binding found in brain regions implicated in the actions of cannabis (83–85). Conversely, CB2 is more abundant in the periphery and expressed primarily in immune cells, including microglia of the CNS (86), and is thus believed to primarily play a role in immune function (87). Recent evidence, however, also supports a role for CB2 in a variety of neurological processes, such as anxiety, pain, and addiction (88–91). It is important to note that while this review will focus on activity at CB receptors, endocannabinoids also interact with various ligand-gated ion channels (e.g., vanilloid receptor type 1 channels) as well as other GPCRs, such as GPR55 (92, 93).
The discovery of cannabinoid receptors was followed shortly after by the identification of their primary endogenous ligands – N-arachidonylethanolamine (anandamide; AEA), a partial agonist at CB1 receptors, and 2-arachidonylglycerol (2-AG), a full agonist at both CB1 and CB2 receptors (94–96). The biosynthesis of AEA is not fully understood, although it is generally agreed that AEA is synthesized from N-arachidonoyl phosphatidylethanolamine (NAPE) in a Ca2+-dependent manner via one of the several possible enzymatic pathways (97, 98). Conversely, both Ca2+-dependent and Ca2+-independent synthesis pathways for 2-AG have been outlined (illustrated in Figure 1). Following synthesis and release, AEA and 2-AG signaling is quickly terminated through cellular reuptake and hydrolysis primarily by the enzymes fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), respectively [FAAH can also hydrolyze 2-AG (99)]. While several additional endocannabinoids have since been discovered, AEA and 2-AG remain the best characterized.
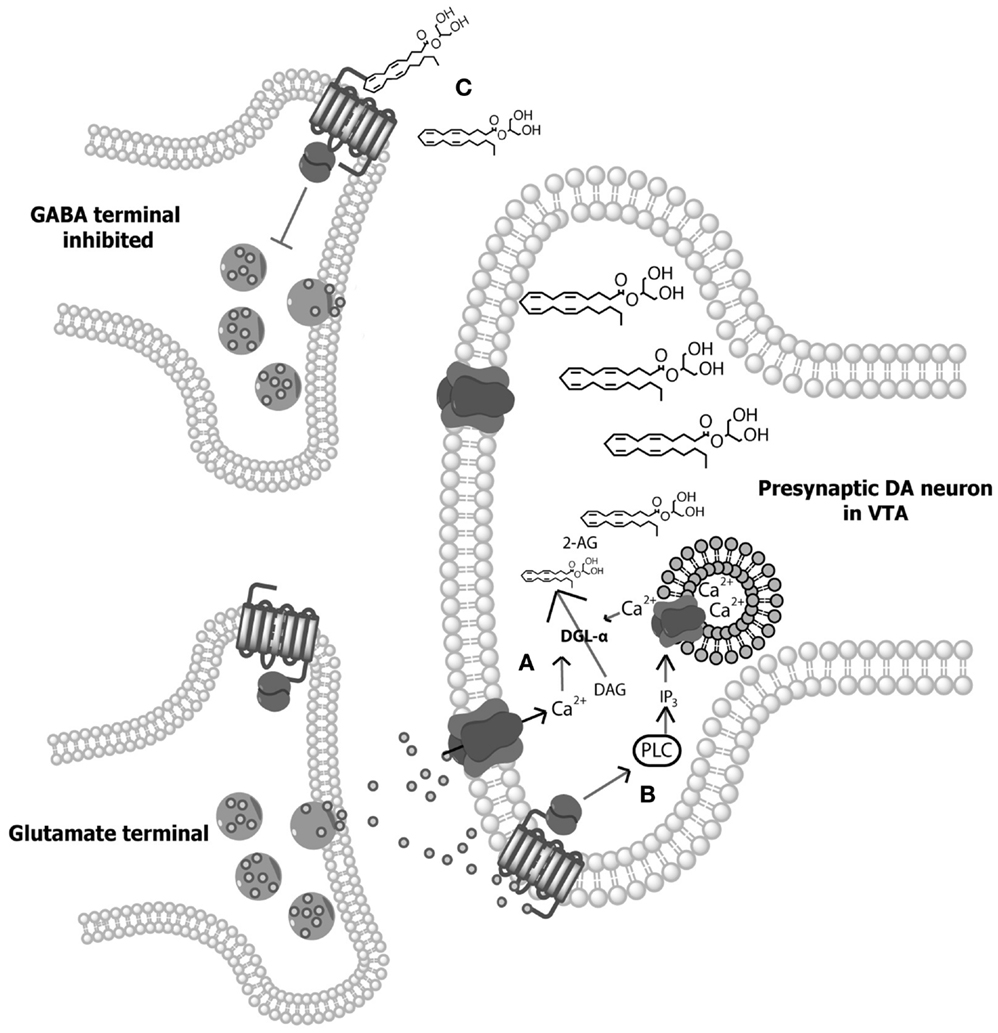
Figure 1. Illustration of 2-arachidonylglycerol (2-AG) synthesis. (A) Depolarization-induced Ca2+ influx within dopamine (DA) neurons of the ventral tegmental area (VTA) results in the hydrolysis of 1,2-diacylglycerol (DAG) by DGL-α and DGL-β lipases to form 2-AG (98, 100). (B) Alternatively, activation of Gq/11 protein-coupled receptors (e.g., group 1 metabotropic glutamate receptors) directly stimulate phospholipase-Cβ (PLC), resulting in the hydrolysis of membrane phosphate phosphatidylinositol 4,5-bisphosphate (PIP2) to DAG, allowing for subsequent hydrolysis of DAG to 2-AG (101–103). In addition, Ca2+-dependent and GPCR-dependent 2-AG synthesis can co-occur to synergistically produce high concentrations of 2-AG (104, 105). (C) Following on-demand synthesis, 2-AG then diffuses from the postsynaptic DA neurons and binds with CB1 receptors on presynaptic gamma- aminobutyric acid (GABA) cells, inhibiting GABA release and thereby disinhibiting DAergic cell activity.
The most prominent physiological role for endocannabinoids is as synaptic retrograde messengers – molecules that transmit messages from post- to presynaptic cells. Indeed, both AEA and 2-AG signal retrogradely. Like all known endocannabinoids, AEA and 2-AG are lipid molecules, which in contrast to classic neurotransmitters, are synthesized and released from neurons “on demand” upon stimulation (106, 107). These lipophilic messengers diffuse through the postsynaptic membrane and interact with cannabinoid receptors of nearby cells. Both CB1 and CB2 receptors are Gi/o-coupled receptors; therefore, their activation results in a decrease in cAMP activity within the cell and subsequent inhibition of neurotransmitter release, resulting in negative feedback to presynaptic cells (108–110). When Ca2+-mediated, this process has been termed depolarization-induced suppression of inhibition (DSI) or excitation (DSE) depending on the neurotransmitter system being inhibited (111), and works to modulate synaptic plasticity. Research on the individual functions of AEA and 2-AG suggests that 2-AG is the principal endocannabinoid involved in DSI- and DSE-induced plasticity (109).
Endocannabinoid Modulation of Dopamine Transmission
Like the discovery of the endocannabinoid system, the ability of endocannabinoids to modulate mesolimbic DA transmission was uncovered through investigations into the effects of THC. All drugs of abuse increase DA neurotransmission in the NAcc (112, 113) and although some early studies exploring a role for DA in cannabinoid abuse potential yielded incongruent results [e.g., Ref. (114, 115)], today a large body of evidence suggests that marijuana is no exception. Indeed, cannabinoid administration enhances extracellular DA concentrations in the ventral striatum (116–119). Further, this effect is dependent on CB1 receptor signaling, as pretreatment with the CB1 receptor antagonist/inverse agonist SR141716A (rimonabant) blocks the ability of THC, or the synthetic cannabinoid receptor agonist WIN 55,212-2 (WIN), to enhance striatal DA levels (119). Seminal single-unit recording data from French et al. (120) show that cannabinoids enhance extracellular DA concentrations in the NAcc through increasing both the baseline firing rates and burst frequency of midbrain DA neurons. These enhancements in VTA DA cell firing are also CB1-dependent as they are attenuated by rimonabant (121).
Interestingly, while midbrain DA neurons release endocannabinoids during phasic activation, they do not express CB1 receptors (83, 109, 122, 123). This suggests that cannabinoids excite VTA DA cells via an indirect mechanism. Aside from DAergic cell bodies, the VTA also contains GABAergic neurons that inhibit midbrain DA activity through interaction with GABAA receptors on DA cells (8). The application of the GABAA agonist bicuculine to VTA-containing slices enhances DAergic cell activity (124). Thus, cannabinoids may increase DA neuron burst firing through the inhibition of VTA GABA cells, resulting in disinhibition of midbrain DA neurons (109, 110). In support of this explanation, Szabo et al. (125) found that WIN reduces electrically evoked GABAA-mediated inhibitory postsynaptic currents in DA neurons of VTA slices, and these effects are blocked by rimonabant. These data led Lupica and Riegel (110, 123) to propose a model of endocannabinoid-dopamine system interaction wherein enhanced activation of VTA DA neurons promotes release of endocannabinoids, which then activate presynaptic CB1 receptors on GABA terminals, resulting in DSI (illustrated in Figure 1). In line with this model, rimonabant administration attenuates the typical enhancement of DA transient concentrations in the NAcc seen following cocaine administration (64).
The Role of Endocannabinoids in Cue-Motivated Behaviors
As discussed above, burst firing of DAergic neurons plays an integral role in stimulus-response learning (39, 55, 58, 59). Therefore, given the well-documented ability of endocannabinoids to modulate phasic DA signaling, it stands to reason that endocannabinoids are also involved in modulating incentive motivation. Much research utilizing cue-induced reinstatement of operant behaviors illustrates the involvement of the endocannabinoid system in various aspects of cue-induced responding. In this model, an established operant behavior is extinguished and then reinstated following the presentation of stimuli previously paired with reinforcer availability. Thus incentive cues work to reinvigorate reward-seeking in the absence of reward. Utilization of this model shows that while systemic administration of rimonabant does not affect operant responding for palatable stimuli (126), it attenuates cue-induced reinstatement of food-seeking behavior (127–129). Given that rimonabant also blocks the development of conditioned place preference for food (130), these data suggest that endocannabinoid signaling at CB1 receptors modulates the incentive value of food-associated stimuli. Indeed, the CB1 receptor antagonist AM251 attenuates cue-maintained responding for food under a second-order schedule of reinforcement (131). These effects do not seem to be reinforcer specific as endocannabinoid signaling is critical for cue-induced reinstatement of drug-seeking behavior. Systemic rimonabant attenuates cue-induced reinstatement of cocaine (126), heroin (132), methamphetamine (133), alcohol (134), nicotine (127), and THC (135). Further, the effects of CB1 antagonism on cue-induced reinstatement of reward seeking may be dependent on DAergic mechanisms, as DA antagonism also blocks cue-induced reinstatement of reward seeking behavior (136, 137).
In recent research from our laboratory, Oleson et al. (138) employed an intracranial self-stimulation (ICSS) task along with FSCV to investigate endocannabinoid-mediated disinhibition of cue-evoked DA. In this procedure, a light and tone cue signaled reinforcer availability and each operant lever press resulted in the delivery of electrical BSR to the VTA. This work replicated previous findings showing that over the course of operant training as task performance improves, NAcc DA transients time lock to cues that predicted reward (in this case BSR) availability (139). However, either intravenous (i.v.) or intra-VTA delivery of rimonabant dose-dependently increased response latency while simultaneously disrupting cue-evoked DA transients within the ventral striatum, supporting a role for specifically midbrain DAergic activity in the encoding of reward-related cues. Further, similar results were found in rats responding for food reward, suggesting that the involvement of endocannabinoids in cued responding is not reinforcer specific. Next, URB597, a FAAH inhibitor, and JZL184, an inhibitor of MAGL, were utilized to increase levels of either AEA or 2-AG, respectively. While pretreatment with URB597 (i.v.) had no effect on cued ICSS responding, both i.v. and intra-VTA administration of JZL184 decreased response latency and enhanced cue-evoked accumbal DA transmission. Further, the effects of JZL184 on ICSS responding and DAergic transmission were blocked by pretreatment with a sub-threshold dose of rimonabant, suggesting efficacy through a CB1-dependent mechanism. Taken together, these data indicate that endocannabinoids, specifically 2-AG, modulate DAergic encoding of environmental cues to control reinforcement-directed behaviors.
Endocannabinoids and Time as a Discriminative Cue
While the perception and processing of external environmental stimuli directs reward-seeking behaviors, organisms also rely on interoceptive cues (e.g., hunger or thirst) in deciding how to interact with their environment. One such internal signal, which has recently received a great deal of attention, is time estimation. Indeed, organisms rely on internal “biological clocks” to coordinate behaviors from the microsecond processing of fluid movements to the daily rhythms of the sleep–wake cycle. Of particular relevance to reward-motivated behaviors is the timing of intervals in the seconds-to-minutes range involved in a number of fundamental behaviors such as reward seeking and decision making (140). Like the processing of external reward-predictive stimuli, internal processing of temporal information is mediated by DAergic systems. Disorders that result in disruption of DA function, such as Parkinson’s disease (141, 142) or Huntington’s disease (140), or the administration of DA antagonists (e.g., haloperidol) slows the internal clock resulting in the perception that time is passing much faster (in comparison to internal temporal representations) and consequent overestimation of interval time (143–145). Conversely, disorders associated with augmented DA levels [e.g., schizophrenia (146)], or the administration of drugs that agonize DAergic signaling (e.g., cocaine and amphetamine) speeds up the internal clock leading to the perception that time is passing more slowly and resulting in underestimation of interval time (147–149). Similarly, cannabis users report a slowing of the subjective experience of time (150, 151), presumably related to the ability of cannabinoids to enhance DA transmission. These findings have been replicated in animals with both THC and WIN administration decreasing time sensitivity in both temporal discrimination (152) and estimation (153) tasks. Importantly, Han and Robinson (153) found that administration of rimonabant disrupts interval estimation and delays operant responding, suggesting a role for endocannabinoids in interval timing.
Oleson et al. (154) utilized a fixed-interval (FI) schedule of food reinforcement to uncover the role of endocannabinoid modulation of phasic DA signaling in interval timing. In FI schedules, operant behaviors are reinforced on the basis of time (155). These schedules are known to produce a behavioral pattern known as an FI “scallop,” wherein rates of responding accelerate over the course of the interval until reaching a peak response rate just before reward delivery (155). In this investigation, Oleson et al. (154) showed that phasic DA release in the mouse NAcc is inversely related to interval time; i.e., the start of the interval is characterized by a high frequency of phasic DA release events (resulting in enhanced DA concentration) which then gradually decrease in frequency as the interval continues, finally reaching a DA concentration minimum at interval terminus. These data thus support a role for DA in encoding of the principal cue predicting reinforcer availability during FI schedules – time. The endocannabinoid system was subsequently shown to modulate these effects as systemic administration of WIN dose-dependently increased the pattern of DA release and accelerated the temporal pattern of responding. These results were mimicked by intraperitoneal (i.p.) administration of JZL184, but not by i.p. URB597. Further, the effects of JZL184 were CB1-dependent as pretreatment with AM251 blocked 2-AG-induced increases in DA concentration as well as elevations in response rate and thus normalized behavior to the typical FI scallop response pattern. Although both DAergic and endocannabinoid systems have been independently implicated in the temporal control of behavior (153, 156, 157), these data represent the first report showing how phasic striatal DA may work to signal interval duration and the crucial role for endocannabinoid signaling in the encoding of this interoceptive time cue.
Oleson et al. (154) posit that the high DA concentrations seen at the start of the interval and the low DA concentrations observed at interval terminus, promote reinforcement-driven motivation resulting in the initiation of regulated operant responding and increased lever pressing, respectively. Conversely, moderate levels of DA, characteristic of the middle of the interval, promote engagement in adjunctive behaviors – behaviors that do not result in reinforcement delivery, measured as inactive lever presses. Their data align well with current theories on basal ganglia function (158). These theories suggest that high concentration DA surges arriving in the NAcc activate D1 receptors on medium spiny neurons (MSNs) comprising the direct pathway and promote action sequences (e.g., lever pressing). Baseline moderate concentration DA signals, however, are believed to activate the indirect pathway through interaction with D2 receptors on MSNs and work to inhibit primary reward-directed action sequences in favor of alternative, perhaps exploratory, adjunctive behaviors (24, 159). Thus, fluctuations in NAcc DA concentration direct reward-related behavior, likely through the modulation of striatal afferent inputs (160, 161). Enhancement of CB1 activation through administration of WIN or JZL184 increased both NAcc DA concentration and response rate, suggesting that activation of the endocannabinoid system drives direct pathway activation and primary reward seeking. Interestingly, both enhancement of 2-AG transmission (with JZL184) and disruption of CB1 activation (with AM251) attenuated adjunctive behaviors, supporting a role for the endocannabinoid system, not only in the direction of primary-reward driven behaviors governed by environmental cues but also the directions of adjunctive behaviors when environmental cues dictate that reward is not available. However, in opposition to this view, a recent report by Cui et al. (162) suggests a departure from the classical interpretation of basal ganglia function and supports a role for MSNs of both the direct and indirect pathway in action initiation. In this investigation, Cui et al. (162) utilized Cre-dependent viral expression of genetically encoded calcium indicators in transgenic mice along with time-correlated single photon counting to individually quantify D1-expressing and D2-expressing striatal MSN activation during an operant lever pressing task for food. They found that MSNs of both pathways were transiently activated just prior to the initiation of contralateral movements and quiescent during periods when the animal was not moving, suggesting that activation of both the direct and the indirect pathway promote behavioral output. Therefore, it may be that over a fixed time interval, fluctuation in extracellular DA preferentially excites individual cells within both pathways, which work together to coordinate either primary reward seeking or adjunctive behaviors during FI schedules.
The Striatal Beat Frequency Model and Endocannabinoid Modulation of Timing
The question remains, however, how does the DAergic system signal the occurrence of a distinct temporal window in order to represent an interoceptive time cue? One convincing model on the neurobiology of interval timing is the striatal beat frequency (SBF) model (148, 163). SBF postulates that cortico-striatal circuits encode interval durations of seconds to minutes, and these circuits are coordinated by midbrain DAergic input. Within the frontal cortex, neurons oscillating in the alpha range (8–13 Hz) act as an internal clock (164). At interval onset, the presentation of a distinct conditioned stimulus results in phasic firing of midbrain DA neurons and these transient surges of extracellular DA synchronize oscillating neurons in the frontal cortex, likely through inhibition of desynchronized cells (163). However, briefly after synchronization, cortical neurons begin to fall out of phase with one another, returning to their inherent individual periodicities. Thus, distinct interval durations are marked by unique patterns of neuronal ensembles firing in-phase. These cortical oscillators synapse onto MSNs of the striatum that function as “coincidence detectors” – connecting distinct patterns of oscillator activity with the occurrence of external stimuli (e.g., reward delivery). Striatal MSNs are uniquely situated to detect coincident neuronal activity with each MSN receiving input from 10,000 to 30,000 different thalamic and cortical neurons (165). Further, striatal MSNs exhibit both highly polarized “down” states (−90 mV) and less polarized “up” states (approximately −60 mV) (166) with transitions from down to up states requiring either VTA DA input (26, 167) or excitatory glutamatergic input from other cortical or subcortical structures (166, 168). High levels of DAergic input to the striatum at interval onset are hypothesized to “clear out” irrelevant information within the coincidence detector by hyperpolarizing striatal cells into their down state (148). Conversely, phasic surges of extracellular DA within the striatum at reward delivery bring striatal cells into their up state and promote LTP at active cortico-striatal synapses (27, 163, 169). Therefore, the pattern of cortical inputs spiking at the time of reinforcement, which represents a unique population code for the time elapsed since interval initiation, will undergo Hebbian strengthening. LTP at these striatal synapses allows the organism to learn specific interval durations and initiate reward seeking when a matching pattern of cortical efferent activity is encountered.
While the SBF model represents a neurobiologically plausible framework within which to examine interval timing, there are inconsistencies in this model. For example: how does phasic DA at cue onset hyperpolarize striatal cells given that D1 stimulation is excitatory (i.e., Gs-coupled)? One possibility is through collateral inhibition wherein D1-mediated excitation in specific MSNs leads to a net inhibition of striatal cells through a lateral inhibitory feedback network (170–172); however, this has yet to be explored within interval timing tasks. Further, there has been no direct voltammetric assessment of striatal DA levels and consequent induction of striatal LTP mechanisms.
The voltammetric data presented by Oleson et al. (154) lend support to the SBF model. In their investigation, extracellular DA is high at the start of the interval, providing a mechanism through which NAcc cells may be hyperpolarized (perhaps through collateral-driven lateral inhibition) and the coincidence detector reset. As the interval progresses DA levels gradually decrease, likely releasing striatal cells from collateral feedback inhibition and allowing for DA transient-induced cortico-striatal LTP. It remains unclear, however, how DA levels gradually decrease as the interval continues. This may occur through striatal cell ramping activity. Indeed, a subset of NAcc neurons display a ramping activity pattern during interval estimation wherein firing rates linearly increase from CS presentation and peak at the time of reward expectation (173, 174). The accumbens sends direct inhibitory input to the VTA (175), thus ramping activity may result in progressive DA decline from CS presentation (176). Data from Oleson et al. (154), however, do not determine if NAcc DA activity is integral for interval estimation, but, rather, provide evidence for a correlation between accumbal phasic DA activity and interval estimation. Future investigations are required to determine if phasic NAcc DA release is required for interval estimation.
The SBF model suggests a specific role for the dorsal striatum in temporal processing, citing the ability of dorsal, but not ventral, striatal lesions to impair temporal control (177, 178). However, lesions of the NAcc core disrupt timing of Pavlovian responses, as evidenced by decreased approach to a food receptacle at CS+ presentation (179). Further, while Galtress and Kirkpatrick (180) found that accumbal lesions do not disrupt the tracking of temporal windows in a peak-interval (PI) timing procedure, animals do exhibit a decreased ability to modify behavior when an expected reward is not delivered at interval terminus. PI is an extension of an FI schedule with the addition of non-reinforced probe trials which result in Gaussian response curves characterized by increased operant responding up to the point of expected reward followed by a gradual decrease in responding. Thus, the NAcc core likely functions to integrate temporal cues specifically with reward value and availability. Given that responding during an FI combines both temporal estimation and expectations regarding reinforcer value, the role of the NAcc core in operant responding during FI schedules of reinforcement merits more examination. Finally, it should be noted that while recordings of phasic NAcc DA activity support the SBF model, no voltammetric recordings during FI responding have been taken in the frontal cortex due to the high levels of cortical noradrenergic innervation. However, recent optogenetic investigations show that disruption of D1 receptor transmission within the prefrontal cortex impairs performance on an FI task (157), supporting an integral role for phasic DA within the frontal cortex in interval timing.
The ability of cannabinoids, as well as other drugs that augment DAergic signaling, to enhance internal clock speed has been well documented (181–184), however, the neurobiological mechanisms underlying this effect remains unclear. Within the framework of the SBF model, enhancement of phasic DA following cannabinoid administration may affect time perception through agonizing DA transmission within the striatum. Indeed, selective manipulation of DA levels within the ventral striatum disrupts performance in timing-dependent operant tasks (185–187). Further, direct administration of DA into the NAcc enhances internal clock speed (188). Increased phasic DAergic activity in the NAcc core, as seen following WIN administration (154), may result in induction of LTP at cortico-striatal synapses that are active much earlier in the interval than those active at reinforcer delivery. This abnormal “stamping in” of premature interval estimations would thus promote reward seeking prior to interval terminus, likely through activation of the basal ganglia’s direct pathway [but see Ref. (162)] (Figure 2). However, future investigations are necessary to determine if cortico-striatal LTP adheres to specific patterns during interval estimation tasks, and examine how these patterns are changed following DAergic agonist administration.
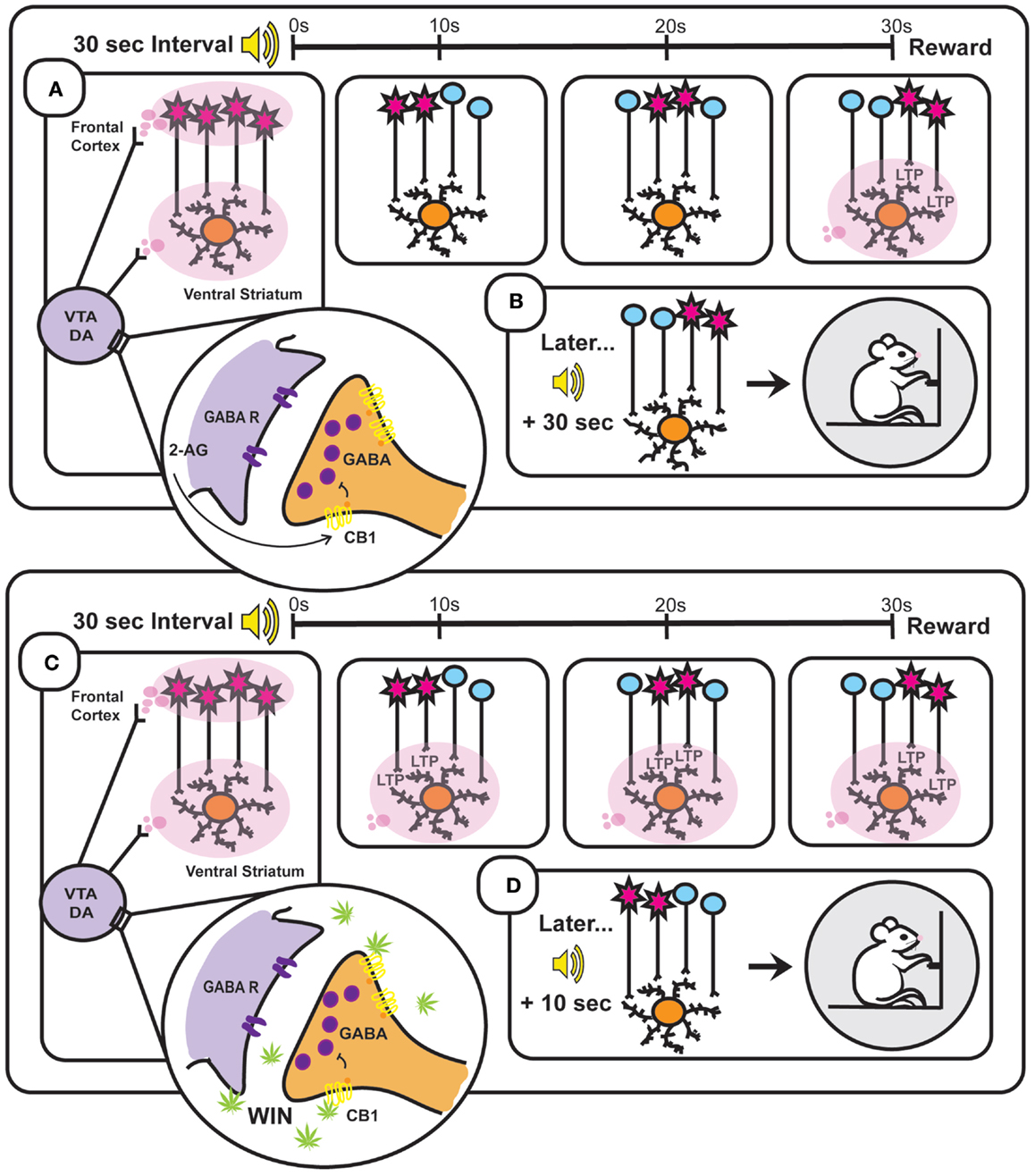
Figure 2. Based on the striatal beat frequency model of interval timing – a schematic representation of the neurobiology underlying interval timing during a typical 30 s fixed interval (A,B) and during a 30 s fixed interval following administration of the synthetic cannabinoid WIN 55,212-2 (WIN) (C,D). (A) At interval onset, phasic dopamine (DA) transmission resets the internal clock through synchronization of frontal cortical oscillators [depicted in (A) as simultaneously firing cells and illustrated as red cell bodies] and clears out the coincidence detector (ventral striatum). These phasic signals arise through burst firing of ventral tegmental area (VTA) DA cells, which is facilitated by endocannabinoid (2-AG)-mediated suppression of GABA release onto VTA DA neurons. As the 30 s interval progresses (illustrated in the three panels to the right depicting time points at 10, 20, and 30 s), the once synchronized cortical oscillators fall out of phase with one another at a reliable rate. Reward delivery at interval terminus results in phasic DA transmission within the ventral striatum that enhances LTP at active cortico-striatal synapses. (B) Later when the same (in this case auditory) stimulus signals interval onset, cortical oscillators exhibit characteristic periodicities and when the previously strengthened synaptic pattern active at interval terminus is encountered again (30 s after interval onset) its activation will promote reward seeking. (C) WIN administration results in phasic activation of VTA DA neurons through binding to CB1 receptors on VTA GABAergic neurons and thereby disinhibiting VTA DA transmission. Drug-induced aberrant DAergic activation throughout the interval induces LTP at cortico-striatal synapses active prior to reward delivery (illustrated in the three panels to the right depicting time points at 10, 20, and 30 s). (D) This Hebbian strengthening of synaptic activity characteristic of earlier time points within the interval promotes premature reward seeking following subsequent cue presentation.
A greater understanding of the role of phasic DA in interval timing is integral to the study of drug addiction. Individuals that are more sensitive to the time-altering effects of drug administration are also more sensitive to the experience of stimulant-induced euphoria, suggesting that similar brain pathways maintain internal clock speed and drug reward (189). Indeed, drugs of abuse increase extracellular DA within the ventral striatum and have the ability to hasten internal clock speed. Therefore, augmentation of internal clock representation may underlie a path through which drugs alter stimulus-reward associations and promote inappropriate reward seeking, such as that seen in delay discounting paradigms. Delay discounting is a maladaptive decision-making strategy characteristic to addiction wherein individuals show a preference for smaller/immediate over larger/delayed rewards. This temporal shift in reward seeking is generally considered an indication of impulsivity, but one could also imagine delay discounting to result from augmentation of internal clock representations. Certainly, a number of human studies indicate a positive correlation between substance abuse, including marijuana abuse, and measures of delay discounting (190–196). Investigations into the neurobiology of this phenomenon implicate alterations in striatal DA activity (197, 198), suggesting a link between drug-induced enhancement of phasic DA signaling and temporally biased reward seeking. Ostlund et al. (199) showed that repeated cocaine exposure enhances phasic DA transmission and increases cue-evoked food seeking. Thus, enhanced accumbal DA in response to reward-predictive cues following drug use, may, through interaction with the proposed SBF timing neurocircuitry, accelerate the internal clock and promote the choice of immediate over delayed rewards. Importantly, Hernandez et al. (198) showed that systemic pretreatment with rimonabant attenuates cocaine-induced delay discounting. These findings support a role for endocannabinoid-modulated enhancement of phasic DA transmission following drug use and subsequent temporal shifts in reward seeking resulting from an enhancement of internal clock speed. However, it remains unclear if decision making during delay discounting paradigms occurs within the same timescale as that of interval estimation, or relies on strategies adopted prior to task onset.
Conclusion
A wide body of evidence supports a role for endocannabinoid modulation of phasic midbrain DAergic activity. Phasic burst activation of DA neurons can be measured as transient fluctuations of extracellular DA in terminal regions of the mesolimbic system (e.g., the NAcc) using FSCV. In congruence with reward prediction error, following repeated stimulus-reward pairings, reward-predictive cues result in burst firing of mesolimbic DA neurons and transient increases in NAcc DA levels. This transient burst of striatal DA likely promotes maintenance of reward-seeking behaviors through activation of the basal ganglia pathways. In addition to tracking of external reward-related cues, phasic changes in striatal DA encode interoceptive cues, such as interval time that allow for the coordination of goal-directed responding. In support of this view, drugs that enhance striatal DA levels also increase individuals’ internal clock thereby resulting in the perception that time is passing more slowly. Further, animal studies show that administration of the CB1/CB2 agonist WIN or the 2-AG degradation inhibitor JZL184 enhances NAcc core DA levels and promotes premature reward seeking in a FI task. The precise neurobiological mechanism(s) through which these effects occur, however, remains unknown. The SBF model of interval timing provides a unique framework with which to examine the role of phasic DA in interval timing. SBF posits that striatal DA fluctuations mediate interval timing through selectively promoting LTP at cortico-striatal synapses active at specific interval durations. Thus, DAergic disruption of interval timing may occur through aberrant induction of LTP at cortico-striatal synapses representative of premature interval lengths. Given the ability of drugs of abuse to potently augment NAcc DA levels, it is not surprising that drug administration also disrupts interval estimation. Alterations in interval timing may also underlie addiction-related behaviors such as delay discounting, which is enhanced by endocannabinoid-mediated increases in striatal DA. A conserved mechanism in interval timing and impulsive choice suggests that following drug use, enhanced delayed discounting is consequential to augmentation of one’s internal clock, which results in temporal-biasing of reward-seeking at the presentation of reward-related cues.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Dr. Erik Oleson for the preparation of Figure 1.
References
1. Bindra D. Neuropsychological interpretation of the effects of drive and incentive motivation on general activity and instrumental behavior. Psychol Rev (1968) 75:1–22. doi: 10.1037/h0025306
3. Toates F. The interaction of cognitive and stimulus-response processes in the control of behaviour. Neurosci Biobehav Rev (1998) 22(1):59–83. doi:10.1016/S0149-7634(97)00022-5
4. Rodin J. Current status of the internal-external hypothesis for obesity: what went wrong? Am Psychol (1981) 36(4):361–72. doi:10.1037/0003-066X.36.4.361
6. Shaham Y, Shalev U, Lu L, De Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (2003) 168(1–2):3–20. doi:10.1007/s00213-002-1224-x
7. Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Rev (1998) 28(3):309–69. doi:10.1016/S0165-0173(98)00019-8
8. Swanson LW. The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull (1982) 9(1–6):321–53. doi:10.1016/0361-9230(82)90145-9
9. Berridge KC. Liking’ and ‘wanting’ food rewards: brain substrates and roles in eating disorders. Physiol Behav (2009) 97(5):537–50. doi:10.1016/j.physbeh.2009.02.044
10. Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Rev (2007) 56(1):27–78. doi:10.1016/j.brainresrev.2007.05.004
11. Taber KH, Black DN, Porrino LJ, Hurley RA. Neuroanatomy of dopamine: reward and addiction. J Neuropsychiatry Clin Neurosci (2012) 24(1):1–4. doi:10.1176/appi.neuropsych.24.1.1
12. Wise RA, Rompre PP. Brain dopamine and reward. Annu Rev Psychol. (1989) 40:191–225. doi:10.1146/annurev.ps.40.020189.001203
13. Di Chiara G, Bassareo V, Fenu S, De Luca MA, Spina L, Cadoni C, et al. Dopamine and drug addiction: the nucleus accumbens shell connection. Neuropharmacology (2004) 47(Suppl 1):227–41. doi:10.1016/j.neuropharm.2004.06.032
14. Hernandez L, Hoebel BG. Food reward and cocaine increase extracellular dopamine in the nucleus accumbens as measured by microdialysis. Life Sci (1988) 42(18):1705–12. doi:10.1016/0024-3205(88)90036-7
15. Hernandez L, Hoebel BG. Feeding and hypothalamic stimulation increase dopamine turnover in the accumbens. Physiol Behav (1988) 44(4–5):599–606. doi:10.1016/0031-9384(88)90324-1
16. Hoebel BG, Hernandez L, Schwartz DH, Mark GP, Hunter GA. Microdialysis studies of brain norepinephrine, serotonin, and dopamine release during ingestive behavior. Theoretical and clinical implications. Ann N Y Acad Sci. (1989) 575:171–91. doi:10.1111/j.1749-6632.1989.tb53242.x
17. Yoshida M, Yokoo H, Mizoguchi K, Kawahara H, Tsuda A, Nishikawa T, et al. Eating and drinking cause increased dopamine release in the nucleus accumbens and ventral tegmental area in the rat: measurement by in vivo microdialysis. Neurosci Lett (1992) 139(1):73–6. doi:10.1016/0304-3940(92)90861-Z
18. Ettenberg A, Camp CH. A partial reinforcement extinction effect in water-reinforced rats intermittently treated with haloperidol. Pharmacol Biochem Behav (1986) 25(6):1231–5. doi:10.1016/0091-3057(86)90117-6
19. Ettenberg A, Camp CH. Haloperidol induces a partial reinforcement extinction effect in rats: implications for a dopamine involvement in food reward. Pharmacol Biochem Behav (1986) 25(4):813–21. doi:10.1016/0091-3057(86)90117-6
20. Salamone JD, Correa M, Mingote S, Weber SM. Nucleus accumbens dopamine and the regulation of effort in food-seeking behavior: implications for studies of natural motivation, psychiatry, and drug abuse. J Pharmacol Exp Ther (2003) 305(1):1–8. doi:10.1124/jpet.102.035063
21. Zis AP, Fibiger HC. Neuroleptic-induced deficits in food and water regulation: similarities to the lateral hypothalamic syndrome. Psychopharmacology (1975) 43(1):63–8. doi:10.1007/BF00437616
22. Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci (1984) 4(11):2877–90.
23. Cooper DC. The significance of action potential bursting in the brain reward circuit. Neurochem Int (2002) 41(5):333–40. doi:10.1016/S0197-0186(02)00068-2
24. Dreyer JK, Herrik KF, Berg RW, Hounsgaard JD. Influence of phasic and tonic dopamine release on receptor activation. J Neurosci (2010) 30(42):14273–83. doi:10.1523/JNEUROSCI.1894-10.2010
25. Grace AA. Regulation of spontaneous activity and oscillatory spike firing in rat midbrain dopamine neurons recorded in vitro. Synapse (1991) 7(3):221–34. doi:10.1002/syn.890070307
26. Goto Y, Grace A. Dopaminergic modulation of limbic and cortical drive of nucleus accumbens in goal-directed behavior. Nat Neurosci (2005) 8(6):805–12. doi:10.1038/nn1471
27. Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science (2008) 321(5890):848–51. doi:10.1126/science.1160575
28. Grace A, Floresco SB, Goto Y, Lodge DJ. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci. (2007) 30(5):220–7. doi:10.1016/j.tins.2007.03.003
29. Romo R, Schultz W. Dopamine neurons of the monkey midbrain: contingencies of responses to active touch during self-initiated arm movements. J Neurophysiol (1990) 63(3):592–606.
31. Ljungberg T, Apicella P, Schultz W. Responses of monkey dopamine neurons during learning of behavioral reactions. J Neurophysiol (1992) 67(1):145–63.
32. Freeman AS, Meltzer LT, Bunney BS. Firing properties of substantia nigra dopaminergic neurons in freely moving rats. Life Sci (1985) 36(20):1983–94. doi:10.1016/0024-3205(85)90448-5
33. Freeman AS, Bunney BS. Activity of A9 and A10 dopaminergic neurons in unrestrained rats: further characterization and effects of apomorphine and cholecystokinin. Brain Res (1987) 405(1):46–55.
34. Horvitz JC. Mesolimbocortical and nigrostriatal dopamine responses to salient non-reward events. Neuroscience (2000) 96(4):651–6. doi:10.1016/S0306-4522(00)00019-1
35. Rebec GV, Christensen JR, Guerra C, Bardo MT. Regional and temporal differences in real-time dopamine efflux in the nucleus accumbens during free-choice novelty. Brain Res (1997) 776(1–2):61–7.
36. Wightman RM, Robinson DL. Transient changes in mesolimbic dopamine and their association with ‘reward’. J Neurochem (2002) 82(4):721–35. doi:10.1046/j.1471-4159.2002.01005.x
37. Tsai H, Zhang F, Adamantidis A, Stuber GD, Bonci A, de Lecea L, et al. Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science (2009) 324(5930):1080–4. doi:10.1126/science.1168878
38. Nicola SM. The flexible approach hypothesis: unification of effort and cue-responding hypotheses for the role of nucleus accumbens dopamine in the activation of reward-seeking behavior. J Neurosci (2010) 30(49):16585–600. doi:10.1523/JNEUROSCI.3958-10.2010
39. Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, et al. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc Natl Acad Sci USA. (2009) 106(18):7281–8. doi:10.1073/pnas.0813415106
40. Garris PA, Kilpatrick M, Bunin MA, Michael D, Walker QD, Wightman RM. Dissociation of dopamine release in the nucleus accumbens from intracranial self-stimulation. Nature (1999) 398(6722):67–9. doi:10.1038/18019
41. Bito L, Davson H, Levin E, Murray M, Snider N. The concentrations of free amino acids and other electrolytes in cerebrospinal fluid, in vivo dialysate of brain, and blood plasma of the dog. J Neurochem (1966) 13(11):1057–67. doi:10.1111/j.1471-4159.1966.tb04265.x
42. Westerink BH. Brain microdialysis and its application for the study of animal behaviour. Behav Brain Res (1995) 70(2):103–24. doi:10.1016/0166-4328(95)80001-8
43. Radhakishun FS, van Ree JM, Westerink BH. Scheduled eating increases dopamine release in the nucleus accumbens of food-deprived rats as assessed with on-line brain dialysis. Neurosci Lett (1988) 85(3):351–6. doi:10.1016/0304-3940(88)90591-5
44. Church WH, Justice JB, Byrd LD. Extracellular dopamine in rat striatum following uptake inhibition by cocaine, nomifensine and benztropine. Eur J Pharmacol (1987) 139(3):345–8. doi:10.1016/0014-2999(87)90592-9
45. Hurd YL, Weiss F, Koob GF, Ungerstedt U. Cocaine reinforcement and extracellular dopamine overflow in rat nucleus accumbens: an in vivo microdialysis study. Brain Res (1989) 498(1):199–203.
46. Hurd YL, Weiss F, Koob G, Ungerstedt U. The influence of cocaine self-administration on in vivo dopamine and acetylcholine neurotransmission in rat caudate-putamen. Neurosci Lett (1990) 109(1–2):227–33. doi:10.1016/0304-3940(90)90568-T
47. Pettit HO, Justice JB. Dopamine in the nucleus accumbens during cocaine self-administration as studied by in vivo microdialysis. Pharmacol Biochem Behav (1989) 34(4):899–904. doi:10.1016/0091-3057(89)90291-8
48. Pettit HO, Justice JB. Effect of dose on cocaine self-administration behavior and DA levels in the nucleus accumbens. Brain Res (1991) 539(1):94–102.
49. Weiss F, Paulus MP, Lorang MT, Koob GF. Increases in extracellular dopamine in the nucleus accumbens by cocaine are inversely related to basal levels: effects of acute and repeated administration. J Neurosci (1992) 12(11):4372–80.
50. Wise RA, Leone P, Rivest R, Leeb K. Elevations of nucleus accumbens dopamine and DOPAC levels during intravenous heroin self-administration. Synapse (1995) 21(2):140–8. doi:10.1002/syn.890210207
51. Robinson DL, Venton BJ, Heien ML, Wightman RM. Detecting subsecond dopamine release with fast-scan cyclic voltammetry in vivo. Clin Chem (2003) 49(10):1763–73. doi:10.1373/49.10.1763
52. Millar J, Stamford JA, Kruk ZL, Wightman RM. Electrochemical, pharmacological and electrophysiological evidence of rapid dopamine release and removal in the rat caudate nucleus following electrical stimulation of the median forebrain bundle. Eur J Neurosci (1985) 109(3):341–8.
53. Budygin EA, Phillips PE, Robinson DL, Kennedy AP, Gainetdinov RR, Wightman RM. Effect of acute ethanol on striatal dopamine neurotransmission in ambulatory rats. J Pharmacol Exp Ther (2001) 297(1):27–34.
54. Garris PA, Christensen JR, Rebec GV, Wightman RM. Real-time measurement of electrically evoked extracellular dopamine in the striatum of freely moving rats. J Neurochem. (1997) 68(1):152–61. doi:10.1046/j.1471-4159.1997.68010152.x
55. Brown HD, McCutcheon JE, Cone J, Ragozzino ME, Roitman MF. Primary food reward and reward-predictive stimuli evoke different patterns of phasic dopamine signaling throughout the striatum. Eur J Neurosci (2011) 34(12):1997–2006. doi:10.1111/j.1460-9568.2011.07914.x
56. Flagel SB, Clark J, Robinson TE, Mayo L, Czuj A, Willuhn I. A selective role for dopamine in stimulus-reward learning. Nature (2011) 469(7328):53–7. doi:10.1038/nature09588
57. Roitman MF, Stuber GD, Phillips PE, Wightman RM, Carelli RM. Dopamine operates as a subsecond modulator of food seeking. J Neurosci (2004) 24(6):1265–71. doi:10.1523/JNEUROSCI.3823-03.2004
58. Roitman MF, Wheeler RA, Wightman RM, Carelli RM. Real-time chemical responses in the nucleus accumbens differentiate rewarding and aversive stimuli. Nat Neurosci (2008) 11(12):1376–7. doi:10.1038/nn.2219
59. Sunsay C, Rebec GV. Real-time dopamine efflux in the nucleus accumbens core during Pavlovian conditioning. Behav Neurosci (2008) 122(2):358–67. doi:10.1037/0735-7044.122.2.358
60. Hyland BI, Reynolds JN, Hay J, Perk CG, Miller R. Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience (2002) 114(2):475–92. doi:10.1016/S0306-4522(02)00267-1
61. Nishino H, Ono T, Muramoto K, Fukuda M, Sasaki K. Neuronal activity in the ventral tegmental area (VTA) during motivated bar press feeding in the monkey. Brain Res (1987) 413(2):302–13.
62. Nakazato T. Striatal dopamine release in the rat during a cued lever-press task for food reward and the development of changes over time measured using high-speed voltammetry. Exp Brain Res (2005) 166(1):137–46. doi:10.1007/s00221-005-2345-3
63. Phillips PE, Stuber GD, Heien ML, Wightman RM, Carelli RM. Subsecond dopamine release promotes cocaine seeking. Nature (2003) 422(6932):614–8. doi:10.1038/nature01476
64. Cheer JF, Wassum KM, Sombers LA, Heien ML, Ariansen JL, Aragona BJ, et al. Phasic dopamine release evoked by abused substances requires cannabinoid receptor activation. J Neurosci (2007) 27(4):791–5. doi:10.1523/JNEUROSCI.4152-06.2007
65. Oleson EB, Gentry RN, Chioma VC, Cheer JF. Subsecond dopamine release in the nucleus accumbens predicts conditioned punishment and its successful avoidance. J Neurosci (2012) 32(42):14804–8. doi:10.1523/JNEUROSCI.3087-12.2012
66. McCutcheon JE, Ebner SR, Loriaux AL, Roitman MF. Encoding of aversion by dopamine and the nucleus accumbens. Front Neurosci. (2012) 6:137. doi:10.3389/fnins.2012.00137
67. Schultz W. Behavioral dopamine signals. Trends Neurosci. (2007) 30(5):203–10. doi:10.1016/j.tins.2007.03.007
68. Forster GL, Blaha CD. Pedunculopontine tegmental stimulation evokes striatal dopamine efflux by activation of acetylcholine and glutamate receptors in the midbrain and pons of the rat. Eur J Neurosci (2003) 17(4):751–62. doi:10.1046/j.1460-9568.2003.02511.x
69. Omelchenko N, Sesack SR. Laterodorsal tegmental projections to identified cell populations in the rat ventral tegmental area. J Comp Neurol (2005) 483(2):217–35. doi:10.1002/cne.20417
70. Phillipson OT. Afferent projections to the ventral tegmental area of Tsai and interfascicular nucleus: a horseradish peroxidase study in the rat. J Comp Neurol (1979) 187(1):117–43. doi:10.1002/cne.901870106
71. Grace AA, Onn SP. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J Neurosci (1989) 9(10):3463–81.
72. Sanghera MK, Trulson ME, German DC. Electrophysiological properties of mouse dopamine neurons: in vivo and in vitro studies. Neuroscience (1984) 12(3):793–801. doi:10.1016/0306-4522(84)90171-4
73. Charlety PJ, Grenhoff J, Chergui K, De la Chapelle B, Buda M, Svensson TH, et al. Burst firing of mesencephalic dopamine neurons is inhibited by somatodendritic application of kynurenate. Acta Physiol Scand (1991) 142(1):105–12. doi:10.1111/j.1748-1716.1991.tb09134.x
74. Overton PG, Clark D. Burst firing in midbrain dopaminergic neurons. Brain Res Rev (1997) 25(3):312–34. doi:10.1016/S0165-0173(97)00039-8
75. Engberg G, Kling-Petersen T, Nissbrandt H. GABAB-receptor activation alters the firing pattern of dopamine neurons in the rat substantia nigra. Synapse (1993) 15(3):229–38. doi:10.1002/syn.890150308
76. Ameri A. The effects of cannabinoids on the brain. Prog Neurobiol. (1999) 58(4):315–48. doi:10.1016/S0301-0082(98)00087-2
77. Fattore L, Melis M, Fadda P, Pistis M, Fratta W. The endocannabinoid system and nondrug rewarding behaviours. Exp Neurol (2010) 224(1):23–36. doi:10.1016/j.expneurol.2010.03.020
78. Maldonado R, Valverde O, Berrendero F. Involvement of the endocannabinoid system in drug addiction. Trends Neurosci. (2006) 29(4):225–32. doi:10.1016/j.tins.2006.01.008
79. Solinas M, Goldberg SR, Piomelli D. The endocannabinoid system in brain reward processes. Br J Pharmacol. (2008) 154(2):369–83. doi:10.1038/bjp.2008.130
80. Devane WA, Dysarz FA, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol (1988) 34(5):605–13.
81. Matsuda LA, Bonner TI, Lolait SJ. Cannabinoid receptors: which cells, where, how, and why? NIDA Res Monogr (1992) 126:48–56.
82. Munro S, Thomas KL, Abu Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature (1993) 365(6441):61–5. doi:10.1038/365061a0
83. Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci (1991) 11(2):563–83.
84. Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. International union of pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev (2002) 54(2):161–202. doi:10.1124/pr.54.2.161
85. Jansen EM, Haycock DA, Ward SJ, Seybold VS. Distribution of cannabinoid receptors in rat brain determined with aminoalkylindoles. Brain Res (1992) 575(1):93–102.
86. Nuñez E, Benito C, Pazos MR, Barbachano A, Fajardo O, González S, et al. Cannabinoid CB2 receptors are expressed by perivascular microglial cells in the human brain: an immunohistochemical study. Synapse (2004) 53(4):208–13. doi:10.1002/syn.20050
87. Cabral GA, Raborn ES, Griffin L, Dennis J, Marciano-Cabral F. CB2 receptors in the brain: role in central immune function. Br J Pharmacol (2008) 153(2):240–51. doi:10.1038/sj.bjp.0707584
88. Adamczyk P, Miszkiel J, McCreary AC, Filip M, Papp M, Przegalinski E. The effects of cannabinoid CB1, CB2 and vanilloid TRPV1 receptor antagonists on cocaine addictive behavior in rats. Brain Res (2012) 1444:45–54. doi:10.1016/j.brainres.2012.01.030
89. Flake NM, Zweifel LS. Behavioral effects of pulp exposure in mice lacking cannabinoid receptor 2. J Endod (2012) 38(1):86–90. doi:10.1016/j.joen.2011.09.015
90. Onaivi ES. Neuropsychobiological evidence for the functional presence and expression of cannabinoid CB2 receptors in the brain. Neuropsychobiology (2006) 54(4):231–46. doi:10.1159/000100778
91. Xi ZX, Peng X, Li X, Song R, Zhang HY, Liu QR, et al. Brain cannabinoid CBâ‚‚ receptors modulate cocaine’s actions in mice. Nat Neurosci (2011) 14(9):1160–6. doi:10.1038/nn.2874
92. Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson NO, Leonova J, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol (2007) 152(7):1092–101. doi:10.1038/sj.bjp.0707460
93. Szallasi A, Di Marzo V. New perspectives on enigmatic vanilloid receptors. Trends Neurosci. (2000) 23(10):491–7. doi:10.1016/S0166-2236(00)01630-1
94. Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science (1992) 258(5090):1946–9. doi:10.1126/science.1470919
95. Mechoulam R, Ben Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol (1995) 50(1):83–90. doi:10.1016/0006-2952(95)00109-D
96. Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, et al. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. (1995) 215(1):89–97. doi:10.1006/bbrc.1995.2437
97. Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D. Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J Neurosci (1996) 16(12):3934–42.
98. Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci (2003) 4(11):873–84. doi:10.1038/nrn1247
99. Goparaju SK, Ueda N, Yamaguchi H, Yamamoto S. Anandamide amidohydrolase reacting with 2-arachidonoylglycerol, another cannabinoid receptor ligand. FEBS Lett (1998) 422(1):69–73. doi:10.1016/S0014-5793(97)01603-7
100. Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature (1997) 388(6644):773–8. doi:10.1038/42015
101. Haj-Dahmane S, Shen R. The wake-promoting peptide orexin-B inhibits glutamatergic transmission to dorsal raphe nucleus serotonin neurons through retrograde endocannabinoid signaling. J Neurosci (2005) 25(4):896–905. doi:10.1523/JNEUROSCI.3258-04.2005
102. Kortleven C, Bruneau LC, Trudeau L. Neurotensin inhibits glutamate-mediated synaptic inputs onto ventral tegmental area dopamine neurons through the release of the endocannabinoid 2-AG. Neuropharmacology (2012) 63(6):983–91. doi:10.1016/j.neuropharm.2012.07.037
103. Maejima T, Ohno-Shosaku T, Kano M. Endogenous cannabinoid as a retrograde messenger from depolarized postsynaptic neurons to presynaptic terminals. Neurosci Res (2001) 40(3):205–10. doi:10.1016/S0168-0102(01)00241-3
104. Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, et al. Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron (2005) 45(2):257–68. doi:10.1016/j.neuron.2005.01.004
105. Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev (2009) 89(1):309–80. doi:10.1152/physrev.00019.2008
106. Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev (2003) 83(3):1017–66.
107. Ohno-Shosaku T, Kano M. Endocannabinoid-mediated retrograde modulation of synaptic transmission. Curr Opin Neurobiol. (2014) 29C:1–8. doi:10.1016/j.conb.2014.03.017
108. Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. (2002) 68(4):247–86. doi:10.1016/S0301-0082(02)00080-1
109. Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci (2004) 24(1):53–62. doi:10.1523/JNEUROSCI.4503-03.2004
110. Riegel AC, Lupica CR. Independent presynaptic and postsynaptic mechanisms regulate endocannabinoid signaling at multiple synapses in the ventral tegmental area. J Neurosci (2004) 24(49):11070–8. doi:10.1523/JNEUROSCI.3695-04.2004
111. Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron (2001) 29(3):729–38. doi:10.1016/S0896-6273(01)00247-1
112. Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA (1988) 85(14):5274–8. doi:10.1073/pnas.85.14.5274
113. Wise RA. Brain reward circuitry: insights from unsensed incentives. Neuron (2002) 36(2):229–40. doi:10.1016/S0896-6273(02)00965-0
114. Castañeda E, Moss DE, Oddie SD, Whishaw IQ. THC does not affect striatal dopamine release: microdialysis in freely moving rats. Pharmacol Biochem Behav (1991) 40(3):587–91. doi:10.1016/0091-3057(91)90367-B
115. Gardner EL, Lowinson JH. Marijuana’s interaction with brain reward systems: update 1991. Pharmacol Biochem Behav (1991) 40(3):571–80. doi:10.1016/0091-3057(91)90365-9
116. Cheer JF, Wassum KM, Heien ML, Phillips PE, Wightman RM. Cannabinoids enhance subsecond dopamine release in the nucleus accumbens of awake rats. J Neurosci (2004) 24(18):4393–400. doi:10.1523/JNEUROSCI.0529-04.2004
117. Chen JP, Paredes W, Lowinson JH, Gardner EL. Strain-specific facilitation of dopamine efflux by delta 9-tetrahydrocannabinol in the nucleus accumbens of rat: an in vivo microdialysis study. Neurosci Lett (1991) 129(1):136–80. doi:10.1016/0304-3940(91)90739-G
118. Malone DT, Taylor DA. Modulation by fluoxetine of striatal dopamine release following Delta9-tetrahydrocannabinol: a microdialysis study in conscious rats. Br J Pharmacol. (1999) 128(1):21–6.
119. Tanda G, Pontieri FE, Di Chiara G. Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science (1997) 276(5321):2048–50. doi:10.1126/science.276.5321.2048
120. French ED, Dillon K, Wu X. Cannabinoids excite dopamine neurons in the ventral tegmentum and substantia nigra. Neuroreport (1997) 8(3):649–52. doi:10.1097/00001756-199702100-00014
121. Gessa GL, Melis M, Muntoni AL, Diana M. Cannabinoids activate mesolimbic dopamine neurons by an action on cannabinoid CB1 receptors. Eur J Pharmacol (1998) 341(1):39–44. doi:10.1016/S0014-2999(97)01442-8
122. Julian MD, Martin AB, Cuellar B, Rodriguez De Fonseca F, Navarro M, Moratalla R, et al. Neuroanatomical relationship between type 1 cannabinoid receptors and dopaminergic systems in the rat basal ganglia. Neuroscience (2003) 119(1):309–18. doi:10.1016/S0306-4522(03)00070-8
123. Lupica CR, Riegel AC. Endocannabinoid release from midbrain dopamine neurons: a potential substrate for cannabinoid receptor antagonist treatment of addiction. Neuropharmacology (2005) 48(8):1105–16. doi:10.1016/j.neuropharm.2005.03.016
124. Cheer JF, Marsden CA, Kendall DA, Mason R. Lack of response suppression follows repeated ventral tegmental cannabinoid administration: an in vitro electrophysiological study. Neuroscience (2000) 99(4):661–7. doi:10.1016/S0306-4522(00)00241-4
125. Szabo B, Siemes S, Wallmichrath I. Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Eur J Neurosci (2002) 15(12):2057–61. doi:10.1046/j.1460-9568.2002.02041.x
126. De Vries TJ, Shaham Y, Homberg JR, Crombag H, Schuurman K, Dieben J, et al. A cannabinoid mechanism in relapse to cocaine seeking. Nat Med (2001) 7(10):1151–4. doi:10.1038/nm1001-1151
127. De Vries TJ, de Vries W, Janssen MC, Schoffelmeer AN. Suppression of conditioned nicotine and sucrose seeking by the cannabinoid-1 receptor antagonist SR141716A. Behav Brain Res (2005) 161(1):164–8. doi:10.1016/j.bbr.2005.02.021
128. Hernandez G, Cheer JF. Extinction learning of rewards in the rat: is there a role for CB1 receptors? Psychopharmacology (2011) 217(2):189–97. doi:10.1007/s00213-011-2275-7
129. Ward SJ, Walker EA, Dykstra LA. Effect of cannabinoid CB1 receptor antagonist SR141716A and CB1 receptor knockout on cue-induced reinstatement of Ensure and corn-oil seeking in mice. Neuropsychopharmacology (2007) 32(12):2592–600. doi:10.1038/sj.npp.1301384
130. Chaperon F, Thiébot MH. Behavioral effects of cannabinoid agents in animals. Crit Rev Neurobiol (1999) 13(3):243–81.
131. Wing VC, Shoaib M. A second-order schedule of food reinforcement in rats to examine the role of CB1 receptors in the reinforcement-enhancing effects of nicotine. Addict Biol. (2010) 15(4):380–92. doi:10.1111/j.1369-1600.2009.00203.x
132. De Vries TJ, Homberg JR, Binnekade R, Raasø H, Schoffelmeer AN. Cannabinoid modulation of the reinforcing and motivational properties of heroin and heroin-associated cues in rats. Psychopharmacology (2003) 168(1–2):164–9. doi:10.1007/s00213-003-1422-1
133. Anggadiredja K, Nakamichi M, Hiranita T, Tanaka H, Shoyama Y, Watanabe S, et al. Endocannabinoid system modulates relapse to methamphetamine seeking: possible mediation by the arachidonic acid cascade. Neuropsychopharmacology (2004) 29(8):1470–8. doi:10.1038/sj.npp.1300454
134. Economidou D, Mattioli L, Cifani C, Perfumi M, Massi M, Cuomo V, et al. Effect of the cannabinoid CB1 receptor antagonist SR-141716A on ethanol self-administration and ethanol-seeking behaviour in rats. Psychopharmacology (2006) 183(4):394–403. doi:10.1007/s00213-005-0199-9
135. Justinova Z, Munzar P, Panlilio LV, Yasar S, Redhi GH, Tanda G, et al. Blockade of THC-seeking behavior and relapse in monkeys by the cannabinoid CB(1)-receptor antagonist rimonabant. Neuropsychopharmacology (2008) 33(12):2870–7. doi:10.1038/npp.2008.21
136. Ball KT, Combs TA, Beyer DN. Opposing roles for dopamine D1- and D2-like receptors in discrete cue-induced reinstatement of food seeking. Behav Brain Res (2011) 222(2):390–3. doi:10.1016/j.bbr.2011.03.064
137. Ostlund SB, Maidment NT. Dopamine receptor blockade attenuates the general incentive motivational effects of noncontingently delivered rewards and reward-paired cues without affecting their ability to bias action selection. Neuropsychopharmacology (2012) 37(2):508–19. doi:10.1038/npp.2011.217
138. Oleson EB, Beckert MV, Morra JT, Lansink CS, Cachope R, Abdullah RA, et al. Endocannabinoids shape accumbal encoding of cue-motivated behavior via CB1 receptor activation in the ventral tegmentum. Neuron (2012) 73(2):360–73. doi:10.1016/j.neuron.2011.11.018
139. Day J, Roitman MF, Wightman RM, Carelli RM. Associative learning mediates dynamic shifts in dopamine signaling in the nucleus accumbens. Nat Neurosci (2007) 10(8):1020–8. doi:10.1038/nn1923
140. Buhusi CV, Meck WH. What makes us tick? Functional and neural mechanisms of interval timing. Nat Rev Neurosci (2005) 6(10):755–65. doi:10.1038/nrn1764
141. Harrington DL, Haaland KY. Sequencing in Parkinson’s disease. Abnormalities in programming and controlling movement. Brain (1991) 114(1A):99–115.
142. Malapani C, Rakitin B, Levy R, Meck WH, Deweer B, Dubois B, et al. Coupled temporal memories in Parkinson’s disease: a dopamine-related dysfunction. J Cogn Neurosci. (1998) 10(3):316–31. doi:10.1162/089892998562762
143. MacDonald CJ, Meck WH. Differential effects of clozapine and haloperidol on interval timing in the supraseconds range. Psychopharmacology (2005) 182(2):232–44. doi:10.1007/s00213-005-0074-8
144. Meck WH, Church RM. A mode control model of counting and timing processes. J Exp Psychol Anim Behav Process (1983) 9(3):320–34. doi:10.1037/0097-7403.9.3.320
145. Coull JT, Cheng R, Meck WH. Neuroanatomical and neurochemical substrates of timing. Neuropsychopharmacology (2011) 36(1):3–25. doi:10.1038/npp.2010.113
146. Andreasen NC. Understanding the causes of schizophrenia. N Engl J Med (1999) 340(8):645–7. doi:10.1056/NEJM199902253400811
147. Matell MS, King GR, Meck WH. Differential modulation of clock speed by the administration of intermittent versus continuous cocaine. Behav Neurosci (2004) 118(1):150–6. doi:10.1037/0735-7044.118.1.150
148. Matell MS, Meck WH. Neuropsychological mechanisms of interval timing behavior. Bioessays (2000) 22(1):94–103. doi:10.1002/(SICI)1521-1878(200001)22:1<94::AID-BIES14>3.0.CO;2-E
149. Meck WH. Selective adjustment of the speed of internal clock and memory processes. J Exp Psychol Anim Behav Process. (1983) 9(2):171–201.
150. Hicks RE, Gualtieri CT, Mayo JP, Perez-Reyes M. Cannabis, atropine, and temporal information processing. Neuropsychobiology (1984) 12(4):229–37. doi:10.1159/000118144
151. Tinklenberg JR, Kopell BS, Melges FT, Hollister LE. Marihuana and alcohol, Time production and memory functions. Arch Gen Psychiatry (1972) 27(6):812–5. doi:10.1001/archpsyc.1972.01750300074013
152. Crystal JD, Maxwell KW, Hohmann AG. Cannabinoid modulation of sensitivity to time. Behav Brain Res (2003) 144(1–2):57–66. doi:10.1016/S0166-4328(03)00062-7
153. Han CJ, Robinson JK. Cannabinoid modulation of time estimation in the rat. Behav Neurosci (2001) 115(1):243–6. doi:10.1037/0735-7044.115.1.243
154. Oleson EB, Cachope R, Fitoussi A, Tsutsui K, Wu S, Gallegos JA, et al. Cannabinoid receptor activation shifts temporally engendered patterns of dopamine release. Neuropsychopharmacology (2014) 39(6):1441–52. doi:10.1038/npp.2013.340
155. Dews PB. The theory of fixed-interval responding. In: Schoenfeld WN, editor. The Theory of Reinforcement Schedules. New York, NY: Appleton Century Crofts (1970). p. 43–61.
156. Pattij T, Wiskerke J, Schoffelmeer AN. Cannabinoid modulation of executive functions. Eur J Pharmacol (2008) 585(2–3):458–63. doi:10.1016/j.ejphar.2008.02.099
157. Narayanan NS, Land B, Solder JE, Deisseroth K, DiLeone RJ. Prefrontal D1 dopamine signaling is required for temporal control. Proc Natl Acad Sci USA (2012) 109(50):20726–31. doi:10.1073/pnas.1211258109
158. Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. (1989) 12(10):366–75. doi:10.1016/0166-2236(89)90074-X
159. Kravitz AV, Tye LD, Kreitzer AC. Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci (2012) 15(6):816–8. doi:10.1038/nn.3100
160. Brady AM, O’Donnell P. Dopaminergic modulation of prefrontal cortical input to nucleus accumbens neurons in vivo. J Neurosci (2004) 24(5):1040–9. doi:10.1523/JNEUROSCI.4178-03.2004
161. Floresco SB, Blaha CD, Yang CR, Phillips AG. Modulation of hippocampal and amygdalar-evoked activity of nucleus accumbens neurons by dopamine: cellular mechanisms of input selection. J Neurosci (2001) 21(8):2851–60.
162. Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, et al. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature (2013) 494(7436):238–42. doi:10.1038/nature11846
163. Matell MS, Meck WH. Cortico-striatal circuits and interval timing: coincidence detection of oscillatory processes. Brain Res Cogn Brain Res. (2004) 21(2):139–70. doi:10.1016/j.cogbrainres.2004.06.012
164. Matell MS, Meck WH, Nicolelis MA. Interval timing and the encoding of signal duration by ensembles of cortical and striatal neurons. Behav Neurosci (2003) 117(4):760–73. doi:10.1037/0735-7044.117.4.760
165. Groves PM, Garcia-Munoz M, Linder JC, Manley MS, Martone ME, Young SJ. Elements of the intrinsic organization and information processing in the neostriatum. In: Houk JC, Davis JL, Beiser DG, editors. Models of Information Processing in the Basal Ganglia. Cambridge: MIT Press (1995). p. 51–96.
166. O’Donnell P, Grace AA. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci (1995) 15(5):3622–39.
167. Gruber AJ, Solla SA, Surmeier DJ, Houk JC. Modulation of striatal single units by expected reward: a spiny neuron model displaying dopamine-induced bistability. J Neurophysiol (2003) 90(2):1095–114. doi:10.1152/jn.00618.2002
168. McGinty VB, Grace A. Timing-dependent regulation of evoked spiking in nucleus accumbens neurons by integration of limbic and prefrontal cortical inputs. J Neurophysiol (2009) 101(4):1823–35. doi:10.1152/jn.91162.2008
169. Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci (2007) 30(5):211–9. doi:10.1016/j.tins.2007.03.001
170. Groves PM. A theory of the functional organization of the neostriatum and the neostriatal control of voluntary movement. Brain Res (1983) 286(2):109–32. doi:10.1016/0165-0173(83)90011-5
171. Wickens JR, Alexander ME, Miller R. Two dynamic modes of striatal function under dopaminergic-cholinergic control: simulation and analysis of a model. Synapse (1991) 8(1):1–12. doi:10.1002/syn.890080102
172. Wickens JR, Kotter R, Alexander ME. Effects of local connectivity on striatal function: stimulation and analysis of a model. Synapse (1995) 20(4):281–98. doi:10.1002/syn.890200402
173. Carelli RM, Deadwyler SA. Cellular mechanisms underlying reinforcement-related processing in the nucleus accumbens: electrophysiological studies in behaving animals. Pharmacol Biochem Behav (1997) 57(3):495–504. doi:10.1016/S0091-3057(96)00442-X
174. Peoples LL, West MO. Phasic firing of single neurons in the rat nucleus accumbens correlated with the timing of intravenous cocaine self-administration. J Neurosci (1996) 16(10):3459–73.
175. Usuda I, Tanaka K, Chiba T. Efferent projections of the nucleus accumbens in the rat with special reference to subdivision of the nucleus: biotinylated dextran amine study. Brain Res (1998) 797(1):73–93.
176. Vitay J, Hamker FH. Timing and expectation of reward: a neuro-computational model of the afferents to the ventral tegmental area. Front Neurorobot. (2014) 8:4. doi:10.3389/fnbot.2014.00004
177. Kurti AN, Matell MS. Nucleus accumbens dopamine modulates response rate but not response timing in an interval timing task. Behav Neurosci (2011) 125(2):215–25. doi:10.1037/a0022892
178. Meck WH. Neuroanatomical localization of an internal clock: a functional link between mesolimbic, nigrostriatal, and mesocortical dopaminergic systems. Brain Res (2006) 1109(1):93–107. doi:10.1016/j.brainres.2006.06.031
179. Singh T, McDannald MA, Takahashi YK, Haney RZ, Cooch NK, Lucantonio F, et al. The role of the nucleus accumbens in knowing when to respond. Learn Mem (2011) 18(2):85–7. doi:10.1101/lm.2008111
180. Galtress T, Kirkpatrick K. The role of the nucleus accumbens core in impulsive choice, timing, and reward processing. Behav Neurosci (2010) 124(1):26–43. doi:10.1037/a0018464
181. Buhusi CV, Meck WH. Differential effects of methamphetamine and haloperidol on the control of an internal clock. Behav Neurosci (2002) 116(2):291–7. doi:10.1037/0735-7044.116.2.291
182. Lichtman AH, Martin BR. Cannabinoid tolerance and dependence. Handb Exp Pharmacol (2005) 168:691–717. doi:10.1007/3-540-26573-2_24
183. Nalwa V, Rao PS. Comparison of the effects of d-amphetamine on FI and DRL schedule performance of self-stimulating rats. Clin Exp Pharmacol Physiol (1984) 11(3):261–7. doi:10.1111/j.1440-1681.1984.tb00263.x
184. Sewell RA, Schnakenberg A, Elander J, Radhakrishnan R, Williams A, Skosnik PD, et al. Acute effects of THC on time perception in frequent and infrequent cannabis users. Psychopharmacology (2013) 226(2):401–13. doi:10.1007/s00213-012-2915-6
185. Cory-Slechta DA, Pazmino R, Bare C. The critical role of nucleus accumbens dopamine systems in the mediation of fixed interval schedule-controlled operant behavior. Brain Res (1997) 764(1–2):253–6.
186. Cory-Slechta DA, O’Mara DJ, Brockel BJ. Nucleus accumbens dopaminergic medication of fixed interval schedule-controlled behavior and its modulation by low-level lead exposure. J Pharmacol Exp Ther (1998) 286(2):794–805.
187. Robbins TW, Roberts DC, Koob GF. Effects of d-amphetamine and apomorphine upon operant behavior and schedule-induced licking in rats with 6-hydroxydopamine-induced lesions of the nucleus accumbens. J Pharmacol Exp Ther (1983) 224(3):662–73.
188. Neill DB, Herndon JG. Anatomical specificity within rat striatum for the dopaminergic modulation of DRL responding and activity. Brain Res (1978) 153(3):529–38.
189. Lake JI, Meck WH. Differential effects of amphetamine and haloperidol on temporal reproduction: dopaminergic regulation of attention and clock speed. Neuropsychologia (2013) 51(2):284–92. doi:10.1016/j.neuropsychologia.2012.09.014
190. Bickel WK, Odum AL, Madden GJ. Impulsivity and cigarette smoking: delay discounting in current, never, and ex-smokers. Psychopharmacology (1999) 146(4):447–54. doi:10.1007/PL00005490
191. Coffey SF, Gudleski GD, Saladin ME, Brady KT. Impulsivity and rapid discounting of delayed hypothetical rewards in cocaine-dependent individuals. Exp Clin Psychopharmacol. (2003) 11(1):18–25. doi:10.1037/1064-1297.11.1.18
192. Madden GJ, Petry NM, Badger GJ, Bickel WK. Impulsive and self-control choices in opioid-dependent patients and non-drug-using control participants: drug and monetary rewards. Exp Clin Psychopharmacol. (1997) 5(3):256–62. doi:10.1037/1064-1297.5.3.256
193. Mitchell JM, Fields HL, D’Esposito M, Boettiger CA. Impulsive responding in alcoholics. Alcohol Clin Exp Res (2005) 29(12):2158–69. doi:10.1097/01.alc.0000191755.63639.4a
194. Peters EN, Petry NM, Lapaglia DM, Reynolds B, Carroll KM. Delay discounting in adults receiving treatment for marijuana dependence. Exp Clin Psychopharmacol (2013) 21(1):46–54. doi:10.1037/a0030943
195. Petry NM, Kirby KN, Kranzler HR. Effects of gender and family history of alcohol dependence on a behavioral task of impulsivity in healthy subjects. J Stud Alcohol (2002) 63(1):83–90.
196. Vuchinich RE, Simpson CA. Hyperbolic temporal discounting in social drinkers and problem drinkers. Exp Clin Psychopharmacol (1998) 6(3):292–305. doi:10.1037/1064-1297.6.3.292
197. Day J, Jones JL, Carelli RM. Nucleus accumbens neurons encode predicted and ongoing reward costs in rats. Eur J Neurosci (2011) 33(2):308–21. doi:10.1111/j.1460-9568.2010.07531.x
198. Hernandez G, Oleson EB, Gentry RN, Abbas Z, Bernstein DL, Arvanitogiannis A, et al. Endocannabinoids promote cocaine-induced impulsivity and its rapid dopaminergic correlates. Biol Psychiatry (2014) 75(6):487–98. doi:10.1016/j.biopsych.2013.09.005
Keywords: dopamine, cannabinoids, reward-seeking behavior, cues, endocannabinoids, fixed interval
Citation: Wenzel JM and Cheer JF (2014) Endocannabinoid-dependent modulation of phasic dopamine signaling encodes external and internal reward-predictive cues. Front. Psychiatry 5:118. doi: 10.3389/fpsyt.2014.00118
Received: 02 July 2014; Paper pending published: 21 July 2014;
Accepted: 13 August 2014; Published online: 01 September 2014.
Edited by:
Steven R. Laviolette, University of Western Ontario, CanadaReviewed by:
Sulev Kõks, University of Tartu, EstoniaSteven R. Laviolette, University of Western Ontario, Canada
Copyright: © 2014 Wenzel and Cheer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph F. Cheer, Department of Anatomy and Neurobiology, University of Maryland School of Medicine, 20 Penn Street, Baltimore, MD 21201, USA e-mail:amNoZWUwMDFAdW1hcnlsYW5kLmVkdQ==