 Fulvio Plescia1
Fulvio Plescia1 Anna Brancato1
Anna Brancato1 Rosa Anna Maria Marino1
Rosa Anna Maria Marino1 Carlotta Vita1
Carlotta Vita1 Michele Navarra2
Michele Navarra2 Carla Cannizzaro1*
Carla Cannizzaro1*- 1Department of Sciences for Health Promotion and Mother and Child Care “Giuseppe D’Alessandro”, University of Palermo, Palermo, Italy
- 2Department of Drug Sciences and Products for Health, University of Messina, Messina, Italy
Acetaldehyde (ACD), the first alcohol metabolite, plays a pivotal role in the rewarding, motivational, and addictive properties of the parental compound. Many studies have investigated the role of ACD in mediating neurochemical and behavioral effects induced by alcohol administration, but very little is known about the modulation of neuropeptide systems following ACD intoxication and withdrawal. Indeed, the neuropeptide Y (NPY) system is altered during alcohol withdrawal in key regions for cerebrocortical excitability and neuroplasticity. The primary goal of this research was to investigate the effects of ACD intoxication and withdrawal by recording rat behavior and by measuring NPY immunoreactivity in hippocampus and NAcc, two brain regions mainly involved in processes which encompass neuroplasticity in alcohol dependence. Furthermore, on the basis of the involvement of endocannabinoidergic system in alcohol and ACD reinforcing effects, the role of the selective CB1 receptor antagonist AM281 in modulating NPY expression during withdrawal was assessed. Our results indicate that (i) ACD intoxication induced a reduction in NPY expression in hippocampus and NAcc; (ii) symptoms of physical dependence, similar to alcohol’s, were scored at 12 h from the last administration of ACD; and (iii) NPY levels increased in early and prolonged acute withdrawal in both brain regions examined. The administration of AM281 was able to blunt signs of ACD-induced physical dependence, to modulate NPY levels, and to further increase NPY expression during ACD withdrawal both in hippocampus and NAcc. In conclusion, the present study shows that complex plastic changes take place in NPY system during ACD intoxication and subsequent withdrawal in rat hippocampal formation and NAcc. The pharmacological inhibition of CB1 signaling could counteract the neurochemical imbalance associated with ACD, and alcohol withdrawal, likely boosting the setting up of homeostatic functional recovery.
Introduction
Acetaldehyde (ACD), the first oxidation product of alcohol, is one of the mediators of the peripheral and central effects of alcohol (1–5), in particular playing a main role in the rewarding, motivational, and addictive properties of the parental compound (6–8). Although not fully investigated, ACD reinforcing properties are likely due to its capability to affect the dopaminergic and endocannabinoidergic systems. As alcohol, ACD is able to induce and maintain an operant drinking behavior and relapse following repeated forced abstinence (6) and to increase the firing rate, spikes/burst, and burst firing of VTA neurons (9–11); furthermore, the pharmacological manipulation of dopaminergic D2 and endocannabinoidergic CB1 receptors decreases its motivational and incentive value (8, 12). As largely described, after abrupt suspension of long-term repetitive consumption, alcohol withdrawal syndrome reflects severe neuro-adaptation of membrane and intracellular molecular targets that results in disruption and perturbation in neurotransmitter and neuropeptide systems (13–15). Among them, CRH and neuropeptide Y (NPY) have been mainly evoked as responsible for the affective and somatic components of alcohol withdrawal (16, 17). In particular, NPY, a 36-amino acid peptide neuromodulator largely distributed in the central nervous system, is implicated in a wide range of functions including feeding, anxiety, seizures, circadian rhythms, memory, and cardiovascular regulation (18–21), besides its involvement in the neuronal mechanisms of alcohol consumption (22, 23). Indeed, lower levels of NPY-IR in hippocampus, amygdala, and frontal cortex have been reported in selectively bred alcohol-preferring rats compared to non-preferring rats (24), as well as lower expression in NPY protein in NAcc has been measured in C57BL/6J mice, that innately consume larger amount of alcohol (25). On the other hand, intracerebroventricular infusion of NPY produces electrophysiological effects similar to those of alcohol in rats (26); consistently, NPY-deficient mice drink more alcohol compared with wild-type mice, whereas mice over-expressing NPY display a lower preference for alcohol (27, 28). Additionally, NPY plays a central role in the modulation of neuronal excitability mainly in cortex and hippocampus (29–32), where NPY is mostly co-localized with γ-aminobutyric acid within interneurons (33–35). Cerebrocortical excitability is altered during the development of alcohol tolerance and dependence, and greatly enhanced during alcohol withdrawal (36, 37). Notably, intracerebroventricular administration of NPY attenuates symptoms of alcohol withdrawal in rats, probably due to presynaptical inhibition of glutamate release (29, 38, 39). Recent data have identified NPY as a promoter of hippocampal neurogenesis since it is able to enhance cell proliferation and promote neuronal differentiation in adult mice (40–42). Adult neurogenesis occurs constitutively in the subgranular zone of the hippocampal dentate gyrus and in the subventricular zone of the walls of the lateral ventricles, adjacent to the ventral striatum (43–45). The modulation of NPYergic system seems to be involved in the regulation of alcohol-induced reactive neurogenesis (46, 47). Thus, the assessment of ACD activity on NPY expression in brain areas closely linked to the neurogenic niches may be helpful to clarify its role as a mediator of alcohol effects on brain neuroplasticity. In this study, ACD was administered according to a binge model previously characterized to induce tolerance and physical dependence to alcohol (48–51). The effects of ACD intoxication and withdrawal were investigated by recording rat behavior, and by measuring NPY expression in the hippocampus and ventral striatum, two of the brain regions mainly involved in processes which encompass neuroplasticity in alcohol dependence (46). Moreover, since the endocannabinoidergic system plays a relevant role in the reinforcing effects of alcohol (52–55), and also in the development of alcohol tolerance and withdrawal (56), the present research aimed at the evaluation of the effect of a selective CB1 receptor antagonist AM281 on NPY expression during withdrawal. Indeed, an interplay between endocannabinoidergic system and NPY expression and release has been demonstrated in the hypothalamus (57, 58), but so far, no data exist on a functional correlation between ACD, NPY, and endocannabinoids in the brain.
Materials and Methods
Animals
Adult male Wistar rats, weighing 250–300 g, were used in this study. Animals were housed two per cage and maintained on a 12 h light/dark cycle, on temperature (22 ± 2°C) and humidity (55 ± 5%) controlled conditions, with ad libitum access to food and water. All efforts were made to minimize suffering and number of animals used. Experimental procedures were in strict accordance with Italian legislation dealing with research on experimental animals (D.L. 116/92) and European Council Directive (2010/63/EU) on animals used for scientific purposes.
Drugs and Pharmacological Treatment
Acetaldehyde 99.98% (Sigma-Aldrich, Milan, Italy) was daily diluted with tap water to a final concentration of 8% v/v; each intragastric ACD administration provided 450 mg/kg. The selective CB1 receptor antagonist AM281 (Sigma-Aldrich, Milan, Italy) was suspended in saline solution containing 3% Tween 80 and administered i.p. at 2.5 mg/kg, in ACD and CTR animals at day 5, 3 and 12 h after the last intragastric administration [modified from Ref. (59)]. The dose of AM281 used in this study was chosen to avoid any aspecific effect (60, 61).
Binge ACD Treatment
Rats received intragastric infusions of ACD (450 mg/kg) by gavage, five times daily (7 a.m., 11 a.m., 3 p.m., 7 p.m., and 11 p.m.) for 4 days, in order to induce intoxication and withdrawal syndrome (49). The control group received intragastric infusions of water, according to the time schedule of the protocol. Behavioral signs of ACD intoxication were observed according to the severity scale of Majchrowicz (49). During the intoxication paradigm, ACD treatment was individually adjusted to reach an intoxication score between 3 and 5 (Ataxia 2–LRR) on each entire day (51) to avoid lethal toxicity (seven animals died during the protocol, before the assessments.).
Acetaldehyde-binge treated animals and controls were allocated in the following experimental groups according to the assigned procedure: ACD/T1 (n = 6) and CTR/T1 (n = 6) were decapitated 1 h after the last intragastric administration; ACD/T16 (n = 6), CTR/T16 (n = 6), ACD + AM281/T16 (n = 6), and CTR + AM281/T16 (n = 6) were evaluated for the behavioral signs of withdrawal at T12, and were decapitated at 16 h after the last intragastric infusion; ACD/T72 (n = 6), CTR/T72 (n = 6), ACD + AM281/T72 (n = 6), and CTR + AM281/T72 (n = 6) were decapitated 72 h after the last intragastric administration.
Behavioral Observations of Withdrawal
At 12 h after the last infusion of ACD (T12), an observer blinded to the treatment assessed the severity of physical dependence considering the following signs: general hyperactivity, irritability, tail tremors, tail stiffness, general tremor, spasticity, wet (dog) shakes, and spontaneous convulsive seizures (62). Each sign was assigned a score of 0–3 (0 = not present, 1 = slight, 2 = moderate, and 3 = severe). The sum of these scores (0–24) was used as a quantitative measurement of the severity of the withdrawal reaction, the “total withdrawal score.”
Immunohistochemical Detection of NPY
After decapitation, brains were rapidly removed, frozen on dry ice, and stored at −80°C. Coronal serial sections (20 μm) from frozen rat brains were cut on a cryomicrotome from plate 29 to 36, corresponding to dorsal hippocampus, and from plate 10 to 14, equivalent to nucleus accumbens, according to the atlas of Paxinos and Watson (63). Sections were thaw-mounted onto Superfrost glass slides, dried on a hotplate, and processed for NPY immunohistochemical analysis using a commercially available NPY immunohistochemistry staining kit (D.B.A., Italy). The total number of NPY-positive neurons in each target brain region was achieved by counting the number of positive cells of labeled cell bodies determined with cresyl violet staining. Coronal brain sections were further divided into different quadrants: hippocampus in CA1, CA2, CA3, and DG, nucleus accumbens in shell and core. The counterstained sections were placed under the microscope, and the number of positive cells was counted manually in all quadrants. Each labeled cell was viewed under bright-field illumination using a 100× objective (Meiji Techno, Japan). Real-time microscopic images were captured by a video camera, digitized, and displayed on a monitor. Two repeated measurements by two different experimenters were performed bilaterally in three adjacent sections per animal in the brain regions of interest (63).
Statistical Analysis
The differences in total withdrawal score between the groups were assessed by the Kruskal–Wallis analysis of variance (ANOVA) followed by the Dunn post hoc test. A two-way ANOVA was conducted on the number of NPY-positive neurons as dependent variables, with treatment (control, ACD, AM281) as the between-subjects factor and “time” or “brain Area” as within-subjects factor. When necessary, simple main effects and post hoc comparison were calculated with Bonferroni post-test (α = 5). Values were considered statistically significant when p < 0.05. All data are presented as mean (S.D.). Statistical analysis was conducted by using a GraphPad Prism software 6.1 (GraphPad Software, San Diego, CA, USA) on data from all experimental animals used.
Results
Behavioral Observations of Withdrawal
In order to investigate spontaneous withdrawal behavior, rats were observed at 12 h from the last ACD intragastric administration and scored for general hyperactivity, irritability, tail tremors, tail stiffness, general tremor, spasticity, wet (dog) shakes, and spontaneous convulsive seizures. ACD-treated animals showed discrete behavioral signs of withdrawal, and among them, general hyperactivity, irritability, and spasticity were recorded more frequently. Somatic dependence symptoms persisted until 16 h and were absent when the animals were observed again at 36 h. According to the score assigned to each behavioral sign, the mean total withdrawal score in ACD rats was of 11.67 ± 1.63 (Figure 1). Results of a Kruskal–Wallis test, performed on each behavioral score and on total withdrawal score, including “treatment” as the between-subjects factor, showed significant differences among the experimental groups (general hyperactivity: p < 0.00; irritability: p < 0.001; tail tremors: p < 0.01; tail stiffness: p < 0.001; general tremor: p < 0.01; spasticity: p < 0.001). Dunn’s post hoc analysis, highlighted a significantly higher presence of individual and total withdrawal symptoms in ACD group, with respect to CTR, while individual and total withdrawal score of ACD-AM281 rats was non-statistically different than controls’. Results of Dunn’s post hoc analysis are showed in Table 1.
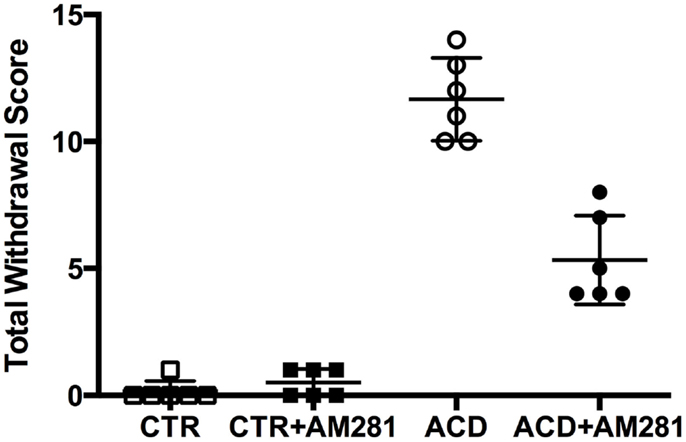
Figure 1. Total withdrawal score. (□) CTR, (■) CTR + AM281, (○) ACD, and (●) ACD + AM281.

Table 1. Behavioral observations of withdrawal.
Quantification of NPY-Positive Neurons
Hippocampus
The number of NPY-positive neurons was evaluated in the hippocampus as a whole. The results of a two-way ANOVA including “ACD treatment” as the between-subjects factor and “time” as within-subjects factor showed a significant effect of time, treatment, and their interaction [F(2, 66) = 223.65, p < 0.0001; F(1, 66) = 204.16, p < 0.0001; F(2, 66) = 220.28, p < 0.0001]. Bonferroni post hoc analysis showed a significant reduction in NPY-positive neurons in ACD group at T1 (t = 6.729, p < 0.001) and an increase in NPY-positive neurons at T16 (t = 8.526, p < 0.001) and T72 (t = 22.95, p < 0.001) when compared to CTR group. NPY expression was also analyzed within ACD group in order to reveal time-related differences in relative NPY-positive neurons expression during withdrawal (Figure 2). The results of a two-way ANOVA showed a significant effect of time, treatment, and their interaction [F(2, 66) = 233.09, p < 0.0001; F(1,66) = 212.78, p < 0.0001; F(2, 66) = 229.57, p < 0.0001]. Bonferroni post hoc analysis showed that NPY expression increased at T16 (t = 15.41, p < 0.001), with respect toT1, and at T72 (t = 15.01, p < 0.001) compared to T16 (Figure 3). A detailed analysis of NPY expression in the hippocampal sub-regions was also carried out. The results of a two-way ANOVA performed, respectively, at T1, at T16, and at T72, including “ACD treatment” as the between-subjects factor and “sub-regional NPY expression” as within-subjects factor showed a significant effect of sub-regional NPY expression [F(3, 88) = 1768.82, p < 0.0001; F(3,88) = 233.30, p < 0.0001; F(3, 88) = 24.30, p < 0.0001], treatment [F(1, 88) = 1284.10, p < 0.0001; F(1,88) = 160.9, p < 0.0001; F(1,88) = 636.3, p < 0.0001] and their interaction [F(3,88) = 298.08, p < 0.0001; F(3,88) = 23.75, p < 0.0001; F(2,88) = 54.12, p < 0.0001). Bonferroni post hoc analysis showed a decrease in the number of NPY-positive cells at T1 in ACD group in all the hippocampal sub-regions, while an increase in NPY expression was observed at T16 and T72, compared to controls (Table 2).
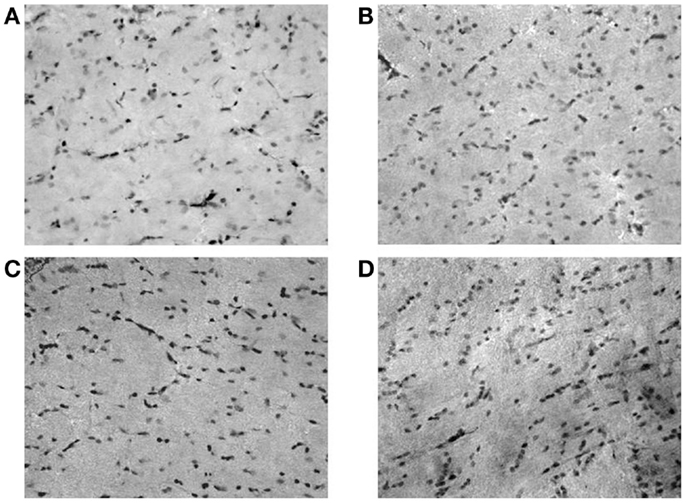
Figure 2. Microphotographs of neuropeptide Y (NPY)-positive neurons in rat hippocampus (dentate gyrus); (A) controls; (B) ACD rats at T1; (C) ACD rats at T16; and (D) ACD rats at T72. The specific labeling is observed under bright-field illumination using a 100× objective.
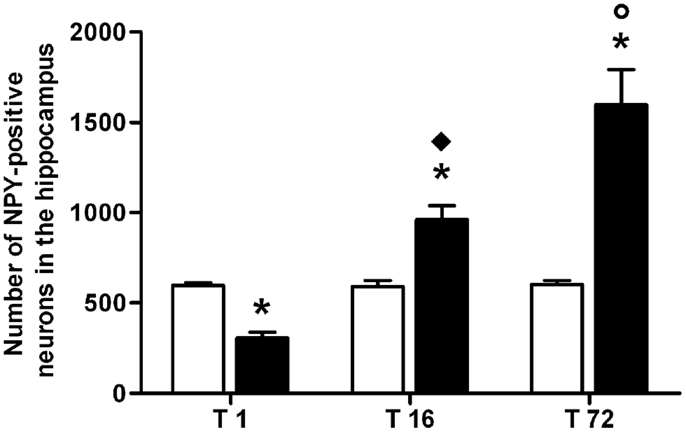
Figure 3. Average number of neuropeptide Y (NPY)-positive neurons in the hippocampus of ACD and control rats at different times following the last intragastric infusion. Each value represents the mean ± S.D. of 12 sections for each experimental condition. (□) CTR, (■) ACD. *p < 0.001 vs CTR, ♦p < 0.001 vs T1, ○p < 0.001 vs T16.
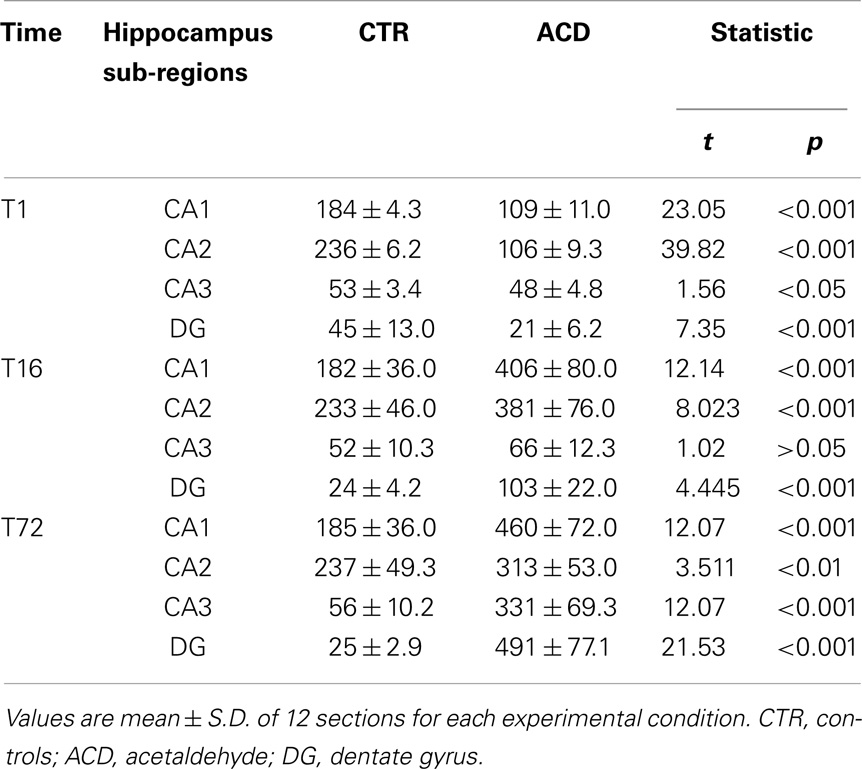
Table 2. Number of NPY-positive neurons in different sub-regions of the hippocampus, in ACD and in control rats, at different times following the last intragastric infusion (T1, T16, T72).
Effects of AM281 on hippocampal NPY expression
In order to assess the involvement of CB1 signaling on the modulation of NPY-positive neurons expression in hippocampus, statistical analysis by a two-way ANOVA was performed on the effect of the CB1 antagonist AM281 both in ACD group and in controls. Our results showed significant effects of time, treatment, and their interaction in ACD rats [F(1, 44) = 422.31, p < 0.0001; F(1,44) = 34.86, p < 0.0001; F(1, 44) = 15.29, p < 0.0001]. Bonferroni post hoc analysis showed that AM281 was able to induce an increase in the number of NPY-positive neurons at 16 h (t = 6.940, p < 0.001) in ACD group compared to respective controls (Figure 4A). In control animals, a two-way ANOVA including “AM281 treatment” as the between-subjects factor and “time” as within-subjects factor revealed a significant effect of time, treatment, and their interaction [F(1, 44) = 9.24, p < 0.0040; F(1,44) = 106.99, p < 0.0001; F(1, 44) = 6.32, p < 0.0156]. Bonferroni post hoc analysis showed that AM281 was able to induce an increase in the number of NPY-positive neurons at T16 (t = 5.536, p < 0.001) and T72 (t = 9.092, p < 0.001) in CTR group (Figure 4B).
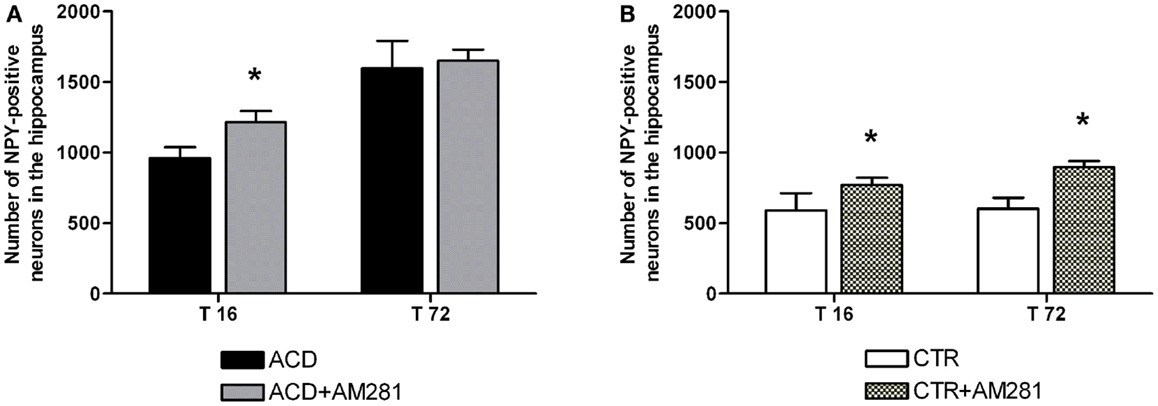
Figure 4. Effect of AM281 treatment on the number of hippocampus neuropeptide Y (NPY)-positive neurons in ACD rats (A) and in controls (B). Each value represents the mean ± S.D. of 12 sections for each experimental condition. *p < 0.001 vs respective controls.
Nucleus accumbens
Results from a two-way ANOVA including “ACD treatment” as the between-subjects factor and “time” as within-subjects factor performed on the number of NPY-positive neurons in NAcc, at different time points, showed a significant effect of time, treatment, and their interaction [F(2, 66) = 185.97, p < 0.0001; F(1,66) = 139.60, p < 0.0001; F(2, 66) = 176.71, p < 0.0001]. Bonferroni post hoc analysis showed a significant reduction in NPY-positive neurons in ACD group at T1 (t = 5.036, p < 0.001) and an increase in NPY-positive neurons at T16 (t = 3.995, p < 0.001) and T72 (t = 21.14, p < 0.001), when compared to CTR group (Figure 5). NPY expression was also analyzed within ACD group in order to reveal time-related differences in relative NPY-positive neurons during withdrawal. The results of a two-way ANOVA showed a significant effect of time, treatment, and their interaction [F(2, 66) = 195.97, p < 0.0001; F(1,66) = 134.59, p < 0.0001; F(2, 66) = 176.57, p < 0.0001]. Bonferroni post hoc analysis showed that NPY expression increased at T16 (t = 8.788, p < 0.001), with respect to T1, and at T72 (t = 17.65, p < 0.001) compared to T16 (Figure 6). When NPY expression was evaluated in accumbal sub-regions shell and core, a two-way ANOVA performed, respectively, at T1, at T16, and at T72, including “ACD treatment” as the between-subjects factor and “sub-regional NPY expression” as within-subjects factor showed a significant effect of sub-regional NPY expression [F(1, 44) = 199.59, p < 0.0001; F(1,44) = 16.67, p < 0.0002; F(1, 44) = 5.11, p < 0.028], treatment [F(1, 44) = 353.82, p < 0.0001; F(1,44) = 156.94, p < 0.0001; F(1,44) = 257.57, p < 0.0001] and their interaction [F(1,44) = 7.10, p < 0.0001; F(1,44) = 57.90, p < 0.0001; F(1,44) = 24.45, p < 0.0001]. Bonferroni post hoc analysis showed a decrease in the number of NPY-positive cells at T1 in ACD group in shell and core, while an increase in NPY expression was observed at T16 and T72, compared to controls (Table 3). In order to evaluate whether ACD treatment could differentially affect NPY expression in the shell of the NAcc, with respect to core, a two-way ANOVA performed on “sub-regional NPY expression” as the between-subjects factor” and “time” as within-subjects factor, displayed a significant effect of sub-regional NPY expression [F(2,66) = 1475.13, p < 0.0001], time [F(1,66) = 23.86, p < 0.0001] and their interaction [F(2,66) = 37.10, p < 0.0001]. Bonferroni post hoc analysis showed that NPY expression decreased at T1 (t = 4.036, p < 0.001) and increase at T16 (t = 4.890, p < 0.001) and T72 (t = 7.606, p < 0.001) in shell when compared to core in ACD-treated rats.
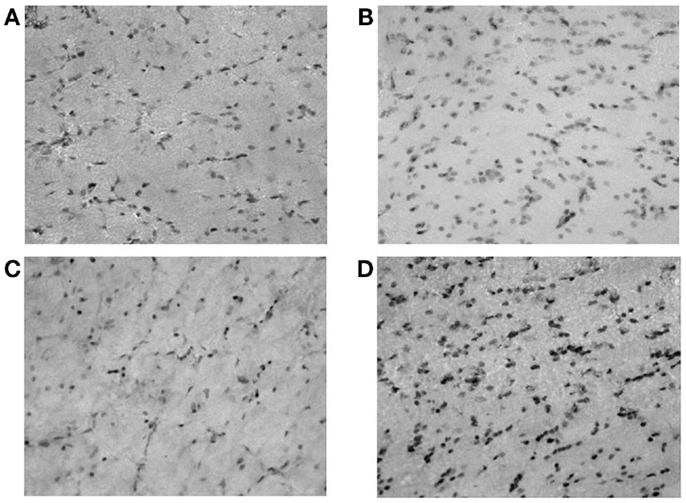
Figure 5. Microphotographs of neuropeptide Y (NPY)-positive neurons in rat nucleus accumbens (shell); (A) controls; (B) ACD rats at T1; (C) ACD rats at T16; and (D) ACD rats at T72. The specific labeling is observed under bright-field illumination using a 100× objective.
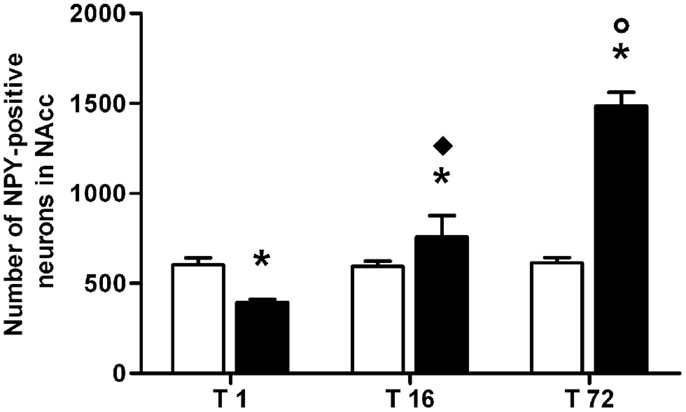
Figure 6. Average number of neuropeptide Y (NPY)-positive neurons detected in the nucleus accumbens (NAcc) during acetaldehyde intoxication and withdrawal. Each value represents the mean ± S.D. of 12 sections for each experimental condition. (□) CTR, (■) ACD. *p < 0.001 vs CTR, ♦p < 0.001 vs T1, ○p < 0.001 vs T16.
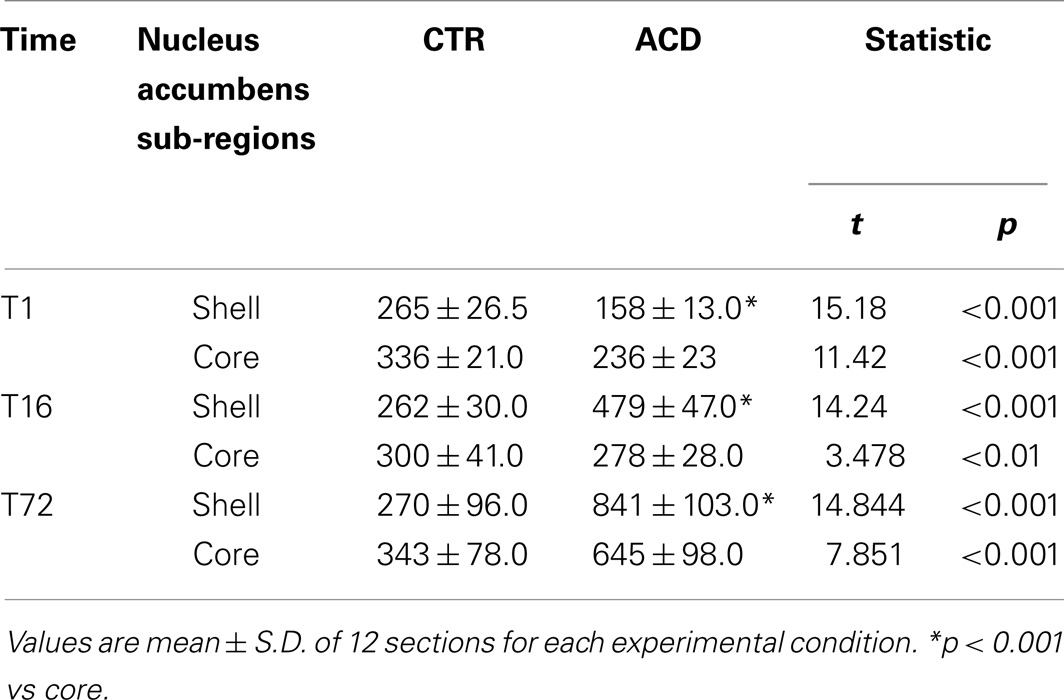
Table 3. Number of NPY-positive neurons expression in nucleus accumbens shell and core, in ACD and in control rats at different times following the last intragastric infusion (T1, T16, T72).
Effects of AM281 on accumbal NPY expression
In order to assess the involvement of CB1 signaling in the modulation of NPY-positive neurons expression in NAcc, statistical analysis by a two-way ANOVA was performed on the effects exerted by the CB1 antagonist AM281 both in ACD group and in controls. Our results showed significant effects of time, treatment, and their interaction [F(1, 44) = 489.21, p < 0.0001; F(1,44) = 6.24, p < 0.0163; F(1, 44) = 4.46, p < 0.043]. Bonferroni post hoc analysis displayed that AM281 was able to induce an increase in the number of NPY-positive neurons at T72 (t = 3.260, p < 0.01) in ACD group compared to respective controls (Figure 7A). Furthermore, when CTR animals received the selective cannabinoid antagonist, statistical analysis performed by a two-way ANOVA including “AM281 treatment” as the between-subjects factor and “time” as within-subjects factor revealed a significant effect of time, treatment, and their interaction [F(1, 44) = 25.74, p < 0.0001; F(1,44) = 41.33, p < 0.0001; F(1, 44) = 20.69, p < 0.0001]. Bonferroni post hoc analysis showed that AM281 was able to induce an increase in the number of NPY-positive neurons at T72 (t = 7.762, p < 0.001) in CTR group, compared to respective controls (Figure 7B).
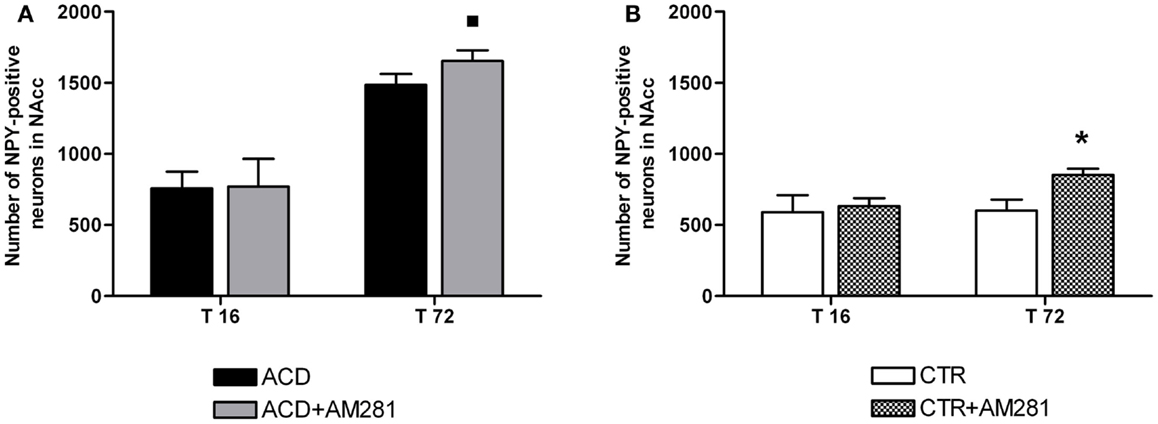
Figure 7. Effect of AM281 treatment on the number of nucleus accumbens neuropeptide Y (NPY)-positive neurons in ACD rats (A) and in controls (B). Each value represents the mean ± S.D. of 12 sections for each experimental condition. *p < 0.001; ■p < 0.01 vs respective controls.
Discussion
In the current study, the primary goal was to verify if ACD, the first metabolite of alcohol, is able to produce alterations in NPY protein levels in hippocampus and ventral striatum neurons, as already shown for alcohol (51, 64–66). The major result is that a 4-day long ACD binge treatment modulates NPY expression as a result of ACD intoxication but also as a consequence of early and prolonged, acute withdrawal. In particular, a significant decrease in NPY-positive neurons was observed 1 h after the last ACD infusion both in hippocampus and in NAcc; moreover, 16 and 72 h following the last ACD administration, a relevant increase in NPY expression was reported in the same areas. As recorded following alcohol binge treatment, ACD intoxication produced somatic withdrawal features that were observed at 12 h from the last ACD administration, started to decline at 16 h, and disappeared at 36 h abstinence. ACD-induced physical dependence did not display all the classical signs observed in alcohol withdrawal; indeed, general hyperactivity, irritability, tail tremors, tail stiffness, general tremor, and spasticity were recorded and scored, but they reached a lower severity than in alcohol withdrawal (49, 50, 62); wet dog shakes and spontaneous convulsive seizures were not observed in these experimental conditions. Several factors can contribute to the explanation of this evidence that, as far as we know, has never been reported before. As alcohol, peripherally administered ACD does reach the brain, due to its capability of overwhelming the metabolic barrier constituted by epithelial aldehyde dehydrogenase, a low Km ACD-oxidizing enzyme expressed in the gastrointestinal tract (67, 68). Moreover, high blood ACD concentration can saturate the moderate aldehyde dehydrogenase activity of the BBB capillaries, enter the brain and exert central activity (69–72). Indeed, ACD itself is able to interact with channels and receptors producing relevant alterations in discrete neurotransmitter systems (9, 73–75). Therefore, although ACD does not share the same pharmacodynamic and pharmacokinetic properties of alcohol, its involvement as a mediator of alcohol-induced dependence cannot be ruled out. ACD-induced reduction in NPY expression was observed in all the hippocampal sub-regions, with a prominent effect in DG and CA1 where NPY is expressed in the basket and the granule cells (64). NPY is known to inhibit excitatory transmission by reducing glutamate release (29); indeed, mice lacking NPY are more susceptible to the epileptogenic effect exerted by pentylenetetrazole and kainate (76, 77). In accordance, the reduction in NPY levels observed in this study in the hippocampus, as a consequence of the effect of ACD binge treatment, could be associated to a dampened inhibitory transmission that contributes to the hyperexcitable state that follows physical dependence. The reduction in NPY levels in NAcc following ACD intoxication is consistent with data showing that excessive alcohol drinking behavior is related to lower expression in NPY protein in NAcc in C57BL/6J mice, which innately consume larger amount of alcohol. Conversely, mice showing lower intake of alcohol display higher expression of NPY neurons in the same area. Alcohol drinking behavior has been related to profound modifications in the transductional processes in NAcc neurons that are correlated to lower expression of NPY gene (25). Our data fall into agreement with these findings pointing to a prominent role of NPY in modulating the rewarding, reinforcing, and motivational responses in the mesolimbic system (78, 79). Notably, a dramatic, and time dependent, increase in NPY levels was observed at 16 and 72 h of withdrawal in all the hippocampal sub-regions, primarily in DG and CA1, as well as in NAcc. Alcohol withdrawal is characterized by a great perturbation of the homeostatic systems that in fact leads to the appearance of profound signs of physical dependence. Therefore, it is reasonable to hypothesize that the increase in NPY protein levels in hippocampus, in particular in CA1 and DG, primarily represents a compensatory mechanism, which allows the organism to limit the intensity and duration of hippocampal hyperexcitability. Accordingly, intracerebroventricular administration of NPY has been shown to reduce alcohol withdrawal seizures, after a 4-day alcohol treatment similar to our ACD binge protocol (38). Interestingly, DG is one of the brain regions where neurogenesis of adult neurons occurs, together with the subventricular zone of the lateral ventricles, that in human beings supplies new interneurons to the adjacent striatum (80).
Neurogenesis is a neuronal activity driven process involved in stress-mediated behavioral responses, mood control, and certain forms of learning and memory; NPY has been reported to be a promoter of this form of neuronal plasticity in terms of enhanced proliferation and differentiation in DG neurons in adult mice (40, 41). Hence, the increase in NPY levels, following acute withdrawal observed in this study, could induce hippocampal and subventricular neurogenesis and neural homeostasis as a compensatory mechanism toward ACD-induced loss of inhibitory control and neuronal damage, as also reported following alcohol intoxication (51, 81, 82). Moreover, NPY enhancement in NAcc suggests a direct interaction between dopamine and NPY and in particular the set up of a recovery process in the mesolimbic dopaminergic transmission, so that an increase in NPY levels could boost the activity of TH-positive neurons and increase extracellular dopamine release (83). These findings lead to focus on NPY as a crucial player in the modulation of accumbal dopamine transmission. On the other hand, reports on nigrostriatal regulation in human brain with Parkinson’s disease show that NPY mRNA expression rises as a consequence of a dampened dopaminergic tone (84). The hypodopaminergic state is a neurochemical and functional feature of withdrawal after the chronic exposure to several drugs of abuse, including alcohol (85). Therefore, we hypothesize that the rise in NPY levels in the ventral striatum during early and prolonged acute withdrawal, besides a putative role in promoting neuroplasticity, could contribute to the slow, long transition phase that takes to the reinstatement of the dopaminergic tone, which is compromised during ACD treatment. A very few reports examine the expression of NPY in the striatum following alcohol intoxication, and most of them describe a modulation of NPY in Nacc shell (86). In this study, we observed a modulation of NPY expression in both the accumbal sub-regions, although the increase in NPY levels was prominent in NAcc shell, the accumbal sub-region primarily involved in adaptive neuromechanisms underlying the onset of addiction.
The second major finding of this study is the evidence of an interplay between NPY expression and CB1 signaling in rat hippocampus and NAcc. Endocannabinoids can be released in NAcc (87) and VTA, where they modulate the excitatory and inhibitory inputs that control mesolimbic pathways by acting as retrograde messengers on CB1 receptors (88–92). In this study, the administration of AM281, a selective CB1 antagonist, avoid of aspecific effects, or partial agonist activity produced a significant increase in NPY-positive neurons both in ACD group and in controls. Indeed, injected before the onset of the withdrawal syndrome, AM281 was able to reduce the behavioral signs that follow ACD treatment suspension. This was accompanied by a further increase in the number of NPY-positive neurons both in hippocampus and in NAcc, measured, respectively, at early and prolonged acute withdrawal. Little is known about the functional role of endocannabinoids on NPY system. It has been reported that anandamide and 2-arachidonoylglycerol, through CB1 receptor signaling, dose dependently downregulate NPY mRNA levels (58). The activation of the CB1 signal is associated with the inhibition of PKA and CREB phosphorylation (93). Indeed, the reduction in pCREB could explain the decrease in NPY levels observed, in that NPY gene is a cAMP-inducible element. Moreover, during a binge alcohol treatment, an increase in CREB mRNA and pCREB-IR has been reported at 24 and 72 h withdrawal, when higher NPY expression was also found in the basket cells of the hippocampus (64); this is consistent with our finding and prompts to suggest an inverse correlation between endocannabinoids and NPY expression in the area analyzed. The modulation by AM281 appears to be region- and time-dependent; NPY expression in ACD binge treated rats is increased in NAcc at 3 days of withdrawal, while in the hippocampus it rises at 16 h of withdrawal. At present, this different pattern of expression is difficult to explain; however, a peculiar sensitivity of NPY system to CB1 signaling in NAcc, along with the impact of intracellular and molecular disarrangements linked to withdrawal and the onset of the processes of recovery, cannot be ruled out. In conclusion, the present study shows that complex plastic changes take place in NPY system during ACD binge treatment and subsequent withdrawal in the rat hippocampal formation and NAcc; we hypothesize that ACD binge treatment increases endocannabinoidergic transmission, similar to alcohol (94), thus resulting in a downregulation of NPY system that plays a role in physical dependence and in the onset of withdrawal syndrome. During early and prolonged acute withdrawal, NPY expression progressively rises, likely as a consequence of the decreased endocannabinoidergic tone, thus contributing to the control of neuronal hyperexcitability and of the disarrangement in the mesolimbic system. The pharmacological inhibition of CB1 signaling could be effective in counteracting the neurochemical imbalance associated with ACD and alcohol withdrawal, likely boosting the setting up of homeostatic functional recovery.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Correa M, Arizzi MN, Betz A, Mingote S, Salamone JD. Open field locomotor effects in rats after intraventricular injections of ethanol and the ethanol metabolites acetaldehyde and acetate. Brain Res Bull (2003) 62(3):197–202. doi:10.1016/j.brainresbull.2003.09.013
2. Correa M, Arizzi MN, Betz A, Mingote S, Salamone JD. Locomotor stimulant effects of intraventricular injections of low doses of ethanol in rats: acute and repeated administration. Psychopharmacology (Berl) (2003) 170(4):368–75. doi:10.1007/s00213-003-1557-0
3. Diana M, Peana AT, Sirca D, Lintas A, Melis M, Enrico P. Crucial role of acetaldehyde in alcohol activation of the mesolimbic dopamine system. Ann N Y Acad Sci (2008) 1139:307–17. doi:10.1196/annals.1432.009
4. Cannizzaro C, La Barbera M, Plescia F, Cacace S, Tringali G. Ethanol modulates corticotropin releasing hormone release from the rat hypothalamus: does acetaldehyde play a role? Alcohol Clin Exp Res (2010) 34(4):588–93. doi:10.1111/j.1530-0277.2009.01127.x
5. Cannizzaro C, Plescia F, Cacace S. Role of acetaldehyde in alcohol addiction: current evidence and future perspectives. Malta Med J (2011) 23:27.
6. Cacace S, Plescia F, Barberi I, Cannizzaro C. Acetaldehyde oral self-administration: evidence from the operant-conflict paradigm. Alcohol Clin Exp Res (2012) 36(7):1278–87. doi:10.1111/j.1530-0277.2011.01725.x
7. Correa M, Salamone JD, Segovia KN, Pardo M, Longoni R, Spina L, et al. Piecing together the puzzle of acetaldehyde as a neuroactive agent. Neurosci Biobehav Rev (2012) 36(1):404–30. doi:10.1016/j.neubiorev.2011.07.009
8. Plescia F, Brancato A, Marino RA, Cannizzaro C. Acetaldehyde as a drug of abuse: insight into AM281 administration on operant-conflict paradigm in rats. Front Behav Neurosci (2013) 7:64. doi:10.3389/fnbeh.2013.00064
9. Melis M, Enrico P, Peana AT, Diana M. Acetaldehyde mediates alcohol activation of the mesolimbic dopamine system. Eur J Neurosci (2007) 26(10):2824–33. doi:10.1111/j.1460-9568.2007.05887.x
10. Foddai M, Dosia G, Spiga S, Diana M. Acetaldehyde increases dopaminergic neuronal activity in the VTA. Neuropsychopharmacology (2004) 29(3):530–6. doi:10.1038/sj.npp.1300326
11. Enrico P, Sirca D, Mereu M, Peana AT, Lintas A, Golosio A, et al. Acetaldehyde sequestering prevents ethanol-induced stimulation of mesolimbic dopamine transmission. Drug Alcohol Depend (2009) 100(3):265–71. doi:10.1016/j.drugalcdep.2008.10.010
12. Brancato A, Plescia F, Marino RA, Maniaci G, Navarra M, Cannizzaro C. Involvement of dopamine D2 receptors in addictive-like behaviour for acetaldehyde. PLoS One (2014) 9(6):e99454. doi:10.1371/journal.pone.0099454
13. Heilig M, Egli M, Crabbe JC, Becker HC. Acute withdrawal, protracted abstinence and negative affect in alcoholism: are they linked? Addict Biol (2010) 15(2):169–84. doi:10.1111/j.1369-1600.2009.00194.x
14. Roberto M, Gilpin NW, Siggins GR. The central amygdala and alcohol: role of γ-aminobutyric acid, glutamate, and neuropeptides. Cold Spring Harb Perspect Med (2012) 2(12):a012195. doi:10.1101/cshperspect.a012195
15. De Witte P, Pinto E, Ansseau M, Verbanck P. Alcohol and withdrawal: from animal research to clinical issues. Neurosci Biobehav Rev (2003) 27(3):189–97. doi:10.1016/S0149-7634(03)00030-7
16. Hershon HI. Alcohol withdrawal symptoms and drinking behavior. J Stud Alcohol (1977) 38(5):953–71.
17. Thorsell A. Brain neuropeptide Y and corticotropin-releasing hormone in mediating stress and anxiety. Exp Biol Med (Maywood) (2010) 235(10):1163–7. doi:10.1258/ebm.2010.009331
18. Heilig M, Widerlöv E. Neurobiology and clinical aspects of neuropeptide Y. Crit Rev Neurobiol (1995) 9(2–3):115–36.
19. Munglani R, Hudspith MJ, Hunt SP. The therapeutic potential of neuropeptide Y. Analgesic, anxiolytic and antihypertensive. Drugs (1996) 52(3):371–89. doi:10.2165/00003495-199652030-00004
20. Palmiter RD, Erickson JC, Hollopeter G, Baraban SC, Schwartz MW. Life without neuropeptide Y. Recent Prog Horm Res (1998) 53:163–99.
21. Wettstein JG, Earley B, Junien JL. Central nervous system pharmacology of neuropeptide Y. Pharmacol Ther (1995) 65(3):397–414. doi:10.1016/0163-7258(95)98598-K
22. Ehlers CL, Somes C, Cloutier D. Are some of the effects of ethanol mediated through NPY? Psychopharmacology (Berl) (1998) 139(1–2):136–44. doi:10.1007/s002130050698
23. Kelley SP, Nannini MA, Bratt AM, Hodge CW. Neuropeptide-Y in the paraventricular nucleus increases ethanol self-administration. Peptides (2001) 22(3):515–22. doi:10.1016/S0196-9781(01)00361-8
24. Ehlers CL, Li TK, Lumeng L, Hwang BH, Somes C, Jimenez P, et al. Neuropeptide Y levels in ethanol-naive alcohol-preferring and nonpreferring rats and in Wistar rats after ethanol exposure. Alcohol Clin Exp Res (1998) 22(8):1778–82. doi:10.1111/j.1530-0277.1998.tb03979.x
25. Misra K, Pandey SC. Differences in basal levels of CREB and NPY in nucleus accumbens regions between C57BL/6 and DBA/2 mice differing in inborn alcohol drinking behavior. J Neurosci Res (2003) 74(6):967–75. doi:10.1002/jnr.10831
26. Ehlers CL, Somes C, Lopez A, Kirby D, Rivier JE. Electrophysiological actions of neuropeptide Y and its analogs: new measures for anxiolytic therapy? Neuropsychopharmacology (1997) 17(1):34–43. doi:10.1016/S0893-133X(97)00001-8
27. Thiele TE, Marsh DJ, Ste Marie L, Bernstein IL, Palmiter RD. Ethanol consumption and resistance are inversely related to neuropeptide Y levels. Nature (1998) 396(6709):366–9. doi:10.1038/24614
28. Thiele TE, Miura GI, Marsh DJ, Bernstein IL, Palmiter RD. Neurobiological responses to ethanol in mutant mice lacking neuropeptide Y or the Y5 receptor. Pharmacol Biochem Behav (2000) 67(4):683–91. doi:10.1016/S0091-3057(00)00413-5
29. Bijak M. Neuropeptide Y reduces epileptiform discharges and excitatory synaptic transmission in rat frontal cortex in vitro. Neuroscience (2000) 96:487–94. doi:10.1016/S0306-4522(99)00594-1
30. Klemp K, Woldbye DP. Repeated inhibitory effects of NPY on hippocampal CA3 seizures and wet dog shakes. Peptides (2001) 22(3):523–7. doi:10.1016/S0196-9781(01)00345-X
31. Vezzani A, Civenni G, Rizzi M, Monno A, Messali S, Samanin R. Enhanced neuropeptide Y release in the hippocampus is associated with chronic seizure susceptibility in kainic acid treated rats. Brain Res (1994) 660(1):138–43. doi:10.1016/0006-8993(94)90847-8
32. Vezzani A, Sperk G, Colmers WF. Neuropeptide Y: emerging evidence for a functional role in seizure modulation. Trends Neurosci (1999) 22(1):25–30. doi:10.1016/S0166-2236(98)01284-3
33. Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus (1996) 6(4):347–470. doi:10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I
34. Hendry SH, Jones EG, DeFelipe J, Schmechel D, Brandon C, Emson PC. Neuropeptide-containing neurons of the cerebral cortex are also GABAergic. Proc Natl Acad Sci U S A (1984) 81(20):6526–30. doi:10.1073/pnas.81.20.6526
35. Milner TA, Veznedaroglu E. Ultrastructural localization of neuropeptide Y-like immunoreactivity in the rat hippocampal formation. Hippocampus (1992) 2(2):107–25. doi:10.1002/hipo.450020204
36. Chandler LJ, Newsom H, Sumners C, Crews F. Chronic ethanol exposure potentiates NMDA excitotoxicity in cerebral cortical neurons. J Neurochem (1993) 60(4):1578–81. doi:10.1111/j.1471-4159.1993.tb03326.x
37. Obernier JA, Bouldin TW, Crews FT. Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol Clin Exp Res (2002) 26(4):547–57. doi:10.1111/j.1530-0277.2002.tb02573.x
38. Woldbye DP, Ulrichsen J, Haugbøl S, Bolwig TG. Ethanol withdrawal in rats is attenuated by intracerebroventricular administration of neuropeptide Y. Alcohol Alcohol (2002) 37(4):318–21. doi:10.1093/alcalc/37.4.318
39. Greber S, Schwarzer C, Sperk G. Neuropeptide Y inhibits potassium-stimulated glutamate release through Y2 receptors in rat hippocampal slices in vitro. Br J Pharmacol (1994) 113(3):737–40. doi:10.1111/j.1476-5381.1994.tb17055.x
40. Decressac M, Wright B, David B, Tyers P, Jaber M, Barker RA, et al. Exogenous neuropeptide Y promotes in vivo hippocampal neurogenesis. Hippocampus (2011) 21(3):233–8. doi:10.1002/hipo.20765
41. Howell OW, Doyle K, Goodman JH, Scharfman HE, Herzog H, Pringle A, et al. Neuropeptide Y stimulates neuronal precursor proliferation in the post-natal and adult dentate gyrus. J Neurochem (2005) 93(3):560–70. doi:10.1111/j.1471-4159.2005.03057.x
42. Malva JO, Xapelli S, Baptista S, Valero J, Agasse F, Ferreira R, et al. Multifaces of neuropeptide Y in the brain – neuroprotection, neurogenesis and neuroinflammation. Neuropeptides (2012) 46(6):299–308. doi:10.1016/j.npep.2012.09.001
43. Altman J, Das GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol (1965) 124(3):319–35. doi:10.1002/cne.901240303
44. Doetsch F, García-Verdugo JM, Alvarez-Buylla A. Regeneration of a germinal layer in the adult mammalian brain. Proc Natl Acad Sci U S A (1999) 96(20):11619–24. doi:10.1073/pnas.96.20.11619
45. Taupin P, Gage FH. Adult neurogenesis and neural stem cells of the central nervous system in mammals. J Neurosci Res (2002) 69(6):745–9. doi:10.1002/jnr.10378
46. Chambers RA. Adult hippocampal neurogenesis in the pathogenesis of addiction and dual diagnosis disorders. Drug Alcohol Depend (2013) 130(1–3):1–12. doi:10.1016/j.drugalcdep.2012.12.005
47. Geil CR, Hayes DM, McClain JA, Liput DJ, Marshall SA, Chen KY, et al. Alcohol and adult hippocampal neurogenesis: promiscuous drug, wanton effects. Prog Neuropsychopharmacol Biol Psychiatry (2014) 54C:103–13. doi:10.1016/j.pnpbp.2014.05.003
48. Knapp DJ, Crews FT. Induction of cyclooxygenase-2 in brain during acute and chronic ethanol treatment and ethanol withdrawal. Alcohol Clin Exp Res (1999) 23(4):633–43. doi:10.1111/j.1530-0277.1999.tb04165.x
49. Majchrowicz E. Induction of physical dependence upon ethanol and the associated behavioral changes in rats. Psychopharmacologia (1975) 43(3):245–54. doi:10.1007/BF00429258
50. Penland S, Hoplight B, Obernier J, Crews FT. Effects of nicotine on ethanol dependence and brain damage. Alcohol (2001) 24(1):45–54. doi:10.1016/S0741-8329(01)00142-2
51. Olling JD, Ulrichsen J, Haugbøl S, Glenthøj B, Hemmingsen R, Woldbye DP. Decreased gene expression of neuropeptide Y and its receptors in hippocampal regions during ethanol withdrawal in rats. Neurosci Lett (2007) 424(3):160–4. doi:10.1016/j.neulet.2007.07.050
52. Serrano A, Parsons LH. Endocannabinoid influence in drug reinforcement, dependence and addiction-related behaviors. Pharmacol Ther (2011) 132(3):215–41. doi:10.1016/j.pharmthera.2011.06.005
53. Cippitelli A, Bilbao A, Hansson AC, del Arco I, Sommer W, Heilig M, et al. Cannabinoid CB1 receptor antagonism reduces conditioned reinstatement of ethanol-seeking behavior in rats. Eur J Neurosci (2005) 21(8):2243–51. doi:10.1111/j.1460-9568.2005.04056.x
54. Hungund BL, Basavarajappa BS, Vadasz C, Kunos G, Rodriguez de Fonseca F, Colombo G, et al. Ethanol, endocannabinoids, and the cannabinoidergic signaling system. Alcohol Clin Exp Res (2002) 26(4):565–74. doi:10.1111/j.1530-0277.2002.tb02575.x
55. Wang L, Liu J, Harvey-White J, Zimmer A, Kunos G. Endocannabinoid signaling via cannabinoid receptor 1 is involved in ethanol preference and its age-dependent decline in mice. Proc Natl Acad Sci U S A (2003) 100(3):1393–8. doi:10.1073/pnas.0336351100
56. Erdozain AM, Callado LF. Involvement of the endocannabinoid system in alcohol dependence: the biochemical, behavioral and genetic evidence. Drug Alcohol Depend (2011) 117(2–3):102–10. doi:10.1016/j.drugalcdep.2011.02.003
57. Gamber KM, Macarthur H, Westfall TC. Cannabinoids augment the release of neuropeptide Y in the rat hypothalamus. Neuropharmacology (2005) 49(5):646–52. doi:10.1016/j.neuropharm.2005.04.017
58. Cottone E, Guastalla A, Pomatto V, Campantico E, Palermo F, Magni AM, et al. Interplay of the endocannabinoid system with neuropeptide Y and corticotropin-releasing factor in the goldfish forebrain. Ann N Y Acad Sci (2009) 1163:372–5. doi:10.1111/j.1749-6632.2009.04432.x
59. Rubio M, Fernández-Ruiz J, de Miguel R, Maestro B, Michael Walker J, Ramos JA. CB1 receptor blockade reduces the anxiogenic-like response and ameliorates the neurochemical imbalances associated with alcohol withdrawal in rats. Neuropharmacology (2008) 54(6):976–88. doi:10.1016/j.neuropharm.2008.02.005
60. Rutkowska M, Jamontt J, Gliniak H. Effects of cannabinoids on the anxiety-like response in mice. Pharmacol Rep (2006) 58(2):200–6.
61. Cosenza M, Gifford AN, Gatley SJ, Pyatt B, Liu Q, Makriyannis A, et al. Locomotor activity and occupancy of brain cannabinoid CB1 receptors by the antagonist/inverse agonist AM281. Synapse (2000) 38(4):477–82. doi:10.1002/1098-2396(20001215)38:4<477::AID-SYN13>3.0.CO;2-Y
62. Faingold CL. The Majchrowicz binge alcohol protocol: an intubation technique to study alcohol dependence in rats. Curr Protoc Neurosci (2008) Chapter 9:Unit 9.28. doi:10.1002/0471142301.ns0928s44
63. Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 4th ed. San Diego, CA: Academic Press (1998).
64. Bison S, Crews F. Alcohol withdrawal increases neuropeptide Y immunoreactivity in rat brain. Alcohol Clin Exp Res (2003) 27(7):1173–83. doi:10.1097/01.ALC.0000075827.74538.FE
65. Olling JD, Ulrichsen J, Christensen DZ, Woldbye DP. Complex plastic changes in the neuropeptide Y system during ethanol intoxication and withdrawal in the rat brain. J Neurosci Res (2009) 87(10):2386–97. doi:10.1002/jnr.22049
66. Olling JD, Ulrichsen J, Correll M, Woldbye DP. Gene expression in the neuropeptide Y system during ethanol withdrawal kindling in rats. Alcohol Clin Exp Res (2010) 34(3):462–70. doi:10.1111/j.1530-0277.2009.01110.x
67. Westerlund M, Belin AC, Felder MR, Olson L, Galter D. High and complementary expression patterns of alcohol and aldehyde dehydrogenases in the gastrointestinal tract: implications for Parkinson’s disease. FEBS J (2007) 274(5):1212–23. doi:10.1111/j.1742-4658.2007.05665.x
68. Peana AT, Muggironi G, Diana M. Acetaldehyde-reinforcing effects: a study on oral self administration behavior. Front Psychiatry (2010) 1:23. doi:10.3389/fpsyt.2010.00023
69. Zimatkin SM. Histochemical study of aldehyde dehydrogenase in the rat CNS. J Neurochem (1991) 56(1):1–11. doi:10.1111/j.1471-4159.1991.tb02555.x
70. Hoover DJ, Brien JF. Acetaldehyde concentration in rat blood and brain during the calcium carbimide – ethanol interaction. Can J Physiol Pharmacol (1981) 59(1):65–70. doi:10.1139/y81-011
71. Westcott JY, Weiner H, Shultz J, Myers RD. In vivo acetaldehyde in the brain of the rat treated with ethanol. Biochem Pharmacol (1980) 29(3):411–7. doi:10.1016/0006-2952(80)90521-3
72. Quertemont E, Grant KA, Correa M, Arizzi MN, Salamone JD, Tambour S, et al. The role of acetaldehyde in the central effects of ethanol. Alcohol Clin Exp Res (2005) 29:221–34. doi:10.1097/01.ALC.0000156185.39073.D2
73. Bang S, Kim KY, Yoo S, Kim YG, Hwang SW. Transient receptor potential A1 mediates acetaldehyde-evoked pain sensation. Eur J Neurosci (2007) 26(9):2516–23. doi:10.1111/j.1460-9568.2007.05882.x
74. Handlechner AG, Hermann A, Fuchs R, Weiger TM. Acetaldehyde-ethanol interactions on calcium-activated potassium (BK) channels in pituitary tumor (GH3) cells. Front Behav Neurosci (2013) 7:58. doi:10.3389/fnbeh.2013.00058
75. Mascia MP, Maiya R, Borghese CM, Lobo IA, Hara K, Yamakura T, et al. Does acetaldehyde mediate ethanol action in the central nervous system? Alcohol Clin Exp Res (2001) 25(11):1570–5. doi:10.1111/j.1530-0277.2001.tb02163.x
76. Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature (1996) 381:415–21. doi:10.1038/381415a0
77. Baraban SC, Hollopeter G, Erickson JC, Schwartzkroin PA, Palmiter RD. Knock-out mice reveal a critical antiepileptic role for neuropeptide Y. J Neurosci (1997) 17:8927–36.
78. McBride WJ. Central nucleus of the amygdala and the effects of alcohol and alcohol-drinking behavior in rodents. Pharmacol Biochem Behav (2002) 71(3):509–15. doi:10.1016/S0091-3057(01)00680-3
79. Koob GF. Neuroadaptive mechanisms of addiction: studies on the extended amygdala. Eur Neuropsychopharmacol (2003) 13(6):442–52. doi:10.1016/j.euroneuro.2003.08.005
80. Ernst A, Alkass K, Bernard S, Salehpour M, Perl S, Tisdale J, et al. Neurogenesis in the striatum of the adult human brain. Cell (2014) 156(5):1072–83. doi:10.1016/j.cell.2014.01.044
81. Mandyam CD. The interplay between the hippocampus and amygdala in regulating aberrant hippocampal neurogenesis during protracted abstinence from alcohol dependence. Front Psychiatry (2013) 4:61. doi:10.3389/fpsyt.2013.00061
82. Pawlak R, Skrzypiec A, Sulkowski S, Buczko W. Ethanol-induced neurotoxicity is counterbalanced by increased cell proliferation in mouse dentate gyrus. Neurosci Lett (2002) 327(2):83–6. doi:10.1016/S0304-3940(02)00369-5
83. Sørensen G, Wegener G, Hasselstrøm J, Hansen TV, Wörtwein G, Fink-Jensen A, et al. Neuropeptide Y infusion into the shell region of the rat nucleus accumbens increases extracellular levels of dopamine. Neuroreport (2009) 20(11):1023–6. doi:10.1097/WNR.0b013e32832d4848
84. Cannizzaro C, Tel BC, Rose S, Zeng BY, Jenner P. Increased neuropeptide Y mRNA expression in striatum in Parkinson’s disease. Brain Res Mol Brain Res (2003) 110(2):169–76. doi:10.1016/S0169-328X(02)00555-7
85. Diana M. The dopamine hypothesis of drug addiction and its potential therapeutic value. Front Psychiatry (2011) 2:64. doi:10.3389/fpsyt.2011.00064
86. Desai SJ, Upadhya MA, Subhedar NK, Kokare DM. NPY mediates reward activity of morphine, via NPY Y1 receptors, in the nucleus accumbens shell. Behav Brain Res (2013) 247:79–91. doi:10.1016/j.bbr.2013.03.018
87. Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A (2002) 99(12):8384–8. doi:10.1073/pnas.122149199
88. Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci (2004) 24(1):53–62. doi:10.1523/JNEUROSCI.4503-03.2004
89. Riegel AC, Lupica CR. Independent presynaptic and postsynaptic mechanisms regulate endocannabinoid signaling at multiple synapses in the ventral tegmental area. J Neurosci (2004) 24(49):11070–8. doi:10.1523/JNEUROSCI.3695-04.2004
90. Herkenham M, Lynn AB, de Costa BR, Richfield EK. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res (1991) 547(2):267–74. doi:10.1016/0006-8993(91)90970-7
91. Glass M, Felder CC. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci (1997) 17(14):5327–33.
92. Childers SR, Breivogel CS. Cannabis and endogenous cannabinoid systems. Drug Alcohol Depend (1998) 51(1–2):173–87. doi:10.1016/S0376-8716(98)00075-1
93. Basavarajappa BS, Nagre NN, Xie S, Subbanna S. Elevation of endogenous anandamide impairs LTP, learning, and memory through CB1 receptor signaling in mice. Hippocampus (2014) 24(7):808–18. doi:10.1002/hipo.22272
Keywords: acetaldehyde withdrawal, neuropeptide Y expression, endocannabinoidergic system, hippocampus, nucleus accumbens
Citation: Plescia F, Brancato A, Marino RAM, Vita C, Navarra M and Cannizzaro C (2014) Effect of acetaldehyde intoxication and withdrawal on NPY expression: focus on endocannabinoidergic system involvement. Front. Psychiatry 5:138. doi: 10.3389/fpsyt.2014.00138
Received: 01 August 2014; Accepted: 18 September 2014;
Published online: 01 October 2014.
Edited by:
Marco Diana, University of Sassari, ItalyReviewed by:
Andrea Cippitelli, Torrey Pines Institute for Molecular Studies, USAMerce Correa, Universitat Jaume I, Spain
Copyright: © 2014 Plescia, Brancato, Marino, Vita, Navarra and Cannizzaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carla Cannizzaro, Department of Sciences for Health Promotion and Mother and Child Care “Giuseppe D’Alessandro”, University of Palermo, Via del Vespro 133, Palermo 90127, Italy e-mail:Y2FybGEuY2Fubml6emFyb0B1bmlwYS5pdA==;Y2FybGEuY2Fubml6emFyb0BrYXRhbWFpbC5jb20=