 Rachel D. Moloney
Rachel D. Moloney Siobhain M. O’Mahony
Siobhain M. O’Mahony Timothy G. Dinan
Timothy G. Dinan John F. Cryan
John F. Cryan- 1Laboratory of Neurogastroenterology, Alimentary Pharmabiotic Centre, Biosciences Institute, University College Cork, Cork, Ireland
- 2Department of Anatomy and Neuroscience, University College Cork, Cork, Ireland
- 3Department of Psychiatry, University College Cork, Cork, Ireland
Visceral pain is a global term used to describe pain originating from the internal organs, which is distinct from somatic pain. It is a hallmark of functional gastrointestinal disorders such as irritable-bowel syndrome (IBS). Currently, the treatment strategies targeting visceral pain are unsatisfactory, with development of novel therapeutics hindered by a lack of detailed knowledge of the underlying mechanisms. Stress has long been implicated in the pathophysiology of visceral pain in both preclinical and clinical studies. Here, we discuss the complex etiology of visceral pain reviewing our current understanding in the context of the role of stress, gender, gut microbiota alterations, and immune functioning. Furthermore, we review the role of glutamate, GABA, and epigenetic mechanisms as possible therapeutic strategies for the treatment of visceral pain for which there is an unmet medical need. Moreover, we discuss the most widely described rodent models used to model visceral pain in the preclinical setting. The theory behind, and application of, animal models is key for both the understanding of underlying mechanisms and design of future therapeutic interventions. Taken together, it is apparent that stress-induced visceral pain and its psychiatric comorbidities, as typified by IBS, has a multifaceted etiology. Moreover, treatment strategies still lag far behind when compared to other pain modalities. The development of novel, effective, and specific therapeutics for the treatment of visceral pain has never been more pertinent.
Introduction
Visceral pain is a severe form of pain that can be debilitating for the patient. Moreover, it affects a significant proportion of the population with up to 25% of people reporting visceral pain at any one time. Development of novel therapeutics is hindered by a lack of detailed knowledge of the underlying mechanisms, however progress is being made in this regard. The use of animal models has proved crucial in the advancement of our knowledge of what really is going on in visceral pain. This review aims to highlight the current state of play in the context of both preclinical and clinical research in the area of visceral pain. This review covers a broad range of research and as such in-depth details of studies is not included but is cited appropriately throughout. This review will summarize what is already known in the field and elude to future avenues yet to be explored in visceral pain research.
Visceral Pain
Visceral pain is by definition, pain sensed as arising from the internal organs of the body (1). The pain may be described as sickening, deep, squeezing, and dull. Moreover, some organs are more sensitive to visceral pain than others (2). Diseases or disorders effecting certain organs such as the liver, lungs, or kidneys are commonly not associated with any overt symptoms of pain per se but mainly symptoms that are due to altered functioning of the organ itself. Conversely, other organs are far more sensitive to damage and can elicit excruciating pain. These organs include the stomach, bladder, and ureters (2, 3).
There are multiple etiologies for pain sensed in the internal organs, including: inflammation (acute and chronic), disruption of normal mechanical processes, neoplasms (benign or malignant), alterations in neurotransmission from the viscera, and ischemia (4–8).
Interestingly, visceral pain is intriguing in that pain is commonly felt in sites distant from the location of the organ itself. This referred pain, as it is known, is a key feature of visceral pain and is used by many clinicians in the diagnosis of certain diseases (1, 3). The pattern of pain sensation in referred pain can be similar across multiple organs and disease types, i.e., disorders of the gut, bladder, and other viscera are sensed as global abdominal pain, pelvic pain, or back pain, with specific localization very difficult to identify (3, 9, 10).
Visceral pain is the most common form of pain reported in the clinic and is the most common form of pain produced by disease (1). Although visceral pain is experienced by 25% of the population at any one time (11), in many cases it is insufficiently treated as it still remains to be considered as just a symptom of an underlying disease and not a disease in its own right. Over the last decades, the unsatisfactory treatment of visceral pain has led to an immense economic and personal cost, with patients experiencing a reduced quality of life and increased work absenteeism with escalating healthcare costs (12, 13). However, more recent literature suggests that novel pharmaco-therapeutic targets such as linaclotide (14) and μ-opioid receptor agonists and antagonists, selective κ-opioid receptor agonists, anti-inflammatory drugs, serotonergic agents, bile acid modulators, and intestinal bile acid transporters are performing well in clinical trials (15). To build momentum on these advances in clinical treatments, we must strive to enhance our understanding of the underlying mechanisms of visceral pain to aid future development of novel therapeutics. To fully appreciate the complexity of visceral pain processing, we must first understand the characteristics and neurobiology of this pain modality.
Characteristics of Visceral Pain
As mentioned earlier, visceral pain perception and psychological processing is divergent to that of somatic pain (1). Importantly, there are clear distinctions which set visceral pain aside from all other pain modalities. These clinical features are crucial for the understanding of this complex physiological process. The characteristics of visceral pain were first outlined by Cervero and Laird (1) and have advanced our appreciation of this complex phenomenon. These characteristics are summarized in Table 1.

Table 1. Characteristics of visceral pain [adapted from Cervero and Laird (1)].
Types of Visceral Pain
Visceral pain is the pain associated with a wide variety of disorders including gallstones, acute pancreatitis, acute appendicitis, diverticulitis, painful functional bowel syndromes such as irritable-bowel syndrome (IBS) and functional dyspepsia (FD), inflammatory bowel disease (IBD), gastroesophageal reflux disease (GERD), interstitial cystitis/bladder pain syndrome (IC/BPS), male chronic pelvic pain syndrome, and gynecological pain [endometriosis, vulvodynia, menstrual pain, polycystic ovary syndrome (PCOS)] (16–20). Moreover, and less commonly known is that visceral pain also encompasses chronic chest pain and colic (21, 22).
Appreciating this wide array of disorders has allowed us to understand the complex nature of the pathophysiology of visceral pain but have also induced considerable hurdles when aiming to fully understand the distinct subset of molecular mechanisms which underpin this phenomenon. For the remainder of this review, we will specifically discuss gastrointestinal (GI) visceral pain.
Neuroanatomy of Visceral Pain
Knowing that the clinical characteristics of visceral pain are distinct from that of somatic pain, it implies that the neurobiology must also be distinct between these modalities. Indeed this appears to be the case with numerous groups reporting both anatomical and physiological differences (23–26).
Pathways for visceral sensation are diffusely organized both peripherally and centrally. Communication of sensory information from the GI tract to the central nervous system (CNS) occurs via vagal, pelvic, and splanchnic nerve pathways (27–29). Vagal afferents innervate the GI tract from the esophagus to the transverse colon (8). Pelvic nerves innervate the remaining parts of the colon and rectum (8). A smaller group of afferents called the splanchnic afferents whose cell bodies arise from the thoracolumbar region of the spinal cord innervate the whole GI tract (27), Figure 1.
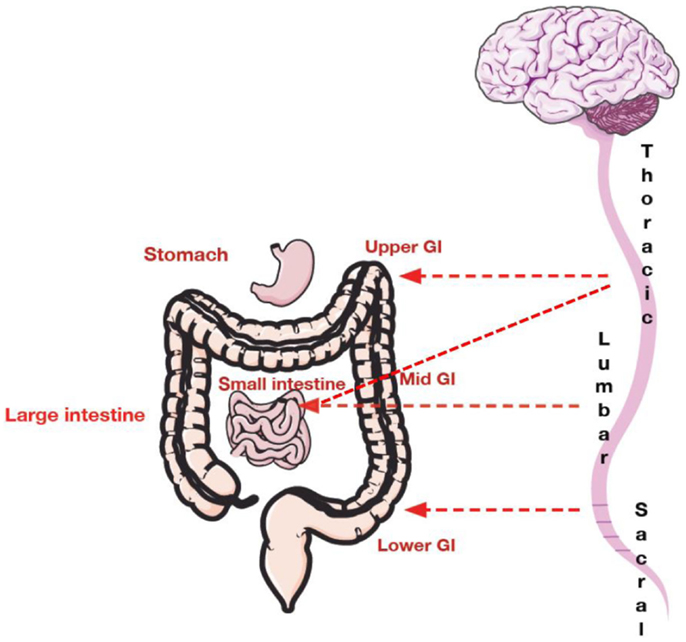
Figure 1. Spinal innervation of the gastrointestinal tract. The upper GI tract (including esophagus and stomach) is innervated with thoracic and lumbar afferents. The mid GI tract composed of the small intestine is innervated by both thoracic and lumbar afferents. The mid to lower GI tract including the large intestine is innervated by lower lumbar afferents and upper sacral afferents. The pelvic region is innervated by sacral afferents.
More specifically, primary afferent nerve fibers innervating the viscera, project into the CNS via three pathways: (1) the vagus nerve and its branches; (2) sympathetic pathways; and (3) the pelvic nerve (parasympathetic pathways) and its branches (26). Primary afferents signaling to the CNS reside primarily in the vagal nodose ganglion, which project to the nucleus tractus solitarius (NTS) located within the medulla of the brainstem (8), and in the T2–L2 and S1–5 dorsal root ganglia (30).
Visceral primary afferents have been demonstrated to enter the spinal cord and form synapses with dorsal horn neurons ipsilateral and contralateral to the site of entry. The result is extensive, diffuse CNS activation (31, 32). These axons form the postsynaptic dorsal column pathway. Interestingly, the dorsal column in itself has been shown to relay visceral nociceptive information and is now thought of as a visceral pain pathway in its own right. Numerous clinical studies have shown that lesioning the fibers of the dorsal columns significantly relieves pain and decreases analgesic requirements in patients suffering from cancer originating in the visceral organs (33, 34).
Visceral afferents terminate in laminae I, II(outer), V–VII and X in the spinal cord. Laminae I and V form part of the spinothalamic tract, laminae VII and X constitute part of the dorsal column pathway (35, 36).
Second-order processing of visceral stimuli occurs at spinal segments and brainstem sites receiving primary afferent input. Figure 2 depicts the principal visceral afferent pathways projecting to the spinal cord, and then ascending to the thalamus and midbrain. These are the spinothalamic, spinoreticular, and spinomesencephalic tracts (37, 38). The spinothalamic tract transmits sensory information from the spinal cord to the reticular formation in the brainstem and terminates in the thalamus (medial and posterior) at which point the thalamocortical fibers project to the primary somatosensory cortex (38). This tract is responsible for sensory discrimination and localization of painful stimuli and thus the reflexive, affective, and motivational properties of noxious stimulation (38, 39).
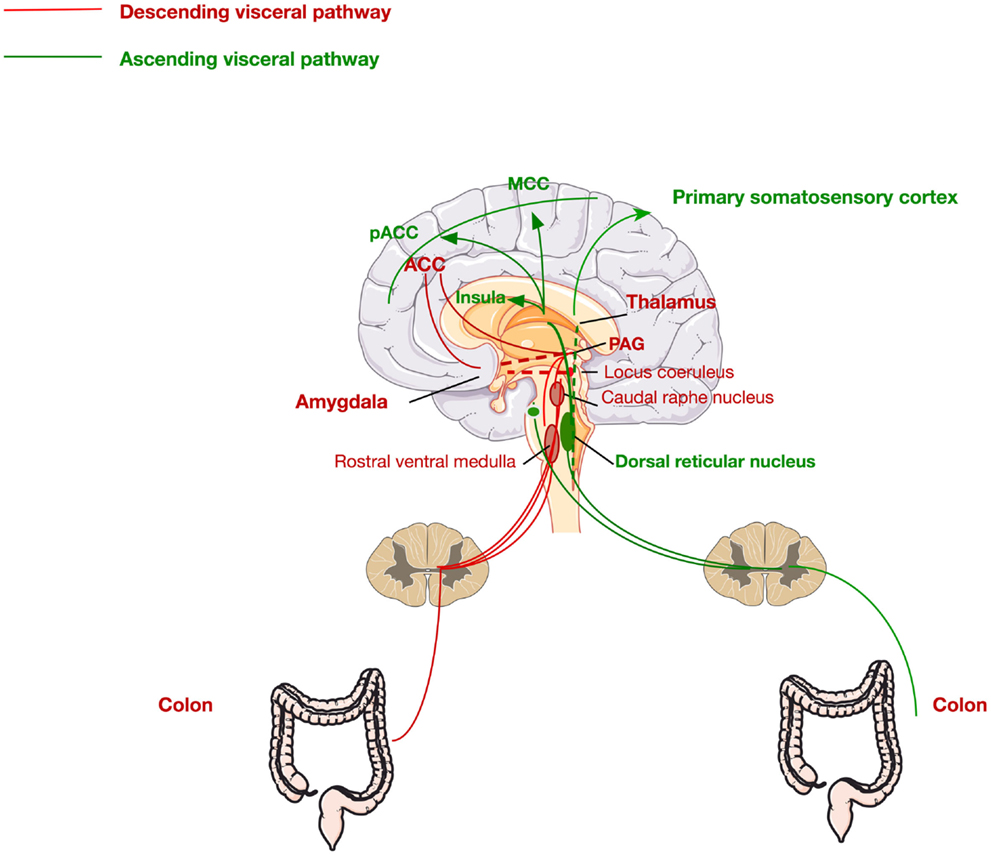
Figure 2. Ascending and descending pathways mediating visceral pain sensation. The ascending pathway for visceral pain perception from the periphery through the dorsal root ganglia via the dorsal reticular nucleus to the primary somatosensory cortex, insula, pregenual anterior cingulate cortex (pACC), and the midcingulate cortex (MCC). The descending pathway is mediated via signals from the ACC, thalamus, and amygdala to the periaqueductal gray (PAG), locus coeruleus, and raphe nucleus, returning via the rostral ventral medulla to the colon.
The brain regions innervated by these pathways, more commonly known as the pain matrix, which are activated in response to colorectal stimulation include the prefrontal cortex (dorsolateral), the insula, the thalamus, the amygdala, and the anterior cingulate cortex (ACC) (38). The ACC is a critical component in the pain matrix and is functionally divided into two discrete regions, the perigenual ACC and the midcingulate cortex (MCC), with the former involved in affect and the latter in behavioral response modification (38, 40). Moreover, the amygdala, in particular, the central nucleus of the amygdala (CeA), is a critical region in the limbic system and the pain matrix. The CeA integrates many signals for the processing of painful stimuli. It receives input from the brainstem, as well as more complex information from the thalamus and the cerebral cortex. Moreover, it has also been shown to have direct synaptic interactions with locus coeruleus (LC) neurons (41), highlighting a clear role of stress pathways in the development of visceral pain. Furthermore, the amygdala is also known to receive afferents from the spinal cord through the spino-(trigemino-)parabrachio-amygdaloid pathway (42). It is also involved in the descending pain pathway and is involved in the emotional, affective, and cognitive functions of pain processing. Numerous preclinical studies have shown the important role of the amygdala in pain processing (43–52).
This multicomponent integration of nociceptive information explains the variability in the experience and reporting of visceral pain (37, 53) and thus the difficulty in development of effective pharmacological treatments. Interestingly, diffuse noxious inhibitory controls (DNIC) are a phenomenon more commonly referred to as “pain inhibiting pain” whereby a painful stimulus is applied to a part of the body, distant from the actual site of pain, thus inhibiting neurones within the dorsal horn of the spinal cord that are actively responding to chronic (unexplained) pain as seen in visceral hypersensitivity (54). DNIC is frequently used to quantify the central pain sensitization in chronic pain patients such as in the case of IBS (55–58). IBS patients consistently show a deficit in DNIC which correlates with symptom severity. These findings elude to the hypothesis that chronic pain patients are not only hyper-sensitive to pain but they also demonstrate reduced DNIC, possibly because of dysfunction of endogenous pain inhibition systems (55).
Biochemical Mediators of Visceral Pain
Neurotransmitters, cytokines, and other mediators such as peptides and neuropeptides are thought to mediate visceral nociceptive signals from the periphery to the central pathways. Indeed, mediators released during peripheral inflammation and injury, are thought to influence spinal nociceptors resulting in increased nociceptive activity and central sensitization (27). For example, the inflammation and irritation associated with bladder infections is believed to cause the release of glutamate that sensitizes visceral primary afferents (59). Moreover, glutamatergic signaling in particular, metabotropic glutamate (mGlu) receptors and glutamate reuptake are fast becoming attractive areas of research in the context of visceral pain (60, 61). However, there are a whole host of other mediators and receptors that are involved in visceral pain processing including; neurotransmitter receptors [acetylcholine nicotinic receptors (62), cannabinoid receptors (63), opioid receptors (64, 65), GABAA, GABAB, and GABAC receptors (66, 67), glutamate (ionotropic) receptors (68), glutamate (metabotropic) receptors (69, 70), glucocorticoid receptors (47, 71)], inflammatory receptors (bradykinin receptors, cholecystokinin receptors, cytokine receptors, leukotriene receptors, prostanoid receptors, tachykinin receptors, nitric oxide signaling, cyclooxygenase, lipoxygenase) (72, 73) and ion channel receptors [transient receptor potential vanilloid (TRPV) (74), purinergic (P2X) receptors (75), voltage-gated calcium channels (CaV), voltage-gated potassium (KV) channels, voltage-gated sodium (NaV) channels] (76). Due to the vast array of mediators and pathways, a clear pathophysiology of visceral pain remains to be elucidated thus hindering drug development.
Treatment of Visceral Pain
Although most visceral pain disorders are not life-threatening (non-cancer pain), they have a considerable negative impact on the quality of lives of patients with increased psychological distress, increased work absenteeism and both sleep and sexual dysfunction (77, 78). There are currently no pharmacological treatments on the market specifically for visceral pain. This leads to persistent bouts of discomfort and possible debilitation for the patient but also results in recurring visits to clinicians, associated with a significant economic burden on both the patient and healthcare services (12). Many patients are treated with multiple drug combinations to no avail. This void of effective analgesics in the context of visceral pain is frustrating for all parties involved. The drive to develop new analgesics has started right back to the basic molecular mechanisms of which very little is known (60, 79). Basic science and animal models have proved crucial in this effort for future developments of novel visceral analgesics.
In the clinical setting, treatment of visceral pain is extremely difficult. This is due to its complex nature, in that individuals can have many different triggering factors of their visceral pain and with no known cause, effective treatment strategies are difficult to identify. Treatment can differ from patient to patient and indeed treatment of the same patient over time may also change. As a result of this, a wide variety of pharmacological tools are used including a variety of analgesics [opioids (80), non-steroidal anti-inflammatory drugs (NSAIDS) (81), benzodiazepines (82)] and others (antibiotics, laxatives, serotonin modulators) (83, 84). Moreover, patients may also be treated with antispasmodics, particularly in the case of GI visceral pain and anti-depressants as well as others (84). Due to this heterogeneous pharmacological treatment profile, there are numerous and serious side effects including constipation, sedation, vomiting, tolerance, dependence, and addiction. However, none of the above are specific for the treatment of visceral pain per se, and mainly target some other features associated with chronic pain in general. However, in recent years, promising findings are emerging from both preclinical and clinical research which are nicely reviewed in Ref. (15, 83, 85, 86).
As with all chronic/severe pain disorders, opioids form the core of pain management for visceral pain conditions. However, as mentioned earlier, this class of analgesics are associated with the most serious side effects particularly over chronic use ranging from constipation (87) to analgesic tolerance (88–90) and nociceptive sensitization (91). More worryingly is the development of opioid-induced hyperalgesia after periods of prolonged opioid use (92–94).
Over the counter analgesics are routinely used by patients suffering with visceral pain. These include NSAIDS (aspirin, ibuprofen) and paracetamol. Again as with the case of most pharmacological agents used in the treatment of visceral pain, these agents do not specifically target visceral pain and only provide mild pain relief. Moreover, as these compounds are available over the counter they can be abused by consumers and carry a host of side effects not least GI bleeding, increased stomach acid, liver disease, liver failure, and even death in some cases (95).
Serotonergic compounds such as tegaserod (5-HT4 agonist) and alosetron (5-HT3 antagonist) (96–102) have been the main route of treatment for a range of visceral pain conditions, in particular IBS. However, tegaserod has since been removed from the market due to significant cardiovascular effects as outlined by the FDA. Alosetron was also withdrawn from the market in 2000 due to life-threatening adverse GI effects, however, in 2002, it was reintroduced but its availability and use were dramatically restricted.
Recent evidence from preclinical studies has pointed to a role of the transient receptor potential (TRP) channel family in the pathophysiology of visceral pain, which may lead to future development of novel therapeutics (103, 104). Moreover, both preclinical and clinical data have shown analgesic efficacy of pregabalin and gabapentin in acute and chronic visceral pain conditions, acting both at spinal and supra-spinal levels in particular at the level of the rostral ventromedial medulla (RVM) (60, 105–108). Furthermore, the efficacy of mGlu receptors and also glial glutamate transporters have revealed themselves to be very promising targets (59, 61, 70, 109–115).
As mentioned previously, there is an unmet need for effective pharmacological therapeutics in the context of visceral pain. However, there are still major challenges to be met, not least in the fact that we still lack a clear understanding of the etiopathogenesis of visceral pain disorders.
Comorbidities of Visceral Pain
Psychiatric disturbances are the most frequent comorbidity of visceral pain (116–125). In particular, anxiety and depression are the most commonly reported comorbidities. This complex link between visceral sensation and psychological perceptions are mediated via the brain–gut axis.
Moreover, stress-related changes in bowel habit can attest to the fact that the brain can influence gut function and sensation (126). Several clinical studies have suggested that psychosocial comorbidity is a major contributor to the severity and impact on quality of life of visceral pain disorders such as IBS and somatic pain disorders such as fibromyalgia (127–131). These findings are reinforced by a considerable volume of experimental research that links stress, anxiety, and depression to altered GI sensory and motor function as well as altered pain processing (8, 132–138). Indeed, successful management of patients with visceral pain disorders requires careful attention to these psychosocial factors, often in consultation with mental health professionals.
Pathophysiology of Visceral Pain
The etiology of visceral pain is most likely multi-factorial involving biological, psychological, and social factors leading many to describe visceral pain as a biopsychosocial disorder. Due to the array of comorbidities associated with visceral pain, it is clear that both the brain and viscera play significant roles through the brain–gut axis (139–142). Moreover, the emerging role of the gut microbiota on brain signaling has now led to the concept of the microbiota–brain–gut axis (143–145). Numerous other pathways and systems feed into this complex network of communication, including the stress axis and immune system. The role of the amygdala in IBS has been extensively reviewed in the context of brain–gut axis communication (146) and numerous clinical and preclinical trials have also highlighted an important contribution of the amygdala to visceral pain processing and IBS (52, 147, 148). Here, we discuss the pathophysiology of visceral pain in terms of signaling along the microbiota–brain–gut axis, the hypothalamic–pituitary–adrenal (HPA) axis, and the immune system. Many other factors such as gender, genetics, and epigenetics are implicated in these pathways which not only exacerbate visceral hyperalgesia but may also be predisposing factors for the development of visceral hypersensitivity in later life. Furthermore, we also discuss possible future targets for visceral analgesia including the modulation of mGlu receptors in addition to glial glutamate transporters and histone deacetylation.
Stress
Stress was first described by Hans Selye almost 80 years ago, and is defined as an acute threat to the homeostasis of an organism (149). Stressors can be in the form of physical threats such as an adverse event, or psychological stressor, such as anticipation of a threat. Exposure to these stressors will elicit a sequence of physiological, emotional, and behavioral reactions that allow one to cope adequately with the situation (150–152). Behavioral effects of the stress response include increased awareness, improved cognition, and altered pain sensitivity. Physiological adaptations include increased cardiovascular function, enhanced respiratory rate, and altered metabolism, along with inhibition of feeding, digestion, growth, reproduction, and immunity (153, 154).
The complexity of the sequence of responses to stress involves a range of systems including endocrine, nervous, and immune systems. The efficiency of this response ensures that the correct behavioral and physiological changes occur so that the individual responds appropriately to the stressor presented and improves the individual’s chance of survival (155, 156). Understandably, due to the considerable complexity of the stress response, a host of regulatory mechanisms are at play to ensure the stress response is tightly regulated and does not become pathogenic to the host. These regulatory mechanisms are present at all levels of the stress response but particularly so at the neuronal and endocrine level which function to tightly regulate this adaptive process (157). However, the body can also elicit maladaptive changes in brain structure and function in response to chronic and uncontrollable stressors, thus, leaving long-lasting signatures on global wellbeing (158–160).
Dysregulation of the stress response has been associated with a plethora of disorders and diseases including chronic and visceral pain, autoimmune diseases, hypertension, affective disorders, and major depression (8, 161). Deciphering the pathogenesis of such disorders and their aberrant regulatory mechanisms will aid future therapeutic strategies for treatment and prophylaxis of stress-related disorders including stress-induced visceral hypersensitivity (154).
The Hypothalamic–Pituitary–Adrenal Axis
The HPA axis is the main stress axis in mammals and its anatomical structures are located both in the CNS and in the periphery. The major components of the stress axis are localized in the paraventricular nucleus (PVN) of the hypothalamus, the pituitary gland (anterior lobe), and the adrenal gland (154). In response to stress, corticotrophin-releasing hormone (CRH) is released and travels to the anterior pituitary gland where binding of CRH to CRH receptors (CRHR1 and CRHR2) leads to the release of adrenocorticotropic hormone (ACTH) into systemic circulation. ACTH targets the adrenal cortex to produce and secrete glucocorticoids (154). Glucocorticoids are the main effector molecules of the stress response and regulate the physiological adaptations through binding to their intracellular receptors (162, 163). Dysregulation of the HPA axis via inadequate or excessive activation, is thought to contribute to the development of a wide array of pathologies (154, 162, 164). Indeed CRH and its receptors have been extensively investigated in the context of stress and visceral pain (165–177). Taken together, it is apparent that stringent regulation of the HPA axis is essential to a normal adaptive and efficient stress response.
Stress and Irritable-Bowel Syndrome
The association between stress and psychiatric disorders, is well known, however, what exact molecular changes occur to underpin this increased vulnerability to disease is on-going (145, 178). Psychiatric disorders in addition to stressful life events are predisposing factors for the development of functional gastrointestinal disorders (FGIDs) such as IBS. IBS is one of the most common FGIDs with an estimated prevalence of 10–20% (179). Symptoms include abdominal pain, altered stool consistency and frequency, bloating, and distension. The pathophysiology of IBS has implicated stress as one the most significant risk factors for the development of the disorder (180–183). Stress at different stages throughout life, and especially early in life, can have deleterious effects on both psychological wellbeing and GI function of the host (Figure 5). Figure 5 depicts the roles of stress (vulnerability, trigger, perpetuating) in IBS pathophysiology at critical points throughout life.
Over the last decade, there has been an abundance of reports implicating stress in the onset or exacerbation of symptoms of IBS (182, 184–186). Moreover, in a preclinical setting, animal models of IBS are predominantly stress-based models (187) aimed at elucidating biomarkers of this complex biopsychosocial disorder. If we look at stress throughout our life, some critical developmental windows such as early life and adolescence are associated with the development of a wide variety of disorders, not least visceral pain disorders such as IBS. Stress, particularly during early life can manifest in many different forms including physical trauma, loss of a parent, abuse (physical/sexual), all of which have been associated with an increased risk of developing FGIDs later in life (188, 189). Furthermore, acute stressors such as sexual abuse, rape, traumatic event (near fatal event), and warfare are also risk factors for the development of IBS (122). Individuals responses to stress vary, a phenomenon thought to be based both on genetic and epigenetic mechanisms (180). The area of stress susceptibility and stress resilience is of interest across all areas of psychiatry (190, 191) and also in the context of comorbidities such as chronic pain disorders (192). Chronic stress may alter individual’s responses and play a strong role in symptom exacerbation. For example, psychosocial stressors in the form of sustained, threatening life events have been associated with onset and symptom exacerbation in IBS (181, 182, 193–195). Moreover, chronic stress has also been shown to induce adaptive changes in neuronal circuitry at the level of the CRH receptor 1 expression in the PVN of the rat hypothalamus, with reduced CRHR1 expression in comparison to acutely stressed rats after repeated homotypic stress exposures (196).
Differing coping strategies employed by patients, have also been shown to have a profound effect on symptom severity due in part to adaptation to stress (197–200). These studies highlight the high degree of catastrophizing or maladaptive coping within IBS groups. Indeed, positive coping strategies were shown to have positive effects on perceived stress levels and symptoms, however biomarkers such as cortisol remained unchanged. Furthermore, other functional somatic syndromes such as fibromyalgia are also known to exacerbate IBS symptoms (201, 202).
Psychological stressors are not the only risk factor for the development of IBS, but also physical stressors such as infection. Recent evidence from a systematic review and meta-analysis demonstrated that there was a sixfold increase in the risk of developing IBS after GI infection. Moreover, this enhanced risk remained elevated for at least 2–3 years post-infection (203, 204). The vicious cycle that stress plays in terms of vulnerability, trigger and perpetuation is most notable in patients who have developed IBS and associate particular situations, surroundings, or scenarios as provoking factors. This type of fear conditioning plays a key role in triggering stress responses to situations and contexts that by themselves are not threatening or stressful (205). In the context of IBS patients, this forward drive of conditioned fear-responses to both physical stimuli (infection) or contextual stimuli (location of stressful event) may be a significant factor in symptom chronicity (182). Using this knowledge and applying it to animal models will be discussed in later sections (206).
Microbiota and Visceral Pain
The Microbiota–Brain–Gut Axis
The bidirectional signaling network that exists between the GI tract and the brain is vital for maintaining homeostasis and is regulated at the neural [both central and enteric nervous systems (ENS)], hormonal, and immunological levels. Perturbation of these systems results in alterations in the stress response and overall behavior (143, 207). The high rate of psychiatric comorbidities with GI disorders and vice versa (208–210) are further evidence of the importance of this network of communication. The brain–gut axis is by no means a new discovery however its role in many disorders outside of gastroenterology has become an area of intense research. Moreover, advances in biomedical research have allowed us to elucidate the role of the gut microbiota community in signaling along this axis, which is now more commonly referred to as the microbiota–brain–gut axis (Figure 3). The impact of the microbiota–brain–gut axis has become a rapidly advancing research topic encompassing broad domains of biomedical research including neuroscience, psychiatry, gastroenterology, and microbiology.
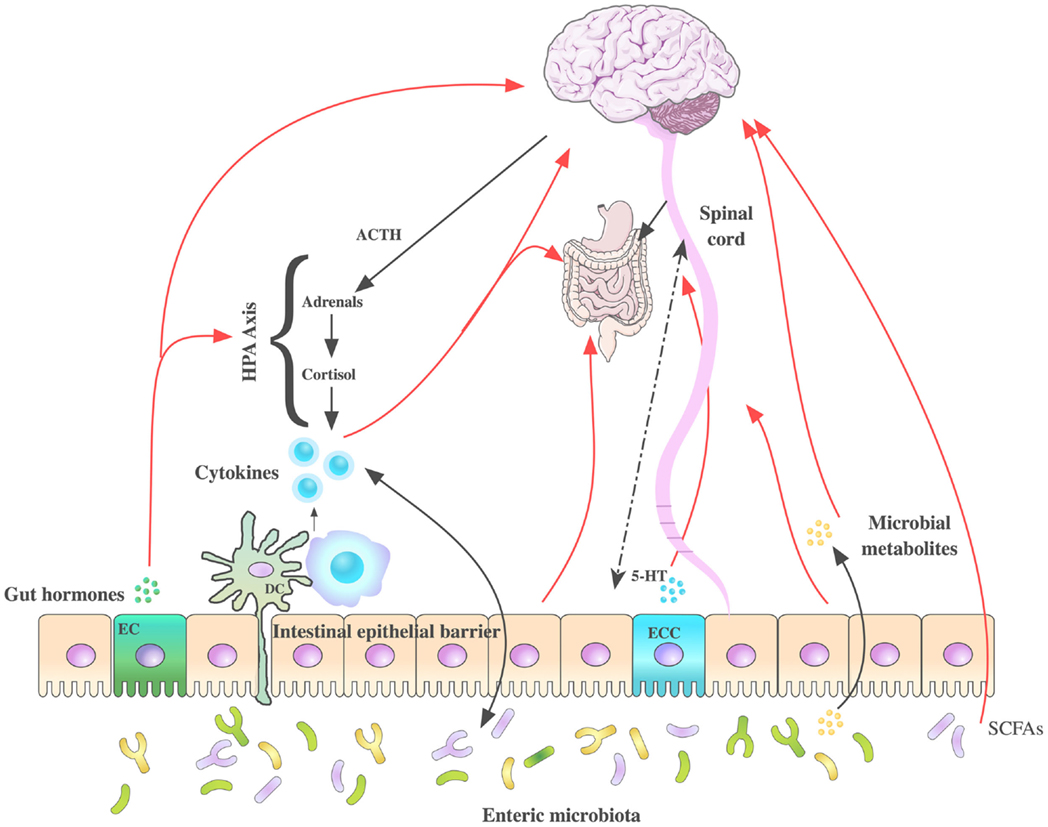
Figure 3. Routes of communication along the microbiota– brain–gut axis. Several pathways have been proposed to understand the communication between the intestinal microbiota and brain function, some of which have been summarized in this figure. These include neuroendocrine (hypothalamic–pituitary–adrenal axis), immune system (neuromodulating cytokines), enteric nervous system, autonomic nervous systems (vagus nerve), and spinal afferents. 5-hydroxytryptamine (5-HT) is produced by enterochromaffin cells in the GI tract. Gut microbes produce tryptophan-related metabolites, gut hormones, short chain fatty acids (SCFAs), and neurometabolites GABA, noradrenaline, and dopamine potentially modulating CNS function. Stress can influence the microbial composition of the intestine through the release of stress hormones (corticosterone/cortisol) or sympathetic neurotransmitters that in turn can influence gut physiology and alter the microflora balance. DC, dendritic cell; EC, enteroendocrine cells; ECC, enterochromaffin cells.
The classical construct of the brain–gut axis, describes the way in which complex bidirectional signals can transmit between the CNS and the GI tract. This axis is critical for preserving gut homeostasis, dysregulation of which has been implicated in an array of disorders and disease states including gut inflammation, chronic abdominal pain syndromes, and eating disorders (143–145, 210–217). The brain–gut axis is responsible for some of our most basic functions such as facilitating the central regulation of digestive function. Indeed, the role of this axis is a well-developed concept in the area of food intake and satiety (218, 219) and more recently obesity (220, 221).
Moreover, the role of the microbiota–brain–gut axis in the stress response via modulation of the HPA axis has been investigated by numerous independent research groups (222–228). Furthermore, many forms of psychological stress have been shown to alter the gut microbiota fingerprint with many prebiotics and probiotics showing beneficial effects against the negative impact of stress. Thus, it is now acknowledged that the gut microbiota themselves are critical mediators of information along this bidirectional communication network (145, 207, 211, 213, 229, 230).
Indeed, in IBS patient cohorts, numerous independent research groups have shown distinct gut microbiota populations when compared to healthy controls (231–236). This was recently reviewed by Mayer and colleagues (237). Moreover, probiotic interventions appear to be beneficial to IBS patients (238).
Manipulation of the gut microbiota through the use of probiotic and prebiotics treatments have shown that by augmenting so-called “good bacteria” such as Bifidobacteria and Lactobacillus, in the gut, visceral hypersensitivity can be reversed in preclinical models. A mixture of eight probiotic bacteria strains (VSL#3) was shown to have protective effects against development of visceral hypersensitivity in the neonatal maternal separation model. Moreover, TPH1, tryptophan hydroxylase 1, the gene for the enzyme responsible for synthesizing serotonin, a key neurotransmitter involved in IBS treatment, was markedly up-regulated by neonatal maternal separation and this effect was reversed by VSL#3 intervention (239). Moreover, the same cocktail of probiotics was shown to prevent visceral hypersensitivity induced by inflammation via intra-colonic instillation of 4% acetic acid when given prophylactically (240). Bifidobacterium species particularly, Bifidobacterium infantis 35624 has been shown to be particularly effective at ameliorating visceral hyperalgesia in both stress-induced visceral hypersensitivity and colitis (241–243). Moreover, Lactobacillus species have also displayed efficacy in visceral pain models (244–247).
Furthermore, antibiotic-induced visceral hypersensitivity again underpins a role of the gut microbiota in the pathophysiology of visceral pain (244, 248). Interestingly, rifaximin, a semisynthetic, non-absorbable antibiotic that demonstrates no clinically relevant bacterial resistance has also shown positive effects in the treatment of IBS (249–257). These findings may seem contradictory, however, rifaximin is particularly efficacious in cases of small bowel bacterial overgrowth found in IBS patients. These findings add to the growing literature that microbiota dysfunction may be a key player in the pathophysiology of IBS and may lead to future novel therapeutic interventions.
Immune System and Visceral Pain
The immune system and thus inflammation have long been associated with psychiatric disorders, in particular, depression (258–260) and chronic pain disorders (261). Depression is a common comorbidity of visceral pain, as discussed earlier, so it is not surprising that a common mechanism such as neuroinflammation may be at play. The immune system is a critical component of the microbiota–brain–gut axis and plays vital roles in maintaining homeostasis in the nervous systems and GI tract (262). Moreover, direct communication occurs between the immune system and the HPA axis, autonomic nervous system (ANS) and ENS (263–267). These integrated pathways are all known to be involved in the pathophysiology of visceral pain, and thus it is not unforeseen that the immune system plays a key role in the development of visceral hypersensitivity.
Microglia and Visceral Pain
Microglia represent the first line of defense for the CNS, acting as a sensor for pathological events (268). The process of central sensitization and the subsequent changes in synaptic plasticity has long been thought to play a major role in nociceptive processes both in the context of chronic pain as well as acute, in both somatic and visceral modalities (269, 270). In the last decade, the role of microglia, both spinal and supra-spinal, has become an area of interest in the context of nociception (271–273). In animal models of both inflammatory and neuropathic pain, activation of microglia is a key step in the onset and maintenance of hypersensitivity and allodynia (274–279).
The role of spinal microglia in visceral pain has only recently been addressed and reviewed nicely by Lu (269). Saab and colleagues were first to report increased activated microglia in a rat model of chronic visceral hyperalgesia, namely the neonatal colon irritation model (280). Moreover, minocycline, a second-generation tetracycline compound known to interrupt microglia activation and its associated pro-inflammatory response, reversed the visceral hypersensitivity in adulthood (269, 280).
Furthermore, enhanced microglia activation was reported in the hippocampus, in a model of trinitrobenzene sulfonic acid (TNBS)-induced colitis, concomitant with increased tumor necrosis factor-α (281). However, this study did not assess nociceptive behavior, but one could predict visceral hyperalgesia as observed by others in the TNBS-model (282). More recently, it was shown that the activation of spinal microglia plays a critical role in the initiation and maintenance of visceral hypersensitivity in the TNBS-induced chronic pancreatitis (CP) rat model (283). Intrathecal injection of minocycline attenuated visceral hypersensitivity and phospho-p38 levels in CP rats.
In other non-inflammatory models of visceral hyperalgesia, it has also been shown that chronic psychological stress leads to microglia activation in the lumbar spinal cord (284). Moreover, this stress-induced increase in activated microglia could be blocked by SB203580, a p38 inhibitor or minocycline. Other compounds known to alter the microglia phenotype such as FKN, a microglia activator, was shown to induce visceral hypersensitivity in naïve rats when administered spinally, an effect which was blocked by minocycline (269), thus adding further evidence to support the role of spinal microglia in mediating stress-induced visceral hypersensitivity (269). Taken together, these studies suggest that microglia may play an important role in the pathogenesis of visceral pain.
However, it is also pertinent to note that exact role of inflammation and indeed micro-inflammation still remains controversial in the context of IBS. Indeed, no effect of anti-inflammatory drugs has been shown in IBS such as prednisolone (285).
Toll-Like Receptors as Novel Therapeutic Targets for Visceral Analgesia
Toll-like receptors (TLRs) are a family of pattern-recognition receptors of the innate immune system (Figure 4). There are 10 human TLRs and 12 mouse TLRs (286). TLR signaling consists of at least two distinct pathways: (1) the MyD88-dependent pathway, which leads to a pro-inflammatory phenotype, and (2) the MyD88-independent pathway, which leads to the production of interferon-β and the maturation of dendritic cells. The MyD88-dependent pathway is common to all TLRs, except TLR3 (287). TLRs are important players in the maintenance of mucosal and commensal homeostasis within the gut via innate host defense mechanisms. Intestinal dysbiosis and inflammation underlie several disease states affecting the GI tract such as IBS and IBD (288, 289). Indeed, reports have shown the involvement of peripheral TLR4 in patients suffering from IBS (290, 291) and in animal models of visceral pain (289, 292, 293). Moreover, the presence of TLR4 in the ENS and in the dorsal root ganglia indicate a role for TLR4 in the transmission of sensory information from the GI tract (294, 295). Furthermore, TLR4 is also expressed within the CNS, predominately in microglia (296), which have been discussed above and their role in visceral pain. Moreover, it is now emerging as a possible therapeutic target for neuropathic pain (297). Taken together, these findings suggest TLR4 as a promising novel target for the treatment of visceral pain.
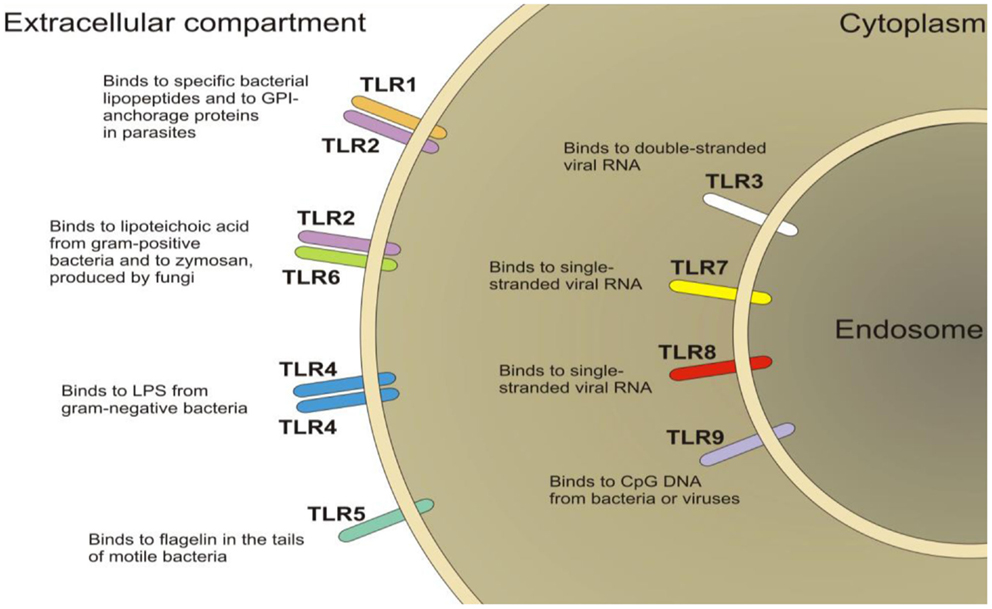
Figure 4. Schematic of the localization of toll-like receptors (TLRs). TLRs are located on the plasma membrane (TLR1, TLR2, TLR4, TLR5, TLR6, TLR10) with the exception of TLR3, TLR7, TLR8, and TLR9, which are localized in the endosomal compartment.
Interaction of TLRs and Opioid Receptors
In recent years, it has been shown using in vivo, in vitro, and in silico techniques that members of each structural class of opioids (μ, κ, δ) activate TLR4 (298). Moreover, opioid antagonists such as naloxone and naltrexone non-stereoselectively block TLR4 signaling (299, 300). Modulation of TLR4 expression/function by acute blockade of TLR4, genetic knockout of TLR4, or blockade of TLR4 downstream signaling each lead to a potentiation of the magnitude and duration of opioid analgesia (301). These effects are thought to be mediated at both spinal and supra-spinal sites (299). Given the breadth of opioids now documented to interact with TLR4, many off-target opioid effects previously attributed to unilateral opioid action at classical neuronal opioid receptors might in fact result, at least in part, from the duality of opioid actions at TLR4 (301).
Gender and Visceral Pain
Sex differences in pain sensitivity has been a topic of debate for many years and was recently reviewed by Mogil (302). More recently, gender differences in visceral pain and in particular IBS have been reviewed in Ref. (303) and indeed the contribution of sex hormones (304). Moreover, we can now appreciate that gender differences are also apparent in analgesic response (305). Many forms of visceral pain due to their nature are especially prevalent in women, i.e., pain associated with reproductive function (menstruation pains, pain of child birth, or postmenopausal pelvic pain). IBS is a disorder predominated by females (female to male ratio ~ 2:1) which is in parallel with women also being more susceptible to stress-related disorders (306, 307). Indeed, many studies have reported sex differences in the stress response itself and stress-induced pain modulation (8, 308). However, in the case of animal studies, the picture is not as clear. This is due in part to a bias for using male rodents in most preclinical studies addressing the role of stress modulation on visceral hypersensitivity (134, 135, 289, 309–311).
Sex/gender can influence the brain–gut axis, at an array of sites which logically will effect subsequent clinical outcomes including response to behavioral and drug therapies in patients suffering with visceral hypersensitivity (312). A plethora of factors including mood, stress, gender role, hormones, as well as inflammatory mediators modulate the brain–gut axis (313). As a result, the explanation for gender-related differences in visceral pain is likely multi-factorial involving environmental, psychological, and biological (sex) influences (312, 314, 315).
Studying gender differences in any context comes with significant difficulties, not least in the context of visceral pain disorders. Due to the greater number of women who are diagnosed with these disorders, there is often insufficient power to detect differences in the etiologic factors and treatment response by gender or sex. For example, several studies examining the effectiveness of cognitive behavioral therapy in reducing symptom distress have included only women (316, 317) or were disproportionately composed of women (318). In particular, the smaller number of men in drug trials leaves many studies underpowered to examine gender differences (313).
The variability in reproductive hormones such as estrogen and progesterone, across the menstrual cycle as well as significantly reduced ovarian function during and after menopause may explain changes in GI motility and visceral sensitivity (319). This has been reported in several studies documenting the impact of menstrual cycle on GI symptom reporting including visceral pain (320–323). The most consistent finding is that, many women with and without FGIDs experience an increase in GI symptoms including visceral pain during the late luteal and menses phases of the cycle relative to other cycle phases (313, 323–325). To add to the complexity, is the addition of pharmacological agents known to impact on the menstrual cycle and associated functions, such as oral contraceptive use and hormone replacement therapy. Moreover, the presence of other reproductive organ problems unique to women such as dysmenorrhea can also confound such studies.
It has been well established that adult females have higher basal and stress levels of ACTH and corticosterone than do males (326–330). Moreover, there is convergent lines of evidence reporting that gonadal hormones, specifically the estrogens, are important regulators of the HPA axis. Indeed, estrogen receptors α and β themselves have been shown to increase CRH expression (8, 331, 332). Females have higher CRH, ACTH, and corticosterone levels during proestrus (333), the phase of the cycle in which estradiol levels are highest, than during other phases of the estrous cycle (334–336). Fluctuations in gonadal hormones specifically, estradiol, during the menstrual cycle can also lead to changes in the neurotransmitter systems, in particular, the serotonergic and glutamatergic systems. Interactions between these systems have implications in the etiology and treatment of stress-related disorders and pain circuitry (142, 307, 337–340). Moreover, gonadal hormones have been shown to significantly affect visceral sensitivity in animal models, however some conflicting results have also shown no effect again highlighting the complexity of sex differences in pain processing both in a preclinical and clinical setting (173, 341–344). Interestingly, the gender of the experimenter was also shown to have a role in pain responses in preclinical models (345). Taken together, it is clear that the role of gender and/or sex in the pathophysiology of visceral pain remains a complex phenomenon which requires further investigation.
Glutamate and Visceral Pain
Ionotropic Glutamate Receptors and Visceral Pain
N-Methyl-D-aspartate (NMDA) receptors are not only known for their important role in excitatory synaptic transmission but also for their role in pain (346). Glutamate found in vagal and spinal afferents contributes to nociceptive signaling via NMDA and non-NMDA receptors (347). Though evidence for direct links between glutamate and visceral nociception is lacking, NMDA receptor antagonists have been shown to reduce the response of vagal and pelvic afferents in the colon and other viscera to mechanical stimuli (348). A study by Coutinho and colleagues (349) investigated the role of glutamate receptors in the RVM in visceral hyperalgesia using a colonic irritation model. They found that activation of NMDA receptors by colonic inflammation facilitated visceral hyperalgesia while non-NMDA receptors mediate inhibitory effects (349). Moreover, NMDA receptors may also be involved in the transmission of visceral nociception in the non-inflamed gut (350). It was observed that activated peripheral NMDA receptors in the colon caused the release of pro-inflammatory peptides, calcitonin gene-related peptide (CGRP) and substance-P, which have significant roles in the mediation of chronic and severe pain (350).
Metabotropic Glutamate Receptors and Visceral Pain
Metabotropic glutamate receptors with the exception of mGlu6 receptor are expressed in all areas of the pain matrix from the spinal cord to supra-spinal sites. The action of mGlu receptors can be pro-nociceptive or anti-nociceptive depending on the subtype and site of action (351).
In the last decade, the role of mGlu receptors has gained attention in the realm of visceral pain. To our knowledge, the first study explicitly investigating the role of these receptors in visceral nociceptive processes was performed by Chen et al. (352). They found that antagonism of group 1 mGlu receptors with LY393053 reduced nociceptive behaviors in the mouse acetic acid writhing test. Furthermore, administration of a group 1 mGlu receptor agonist (S)-3,5-dihydroxyphenylglycine (DHPG) into the CeA by microdialysis increased the responses to innocuous visceral stimulation; an effect that was reversed by a reactive oxygen species (ROS) scavenger phenyl-N-t-butyl nitrone (PBN) and a superoxide dismutase mimetic (TEMPOL). In the same study, mGlu receptor 1 antagonist LY367385 was also found to decrease the responses to visceral stimulation (353).
Moreover, work by Lindstrom and colleagues investigated specifically the role of mGlu5 receptor in a rat model of visceral hypersensitivity (111). Here they found that mGlu5 receptor antagonism with MPEP [2-Methyl-6-(phenylethynyl)-pyridine] and MTEP [3-(2-Methyl-1,3-thiazol-4-yl)ethynyl]pyridine was sufficient to reduce the visceromotor response (VMR) in conscious Sprague-Dawley (SD) rats without altering colonic compliance. Moreover, mGlu5 receptor antagonism reduced colorectal distension (CRD)-evoked increases in heart rate and blood pressure (111). In this study, the effects seen could not be conclusively due to a peripheral or central site of action. Indeed, it has also been found that noxious colonic stimulation increases the number of Fos-positive neurons in the dorsal horn of the thoracic and lumbar spinal cord. Moreover, pre-treatment with MPEP significantly attenuated this (354).
Visceral pain originating from the bladder has also been shown to be mediated via mGlu receptors; specifically mGlu receptor 5 activation in the CeA induces bladder pain sensitization by increasing CeA output, an effect that was reversed by intra-amygdala MPEP treatment and lentivirus-mediated conditional disruption of mGlu5 receptor in the CeA (70).
Glutamate Transporters and Visceral Pain
Due to the negative side effects (psychomimetic activity) seen with modulation of ionotropic receptors, compounds which target these receptors are not suitable for long-term treatment of pain. To redress this and in the drive to develop better analgesics, other mechanisms such as glutamate reuptake may provide more effective treatments in controlling glutamate neurotransmission and thereby exert anti-nociceptive effects (109).
One of the first clear demonstrations that glutamate transport [via excitatory amino-acid transporters (EAATs)] may be implicated in pain processing was performed by Liaw and colleagues where they showed that selective inhibition of glutamate transporters with dl-threo-β-benzyloxyaspartate (TBOA) and dihydrokainate (DHK) produced a dose-dependent spontaneous nociceptive behavior response. These behaviors included licking, shaking, and caudally directed biting (355). Moreover, TBOA administered intrathecally was also found to induce visceral nociceptive behaviors in naïve rats (114). To further unravel the role of glutamate transport in visceral nociception, in particular stress-induced visceral hypersensitivity, the neuroprotective drug riluzole known to activate glutamate reuptake, was investigated to see whether it could attenuate visceral hypersensitivity induced by maternal separation of rats. It was found that riluzole reduces visceral hypersensitivity in stressed animals only, having no effect in non-separated animals. Moreover, EAAT-1 expression was also found to be reduced in the lumbar region of the spinal cord in hyper-sensitive animals. As mentioned previously, riluzole does not affect visceral sensitivity in naïve animals, further emphasizing the role of glutamate transport in pathological pain (114).
EAAT-2 is the main glial transporter for glutamate reuptake and its experimental over-expression in animal models has recently been found to be effective in reducing visceral pain (59, 112, 113). A study by Lin and colleagues (112) found the over-expression of EAAT-2 with the use of cephalosporin antibiotic ceftriaxone, in wildtype mice, attenuated the visceral nociceptive response to CRD. Moreover, similar effects were seen in the EAAT-2 overexpressing transgenic mouse (112). To further support these findings, systemic and intrathecal administration of DHK, a selective EAAT-2 inhibitor blocks the ceftriaxone-induced attenuation of visceral pain (112, 113). In subsequent studies performed by the same group assessing the efficacy of glutamate reuptake in animal models of colitis, it was shown that adeno-associated virus-mediated EAAT-2 over-expression was effective to mitigate VMR to 60 mmHg CRD (113). Interestingly, overexpression of EAAT-2 has been shown to reduce bladder nociception (59). However, colon irritation may affect afferents innervating the bladder thus giving a plausible mechanism for cross-organ sensitization (59). In the same study, it was also found that enhanced expression of EAAT-2 by ceftriaxone also reduced the VMR to bladder distension caused by colonic irritation (59).
As stated earlier, stress is one of the main predisposing factors for the development of visceral pain. Interestingly, glutamate transport has also been shown to be altered due to stress, with both early-life stress and stress in adulthood both showing significant alteration in EAAT expression (114, 115). Taken together, these findings provide evidence for an important role of glutamate transport in visceral pain states (271, 356). Moreover, glutamatergic signaling is critical to the process of central sensitization. This term describes the way in which excessive glutamatergic function within neurons and circuits, in particular pain circuits, leads to increases in membrane excitability and synaptic efficacy as well as reduced inhibition. These changes in synaptic function manifest as altered plasticity within the somatosensory nervous system in response to activity, inflammation, and neural injury (357).
GABA Receptors and Visceral Pain
γ-Aminobutyric acid (GABA), the major inhibitory neurotransmitter in the CNS, plays an important role in anti-nociception. GABA is the key player acting at inhibitory synapses where it exerts its effect by binding to it respective receptors both pre- and post-synaptically. This binding causes conformational change and subsequent opening of the ion channel. The direction of the flow of ions (in/out) and their charge (+/−) result in a negative change in the transmembrane potential, causing hyperpolarization. To date, two classes of GABA receptors are known: (1) GABAA receptors are ligand-gated ion channels, and (2) GABAB receptors are metabotropic receptors, which are G protein-coupled receptors.
Pharmacological modulation of the GABAA receptor through the use of agonists and antagonists indicate that modulation of these circuits within the spinal cord has important implications for pain processing (358–360). However, these analgesic effects particularly seen with benzodiazepines may not be purely anti-nociceptive actions and maybe more non-specific centrally mediated effects.
GABA’s anti-nociceptive effects are thought to be mediated by GABAB receptors, which are ideally located in both the brain and spinal cord with ubiquitous expression. Moreover, GABAB receptors have also been implicated in a whole host of GI functions, such as altering GI motility and visceral sensation (361). The mostly widely used GABAB receptor agonist, baclofen has been shown to produce anti-nociception in numerous rat models of visceral pain (67, 362–365). Moreover, subcutaneous injection of baclofen was shown to prevent behavioral responses to bladder pain (362). Likewise, intrathecal administration of baclofen increases the sensitivity threshold to CRD (363). Furthermore, intravenous administration of baclofen attenuated CRD-evoked increases in arterial blood pressure and heart rate (67, 364).
Intraperitoneal injection of baclofen was shown to alter the expression of early immediate genes such as c-fos in the lumbosacral spinal cord after intra-colonic mustard oil-induced inflammation (366, 367). Moreover, it has also been shown via electrophysiological examination that GABAB receptor agonists have the ability to modulate responses of vagal mucosal and muscle afferents innervating particular parts of the GI tract, specifically, the esophagus and proximal stomach (368–370). More recently, it has also been shown that baclofen dose-dependently attenuates responses of mechanosensitive pelvic nerve afferents to noxious CRD (367, 371). This data provide evidence that GABAB receptor agonists may exert their positive effects by acting at peripheral sites.
However, it is important to note the effects of GABAB receptor agonism, other than its anti-nociceptive effect, are undesirable centrally mediated effects, including sedation, respiratory depression, drug tolerance, and motor deficiency, thus, its potential therapeutic use could be significantly curtailed (367). Interestingly, GABA receptor pharmacology is complex with dimerization required for many subunits to form a functional receptor. Moreover, a number of splice variants of the GABAB1 subunit (GABAB1a, GABAB1b, GABAB1c, and GABAB1d) have been identified both in rat and human tissues, and have been found to be differentially expressed depending on the tissue type (372, 373).
Indeed GABA analogs such as gabapentin and pregabalin have also shown efficacy in preclinical models of visceral hypersensitivity (105, 106, 108, 374–377) and moreover, pregabalin has undergone clinical trials for painful CP (107). However, these compounds may exert their effects indirectly in the GABAergic system, as their main mode of action is on α2δ subunits of CaV (375). The emergence of positive allosteric modulators of GABAB may provide a novel therapeutic target for treatment of visceral pain disorders (66).
Genetic and Epigenetic Regulation of Visceral Pain
Over the last two decades, the field of pain genetics has explored the influence of genetic make-up on pain perception and processing. Recent work by Camilleri and others has described the role of genetics in IBS (378–384). Genome-wide association (GWA) studies amongst others have led to the elucidation of specific genetic alterations in IBS such as Nav1.5 (385), GPBAR1 (G protein-coupled bile acid receptor 1) (386), KDELR2 (KDEL endoplasmic reticulum protein retention receptor-2) and GRID2IP [glutamate receptor, ionotropic, delta 2 (Grid2) interacting protein] (381), NXPH1 (neurexophilin 1), and CDC42 (cell division control protein 42 homolog) (387). These studies are illustrative examples of how research in the genetic area can contribute to the achievement of better general knowledge in the visceral pain field.
Strain differences have been shown in many behavioral phenotypes including anxiety and depression (388, 389). Moreover, differences due to background strain have also been shown for somatic nociception (390–392). However, there is a dearth of information regarding strain differences in basal visceral sensitivity as opposed to inflammatory-induced visceral sensitivity. To our knowledge, the Wistar Kyoto (WKY) rat strain and the Flinders Sensitive Line rat strain are the only well-validated models of genetic predisposition to visceral hypersensitivity (132, 393, 394). This highlights the need for more comprehensive testing in particular assessment of visceral pain in animal models and elucidating the underlying genetic mechanisms. Moreover, the field of pain epigenetics has progressed our simplistic view of one gene, one protein, one function, to a more complex view of gene–environment interactions.
The term epigenetics refers to processes that lead to stable and/or heritable changes in gene function without any concomitant DNA sequence changes (395). Examples include DNA methylation, histone modification, and chromatin remodeling. The vast majority of work investigating epigenetic mechanisms in pain processing center around histone acetylation and DNA methylation. Pharmacological interference with the process of histone acetylation can affect pain behavior, with both systemic and intrathecal administration of histone deacetylase (HDAC) inhibitors having analgesic effects in models of inflammatory pain (396–398). In one study, this effect was shown to be mediated by expression changes of the mGlu2 receptor in both dorsal root ganglia (DRG) and spinal cord (399). Indeed, histone acetylation has been implicated in stress-induced visceral hypersensitivity and HDAC inhibitors have also shown efficacy in a model of visceral pain (69, 71, 400–402). The hypothesis that IBS could be transferred to future generations has recently been investigated (403) and discussed (404).
Similar influences could be shown in the case of DNA methylation and its reader molecule MeCP2. The methyl binding protein MeCP2 has been shown to promote abnormal up-regulation of a group of genes in inflammatory pain conditions. In rats, its usually repressive function appears to be curtailed through phosphorylation after injection of Complete Freund’s adjuvant (CFA) into the ankle joint (405), an effect thought to be partly dependent on intact descending serotonergic input into the spinal dorsal horn (398, 406). Recently, it was shown that chronic stress was associated with increased methylation of the Nr3c1 (glucocorticoid nuclear receptor) promoter and reduced expression of this gene in L6–S2 region of the spinal cord which was associated with visceral hypersensitivity in rat model (401).
Finally, recent speculation has implicated that the mechanisms and indeed pathways by which the gut microbiota may communicate with the CNS may be due in part to epigenetic processes (407).
Experimental Models of Visceral Pain
Colorectal Distension
Colorectal distension is the most widely used method to assess visceral sensation both preclinically and clinically. This technique involves insertion of a balloon into the colorectal cavity of the human subject or animal, and with the aid of a barostat, distending in a repeated or ascending phasic manner. Although the main premise of the technique is essentially identical, the way in which it is performed varies between laboratories and researchers. This can be due to numerous factors including the model employed; whether it be human, rat, or mouse, the parameter of interest; baseline visceral sensation, pain threshold, and tolerance to painful stimuli. CRD has been characterized in humans, rats, and mice with the bulk of the work being performed in rats due to ease of use and robust reproducibility (408).
Monitoring of Visceral Pain in Rodents
In 1988, Ness and Gebhart were the first to describe a technique used to assess visceral sensitivity in the preclinical setting. The technique was based on the assessment of pseudoaffective and behavioral responses to controlled isobaric distensions of the GI tract. This has now become the mainstay for assessing visceral pain both clinically and preclinically (409). CRD in rats induces an array of autonomic and behavioral outputs termed pseudoaffective reflexes. These reflexes include; alterations in blood pressure and heart rate, passive avoidance behaviors, and contraction of the abdominal musculature (8), the latter of which is more commonly referred to as the VMR. VMR is the most widely used parameter of the visceral pain response (8, 408).
In the last decade, the use of electromyographic (EMG) signals and its numerous applications have allowed many to assess visceral sensitivity in conscious animals. This procedure allows recording from electrodes which are implanted in the musculature and externalized through the skin, primarily on the abdomen or neck (410–412). They can also be connected to radiotelemetric implants in the abdominal cavity (413, 414). Moreover, the development of manometric recordings measuring changes in the pressure of the balloon inserted into the GI tract, have also allowed for VMR to be assessed in freely moving animals (134, 175, 311, 408, 415, 416). Other groups have implemented other indirect approaches such as manual scoring of the abdominal withdrawal reflex (408, 417), operant behavioral assays (409), and functional brain imaging tools such as functional MRI (44, 418).
Animal Models of Stress-Induced Visceral Pain
Stressful episodes during critical windows of development can have long-lasting effects on the host. Stress occurring during the perinatal period has been linked to the development of psychiatric disorders such as schizophrenia and autism spectrum disorders (419, 420). The early postnatal period is a stress hypo-responsive period during which time there is an intense phase of neuronal growth and myelination (421). Stress during this critical time point has been linked to the development of both somatic and psychiatric phenotypes in preclinical models, including IBS (135, 422, 423). The adolescent period is also a time of neuronal restructuring and is fundamental to precise development of the CNS (215). This developmental period is also the peak time for the onset of numerous psychiatric disorders including schizophrenia, substance abuse, and mood disorders (424). Stress in adulthood can have profound effects on its host. Both acute and chronic stressors can elicit detrimental impacts on physical as well as mental wellbeing. Taken together, it is clear that stress at critical points during our life time can have lifelong effects on the host. Overcoming the effects of these stressors is based on the individual’s HPA axis ability to adapt and overcome such insults, however, in the genetically predisposed individual, this feat maybe too high. Aberrant development of the HPA axis can prove detrimental and exhibit itself in the form of both psychiatric and somatic disorders. The importance of animal models in the search for underlying molecular mechanisms and future development of novel therapeutics has never been more pertinent (8, 425–431). Here, we review the current stress-based models in the context of IBS (Figure 5).
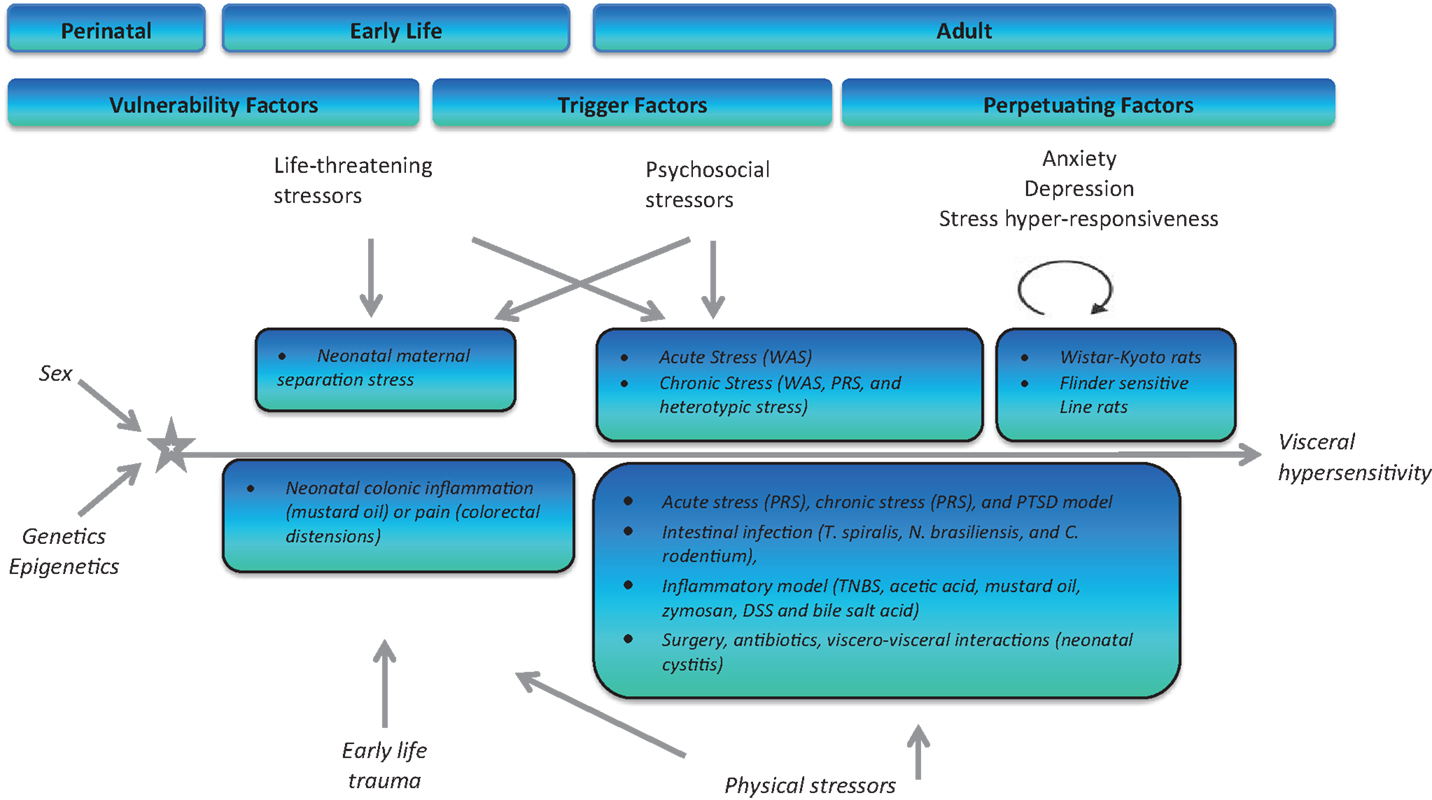
Figure 5. Schematic representation of both physical and psychological stressors used in the generation of animal models for stress-induced visceral hypersensitivity. WAS, water-avoidance stress; PRS, partial restraint stress; TNBS, trinitro benzene sulfonic acid; DSS, dextran sodium sulfate.
Experimental models of stress and stressful events have been developed bearing in mind the critical windows for HPA axis development and maturation. Animal models have been specifically developed to target different periods throughout the lifespan to assess (1) vulnerability, (2) triggering, and (3) perpetuating influences of stress and future development of IBS (425). Early-life stressors in the form of maternal neglect (maternal separation model) or injury (colonic irritation) can increase an individual’s risk to develop IBS and other disorders in adulthood (425). During adulthood, life-threatening stressors (rape, warfare), psychological stressors (acute and chronic stress), or physical stressors (in the form of intestinal dysbiosis due to infection, inflammation, antibiotic usage, and surgery) have all been described and documented as triggering factors to the development of an IBS-type phenotype in rats and mice (425). Finally, the ever expanding catalog of rodent strains and transgenic models has allowed us to use specific strains/knockouts known to exhibit various levels of stress responsivity (Wistar Kyoto and Flinders Sensitive Line) to mimic the influence of genetic and perpetuating factors on the development, severity and duration of IBS symptoms (425).
Genetic Stress Models of Visceral Pain
Mood disorders are commonly comorbid with IBS (432–434). Genetic predisposition to such disorders has been implicated in the development of IBS in later life. These genetic factors may not lead directly to the development of IBS per se but may indirectly, through heightened stress responsivity, cause altered GI function and IBS symptomatology. The availability of a catalog of rodent strains has led to the advent of genetic studies assessing the exact contribution of genetic factors to disease presentation and progression. Moreover, the use of transgenic rodent strains allows us to specifically investigate single genes implicated in disease pathology.
Using different rat strains of known levels of baseline anxiety: low-anxiety SD and Fisher-344 (F344), and high-anxiety WKY rats, Gunter and colleagues were able to demonstrate a link between anxiety and visceral hypersensitivity. Specifically, high-anxiety WKY animals had increased response to CRD compared to low-anxiety strains (132). Moreover, WKY rats exhibited an exaggerated response to acetic acid instillation into the colon, a peripheral sensitization model, compared with low-anxiety strains, SD and F344 (132).
The role of the stress response and the development of IBS was again shown to be intrinsically linked when CRHR1 (CRHR1−/−) knockout animals were shown to have an altered VMR to CRD. VMR to CRD was only observed at the highest distension pressure (60 mmHg). Moreover, pharmacological CRHR1 antagonism decreased the VMR to CRD in CRHR1+/+ mice (167).
Small interfering RNA (siRNA) technologies have now become one of the most widely used approaches to selectively suppress gene expression and is a powerful tool to assess gene function. This technology has been used extensively to suppress stress-related genes in specific brain areas and models are continuously being developed (435–440). Whilst genetically modified mice have been the mainstay of behavioral genetics to date there is a growing utility for genetically modified rats (441). Moreover, other genetic tools such as optogenetic and Designer Receptor Exclusively Activated by Designer Drugs (DREADD)-based manipulations allows for both species to be used in the future (442).
Animal Models of Early-Life Stress-Induced Visceral Pain
Maternal separation stress model
Stress in early life is a well-established risk factor for the development of psychiatric and somatic disorders in later life. The biopsychosocial model of IBS pathophysiology implicates adverse early-life events and childhood traumas such sexual abuse, neglect, loss of a family member, or a life-threatening situation. These factors have been linked to enhanced vulnerability of individuals to develop stress-related affective disorders such as depression and anxiety. Moreover, these individuals are also at a higher risk for developing FGIDs such as IBS and visceral pain (181, 443).
To model these environmental factors in rodent models, the maternal separation model was constructed (Figure 6) (444). Briefly, this model involves removing new-born pups from the dam, during the critical HPA axis hypo-responsive phase in the early postnatal period. Most commonly, pups are removed for 2–3 h per day during the first 2 weeks of life from postnatal days 1–2 to postnatal day 12–14 (108, 188, 445, 446). This interruption in the normal maternal environment, leads to a stress response not only in the pups but also in the dams. As a result of this maternal stress response, the maternal care given to the pups is altered. In the last 20 years, the critical importance of maternal care has been researched extensively no more so in the area of epigenetics (447–449). Alterations in maternal care has been shown to have effects on the development and function of the HPA axis. Moreover, the impaired stress response is thought to underlie the deficits seen in different behavioral domains, in particular, both cognitive and emotional modalities (447). This disruption of the normal maternal environment and dam–offspring interaction can subsequently affect the quality of the maternal care received by the pups. Indeed, this model of early-life stress results in long-lasting changes to the offspring’s CNS, at all levels including altered expression, neurochemistry, electrophysiology, and morphology (8, 450).
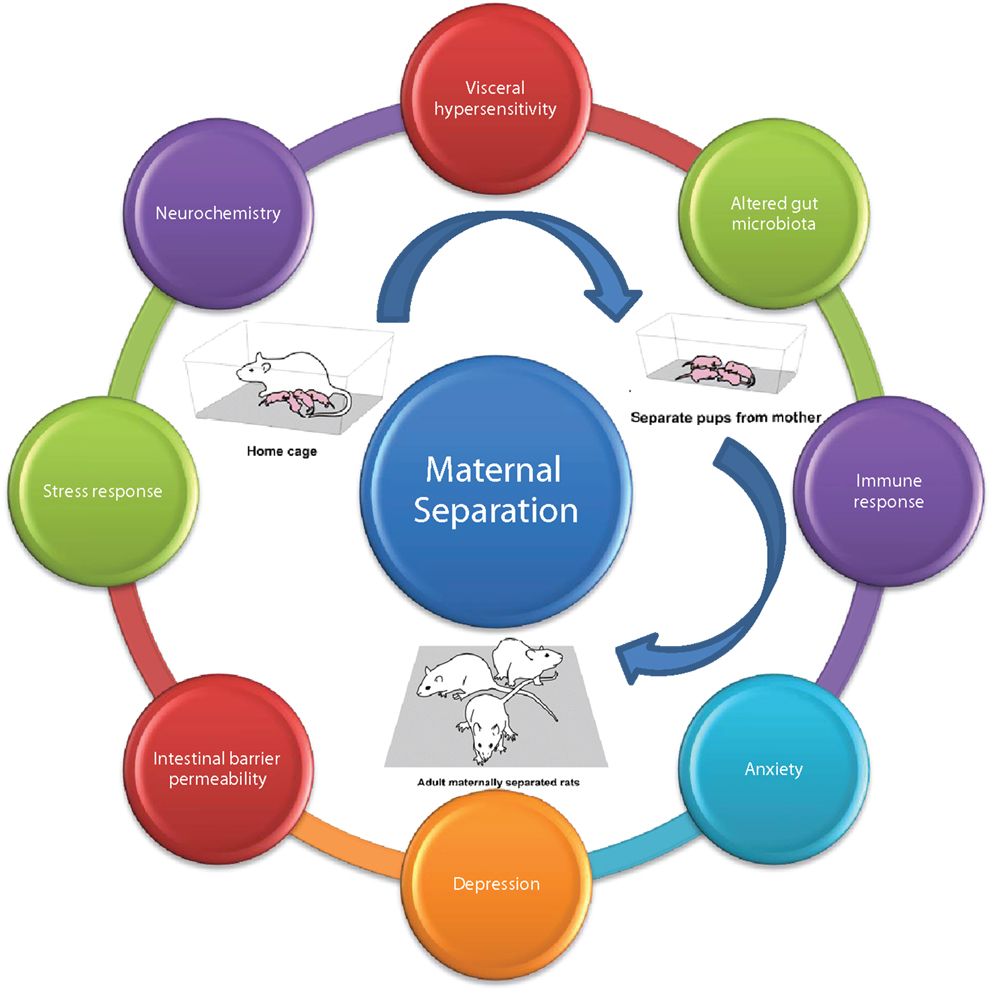
Figure 6. Maternal separation model of brain–gut axis dysfunction. Adult rodents subjected to maternal separation in early life develop the characteristic MS phenotype; typified by altered; visceral sensation, microbiota, immune response, anxiety, depression, intestinal permeability, stress response, neurochemistry adapted from Ref. (108).
When animals are allowed to grow to maturity, a behavioral phenotype is present, characterized by visceral hypersensitivity, increased anxiety and depressive behaviors, altered stress responsivity, altered neurochemistry specifically serotonergic neurotransmission, enhanced immune response, altered gut microbiota profile, and disruption of the intestinal barrier (108). We have previously shown that adult male rats previously subjected to maternal separation exhibit visceral hypersensitivity to CRD (135). Moreover, the effects of MS are exacerbated by exposure to an acute stressor specifically 1 h of water-avoidance stress (WAS) (451, 452).
The maternal separation model is sensitive to many factors such as sex differences, with the abundance of studies being performed in male rodents, however clinically the preponderance of female to male IBS patients is 3:1. It is unclear from the literature that the MS procedure induces the same robust phenotype in female rodents. Moreover, the protocol of separation itself has also proved crucial. Specifically, sex-dependent effects on VMR were evident when the separation was performed by the removal of an entire or half litter from the home cage (446). Males that were exposed to whole litter separation and females that were exposed to both whole litter and half litter separation developed visceral allodynia and hypersensitivity to CRD (446). Moreover, when males underwent an additional acute stress, this did not modify the CRD response. On the other hand, when females were exposed to an additional acute stress, they exhibited an exacerbated response to CRD (8, 446). The rat MS model is the most commonly used rodent model of IBS and has proved crucial in delineating the underlying mechanisms as well as testing novel therapeutic strategies.
Although the maternal separation model is well established and characterized in rats, its utility in mouse strains has proved difficult to replicate. Data from our lab and others suggest that maternal separation stress alone is insufficient to induce a robust, reproducible behavioral phenotype (453, 454). However, when maternal separation stress is combined with unpredictable maternal stress, a more overt behavioral phenotype is exhibited (310, 455).
Neonatal Irritation Models
Early-life stress not only in the form of psychological stress but also in the context of physical stress has been shown to be a valid preclinical model of IBS pathophysiology. The neonatal intestine when exposed to mechanical and chemical stressors results in a pro-inflammatory phenotype with mucosal inflammation and tissue irritation. Daily irritation of the neonatal colon by mechanical irritation (daily noxious CRD) or chemical irritation (daily intra-colonic injection of mustard oil) increases pain behaviors in response to CRD from adolescence to adulthood (417, 456). Moreover, this heightened pain response was accompanied with decreases in exploratory behavior, indicative of an anxiety-phenotype. The findings described in these studies implies that irritation of the neonatal gut, be it via mechanical or chemical means, can lead to the process of central sensitization due in part to sensitized peripheral afferents (8, 417, 456). These behavioral outputs are in parallel with IBS in the clinical scenario; visceral hypersensitivity with comorbid anxiety.
Animal Models of Adult Stress-Induced Visceral Pain
Acute stress models
Acute stressors in adulthood can lead to an immediate behavioral phenotype. The most widely used acute stress paradigms to model IBS preclinically are WAS and restraint stress. The WAS paradigm was originally developed by Bonaz and Tache and Enck et al. (457, 458) to assess stress-related alterations in gut motility and motor function. Briefly, rodents are placed on a small platform raised slightly above water level. This stressor is based on the aversive environment of surrounding water. The WAS paradigm has been used extensively to assess the impact of psychological stress on visceral pain modulation (8, 311). Data from the literature show that 1 h WAS was sufficient to induce a delayed visceral hypersensitivity to CRD, in male Wistar rats (459). Moreover, others have shown that chronic WAS-induced effects are thought to be modulated at an epigenetic level, with HDAC inhibitors shown to reverse WAS-induced hyperalgesia (400).
Moreover, other forms of acute stress such as restraint stress for 2 h, was also found to induce heightened VMR to CRD in male (460) and female Wistar rats (446). The time spent in an acute stress situation has proven crucial to the development of the IBS phenotype (461). Acute stressors of longer duration >2 h tend to be susceptible to the animals habituating to the stressful environment and this can confound the behavioral outputs. Moreover, WAS was recently shown to induce both hyperalgesia and analgesia depending on the duration and number of stress sessions (311, 462). Transient stressors generally trigger adaptive responses and this type of model mimics the stress-related hypersensitivity to CRD as reported in IBS patients.
Chronic mild stress
Daily life stressors affect individuals in different ways. Persistent mild stressors can accumulate and potentiate the effects of stress on the host. Modeling chronic daily stress in rodents is achieved through the chronic mild stress (CMS) paradigm. Convergent lines of evidence suggests that high levels of chronic daily stress can impact on the intensity and severity of visceral pain symptoms (181, 463–466). In light of this, an array of rodent models involving unpredictable chronic and intermittent exposure to variety of stressors have recently been developed (8). Following on from the previous section, WAS was in fact one of the first chronic stress models to be adapted to the study of visceral hypersensitivity (467). Early data proved promising with studies showing that a 1 h daily WAS, for 10 consecutive day protocol was sufficient to induce visceral hypersensitivity to CRD in male Wistar rats (410, 468). This effect was long-lasting and persistent up to 30 days after the end of the stress. One of the confounds of this study was based on the methodology used to assess VMR. EMG recordings were performed however this involves surgical implantation of electrodes and ensuing single housing of animals, to avoid injury (410), which in itself could be described as a stressor. Indeed, when VMR was assessed using intraluminal colonic solid-state manometry, both male and female naïve Wistar rats exposed to WAS developed a reduced response to CRD referred to as visceral analgesia (311). This findings appear to be in direct contrast to each other and highlight the challenges of developing models of stress-induced visceral hypersensitivity.
Moreover, the picture becomes even more complex when we consider mouse models. Indeed, chronic WAS in C57Bl/6 mice has shown many varied effects including visceral hyperalgesia (311), visceral analgesia (311), or to have no influence on the VMR (469). Many other factors may be of course at play such as the conditions prior to CRD including surgery and housing environment, and these factors are dependent on the route of VMR assessment (311). Taken together, it becomes clear that the basal environment and state of the animals prior to stress exposure can impact their response to the stressor itself (8).
Furthermore, the nature or type of stressor is also crucial, with habituation known to occur when animals are repeatedly exposed to the same type of stressor, a phenomenon thought to be mediated via oxytocin (470) and endocannabinoid pathways (471). Moreover, these pathways are indeed themselves also important in pain processing (472, 473). Taking this knowledge on board, that homotypic stressors may/do lead to habituation, more recent models employ stressors of different natures. These heterotypic stressors such as cold restraint stress, WAS, or forced swimming were found to induce immediate but not long-term visceral hyperalgesia as monitored by EMG recordings at 8 h, 24 h, and 7 days after the cessation of the stress paradigm in male Wistar rats (474). Developing the model further, with more multifaceted paradigms and longer lasting stress sessions (134, 475, 476) causes a behavioral phenotype in rodents that mimics aspects of depression. This more recently developed model may prove useful when assessing the mechanisms of chronic visceral pain comorbid with depression-like symptoms (432). Taken together, it is clear that many other factors outside of the stress model can have significant effects on the parameter of interest and thus limit the models utility (477). Factors such as the sex and strain of the animal model, housing conditions before, during and after the stress paradigm and diurnal variation are all known to alter sensitivity to stress (477). From the literature, what we have learned is that variety within the stress paradigm itself (time, type of stressor) appears crucial to inducing stress-induced effects and preventing habituation (470, 478–480). All of these facets could also in themselves potentially alter the influence of CMS paradigms on the visceral pain sensitivity, as recently assessed by Larauche et al. (311).
Chronic psychosocial stress
Daily stressful events are commonplace in modern society. However, when modeling stress in rodent models, the stressors most often occur in a novel environment and not in the animals’ home cage. This has been a cause for critique with some of the most widely used stress models. The chronic social defeat and overcrowding paradigm was designed to overcome this obstacle and thus animals undergo stress scenarios in their home cage. The paradigm is based on the unpredictable nature of life’s stressors. Sessions of resident intruder social defeat and cage overcrowding are randomized so as to occur at different times of the day, in an unpredictable manner and for a chronic period, 19 days in total. We have recently shown this model to be an effective preclinical model to mimic many of the key phenotypes in the IBS population, specifically a heightened response to CRD (134) and anxiety- and depression-related behaviors (481).
Moreover, Reber and colleagues (482) have shown that this model of chronic psychosocial stress robustly enhances GI dysfunction in a mouse model of colitis. Furthermore, other chronic psychosocial stressors such as chronic overcrowding when applied to rats also induces a heightened sensitivity to CRD concomitant with enhanced HPA axis activity and intestinal mucosal inflammation (483). Taken together, these studies demonstrate that this model of chronic psychosocial stress may have multiple effects across the brain–gut axis resulting in an IBS phenotype.
Conditioned fear-induced stress model
There is increasing evidence of augmented prevalence of GI symptoms, including visceral pain and IBS in patients suffering from post-traumatic stress disorder (PTSD) (122, 484–487). Indeed, it has been shown that in some individuals, the experience of stress can prime such individuals to respond differently to subsequent stressful events. Over the last decade, Stam and colleagues have investigated this in preclinical models, particularly rats. They have shown that short exposures to shocks or a social confrontation environment with a predator or aggressive conspecific animal induces long-lasting conditioned fear-responses to trauma-related cues (425). Moreover, these animals exhibit a generalized behavioral sensitization to novel stressful stimuli that is persistent and may intensify over time (488–491). Furthermore, this group have also shown that 2 weeks after a single session of foot shocks, repeated CRD causes increased cardiovascular reflexes in pre-shocked rats when compared to their non-shocked controls (489). Remarkably, female rats appear to exhibit an alternative pattern of sensitized behavioral responsiveness to the same challenge, again highlighted the strong role of sex differences in visceral pain processing (492). This specific rodent model mimics clinical features of a subset of IBS patients exhibiting stress-related visceral hypersensitivity due to PTSD. However, it is important to note that the findings presented above are mainly characterized by a single group of investigators, thus the reliability of the model needs further independent confirmation.
Physical stressors
Post-infectious models of visceral pain. A significant proportion of IBS cases occur after an illness particularly an infection of the GI tract. This is reported to be as much as 10% of patients with IBS (493). Interestingly, the proportion of people who suffer from an intestinal infection who will then go on to develop IBS ranges from 3 to 36% and appears to be dependent upon the infecting organism. Moreover, the psychological state of the individual at the time of infection appears to also play a critical role in the development of IBS symptoms (494). Infections of bacterial origin when compared to the short-lived effects of viral gastroenteritis appear to be more long-lasting and persistent (494). A transient Trichinella spiralis infection was shown to induce sustained visceral hypersensitivity in a mouse model (5, 495). Moreover, similar findings were found in a rat model of Nippostrongylus brasiliensis infection (496). Although the vast majority of human post-infectious hypersensitivity symptoms are observed after bacterial infection with Salmonella, Escherichia coli, Shigella, or Campylobacter, there has been limited animal models of this type of visceral hypersensitivity (497, 498).
Post-inflammatory models of visceral pain. Inflammation is one of the leading causes/mechanisms thought to underpin IBS and its associated symptomatology (499–502). However, this remains a controversial topic and others have reviewed this previously (503, 504). Moreover, this symptom set appears to be common in patients of inflammatory bowel disorders also such as those in remission from ulcerative colitis (505). Indeed, many clinical cases of IBD switch to IBS cases and this occurs in 30–50% of patients (506). Inflammatory pain models have been well developed in somatic pain assays and to some extinct in visceral pain models. Indeed, an array of chemical irritants have been used in preclinical models to induce colonic inflammation and resultant visceral hypersensitivity (8). In rats, mustard oil (507, 508), acetic acid (509), and zymosan (510, 511) have been shown to induce short-term visceral hypersensitivity due in part to colonic inflammation. Moreover, other compounds such as TNBS induce severe colonic inflammation and visceral hypersensitivity when administered intra-colonically (512, 513). Interestingly, in some models of inflammatory-induced visceral hypersensitivity, the phenotype can re-emerge days or weeks after the initial inflammatory response. Moreover this re-emergence is not associated with any inflammatory biomarkers (512, 514). Importantly, the experimental design appears to play a major role in the application and effectiveness of such models. Models, such as the dextran sodium sulfate (DSS)-induced colitis model has provided us with some surprising findings where animals exhibited an increased response to CRD on day 5 or day 60 after the induction of colitis in male Balb/c mice. However, chronic DSS colitis was not associated with changes in VMR to CRD (515). However, contrasting findings have also been reported by other groups, whereby DSS colitis failed to induce any alterations in VMR or visceral sensitivity at all time points tested in either C57Bl/6 or Balb/c mice (516). One can speculate on the many possible reasons for these observed differences, however, what they do suggest is that inflammation on its own may not be sufficient to induce visceral hypersensitivity and that the nature and severity of the inflammatory stimulus and their combined effects may determine when/whether post-inflammatory hypersensitivity will result if at all (8, 512). Moreover, as is seen in post-infectious visceral hypersensitivity, psychological state may also play a role (517). Indeed this was investigated by Larsson et al. (469), who demonstrated that prior exposure of animals to a stressor, be it either psychological or psychosocial in origin, revealed an enhanced susceptibility to colitis and an aggravated colonic inflammatory response (482, 518, 519). Furthermore, this was also associated with a heightened susceptibility to recurrence of colonic inflammation even though the colitis had dissipated (520, 521). Similarly, a prior bout of colitis was sufficient to leave the colon in a state more susceptible to the negative effects of stress (522). However, the complex association between colitis, stress, and visceral pain response remains contentious as stress has also been found to both exacerbate or indeed to have no effect on post-inflammatory visceral sensitivity in rats (523) and in mice (8, 469).
Future Directions
Microbiota Manipulations
The role of the microbiota–brain–gut axis on health and disease is an area of biomedicine that is receiving much media attention of late. The complex communication between the gut microbial population and the CNS has far reaching implications not least in the area of visceral pain and psychiatric comorbidities. Modulation of the gut microbiota via prebiotic/probiotic treatment has been shown to have positive effects on visceral pain behaviors as discussed earlier (222, 240–242). Antibiotic treatment has long been known to alter GI function, effects which are reversed upon probiotic treatment (524). Thus, it is logical that antibiotic-induced visceral pain may be a future model used to investigate the underlying mechanism of such a phenomenon. Indeed work from our own lab has recently demonstrated that early-life antibiotic-induced disruption of the colonizing microbiota is associated with visceral hypersensitivity and altered spinal signaling in adulthood. These results indicate that a temporary alteration in the composition of the GI microbiota during a crucial time-window in the neonatal rat has a long-lasting impact on nociceptive pathways and that the developing pain systems are subject to modulation by the microbiota (248).
Moreover, intriguing studies to assess the exact contribution of the gut microbiota to the development of visceral hypersensitivity and IBS can be achieved through fecal transplantation studies whereby fecal matter from donor IBS patients can be used to inoculate recipient animals and subsequent behaviors can be assessed. This paradigm has already shown its merit in transferring other behaviors specifically anxiety-related behaviors in a mouse model (525).
Conclusion
An effective stress response is critical to the survival of any living creature. The ability to sense changes in the environment and adapt accordingly is quintessence to survival. However, the host’s reaction to perceived stress can sometimes become disturbed leading to an over-exaggerated response. Stress has been implicated in and associated with a plethora of somatic disorders; ulcers, migraine, hypertension, and psychiatric disorders; anxiety, depression, PTSD. Here, we specifically reviewed the literature in the context of stress as a major risk factor for the development of IBS and visceral pain. The mechanism by which stress impacts on normal physiology to cause such devastating disorders still remains unclear. Numerous mechanisms have been postulated including alterations in plasticity, neurogenesis, catecholaminergic neurotransmission, and more recently gut microbiota. Modern day life exposes us all to stressors of varying types (psychological/physical) and severities (acute/chronic) and as such the search for novel pharmaco-therapeutics has never been more pertinent.
Despite the ever growing body of literature, the exact mechanisms underlying visceral pain still remain less well understood than that of somatic pain. Central sensitization of primary sensory afferents is an important underlying mechanism for both somatic and visceral hypersensitivity and hyperalgesia. One of the hurdles in understanding the mechanisms of stress-induced visceral pain is that visceral pain response is experienced differentially depending on numerous factors, including the time at which stress was applied, duration of stress, sex differences, and genetic background amongst others. Psychological stress at any point during our lifetime can lead to permanent alterations of the HPA axis, the descending pain modulatory system, the immune system, and the gut microbiota, all of which can be manifested as chronic visceral hypersensitivity. Mechanisms by which physical stress such as infections, mediate visceral hypersensitivity are likely to be different to that of psychological stress and may involve altered immune system functioning. Considering the diverse mechanisms of visceral pain, the development of treatment strategies and therapeutic interventions will rely on good animal models, all of which have been reviewed here. It is clear that that the drive to develop clinically relevant models and thus design new novel therapeutics has never been more pertinent.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to gratefully acknowledge financial support from Science Foundation Ireland in the form of a center grant (Alimentary Pharmabiotic Centre Grant Number SFI/12/RC/2273). The authors would also like to acknowledge Dr. Yuliya Borre and Dr. Anna Golubeva for assistance in creating the schematic figures.
References
1. Cervero F, Laird JM. Visceral pain. Lancet (1999) 353:2145–8. doi:10.1016/S0140-6736(99)01306-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Robinson DR, Gebhart GF. Inside information: the unique features of visceral sensation. Mol Interv (2008) 8:242–53. doi:10.1124/mi.8.5.9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Giamberardino MA. Recent and forgotten aspects of visceral pain. Eur J Pain (1999) 3:77–92. doi:10.1053/eujp.1999.0117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Giamberardino MA. Visceral pain model, kidney stone pain. In: Schmidt RF, Willis WD, editors. Encyclopedia of Pain. Berlin: Springer (2007). p. 2626–9.
5. Bercik P, Wang L, Verdu EF, Mao YK, Blennerhassett P, Khan WI, et al. Visceral hyperalgesia and intestinal dysmotility in a mouse model of postinfective gut dysfunction. Gastroenterology (2004) 127:179–87. doi:10.1053/j.gastro.2004.04.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Caraceni A, Martini C, Zecca E, Portenoy RK. Breakthrough pain characteristics and syndromes in patients with cancer pain. An international survey. Palliat Med (2004) 18:177–83. doi:10.1191/0269216304pm890oa
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Stoney RJ, Reilly LM. Chronic visceral ischemia. An often overlooked cause of abdominal pain. Postgrad Med (1983) 74:111–8.
8. Larauche M, Mulak A, Tache Y. Stress and visceral pain: from animal models to clinical therapies. Exp Neurol (2012) 233:49–67. doi:10.1016/j.expneurol.2011.04.020
9. Giamberardino MA, Vecchiet L. Visceral pain, referred hyperalgesia and outcome: new concepts. Eur J Anaesthesiol Suppl (1995) 10:61–6.
10. Laird JM, Martinez-Caro L, Garcia-Nicas E, Cervero F. A new model of visceral pain and referred hyperalgesia in the mouse. Pain (2001) 92:335–42. doi:10.1016/S0304-3959(01)00275-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Halder SL, Mcbeth J, Silman AJ, Thompson DG, Macfarlane GJ. Psychosocial risk factors for the onset of abdominal pain. Results from a large prospective population-based study. Int J Epidemiol (2002) 31:1219–25. doi:10.1093/ije/31.6.1219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Hillila MT, Farkkila NJ, Farkkila MA. Societal costs for irritable bowel syndrome – a population based study. Scand J Gastroenterol (2010) 45:582–91. doi:10.3109/00365521003637211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Faresjo A, Grodzinsky E, Hallert C, Timpka T. Patients with irritable bowel syndrome are more burdened by co-morbidity and worry about serious diseases than healthy controls – eight years follow-up of IBS patients in primary care. BMC Public Health (2013) 13:832. doi:10.1186/1471-2458-13-832
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Yu SW, Rao SS. Advances in the management of constipation-predominant irritable bowel syndrome: the role of linaclotide. Therap Adv Gastroenterol (2014) 7:193–205. doi:10.1177/1756283X14537882
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Barboza JL, Talley NJ, Moshiree B. Current and emerging pharmacotherapeutic options for irritable bowel syndrome. Drugs (2014) 74:1849–70. doi:10.1007/s40265-014-0292-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Vercellini P, Somigliana E, Vigano P, Abbiati A, Barbara G, Fedele L. Chronic pelvic pain in women: etiology, pathogenesis and diagnostic approach. Gynecol Endocrinol (2009) 25:149–58. doi:10.1080/09513590802549858
18. Farmer AD, Aziz Q. Gut pain & visceral hypersensitivity. Br J Pain (2013) 7:39–47. doi:10.1177/2049463713479229
19. Farmer AD, Aziz Q. Visceral pain hypersensitivity in functional gastrointestinal disorders. Br Med Bull (2009) 91:123–36. doi:10.1093/bmb/ldp026
20. Handa M, Nukina H, Ando K, Kubo C. What does pain or discomfort in irritable bowel syndrome mean? Dig Dis Sci (2004) 49:575–8. doi:10.1023/B:DDAS.0000026301.47638.92
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Fass R, Achem SR. Noncardiac chest pain: epidemiology, natural course and pathogenesis. J Neurogastroenterol Motil (2011) 17:110–23. doi:10.5056/jnm.2011.17.2.110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Giamberardino MA, Valente R, de Bigontina P, Vecchiet L. Artificial ureteral calculosis in rats: behavioural characterization of visceral pain episodes and their relationship with referred lumbar muscle hyperalgesia. Pain (1995) 61:459–69. doi:10.1016/0304-3959(94)00208-V
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Cervero F. Visceral versus somatic pain: similarities and differences. Dig Dis (2009) 27(Suppl 1):3–10. doi:10.1159/000268115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Dunckley P, Wise RG, Fairhurst M, Hobden P, Aziz Q, Chang L, et al. A comparison of visceral and somatic pain processing in the human brainstem using functional magnetic resonance imaging. J Neurosci (2005) 25:7333–41. doi:10.1523/JNEUROSCI.1100-05.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Dunckley P, Wise RG, Aziz Q, Painter D, Brooks J, Tracey I, et al. Cortical processing of visceral and somatic stimulation: differentiating pain intensity from unpleasantness. Neuroscience (2005) 133:533–42. doi:10.1016/j.neuroscience.2005.02.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Vermeulen W, de Man JG, Pelckmans PA, de Winter BY. Neuroanatomy of lower gastrointestinal pain disorders. World J Gastroenterol (2014) 20:1005–20. doi:10.3748/wjg.v20.i4.1005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Grundy D. Neuroanatomy of visceral nociception: vagal and splanchnic afferent. Gut (2002) 51(Suppl 1):i2–5. doi:10.1136/gut.51.suppl_1.i2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Anand P, Aziz Q, Willert R, van Oudenhove L. Peripheral and central mechanisms of visceral sensitization in man. Neurogastroenterol Motil (2007) 19:29–46. doi:10.1111/j.1365-2982.2006.00873.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Almeida TF, Roizenblatt S, Tufik S. Afferent pain pathways: a neuroanatomical review. Brain Res (2004) 1000:40–56. doi:10.1016/j.brainres.2003.10.073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Johnson AC, Greenwood-van Meerveld B. Stress-induced pain: a target for the development of novel therapeutics. J Pharmacol Exp Ther (2014) 351:327–35. doi:10.1124/jpet.114.218065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Ness TJ. Evidence for ascending visceral nociceptive information in the dorsal midline and lateral spinal cord. Pain (2000) 87:83–8. doi:10.1016/S0304-3959(00)00272-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Camilleri M, Coulie B, Tack JF. Visceral hypersensitivity: facts, speculations, and challenges. Gut (2001) 48:125–31. doi:10.1136/gut.48.1.125
33. Palecek J. The role of dorsal columns pathway in visceral pain. Physiol Res (2004) 53(Suppl 1):S125–30. doi:10.1016/S0304-3959(03)00075-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Willis WD, Al-Chaer ED, Quast MJ, Westlund KN. A visceral pain pathway in the dorsal column of the spinal cord. Proc Natl Acad Sci U S A (1999) 96:7675–9. doi:10.1073/pnas.96.14.7675
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Rustioni A, Hayes NL, O’Neill S. Dorsal column nuclei and ascending spinal afferents in macaques. Brain (1979) 102:95–125. doi:10.1093/brain/102.1.95
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Bennett GJ, Seltzer Z, Lu GW, Nishikawa N, Dubner R. The cells of origin of the dorsal column postsynaptic projection in the lumbosacral enlargements of cats and monkeys. Somatosens Res (1983) 1:131–49. doi:10.3109/07367228309144545
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Drossman DA. Functional abdominal pain syndrome. Clin Gastroenterol Hepatol (2004) 2:353–65. doi:10.1016/S1542-3565(04)00118-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Sharma A, Lelic D, Brock C, Paine P, Aziz Q. New technologies to investigate the brain-gut axis. World J Gastroenterol (2009) 15:182–91. doi:10.3748/wjg.15.182
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Jones MP, Dilley JB, Drossman D, Crowell MD. Brain-gut connections in functional GI disorders: anatomic and physiologic relationships. Neurogastroenterol Motil (2006) 18:91–103. doi:10.1111/j.1365-2982.2005.00730.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Bush G, Luu P, Posner MI. Cognitive and emotional influences in anterior cingulate cortex. Trends Cogn Sci (2000) 4:215–22. doi:10.1016/S1364-6613(00)01483-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Kravets JL, Reyes BA, Unterwald EM, van Bockstaele EJ. Direct targeting of peptidergic amygdalar neurons by noradrenergic afferents: linking stress-integrative circuitry. Brain Struct Funct (2013) 220:541–58. doi:10.1007/s00429-013-0674-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Jasmin L, Burkey AR, Card JP, Basbaum AI. Transneuronal labeling of a nociceptive pathway, the spino-(trigemino-)parabrachio-amygdaloid, in the rat. J Neurosci (1997) 17:3751–65.
43. Gustafsson JK, Greenwood-van Meerveld B. Amygdala activation by corticosterone alters visceral and somatic pain in cycling female rats. Am J Physiol Gastrointest Liver Physiol (2011) 300:G1080–5. doi:10.1152/ajpgi.00349.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Johnson AC, Myers B, Lazovic J, Towner R, Greenwood-van Meerveld B. Brain activation in response to visceral stimulation in rats with amygdala implants of corticosterone: an FMRI study. PLoS One (2010) 5:e8573. doi:10.1371/journal.pone.0008573
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Johnson AC, Tran L, Schulkin J, Greenwood-van Meerveld B. Importance of stress receptor-mediated mechanisms in the amygdala on visceral pain perception in an intrinsically anxious rat. Neurogastroenterol Motil (2012) 24(479–86):e219. doi:10.1111/j.1365-2982.2012.01899.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Myers B, Dittmeyer K, Greenwood-van Meerveld B. Involvement of amygdaloid corticosterone in altered visceral and somatic sensation. Behav Brain Res (2007) 181:163–7. doi:10.1016/j.bbr.2007.03.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Myers B, Greenwood-van Meerveld B. Divergent effects of amygdala glucocorticoid and mineralocorticoid receptors in the regulation of visceral and somatic pain. Am J Physiol Gastrointest Liver Physiol (2010) 298:G295–303. doi:10.1152/ajpgi.00298.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Myers B, Greenwood-van Meerveld B. Differential involvement of amygdala corticosteroid receptors in visceral hyperalgesia following acute or repeated stress. Am J Physiol Gastrointest Liver Physiol (2012) 302:G260–6. doi:10.1152/ajpgi.00353.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Myers B, Schulkin J, Greenwood-van Meerveld B. Sex steroids localized to the amygdala increase pain responses to visceral stimulation in rats. J Pain (2011) 12:486–94. doi:10.1016/j.jpain.2010.10.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Tran L, Greenwood-van Meerveld B. Lateralized amygdala activation: importance in the regulation of anxiety and pain behavior. Physiol Behav (2012) 105:371–5. doi:10.1016/j.physbeh.2011.08.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Venkova K, Foreman RD, Greenwood-van Meerveld B. Mineralocorticoid and glucocorticoid receptors in the amygdala regulate distinct responses to colorectal distension. Neuropharmacology (2009) 56:514–21. doi:10.1016/j.neuropharm.2008.10.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Felice VD, Gibney SM, Gosselin RD, Dinan TG, O’Mahony SM, Cryan JF. Differential activation of the prefrontal cortex and amygdala following psychological stress and colorectal distension in the maternally separated rat. Neuroscience (2014) 267:252–62. doi:10.1016/j.neuroscience.2014.01.064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Grover M, Drossman DA. Functional abdominal pain. Curr Gastroenterol Rep (2010) 12:391–8. doi:10.1007/s11894-010-0125-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Le Bars D, Villanueva L, Bouhassira D, Willer JC. Diffuse noxious inhibitory controls (DNIC) in animals and in man. Patol Fiziol Eksp Ter (1992) 4:55–65.
55. King CD, Wong F, Currie T, Mauderli AP, Fillingim RB, Riley JL III. Deficiency in endogenous modulation of prolonged heat pain in patients with irritable bowel syndrome and temporomandibular disorder. Pain (2009) 143:172–8. doi:10.1016/j.pain.2008.12.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Heymen S, Maixner W, Whitehead WE, Klatzkin RR, Mechlin B, Light KC. Central processing of noxious somatic stimuli in patients with irritable bowel syndrome compared with healthy controls. Clin J Pain (2010) 26:104–9. doi:10.1097/AJP.0b013e3181bff800
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Bouhassira D, Moisset X, Jouet P, Duboc H, Coffin B, Sabate JM. Changes in the modulation of spinal pain processing are related to severity in irritable bowel syndrome. Neurogastroenterol Motil (2013) 25:623–e468. doi:10.1111/nmo.12123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Wilder-Smith CH, Schindler D, Lovblad K, Redmond SM, Nirkko A. Brain functional magnetic resonance imaging of rectal pain and activation of endogenous inhibitory mechanisms in irritable bowel syndrome patient subgroups and healthy controls. Gut (2004) 53:1595–601. doi:10.1136/gut.2003.028514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Yang M, Roman K, Chen DF, Wang ZG, Lin Y, Stephens RL Jr. GLT-1 overexpression attenuates bladder nociception and local/cross-organ sensitization of bladder nociception. Am J Physiol Renal Physiol (2011) 300:F1353–9. doi:10.1152/ajprenal.00009.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Bradesi S, Herman J, Mayer EA. Visceral analgesics: drugs with a great potential in functional disorders? Curr Opin Pharmacol (2008) 8:697–703. doi:10.1016/j.coph.2008.08.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Blackshaw LA, Page AJ, Young RL. Metabotropic glutamate receptors as novel therapeutic targets on visceral sensory pathways. Front Neurosci (2011) 5:40. doi:10.3389/fnins.2011.00040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Joshi SK, Mikusa JP, Weaver B, Honore P. Morphine and ABT-594 (a nicotinic acetylcholine agonist) exert centrally mediated antinociception in the rat cyclophosphamide cystitis model of visceral pain. J Pain (2008) 9:146–56. doi:10.1016/j.jpain.2007.09.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Fioramonti J, Bueno L. Role of cannabinoid receptors in the control of gastrointestinal motility and perception. Expert Rev Gastroenterol Hepatol (2008) 2:385–97. doi:10.1586/17474124.2.3.385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Hughes PA, Castro J, Harrington AM, Isaacs N, Moretta M, Hicks GA, et al. Increased kappa-opioid receptor expression and function during chronic visceral hypersensitivity. Gut (2014) 63:1199–200. doi:10.1136/gutjnl-2013-306240
65. Gonzalez-Cano R, Merlos M, Baeyens JM, Cendan CM. sigma1 receptors are involved in the visceral pain induced by intracolonic administration of capsaicin in mice. Anesthesiology (2013) 118:691–700. doi:10.1097/ALN.0b013e318280a60a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Hyland NP, Golubeva AV. GABA receptors in the bladder and bowel: therapeutic potential for positive allosteric modulators? Br J Pharmacol (2014). doi:10.1111/bph.12617
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Lindstrom E, Brusberg M, Ravnefjord A, Kakol-Palm D, Pahlman I, Noven A, et al. Oral baclofen reduces visceral pain-related pseudo-affective responses to colorectal distension in rats: relation between plasma exposure and efficacy. Scand J Gastroenterol (2011) 46:652–62. doi:10.3109/00365521.2011.560677
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Zhou L, Huang J, Gao J, Zhang G, Jiang J. NMDA and AMPA receptors in the anterior cingulate cortex mediates visceral pain in visceral hypersensitivity rats. Cell Immunol (2014) 287:86–90. doi:10.1016/j.cellimm.2013.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Cao DY, Bai G, Ji Y, Traub RJ. Epigenetic upregulation of metabotropic glutamate receptor 2 in the spinal cord attenuates oestrogen-induced visceral hypersensitivity. Gut (2014). doi:10.1136/gutjnl-2014-307748
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Crock LW, Kolber BJ, Morgan CD, Sadler KE, Vogt SK, Bruchas MR, et al. Central amygdala metabotropic glutamate receptor 5 in the modulation of visceral pain. J Neurosci (2012) 32:14217–26. doi:10.1523/JNEUROSCI.1473-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Tran L, Schulkin J, Ligon CO, Greenwood-van Meerveld B. Epigenetic modulation of chronic anxiety and pain by histone deacetylation. Mol Psychiatry (2014). doi:10.1038/mp.2014.122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Kawabata A, Kawao N, Kitano T, Matsunami M, Satoh R, Ishiki T, et al. Colonic hyperalgesia triggered by proteinase-activated receptor-2 in mice: involvement of endogenous bradykinin. Neurosci Lett (2006) 402:167–72. doi:10.1016/j.neulet.2006.03.074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Couture R, Harrisson M, Vianna RM, Cloutier F. Kinin receptors in pain and inflammation. Eur J Pharmacol (2001) 429:161–76. doi:10.1016/S0014-2999(01)01318-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Qin HY, Luo JL, Qi SD, Xu HX, Sung JJ, Bian ZX. Visceral hypersensitivity induced by activation of transient receptor potential vanilloid type 1 is mediated through the serotonin pathway in rat colon. Eur J Pharmacol (2010) 647:75–83. doi:10.1016/j.ejphar.2010.08.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Xu GY, Shenoy M, Winston JH, Mittal S, Pasricha PJ. P2X receptor-mediated visceral hyperalgesia in a rat model of chronic visceral hypersensitivity. Gut (2008) 57:1230–7. doi:10.1136/gut.2007.134221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. de Carvalho Rocha HA, Dantas BP, Rolim TL, Costa BA, de Medeiros AC. Main ion channels and receptors associated with visceral hypersensitivity in irritable bowel syndrome. Ann Gastroenterol (2014) 27:200–6.
77. Gralnek IM, Hays RD, Kilbourne A, Naliboff B, Mayer EA. The impact of irritable bowel syndrome on health-related quality of life. Gastroenterology (2000) 119:654–60. doi:10.1053/gast.2000.16484
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Whitehead WE, Burnett CK, Cook EW III, Taub E. Impact of irritable bowel syndrome on quality of life. Dig Dis Sci (1996) 41:2248–53. doi:10.1007/BF02071408
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Mishra S, Singh A, Pandey BL. Current state of pharmacology and therapeutics in irritable bowel syndrome with special reference to brain-gut axis. Int J Basic Clin Pharmacol (2013) 2:122–9. doi:10.5455/2319-2003.ijbcp20130303
80. Holdgate A, Pollock T. Nonsteroidal anti-inflammatory drugs (NSAIDs) versus opioids for acute renal colic. Cochrane Database Syst Rev (2004) 1:Cd004137. doi:10.1002/14651858.CD004137.pub2
81. Morgan G. Beneficial effects of NSAIDs in the gastrointestinal tract. Eur J Gastroenterol Hepatol (1999) 11:393–400. doi:10.1097/00042737-199904000-00006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Leventer SM, Raudibaugh K, Frissora CL, Kassem N, Keogh JC, Phillips J, et al. Clinical trial: dextofisopam in the treatment of patients with diarrhoea-predominant or alternating irritable bowel syndrome. Aliment Pharmacol Ther (2008) 27:197–206. doi:10.1111/j.1365-2036.2007.03566.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Lazaraki G, Chatzimavroudis G, Katsinelos P. Recent advances in pharmacological treatment of irritable bowel syndrome. World J Gastroenterol (2014) 20:8867–85. doi:10.3748/wjg.v20.i27.8867
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Camilleri M, Andresen V. Current and novel therapeutic options for irritable bowel syndrome management. Dig Liver Dis (2009) 41:854–62. doi:10.1016/j.dld.2009.07.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Jadallah KA, Kullab SM, Sanders DS. Constipation-predominant irritable bowel syndrome: a review of current and emerging drug therapies. World J Gastroenterol (2014) 20:8898–909. doi:10.3748/wjg.v20.i27.8898
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Vanuytsel T, Tack JF, Boeckxstaens GE. Treatment of abdominal pain in irritable bowel syndrome. J Gastroenterol (2014) 49:1193–205. doi:10.1007/s00535-014-0966-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Benyamin R, Trescot AM, Datta S, Buenaventura R, Adlaka R, Sehgal N, et al. Opioid complications and side effects. Pain Physician (2008) 11:S105–20.
88. Tsang BK, He Z, Wongchanapai W, Ho IK, Eichhorn JH. Visceral analgesic tolerance to intrathecal butorphanol in rats. Can J Anaesth (1998) 45:1019–23. doi:10.1007/BF03012311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Saito Y, Kaneko M, Kirihara Y, Sakura S, Kosaka Y. Characteristics of tolerance to somatic and visceral antinociception after continuous epidural infusion of morphine in rats. Anesth Analg (1998) 87:1340–5. doi:10.1097/00000539-199812000-00024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Ness TJ, Follett KA. The development of tolerance to intrathecal morphine in rat models of visceral and cutaneous pain. Neurosci Lett (1998) 248:33–6. doi:10.1016/S0304-3940(98)00327-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Lian B, Vera-Portocarrero L, King T, Ossipov MH, Porreca F. Opioid-induced latent sensitization in a model of non-inflammatory viscerosomatic hypersensitivity. Brain Res (2010) 1358:64–70. doi:10.1016/j.brainres.2010.08.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Mao J, Mayer DJ. Spinal cord neuroplasticity following repeated opioid exposure and its relation to pathological pain. Ann N Y Acad Sci (2001) 933:175–84. doi:10.1111/j.1749-6632.2001.tb05823.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Hay JL, White JM, Bochner F, Somogyi AA, Semple TJ, Rounsefell B. Hyperalgesia in opioid-managed chronic pain and opioid-dependent patients. J Pain (2009) 10:316–22. doi:10.1016/j.jpain.2008.10.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Arner S, Rawal N, Gustafsson LL. Clinical experience of long-term treatment with epidural and intrathecal opioids – a nationwide survey. Acta Anaesthesiol Scand (1988) 32:253–9. doi:10.1111/j.1399-6576.1988.tb02725.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Davis MP. Drug management of visceral pain: concepts from basic research. Pain Res Treat (2012) 2012:265605. doi:10.1155/2012/265605
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Kozlowski C, Green A, Grundy D, Boissonade F, Bountra C. The 5-HT3 receptor antagonist alosetron inhibits the colorectal distention induced depressor response and spinal c-fos expression in the anaesthetised rat. Gut (2000) 46:474–80. doi:10.1136/gut.46.4.474
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Mayer EA, Berman S, Derbyshire SW, Suyenobu B, Chang L, Fitzgerald L, et al. The effect of the 5-HT3 receptor antagonist, alosetron, on brain responses to visceral stimulation in irritable bowel syndrome patients. Aliment Pharmacol Ther (2002) 16:1357–66. doi:10.1046/j.1365-2036.2002.01287.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Camilleri M, Mayer E, Drossman D, Heath A, Dukes G, Mcsorley D, et al. Improvement in pain and bowel function in female irritable bowel patients with alosetron, a 5-HT. Aliment Pharmacol Ther (1999) 13:1149–59. doi:10.1046/j.1365-2036.1999.00610.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Degen L, Matzinger D, Merz M, Appel-Dingemanse S, Osborne S, Luchinger S, et al. Tegaserod, a 5-HT4 receptor partial agonist, accelerates gastric emptying and gastrointestinal transit in healthy male subjects. Aliment Pharmacol Ther (2001) 15:1745–51. doi:10.1046/j.1365-2036.2001.01103.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Coffin B, Farmachidi JP, Rueegg P, Bastie A, Bouhassira D. Tegaserod, a 5-HT4 receptor partial agonist, decreases sensitivity to rectal distension in healthy subjects. Aliment Pharmacol Ther (2003) 17:577–85. doi:10.1046/j.1365-2036.2003.01449.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Beglinger C. Tegaserod: a novel, selective 5-HT4 receptor partial agonist for irritable bowel syndrome. Int J Clin Pract (2002) 56:47–51.
102. Liang LX, Zhang Q, Qian W, Hou XH. Antinociceptive property of tegaserod in a rat model of chronic visceral hypersensitivity. Chin J Dig Dis (2005) 6:21–5. doi:10.1111/j.1443-9573.2005.00182.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Ravnefjord A, Brusberg M, Kang D, Bauer U, Larsson H, Lindstrom E, et al. Involvement of the transient receptor potential vanilloid 1 (TRPV1) in the development of acute visceral hyperalgesia during colorectal distension in rats. Eur J Pharmacol (2009) 611:85–91. doi:10.1016/j.ejphar.2009.03.058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Blackshaw LA, Brierley SM, Hughes PA. TRP channels: new targets for visceral pain. Gut (2010) 59:126–35. doi:10.1136/gut.2009.179523
105. Ravnefjord A, Brusberg M, Larsson H, Lindstrom E, Martinez V. Effects of pregabalin on visceral pain responses and colonic compliance in rats. Br J Pharmacol (2008) 155:407–16. doi:10.1038/bjp.2008.259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Million M, Wang L, Adelson DW, Roman F, Diop L, Tache Y. Pregabalin decreases visceral pain and prevents spinal neuronal activation in rats. Gut (2007) 56:1482–4. doi:10.1136/gut.2007.129304
107. Olesen SS, Graversen C, Olesen AE, Frokjaer JB, Wilder-Smith O, van Goor H, et al. Randomised clinical trial: pregabalin attenuates experimental visceral pain through sub-cortical mechanisms in patients with painful chronic pancreatitis. Aliment Pharmacol Ther (2011) 34:878–87. doi:10.1111/j.1365-2036.2011.04802.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. O’Mahony SM, Coelho AM, Fitzgerald P, Lee K, Winchester W, Dinan TG, et al. The effects of gabapentin in two animal models of co-morbid anxiety and visceral hypersensitivity. Eur J Pharmacol (2011) 667:169–74. doi:10.1016/j.ejphar.2011.05.055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Stephens RL Jr. Glutamate transporter activators as anti-nociceptive agents. Eurasian J Med (2011) 43:182–5. doi:10.5152/eajm.2011.39
110. Roman K, Yang M, Stephens RL. Characterization of the visceral antinociceptive effect of glial glutamate transporter GLT-1 upregulation by ceftriaxone. ISRN Pain (2013) 2013:1–10. doi:10.1155/2013/726891
111. Lindstrom E, Brusberg M, Hughes PA, Martin CM, Brierley SM, Phillis BD, et al. Involvement of metabotropic glutamate 5 receptor in visceral pain. Pain (2008) 137:295–305. doi:10.1016/j.pain.2007.09.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Lin Y, Tian G, Roman K, Handy C, Travers JB, Lin CL, et al. Increased glial glutamate transporter EAAT2 expression reduces visceral nociceptive response in mice. Am J Physiol Gastrointest Liver Physiol (2009) 296:G129–34. doi:10.1152/ajpgi.90556.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Lin Y, Roman K, Foust KD, Kaspar BK, Bailey MT, Stephens RL. Glutamate transporter GLT-1 upregulation attenuates visceral nociception and hyperalgesia via spinal mechanisms not related to anti-inflammatory or probiotic effects. Pain Res Treat (2011) 2011:507029. doi:10.1155/2011/507029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Gosselin RD, O’Connor RM, Tramullas M, Julio-Pieper M, Dinan TG, Cryan JF. Riluzole normalizes early-life stress-induced visceral hypersensitivity in rats: role of spinal glutamate reuptake mechanisms. Gastroenterology (2010) 138:2418–25. doi:10.1053/j.gastro.2010.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Bradesi S, Golovatscka V, Ennes HS, Mcroberts JA, Karagiannides I, Bakirtzi K, et al. Role of astrocytes and altered regulation of spinal glutamatergic neurotransmission in stress-induced visceral hyperalgesia in rats. Am J Physiol Gastrointest Liver Physiol (2011) 301:G580–9. doi:10.1152/ajpgi.00182.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Whitehead WE, Palsson OS, Levy RR, Feld AD, Turner M, von Korff M. Comorbidity in irritable bowel syndrome. Am J Gastroenterol (2007) 102:2767–76. doi:10.1111/j.1572-0241.2007.01540.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Whitehead WE, Palsson O, Jones KR. Systematic review of the comorbidity of irritable bowel syndrome with other disorders: what are the causes and implications? Gastroenterology (2002) 122:1140–56. doi:10.1053/gast.2002.32392
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Vandvik PO, Wilhelmsen I, Ihlebaek C, Farup PG. Comorbidity of irritable bowel syndrome in general practice: a striking feature with clinical implications. Aliment Pharmacol Ther (2004) 20:1195–203. doi:10.1111/j.1365-2036.2004.02250.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Vandvik PO, Lydersen S, Farup PG. Prevalence, comorbidity and impact of irritable bowel syndrome in Norway. Scand J Gastroenterol (2006) 41:650–6. doi:10.1080/00365520500442542
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Spetalen S, Sandvik L, Blomhoff S, Jacobsen MB. Rectal visceral sensitivity in women with irritable bowel syndrome without psychiatric comorbidity compared with healthy volunteers. Gastroenterol Res Pract (2009) 2009:130684. doi:10.1155/2009/130684
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Kumano H, Kaiya H, Yoshiuchi K, Yamanaka G, Sasaki T, Kuboki T. Comorbidity of irritable bowel syndrome, panic disorder, and agoraphobia in a Japanese representative sample. Am J Gastroenterol (2004) 99:370–6. doi:10.1111/j.1572-0241.2004.04048.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Irwin C, Falsetti SA, Lydiard RB, Ballenger JC, Brock CD, Brener W. Comorbidity of posttraumatic stress disorder and irritable bowel syndrome. J Clin Psychiatry (1996) 57:576–8. doi:10.4088/JCP.v57n1204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Hillila MT, Siivola MT, Farkkila MA. Comorbidity and use of health-care services among irritable bowel syndrome sufferers. Scand J Gastroenterol (2007) 42:799–806. doi:10.1080/00365520601113927
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Garakani A, Win T, Virk S, Gupta S, Kaplan D, Masand PS. Comorbidity of irritable bowel syndrome in psychiatric patients: a review. Am J Ther (2003) 10:61–7. doi:10.1097/00045391-200301000-00014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Frissora CL, Koch KL. Symptom overlap and comorbidity of irritable bowel syndrome with other conditions. Curr Gastroenterol Rep (2005) 7:264–71. doi:10.1007/s11894-005-0018-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Park HJ, Jarrett M, Cain K, Heitkemper M. Psychological distress and GI symptoms are related to severity of bloating in women with irritable bowel syndrome. Res Nurs Health (2008) 31:98–107. doi:10.1002/nur.20237
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Resendiz-Figueroa FE, Ortiz-Garrido OM, Pulido D, Arcila-Martinez D, Schmulson M. Impact of the anxiety characteristics and depression on clinical aspects and quality of life in patients with irritable bowel syndrome. Rev Gastroenterol Mex (2008) 73:3–10.
128. Mikocka-Walus A, Turnbull D, Moulding N, Wilson I, Andrews JM, Holtmann G. Psychological comorbidity and complexity of gastrointestinal symptoms in clinically diagnosed irritable bowel syndrome patients. J Gastroenterol Hepatol (2008) 23:1137–43. doi:10.1111/j.1440-1746.2007.05245.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Gralnek IM, Hays RD, Kilbourne AM, Chang L, Mayer EA. Racial differences in the impact of irritable bowel syndrome on health-related quality of life. J Clin Gastroenterol (2004) 38:782–9. doi:10.1097/01.mcg.0000140190.65405.fb
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Gupta A, Silman AJ. Psychological stress and fibromyalgia: a review of the evidence suggesting a neuroendocrine link. Arthritis Res Ther (2004) 6:98–106. doi:10.1186/ar1140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Boudewijn van Houdenhove M, Egle U. The role of life stress in fibromyalgia. Curr Rheumatol Rep (2005) 7:365–70. doi:10.1007/s11926-005-0021-z
132. Gunter WD, Shepard JD, Foreman RD, Myers DA, Greenwood-van Meerveld B. Evidence for visceral hypersensitivity in high-anxiety rats. Physiol Behav (2000) 69:379–82. doi:10.1016/S0031-9384(99)00254-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Bravo JA, Dinan TG, Cryan JF. Alterations in the central CRF system of two different rat models of comorbid depression and functional gastrointestinal disorders. Int J Neuropsychopharmacol (2011) 14:666–83. doi:10.1017/S1461145710000994
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Tramullas M, Dinan TG, Cryan JF. Chronic psychosocial stress induces visceral hyperalgesia in mice. Stress (2012) 15:281–92. doi:10.3109/10253890.2011.622816
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. O’Mahony SM, Marchesi JR, Scully P, Codling C, Ceolho AM, Quigley EM, et al. Early life stress alters behavior, immunity, and microbiota in rats: implications for irritable bowel syndrome and psychiatric illnesses. Biol Psychiatry (2009) 65:263–7. doi:10.1016/j.biopsych.2008.06.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Nishiyama H, Mizuta Y, Isomoto H, Takeshima F, Omagari K, Miyahara Y, et al. Chronic visceral hypersensitivity renders defecation more susceptible to stress via a serotonergic pathway in rats. Dig Dis Sci (2004) 49:763–9. doi:10.1023/B:DDAS.0000030086.03506.c7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Geuze E, Westenberg HG, Jochims A, de Kloet CS, Bohus M, Vermetten E, et al. Altered pain processing in veterans with posttraumatic stress disorder. Arch Gen Psychiatry (2007) 64:76–85. doi:10.1001/archpsyc.64.1.76
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Staud R, Domingo M. Evidence for abnormal pain processing in fibromyalgia syndrome. Pain Med (2001) 2:208–15. doi:10.1046/j.1526-4637.2001.01030.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Coss-Adame E, Rao SS. Brain and gut interactions in irritable bowel syndrome: new paradigms and new understandings. Curr Gastroenterol Rep (2014) 16:379. doi:10.1007/s11894-014-0379-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Mayer EA, Chang L, Lembo T. Brain-gut interactions: implications for newer therapy. Eur J Surg Suppl (1998) 582:50–5.
141. Fukudo S, Muranaka M, Nomura T, Satake M. Brain-gut interactions in irritable bowel syndrome: physiological and psychological aspect. Nihon Rinsho (1992) 50:2703–11.
142. Mulak A, Bonaz B. Irritable bowel syndrome: a model of the brain-gut interactions. Med Sci Monit (2004) 10:Ra55–62.
143. Cryan JF, O’Mahony SM. The microbiome-gut-brain axis: from bowel to behavior. Neurogastroenterol Motil (2011) 23:187–92. doi:10.1111/j.1365-2982.2010.01664.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Borre YE, Moloney RD, Clarke G, Dinan TG, Cryan JF. The impact of microbiota on brain and behavior: mechanisms & therapeutic potential. Adv Exp Med Biol (2014) 817:373–403. doi:10.1007/978-1-4939-0897-4_17
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Moloney RD, Desbonnet L, Clarke G, Dinan TG, Cryan JF. The microbiome: stress, health and disease. Mamm Genome (2014) 25:49–74. doi:10.1007/s00335-013-9488-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Bonaz B, Pellissier S, Sinniger V, Clarençon D, Peinnequin A, Canini F. The Irritable Bowel Syndrome: How Stress can Affect the Amygdala Activity and the Brain-Gut Axis. InTech (2012).
147. Labus JS, Dinov ID, Jiang Z, Ashe-Mcnalley C, Zamanyan A, Shi Y, et al. Irritable bowel syndrome in female patients is associated with alterations in structural brain networks. Pain (2014) 155:137–49. doi:10.1016/j.pain.2013.09.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Icenhour A, Langhorst J, Benson S, Schlamann M, Hampel S, Engler H, et al. Neural circuitry of abdominal pain-related fear learning and reinstatement in irritable bowel syndrome. Neurogastroenterol Motil (2015) 27:114–27. doi:10.1111/nmo.12489
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Selye H. A syndrome produced by diverse nocuous agents. 1936. J Neuropsychiatry Clin Neurosci (1936) 10:230–1. doi:10.1176/jnp.10.2.230a
150. Carrasco GA, van de Kar LD. Neuroendocrine pharmacology of stress. Eur J Pharmacol (2003) 463:235–72. doi:10.1016/S0014-2999(03)01285-8
151. Charmandari E, Tsigos C, Chrousos G. Endocrinology of the stress response. Annu Rev Physiol (2005) 67:259–84. doi:10.1146/annurev.physiol.67.040403.120816
152. Habib KE, Gold PW, Chrousos GP. Neuroendocrinology of stress. Endocrinol Metab Clin North Am (2001) 30:695–728. doi:10.1016/S0889-8529(05)70208-5
153. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev (2007) 87:873–904. doi:10.1152/physrev.00041.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Smith SM, Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin Neurosci (2006) 8:383–95.
155. Chrousos GP. Regulation and dysregulation of the hypothalamic-pituitary-adrenal axis. The corticotropin-releasing hormone perspective. Endocrinol Metab Clin North Am (1992) 21:833–58.
156. Whitnall MH. Regulation of the hypothalamic corticotropin-releasing hormone neurosecretory system. Prog Neurobiol (1993) 40:573–629. doi:10.1016/0301-0082(93)90035-Q
157. McEwen BS. The neurobiology of stress: from serendipity to clinical relevance. Brain Res (2000) 886:172–89. doi:10.1016/S0006-8993(00)02950-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. McEwen BS. Brain on stress: how the social environment gets under the skin. Proc Natl Acad Sci U S A (2012) 109(Suppl 2):17180–5. doi:10.1073/pnas.1121254109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Nutt DJ, Malizia AL. Structural and functional brain changes in posttraumatic stress disorder. J Clin Psychiatry (2004) 65(Suppl 1):11–7.
160. Lupien SJ, Mcewen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci (2009) 10:434–45. doi:10.1038/nrn2639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. O’Malley D, Quigley EM, Dinan TG, Cryan JF. Do interactions between stress and immune responses lead to symptom exacerbations in irritable bowel syndrome? Brain Behav Immun (2011) 25:1333–41. doi:10.1016/j.bbi.2011.04.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev (1984) 5:25–44. doi:10.1210/edrv-5-1-25
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Bamberger CM, Schulte HM, Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev (1996) 17:245–61. doi:10.1210/edrv-17-3-245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. McEwen BS, Stellar E. Stress and the individual. Mechanisms leading to disease. Arch Intern Med (1993) 153:2093–101. doi:10.1001/archinte.153.18.2093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Buckley MM, O’Halloran KD, Rae MG, Dinan TG, O’Malley D. Modulation of enteric neurons by interleukin-6 and corticotropin-releasing factor contributes to visceral hypersensitivity and altered colonic motility in a rat model of irritable bowel syndrome. J Physiol (2014) 592:5235–50. doi:10.1113/jphysiol.2014.279968
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Greenwood-van Meerveld B, Johnson AC, Cochrane S, Schulkin J, Myers DA. Corticotropin-releasing factor 1 receptor-mediated mechanisms inhibit colonic hypersensitivity in rats. Neurogastroenterol Motil (2005) 17:415–22. doi:10.1111/j.1365-2982.2005.00648.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Trimble N, Johnson AC, Foster A, Greenwood-van Meerveld B. Corticotropin-releasing factor receptor 1-deficient mice show decreased anxiety and colonic sensitivity. Neurogastroenterol Motil (2007) 19:754–60. doi:10.1111/j.1365-2982.2007.00951.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Tran L, Schulkin J, Greenwood-van Meerveld B. Importance of CRF receptor-mediated mechanisms of the bed nucleus of the stria terminalis in the processing of anxiety and pain. Neuropsychopharmacology (2014) 39:2633–45. doi:10.1038/npp.2014.117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Tache Y, Martinez V, Wang L, Million M. CRF1 receptor signaling pathways are involved in stress-related alterations of colonic function and viscerosensitivity: implications for irritable bowel syndrome. Br J Pharmacol (2004) 141:1321–30. doi:10.1038/sj.bjp.0705760
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Tache Y, Martinez V, Million M, Maillot C. Role of corticotropin releasing factor receptor subtype 1 in stress-related functional colonic alterations: implications in irritable bowel syndrome. Eur J Surg (2002) (Suppl) 587:16–22.
171. Schwetz I, Mcroberts JA, Coutinho SV, Bradesi S, Gale G, Fanselow M, et al. Corticotropin-releasing factor receptor 1 mediates acute and delayed stress-induced visceral hyperalgesia in maternally separated Long-Evans rats. Am J Physiol Gastrointest Liver Physiol (2005) 289:G704–12. doi:10.1152/ajpgi.00498.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Million M, Wang L, Wang Y, Adelson DW, Yuan PQ, Maillot C, et al. CRF2 receptor activation prevents colorectal distension induced visceral pain and spinal ERK1/2 phosphorylation in rats. Gut (2006) 55:172–81. doi:10.1136/gut.2004.051391
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Tache Y, Million M, Nelson AG, Lamy C, Wang L. Role of corticotropin-releasing factor pathways in stress-related alterations of colonic motor function and viscerosensibility in female rodents. Gend Med (2005) 2:146–54. doi:10.1016/S1550-8579(05)80043-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Martinez V, Tache Y. CRF1 receptors as a therapeutic target for irritable bowel syndrome. Curr Pharm Des (2006) 12:4071–88. doi:10.2174/138161206778743637
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Larauche M, Gourcerol G, Wang L, Pambukchian K, Brunnhuber S, Adelson DW, et al. Cortagine, a CRF1 agonist, induces stresslike alterations of colonic function and visceral hypersensitivity in rodents primarily through peripheral pathways. Am J Physiol Gastrointest Liver Physiol (2009) 297:G215–27. doi:10.1152/ajpgi.00072.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Tache Y. Corticotrophin-releasing factor 1 activation in the central amygdale and visceral hyperalgesia. Neurogastroenterol Motil (2015) 27:1–6. doi:10.1111/nmo.12495
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Myers DA, Gibson M, Schulkin J, Greenwood van-Meerveld B. Corticosterone implants to the amygdala and type 1 CRH receptor regulation: effects on behavior and colonic sensitivity. Behav Brain Res (2005) 161:39–44. doi:10.1016/j.bbr.2005.03.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Hornig M. The role of microbes and autoimmunity in the pathogenesis of neuropsychiatric illness. Curr Opin Rheumatol (2013) 25:488–795. doi:10.1097/BOR.0b013e32836208de
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Longstreth GF, Thompson WG, Chey WD, Houghton LA, Mearin F, Spiller RC. Functional bowel disorders. Gastroenterology (2006) 130:1480–91. doi:10.1053/j.gastro.2005.11.061
180. Dinan TG, Cryan J, Shanahan F, Keeling PW, Quigley EM. IBS: an epigenetic perspective. Nat Rev Gastroenterol Hepatol (2010) 7:465–71. doi:10.1038/nrgastro.2010.99
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Elsenbruch S. Abdominal pain in irritable Bowel syndrome: a review of putative psychological, neural and neuro-immune mechanisms. Brain Behav Immun (2011) 25:386–94. doi:10.1016/j.bbi.2010.11.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Mayer EA. The neurobiology of stress and gastrointestinal disease. Gut (2000) 47:861–9. doi:10.1136/gut.47.6.861
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Mayer EA, Naliboff BD, Chang L, Coutinho SV. Stress and irritable bowel syndrome. Am J Physiol Gastrointest Liver Physiol (2001) 280:G519–24.
184. Mayer EA, Craske M, Naliboff BD. Depression, anxiety, and the gastrointestinal system. J Clin Psychiatry (2001) 62(Suppl 8):28–36.
185. McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med (1998) 338:171–9. doi:10.1056/NEJM199801153380307
186. Fukudo S. Stress and visceral pain: focusing on irritable bowel syndrome. Pain (2013) 154(Suppl 1):S63–70. doi:10.1016/j.pain.2013.09.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Tache Y, Martinez V, Million M, Rivier J. Corticotropin-releasing factor and the brain-gut motor response to stress. Can J Gastroenterol (1999) 13(Suppl A):18A–25A.
188. Barreau F, Ferrier L, Fioramonti J, Bueno L. New insights in the etiology and pathophysiology of irritable bowel syndrome: contribution of neonatal stress models. Pediatr Res (2007) 62:240–5. doi:10.1203/PDR.0b013e3180db2949
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Chitkara DK, van Tilburg MA, Blois-Martin N, Whitehead WE. Early life risk factors that contribute to irritable bowel syndrome in adults: a systematic review. Am J Gastroenterol (2008) 103:765–74. doi:10.1111/j.1572-0241.2007.01722.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Haglund ME, Nestadt PS, Cooper NS, Southwick SM, Charney DS. Psychobiological mechanisms of resilience: relevance to prevention and treatment of stress-related psychopathology. Dev Psychopathol (2007) 19:889–920. doi:10.1017/S0954579407000430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Charney DS. Psychobiological mechanisms of resilience and vulnerability: implications for successful adaptation to extreme stress. Am J Psychiatry (2004) 161:195–216. doi:10.1176/appi.ajp.161.2.195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Drossman DA, Whitehead WE, Toner BB, Diamant N, Hu YJB, Bangdiwala SI, et al. What determines severity among patients with painful functional bowel disorders[quest]. Am J Gastroenterol (2000) 95:974–80. doi:10.1111/j.1572-0241.2000.01936.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Dinan TG, Quigley EM, Ahmed SM, Scully P, O’Brien S, O’Mahony L, et al. Hypothalamic-pituitary-gut axis dysregulation in irritable bowel syndrome: plasma cytokines as a potential biomarker? Gastroenterology (2006) 130:304–11. doi:10.1053/j.gastro.2005.11.033
194. Hertig VL, Cain KC, Jarrett ME, Burr RL, Heitkemper MM. Daily stress and gastrointestinal symptoms in women with irritable bowel syndrome. Nurs Res (2007) 56:399–406. doi:10.1097/01.NNR.0000299855.60053.88
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Lee OY. [Psychosocial factors and visceral hypersensitivity in irritable bowel syndrome]. Korean J Gastroenterol (2006) 47:111–9.
196. Bonaz B, Rivest S. Effect of a chronic stress on CRF neuronal activity and expression of its type 1 receptor in the rat brain. Am J Physiol (1998) 275:R1438–49.
197. Seres G, Kovacs Z, Kovacs A, Kerekgyarto O, Sardi K, Demeter P, et al. Different associations of health related quality of life with pain, psychological distress and coping strategies in patients with irritable bowel syndrome and inflammatory bowel disorder. J Clin Psychol Med Settings (2008) 15:287–95. doi:10.1007/s10880-008-9132-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. Minderhoud IM, Oldenburg B, Wismeijer JA, van Berge Henegouwen GP, Smout AJ. IBS-like symptoms in patients with inflammatory bowel disease in remission; relationships with quality of life and coping behavior. Dig Dis Sci (2004) 49:469–74. doi:10.1023/B:DDAS.0000020506.84248.f9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. Jones MP, Wessinger S, Crowell MD. Coping strategies and interpersonal support in patients with irritable bowel syndrome and inflammatory bowel disease. Clin Gastroenterol Hepatol (2006) 4:474–81. doi:10.1016/j.cgh.2005.12.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
200. Deechakawan W, Cain KC, Jarrett ME, Burr RL, Heitkemper MM. Effect of self-management intervention on cortisol and daily stress levels in irritable bowel syndrome. Biol Res Nurs (2013) 15:26–36. doi:10.1177/1099800411414047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
201. Warren JW, Langenberg P, Clauw DJ. The number of existing functional somatic syndromes (FSSs) is an important risk factor for new, different FSSs. J Psychosom Res (2013) 74:12–7. doi:10.1016/j.jpsychores.2012.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
202. Tremolaterra F, Gallotta S, Morra Y, Lubrano E, Ciacci C, Iovino P. The severity of irritable bowel syndrome or the presence of fibromyalgia influencing the perception of visceral and somatic stimuli. BMC Gastroenterol (2014) 14:182. doi:10.1186/1471-230X-14-182
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
203. Thabane M, Kottachchi DT, Marshall JK. Systematic review and meta-analysis: the incidence and prognosis of post-infectious irritable bowel syndrome. Aliment Pharmacol Ther (2007) 26:535–44. doi:10.1111/j.1365-2036.2007.03399.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
204. Thabane M, Marshall JK. Post-infectious irritable bowel syndrome. World J Gastroenterol (2009) 15:3591–6. doi:10.3748/wjg.15.3591
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
205. Fendt M, Fanselow MS. The neuroanatomical and neurochemical basis of conditioned fear. Neurosci Biobehav Rev (1999) 23:743–60. doi:10.1016/S0149-7634(99)00016-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
206. Mayer EA, Naliboff BD, Chang L. Basic pathophysiologic mechanisms in irritable bowel syndrome. Dig Dis (2001) 19:212–8. doi:10.1159/000050682
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
207. Rhee SH, Pothoulakis C, Mayer EA. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat Rev Gastroenterol Hepatol (2009) 6:306–14. doi:10.1038/nrgastro.2009.35
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
208. Camara RJ, Ziegler R, Begre S, Schoepfer AM, von Kanel R, Swiss Inflammatory Bowel Disease Cohort Study, G. The role of psychological stress in inflammatory bowel disease: quality assessment of methods of 18 prospective studies and suggestions for future research. Digestion (2009) 80:129–39. doi:10.1159/000226087
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
209. Mawdsley JE, Rampton DS. The role of psychological stress in inflammatory bowel disease. Neuroimmunomodulation (2006) 13:327–36. doi:10.1159/000104861
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
210. Bonaz BL, Bernstein CN. Brain-gut interactions in inflammatory bowel disease. Gastroenterology (2013) 144:36–49. doi:10.1053/j.gastro.2012.10.003
211. Grenham S, Clarke G, Cryan JF, Dinan TG. Brain-gut-microbe communication in health and disease. Front Physiol (2011) 2:94. doi:10.3389/fphys.2011.00094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
212. Aziz Q, Thompson DG. Brain-gut axis in health and disease. Gastroenterology (1998) 114:559–78. doi:10.1016/S0016-5085(98)70540-2
213. Mayer EA. Gut feelings: the emerging biology of gut-brain communication. Nat Rev Neurosci (2011) 12:453–66. doi:10.1038/nrn3071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
214. Davari S, Talaei SA, Alaei H, Salami M. Probiotics treatment improves diabetes-induced impairment of synaptic activity and cognitive function: behavioral and electrophysiological proofs for microbiome-gut-brain axis. Neuroscience (2013) 240:287–96. doi:10.1016/j.neuroscience.2013.02.055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
215. Borre YE, O’Keeffe GW, Clarke G, Stanton C, Dinan TG, Cryan JF. Microbiota and neurodevelopmental windows: implications for brain disorders. Trends Mol Med (2014) 20:509–18. doi:10.1016/j.molmed.2014.05.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
216. Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci (2012) 13:701–12. doi:10.1038/nrn3346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
217. Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science (2012) 336:1262–7. doi:10.1126/science.1223813
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
218. Sam AH, Troke RC, Tan TM, Bewick GA. The role of the gut/brain axis in modulating food intake. Neuropharmacology (2012) 63:46–56. doi:10.1016/j.neuropharm.2011.10.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
219. Schellekens H, Finger BC, Dinan TG, Cryan JF. Ghrelin signalling and obesity: at the interface of stress, mood and food reward. Pharmacol Ther (2012) 135:316–26. doi:10.1016/j.pharmthera.2012.06.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
220. Ellekilde M, Selfjord E, Larsen CS, Jakesevic M, Rune I, Tranberg B, et al. Transfer of gut microbiota from lean and obese mice to antibiotic-treated mice. Sci Rep (2014) 4:5922. doi:10.1038/srep05922
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
221. Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell (2014) 158:705–21. doi:10.1016/j.cell.2014.05.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
222. Clarke G, Cryan JF, Dinan TG, Quigley EM. Review article: probiotics for the treatment of irritable bowel syndrome – focus on lactic acid bacteria. Aliment Pharmacol Ther (2012) 35:403–13. doi:10.1111/j.1365-2036.2011.04965.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
223. Galley JD, Nelson MC, Yu Z, Dowd SE, Walter J, Kumar PS, et al. Exposure to a social stressor disrupts the community structure of the colonic mucosa-associated microbiota. BMC Microbiol (2014) 14:189. doi:10.1186/1471-2180-14-189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
224. de Palma G, Collins SM, Bercik P, Verdu EF. The microbiota-gut-brain axis in gastrointestinal disorders: stressed bugs, stressed brain or both? J Physiol (2014) 592:2989–97. doi:10.1113/jphysiol.2014.273995
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
225. Crumeyrolle-Arias M, Jaglin M, Bruneau A, Vancassel S, Cardona A, Dauge V, et al. Absence of the gut microbiota enhances anxiety-like behavior and neuroendocrine response to acute stress in rats. Psychoneuroendocrinology (2014) 42:207–17. doi:10.1016/j.psyneuen.2014.01.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
226. Ait-Belgnaoui A, Colom A, Braniste V, Ramalho L, Marrot A, Cartier C, et al. Probiotic gut effect prevents the chronic psychological stress-induced brain activity abnormality in mice. Neurogastroenterol Motil (2014) 26:510–20. doi:10.1111/nmo.12295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
227. Xu D, Gao J, Gillilland M III, Wu X, Song I, Kao JY, et al. Rifaximin alters intestinal bacteria and prevents stress-induced gut inflammation and visceral hyperalgesia in rats. Gastroenterology (2014) 146:484.e–96.e. doi:10.1053/j.gastro.2013.10.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
228. Scott LV, Clarke G, Dinan TG. The brain-gut axis: a target for treating stress-related disorders. Mod Trends Pharmacopsychiatri (2013) 28:90–9. doi:10.1159/000343971
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
229. Collins SM, Surette M, Bercik P. The interplay between the intestinal microbiota and the brain. Nat Rev Microbiol (2012) 10:735–42. doi:10.1038/nrmicro2876
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
230. Bercik P. The microbiota-gut-brain axis: learning from intestinal bacteria? Gut (2011) 60:288–9. doi:10.1136/gut.2010.226779
231. Jeffery IB, O’Toole PW, Ohman L, Claesson MJ, Deane J, Quigley EM, et al. An irritable bowel syndrome subtype defined by species-specific alterations in faecal microbiota. Gut (2012) 61:997–1006. doi:10.1136/gutjnl-2011-301501
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
232. Matto J, Maunuksela L, Kajander K, Palva A, Korpela R, Kassinen A, et al. Composition and temporal stability of gastrointestinal microbiota in irritable bowel syndrome – a longitudinal study in IBS and control subjects. FEMS Immunol Med Microbiol (2005) 43:213–22. doi:10.1016/j.femsim.2004.08.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
233. Malinen E, Rinttila T, Kajander K, Matto J, Kassinen A, Krogius L, et al. Analysis of the fecal microbiota of irritable bowel syndrome patients and healthy controls with real-time PCR. Am J Gastroenterol (2005) 100:373–82. doi:10.1111/j.1572-0241.2005.40312.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
234. Kassinen A, Krogius-Kurikka L, Makivuokko H, Rinttila T, Paulin L, Corander J, et al. The fecal microbiota of irritable bowel syndrome patients differs significantly from that of healthy subjects. Gastroenterology (2007) 133:24–33. doi:10.1053/j.gastro.2007.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
235. Lyra A, Rinttila T, Nikkila J, Krogius-Kurikka L, Kajander K, Malinen E, et al. Diarrhoea-predominant irritable bowel syndrome distinguishable by 16S rRNA gene phylotype quantification. World J Gastroenterol (2009) 15:5936–45. doi:10.3748/wjg.15.5936
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
236. Tana C, Umesaki Y, Imaoka A, Handa T, Kanazawa M, Fukudo S. Altered profiles of intestinal microbiota and organic acids may be the origin of symptoms in irritable bowel syndrome. Neurogastroenterol Motil (2010) 22(512–9):e114–5. doi:10.1111/j.1365-2982.2009.01427.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
237. Mayer EA, Savidge T, Shulman RJ. Brain-gut microbiome interactions and functional bowel disorders. Gastroenterology (2014) 146:1500–12. doi:10.1053/j.gastro.2014.02.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
238. Theodorou V, Ait Belgnaoui A, Agostini S, Eutamene H. Effect of commensals and probiotics on visceral sensitivity and pain in irritable bowel syndrome. Gut Microb (2014) 5:430–6. doi:10.4161/gmic.29796
239. Distrutti E, Cipriani S, Mencarelli A, Renga B, Fiorucci S. Probiotics VSL#3 protect against development of visceral pain in murine model of irritable bowel syndrome. PLoS One (2013) 8:e63893. doi:10.1371/journal.pone.0063893
240. Dai C, Guandalini S, Zhao DH, Jiang M. Antinociceptive effect of VSL#3 on visceral hypersensitivity in a rat model of irritable bowel syndrome: a possible action through nitric oxide pathway and enhance barrier function. Mol Cell Biochem (2012) 362:43–53. doi:10.1007/s11010-011-1126-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
241. McKernan DP, Fitzgerald P, Dinan TG, Cryan JF. The probiotic Bifidobacterium infantis 35624 displays visceral antinociceptive effects in the rat. Neurogastroenterol Motil (2010) 22(1029–35):e268. doi:10.1111/j.1365-2982.2010.01520.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
242. Johnson AC, Greenwood-van Meerveld B, Mcrorie J. Effects of Bifidobacterium infantis 35624 on post-inflammatory visceral hypersensitivity in the rat. Dig Dis Sci (2011) 56:3179–86. doi:10.1007/s10620-011-1730-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
243. Agostini S, Goubern M, Tondereau V, Salvador-Cartier C, Bezirard V, Leveque M, et al. A marketed fermented dairy product containing Bifidobacterium lactis CNCM I-2494 suppresses gut hypersensitivity and colonic barrier disruption induced by acute stress in rats. Neurogastroenterol Motil (2012) 24:376–e172. doi:10.1111/j.1365-2982.2011.01865.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
244. Verdu EF, Bercik P, Verma-Gandhu M, Huang XX, Blennerhassett P, Jackson W, et al. Specific probiotic therapy attenuates antibiotic induced visceral hypersensitivity in mice. Gut (2006) 55:182–90. doi:10.1136/gut.2005.066100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
245. Kamiya T, Wang L, Forsythe P, Goettsche G, Mao Y, Wang Y, et al. Inhibitory effects of Lactobacillus reuteri on visceral pain induced by colorectal distension in Sprague-Dawley rats. Gut (2006) 55:191–6. doi:10.1136/gut.2005.070987
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
246. Rousseaux C, Thuru X, Gelot A, Barnich N, Neut C, Dubuquoy L, et al. Lactobacillus acidophilus modulates intestinal pain and induces opioid and cannabinoid receptors. Nat Med (2007) 13:35–7. doi:10.1038/nm1521
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
247. Eutamene H, Lamine F, Chabo C, Theodorou V, Rochat F, Bergonzelli GE, et al. Synergy between Lactobacillus paracasei and its bacterial products to counteract stress-induced gut permeability and sensitivity increase in rats. J Nutr (2007) 137:1901–7.
248. O’Mahony SM, Felice VD, Nally K, Savignac HM, Claesson MJ, Scully P, et al. Disturbance of the gut microbiota in early-life selectively affects visceral pain in adulthood without impacting cognitive or anxiety-related behaviors in male rats. Neuroscience (2014) 277:885–901. doi:10.1016/j.neuroscience.2014.07.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
249. Saadi M, McCallum RW. Rifaximin in irritable bowel syndrome: rationale, evidence and clinical use. Ther Adv Chronic Dis (2013) 4:71–5. doi:10.1177/2040622312472008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
250. Schoenfeld P, Pimentel M, Chang L, Lembo A, Chey WD, Yu J, et al. Safety and tolerability of rifaximin for the treatment of irritable bowel syndrome without constipation: a pooled analysis of randomised, double-blind, placebo-controlled trials. Aliment Pharmacol Ther (2014) 39:1161–8. doi:10.1111/apt.12735
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
251. Schmulson M, Bielsa MV, Carmona-Sanchez R, Hernandez A, Lopez-Colombo A, LOPEZ Vidal Y, et al. Microbiota, gastrointestinal infections, low-grade inflammation, and antibiotic therapy in irritable bowel syndrome: an evidence-based review. Rev Gastroenterol Mex (2014) 79:96–134. doi:10.1016/j.rgmx.2014.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
252. Pimentel M, Morales W, Chua K, Barlow G, Weitsman S, Kim G, et al. Effects of rifaximin treatment and retreatment in nonconstipated IBS subjects. Dig Dis Sci (2011) 56:2067–72. doi:10.1007/s10620-011-1728-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
253. Pimentel M, Lembo A, Chey WD, Zakko S, Ringel Y, Yu J, et al. Rifaximin therapy for patients with irritable bowel syndrome without constipation. N Engl J Med (2011) 364:22–32. doi:10.1056/NEJMoa1004409
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
254. Moraru IG, Portincasa P, Moraru AG, Diculescu M, Dumitrascu DL. Small intestinal bacterial overgrowth produces symptoms in irritable bowel syndrome which are improved by rifaximin. A pilot study. Rom J Intern Med (2013) 51:143–7.
255. Menees SB, Maneerattannaporn M, Kim HM, Chey WD. The efficacy and safety of rifaximin for the irritable bowel syndrome: a systematic review and meta-analysis. Am J Gastroenterol (2012) 107:28–35. doi:10.1038/ajg.2011.355
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
256. Jolley J. High-dose rifaximin treatment alleviates global symptoms of irritable bowel syndrome. Clin Exp Gastroenterol (2011) 4:43–8. doi:10.2147/CEG.S18051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
257. Boltin D, Perets TT, Shporn E, Aizic S, Levy S, Niv Y, et al. Rifaximin for small intestinal bacterial overgrowth in patients without irritable bowel syndrome. Ann Clin Microbiol Antimicrob (2014) 13:49. doi:10.1186/s12941-014-0049-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
258. Leonard BE. Impact of inflammation on neurotransmitter changes in major depression: an insight into the action of antidepressants. Prog Neuropsychopharmacol Biol Psychiatry (2014) 48:261–7. doi:10.1016/j.pnpbp.2013.10.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
259. Skaper SD, Facci L, Giusti P. Neuroinflammation, microglia and mast cells in the pathophysiology of neurocognitive disorders: a review. CNS Neurol Disord Drug Targets (2014) 13:1654–66.
260. Leonard BE. The concept of depression as a dysfunction of the immune system. Curr Immunol Rev (2010) 6:205–12. doi:10.2174/157339510791823835
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
261. Marchand F, Perretti M, Mcmahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci (2005) 6:521–32. doi:10.1038/nrn1700
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
262. Bengmark S. Gut microbiota, immune development and function. Pharmacol Res (2013) 69:87–113. doi:10.1016/j.phrs.2012.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
263. Leonard BE. The HPA and immune axes in stress: the involvement of the serotonergic system. Eur Psychiatry (2005) 20(Suppl 3):S302–6. doi:10.1016/S0924-9338(05)80180-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
264. Bateman A, Singh A, Kral T, Solomon S. The immune-hypothalamic-pituitary-adrenal axis. Endocr Rev (1989) 10:92–112. doi:10.1210/edrv-10-1-92
265. Genton L, Kudsk KA. Interactions between the enteric nervous system and the immune system: role of neuropeptides and nutrition. Am J Surg (2003) 186:253–8. doi:10.1016/S0002-9610(03)00210-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
266. Hori T, Katafuchi T, Take S, Shimizu N, Niijima A. The autonomic nervous system as a communication channel between the brain and the immune system. Neuroimmunomodulation (1995) 2:203–15. doi:10.1159/000097198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
267. Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987-2007). Brain Behav Immun (2007) 21:736–45. doi:10.1016/j.bbi.2007.03.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
268. Graeber MB. Changing face of microglia. Science (2010) 330:783–8. doi:10.1126/science.1190929
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
269. Lu CL. Spinal microglia: a potential target in the treatment of chronic visceral pain. J Chin Med Assoc (2014) 77:3–9. doi:10.1016/j.jcma.2013.08.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
270. Cervero F. Visceral pain: mechanisms of peripheral and central sensitization. Ann Med (1995) 27:235–9. doi:10.3109/07853899509031965
271. Bradesi S. Role of spinal cord glia in the central processing of peripheral pain perception. Neurogastroenterol Motil (2010) 22:499–511. doi:10.1111/j.1365-2982.2010.01491.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
272. Gosselin RD, Suter MR, Ji RR, Decosterd I. Glial cells and chronic pain. Neuroscientist (2010) 16:519–31. doi:10.1177/1073858409360822
273. Inoue K, Tsuda M. Microglia and neuropathic pain. Glia (2009) 57:1469–79. doi:10.1002/glia.20871
274. Graeber MB, Christie MJ. Multiple mechanisms of microglia: a gatekeeper’s contribution to pain states. Exp Neurol (2012) 234:255–61. doi:10.1016/j.expneurol.2012.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
275. Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci (2007) 10:1361–8. doi:10.1038/nn1992
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
276. Tsuda M, Beggs S, Salter MW, Inoue K. Microglia and intractable chronic pain. Glia (2013) 61:55–61. doi:10.1002/glia.22379
277. Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci (2005) 28:101–7. doi:10.1016/j.tins.2004.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
278. Zhang Y, Li H, Li Y, Sun X, Zhu M, Hanley G, et al. Essential role of toll-like receptor 2 in morphine-induced microglia activation in mice. Neurosci Lett (2011) 489:43–7. doi:10.1016/j.neulet.2010.11.063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
279. Mika J, Zychowska M, Popiolek-Barczyk K, Rojewska E, Przewlocka B. Importance of glial activation in neuropathic pain. Eur J Pharmacol (2013) 716:106–19. doi:10.1016/j.ejphar.2013.01.072
280. Saab CY, Wang J, Gu C, Garner KN, Al-Chaer ED. Microglia: a newly discovered role in visceral hypersensitivity? Neuron Glia Biol (2006) 2:271–7. doi:10.1017/S1740925X07000439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
281. Riazi K, Galic MA, Kuzmiski JB, Ho W, Sharkey KA, Pittman QJ. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc Natl Acad Sci U S A (2008) 105:17151–6. doi:10.1073/pnas.0806682105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
282. Delafoy L, Raymond F, Doherty AM, Eschalier A, Diop L. Role of nerve growth factor in the trinitrobenzene sulfonic acid-induced colonic hypersensitivity. Pain (2003) 105:489–97. doi:10.1016/S0304-3959(03)00266-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
283. Liu PY, Lu CL, Wang CC, Lee IH, Hsieh JC, Chen CC, et al. Spinal microglia initiate and maintain hyperalgesia in a rat model of chronic pancreatitis. Gastroenterology (2012) 142(165–173):e2. doi:10.1053/j.gastro.2011.09.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
284. Bradesi S, Svensson CI, Steinauer J, Pothoulakis C, Yaksh TL, Mayer EA. Role of spinal microglia in visceral hyperalgesia and NK1R up-regulation in a rat model of chronic stress. Gastroenterology (2009) 136(1339–48):e1–2. doi:10.1053/j.gastro.2008.12.044
285. Dunlop SP, Jenkins D, Neal KR, Naesdal J, Borgaonker M, Collins SM, et al. Randomized, double-blind, placebo-controlled trial of prednisolone in post-infectious irritable bowel syndrome. Aliment Pharmacol Ther (2003) 18:77–84. doi:10.1046/j.1365-2036.2003.01640.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
286. Casanova JL, Abel L, Quintana-Murci L. Human TLRs and Il-1Rs in host defense: natural insights from evolutionary, epidemiological, and clinical genetics. Annu Rev Immunol (2011) 29:447–91. doi:10.1146/annurev-immunol-030409-101335
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
287. Takeda K, Akira S. TLR signaling pathways. Semin Immunol (2004) 16:3–9. doi:10.1016/j.smim.2003.10.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
288. Cario E. Toll-like receptors in inflammatory bowel diseases: a decade later. Inflamm Bowel Dis (2010) 16:1583–97. doi:10.1002/ibd.21282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
289. Tramullas M, Finger BC, Moloney RD, Golubeva AV, Moloney G, Dinan TG, et al. Toll-like receptor 4 regulates chronic stress-induced visceral pain in mice. Biol Psychiatry (2014) 76:340–8. doi:10.1016/j.biopsych.2013.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
290. Brint EK, Macsharry J, Fanning A, Shanahan F, Quigley EM. Differential expression of toll-like receptors in patients with irritable bowel syndrome. Am J Gastroenterol (2011) 106:329–36. doi:10.1038/ajg.2010.438
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
291. McKernan DP, Gaszner G, Quigley EM, Cryan JF, Dinan TG. Altered peripheral toll-like receptor responses in the irritable bowel syndrome. Aliment Pharmacol Ther (2011) 33:1045–52. doi:10.1111/j.1365-2036.2011.04624.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
292. McKernan DP, Nolan A, Brint EK, O’Mahony SM, Hyland NP, Cryan JF, et al. Toll-like receptor mRNA expression is selectively increased in the colonic mucosa of two animal models relevant to irritable bowel syndrome. PLoS One (2009) 4:e8226. doi:10.1371/journal.pone.0008226
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
293. Mbodji K, Torre S, Haas V, Dechelotte P, Marion-Letellier R. Alanyl-glutamine restores maternal deprivation-induced TLR4 levels in a rat neonatal model. Clin Nutr (2011) 30:672–7. doi:10.1016/j.clnu.2011.04.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
294. Barajon I, Serrao G, Arnaboldi F, Opizzi E, Ripamonti G, Balsari A, et al. Toll-like receptors 3, 4, and 7 are expressed in the enteric nervous system and dorsal root ganglia. J Histochem Cytochem (2009) 57:1013–23. doi:10.1369/jhc.2009.953539
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
295. Ochoa-Cortes F, Ramos-Lomas T, Miranda-Morales M, Spreadbury I, Ibeakanma C, Barajas-Lopez C, et al. Bacterial cell products signal to mouse colonic nociceptive dorsal root ganglia neurons. Am J Physiol Gastrointest Liver Physiol (2010) 299:G723–32. doi:10.1152/ajpgi.00494.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
296. Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, et al. Activation of innate immunity in the CNS triggers neurodegeneration through a toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A (2003) 100:8514–9. doi:10.1073/pnas.1432609100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
297. Bettoni I, Comelli F, Rossini C, Granucci F, Giagnoni G, Peri F, et al. Glial TLR4 receptor as new target to treat neuropathic pain: efficacy of a new receptor antagonist in a model of peripheral nerve injury in mice. Glia (2008) 56:1312–9. doi:10.1002/glia.20699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
298. Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, Watkins LR. Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence, and reward. ScientificWorldJournal (2007) 7:98–111. doi:10.1100/tsw.2007.230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
299. Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, et al. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun (2010) 24:83–95. doi:10.1016/j.bbi.2009.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
300. Lewis SS, Hutchinson MR, Rezvani N, Loram LC, Zhang Y, Maier SF, et al. Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1β. Neuroscience (2010) 165:569–83. doi:10.1016/j.neuroscience.2009.10.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
301. Watkins LR, Hutchinson MR, Rice KC, Maier SF. The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci (2009) 30:581–91. doi:10.1016/j.tips.2009.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
302. Mogil JS. Sex differences in pain and pain inhibition: multiple explanations of a controversial phenomenon. Nat Rev Neurosci (2012) 13:859–66. doi:10.1038/nrn3360
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
303. Meleine M, Matricon J. Gender-related differences in irritable bowel syndrome: potential mechanisms of sex hormones. World J Gastroenterol (2014) 20:6725–43. doi:10.3748/wjg.v20.i22.6725
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
304. Mulak A, Tache Y, Larauche M. Sex hormones in the modulation of irritable bowel syndrome. World J Gastroenterol (2014) 20:2433–48. doi:10.3748/wjg.v20.i10.2433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
305. Mogil JS, Bailey AL. Sex and gender differences in pain and analgesia. Prog Brain Res (2010) 186:141–57. doi:10.1016/B978-0-444-53630-3.00009-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
306. Adeyemo MA, Spiegel BM, Chang L. Meta-analysis: do irritable bowel syndrome symptoms vary between men and women? Aliment Pharmacol Ther (2010) 32:738–55. doi:10.1111/j.1365-2036.2010.04409.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
307. Heitkemper MM, Chang L. Do fluctuations in ovarian hormones affect gastrointestinal symptoms in women with irritable bowel syndrome? Gend Med (2009) 6(Suppl 2):152–67. doi:10.1016/j.genm.2009.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
308. Fillingim RB, King CD, Ribeiro-Dasilva MC, Rahim-Williams B, Riley JL III. Sex, gender, and pain: a review of recent clinical and experimental findings. J Pain (2009) 10:447–85. doi:10.1016/j.jpain.2008.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
309. Gui X, Carraway RE, Dobner PR. Endogenous neurotensin facilitates visceral nociception and is required for stress-induced antinociception in mice and rats. Neuroscience (2004) 126:1023–32. doi:10.1016/j.neuroscience.2004.04.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
310. Moloney RD, O’Leary OF, Felice D, Bettler B, Dinan TG, Cryan JF. Early-life stress induces visceral hypersensitivity in mice. Neurosci Lett (2012) 512:99–102. doi:10.1016/j.neulet.2012.01.066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
311. Larauche M, Gourcerol G, Million M, Adelson DW, Tache Y. Repeated psychological stress-induced alterations of visceral sensitivity and colonic motor functions in mice: influence of surgery and postoperative single housing on visceromotor responses. Stress (2010) 13:343–54. doi:10.3109/10253891003664166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
312. Ouyang A, Wrzos HF. Contribution of gender to pathophysiology and clinical presentation of IBS: should management be different in women? Am J Gastroenterol (2006) 101:S602–9. doi:10.1111/j.1572-0241.2006.00975.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
313. Heitkemper M, Jarrett M. Irritable bowel syndrome: does gender matter? J Psychosom Res (2008) 64:583–7. doi:10.1016/j.jpsychores.2008.02.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
314. Lee OY, Mayer EA, Schmulson M, Chang L, Naliboff B. Gender-related differences in IBS symptoms. Am J Gastroenterol (2001) 96:2184–93. doi:10.1111/j.1572-0241.2001.03961.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
315. North CS, Downs D, Clouse RE, Alrakawi A, Dokucu ME, Cox J, et al. The presentation of irritable bowel syndrome in the context of somatization disorder. Clin Gastroenterol Hepatol (2004) 2:787–95. doi:10.1016/S1542-3565(04)00350-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
316. Heitkemper MM, Jarrett ME, Levy RL, Cain KC, Burr RL, Feld A, et al. Self-management for women with irritable bowel syndrome. Clin Gastroenterol Hepatol (2004) 2:585–96. doi:10.1016/S1542-3565(04)00242-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
317. Drossman DA, Toner BB, Whitehead WE, Diamant NE, Dalton CB, Duncan S, et al. Cognitive-behavioral therapy versus education and desipramine versus placebo for moderate to severe functional bowel disorders. Gastroenterology (2003) 125:19–31. doi:10.1016/S0016-5085(03)00669-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
318. Blanchard EB, Lackner JM, Sanders K, Krasner S, Keefer L, Payne A, et al. A controlled evaluation of group cognitive therapy in the treatment of irritable bowel syndrome. Behav Res Ther (2007) 45:633–48. doi:10.1016/j.brat.2006.07.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
319. Anbardan SJ, Daryani NE, Fereshtehnejad SM, Taba Taba, Vakili S, Keramati MR, et al. Gender role in irritable bowel syndrome: a comparison of irritable bowel syndrome module (ROME III) between male and female patients. J Neurogastroenterol Motil (2012) 18:70–7. doi:10.5056/jnm.2012.18.1.70
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
320. Heitkemper MM, Cain KC, Jarrett ME, Burr RL, Hertig V, Bond EF. Symptoms across the menstrual cycle in women with irritable bowel syndrome. Am J Gastroenterol (2003) 98:420–30. doi:10.1111/j.1572-0241.2003.07233.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
321. Altman G, Cain KC, Motzer S, Jarrett M, Burr R, Heitkemper M. Increased symptoms in female IBS patients with dysmenorrhea and PMS. Gastroenterol Nurs (2006) 29:4–11. doi:10.1097/00001610-200601000-00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
322. Bjornsson B, Orvar KB, Theodors A, Kjeld M. [The relationship of gastrointestinal symptoms and menstrual cycle phase in young healthy women]. Laeknabladid (2006) 92:677–82.
323. Whitehead WE, Cheskin LJ, Heller BR, Robinson JC, Crowell MD, Benjamin C, et al. Evidence for exacerbation of irritable bowel syndrome during menses. Gastroenterology (1990) 98:1485–9.
324. Laessle RG, Tuschl RJ, Schweiger U, Pirke KM. Mood changes and physical complaints during the normal menstrual cycle in healthy young women. Psychoneuroendocrinology (1990) 15:131–8. doi:10.1016/0306-4530(90)90021-Z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
325. Kane SV, Sable K, Hanauer SB. The menstrual cycle and its effect on inflammatory bowel disease and irritable bowel syndrome: a prevalence study. Am J Gastroenterol (1998) 93:1867–72. doi:10.1111/j.1572-0241.1998.540_i.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
326. Lesniewska B, Nowak M, Malendowicz LK. Sex differences in adrenocortical structure and function. XXVIII. ACTH and corticosterone in intact, gonadectomised and gonadal hormone replaced rats. Horm Metab Res (1990) 22:378–81.
327. Kitay JI. Sex differences in adrenal cortical secretion in the rat. Endocrinology (1961) 68:818–24. doi:10.1210/endo-68-5-818
328. Kant GJ, Lenox RH, Bunnell BN, Mougey EH, Pennington LL, Meyerhoff JL. Comparison of stress response in male and female rats: pituitary cyclic AMP and plasma prolactin, growth hormone and corticosterone. Psychoneuroendocrinology (1983) 8:421–8. doi:10.1016/0306-4530(83)90021-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
329. Critchlow V, Liebelt RA, Bar-Sela M, Mountcastle W, Lipscomb HS. Sex difference in resting pituitary-adrenal function in the rat. Am J Physiol (1963) 205:807–15.
330. Allen-Rowlands CF, Allen JP, Greer MA, Wilson M. Circadian rhythmicity of ACTH and corticosterone in the rat. J Endocrinol Invest (1980) 3:371–7. doi:10.1007/BF03349373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
331. Chen X-N, Zhu H, Meng Q-Y, Zhou J-N. Estrogen receptor-α and-β regulate the human corticotropin-releasing hormone gene through similar pathways. Brain Res (2008) 1223:1–10. doi:10.1016/j.brainres.2008.05.043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
332. Mulak A, Taché Y. Sex difference in irritable bowel syndrome: do gonadal hormones play a role? Gastroenterol Pol (2010) 17:89.
333. McCormick CM, Linkroum W, Sallinen BJ, Miller NW. Peripheral and central sex steroids have differential effects on the HPA axis of male and female rats. Stress (2002) 5:235–47. doi:10.1080/1025389021000061165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
334. Atkinson HC, Waddell BJ. Circadian variation in basal plasma corticosterone and adrenocorticotropin in the rat: sexual dimorphism and changes across the estrous cycle. Endocrinology (1997) 138:3842–8. doi:10.1210/en.138.9.3842
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
335. Bohler HC Jr, Zoeller RT, King JC, Rubin BS, Weber R, Merriam GR. Corticotropin releasing hormone mRNA is elevated on the afternoon of proestrus in the parvocellular paraventricular nuclei of the female rat. Brain Res Mol Brain Res (1990) 8:259–62. doi:10.1016/0169-328X(90)90025-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
336. Buckingham JC, Dohler KD, Wilson CA. Activity of the pituitary-adrenocortical system and thyroid gland during the oestrous cycle of the rat. J Endocrinol (1978) 78:359–66. doi:10.1677/joe.0.0780359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
337. Linthorst A. Interactions Between Corticotropin-Releasing Hormone and Serotonin: Implications for the Aetiology and Treatment of Anxiety Disorders. Anxiety and Anxiolytic Drugs. Berlin: Springer (2005).
338. Lu CL, Hsieh JC, Dun NJ, Oprea TI, Wang PS, Luo JC, et al. Estrogen rapidly modulates 5-hydroxytrytophan-induced visceral hypersensitivity via GPR30 in rats. Gastroenterology (2009) 137:1040–50. doi:10.1053/j.gastro.2009.03.047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
339. Warnock JK, Clayton AH. Chronic episodic disorders in women. Psychiatr Clin North Am (2003) 26:725–40. doi:10.1016/S0193-953X(03)00042-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
340. Mermelstein PG. Membrane-localised oestrogen receptor alpha and beta influence neuronal activity through activation of metabotropic glutamate receptors. J Neuroendocrinol (2009) 21:257–62. doi:10.1111/j.1365-2826.2009.01838.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
341. Aloisi AM, Affaitati G, Ceccarelli I, Fiorenzani P, Lerza R, Rossi C, et al. Estradiol and testosterone differently affect visceral pain-related behavioural responses in male and female rats. Eur J Pain (2010) 14:602–7. doi:10.1016/j.ejpain.2009.10.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
342. Sapsed-Byrne S, Ma D, Ridout D, Holdcroft A. Estrous cycle phase variations in visceromotor and cardiovascular responses to colonic distension in the anesthetized rat. Brain Res (1996) 742:10–6. doi:10.1016/S0006-8993(96)00989-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
343. Holdcroft A, Sapsed-Byrne S, Ma D, Hammal D, Forsling ML. Sex and oestrous cycle differences in visceromotor responses and vasopressin release in response to colonic distension in male and female rats anaesthetized with halothane. Br J Anaesth (2000) 85:907–10. doi:10.1093/bja/85.6.907
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
344. Ji Y, Tang B, Traub RJ. The visceromotor response to colorectal distention fluctuates with the estrous cycle in rats. Neuroscience (2008) 154:1562–7. doi:10.1016/j.neuroscience.2008.04.070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
345. Sorge RE, Martin LJ, Isbester KA, Sotocinal SG, Rosen S, Tuttle AH, et al. Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nat Methods (2014) 11:629–32. doi:10.1038/nmeth.2935
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
346. Petrenko AB, Yamakura T, Baba H, Shimoji K. The role of N-methyl-D-aspartate (NMDA) receptors in pain: a review. Anesth Analg (2003) 97:1108–16. doi:10.1213/01.ANE.0000081061.12235.55
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
347. Holzer P, Holzer-Petsche U. Tachykinin receptors in the gut: physiological and pathological implications. Curr Opin Pharmacol (2001) 1:583–90. doi:10.1016/S1471-4892(01)00100-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
348. Blackshaw LA, Gebhart GF. The pharmacology of gastrointestinal nociceptive pathways. Curr Opin Pharmacol (2002) 2:642–9. doi:10.1016/S1471-4892(02)00211-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
349. Coutinho SV, Urban MO, Gebhart GF. Role of glutamate receptors and nitric oxide in the rostral ventromedial medulla in visceral hyperalgesia. Pain (1998) 78:59–69. doi:10.1016/S0304-3959(98)00137-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
350. McRoberts JA, Coutinho SV, Marvizon JC, Grady EF, Tognetto M, Sengupta JN, et al. Role of peripheral N-methyl-D-aspartate (NMDA) receptors in visceral nociception in rats. Gastroenterology (2001) 120:1737–48. doi:10.1053/gast.2001.24848
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
351. Goudet C, Magnaghi V, Landry M, Nagy F, Gereau RWT, Pin JP. Metabotropic receptors for glutamate and GABA in pain. Brain Res Rev (2009) 60:43–56. doi:10.1016/j.brainresrev.2008.12.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
352. Chen Y, Bacon G, Sher E, Clark BP, Kallman MJ, Wright RA, et al. Evaluation of the activity of a novel metabotropic glutamate receptor antagonist (±)-2-amino-2-(3-cis and trans-carboxycyclobutyl-3-(9-thioxanthyl)propionic acid) in the in vitro neonatal spinal cord and in an in vivo pain model. Neuroscience (2000) 95:787–93. doi:10.1016/S0306-4522(99)00496-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
353. Ji G, Neugebauer V. Reactive oxygen species are involved in group I mGluR-mediated facilitation of nociceptive processing in amygdala neurons. J Neurophysiol (2010) 104:218–29. doi:10.1152/jn.00223.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
354. Bianchi R, Rezzani R, Borsani E, Rodella L. mGlu5 receptor antagonist decreases Fos expression in spinal neurons after noxious visceral stimulation. Brain Res (2003) 960:263–6. doi:10.1016/S0006-8993(02)03697-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
355. Liaw W, Stephens R, Binns B, Chu Y, Sepkuty J, Johns R, et al. Spinal glutamate uptake is critical for maintaining normal sensory transmission in rat spinal cord. Pain (2005) 115:60–70. doi:10.1016/j.pain.2005.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
356. Tao YX, Gu J, Stephens RL Jr. Role of spinal cord glutamate transporter during normal sensory transmission and pathological pain states. Mol Pain (2005) 1:30. doi:10.1186/1744-8069-1-30
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
357. Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain (2009) 10:895–926. doi:10.1016/j.jpain.2009.06.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
358. Hwang JH, Yaksh TL. The effect of spinal GABA receptor agonists on tactile allodynia in a surgically-induced neuropathic pain model in the rat. Pain (1997) 70:15–22. doi:10.1016/S0304-3959(96)03249-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
359. Rode F, Jensen DG, Blackburn-Munro G, Bjerrum OJ. Centrally-mediated antinociceptive actions of GABA(A) receptor agonists in the rat spared nerve injury model of neuropathic pain. Eur J Pharmacol (2005) 516:131–8. doi:10.1016/j.ejphar.2005.04.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
360. Reichl S, Augustin M, Zahn PK, Pogatzki-Zahn EM. Peripheral and spinal GABAergic regulation of incisional pain in rats. Pain (2012) 153:129–41. doi:10.1016/j.pain.2011.09.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
361. Hyland NP, Cryan JF. A gut feeling about GABA: focus on GABA(B) receptors. Front Pharmacol (2010) 1:124. doi:10.3389/fphar.2010.00124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
362. Abelli L, Conte B, Somma V, Maggi CA, Giuliani S, Meli A. A method for studying pain arising from the urinary bladder in conscious, freely-moving rats. J Urol (1989) 141:148–51.
363. Hara K, Saito Y, Kirihara Y, Yamada Y, Sakura S, Kosaka Y. The interaction of antinociceptive effects of morphine and GABA receptor agonists within the rat spinal cord. Anesth Analg (1999) 89:422–7. doi:10.1213/00000539-199908000-00032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
364. Brusberg M, Ravnefjord A, Martinsson R, Larsson H, Martinez V, Lindstrom E. The GABA(B) receptor agonist, baclofen, and the positive allosteric modulator, CGP7930, inhibit visceral pain-related responses to colorectal distension in rats. Neuropharmacology (2009) 56:362–7. doi:10.1016/j.neuropharm.2008.09.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
365. Liu LS, Shenoy M, Pasricha PJ. The analgesic effects of the GABAB receptor agonist, baclofen, in a rodent model of functional dyspepsia. Neurogastroenterol Motil (2011) 23(356–61):e160–1. doi:10.1111/j.1365-2982.2010.01649.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
366. Lu Y, Westlund KN. Effects of baclofen on colon inflammation-induced Fos, CGRP and SP expression in spinal cord and brainstem. Brain Res (2001) 889:118–30. doi:10.1016/S0006-8993(00)03124-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
367. Sengupta JN. Visceral pain: the neurophysiological mechanism. Handb Exp Pharmacol (2009) 194:31–74. doi:10.1007/978-3-540-79090-7_2
368. Smid SD, Young RL, Cooper NJ, Blackshaw LA. GABA(B)R expressed on vagal afferent neurones inhibit gastric mechanosensitivity in ferret proximal stomach. Am J Physiol Gastrointest Liver Physiol (2001) 281:G1494–501.
369. Partosoedarso ER, Young RL, Blackshaw LA. GABA(B) receptors on vagal afferent pathways: peripheral and central inhibition. Am J Physiol Gastrointest Liver Physiol (2001) 280:G658–68.
370. Page AJ, Blackshaw LA. GABA(B) receptors inhibit mechanosensitivity of primary afferent endings. J Neurosci (1999) 19:8597–602.
371. Sengupta JN, Medda BK, Shaker R. Effect of GABA(B) receptor agonist on distension-sensitive pelvic nerve afferent fibers innervating rat colon. Am J Physiol Gastrointest Liver Physiol (2002) 283:G1343–51. doi:10.1152/ajpgi.00124.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
372. Isomoto S, Kaibara M, Sakurai-Yamashita Y, Nagayama Y, Uezono Y, Yano K, et al. Cloning and tissue distribution of novel splice variants of the rat GABAB receptor. Biochem Biophys Res Commun (1998) 253:10–5. doi:10.1006/bbrc.1998.9706
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
373. Cryan JF, Kaupmann K. Don’t worry ‘B’ happy!: a role for GABA(B) receptors in anxiety and depression. Trends Pharmacol Sci (2005) 26:36–43. doi:10.1016/j.tips.2004.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
374. Houghton LA, Fell C, Whorwell PJ, Jones I, Sudworth DP, Gale JD. Effect of a second-generation alpha2delta ligand (pregabalin) on visceral sensation in hypersensitive patients with irritable bowel syndrome. Gut (2007) 56:1218–25. doi:10.1136/gut.2006.110858
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
375. Gale JD, Houghton LA. Alpha 2 delta (alpha(2)delta) ligands, gabapentin and pregabalin: what is the evidence for potential use of these ligands in irritable bowel syndrome. Front Pharmacol (2011) 2:28. doi:10.3389/fphar.2011.00028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
376. Mozaffari S, Nikfar S, Abdollahi M. Metabolic and toxicological considerations for the latest drugs used to treat irritable bowel syndrome. Expert Opin Drug Metab Toxicol (2013) 9:403–21. doi:10.1517/17425255.2013.759558
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
377. Zhang MM, Liu SB, Chen T, Koga K, Zhang T, Li YQ, et al. Effects of NB001 and gabapentin on irritable bowel syndrome-induced behavioral anxiety and spontaneous pain. Mol Brain (2014) 7:47. doi:10.1186/1756-6606-7-47
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
378. Camilleri M. Genetics and irritable bowel syndrome: from genomics to intermediate phenotype and pharmacogenetics. Dig Dis Sci (2009) 54:2318–24. doi:10.1007/s10620-009-0903-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
379. Camilleri M. Genetics of human gastrointestinal sensation. Neurogastroenterol Motil (2013) 25:458–66. doi:10.1111/nmo.12132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
380. Camilleri M, Busciglio I, Carlson P, Mckinzie S, Burton D, Baxter K, et al. Candidate genes and sensory functions in health and irritable bowel syndrome. Am J Physiol Gastrointest Liver Physiol (2008) 295:G219–25. doi:10.1152/ajpgi.90202.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
381. Ek WE, Reznichenko A, Ripke S, Niesler B, Zucchelli M, Rivera NV, et al. Exploring the genetics of irritable bowel syndrome: a GWA study in the general population and replication in multinational case-control cohorts. Gut (2014). doi:10.1136/gutjnl-2014-307997
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
382. Camilleri M, Carlson P, Acosta A, Busciglio I, Nair AA, Gibbons SJ, et al. RNA sequencing shows transcriptomic changes in rectosigmoid mucosa in patients with irritable bowel syndrome-diarrhea: a pilot case-control study. Am J Physiol Gastrointest Liver Physiol (2014) 306:G1089–98. doi:10.1152/ajpgi.00068.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
383. Swan C, Duroudier NP, Campbell E, Zaitoun A, Hastings M, Dukes GE, et al. Identifying and testing candidate genetic polymorphisms in the irritable bowel syndrome (IBS): association with TNFSF15 and TNFalpha. Gut (2013) 62:985–94. doi:10.1136/gutjnl-2011-301213
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
384. Saito YA. The role of genetics in IBS. Gastroenterol Clin North Am (2011) 40:45–67. doi:10.1016/j.gtc.2010.12.011
385. Beyder A, Mazzone A, Strege PR, Tester DJ, Saito YA, Bernard CE, et al. Loss-of-function of the voltage-gated sodium channel NaV1.5 (channelopathies) in patients with irritable bowel syndrome. Gastroenterology (2014) 146:1659–68. doi:10.1053/j.gastro.2014.02.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
386. Camilleri M, Shin A, Busciglio I, Carlson P, Acosta A, Bharucha AE, et al. Genetic variation in GPBAR1 predisposes to quantitative changes in colonic transit and bile acid excretion. Am J Physiol Gastrointest Liver Physiol (2014) 307:G508–16. doi:10.1152/ajpgi.00178.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
387. Wouters MM, Lambrechts D, Knapp M, Cleynen I, Whorwell P, Agreus L, et al. Genetic variants in CDC42 and NXPH1 as susceptibility factors for constipation and diarrhoea predominant irritable bowel syndrome. Gut (2014) 63:1103–11. doi:10.1136/gutjnl-2013-304570
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
388. Jacobson LH, Cryan JF. Genetic approaches to modeling anxiety in animals. Curr Top Behav Neurosci (2010) 2:161–201. doi:10.1007/7854_2009_31
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
389. Jacobson LH, Cryan JF. Feeling strained? Influence of genetic background on depression-related behavior in mice: a review. Behav Genet (2007) 37:171–213. doi:10.1007/s10519-006-9106-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
390. Lariviere WR, Wilson SG, Laughlin TM, Kokayeff A, West EE, Adhikari SM, et al. Heritability of nociception. III. Genetic relationships among commonly used assays of nociception and hypersensitivity. Pain (2002) 97:75–86. doi:10.1016/S0304-3959(01)00492-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
391. Mogil JS, Wilson SG, Bon K, Lee SE, Chung K, Raber P, et al. Heritability of nociception II. ‘Types’ of nociception revealed by genetic correlation analysis. Pain (1999) 80:83–93. doi:10.1016/S0304-3959(98)00196-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
392. Mogil JS, Wilson SG, Bon K, Lee SE, Chung K, Raber P, et al. Heritability of nociception I: responses of 11 inbred mouse strains on 12 measures of nociception. Pain (1999) 80:67–82. doi:10.1016/S0304-3959(98)00197-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
393. Elsenbruch S, Wang L, Hollerbach S, Schedlowski M, Tougas G. Pseudo-affective visceromotor responses and HPA axis activation following colorectal distension in rats with increased cholinergic sensitivity. Neurogastroenterol Motil (2004) 16:801–9. doi:10.1111/j.1365-2982.2004.00563.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
394. O’Malley D, Julio-Pieper M, O’Mahony SM, Dinan TG, Cryan JF. Differential visceral pain sensitivity and colonic morphology in four common laboratory rat strains. Exp Physiol (2014) 99:359–67. doi:10.1113/expphysiol.2013.076109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
395. Tang WY, Ho SM. Epigenetic reprogramming and imprinting in origins of disease. Rev Endocr Metab Disord (2007) 8:173–82. doi:10.1007/s11154-007-9042-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
396. Bai G, Wei D, Zou S, Ren K, Dubner R. Inhibition of class II histone deacetylases in the spinal cord attenuates inflammatory hyperalgesia. Mol Pain (2010) 6:51. doi:10.1186/1744-8069-6-51
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
397. Chiechio S, Copani A, Zammataro M, Battaglia G, Gereau RWT, Nicoletti F. Transcriptional regulation of type-2 metabotropic glutamate receptors: an epigenetic path to novel treatments for chronic pain. Trends Pharmacol Sci (2010) 31:153–60. doi:10.1016/j.tips.2009.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
398. Denk F, McMahon SB. Chronic pain: emerging evidence for the involvement of epigenetics. Neuron (2012) 73:435–44. doi:10.1016/j.neuron.2012.01.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
399. Chiechio S, Zammataro M, Morales ME, Busceti CL, Drago F, Gereau RWT, et al. Epigenetic modulation of mGlu2 receptors by histone deacetylase inhibitors in the treatment of inflammatory pain. Mol Pharmacol (2009) 75:1014–20. doi:10.1124/mol.108.054346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
400. Tran L, Chaloner A, Sawalha AH, Greenwood van-Meerveld B. Importance of epigenetic mechanisms in visceral pain induced by chronic water avoidance stress. Psychoneuroendocrinology (2013) 38:898–906. doi:10.1016/j.psyneuen.2012.09.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
401. Hong S, Zheng G, Wiley JW. Epigenetic regulation of genes that modulate chronic stress-induced visceral pain in the peripheral nervous system. Gastroenterology (2015) 148:148–57. doi:10.1053/j.gastro.2014.09.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
402. Winston JH, Li Q, Sarna SK. Chronic prenatal stress epigenetically modifies spinal cord BDNF expression to induce sex-specific visceral hypersensitivity in offspring. Neurogastroenterol Motil (2014) 26:715–30. doi:10.1111/nmo.12326
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
403. van den Wijngaard RM, Stanisor OI, van Diest SA, Welting O, Wouters MM, Cailotto C, et al. Susceptibility to stress induced visceral hypersensitivity in maternally separated rats is transferred across generations. Neurogastroenterol Motil (2013) 25:e780–90. doi:10.1111/nmo.12202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
404. Theodorou V. Susceptibility to stress-induced visceral sensitivity: a bad legacy for next generations. Neurogastroenterol Motil (2013) 25:927–30. doi:10.1111/nmo.12260
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
405. Geranton SM, Morenilla-Palao C, Hunt SP. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J Neurosci (2007) 27:6163–73. doi:10.1523/JNEUROSCI.1306-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
406. Geranton SM, Fratto V, Tochiki KK, Hunt SP. Descending serotonergic controls regulate inflammation-induced mechanical sensitivity and methyl-CpG-binding protein 2 phosphorylation in the rat superficial dorsal horn. Mol Pain (2008) 4:35. doi:10.1186/1744-8069-4-35
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
407. Stilling RM, Dinan TG, Cryan JF. Microbial genes, brain & behaviour – epigenetic regulation of the gut-brain axis. Genes Brain Behav (2013) 13:69–86. doi:10.1111/gbb.12109
408. O’Mahony SM, Tramullas M, Fitzgerald P, Cryan JF. Rodent models of colorectal distension. Curr Protoc Neurosci (2012) 61:9.40.1–13. doi:10.1002/0471142301.ns0940s61
409. Ness TJ, Gebhart GF. Colorectal distension as a noxious visceral stimulus: physiologic and pharmacologic characterization of pseudaffective reflexes in the rat. Brain Res (1988) 450:153–69. doi:10.1016/0006-8993(88)91555-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
410. Bradesi S, Schwetz I, Ennes HS, Lamy CM, Ohning G, Fanselow M, et al. Repeated exposure to water avoidance stress in rats: a new model for sustained visceral hyperalgesia. Am J Physiol Gastrointest Liver Physiol (2005) 289:G42–53. doi:10.1152/ajpgi.00500.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
411. Christianson JA, Gebhart GF. Assessment of colon sensitivity by luminal distension in mice. Nat Protoc (2007) 2:2624–31. doi:10.1038/nprot.2007.392
412. Larsson M, Arvidsson S, Ekman C, Bayati A. A model for chronic quantitative studies of colorectal sensitivity using balloon distension in conscious mice – effects of opioid receptor agonists. Neurogastroenterol Motil (2003) 15:371–81. doi:10.1046/j.1365-2982.2003.00418.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
413. Nijsen MJ, Ongenae NG, Coulie B, Meulemans AL. Telemetric animal model to evaluate visceral pain in the freely moving rat. Pain (2003) 105:115–23. doi:10.1016/S0304-3959(03)00170-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
414. Welting O, van den Wijngaard RM, de Jonge WJ, Holman R, Boeckxstaens GE. Assessment of visceral sensitivity using radio telemetry in a rat model of maternal separation. Neurogastroenterol Motil (2005) 17:838–45. doi:10.1111/j.1365-2982.2005.00677.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
415. Arvidsson S, Larsson M, Larsson H, Lindstrom E, Martinez V. Assessment of visceral pain-related pseudo-affective responses to colorectal distension in mice by intracolonic manometric recordings. J Pain (2006) 7:108–18. doi:10.1016/j.jpain.2005.09.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
416. Tammpere A, Brusberg M, Axenborg J, Hirsch I, Larsson H, Lindstrom E. Evaluation of pseudo-affective responses to noxious colorectal distension in rats by manometric recordings. Pain (2005) 116:220–6. doi:10.1016/j.pain.2005.04.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
417. Al-Chaer ED, Kawasaki M, Pasricha PJ. A new model of chronic visceral hypersensitivity in adult rats induced by colon irritation during postnatal development. Gastroenterology (2000) 119:1276–85. doi:10.1053/gast.2000.19576
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
418. Wang Z, Bradesi S, Maarek JM, Lee K, Winchester WJ, Mayer EA, et al. Regional brain activation in conscious, nonrestrained rats in response to noxious visceral stimulation. Pain (2008) 138:233–43. doi:10.1016/j.pain.2008.04.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
419. Marques AH, O’Connor TG, Roth C, Susser E, Bjorke-Monsen AL. The influence of maternal prenatal and early childhood nutrition and maternal prenatal stress on offspring immune system development and neurodevelopmental disorders. Front Neurosci (2013) 7:120. doi:10.3389/fnins.2013.00120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
420. Bale TL, Baram TZ, Brown AS, Goldstein JM, Insel TR, McCarthy MM, et al. Early life programming and neurodevelopmental disorders. Biol Psychiatry (2010) 68:314–9. doi:10.1016/j.biopsych.2010.05.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
421. Morgane PJ, Mokler DJ, Galler JR. Effects of prenatal protein malnutrition on the hippocampal formation. Neurosci Biobehav Rev (2002) 26:471–83. doi:10.1016/S0149-7634(02)00012-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
422. Sampath D, Sabitha KR, Hegde P, Jayakrishnan HR, Kutty BM, Chattarji S, et al. A study on fear memory retrieval and REM sleep in maternal separation and isolation stressed rats. Behav Brain Res (2014) 273C:144–54. doi:10.1016/j.bbr.2014.07.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
423. Takase K, Yamamoto Y, Yagami T. Maternal deprivation in the middle of a stress hyporesponsive period decreases hippocampal calcineurin expression and causes abnormal social and cognitive behaviours in adult male Wistar rats: relevance to negative symptoms of schizophrenia. Behav Brain Res (2012) 232:306–15. doi:10.1016/j.bbr.2012.04.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
424. Paus T, Keshavan M, Giedd JN. Why do many psychiatric disorders emerge during adolescence? Nat Rev Neurosci (2008) 9:947–57. doi:10.1038/nrn2513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
425. Larauche M, Mulak A, Tache Y. Stress-related alterations of visceral sensation: animal models for irritable bowel syndrome study. J Neurogastroenterol Motil (2011) 17:213–34. doi:10.5056/jnm.2011.17.3.213
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
426. Bradesi S, Mayer EA. Experimental models of stress and pain: do they help to develop new therapies? Dig Dis (2009) 27(Suppl 1):55–67. doi:10.1159/000268122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
427. Chaloner A, Greenwood-van Meerveld B. Early life adversity as a risk factor for visceral pain in later life: importance of sex differences. Front Neurosci (2013) 7:13. doi:10.3389/fnins.2013.00013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
428. Mayer EA, Bradesi S, Chang L, Spiegel BM, Bueller JA, Naliboff BD. Functional GI disorders: from animal models to drug development. Gut (2008) 57:384–404. doi:10.1136/gut.2006.101675
429. Holschneider DP, Bradesi S, Mayer EA. The role of experimental models in developing new treatments for irritable bowel syndrome. Expert Rev Gastroenterol Hepatol (2011) 5:43–57. doi:10.1586/egh.10.88
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
430. Qin HY, Wu JC, Tong XD, Sung JJ, Xu HX, Bian ZX. Systematic review of animal models of post-infectious/post-inflammatory irritable bowel syndrome. J Gastroenterol (2011) 46:164–74. doi:10.1007/s00535-010-0321-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
431. Bulmer DC, Grundy D. Achieving translation in models of visceral pain. Curr Opin Pharmacol (2011) 11:575–81. doi:10.1016/j.coph.2011.09.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
432. Folks DG. The interface of psychiatry and irritable bowel syndrome. Curr Psychiatry Rep (2004) 6:210–5. doi:10.1007/s11920-004-0066-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
433. Lee S, Wu J, Ma YL, Tsang A, Guo WJ, Sung J. Irritable bowel syndrome is strongly associated with generalized anxiety disorder: a community study. Aliment Pharmacol Ther (2009) 30:643–51. doi:10.1111/j.1365-2036.2009.04074.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
434. Thijssen AY, Jonkers DM, Leue C, van der Veek PP, Vidakovic-Vukic M, van Rood YR, et al. Dysfunctional cognitions, anxiety and depression in irritable bowel syndrome. J Clin Gastroenterol (2010) 44:e236–41. doi:10.1097/MCG.0b013e3181eed5d8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
435. Bakshi VP, Kalin NH. Corticotropin-releasing hormone and animal models of anxiety: gene-environment interactions. Biol Psychiatry (2000) 48:1175–98. doi:10.1016/S0006-3223(00)01082-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
436. Delic S, Streif S, Deussing JM, Weber P, Ueffing M, Holter SM, et al. Genetic mouse models for behavioral analysis through transgenic RNAi technology. Genes Brain Behav (2008) 7:821–30. doi:10.1111/j.1601-183X.2008.00412.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
437. Deussing JM, Wurst W. Dissecting the genetic effect of the CRH system on anxiety and stress-related behaviour. C R Biol (2005) 328:199–212. doi:10.1016/j.crvi.2005.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
438. Kimura M, Muller-Preuss P, Lu A, Wiesner E, Flachskamm C, Wurst W, et al. Conditional corticotropin-releasing hormone overexpression in the mouse forebrain enhances rapid eye movement sleep. Mol Psychiatry (2010) 15:154–65. doi:10.1038/mp.2009.46
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
439. Kolber BJ, Boyle MP, Wieczorek L, Kelley CL, Onwuzurike CC, Nettles SA, et al. Transient early-life forebrain corticotropin-releasing hormone elevation causes long-lasting anxiogenic and despair-like changes in mice. J Neurosci (2010) 30:2571–81. doi:10.1523/JNEUROSCI.4470-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
440. Lu A, Steiner MA, Whittle N, Vogl AM, Walser SM, Ableitner M, et al. Conditional mouse mutants highlight mechanisms of corticotropin-releasing hormone effects on stress-coping behavior. Mol Psychiatry (2008) 13:1028–42. doi:10.1038/mp.2008.51
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
441. Homberg JR, Lesch KP. Looking on the bright side of serotonin transporter gene variation. Biol Psychiatry (2011) 69:513–9. doi:10.1016/j.biopsych.2010.09.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
442. Ferguson SM, Neumaier JF. Grateful DREADDs: engineered receptors reveal how neural circuits regulate behavior. Neuropsychopharmacology (2012) 37:296–7. doi:10.1038/npp.2011.179
443. Videlock EJ, Adeyemo M, Licudine A, Hirano M, Ohning G, Mayer M, et al. Childhood trauma is associated with hypothalamic-pituitary-adrenal axis responsiveness in irritable bowel syndrome. Gastroenterology (2009) 137:1954–62. doi:10.1053/j.gastro.2009.08.058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
444. O’Mahony SM, Hyland NP, Dinan TG, Cryan JF. Maternal separation as a model of brain-gut axis dysfunction. Psychopharmacology (Berl) (2011) 214:71–88. doi:10.1007/s00213-010-2010-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
445. Plotsky PM, Meaney MJ. Early, postnatal experience alters hypothalamic corticotropin-releasing factor (CRF) mRNA, median eminence CRF content and stress-induced release in adult rats. Brain Res Mol Brain Res (1993) 18:195–200. doi:10.1016/0169-328X(93)90189-V
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
446. Rosztoczy A, Fioramonti J, Jarmay K, Barreau F, Wittmann T, Bueno L. Influence of sex and experimental protocol on the effect of maternal deprivation on rectal sensitivity to distension in the adult rat. Neurogastroenterol Motil (2003) 15:679–86. doi:10.1046/j.1350-1925.2003.00451.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
447. Fish EW, Shahrokh D, Bagot R, Caldji C, Bredy T, Szyf M, et al. Epigenetic programming of stress responses through variations in maternal care. Ann N Y Acad Sci (2004) 1036:167–80. doi:10.1196/annals.1330.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
448. Francis DD, Meaney MJ. Maternal care and the development of stress responses. Curr Opin Neurobiol (1999) 9:128–34. doi:10.1016/S0959-4388(99)80016-6
449. Francis D, Diorio J, Liu D, Meaney MJ. Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science (1999) 286:1155–8. doi:10.1126/science.286.5442.1155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
450. Szyf M, Weaver I, Meaney M. Maternal care, the epigenome and phenotypic differences in behavior. Reprod Toxicol (2007) 24:9–19. doi:10.1016/j.reprotox.2007.05.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
451. Barreau F, Cartier C, Ferrier L, Fioramonti J, Bueno L. Nerve growth factor mediates alterations of colonic sensitivity and mucosal barrier induced by neonatal stress in rats. Gastroenterology (2004) 127:524–34. doi:10.1053/j.gastro.2004.05.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
452. Coutinho SV, Plotsky PM, Sablad M, Miller JC, Zhou H, Bayati AI, et al. Neonatal maternal separation alters stress-induced responses to viscerosomatic nociceptive stimuli in rat. Am J Physiol Gastrointest Liver Physiol (2002) 282:G307–16. doi:10.1152/ajpgi.00240.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
453. Millstein RA, Holmes A. Effects of repeated maternal separation on anxiety- and depression-related phenotypes in different mouse strains. Neurosci Biobehav Rev (2007) 31:3–17. doi:10.1016/j.neubiorev.2006.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
454. Savignac HM, Dinan TG, Cryan JF. Resistance to early-life stress in mice: effects of genetic background and stress duration. Front Behav Neurosci (2011) 5:13. doi:10.3389/fnbeh.2011.00013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
455. Weiss IC, Franklin TB, Vizi S, Mansuy IM. Inheritable effect of unpredictable maternal separation on behavioral responses in mice. Front Behav Neurosci (2011) 5:3. doi:10.3389/fnbeh.2011.00003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
456. Lin C, Al-Chaer ED. Long-term sensitization of primary afferents in adult rats exposed to neonatal colon pain. Brain Res (2003) 971:73–82. doi:10.1016/S0006-8993(03)02358-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
457. Bonaz B, Tache Y. Water-avoidance stress-induced c-fos expression in the rat brain and stimulation of fecal output: role of corticotropin-releasing factor. Brain Res (1994) 641:21–8. doi:10.1016/0006-8993(94)91810-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
458. Enck P, Merlin V, Erckenbrecht JF, Wienbeck M. Stress effects on gastrointestinal transit in the rat. Gut (1989) 30:455–9. doi:10.1136/gut.30.4.455
459. Schwetz I, Bradesi S, Mcroberts JA, Sablad M, Miller JC, Zhou H, et al. Delayed stress-induced colonic hypersensitivity in male Wistar rats: role of neurokinin-1 and corticotropin-releasing factor-1 receptors. Am J Physiol Gastrointest Liver Physiol (2004) 286:G683–91. doi:10.1152/ajpgi.00358.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
460. Gue M, del Rio-Lacheze C, Eutamene H, Theodorou V, Fioramonti J, Bueno L. Stress-induced visceral hypersensitivity to rectal distension in rats: role of CRF and mast cells. Neurogastroenterol Motil (1997) 9:271–9. doi:10.1046/j.1365-2982.1997.d01-63.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
461. Bradesi S, Eutamene H, Garcia-Villar R, Fioramonti J, Bueno L. Acute and chronic stress differently affect visceral sensitivity to rectal distension in female rats. Neurogastroenterol Motil (2002) 14:75–82. doi:10.1046/j.1365-2982.2002.00305.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
462. Larauche M, Mulak A, Kim YS, Labus J, Million M, Tache Y. Visceral analgesia induced by acute and repeated water avoidance stress in rats: sex difference in opioid involvement. Neurogastroenterol Motil (2012) 24:1031–e547. doi:10.1111/j.1365-2982.2012.01980.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
463. Bennett EJ, Tennant CC, Piesse C, Badcock CA, Kellow JE. Level of chronic life stress predicts clinical outcome in irritable bowel syndrome. Gut (1998) 43:256–61. doi:10.1136/gut.43.2.256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
464. Chang L. Review article: epidemiology and quality of life in functional gastrointestinal disorders. Aliment Pharmacol Ther (2004) 20(Suppl 7):31–9. doi:10.1111/j.1365-2036.2004.02183.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
465. Choung RS, Locke GR III, Zinsmeister AR, Schleck CD, Talley NJ. Psychosocial distress and somatic symptoms in community subjects with irritable bowel syndrome: a psychological component is the rule. Am J Gastroenterol (2009) 104:1772–9. doi:10.1038/ajg.2009.239
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
466. Lackner JM, Brasel AM, Quigley BM, Keefer L, Krasner SS, Powell C, et al. The ties that bind: perceived social support, stress, and IBS in severely affected patients. Neurogastroenterol Motil (2010) 22:893–900. doi:10.1111/j.1365-2982.2010.01516.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
467. Yang PC, Jury J, Soderholm JD, Sherman PM, Mckay DM, Perdue MH. Chronic psychological stress in rats induces intestinal sensitization to luminal antigens. Am J Pathol (2006) 168:104–14. doi:10.2353/ajpath.2006.050575
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
468. Hong S, Fan J, Kemmerer ES, Evans S, Li Y, Wiley JW. Reciprocal changes in vanilloid (TRPV1) and endocannabinoid (CB1) receptors contribute to visceral hyperalgesia in the water avoidance stressed rat. Gut (2009) 58:202–10. doi:10.1136/gut.2008.157594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
469. Larsson MH, Miketa A, Martinez V. Lack of interaction between psychological stress and DSS-induced colitis affecting colonic sensitivity during colorectal distension in mice. Stress (2009) 12:434–44. doi:10.1080/10253890802626603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
470. Zheng J, Babygirija R, Bulbul M, Cerjak D, Ludwig K, Takahashi T. Hypothalamic oxytocin mediates adaptation mechanism against chronic stress in rats. Am J Physiol Gastrointest Liver Physiol (2010) 299:G946–53. doi:10.1152/ajpgi.00483.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
471. Patel S, Hillard CJ. Adaptations in endocannabinoid signaling in response to repeated homotypic stress: a novel mechanism for stress habituation. Eur J Neurosci (2008) 27:2821–9. doi:10.1111/j.1460-9568.2008.06266.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
472. Black LV, Ness TJ, Robbins MT. Effects of oxytocin and prolactin on stress-induced bladder hypersensitivity in female rats. J Pain (2009) 10:1065–72. doi:10.1016/j.jpain.2009.04.007
473. Sanson M, Bueno L, Fioramonti J. Involvement of cannabinoid receptors in inflammatory hypersensitivity to colonic distension in rats. Neurogastroenterol Motil (2006) 18:949–56. doi:10.1111/j.1365-2982.2006.00819.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
474. Winston JH, Xu GY, Sarna SK. Adrenergic stimulation mediates visceral hypersensitivity to colorectal distension following heterotypic chronic stress. Gastroenterology (2010) 138(294–304):e3. doi:10.1053/j.gastro.2009.09.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
475. Seo HJ, Ham HD, Jin HY, Lee WH, Hwang HS, Park SA, et al. Chronic administration of monosodium glutamate under chronic variable stress impaired hypothalamic-pituitary-adrenal axis function in rats. Korean J Physiol Pharmacol (2010) 14:213–21. doi:10.4196/kjpp.2010.14.4.213
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
476. Tagliari B, Dos Santos TM, Cunha AA, Lima DD, Delwing D, Sitta A, et al. Chronic variable stress induces oxidative stress and decreases butyrylcholinesterase activity in blood of rats. J Neural Transm (2010) 117:1067–76. doi:10.1007/s00702-010-0445-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
477. Willner P. Validity, reliability and utility of the chronic mild stress model of depression: a 10-year review and evaluation. Psychopharmacology (Berl) (1997) 134:319–29. doi:10.1007/s002130050473
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
478. Babygirija R, Zheng J, Ludwig K, Takahashi T. Central oxytocin is involved in restoring impaired gastric motility following chronic repeated stress in mice. Am J Physiol Regul Integr Comp Physiol (2010) 298:R157–65. doi:10.1152/ajpregu.00328.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
479. Griffiths J, Shanks N, Anisman H. Strain-specific alterations in consumption of a palatable diet following repeated stressor exposure. Pharmacol Biochem Behav (1992) 42:219–27. doi:10.1016/0091-3057(92)90519-L
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
480. Muscat R, Willner P. Suppression of sucrose drinking by chronic mild unpredictable stress: a methodological analysis. Neurosci Biobehav Rev (1992) 16:507–17. doi:10.1016/S0149-7634(05)80192-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
481. Finger BC, Dinan TG, Cryan JF. High-fat diet selectively protects against the effects of chronic social stress in the mouse. Neuroscience (2011) 192:351–60. doi:10.1016/j.neuroscience.2011.06.072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
482. Reber SO, Obermeier F, Straub RH, Falk W, Neumann ID. Chronic intermittent psychosocial stress (social defeat/overcrowding) in mice increases the severity of an acute DSS-induced colitis and impairs regeneration. Endocrinology (2006) 147:4968–76. doi:10.1210/en.2006-0347
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
483. Vicario M, Alonso C, Guilarte M, Serra J, Martinez C, Gonzalez-Castro AM, et al. Chronic psychosocial stress induces reversible mitochondrial damage and corticotropin-releasing factor receptor type-1 upregulation in the rat intestine and IBS-like gut dysfunction. Psychoneuroendocrinology (2012) 37:65–77. doi:10.1016/j.psyneuen.2011.05.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
484. Cohen H, Jotkowitz A, Buskila D, Pelles-Avraham S, Kaplan Z, Neumann L, et al. Post-traumatic stress disorder and other co-morbidities in a sample population of patients with irritable bowel syndrome. Eur J Intern Med (2006) 17:567–71. doi:10.1016/j.ejim.2006.07.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
485. Drossman DA, Leserman J, Nachman G, Li ZM, Gluck H, Toomey TC, et al. Sexual and physical abuse in women with functional or organic gastrointestinal disorders. Ann Intern Med (1990) 113:828–33. doi:10.7326/0003-4819-113-11-828
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
486. Savas LS, White DL, Wieman M, Daci K, Fitzgerald S, Laday Smith S, et al. Irritable bowel syndrome and dyspepsia among women veterans: prevalence and association with psychological distress. Aliment Pharmacol Ther (2009) 29:115–25. doi:10.1111/j.1365-2036.2008.03847.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
487. White DL, Savas LS, Daci K, Elserag R, Graham DP, Fitzgerald SJ, et al. Trauma history and risk of the irritable bowel syndrome in women veterans. Aliment Pharmacol Ther (2010) 32:551–61. doi:10.1111/j.1365-2036.2010.04387.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
488. Rau V, Decola JP, Fanselow MS. Stress-induced enhancement of fear learning: an animal model of posttraumatic stress disorder. Neurosci Biobehav Rev (2005) 29:1207–23. doi:10.1016/j.neubiorev.2005.04.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
489. Stam R. PTSD and stress sensitisation: a tale of brain and body Part 2: animal models. Neurosci Biobehav Rev (2007) 31:558–84. doi:10.1016/j.neubiorev.2007.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
490. Stam R, Akkermans LM, Wiegant VM. Trauma and the gut: interactions between stressful experience and intestinal function. Gut (1997) 40:704–9. doi:10.1136/gut.40.6.704
491. Wang W, Liu Y, Zheng H, Wang HN, Jin X, Chen YC, et al. A modified single-prolonged stress model for post-traumatic stress disorder. Neurosci Lett (2008) 441:237–41. doi:10.1016/j.neulet.2008.06.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
492. Stam R, van Laar TJ, Akkermans LM, Wiegant VM. Variability factors in the expression of stress-induced behavioural sensitisation. Behav Brain Res (2002) 132:69–76. doi:10.1016/S0166-4328(01)00387-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
493. Collins SM, Vallance B, Barbara G, Borgaonkar M. Putative inflammatory and immunological mechanisms in functional bowel disorders. Baillieres Best Pract Res Clin Gastroenterol (1999) 13:429–36. doi:10.1053/bega.1999.0037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
494. Spiller R, Garsed K. Postinfectious irritable bowel syndrome. Gastroenterology (2009) 136:1979–88. doi:10.1053/j.gastro.2009.02.074
495. Long Y, Liu Y, Tong J, Qian W, Hou X. Effectiveness of trimebutine maleate on modulating intestinal hypercontractility in a mouse model of postinfectious irritable bowel syndrome. Eur J Pharmacol (2010) 636:159–65. doi:10.1016/j.ejphar.2010.03.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
496. McLean PG, Picard C, Garcia-Villar R, More J, Fioramonti J, Bueno L. Effects of nematode infection on sensitivity to intestinal distension: role of tachykinin NK2 receptors. Eur J Pharmacol (1997) 337:279–82. doi:10.1016/S0014-2999(97)01275-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
497. Coelho AM, Fioramonti J, Bueno L. Systemic lipopolysaccharide influences rectal sensitivity in rats: role of mast cells, cytokines, and vagus nerve. Am J Physiol Gastrointest Liver Physiol (2000) 279:G781–90.
498. O’Mahony SM, Clarke G, Mckernan DP, Bravo JA, Dinan TG, Cryan JF. Differential visceral nociceptive, behavioural and neurochemical responses to an immune challenge in the stress-sensitive Wistar Kyoto rat strain. Behav Brain Res (2013) 253:310–7. doi:10.1016/j.bbr.2013.07.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
499. Ohman L, Simren M. Pathogenesis of IBS: role of inflammation, immunity and neuroimmune interactions. Nat Rev Gastroenterol Hepatol (2010) 7:163–73. doi:10.1038/nrgastro.2010.4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
500. Akiho H, Ihara E, Nakamura K. Low-grade inflammation plays a pivotal role in gastrointestinal dysfunction in irritable bowel syndrome. World J Gastrointest Pathophysiol (2010) 1:97–105. doi:10.4291/wjgp.v1.i3.97
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
501. Barbara G, de Giorgio R, Stanghellini V, Cremon C, Corinaldesi R. A role for inflammation in irritable bowel syndrome? Gut (2002) 51(Suppl 1):i41–4. doi:10.1136/gut.51.suppl_1.i41
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
502. Collins SM, Piche T, Rampal P. The putative role of inflammation in the irritable bowel syndrome. Gut (2001) 49:743–5. doi:10.1136/gut.49.6.743
503. Keohane J, O’Mahony C, O’Mahony L, O’Mahony S, Quigley EM, Shanahan F. Irritable bowel syndrome-type symptoms in patients with inflammatory bowel disease: a real association or reflection of occult inflammation? Am J Gastroenterol (2010) 105(1788):1789–94. doi:10.1038/ajg.2010.156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
504. Long MD, Drossman DA. Inflammatory bowel disease, irritable bowel syndrome, or what?: a challenge to the functional-organic dichotomy. Am J Gastroenterol (2010) 105:1796–8. doi:10.1038/ajg.2010.162
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
505. Spiller R, Garsed K. Infection, inflammation, and the irritable bowel syndrome. Dig Liver Dis (2009) 41:844–9. doi:10.1016/j.dld.2009.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
506. Barbara G, Cremon C, Stanghellini V. Inflammatory bowel disease and irritable bowel syndrome: similarities and differences. Curr Opin Gastroenterol (2014) 30:352–8. doi:10.1097/MOG.0000000000000070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
507. Ji Y, Tang B, Traub RJ. Modulatory effects of estrogen and progesterone on colorectal hyperalgesia in the rat. Pain (2005) 117:433–42. doi:10.1016/j.pain.2005.07.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
508. Palecek J, Willis WD. The dorsal column pathway facilitates visceromotor responses to colorectal distention after colon inflammation in rats. Pain (2003) 104:501–7. doi:10.1016/S0304-3959(03)00075-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
509. Burton MB, Gebhart GF. Effects of intracolonic acetic acid on responses to colorectal distension in the rat. Brain Res (1995) 672:77–82. doi:10.1016/0006-8993(94)01382-R
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
510. Coutinho SV, Meller ST, Gebhart GF. Intracolonic zymosan produces visceral hyperalgesia in the rat that is mediated by spinal NMDA and non-NMDA receptors. Brain Res (1996) 736:7–15. doi:10.1016/0006-8993(96)00661-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
511. Traub RJ, Murphy A. Colonic inflammation induces fos expression in the thoracolumbar spinal cord increasing activity in the spinoparabrachial pathway. Pain (2002) 95:93–102. doi:10.1016/S0304-3959(01)00381-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
512. Adam B, Liebregts T, Gschossmann JM, Krippner C, Scholl F, Ruwe M, et al. Severity of mucosal inflammation as a predictor for alterations of visceral sensory function in a rat model. Pain (2006) 123:179–86. doi:10.1016/j.pain.2006.02.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
513. Gschossmann JM, Liebregts T, Adam B, Buenger L, Ruwe M, Gerken G, et al. Long-term effects of transient chemically induced colitis on the visceromotor response to mechanical colorectal distension. Dig Dis Sci (2004) 49:96–101. doi:10.1023/B:DDAS.0000011609.68882.3a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
514. Zhou Q, Price DD, Caudle RM, Verne GN. Visceral and somatic hypersensitivity in a subset of rats following Tnbs-induced colitis. Pain (2008) 134:9–15. doi:10.1016/j.pain.2007.03.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
515. Verma-Gandhu M, Verdu EF, Bercik P, Blennerhassett PA, Al-Mutawaly N, Ghia JE, et al. Visceral pain perception is determined by the duration of colitis and associated neuropeptide expression in the mouse. Gut (2007) 56:358–64. doi:10.1136/gut.2006.100016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
516. Larsson MH, Rapp L, Lindstrom E. Effect of DSS-induced colitis on visceral sensitivity to colorectal distension in mice. Neurogastroenterol Motil (2006) 18:144–52. doi:10.1111/j.1365-2982.2005.00736.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
517. Deiteren A, Vermeulen W, Moreels TG, Pelckmans PA, de Man JG, de Winter BY. The effect of chemically induced colitis, psychological stress and their combination on visceral pain in female Wistar rats. Stress (2014) 17:431–44. doi:10.3109/10253890.2014.951034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
518. Reber SO, Obermeier F, Straub RH, Veenema AH, Neumann ID. Aggravation of DSS-induced colitis after chronic subordinate colony (CSC) housing is partially mediated by adrenal mechanisms. Stress (2008) 11:225–34. doi:10.1080/10253890701733351
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
519. Veenema AH, Reber SO, Selch S, Obermeier F, Neumann ID. Early life stress enhances the vulnerability to chronic psychosocial stress and experimental colitis in adult mice. Endocrinology (2008) 149:2727–36. doi:10.1210/en.2007-1469
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
520. Melgar S, Engstrom K, Jagervall A, Martinez V. Psychological stress reactivates dextran sulfate sodium-induced chronic colitis in mice. Stress (2008) 11:348–62. doi:10.1080/10253890701820166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
521. Saunders PR, Miceli P, Vallance BA, Wang L, Pinto S, Tougas G, et al. Noradrenergic and cholinergic neural pathways mediate stress-induced reactivation of colitis in the rat. Auton Neurosci (2006) 124:56–68. doi:10.1016/j.autneu.2005.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
522. Collins SM, Mchugh K, Jacobson K, Khan I, Riddell R, Murase K, et al. Previous inflammation alters the response of the rat colon to stress. Gastroenterology (1996) 111:1509–15. doi:10.1016/S0016-5085(96)70012-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
523. Liebregts T, Adam B, Bertel A, Lackner C, Neumann J, Talley NJ, et al. Psychological stress and the severity of post-inflammatory visceral hyperalgesia. Eur J Pain (2007) 11:216–22. doi:10.1016/j.ejpain.2006.02.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
524. Armuzzi A, Cremonini F, Ojetti V, Bartolozzi F, Canducci F, Candelli M, et al. Effect of Lactobacillus GG supplementation on antibiotic-associated gastrointestinal side effects during Helicobacter pylori eradication therapy: a pilot study. Digestion (2001) 63:1–7. doi:10.1159/000051865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
525. Bercik P, Denou E, Collins J, Jackson W, Lu J, Jury J, et al. The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology (2011) 141:e1–3. doi:10.1053/j.gastro.2011.04.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: visceral pain, stress, psychological, animal models, irritable-bowel syndrome, colorectal distension, microbiota–gut–brain axis
Citation: Moloney RD, O’Mahony SM, Dinan TG and Cryan JF (2015) Stress-induced visceral pain: toward animal models of irritable-bowel syndrome and associated comorbidities. Front. Psychiatry 6:15. doi: 10.3389/fpsyt.2015.00015
Received: 31 October 2014; Accepted: 28 January 2015;
Published online: 16 February 2015.
Edited by:
David A. Slattery, University of Regensburg, GermanyReviewed by:
Yvette France Taché, University of California Los Angeles, USABruno Bonaz, Grenoble Faculty of Medicine and Hospital, France
Copyright: © 2015 Moloney, O’Mahony, Dinan and Cryan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John F. Cryan, University College Cork, Room 386 Western Gateway Building, Western Road, Cork, Ireland e-mail:ai5jcnlhbkB1Y2MuaWU=