 Seong-Wook Kim
Seong-Wook Kim Taesup Cho
Taesup Cho Sukchan Lee
Sukchan Lee- 1Center for Cognition and Sociality, Institute for Basic Science, Daejeon, South Korea
- 2Department of Physiology, Seoul National University College of Medicine, Seoul, South Korea
Schizophrenia is a mental disorder that is characterized by various abnormal symptoms. Previous studies indicate decreased expression of phospholipase C-β1 (PLC-β1) in the brains of patients with schizophrenia. PLC-β1-null (PLC-β1−/−) mice exhibit multiple endophenotypes of schizophrenia. Furthermore, a study of PLC-β1 knockdown in the medial prefrontal cortex of mice has shown a specific behavioral deficit, impaired working memory. These results support the notion that disruption of PLC-β1-linked signaling in the brain is strongly involved in the pathogenesis of schizophrenia. In this review, we broadly investigate recent studies regarding schizophrenia-related behaviors as well as their various clinical and biological correlates in PLC-β1−/− and knockdown mouse models. This will provide a better understanding of the pathological relevance of the altered expression of PLC-β1 in the brains of patients with schizophrenia. Evidence accumulated will shed light on future in-depth studies, possibly in human subjects.
Introduction
Schizophrenia-Related Phenotypes
Schizophrenia is generally characterized by genetic and neurofunctional abnormalities and classified into three major abnormal symptoms – positive, negative, and cognitive symptoms (1–6). Positive symptoms are those that are present in people with schizophrenia, such as delusions, hallucinations, thought disorder, and paranoia (7), whereas negative symptoms are deficits of normal emotional or of other thought processes such as flat affect, avolition, and social withdrawal (8). Cognitive symptoms refer to the difficulties with concentration and memory; examples include short- and long-term memory deficits as well as deficits in attention, planning, and abstract thinking (9–11).
Pathogenesis of Schizophrenia
The pathogenesis of schizophrenia has been previously reported to arise from specific neuronal abnormalities in several brain regions including prefrontal cortex (PFC) (12–14). Neural mechanisms underlying schizophrenia symptoms in several brain areas have been explained by abnormalities in the dopaminergic (15, 16), serotonergic (17, 18), muscarinic (19–22), and glutamatergic signaling (23–25). Of the many mechanisms, the phosphoinositide (PI) signaling, one of the major G-protein-linked pathways operating in the central nervous system (CNS), seems to be a point of convergence for all signaling pathways mentioned above (26, 27). PI signaling pathways are impaired in specific brain regions of patients with neurological (28–31) and psychiatric disorders (32, 33). Altered activity in PI signal pathways has been implicated in impaired cognition, mood, and abnormal behaviors, which is associated with mental disorders including schizophrenia (34, 35).
PI Signaling
PI signaling involves PI-specific phospholipase C (PLC) (36, 37) (Figure 1). PLC hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to produce a pair of second messengers, diacylglycerol (DAG), and inositol 1,4,5-trisphosphate (IP3) (38). In general, DAG activates protein kinase C (PKC), whereas IP3 mobilizes Ca2+ from the intracellular endoplasmic reticulum (ER) stores to the cytoplasm (39). PLC-β is one of the three subtypes of PLC and is distinguished from PLC-γ and PLC-δ by structure and activation mechanisms (40, 41). PLC-β acts through G protein-dependent pathways, and Gq/PLC-β pathway is readily activated by specific neurotransmitter receptors such as the M1, M3, and M5 muscarinic acetylcholine receptors (mAChRs), the group 1 and 5 metabotropic glutamate receptors (mGluRs), and the 2A and 2C subtype of serotonergic receptors (5-HT2AR and 5-HT2CR) (42–46). Protein purification and molecular cloning have identified four PLC-β isoforms: PLC-β1, PLC-β2, PLC-β3, and PLC-β4 (47, 48). Interestingly, each PLC-β isoenzyme has a unique distribution pattern in the brain (49). PLC-β1 expression is relatively abundant in the cerebral cortex although it is widely distributed in many brain areas (45, 49). PLC-β2 is mainly expressed in the white matter, indicating that its expression is in non-neuronal cells (49). PLC-β3 expression is very low throughout the brain (50). Finally, PLC-β4 is highly expressed in the cerebellum and medial septum but is almost negligible in the hippocampus (49–51).
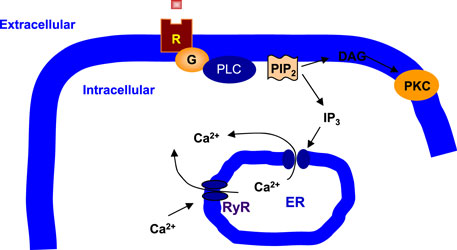
Figure 1. Phospholipase C signaling in neuronal cells. Phospholipase C (PLC) cleaves phosphatidylinositol 4,5-bisphosphate (PIP2), a membrane phospholipid, to generate two second messengers, inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 is water soluble, diffusing through the cytosol to bind to and open a ligand-gated Ca2+ channel, such as ryanodine receptor (RyR) in the endoplasmic reticulum (ER). Thus, stimulation of a receptor (R) linked to this G-alpha (G) is a way to increase Ca2+ inside the cytosol. DAG is lipid soluble and stays in the membrane. It activates protein kinase C (PKC), which, like protein kinase A (PKA), phosphorylates particular target proteins.
Abnormal PLC-β1 expression has been detected in the brains of patients with schizophrenia (52–54). A decrease of PLC-β1 expression in the dorsolateral prefrontal cortex (DLPFC) of the brains of patients with schizophrenia might account for the possible pathogenic involvement of PLC-β1 in schizophrenia patients (54).
In this review, PLC-β1 signaling pathways, especially their hypofunction will be discussed in relation to the pathogenesis of schizophrenia-like symptoms in the rodent medial prefrontal cortex (mPFC) as well as their hypofunction in patients with schizophrenia associated with human DLPFC.
PLC-β1 in Schizophrenia Symptoms
The association of the PLC-β1 with schizophrenia symptoms has been addressed by behavioral studies in PLC-β1−/− mice (55–59). It has been well established that schizophrenia-like animal models exhibit hyperlocomotion (56, 57, 60–64). In the open-field test, for example, PLC–β1−/− mice have shown an increase in locomotor activity (56, 57), a phenotype which was also exhibited in mice with a defect in PLC–β1-relevant signaling. For instance, M1 mAChR knockout results in hyperlocomotion in mice (63, 64), whereas mice lacking M3 and M5 mAChRs show normal locomotor activity (65, 66).
Abnormal social behaviors are regarded as one of the negative symptoms in schizophrenia patients. In mice, barbering behavior, also known as whisker trimming, seems to reflect a cooperative social activity and social dominance (67). Interestingly, PLC–β1−/− mice show a deficit in barbering behaviors, suggesting that PLC–β1 signaling is required for normal social interaction and dominance (56). In addition, the relevance of the lack of nesting behaviors to negative symptoms in patients with schizophrenia has been demonstrated (68, 69). Nest building in mice may represent activities of daily living (70) and a cooperative activity in rodent social behavior (68). Deficits in nest building are related to self-neglect (56) and social withdrawal (71). PLC–β1−/− mice also display significant nesting deficits, addressing negative-like symptoms in rodents.
The lack of sociability is also regarded as a negative symptom in schizophrenia patients (72, 73). A three-chamber procedure was previously used to evaluate sociability by assessing the time spent in each chamber of three divided areas. One area contains a conspecific, but the other area does not. If a mouse spends more time in the chamber with a conspecific, it has higher sociability (74). This procedure has been complemented by measuring sniffing time because sniffing time is more accurate than time in chamber when evaluating sociability (59). The total sniffing time around either a mouse or an object was scored. If the time spent sniffing around an unfamiliar conspecific was greater than an inanimate object, this is indicative of higher sociability. PLC-β1+/+ mice showed normal sociability as they spent more time sniffing around an unfamiliar conspecific than an inanimate object. On the contrary, sniffing time around both a conspecific and inanimate object was decreased in PLC-β1−/− mice. Consequently, PLC-β1−/− mice have lost their interest in the novelty of an unfamiliar conspecific, demonstrating impaired sociability (59).
Cognitive impairment is a core feature of three abnormal symptoms that contribute to the morbidity of schizophrenia. Of cognitive symptoms, sensorimotor gating is essentially a protection mechanism against sensory information overload (75). A disruption of prepulse inhibition (PPI) is a frequently used translational model of abnormal sensorimotor gating (76). The PPI deficits have been detected in some neuropsychiatric disorders including schizophrenia (77–84). Therefore, PPI may at least in part be predictive of certain cognitive functions in schizophrenia (84). Furthermore, PLC-β1−/− mice also display impaired PPI in acoustic startle response (56, 57). Given that schizophrenia patients and PLC-β1−/− mice show impairment in PPI, a measure of sensorimotor gating, PLC-β1 signaling pathways seem to be required for normal sensorimotor gating function.
Impaired working memory is considered as a core cognitive deficit in schizophrenia (85, 86). Delayed non-match to samples (DNMTS) T-maze and Y-maze task have been commonly used to measure working memory function (87, 88). PLC-β1−/− mice exhibit an impairment of working memory in the DNMTS T-maze and Y-maze task (56, 58). Based on the working memory deficits in PLC-β1−/−, PLC-β1 may be a potential target of pharmaceutical intervention to treat cognitive symptoms in schizophrenia.
Human DLPFC and Rodent mPFC
Working memory deficits have been previously reported in a number of neurological and psychiatric disorders including schizophrenia (89–91). Previous studies from human behavioral and functional neuroimaging data during working memory tasks have described deteriorated activation of the DLPFC in patients with schizophrenia (92–94). The human DLPFC, therefore, is important for the working memory process (95, 96). Signals associated with working memory are reduced in the DLPFC of patients with schizophrenia (97, 98). Additionally, deficits in working memory have been shown in human brain injury patients with damage in DLPFC (99, 100). A decrease of both glucose utilization and blood flow during working memory tasks have been observed in the DLPFC of patients with schizophrenia (94, 101–104). Using electroencephalography (EEG) during a working memory test, abnormal brain oscillatory activity has been reported in the frontal theta (4–8 Hz) and alpha (8–12 Hz) frequencies in patients with schizophrenia (105, 106). Human EEG recordings have further revealed abnormal circuitries between the PFC and other brain regions in schizophrenia, such as the temporal lobe and subcortical limbic structures, suggesting that functional connectivity between the PFC and other brain regions play some role in the pathogenesis of schizophrenia (107–109). It has been supported by evidence that alterations in synchronized brain oscillation reflect neuronal changes that lead to schizophrenia (110, 111).
The mouse mPFC is thought to have anatomical and functional homology with the DLPFC in human being (38, 112–114). The mouse mPFC consists of prelimbic (PL) and infralimbic (IL) cortices (38, 112–114). The mouse mPFC receives indirect projections from the dorsal hippocampus (dHPC) and direct afferent inputs from the ventral hippocampus (vHPC) (115, 116). The mouse mPFC has reciprocal connections with the amygdala and other subcortical limbic structures (117). Theta frequency synchrony between the mPFC and dHPC, and/or beta (13–30 Hz) frequency synchrony between the mPFC and mediodorsal thalamic nucleus (MD) are required for working memory (13, 118).
Both cytotoxic lesions and acute inactivations in the rodent mPFC are able to induce most schizophrenia phenotypes including positive, negative, and cognitive-like symptoms, as well as working memory deficits (119, 120). It is also worth noting that mPFC deficits represent a key component of the pathophysiology in patients with schizophrenia (14, 121).
The shRNA-mediated silencing of PLC-β1 in the mPFC, a mouse model that mimics the decrease of PLC-β1 in the DLPFC of patients with schizophrenia, causes an impairment in working memory (59). This effect is specific to working memory; mPFC-specific PLC-β1 knockdown does not have an effect on other behaviors relevant to schizophrenia-related endophenotypes characteristic of PLC-β1−/− mice, such as locomotion, social behaviors, and sensorimotor gating (59).
PLC-β1 Signaling Pathways Underlying Working Memory in the mPFC
A number of biochemical and genetic studies have demonstrated that modulatory neurotransmission in the PFC is required for cognitive functions including working memory (11, 12, 96, 122–125). Dopaminergic neurotransmission in the PFC has been shown to be important for working memory in both animals and human beings (126–134). Regional depletion of PFC dopamine causes profound working memory impairments in monkeys (126) and rats (127). Iontophoretic application of dopamine into the PFC for working memory tasks has revealed an increase of delay-period activity in monkeys (134). During working memory, the dopamine level is transiently increased in the PFC of both rats and human beings (135, 136). Explained briefly, dopamine receptors are divided into two families (D1 and D2 class) based on second messenger coupling and ligand binding (137, 138); D1 class, composed of D1 and D5 dopamine receptors, increases the cAMP levels, whereas D2 class, composed of D2, D3, and D4 receptors, decreases cAMP levels (139, 140). The influence of dopamine in prefrontal neurons is largely mediated by D1 dopamine receptors as they are much more abundant compared to D2 dopamine receptors in PFC pyramidal cells (141). The PFC D1 dopamine receptors are decreased in patients with schizophrenia (133). A D1 antagonist is able to suppress PFC delay-period activity. Similarly, the infusion of D1-specific agonists has further confirmed the importance of D1 dopamine receptors in the PFC with improved working memory (125, 142). Furthermore, the importance of D1 dopamine receptors has been investigated in non-human primates during delayed-response paradigms showing that working memory requires appropriate D1 receptor activation in the DLPFC (126, 129, 130, 142, 143). Taken together, these observations suggest that altered dopamine transmission at D1 receptors in DLPFC could be involved in the pathophysiology of working memory in schizophrenia (90, 95, 144, 145).
In addition to dopamine receptors involved in working memory, other modulatory neurotransmitters, such as norepinephrine (NE) acting through α-adrenergic receptors (146, 147), serotonin through 5-HT2A receptors (124), and acetylcholine through mAChRs (148) have been shown to be involved in working memory. Moderate levels of NE activate α2A adrenergic receptors and result in improved working memory; however, at higher levels, NE activates α1 receptors, resulting in impaired working memory (123, 149, 150). Similarly, it has been proposed that excessive prefrontal NE levels may contribute to the working memory deficits in schizophrenia (122). Investigation of M1 mAChRs knockout mice has provided evidence that M1 mAChRs are critical for the performance of non-matching-to-sample working memory tasks (151).
Several lines of research have indirectly indicated that Gq signaling pathways are implicated in working memory (12, 59, 124, 130). In detail, translocation of the α subunit of Gq proteins from the membrane to a cytosolic fraction can be used as an indicator of activation (152). Gq translocates during the delay period of the match-to-place task for working memory (125), suggesting that the Gq signaling cascade is activated during working memory. Consistently, alterations in prefrontal Gq signalings have been associated with working memory deficits in patients with schizophrenia (153). RGS4, an inhibitor of Gq protein-induced intracellular Ca2+ release, is downregulated in PFC of schizophrenia mouse model (58, 154).
Conclusion and Future Remarks
Although continued investigation is required to fully understand PLC-β1 hypofunction in the pathogenesis of schizophrenia, the results obtained to date suggest some challenges in the treatments for working memory deficits in schizophrenia. As indicated previously, altered expression of PLC-β1 has been detected in several brain regions of patients with schizophrenia including DLPFC (52, 53, 155–158). The major schizophrenia symptoms (1–6), such as negative and cognitive symptoms, have been observed in the behavioral characterization of PLC-β1−/− mice. These results indicate that disrupted PLC-β1 signaling in specific brain regions can be relevant to the pathogenesis of schizophrenia (55–58).
Furthermore, neurotransmitter transmissions such as dopamine through D1 receptors coupled to PLC in the mPFC (129, 141, 159, 160), NE acting through α-adrenoreceptors (146, 147), serotonin through 5-HT2A receptors (124), and acetylcholine through M1 mAChRs (148) are necessary for working memory. The activities of intracellular second messenger pathways linked with PLC in the mPFC are also critical for working memory (161–163). In addition, PLC-β1 knockdown and impaired working memory in the mPFC have been discussed in this review, suggesting that the mPFC-specific PLC-β1 pathways underlying working memory could be different from those for anxiety or other schizophrenia endophenotypes of PLC-β1−/− mice (59).
In conclusion, these results support the notion that the decrease in PLC-β1 expression in the brains of patients with schizophrenia is a pathogenically relevant molecular marker of the disorder (54, 56–59). This interpretation offers new insight into PLC-β1 hypofunction in the pathogenesis of schizophrenia and may aid in a better understanding of the neural mechanisms underlying working memory deficits in schizophrenia. Finally, PLC-β1 is not detected in a peripheral area (164, 165). Thus, PLC-β1 KO could not affect obesity or weight gain. PLC-β1 KO mice are even not hypophagic and lean unlike M3 KO mice (65). Therefore, the PLC-β1-based treatments are necessary for the development of novel psychotherapeutic approaches with reducing metabolic side effects.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Ms. Arie Kim and Mr. Isaac Kim for their excellent editorial assistance. This work was supported by the Institute for Basic Science (IBS-R001-D1), Korea.
References
1. Liddle PF. The symptoms of chronic schizophrenia. A re-examination of the positive-negative dichotomy. Br J Psychiatry (1987) 151:145–51. doi: 10.1192/bjp.151.2.145
2. Barnes TR, Liddle PF. Evidence for the validity of negative symptoms. Mod Probl Pharmacopsychiatry (1990) 24:43–72. doi:10.1159/000418012
3. Kay SR, Sevy S. Pyramidical model of schizophrenia. Schizophr Bull (1990) 16:537–45. doi:10.1093/schbul/16.3.537
4. Arndt S, Alliger RJ, Andreasen NC. The distinction of positive and negative symptoms. The failure of a two-dimensional model. Br J Psychiatry (1991) 158:317–22. doi:10.1192/bjp.158.3.317
5. Lenzenweger MF, Dworkin RH, Wethington E. Examining the underlying structure of schizophrenic phenomenology: evidence for a three-process model. Schizophr Bull (1991) 17:515–24. doi:10.1093/schbul/17.3.515
6. Andreasen NC, Arndt S, Alliger R, Miller D, Flaum M. Symptoms of schizophrenia. Methods, meanings, and mechanisms. Arch Gen Psychiatry (1995) 52:341–51. doi:10.1001/archpsyc.1995.03950170015003
7. Andreasen NC, Olsen S. Negative v positive schizophrenia. Definition and validation. Arch Gen Psychiatry (1982) 39:789–94. doi:10.1001/archpsyc.1982.04290070025006
8. Andreasen NC. Negative symptoms in schizophrenia. Definition and reliability. Arch Gen Psychiatry (1982) 39:784–8. doi:10.1001/archpsyc.1982.04290070020005
9. Green MF. What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry (1996) 153:321–30. doi:10.1176/ajp.153.3.321
10. Cannon TD, Glahn DC, Kim J, Van Erp TG, Karlsgodt K, Cohen MS, et al. Dorsolateral prefrontal cortex activity during maintenance and manipulation of information in working memory in patients with schizophrenia. Arch Gen Psychiatry (2005) 62:1071–80. doi:10.1001/archpsyc.62.10.1071
11. Lesh TA, Niendam TA, Minzenberg MJ, Carter CS. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology (2011) 36:316–38. doi:10.1038/npp.2010.156
12. Lopez-Tellez JF, Lopez-Aranda MF, Navarro-Lobato I, Masmudi-Martin M, Montanez EM, Calvo EB, et al. Prefrontal inositol triphosphate is molecular correlate of working memory in nonhuman primates. J Neurosci (2010) 30:3067–71. doi:10.1523/jneurosci.4565-09.2010
13. Sigurdsson T, Stark KL, Karayiorgou M, Gogos JA, Gordon JA. Impaired hippocampal-prefrontal synchrony in a genetic mouse model of schizophrenia. Nature (2010) 464:763–7. doi:10.1038/nature08855
14. Arnsten AF. Prefrontal cortical network connections: key site of vulnerability in stress and schizophrenia. Int J Dev Neurosci (2011) 29:215–23. doi:10.1016/j.ijdevneu.2011.02.006
15. Seeman P, Lee T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science (1975) 188:1217–9. doi:10.1126/science.1145194
16. Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science (1976) 192:481–3. doi:10.1126/science.3854
17. Scarr E, Copolov DL, Dean B. A proposed pathological model in the hippocampus of subjects with schizophrenia. Clin Exp Pharmacol Physiol (2001) 28:70–3. doi:10.1046/j.1440-1681.2001.03400.x
18. Lopez-Figueroa AL, Norton CS, Lopez-Figueroa MO, Armellini-Dodel D, Burke S, Akil H, et al. Serotonin 5-HT1A, 5-HT1B, and 5-HT2A receptor mRNA expression in subjects with major depression, bipolar disorder, and schizophrenia. Biol Psychiatry (2004) 55:225–33. doi:10.1016/j.biopsych.2003.09.017
19. Bennett JP Jr, Enna SJ, Bylund DB, Gillin JC, Wyatt RJ, Snyder SH. Neurotransmitter receptors in frontal cortex of schizophrenics. Arch Gen Psychiatry (1979) 36:927–34. doi:10.1001/archpsyc.1979.01780090013001
20. Watanabe S, Nishikawa T, Takashima M, Toru M. Increased muscarinic cholinergic receptors in prefrontal cortices of medicated schizophrenics. Life Sci (1983) 33:2187–96. doi:10.1016/0024-3205(83)90290-4
21. Dean B, Crook JM, Opeskin K, Hill C, Keks N, Copolov DL. The density of muscarinic M1 receptors is decreased in the caudate-putamen of subjects with schizophrenia. Mol Psychiatry (1996) 1:54–8.
22. Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Towards a muscarinic hypothesis of schizophrenia. Mol Psychiatry (2007) 12:232–46. doi:10.1038/sj.mp.4001924
23. Noga JT, Hyde TM, Herman MM, Spurney CF, Bigelow LB, Weinberger DR, et al. Glutamate receptors in the postmortem striatum of schizophrenic, suicide, and control brains. Synapse (1997) 27:168–76. doi:10.1002/(SICI)1098-2396(199711)27:3<168::AID-SYN2>3.3.CO;2-#
24. Ohnuma T, Augood SJ, Arai H, Mckenna PJ, Emson PC. Expression of the human excitatory amino acid transporter 2 and metabotropic glutamate receptors 3 and 5 in the prefrontal cortex from normal individuals and patients with schizophrenia. Brain Res Mol Brain Res (1998) 56:207–17. doi:10.1016/S0169-328X(98)00063-1
25. Nudmamud-Thanoi S, Reynolds GP. The NR1 subunit of the glutamate/NMDA receptor in the superior temporal cortex in schizophrenia and affective disorders. Neurosci Lett (2004) 372:173–7. doi:10.1016/j.neulet.2004.09.035
26. Moolenaar WH. G-protein-coupled receptors, phosphoinositide hydrolysis, and cell proliferation. Cell Growth Differ (1991) 2:359–64.
27. Taylor SJ, Chae HZ, Rhee SG, Exton JH. Activation of the beta 1 isozyme of phospholipase C by alpha subunits of the Gq class of G proteins. Nature (1991) 350:516–8. doi:10.1038/350516a0
28. Dubeau F, Sherwin A, Olivier A, Villemure J, Leblanc R, Quesney LF, et al. Excitatory amino acids modulate phosphoinositide signal transduction in human epileptic neocortex. Epilepsia (1992) 33:255–62. doi:10.1111/j.1528-1157.1992.tb02314.x
29. Wallace MA, Claro E. Transmembrane signaling through phospholipase C in human cortical membranes. Neurochem Res (1993) 18:139–45. doi:10.1007/BF01474676
30. Greenwood AF, Powers RE, Jope RS. Phosphoinositide hydrolysis, G alpha q, phospholipase C, and protein kinase C in post mortem human brain: effects of post mortem interval, subject age, and Alzheimer’s disease. Neuroscience (1995) 69:125–38. doi:10.1016/0306-4522(95)00220-D
31. Shimohama S, Fujimoto S, Matsushima H, Takenawa T, Taniguchi T, Perry G, et al. Alteration of phospholipase C-delta protein level and specific activity in Alzheimer’s disease. J Neurochem (1995) 64:2629–34. doi:10.1046/j.1471-4159.1995.64062629.x
32. Pacheco MA, Stockmeier C, Meltzer HY, Overholser JC, Dilley GE, Jope RS. Alterations in phosphoinositide signaling and G-protein levels in depressed suicide brain. Brain Res (1996) 723:37–45. doi:10.1016/0006-8993(96)00207-7
33. Jope RS, Song L, Powers RE. Cholinergic activation of phosphoinositide signaling is impaired in Alzheimer’s disease brain. Neurobiol Aging (1997) 18:111–20. doi:10.1016/S0197-4580(96)00205-9
34. Yao JK, Yasaei P, Van Kammen DP. Increased turnover of platelet phosphatidylinositol in schizophrenia. Prostaglandins Leukot Essent Fatty Acids (1992) 46:39–46. doi:10.1016/0952-3278(92)90057-P
35. McNamara RK, Ostrander M, Abplanalp W, Richtand NM, Benoit SC, Clegg DJ. Modulation of phosphoinositide-protein kinase C signal transduction by omega-3 fatty acids: implications for the pathophysiology and treatment of recurrent neuropsychiatric illness. Prostaglandins Leukot Essent Fatty Acids (2006) 75:237–57. doi:10.1016/j.plefa.2006.07.009
36. Bennett CF, Balcarek JM, Varrichio A, Crooke ST. Molecular cloning and complete amino-acid sequence of form-I phosphoinositide-specific phospholipase C. Nature (1988) 334:268–70. doi:10.1038/334268a0
37. Crooke ST, Bennett CF. Mammalian phosphoinositide-specific phospholipase C isoenzymes. Cell Calcium (1989) 10:309–23. doi:10.1016/0143-4160(89)90057-2
38. Exton JH. Regulation of phosphoinositide phospholipases by hormones, neurotransmitters, and other agonists linked to G proteins. Annu Rev Pharmacol Toxicol (1996) 36:481–509. doi:10.1146/annurev.pa.36.040196.002405
39. Nishizuka Y. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature (1988) 334:661–5. doi:10.1038/334661a0
40. Suh PG, Ryu SH, Moon KH, Suh HW, Rhee SG. Cloning and sequence of multiple forms of phospholipase C. Cell (1988) 54:161–9. doi:10.1016/0092-8674(88)90548-X
41. Jhon DY, Lee HH, Park D, Lee CW, Lee KH, Yoo OJ, et al. Cloning, sequencing, purification, and Gq-dependent activation of phospholipase C-beta 3. J Biol Chem (1993) 268:6654–61.
42. Carter HR, Wallace MA, Fain JN. Activation of phospholipase C in rabbit brain membranes by carbachol in the presence of GTP gamma S; effects of biological detergents. Biochim Biophys Acta (1990) 1054:129–35. doi:10.1016/0167-4889(90)90214-X
43. Gutkind JS, Novotny EA, Brann MR, Robbins KC. Muscarinic acetylcholine receptor subtypes as agonist-dependent oncogenes. Proc Natl Acad Sci U S A (1991) 88:4703–7. doi:10.1073/pnas.88.11.4703
44. Salles J, Wallace MA, Fain JN. Modulation of the phospholipase C activity in rat brain cortical membranes by simultaneous activation of distinct monoaminergic and cholinergic muscarinic receptors. Brain Res Mol Brain Res (1993) 20:111–7. doi:10.1016/0169-328X(93)90115-6
45. Kim D, Jun KS, Lee SB, Kang NG, Min DS, Kim YH, et al. Phospholipase C isozymes selectively couple to specific neurotransmitter receptors. Nature (1997) 389:290–3. doi:10.1038/38508
46. Hannan AJ, Blakemore C, Katsnelson A, Vitalis T, Huber KM, Bear M, et al. PLC-beta1, activated via mGluRs, mediates activity-dependent differentiation in cerebral cortex. Nat Neurosci (2001) 4:282–8. doi:10.1038/85132
47. Lee CW, Park DJ, Lee KH, Kim CG, Rhee SG. Purification, molecular cloning, and sequencing of phospholipase C-beta 4. J Biol Chem (1993) 268:21318–27.
48. Rhee SG, Bae YS. Regulation of phosphoinositide-specific phospholipase C isozymes. J Biol Chem (1997) 272:15045–8. doi:10.1074/jbc.272.24.15045
49. Watanabe M, Nakamura M, Sato K, Kano M, Simon MI, Inoue Y. Patterns of expression for the mRNA corresponding to the four isoforms of PLCb in mouse brain. Eur J Neurosci (1998) 10(6):2016–25. doi:10.1046/j.1460-9568.1998.00213.x
50. Tanaka O, Kondo H. Localization of mRNAs for three novel members (beta 3, beta 4 and gamma 2) of phospholipase C family in mature rat brain. Neurosci Lett (1994) 182:17–20. doi:10.1016/0304-3940(94)90194-5
51. Shin J, Gireesh G, Kim SW, Kim DS, Lee S, Kim YS, et al. Phospholipase C beta 4 in the medial septum controls cholinergic theta oscillations and anxiety behaviors. J Neurosci (2009) 29:15375–85. doi:10.1523/jneurosci.3126-09.2009
52. Lin XH, Kitamura N, Hashimoto T, Shirakawa O, Maeda K. Opposite changes in phosphoinositide-specific phospholipase C immunoreactivity in the left prefrontal and superior temporal cortex of patients with chronic schizophrenia. Biol Psychiatry (1999) 46:1665–71. doi:10.1016/S0006-3223(99)00036-0
53. Shirakawa O, Kitamura N, Lin XH, Hashimoto T, Maeda K. Abnormal neurochemical asymmetry in the temporal lobe of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2001) 25:867–77. doi:10.1016/S0278-5846(01)00149-X
54. Udawela M, Scarr E, Hannan AJ, Thomas EA, Dean B. Phospholipase C beta 1 expression in the dorsolateral prefrontal cortex from patients with schizophrenia at different stages of illness. Aust N Z J Psychiatry (2011) 45:140–7. doi:10.3109/00048674.2010.533364
55. Shin J, Kim D, Bianchi R, Wong RK, Shin HS. Genetic dissection of theta rhythm heterogeneity in mice. Proc Natl Acad Sci U S A (2005) 102:18165–70. doi:10.1073/pnas.0505498102
56. Koh HY, Kim D, Lee J, Lee S, Shin HS. Deficits in social behavior and sensorimotor gating in mice lacking phospholipase Cbeta1. Genes Brain Behav (2007) 7:120–8. doi:10.1111/j.1601-183X.2007.00351.x
57. McOmish CE, Burrows E, Howard M, Scarr E, Kim D, Shin HS, et al. Phospholipase C-beta1 knockout mice exhibit endophenotypes modeling schizophrenia which are rescued by environmental enrichment and clozapine administration. Mol Psychiatry (2008) 13:661–72. doi:10.1038/sj.mp.4002046
58. McOmish CE, Burrows EL, Howard M, Hannan AJ. PLC-beta1 knockout mice as a model of disrupted cortical development and plasticity: behavioral endophenotypes and dysregulation of RGS4 gene expression. Hippocampus (2008) 18:824–34. doi:10.1002/hipo.20443
59. Kim SW, Seo M, Kim DS, Kang M, Kim YS, Koh HY, et al. Knockdown of phospholipase C-beta1 in the medial prefrontal cortex of male mice impairs working memory among multiple schizophrenia endophenotypes. J Psychiatry Neurosci (2015) 40:78–88. doi:10.1503/jpn.130285
60. Andreasen NC, Flaum M, Swayze VW II, Tyrrell G, Arndt S. Positive and negative symptoms in schizophrenia. A critical reappraisal. Arch Gen Psychiatry (1990) 47:615–21. doi:10.1001/archpsyc.1990.01810190015002
61. Andreasen NC. Symptoms, signs, and diagnosis of schizophrenia. Lancet (1995) 346:477–81. doi:10.1016/S0140-6736(95)91325-4
62. Carlsson A. A paradigm shift in brain research. Science (2001) 294:1021–4. doi:10.1126/science.1066969
63. Gerber DJ, Sotnikova TD, Gainetdinov RR, Huang SY, Caron MG, Tonegawa S. Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice. Proc Natl Acad Sci U S A (2001) 98:15312–7. doi:10.1073/pnas.261583798
64. Miyakawa T, Yamada M, Duttaroy A, Wess J. Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J Neurosci (2001) 21:5239–50.
65. Yamada M, Miyakawa T, Duttaroy A, Yamanaka A, Moriguchi T, Makita R, et al. Mice lacking the M3 muscarinic acetylcholine receptor are hypophagic and lean. Nature (2001) 410:207–12. doi:10.1038/35065604
66. Thomsen M, Wortwein G, Fink-Jensen A, Woldbye DP, Wess J, Caine SB. Decreased prepulse inhibition and increased sensitivity to muscarinic, but not dopaminergic drugs in M5 muscarinic acetylcholine receptor knockout mice. Psychopharmacology (Berl) (2007) 192:97–110. doi:10.1007/s00213-006-0682-y
67. Strozik E, Festing MF. Whisker trimming in mice. Lab Anim (1981) 15:309–12. doi:10.1258/002367781780953040
68. Crawley JN. Designing mouse behavioral tasks relevant to autistic-like behaviors. Ment Retard Dev Disabil Res Rev (2004) 10:248–58. doi:10.1002/mrdd.20039
69. Takahata K, Kato M. [Neural mechanism underlying autistic savant and acquired savant syndrome]. Brain Nerve (2008) 60:861–9.
70. Galasko D, Schmitt F, Thomas R, Jin S, Bennett D. Detailed assessment of activities of daily living in moderate to severe Alzheimer’s disease. J Int Neuropsychol Soc (2005) 11:446–53. doi:10.1017/S1355617705050502
71. Powell CM, Miyakawa T. Schizophrenia-relevant behavioral testing in rodent models: a uniquely human disorder? Biol Psychiatry (2006) 59:1198–207. doi:10.1016/j.biopsych.2006.05.008
72. Hamilton NG, Ponzoha CA, Cutler DL, Weigel RM. Social networks and negative versus positive symptoms of schizophrenia. Schizophr Bull (1989) 15:625–33. doi:10.1093/schbul/15.4.625
73. Bellack AS, Morrison RL, Wixted JT, Mueser KT. An analysis of social competence in schizophrenia. Br J Psychiatry (1990) 156:809–18. doi:10.1192/bjp.156.6.809
74. Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, et al. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav (2004) 3:287–302. doi:10.1111/j.1601-1848.2004.00076.x
75. Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl) (2001) 156:117–54. doi:10.1007/s002130100811
76. Turetsky BI, Calkins ME, Light GA, Olincy A, Radant AD, Swerdlow NR. Neurophysiological endophenotypes of schizophrenia: the viability of selected candidate measures. Schizophr Bull (2007) 33:69–94. doi:10.1093/schbul/sbl060
77. Swerdlow NR, Benbow CH, Zisook S, Geyer MA, Braff DL. A preliminary assessment of sensorimotor gating in patients with obsessive compulsive disorder. Biol Psychiatry (1993) 33:298–301. doi:10.1016/0006-3223(93)90300-3
78. Swerdlow NR, Paulsen J, Braff DL, Butters N, Geyer MA, Swenson MR. Impaired prepulse inhibition of acoustic and tactile startle response in patients with Huntington’s disease. J Neurol Neurosurg Psychiatry (1995) 58:192–200. doi:10.1136/jnnp.58.2.192
79. Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) (2001) 156:234–58. doi:10.1007/s002130100810
80. Perry W, Minassian A, Feifel D, Braff DL. Sensorimotor gating deficits in bipolar disorder patients with acute psychotic mania. Biol Psychiatry (2001) 50:418–24. doi:10.1016/S0006-3223(01)01184-2
81. Swerdlow NR, Karban B, Ploum Y, Sharp R, Geyer MA, Eastvold A. Tactile prepuff inhibition of startle in children with Tourette’s syndrome: in search of an “fMRI-friendly” startle paradigm. Biol Psychiatry (2001) 50:578–85. doi:10.1016/S0006-3223(01)01164-7
82. Ludewig S, Ludewig K, Geyer MA, Hell D, Vollenweider FX. Prepulse inhibition deficits in patients with panic disorder. Depress Anxiety (2002) 15:55–60. doi:10.1002/da.10026
83. Perry W, Minassian A, Lopez B, Maron L, Lincoln A. Sensorimotor gating deficits in adults with autism. Biol Psychiatry (2007) 61:482–6. doi:10.1016/j.biopsych.2005.09.025
84. Swerdlow NR, Weber M, Qu Y, Light GA, Braff DL. Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology (Berl) (2008) 199:331–88. doi:10.1007/s00213-008-1072-4
85. Elvevag B, Goldberg TE. Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol (2000) 14:1–21. doi:10.1615/CritRevNeurobiol.v14.i1.10
86. Hanlon FM, Weisend MP, Hamilton DA, Jones AP, Thoma RJ, Huang M, et al. Impairment on the hippocampal-dependent virtual Morris water task in schizophrenia. Schizophr Res (2006) 87:67–80. doi:10.1016/j.schres.2006.05.021
87. Dias R, Aggleton JP. Effects of selective excitotoxic prefrontal lesions on acquisition of nonmatching- and matching-to-place in the T-maze in the rat: differential involvement of the prelimbic-infralimbic and anterior cingulate cortices in providing behavioural flexibility. Eur J Neurosci (2000) 12:4457–66. doi:10.1046/j.0953-816X.2000.01323.x
88. Kellendonk C, Simpson EH, Polan HJ, Malleret G, Vronskaya S, Winiger V, et al. Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron (2006) 49:603–15. doi:10.1016/j.neuron.2006.01.023
89. Morris RG, Baddeley AD. Primary and working memory functioning in Alzheimer-type dementia. J Clin Exp Neuropsychol (1988) 10:279–96. doi:10.1080/01688638808408242
90. Goldman-Rakic PS. Working memory dysfunction in schizophrenia. J Neuropsychiatry Clin Neurosci (1994) 6:348–57. doi:10.1176/jnp.6.4.348
91. Winograd-Gurvich C, Fitzgerald PB, Georgiou-Karistianis N, Bradshaw JL, White OB. Negative symptoms: a review of schizophrenia, melancholic depression and Parkinson’s disease. Brain Res Bull (2006) 70:312–21. doi:10.1016/j.brainresbull.2006.06.007
92. Tan HY, Sust S, Buckholtz JW, Mattay VS, Meyer-Lindenberg A, Egan MF, et al. Dysfunctional prefrontal regional specialization and compensation in schizophrenia. Am J Psychiatry (2006) 163:1969–77. doi:10.1176/appi.ajp.163.11.1969
93. Van Snellenberg JX. Working memory and long-term memory deficits in schizophrenia: is there a common substrate? Psychiatry Res (2009) 174:89–96. doi:10.1016/j.pscychresns.2009.04.001
94. Henseler I, Falkai P, Gruber O. Disturbed functional connectivity within brain networks subserving domain-specific subcomponents of working memory in schizophrenia: relation to performance and clinical symptoms. J Psychiatr Res (2010) 44:364–72. doi:10.1016/j.jpsychires.2009.09.003
95. Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry (1991) 148:1474–86. doi:10.1176/ajp.148.11.1474
96. Abi-Dargham A, Mawlawi O, Lombardo I, Gil R, Martinez D, Huang Y, et al. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci (2002) 22:3708–19.
97. Smith EE, Jonides J. Working memory: a view from neuroimaging. Cogn Psychol (1997) 33:5–42. doi:10.1006/cogp.1997.0658
98. D’Esposito M, Postle BR. The dependence of span and delayed-response performance on prefrontal cortex. Neuropsychologia (1999) 37:1303–15. doi:10.1016/S0028-3932(99)00021-4
99. Mull BR, Seyal M. Transcranial magnetic stimulation of left prefrontal cortex impairs working memory. Clin Neurophysiol (2001) 112:1672–5. doi:10.1016/S1388-2457(01)00606-X
100. Barbey AK, Koenigs M, Grafman J. Dorsolateral prefrontal contributions to human working memory. Cortex (2012) 49:1195–205. doi:10.1016/j.cortex.2012.05.022
101. Ingvar DH, Franzen G. Abnormalities of cerebral blood flow distribution in patients with chronic schizophrenia. Acta Psychiatr Scand (1974) 50:425–62. doi:10.1111/j.1600-0447.1974.tb09707.x
102. Buchsbaum MS, Nuechterlein KH, Haier RJ, Wu J, Sicotte N, Hazlett E, et al. Glucose metabolic rate in normals and schizophrenics during the Continuous Performance Test assessed by positron emission tomography. Br J Psychiatry (1990) 156:216–27. doi:10.1192/bjp.156.2.216
103. Honey GD, Bullmore ET, Soni W, Varatheesan M, Williams SC, Sharma T. Differences in frontal cortical activation by a working memory task after substitution of risperidone for typical antipsychotic drugs in patients with schizophrenia. Proc Natl Acad Sci U S A (1999) 96:13432–7. doi:10.1073/pnas.96.23.13432
104. Meyer-Lindenberg A, Miletich RS, Kohn PD, Esposito G, Carson RE, Quarantelli M, et al. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci (2002) 5:267–71. doi:10.1038/nn804
105. Schmiedt C, Brand A, Hildebrandt H, Basar-Eroglu C. Event-related theta oscillations during working memory tasks in patients with schizophrenia and healthy controls. Brain Res Cogn Brain Res (2005) 25:936–47. doi:10.1016/j.cogbrainres.2005.09.015
106. Bachman P, Kim J, Yee CM, Therman S, Manninen M, Lonnqvist J, et al. Abnormally high EEG alpha synchrony during working memory maintenance in twins discordant for schizophrenia. Schizophr Res (2008) 103:293–7. doi:10.1016/j.schres.2008.04.006
107. Ford JM, Mathalon DH, Whitfield S, Faustman WO, Roth WT. Reduced communication between frontal and temporal lobes during talking in schizophrenia. Biol Psychiatry (2002) 51:485–92. doi:10.1016/S0006-3223(01)01335-X
108. Lawrie SM, Buechel C, Whalley HC, Frith CD, Friston KJ, Johnstone EC. Reduced frontotemporal functional connectivity in schizophrenia associated with auditory hallucinations. Biol Psychiatry (2002) 51:1008–11. doi:10.1016/S0006-3223(02)01316-1
109. Meyer-Lindenberg AS, Olsen RK, Kohn PD, Brown T, Egan MF, Weinberger DR, et al. Regionally specific disturbance of dorsolateral prefrontal-hippocampal functional connectivity in schizophrenia. Arch Gen Psychiatry (2005) 62:379–86. doi:10.1001/archpsyc.62.4.379
110. Lewis DA, Gonzalez-Burgos G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology (2008) 33:141–65. doi:10.1038/sj.npp.1301563
111. Uhlhaas PJ, Roux F, Singer W, Haenschel C, Sireteanu R, Rodriguez E. The development of neural synchrony reflects late maturation and restructuring of functional networks in humans. Proc Natl Acad Sci U S A (2009) 106:9866–71. doi:10.1073/pnas.0900390106
112. Uylings HB, van Eden CG. Qualitative and quantitative comparison of the prefrontal cortex in rat and in primates, including humans. Prog Brain Res (1990) 85:31–62. doi:10.1016/S0079-6123(08)62675-8
113. Brown VJ, Bowman EM. Rodent models of prefrontal cortical function. Trends Neurosci (2002) 25:340–3. doi:10.1016/S0166-2236(02)02164-1
114. Uylings HB, Groenewegen HJ, Kolb B. Do rats have a prefrontal cortex? Behav Brain Res (2003) 146:3–17. doi:10.1016/j.bbr.2003.09.028
115. Hoover WB, Vertes RP. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct (2007) 212:149–79. doi:10.1007/s00429-007-0150-4
116. Parent MA, Wang L, Su J, Netoff T, Yuan LL. Identification of the hippocampal input to medial prefrontal cortex in vitro. Cereb Cortex (2010) 20:393–403. doi:10.1093/cercor/bhp108
117. Vertes RP. Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse (2004) 51:32–58. doi:10.1002/syn.10279
118. Parnaudeau S, O’Neill PK, Bolkan SS, Ward RD, Abbas AI, Roth BL, et al. Inhibition of mediodorsal thalamus disrupts thalamofrontal connectivity and cognition. Neuron (2013) 77:1151–62. doi:10.1016/j.neuron.2013.01.038
119. Touzani K, Puthanveettil SV, Kandel ER. Consolidation of learning strategies during spatial working memory task requires protein synthesis in the prefrontal cortex. Proc Natl Acad Sci U S A (2007) 104:5632–7. doi:10.1073/pnas.0611554104
120. Adhikari A, Topiwala MA, Gordon JA. Single units in the medial prefrontal cortex with anxiety-related firing patterns are preferentially influenced by ventral hippocampal activity. Neuron (2011) 71:898–910. doi:10.1016/j.neuron.2011.07.027
121. Barch DM, Carter CS, Braver TS, Sabb FW, Macdonald A III, Noll DC, et al. Selective deficits in prefrontal cortex function in medication-naive patients with schizophrenia. Arch Gen Psychiatry (2001) 58:280–8. doi:10.1001/archpsyc.58.3.280
122. Friedman JI, Temporini H, Davis KL. Pharmacologic strategies for augmenting cognitive performance in schizophrenia. Biol Psychiatry (1999) 45:1–16. doi:10.1016/S0006-3223(98)00287-X
123. Arnsten AF. Through the looking glass: differential noradenergic modulation of prefrontal cortical function. Neural Plast (2000) 7:133–46. doi:10.1155/np.2000.133
124. Williams GV, Rao SG, Goldman-Rakic PS. The physiological role of 5-HT2A receptors in working memory. J Neurosci (2002) 22:2843–54.
125. Runyan JD, Dash PK. Distinct prefrontal molecular mechanisms for information storage lasting seconds versus minutes. Learn Mem (2005) 12:232–8. doi:10.1101/lm.92405
126. Brozoski TJ, Brown RM, Rosvold HE, Goldman PS. Cognitive deficit caused by regional depletion of dopamine in prefrontal cortex of rhesus monkey. Science (1979) 205:929–32. doi:10.1126/science.112679
127. Simon H, Scatton B, Le Moal M. Definitive disruption of spatial delayed alternation in rats after lesions in the ventral mesencephalic tegmentum. Neurosci Lett (1979) 15:319–24. doi:10.1016/0304-3940(79)96133-0
128. Sawaguchi T, Matsumura M, Kubota K. Dopamine enhances the neuronal activity of spatial short-term memory task in the primate prefrontal cortex. Neurosci Res (1988) 5:465–73. doi:10.1016/0168-0102(88)90030-2
129. Sawaguchi T, Goldman-Rakic PS. D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science (1991) 251:947–50. doi:10.1126/science.1825731
130. Sawaguchi T, Goldman-Rakic PS. The role of D1-dopamine receptor in working memory: local injections of dopamine antagonists into the prefrontal cortex of rhesus monkeys performing an oculomotor delayed-response task. J Neurophysiol (1994) 71:515–28.
131. Williams GV, Goldman-Rakic PS. Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature (1995) 376:572–5. doi:10.1038/376572a0
132. Murphy BL, Arnsten AF, Goldman-Rakic PS, Roth RH. Increased dopamine turnover in the prefrontal cortex impairs spatial working memory performance in rats and monkeys. Proc Natl Acad Sci U S A (1996) 93:1325–9. doi:10.1073/pnas.93.3.1325
133. Okubo Y, Suhara T, Suzuki K, Kobayashi K, Inoue O, Terasaki O, et al. Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature (1997) 385:634–6. doi:10.1038/385634a0
134. Sawaguchi T. The effects of dopamine and its antagonists on directional delay-period activity of prefrontal neurons in monkeys during an oculomotor delayed-response task. Neurosci Res (2001) 41:115–28. doi:10.1016/S0168-0102(01)00270-X
135. Phillips AG, Ahn S, Floresco SB. Magnitude of dopamine release in medial prefrontal cortex predicts accuracy of memory on a delayed response task. J Neurosci (2004) 24:547–53. doi:10.1523/jneurosci.4653-03.2004
136. Aalto S, Bruck A, Laine M, Nagren K, Rinne JO. Frontal and temporal dopamine release during working memory and attention tasks in healthy humans: a positron emission tomography study using the high-affinity dopamine D2 receptor ligand [11C]FLB 457. J Neurosci (2005) 25:2471–7. doi:10.1523/jneurosci.2097-04.2005
137. Stoof JC, Kebabian JW. Opposing roles for D-1 and D-2 dopamine receptors in efflux of cyclic AMP from rat neostriatum. Nature (1981) 294:366–8. doi:10.1038/294366a0
138. Stoof JC, Kebabian JW. Two dopamine receptors: biochemistry, physiology and pharmacology. Life Sci (1984) 35:2281–96. doi:10.1016/0024-3205(84)90519-8
139. Nowak JZ, Sek B, Schorderet M. Bidirectional regulation of cAMP generating system by dopamine-D1 and D2-receptors in the rat retina. J Neural Transm Gen Sect (1990) 81:235–40. doi:10.1007/BF01245045
140. Svenningsson P, Lindskog M, Ledent C, Parmentier M, Greengard P, Fredholm BB, et al. Regulation of the phosphorylation of the dopamine- and cAMP-regulated phosphoprotein of 32 kDa in vivo by dopamine D1, dopamine D2, and adenosine A2A receptors. Proc Natl Acad Sci U S A (2000) 97:1856–60. doi:10.1073/pnas.97.4.1856
141. Lidow MS, Goldman-Rakic PS, Gallager DW, Rakic P. Distribution of dopaminergic receptors in the primate cerebral cortex: quantitative autoradiographic analysis using [3H]raclopride, [3H]spiperone and [3H]SCH23390. Neuroscience (1991) 40:657–71. doi:10.1016/0306-4522(91)90003-7
142. Arnsten AF, Cai JX, Murphy BL, Goldman-Rakic PS. Dopamine D1 receptor mechanisms in the cognitive performance of young adult and aged monkeys. Psychopharmacology (Berl) (1994) 116:143–51. doi:10.1007/BF02245056
143. Arnsten AF, Goldman-Rakic PS. Noise stress impairs prefrontal cortical cognitive function in monkeys: evidence for a hyperdopaminergic mechanism. Arch Gen Psychiatry (1998) 55:362–8. doi:10.1001/archpsyc.55.4.362
144. Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry (1987) 44:660–9. doi:10.1001/archpsyc.1987.01800190080012
145. Goldman-Rakic PS, Muly EC III, Williams GV. D(1) receptors in prefrontal cells and circuits. Brain Res Brain Res Rev (2000) 31:295–301. doi:10.1016/S0165-0173(99)00045-4
146. Arnsten AF, Goldman-Rakic PS. Alpha 2-adrenergic mechanisms in prefrontal cortex associated with cognitive decline in aged nonhuman primates. Science (1985) 230:1273–6. doi:10.1126/science.2999977
147. Li BM, Mei ZT. Delayed-response deficit induced by local injection of the alpha 2-adrenergic antagonist yohimbine into the dorsolateral prefrontal cortex in young adult monkeys. Behav Neural Biol (1994) 62:134–9. doi:10.1016/S0163-1047(05)80034-2
148. Granon S, Poucet B, Thinus-Blanc C, Changeux JP, Vidal C. Nicotinic and muscarinic receptors in the rat prefrontal cortex: differential roles in working memory, response selection and effortful processing. Psychopharmacology (Berl) (1995) 119:139–44. doi:10.1007/BF02246154
149. Franowicz JS, Arnsten AF. The alpha-2a noradrenergic agonist, guanfacine, improves delayed response performance in young adult rhesus monkeys. Psychopharmacology (Berl) (1998) 136:8–14. doi:10.1007/s002130050533
150. Mao ZM, Arnsten AF, Li BM. Local infusion of an alpha-1 adrenergic agonist into the prefrontal cortex impairs spatial working memory performance in monkeys. Biol Psychiatry (1999) 46:1259–65. doi:10.1016/S0006-3223(99)00139-0
151. Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, et al. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci (2003) 6:51–8. doi:10.1038/nn992
152. Arthur JM, Collinsworth GP, Gettys TW, Raymond JR. Agonist-induced translocation of Gq/11alpha immunoreactivity directly from plasma membrane in MDCK cells. Am J Physiol (1999) 276:F528–34.
153. Lidow MS. Calcium signaling dysfunction in schizophrenia: a unifying approach. Brain Res Brain Res Rev (2003) 43:70–84. doi:10.1016/j.brainresrev.2003.09.001
154. Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P. Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry (2001) 6:293–301. doi:10.1038/sj.mp.4000866
155. Peruzzi D, Aluigi M, Manzoli L, Billi AM, Di Giorgio FP, Morleo M, et al. Molecular characterization of the human PLC beta1 gene. Biochim Biophys Acta (2002) 1584:46–54. doi:10.1016/S1388-1981(02)00269-X
156. Arinami T, Ohtsuki T, Ishiguro H, Ujike H, Tanaka Y, Morita Y, et al. Genomewide high-density SNP linkage analysis of 236 Japanese families supports the existence of schizophrenia susceptibility loci on chromosomes 1p, 14q, and 20p. Am J Hum Genet (2005) 77:937–44. doi:10.1086/498122
157. du Bois TM, Deng C, Huang XF. Membrane phospholipid composition, alterations in neurotransmitter systems and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2005) 29:878–88. doi:10.1016/j.pnpbp.2005.04.034
158. Lo Vasco VR, Cardinale G, Polonia P. Deletion of PLCB1 gene in schizophrenia-affected patients. J Cell Mol Med (2012) 16:844–51. doi:10.1111/j.1582-4934.2011.01363.x
159. Didriksen M. Effects of antipsychotics on cognitive behaviour in rats using the delayed non-match to position paradigm. Eur J Pharmacol (1995) 281:241–50. doi:10.1016/0014-2999(95)00242-D
160. Aultman JM, Moghaddam B. Distinct contributions of glutamate and dopamine receptors to temporal aspects of rodent working memory using a clinically relevant task. Psychopharmacology (Berl) (2001) 153:353–64. doi:10.1007/s002130000590
161. Gorelova NA, Yang CR. Dopamine D1/D5 receptor activation modulates a persistent sodium current in rat prefrontal cortical neurons in vitro. J Neurophysiol (2000) 84:75–87.
162. Dong Y, White FJ. Dopamine D1-class receptors selectively modulate a slowly inactivating potassium current in rat medial prefrontal cortex pyramidal neurons. J Neurosci (2003) 23:2686–95.
163. Adlersberg M, Hsiung SC, Glickstein SB, Liu KP, Tamir H, Schmauss C. Regulation of dopamine D-receptor activation in vivo by protein phosphatase 2B (calcineurin). J Neurochem (2004) 90:865–73. doi:10.1111/j.1471-4159.2004.02562.x
164. Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, et al. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol (2010) 28:1248–50. doi:10.1038/nbt1210-1248
Keywords: schizophrenia, PLC-β1, schizophrenia endophenotypes, mPFC, DLPFC
Citation: Kim S-W, Cho T and Lee S (2015) Phospholipase C-β1 Hypofunction in the Pathogenesis of Schizophrenia. Front. Psychiatry 6:159. doi: 10.3389/fpsyt.2015.00159
Received: 27 August 2015; Accepted: 26 October 2015;
Published: 17 November 2015
Edited by:
Blazej Misiak, Wroclaw Medical University, PolandReviewed by:
Madhara Udawela, The Florey Institute of Neuroscience and Mental Health, AustraliaSergej Nadalin, University of Rijeka, Croatia
Copyright: © 2015 Kim, Cho and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seong-Wook Kim, c2Vvbmd3b29rMDIwNUBnbWFpbC5jb20=
†Seong-Wook Kim and Taesup Cho have contributed equally to this work.