 Nawei Sun
Nawei Sun Jay A. Tischfield1,2
Jay A. Tischfield1,2 Robert A. King
Robert A. King Gary A. Heiman
Gary A. Heiman- 1Department of Genetics, Rutgers University, Piscataway, NJ, USA
- 2Human Genetics Institute of New Jersey, Piscataway, NJ, USA
- 3Child Study Center, Yale School of Medicine, New Haven, CT, USA
Tourette’s disorder (TD) is a highly heritable neurodevelopmental disorder with complex genetic architecture and unclear neuropathology. Disruptions of particular genes have been identified in subsets of TD patients. However, none of the findings have been replicated, probably due to the complex and heterogeneous genetic architecture of TD that involves both common and rare variants. To understand the etiology of TD, functional analyses are required to characterize the molecular and cellular consequences caused by mutations in candidate genes. Such molecular and cellular alterations may converge into common biological pathways underlying the heterogeneous genetic etiology of TD patients. Herein, we review specific genes implicated in TD etiology, discuss the functions of these genes in the mammalian central nervous system and the corresponding behavioral anomalies exhibited in animal models, and importantly, review functional analyses that can be performed to evaluate the role(s) that the genetic disruptions might play in TD. Specifically, the functional assays include novel cell culture systems, genome editing techniques, bioinformatics approaches, transcriptomic analyses, and genetically modified animal models applied or developed to study genes associated with TD or with other neurodevelopmental and neuropsychiatric disorders. By describing methods used to study diseases with genetic architecture similar to TD, we hope to develop a systematic framework for investigating the etiology of TD and related disorders.
Introduction
Tourette’s Disorder (TD) is a childhood-onset neurodevelopment disorder characterized by the presence of both motor and vocal tics. Prevalence ranges from 1–3% and is found across many ethnic groups around the world (1). However, a recent meta-analysis of previous TD prevalence studies re-estimates the population prevalence of TD to be 0.3–0.9% (2). Males are affected three to four times more often than females (3–5). A high percentage of TD patients have comorbid conditions, most commonly attention-deficit/hyperactivity disorder (ADHD) and obsessive–compulsive disorder (OCD) and to a lesser extent autism spectrum disorders (ASDs).
Consistent evidence from family and twin studies suggest a significant genetic contribution to TD, most likely the result of complex and heterogeneous inheritance involving both common and rare variants, though most of specific findings still require replication. The neurobiological basis of TD is not well understood, but appears to involve alterations in the development, structure, and/or functioning of cortico-striato-thalamo-cortical (CSTC) circuits (6). Specific genes have been found to be associated with TD. It is unclear if mutations in these genes cause TD and, if so, how these alterations affect the function or structural development of the nervous system. Our focus is to review the neurobiology of TD, describe the biological functions of those genes previously associated with TD, and discuss the various functional analyses that are required for evaluating and establishing the pathogenicity of these putative genetic causal variants for TD.
Neurobiology of Tourette’s Disorder
Alterations of the CSTC circuits are considered as the neuropathological basis of tic generation (6, 7). These alterations are apparent in functional and structural imaging studies (8), histopathological studies of specific neuronal populations (9), and defective inhibition in various electroneurophysiological experimental paradigms (10, 11). In addition to the male predominance, the developmental features of TD pose an explanatory challenge, with tics usually not appearing till 4–6 years of age and most often, but not always, improving spontaneously by late adolescence.
Neurotransmitter pathways that modulate the activity and the output of the CSTC circuits in the basal ganglia have been the focus of intensive investigation, driven in part by the quest for more effective pharmacological interventions. The most supported neurotransmitter dysregulation hypothesis in TD involves the hyperactivity or imbalance of the dopamine signaling in the striatum (12). Within the basal ganglia, dopamine is released to the striatum by dopaminergic neurons originating from the substantia nigra. In the striatum, the effect of dopamine on subsequent neural signal transmission is modulated by the striatal medium spiny neurons expressing either D1 or D2-like dopamine receptors (13). The dopamine hypothesis is supported by the clinical observation that dopamine D2 receptor antagonists effectively reduced tics in some TD patients (14, 15). Also, dopamine pathway dysregulation was reported in post-mortem TD brain samples (16, 17) and in living TD patients’ brain (12, 18).
Due to their excitatory and inhibitory effects within CSTC circuits, glutamatergic and GABAergic pathways have also been studied in TD. In post-mortem samples, lower levels of glutamate in subcortical brain regions were reported (19). However, it is unclear whether TD is associated with hyper- or hypo-glutamate levels (20). For the GABAergic pathway, an altered number and distribution of striatal GABAergic neurons were described in TD post-mortem brain samples (9, 21).
Disrupted serotonin signaling has been implicated in OCD, a common comorbid condition among TD patients. Selective serotonin reuptake inhibitors (SSRIs) have proven effective in reducing OCD symptoms (22) and are also used to treat TD patients with comorbid OCD (23). Interestingly, sequence variants at the serotonin transporter (SERT) gene were found in both OCD and TD patients (24), suggesting alterations in the serotonin pathway as one possible mechanism in the etiology of TD.
Until the recent discovery of a dominant negative non-sense mutation in the HDC gene co-segregating with TD in a large family, histaminergic (HA) neurotransmission was not considered a top candidate for TD etiology (25). However, other findings provide additional support for the involvement of HA neurotransmission in TD. For example, single nucleotide polymorphisms (SNPs) within the HDC gene region showed association with TD (26). Also, rare copy number variants (CNVs) found in TD patients were enriched in chromosomal regions harboring HA pathway genes (27). Furthermore, mice lacking the Hdc gene exhibited tic-like behaviors (28). Even though no evidence showed direct actions of serotonin and histamine on movements, it is proposed that serotonin and histamine pathways might indirectly regulate movements by modulating the dopamine system in the substantia nigra. In particular, both serotoninergic and HA innervations are observed in the substantia nigra (29, 30). Also, serotonin and histamine receptors are expressed on nigrostriatal dopaminergic neurons (30, 31).
Aside from the neurotransmitter dysregulation hypothesis in TD, developmental and neuroimmunological findings also provide a context for assessing the relevance of potential gene findings. Altered distribution of parvalbumin interneurons (21) and reduced numbers of parvalbumin and cholinergic interneurons in basal ganglia were observed in the post-mortem brain samples of TD patients (9), suggesting another possible, perhaps developmental, mechanism for alterations of CSTS circuits. Additionally, a dysregulated brain-immune system involving microglia cells was suggested to contribute to TD (32). Gene expressions of inflammatory factors were examined using post-mortem basal ganglia samples from TD patients and controls (33). An elevated expression of the CD45 gene was observed in TD patients even though the elevation was not statistically significant. CD45 is a surface marker of microglia and its expression is increased due to the activation of microglia. In another study, transcriptome analysis of post-mortem striatum of TD patients and controls revealed upregulation of microglia-related genes (34).
Genes Disrupted in Patients with Tourette’s Disorder
In this section, we will review 15 genes that have been associated in TD (Table 1); to suggest how we might move beyond association to establish a role in TD pathogenesis, we will examine what is known about the biological effects of these genes. We group these genes into several categories: (1) neurite outgrowth: SLITRK1; (2) histamine pathway: HDC; (3) serotonin pathway: SERT, HTR1A HTR2B; (4) glutamate pathway: SLC1A3; (5) synaptic signal transduction and cell-adhesion pathway: NLGN4, CDH2, CNTNAP2/CASPR2, DPP6; (6) mitochondrial functions: IMMP2L, MRPL3; and (7) genes associated with other diseases: DNAJC13 [Parkinson’s disease (PD)], OFCC1 (orofacial clefts), and HCRTR2 or OX2R (excessive daytime sleepiness). The diverse functions of these genes – ranging from neurotransmitter synthesis, neuronal migration, synaptic plasticity, cell adhesion, and protein transportation and synthesis – highlight the complexity of unraveling the pathogenesis of TD. However, in addition to the genetic disruptions discussed here, large structural variations, for example copy number variations (CNVs), have also been investigated in TD patients. Genes disrupted by these structural variants have been discovered and indicated as potential TD associated genes (35–37).
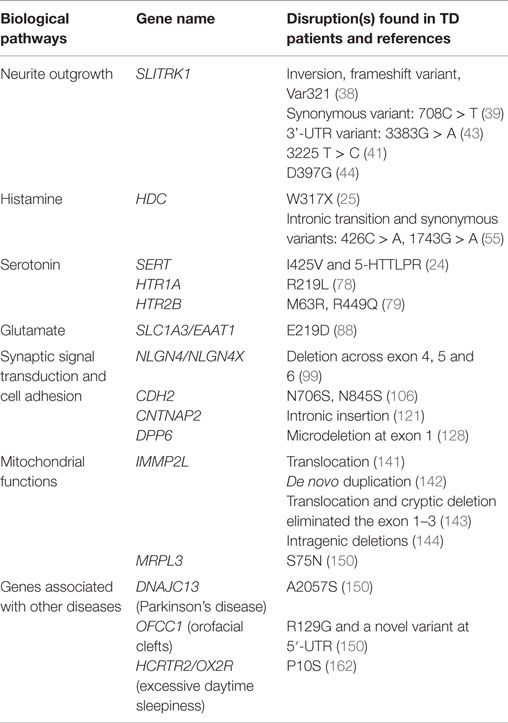
Table 1. Genes disrupted in TD.
Neurite Outgrowth
SLIT and NTRK-Like Family, Member 1 (SLITRK1)
In a TD patient with comorbid ADHD, a de novo chromosome 13 inversion was identified (38). The SLITRK1 gene was mapped close to the breakpoints. Targeted sequencing of the SLITRK1 gene identified a non-coding variant (var321) and a frameshift mutation (38) in another 174 unrelated TD patients but not in a large control sample. The frameshift mutation led to impaired dendrite growth in neurons and the var321 variant may cause reduced SLITRK1 protein expression (38). While the var321 and additional novel variants within SLITRK1 were found in other TD patients, these associations were not replicated in subsequent studies (39–49).
Members of the SLITRK protein family are transmembrane proteins. They are structurally homologous to the SLIT and the TRK proteins, which are involved in axon guidance pathway and neurodevelopment (50). The SLITRK1 gene is highly expressed in developing and mature neuronal tissues and promotes neurite outgrowth in culture (51). The SLITRK1 protein is localized to the post-synaptic densities and has been hypothesized to affect synapse formation at excitatory synapses through interactions with the pre-synaptic cellular adhesion molecule LAR-RPTP (52, 53). In Slitrk1 knockout mice, although stereotypic behaviors were not observed, the mice exhibited anxiety-like and depression-like behaviors, which were attenuated by chemicals modulating noradrenergic neurotransmission (54). Neurochemical abnormalities were also detected in Slitrk1 knockout mice: norepinephrine and its metabolites were significantly increased in the prefrontal cortex and the nucleus accumbens while choline and acetylcholine levels were significantly lower in the striatum (54). Taken together, the evidence suggests that the SLITRK1 gene might play a role in neurochemical modulation.
Histamine Pathway
Histidine Decarboxylase
Histamine neurotransmission was first linked to the etiology of TD when a rare non-sense mutation within the HDC gene was discovered in a multiplex family in which the father and all eight children were diagnosed with TD. The mutation resulted from a G to A transition at the ninth exon of the HDC gene and led to a premature stop codon (W317X) (25). The heterozygous W317X mutation co-segregated with all affected individuals in this family. The W317X mutation was not found in 3360 unrelated individuals unaffected with TD or another 720 TD patients, suggesting this is a very rare cause for TD. In vitro enzymatic assay demonstrated that the truncated protein produced by the mutation lost histidine decarboxylase (HDC) activity and had a dominant negative effect on the activity of wild-type HDC protein. After the initial finding, more TD patients were screened for mutations in the HDC gene in different studies. Only an intronic variant and two synonymous variants were identified in a study involving 120 TD patients (55). However, an association of the HDC gene and TD phenotypes was reported in a study including 520 TD nuclear families (26). Also, rare, genic CNVs identified in 460 TD patients were enriched for HA pathway genes (27), supporting the potential involvement of the histamine pathway in TD etiology.
In the adult human central nervous system, the HDC gene is exclusively expressed in the soma and axon varicosities of HA neurons mostly originating from the tuberomammillary nucleus in the posterior hypothalamic region of the brain (30). The HDC homodimer converts l-histidine into histamine in the soma of HA neurons. Histamine-containing neuronal fibers are seen in many brain areas in rodents and human including cerebral cortex (56, 57). Therefore, loss-of-function mutations at the HDC gene will likely cause a lack of histamine in the widespread brain regions receiving HA innervation. In addition to serving as a neurotransmitter, histamine is a neuromodulator, inhibiting dopamine release by striatal dopaminergic neurons through binding to the H3 receptors expressed on these neurons in mice (58). Given the hypothesis that hyperactivity of nigrostriatal dopaminergic neurons is responsible for tic generation (12), it is reasonable that histamine dysregulation may contribute to TD.
Since HDC protein functions as a homodimer (59), individuals harboring the W317X mutation have approximately 25% residual HDC activity remaining compared to the healthy controls. Therefore, the Hdc knockout mice may recapitulate the behavioral outcomes caused by the W317X mutation in humans. As expected, the Hdc knockout mice show tic-like stereotypic behaviors after psychostimulant administration and reduced pre-pulse inhibition (28). Interestingly, the striatal dopamine level was increased in the Hdc knockout mice during the dark cycle, which could be decreased by administration of histamine in the knockout mice. Also, higher levels of dopamine D2 receptor occupancy were found in the basal ganglia of TD patients carrying the W317X mutations, the Hdc knockout mice and the Hdc heterozygous mice, indicating that the dopamine release in the basal ganglia of the brain might be disinhibited due to histamine depletion (28). Taken together, parallel studies in TD subjects and mice demonstrated that lack of histamine results in dopamine dysregulation, providing a potential mechanism for the proposed role of dopamine disruption in TD (12).
Serotonin Pathway
Serotonin Transporter (SLC6A4 or SERT)
Given the effectiveness of the SSRIs in reducing OCD symptoms, the SERT gene has been studied as candidate gene for OCD (60–64). Serotonin-transporter-linked polymorphic region (5-HTTLPR) polymorphisms and a gain-of-function missense mutation I425V have been associated with OCD (65–69). Sequence variants of the SERT gene were first associated with TD in a two-generation pedigree (24). In this family, the heterozygous “long” 5-HTTLPR variant and the I425V mutation perfectly segregated with TD individuals in a dominant pattern. The “long” 5-HTTLPR produces higher SERT mRNA level compared to the “short” 5-HTTLPR (70). The I425V mutation results in constitutive activation of the SERT protein whose activity is regulated by cGMP-dependent protein kinase (71). Therefore, carrying both “long” 5-HTTLPR and the I425V mutation is expected to have a synergistic effect that increases the expression of SERT mRNA and increases the amount of activated SERT protein.
In the mammalian central nervous system, the SERT gene is primarily expressed in the serotonergic neurons that originate from the raphe nucleus in the hindbrain and project widely to other parts of the nervous system, descending to the spinal cord and ascending to the forebrain (72). The human SERT protein is a transmembrane protein (73, 74). Cell surface expression of SERT protein can be regulated by SERT antagonists and substrates (75). The serotonergic axons can innervate and regulate other neurotransmission systems. For example, serotonin can facilitate or inhibit dopamine release in the striatum in a direct or indirect manner (76). Therefore, dysregulation of SERT expression on the plasma membrane may affect dopamine transmission (77). So far, no other sequence variants in the SERT gene have been associated with TD and no corresponding transgenic mice are available for in vivo studies.
Serotonin Receptor 1A (HTR1A) and Serotonin Receptor 2B (HTR2B)
In addition to the SERT gene, other serotonin pathway genes have been examined in TD patients. A missense mutation causing an amino acid change from arginine to leucine was identified in the serotonin receptor 1A gene (HTR1A) in one TD patient. However, the mutation was not predicted to change the receptor activity (78). Two novel non-synonymous missense variants and three known SNPs in the serotonin receptor 2B (HTR2B) gene were also found in 132 Caucasian and 128 Chinese Han TD individuals, though the associations were not statistically significant (79).
There are currently 14 known serotonin receptors and these are categorized into seven classes (80, 81). The HTR1A and HTR2B, both of which are G protein-coupled receptors, belong to class I and class II, respectively (81). The HTR1A receptor is highly expressed in the brain. Lower brain expression of the HTR1A receptor has been associated with mood disorders in humans. Htr1a knockout mice exhibit depression- and anxiety-like behaviors and have been used for antidepressant drug screening (82, 83). The HTR1A receptors are located at both pre-synaptic and post-synaptic neurons in the CNS. Activation of the pre-synaptic HTR1A receptors on the serotonergic neurons leads to inhibition of serotonergic neuron firing and reduced serotonin release whereas activation of the post-synaptic HTR1A can modulate the release of other neurotransmitters (84). Compared to the HTR1A receptor, the role that HTR2B plays in the CNS is not well understood. However, there is evidence suggesting that HTR2B may regulate SERT activity by phosphorylating SERT protein (85). In mice, the HTR2B receptor may be involved in modulating serotonin release from the serotonergic neurons. (86). Taken together, dysfunction of the HTR1A or the HTR2B receptor might lead to abnormal serotonin release in the CNS.
Glutamate Pathway
Excitatory Amino Acid Transporter 1 (SLC1A3 or EAAT1)
Altered cortico-striatal-thalamo-cortical (CSTC) circuitry is believed to provide the neurobiological basis for TD (7, 87). Glutamate is the major excitatory neurotransmitter in CSTS circuitry. A missense mutation (E219D) in the glutamate transporter gene (SLC1A3) was associated with TD in a case-control study (88). In the same study, cells transfected with the E219D mutant glutamate transporter gene exhibited increased glutamate uptake activity compared to cells transfected with the wild-type gene. The proposed mechanism for the increased glutamate uptake activity was elevated glutamate transporter expression at the plasma membrane due to the E219D mutation. However, whether TD might be associated with hypo- or hyper-glutamate activity is still controversial.
One of the five subtypes of glutamate transporters, EAAT1, is responsible for the reuptake of the excitatory neurotransmitter, l-Glutamate, from the synapses back into cells. Dysfunction of glutamate transporters may lead to imbalanced extracellular glutamate levels, further affecting downstream glutamate neurotransmission or causing glutamate excitotoxicity to neurons (89, 90). EAAT1 is primarily expressed in glial cells. Regionally, EAAT1 proteins are found in neocortex, striatum, cerebellum, and spinal cord (91). Eaat1 knockout mice showed hyperactivity and reduced acoustic startle response compared with the wild-type mice (92) but did not exhibit the altered prepulse inhibition behavior, which has been found in TD patients (93). No Eaat1 gain-of-function mutant mice are available to test the “hypo-glutamate activity” hypothesis in TD.
Synaptic Signal Transduction and Cell-Adhesion Pathways
Neuroligin 4, X Linked (NLGN4 or NLGN4X)
Mutations in the neuroligin (NLGN) family members have been identified in patients with neuropsychiatric disorders such as ASD (94–97) and schizophrenia (98). A small deletion in the NLGN4 gene was detected in a mother and her two sons (99), one of whom was diagnosed with autism while the other was diagnosed with TD and ADHD. Their mother, who also carried the deletion, had a learning disability, depression, and anxiety. The deletion spanned exon 4, 5, and 6 of the NLGN4 gene, resulting in a truncated protein. No other known genes were affected by the deletion.
Neuroligins are cell adhesion molecules located on the plasma membrane of the pre-synaptic and post-synaptic neurons. By interacting with neurexins, another family of cell adhesion proteins, NLGNs modulate proper signal transmission between pre-synaptic and post-synaptic neurons (100).Reduced expression of NLGNs in cultured neurons or mice cause deficits in synaptic maturation and plasticity (101, 102). Nlgn4 knockout mice have been studied at both the behavioral and cellular levels. Because Nlgn4 gene disruptions had been associated with ASD, the Nlgn4 knockout mice were tested for ASD-like behaviors. As expected, Nlgn4 knockout mice exhibited deficits in social interactions and reduced ultrasonic vocalizations compared to the wild-type mice (103). In another study, the Nlgn4 knockout mice displayed reduced neural network response upon stimulation in both excitatory and inhibitory circuits (104). More interestingly, the Nlgn4 knockout mice also showed stereotypic repetitive behaviors and increased obsessive compulsive like behaviors (105), supporting the possibility that disruption of the NLGN4 gene might play a role in TD or related disorders.
Cadherin 2, Type1, N-Cadherin (CDH2)
Cadherin 2 (CDH2), also known as N-Cadherin, is another cell adhesion protein that has been associated with TD. CDH2 participates in neuron–neuron communication and in synaptogenesis. In a recent study, exons of the CDH2 gene were sequenced in 160 OCD probands and 160 controls (106). Four variants in the CDH2 gene were identified in subjects with OCD or TD. Two mutations were of particular interest: the N706S and the N845S variants. N706S is a rare and novel mutation close to the predicted proteolytic cleavage site of the CDH2 protein while the N845S variant is located at the β-catenin interacting region. Both mutations reduced the CDH2 protein level by more than 50% in transfected HEK293 cells (106), suggesting possible adhesion deficits in cells.
Cadherin 2 is a calcium-dependent cell–cell adhesion glycoprotein. CDH2 primarily mediates neurite outgrowth and axon guidance of neurons on myotubes (107) and on the surface of astrocytes (108, 109). The cytosolic domain of the CDH2 protein forms complexes with catenin proteins (110), and these complexes regulate synaptogenesis in both pre- and post-synaptic neurons (111, 112). Additionally, the cleaved C-terminal domain of the CDH2 is a repressor of CBP/CREB-mediated transcription whose target genes are critical in neural development and plasticity (113). The complete knockout of the Cdh2 gene is an embryonic lethal in mice whereas Cdh2 heterozygous null mice do not exhibit obvious morphological defects during development (114). Conditional knockout of the Cdh2 gene in the cerebral cortex of mice caused disrupted cortical structure (115). Thus far, behavioral analyses in the Cdh2+/− mice or Cdh2 conditional knockout mice have not been conducted.
Contactin-Associated Protein-Like 2 (CNTNAP2/CASPR2)
Variants in the CNTNAP2 gene have been associated with ASD in several family based studies (116–119). Also, putative deleterious mutations were found in the CNTNAP2 gene in ASD patients (120). In one family, an insertion on chromosome 7p35–p36, disrupting intron 8 of the CNTNAP2 gene, was shared by a father and his two children, all three of whom were diagnosed with TD (121). However, a translocation disrupting intron 11 of CNTNAP2 did not cause TD phenotypes in another three generation family (122).
CNTNAP2, a transmembrane protein, belongs to the neurexin superfamily and is highly expressed in neurons and localized to the axonal membrane of the juxtaparanodal region. The CNTNAP2 protein interacts with clustered Shaker-type potassium channels and plays an important role in the axon-glia septate-like junction (123). It is required for normal action potential propagation along myelinated axons of the neurons (124), and it has been hypothesized that malfunctions of the CNTNAP2 protein leads to deficits of electric signal transduction between neurons (121, 125, 126).
Behavioral assessments of Cntnap2 knockout mice led to stereotypic motor movements (127). Interestingly, a reduced number of GABAergic interneurons were reported in the striatum of the Cntnap2 knockout mice, which is consistent with previous post-mortem studies showing a reduction in the number of striatal GABAergic interneurons in TD patients (9, 21). Therefore, understanding the molecular mechanism of neuronal loss caused by disruption of CNTNAP2 may help to pinpoint biological pathways altered in TD.
Dipeptidyl-Peptidase 6
A heterozygous microdeletion at the first exon of the DPP6 gene was identified in a boy with TD as well as the boy’s father and paternal uncle both of whom were diagnosed with tic disorder and ADHD (128). The microdeletion led to decreased DPP6 mRNA level in the boy’s blood cells. DPP6 has also been associated with other neuropsychiatric disorders such as ASD (95, 129) and schizophrenia (130).
Dipeptidyl-peptidase 6 (DPP6) is a transmembrane protein belonging to a family of serine proteases. However, DPP6 does not have protease activity (131). DPP6’s expression is enriched in the brain and different isoforms have different distributions in the brain. (132). DPP6 proteins interact with A-type voltage-gated potassium channels, specifically the Kv4 subunit (133, 134). The A-type potassium channels participate in the modulation of dendritic signal transmission (135, 136). Moreover, the A-type potassium channel was reported to control the tonic dopamine release by substantia nigra dopaminergic neurons (137). Even though DPP6 has no peptidase activity, it may have novel functions and play essential roles in Kv4 intracellular trafficking, membrane expression, and proper function of the A-type potassium channels (138). Dpp6 knockout mice show abnormal synaptogenesis (139), and knocking down the Dpp6 gene specifically in the mouse brain caused impaired learning and memory abilities (140).
Mitochondria Functions
Inner Mitochondrial Membrane Peptidase 2 Like
A familial translocation segregating with TD or tics (141) and a de novo duplication in a TD patient with other developmental and mental phenotypes implicated IMMP2L as a possible TD candidate gene (142). This was the first mitochondria-related gene in TD. Subsequently, a cryptic deletion eliminating exons 1–3 of the IMMP2L gene and 21 other genes was identified in a TD patient with learning and speech difficulties (143). Also, a case-control study of copy number variations reported intragenic deletions at the IMMP2L gene in seven TD patients (144). Among the seven, three deletions at intron 3 led to production of a shorter IMMP2L mRNA transcript due to alternative splicing. In the same study, the expression of both the short and the long transcripts was examined by reverse transcription PCR in 19 regions of the human brain. While the long transcript was ubiquitously expressed in all 19 brain regions, the short transcript was selectively expressed at a lower level in only 10 brain regions, suggesting that the short transcript might have a more specific role in the human central nervous system (144).
The human inner mitochondrial membrane peptidase 2 like (IMMP2L) protein is one of the catalytic subunits of the inner mitochondrial membrane peptidase (IMP) complex (145, 146). The IMP complex participates in the cleavage of the inner mitochondrial membrane targeting signal sequence from its protein substrates, allowing maturation of the substrates. Loss of any subunit will cause the decomposition of the whole complex (147). Expression and function of the mammalian IMMP2L protein have been studied in animal models and human tissues. Immp2l knockout mice exhibit mitochondrial dysfunction, increased ischemic brain damage, and infertility (148, 149).The human IMMP2L mRNAs are ubiquitously expressed in various tissues except for adult liver and lungs. Unlike other TD associated genes, there is no enriched IMMP2L mRNA expression in the brain compared to other tissues (142). However, linking mitochondrial dysfunction to TD might lead to further speculation about the varied etiology of TD.
Mitochondrial Ribosomal Protein L3
Whole exome sequencing of a multiplex TD family showed three rare novel non-synonymous mutations in the MRPL3, DNAJC13, and the OFCC1 genes (150). The three variants were not found in controls or dbSNPs/1000 Genomes databases. However, a targeted-sequencing study of the same three variants in Han Chinese TD patients did not replicate these findings (151).
Mitochondrial ribosomal protein L3 (MRPL3) is a mitochondrial ribosome protein involved in mitochondrial protein translation (152). Compound heterozygous mutations in the MRPL3 gene were associated with mitochondrial respiratory chain deficiency in a pedigree of French origin (153), but no psychiatric diseases were reported.
Genes Associated with Other Diseases
DnaJ (Hsp40) Homolog, Subfamily C, Member 13 (DNAJC13)
A missense variant (A2057S) in the DNAJC13 gene was found to segregate with TD or chronic tic disorder (CTD) in a multiplex pedigree (150). Subsequently, a novel missense mutation Asn855Ser in the DNAJC13 gene was found in a multi-generation family with PD and in an additional four other PD patients (154). Human DNAJC13 is a membrane-associated protein involved in endocytosis, specifically in the process of early endosome-mediated membrane trafficking (155, 156). Knocking down the DNAJC13 gene in mammalian cells did not cause obvious dysfunction of endocytosis. However, introducing a C-terminus truncated mutant DNAJC13 protein into the cells did affect the normal distribution and formation of early endosomes (156). No DNAJC13 knockout animal model is available.
Orofacial Cleft 1 Candidate 1
After an initial study suggesting that the OFCC1 gene led to orofacial clefts (157), it was later linked to schizophrenia (158). Recently, a genome-wide association study (GWAS) of OCD found a significant association with OFCC1 (159). Sequence variants in OFCC1 have been found in patients with neurodevelopmental or neuropsychiatric disorders: a missense mutation (R129G) segregating with TD and CTD in a multiplex family (150) and a non-sense and a missense mutation were found in a single autism family (160).
The function of the orofacial cleft 1 candidate 1 (OFCC1) protein is unclear but one study suggested that the OFCC1 protein was an interacting partner and methylation substrate of protein arginine methyltransferase 1 (161). However, Ofcc1 knockout mice did not show any behavioral abnormalities (158).
Hypocretin (Orexin) Receptor 2 (HCRTR2/OX2R)
The coding regions of the orexin-1/hypocretin-1 (OX1R) receptor gene, the orexin-2/hypocretin-2 (OX2R) gene, and the prepro-orexin gene were examined in patients with Excessive Daytime Sleepiness, TD, or ADHD and healthy controls (162). A C29T nucleotide change in the OX2R gene producing a Pro10Ser amino acid substitution was detected in only one TD patient with comorbid ADHD. The Pro10Ser variant reduces responsiveness of the Orexin2 receptor to its ligands, Orexin-A, and Orexin-B. The Orexin receptor 2 is a G protein-coupled receptor that participates the regulation of feeding and sleep-wakefulness in mammalian brains (163). Ox2r knockout mice do not show any tic-like behaviors (164).
Functional Analyses of Genes Disrupted in TD
As indicated in the previous section, evidence for TD susceptibility genes exists. The mutations discussed were found in only a small number of individuals with TD, and replication remains elusive. This lack of replication may be due to the extreme locus heterogeneity, similar to what has been found in ASD (165). What evidence beyond stronger statistical association might help establish their potential role in TD pathogenesis? Some of these genes are found within neurobiological pathways that are presumed to be disrupted in TD (e.g., neural signal transmission/modulation) while others are found in novel pathways. Hence, the evidence that these genes are true susceptibility genes remains insufficient. Consequently, more convincing functional studies are needed to determine how variants in these genes could contribute to TD. In this next section, we review the in vitro and in vivo functional studies and techniques (beyond the knock out experiments referred to above) that will likely be useful to evaluate the consequences of mutations found in these presumptive TD susceptibility genes and for discovery of cellular and molecular phenotypes in disease.
Transgenic Mammalian Cell Lines
Functional studies using neuronal cells from individuals with TD can provide insights into the molecular basis of TD and potentially help to clarify the biological pathways altered in TD. However, one of the difficulties is the inability to obtain relevant biomaterials (e.g., neurons or neural stem cells) from affected individuals. Since TD is not a lethal disorder, there is very limited access to neural tissue from individuals with TD, particularly from individuals with a known causal variant.
Transgenic human non-neuronal cell lines have been used to characterize the cellular and molecular phenotypes resulting from specific mutations. Typically, plasmids carrying a wild type or a mutant gene of interest are delivered into the cell lines. Once the protein products of the transgene are expressed in the cells, assays are developed to evaluate the functional consequences of the mutant proteins. For example, in the CDH2 gene, both the wild type and mutant CDH2 genes were cloned into expression plasmids and subsequently delivered into human embryonic kidney (HEK293) cells and reduced expression of mutant CDH2 proteins was reported (106). While easily done, functional studies in non-neuronal cell lines are suboptimal for a variety of reasons. During transgenesis, the gene of interest is often transiently overexpressed or is controlled by a conditional and/or inducible gene expression system (166). Therefore, the level of transgene expression might not faithfully represent the endogenous gene expression level. Also, the expression levels of many genes are tissue-specific (167). This is particularly relevant for genes with multiple transcript isoforms where the isoform expression pattern in transgenic cell lines may not be comparable to patterns in neurons. Furthermore, because transgenic cells would not be expected to have the same gene expression profile as neurons, they may not provide relevant cellular environment for the transgene to execute its genuine neuronal function(s). Neuronal samples from TD patients are therefore preferred for in vitro functional studies but these are very difficult if not impossible to obtain. A recent technological advance, induced pluripotent stem cells (iPSCs), now makes functional studies of neuronal samples with a known causal variant possible.
Induced Pluripotent Stem Cells
Use of iPSC-Derived Neuronal Cells to Model Neuropsychiatric and Neurodevelopmental Disorders
The relatively new reprograming technique that converts human somatic cells into iPSCs allows researchers to model diseases in vitro using patient-derived cells. Since the first generation of iPSCs from mouse fibroblasts (168), the ability to produce differentiated cells from iPSCs has been intensively studied and improved. Recently, iPSC neuronal differentiation has become routine (169). Generating patient-specific neurons carrying mutations in disease candidate genes is invaluable for researchers who wish to study the cell-autonomous effects of mutations and to understand the cellular basis of neurological and neuropsychiatric disorders. To date, no study using iPSC-derived neurons to model TD has been published. However, mutations associated with other neuropsychiatric disorders have been studied in iPSC-derived neurons, and cellular abnormalities have been demonstrated (170–174). For example, Rett syndrome is a neurodevelopmental disorder occurring mainly in females characterized by mental retardation. Loss-of-function mutations in the methyl-CpG binding protein 2 (MeCP2) gene were reported in the majority of Rett Syndrome cases (170). Therefore, neurons with functional null mutation in the MeCP2 gene were generated from the iPSCs of an individual with Rett Syndrome. In culture, the neurons showed smaller soma size (171). Similarly, iPSC-derived neurons have been used to understand ASD. In an individual with ASD, a balanced translocation spanning the transient receptor potential 6 channel (TRPC6) gene was identified. Neurons derived from the iPSCs exhibited decreased TRPC6 expression, altered morphology and reduced Ca2+ influx (172). In schizophrenia, the DISC1 gene has been considered an important risk factor (175) and in iPSC-derived DISC1 mutant neurons, pre-synaptic vesicle release was impaired (173).
Unfortunately, in vitro neural differentiation from iPSCs yields mixed populations of neurons rather than a homogenous population. For diseases with clear neuropathology, pure cultures of the specific neuron types involved in the disorder are preferred in order to recapitulate disease-related cellular phenotypes. For instance, PD is characterized by loss of substantia nigra dopamine neurons (176). Cultures containing a high percentage of dopaminergic neurons were generated from PD patients who carry monogenic mutations (177–180), and defects in cellular functions such as autophagy, mitophagy, oxidative stress response, and dopamine release were found in these neurons. In patients with amyotrophic lateral sclerosis (ALS), motor neurons degenerate. Therefore, motor neurons were derived from iPSCs of ALS patients carrying known casual mutations (174, 181). As hypothesized, the mutant motor neurons exhibited neurite degeneration (181).
Use of Single-Cell Analysis to Overcome Culture Heterogeneity
One method to overcome cell culture heterogeneity is to analyze single cells, all of the same type. Looking at the transcriptome of single cells using microarray or RNA-seq analysis holds promise of detecting gene dysregulation in particular populations of neurons, which might not be identified by analyzing heterogeneous mixtures of cells. For instance, single-cell gene expression analysis of iPSC-derived dopamine neurons from PD patients with a LRKK2 mutation unveiled dysregulation of oxidative stress genes in mutant dopamine neurons (182). In another study, neurons were generated from the iPSCs of Timothy Syndrome patients with a mutation in the CACNA1C gene. Single cells were taken from the culture containing mixed neuron populations, and gene expression was analyzed by microarray. As a result, the distribution of neuron subtypes was altered in the Timothy Syndrome neuronal culture compared to the control cells (183). Compared to microarray, RNA-seq is gaining greater popularity for analyzing the transcriptome of single cells due to its ability to unbiasedly detect any transcript in cells within a broader dynamic range of expression. Generally, there are four important steps to achieve single-cell RNA-seq: (1) single-cell isolation, (2) RNA capture, (3) cDNA synthesis and, (4) next generation sequencing. The microfluidic system is becoming popular for single-cell RNA-seq because it can isolate single cells, lyse the cells, purify RNA, synthesize cDNA or even conduct gene expression analysis all in one run (184, 185).
Use of iPSC-Derived Cerebral Organoids to Model Neuropsychiatric and Neurodevelopmental Disorders
In contrast to PD or ALS, the neuropathology of many neurodevelopmental and neuropsychiatric disorders, such as TD or ASD, is unclear or is heterogeneous (18). As described above, mutations associated with TD indicate dysregulations of various neurotransmitter pathways or of neural circuits involving multiple brain regions. Hence, studying specific type of neurons may not help to explain the pathogenesis of TD. The recently developed “cerebral organoid” cellular system enables the differentiation of iPSCs into a three-dimensional miniature organ in a bioreactor, with minimum external interferences (186). Such self-organized spherical structures resemble the human brain at very early stages of development. In comparison to monolayer neuronal cultures, the cerebral organoids contain more diverse neuronal populations that define distinct brain regions. Also, within the cerebral organoid, neuronal migration and human-specific brain structures (e.g., the outer subventricular zone) were observed (187). Therefore, the cerebral organoid has been used to model neurodevelopmental diseases. For microcephaly, premature neural differentiation was recapitulated in organoids derived from microcephaly patients’ iPSCs (187). In cases of idiopathic ASD, overproduction of GABAergic inhibitory neurons in patient-derived cerebral organoids was reported (188). At the molecular level, the cellular phenotype was explained by overexpression of the transcription factor FOXG1. However, use of cerebral organoids to model neurodevelopmental diseases has limitations. The various “brain regions” in the organoids are fairly disorganized. Therefore, the cerebral organoid would not be suitable to study neural circuits. Furthermore, neuronal cells within the organoids are mostly neural progenitor cells, and their differentiation is restricted by limited growth of the organoids, which in turn is probably due to the lack of internal nutrient and oxygen supply. More importantly, each organoid is “unique” because the self-organization process is random and is not controlled by external factors. This “uniqueness” will generate variation among organoids, which may mask phenotypic differences between normal and patient-derived cells.
Use of Genome Editing to Generate Isogenic Control iPSC Lines
One challenge in identifying the phenotypic effects of a given mutation in iPSC-derived neurons is finding an appropriate control sample. While age, gender, and ethnicity-matched control samples with the wild-type allele are typically available, they are not matched for all of the other common genomic variants. Failure to control for such variability in genetic background can lead to spurious results. The recent technological advance of highly specific genome editing now allows the production of more comparable isogenic controls for functional studies. Several genome editing systems, such as zinc finger nuclease (189, 190), TALEN, and CRISPR-Cas9 (191, 192), are able to reverse the mutation to wild type at one genomic locus at a time in iPSCs. Comparing neuronal cells generated from mutant iPSCs and their edited, isogenic control neuronal cells with the mutation removed allows identification of molecular and cellular changes that are due only to the mutation (180). However, “off target” mutations at unrelated loci inadvertently introduced by editing remain a potentially important technological hurdle (193).
Gene Expression and Gene Network Analyses
The major goal of genomic sequencing of patients with neurodevelopmental and neuropsychiatric disorders is to identify disease-associated mutations. Once such genes are found, systematic approaches including genome-wide gene expression analysis and gene network analyses can be used to implicate common biological pathways altered in patients with different mutant genes.
Gene Expression Analysis
Gene expression analysis, primarily through the RNA-seq approach, aims to quantify transcript level of target genes or of the whole transcriptome in biological samples from patients and healthy controls to identify genes dysregulated in human diseases (194). For neurodevelopmental disorders, post-mortem brain samples are often used for the transcriptomic analyses. The first transcriptomic analysis of TD patients’ post-mortem striatum samples revealed that interneuron disruption might be involved in the pathophysiology of TD (34). However, to evaluate particular mutations, post-mortem samples meeting specific research criteria are usually difficult or virtually impossible to obtain. With the emergence of somatic cell reprograming techniques, iPSC-derived neurons with and without a putative disease-causing mutation can be produced in vitro (195). The transcriptomes of these iPSC-derived neurons can be compared by microarray or RNA sequencing (RNA-seq) (172, 173). To further dissect the cellular phenotype at single neuron level and to detect abnormalities only shown in particular populations of neurons, single-cell transcriptome analysis can also be performed (182, 183). Multiple bioinformatic tools have been developed for RNA-seq data to detect differentially expressed genes (DEGs) from distinct cell types or under different experimental conditions (196–198). Among these RNA-seq analysis methods, none outperforms the others in all aspects. Selecting an optimal method for a study requires an understanding of the benefits and limitations of each method as well as the parameters of the study (196). Once the DEGs between experimental conditions are determined, gene and pathway annotation tools, gene and protein expression and interaction databases can help to explore the gene pathways underlying the disorder. Gene and pathway annotations tools such as IPA1, KEGG (199, 200), DAVID (201, 202), ConsensusPathDB (203) report biological pathways in which DEGs are enriched or reduced and take these into account to predict how these pathways might be affected. However, the data from which these tools were constructed come from non-neuronal samples which could lead to associations not found in neuronal tissues or failure to detect neuronal associations (204). In order to annotate neuronal gene expression in a temporal and spatial manner, human brain gene expression databases, for example, the Allen Human Brain Atlas (205), BrainSpan (206), GTEx (207) were built using microarray and RNA-seq data from post-mortem brain samples. Mapping DEGs identified in neuronal samples of patients with neurodevelopmental disorders to human brain gene expression databases revealed specific brain regions and neural developmental stages that were affected (34, 208). A more detailed human brain gene expression atlas that annotates gene expression at single cell level has been initiated by a group in Stanford University (209). Single-cell RNA-seq was used to analyze neurons from human adult and fetal cerebral cortex and it identified more diverse populations of neurons within the cortical region (209). Constructing a comprehensive human brain gene expression database at single neuron resolution is quite challenging due to limited access to healthy human brain samples and the high cost of single-cell RNA-seq. Therefore, collaborative work with standardized experimental protocols is required.
Gene Network Analysis
Differential gene expression from transcriptome analysis is sample-dependent and tissue-specific. In order to explore the etiology of complex neurodevelopmental disorders such as TD, disease-associated genes can be mapped to gene networks to visualize relationships between disease candidate genes and, further, to pinpoint annotated or novel pathways. The gene networks can be gene co-expression networks (205, 206), gene regulatory networks2, protein–protein interaction networks (210) or networks constructed with combined criteria (203, 211). For example, in ASD, disease-associated genes have been evaluated using spatiotemporal gene co-expression networks constructed from BrainSpan (206) and were found to be enriched in sub-networks that represent specific brain regions and time periods during human brain development (212). Furthermore, ASD-associated mutations identified by previous genetic studies were mapped to a “background network” which scores each pair of human genes based on very comprehensive information about every known human gene (211). Cell types and brain areas affected in ASD were implicated (213). With a growing number of mutations associated with TD, the same approaches could be utilized in TD.
Animal Models
Another approach to the study of the functional effects of a specific mutation in a gene is to use animal models. Most often, a putative disease gene is knocked out or modified in the animal model. Then, the mutant and wild type animals from the same genetic background are compared. (Some such animal knock-out studies of putative TD genes have been described above). Conventional animal models are designed to study a small number of genes, usually one or two genes at a time. If, however, TD is caused by the combined effect of multiple variant genes, multi-transgenic animals, whose genomes are modified at multiple loci, would be required. Although more challenging, generation of such multi-transgenic animals can be achieved by genome editing as well. (214, 215).
In order to model TD in animals, the following criteria should be met: (1) the gene to be studied is strongly associated with the disease; (2) the gene and the neurological component phenotypes involved in TD are relatively well conserved between humans and the animal; (3) the gene is thought to have similar functions in both humans and the model animals; and (4) the disease phenotype can be experimentally characterized in the model animals by biochemical and/or behavioral approaches (216). One mouse model in TD that meets these criteria is HDC. As indicated previously, a rare dominant non-sense mutation W317X in HDC cosegregated with all TD individuals in a two generation pedigree. (25). Subsequently, a study using HDC knockout mice demonstrated behavioral and molecular abnormalities caused by the loss of the HDC activity in the brain (28). The knockout and heterozygous mice showed tic-like stereotypic movements after psychostimulant administration. Also, the striatal dopaminergic pathway was dysregulated due to the HDC deficiency. Specifically, the dysregulation of dopamine receptors in the basal ganglia region of the HDC knockout and heterozygous mice recapitulated the dysregulation of the same types of dopamine receptors in TD patients carrying the W317X mutation.
Summary and Future Directions
Tourette’s disorder is likely caused by a complex multigenic inheritance pattern that includes locus and allelic heterogeneity of both common and rare variants that interact with environmental factors (14, 217, 218). Despite long-standing interest in the genetic contribution to TD, the overall genetic architecture of TD remains elusive. Some of the genes identified as causal of TD are involved in neurotransmitter pathways presumed to be altered in TD, while others are novel. In this respect, the genetics of TD may resemble that of other complex neuropsychiatric disorders. Indeed, there is evidence of some overlap with subsets of similar genes involved in multiple disorders. Furthermore, there also appears to be an increased rate of comorbidity between some such disorders, such as TD and ASD (27, 219). It may also be that current DSM-based psychiatric nosology does not sufficiently “carve nature at its joints,” and that other classification schemes, such as Research Domain Criteria (RDoC)(220–224), might reveal etiologically more coherent groupings of disorders or patients.
International consortiums including the Tourette International Collaborative Genetics (TIC Genetics), the Tourette Syndrome Association International Consortium for Genetics (TSAICG), the European Multicenter Tics in Children Studies (EMTICS), the European Society for the Study of Tourette Syndrome (ESSTS), the Tourette Syndrome Genetics The Southern and Eastern Europe initiative (TSGeneSEE), and sharing repositories (New Jersey Center for Tourette Syndrome Repository) (225, 226) have initiated large collaborations to collect many patient and family samples in an effort to understand the genetics of TD. Continued efforts in gene discovery from large open-access repositories are needed to find additional risk variants.
Author Contributions
All authors wrote, reviewed, and critiqued the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer Fotis Tsetsos and handling Editor Peristera Paschou declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
This study was supported by a Grant from the National Institute of Mental Health (R01MH092293) and the New Jersey Center for Tourette Syndrome and Associated Disorders (NJCTS).
Funding
National Institute of Mental Health (R01MH092293); New Jersey Center for Tourette Syndrome and Associated Disorders (NJCTS) (Center of Excellence for Tourette Syndrome & Associated Disorders Genetics Repository).
Footnotes
References
1. Robertson MM. Diagnosing Tourette syndrome: is it a common disorder? J Psychosom Res (2003) 55(1):3–6. doi:10.1016/S0022-3999(02)00580-9
2. Scharf JM, Miller LL, Gauvin CA, Alabiso J, Mathews CA, Ben-Shlomo Y. Population prevalence of Tourette syndrome: a systematic review and meta-analysis. Mov Disord (2015) 30(2):221–8. doi:10.1002/mds.26089
3. Robertson MM. The Gilles de la Tourette syndrome: the current status. Arch Dis Child Educ Pract Ed (2012) 97(5):166–75. doi:10.1136/archdischild-2011-300585
4. Shaw ZA, Coffey BJ. Tics and tourette syndrome. Psychiatr Clin North Am (2014) 37(3):269–86. doi:10.1016/j.psc.2014.05.001
5. Freeman RD, Fast DK, Burd L, Kerbeshian J, Robertson MM, Sandor P. An international perspective on Tourette syndrome: selected findings from 3,500 individuals in 22 countries. Dev Med Child Neurol (2000) 42(7):436–47. doi:10.1017/S0012162200000839
6. Paschou P, Fernandez TV, Sharp F, Heiman GA, Hoekstra PJ. Genetic susceptibility and neurotransmitters in Tourette syndrome. Int Rev Neurobiol (2013) 112:155–77. doi:10.1016/B978-0-12-411546-0.00006-8
7. Leckman JF, Bloch MH, Smith ME, Larabi D, Hampson M. Neurobiological substrates of Tourette’s disorder. J Child Adolesc Psychopharmacol (2010) 20(4):237–47. doi:10.1089/cap.2009.0118
8. Wang ZS, Maia TV, Marsh R, Colibazzi T, Gerber A, Peterson BS. The neural circuits that generate tics in Tourette’s syndrome. Am J Psychiatry (2011) 168(12):1326–37. doi:10.1176/appi.ajp.2011.09111692
9. Kataoka Y, Kalanithi PSA, Grantz H, Schwartz ML, Saper C, Leckman JF, et al. Decreased number of parvalbumin and cholinergic interneurons in the striatum of individuals with Tourette syndrome. J Comp Neurol (2010) 518(3):277–91. doi:10.1002/cne.22206
10. Priori A, Giannicola G, Rosa M, Marceglia S, Servello D, Sassi M, et al. Deep brain electrophysiological recordings provide clues to the pathophysiology of Tourette syndrome. Neurosci Biobehav Rev (2013) 37(6):1063–8. doi:10.1016/j.neubiorev.2013.01.011
11. Pogorelov V, Xu MY, Smith HR, Buchanan GF, Pittenger C. Corticostriatal interactions in the generation of tic-like behaviors after local striatal disinhibition. Exp Neurol (2015) 265:122–8. doi:10.1016/j.expneurol.2015.01.001
12. Buse J, Schoenefeld K, Munchau A, Roessner V. Neuromodulation in Tourette syndrome: dopamine and beyond. Neurosci Biobehav Rev (2013) 37(6):1069–84. doi:10.1016/j.neubiorev.2012.10.004
13. Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci (2007) 30(5):228–35. doi:10.1016/j.tins.2007.03.008
14. Bloch M, State M, Pittenger C. Recent advances in Tourette syndrome. Curr Opin Neurol (2011) 24(2):119–25. doi:10.1097/WCO.0b013e328344648c
15. Roessner V, Plessen KJ, Rothenberger A, Ludolph AG, Rizzo R, Skov L, et al. European clinical guidelines for Tourette syndrome and other tic disorders. Part II: pharmacological treatment. Eur Child Adolesc Psychiatry (2011) 20(4):173–96. doi:10.1007/s00787-011-0163-7
16. Minzer K, Lee O, Hong JJ, Singer HS. Increased prefrontal D2 protein in Tourette syndrome: a postmortem analysis of frontal cortex and striatum. J Neurol Sci (2004) 219(1–2):55–61. doi:10.1016/j.jns.2003.12.006
17. Yoon DY, Gause CD, Leckman JF, Singer HS. Frontal dopaminergic abnormality in Tourette syndrome: a postmortem analysis. J Neurol Sci (2007) 255(1–2):50–6. doi:10.1016/j.jns.2007.01.069
18. Felling RJ, Singer HS. Neurobiology of tourette syndrome: current status and need for further investigation. J Neurosci (2011) 31(35):12387–95. doi:10.1523/JNEUROSCI.0150-11.2011
19. Anderson GM, Pollak ES, Chatterjee D, Leckman JF, Riddle MA, Cohen DJ. Brain monoamines and amino acids in Gilles de la Tourette’s syndrome: a preliminary study of subcortical regions. Arch Gen Psychiatry (1992) 49(7):584–6. doi:10.1001/archpsyc.1992.01820070078016
20. Singer HS, Morris C, Grados M. Glutamatergic modulatory therapy for Tourette syndrome. Med Hypotheses (2010) 74(5):862–7. doi:10.1016/j.mehy.2009.11.028
21. Kalanithi PS, Zheng W, Kataoka Y, DiFiglia M, Grantz H, Saper CB, et al. Altered parvalbumin-positive neuron distribution in basal ganglia of individuals with Tourette syndrome. Proc Natl Acad Sci U S A (2005) 102(37):13307–12. doi:10.1073/pnas.0502624102
22. Pittenger C, Bloch MH. Pharmacological treatment of obsessive-compulsive disorder. Psychiatr Clin North Am (2014) 37(3):375–91. doi:10.1016/j.psc.2014.05.006
23. Roessner V, Schoenefeld K, Buse J, Bender S, Ehrlich S, Munchau A. Pharmacological treatment of tic disorders and Tourette syndrome. Neuropharmacology (2013) 68:143–9. doi:10.1016/j.neuropharm.2012.05.043
24. Moya PR, Wendland JR, Rubenstein LM, Timpano KR, Heiman GA, Tischfield JA, et al. Common and rare alleles of the serotonin transporter gene, SLC6A4, associated with Tourette’s disorder. Mov Disord (2013) 28(9):1263–70. doi:10.1002/mds.25460
25. Ercan-Sencicek AG, Stillman AA, Ghosh AK, Bilguvar K, O’Roak BJ, Mason CE, et al. l-histidine decarboxylase and Tourette’s syndrome. N Engl J Med (2010) 362(20):1901–8. doi:10.1056/NEJMoa0907006
26. Karagiannidis I, Dehning S, Sandor P, Tarnok Z, Rizzo R, Wolanczyk T, et al. Support of the histaminergic hypothesis in Tourette syndrome: association of the histamine decarboxylase gene in a large sample of families. J Med Genet (2013) 50(11):760–4. doi:10.1136/jmedgenet-2013-101637
27. Fernandez TV, Sanders SJ, Yurkiewicz IR, Ercan-Sencicek AG, Kim YS, Fishman DO, et al. Rare copy number variants in tourette syndrome disrupt genes in histaminergic pathways and overlap with autism. Biol Psychiatry (2012) 71(5):392–402. doi:10.1016/j.biopsych.2011.09.034
28. Castellan Baldan L, Williams KA, Gallezot JD, Pogorelov V, Rapanelli M, Crowley M, et al. Histidine decarboxylase deficiency causes tourette syndrome: parallel findings in humans and mice. Neuron (2014) 81(1):77–90. doi:10.1016/j.neuron.2013.10.052
29. Parent M, Wallman MJ, Gagnon D, Parent A. Serotonin innervation of basal ganglia in monkeys and humans. J Chem Neuroanat (2011) 41(4):256–65. doi:10.1016/j.jchemneu.2011.04.005
30. Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev (2008) 88(3):1183–241. doi:10.1152/physrev.00043.2007
31. Di Giovanni G, Di Matteo V, Esposito E. Serotonin-dopamine interaction: experimental evidence and therapeutic relevance. Preface. Prog Brain Res (2008) 172:ix. doi:10.1016/S0079-6123(08)00931-X
32. Frick LR, Williams K, Pittenger C. Microglial dysregulation in psychiatric disease. Clin Dev Immunol (2013) 2013:608654. doi:10.1155/2013/608654
33. Morer A, Chae W, Henegariu O, Bothwell AL, Leckman JF, Kawikova I. Elevated expression of MCP-1, IL-2 and PTPR-N in basal ganglia of Tourette syndrome cases. Brain Behav Immun (2010) 24(7):1069–73. doi:10.1016/j.bbi.2010.02.007
34. Lennington JB, Coppola G, Kataoka-Sasaki Y, Fernandez TV, Palejev D, Li Y, et al. Transcriptome analysis of the human striatum in Tourette syndrome. Biol Psychiatry (2014). doi:10.1016/j.biopsych.2014.07.018
35. Sundaram SK, Huq AM, Wilson BJ, Chugani HT. Tourette syndrome is associated with recurrent exonic copy number variants. Neurology (2010) 74(20):1583–90. doi:10.1212/WNL.0b013e3181e0f147
36. Nag A, Bochukova EG, Kremeyer B, Campbell DD, Muller H, Valencia-Duarte AV, et al. CNV analysis in Tourette syndrome implicates large genomic rearrangements in COL8A1 and NRXN1. PLoS One (2013) 8(3):e59061. doi:10.1371/journal.pone.0059061
37. Bertelsen B, Stefansson H, Riff Jensen L, Melchior L, Mol Debes N, Groth C, et al. Association of AADAC deletion and Gilles de la Tourette syndrome in a large European cohort. Biol Psychiatry (2015). doi:10.1016/j.biopsych.2015.08.027
38. Abelson JF, Kwan KY, O’Roak BJ, Baek DY, Stillman AA, Morgan TM, et al. Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science (2005) 310(5746):317–20. doi:10.1126/science.1116502
39. Deng H, Le WD, Xie WJ, Jankovic J. Examination of the SLITRK1 gene in Caucasian patients with Tourette syndrome. Acta Neurol Scand (2006) 114(6):400–2. doi:10.1111/j.1600-0404.2006.00706.x
40. Verkerk AJ, Cath DC, van der Linde HC, Both J, Heutink P, Breedveld G, et al. Genetic and clinical analysis of a large Dutch Gilles de la Tourette family. Mol Psychiatry (2006) 11(10):954–64. doi:10.1038/sj.mp.4001877
41. Chou IC, Wan L, Liu SC, Tsai CH, Tsai FJ. Association of the slit and Trk-like 1 gene in Taiwanese patients with Tourette syndrome. Pediatr Neurol (2007) 37(6):404–6. doi:10.1016/j.pediatrneurol.2007.06.017
42. Scharf JM, Moorjani P, Fagerness J, Platko JV, Illmann C, Galloway B, et al. Lack of association between SLITRK1var321 and Tourette syndrome in a large family-based sample. Neurology (2008) 70(16 Pt 2):1495–6. doi:10.1212/01.wnl.0000296833.25484.bb
43. Zimprich A, Hatala K, Riederer F, Stogmann E, Aschauer HN, Stamenkovic M. Sequence analysis of the complete SLITRK1 gene in Austrian patients with Tourette’s disorder. Psychiatr Genet (2008) 18(6):308–9. doi:10.1097/YPG.0b013e3283060f6f
44. Miranda DM, Wigg K, Kabia EM, Feng Y, Sandor P, Barr CL. Association of SLITRK1 to Gilles de la Tourette syndrome. Am J Med Genet B Neuropsychiatr Genet (2009) 150B(4):483–6. doi:10.1002/ajmg.b.30840
45. O’Roak BJ, Morgan TM, Fishman DO, Saus E, Alonso P, Gratacos M, et al. Additional support for the association of SLITRK1 var321 and Tourette syndrome. Mol Psychiatry (2010) 15(5):447–50. doi:10.1038/mp.2009.105
46. Yasmeen S, Melchior L, Bertelsen B, Skov L, Mol Debes N, Tumer Z. Sequence analysis of SLITRK1 for var321 in Danish patients with Tourette syndrome and review of the literature. Psychiatr Genet (2013) 23(3):130–3. doi:10.1097/YPG.0b013e328360c880
47. Inai A, Tochigi M, Kuwabara H, Nishimura F, Kato K, Eriguchi Y, et al. Analysis of SLITRK1 in Japanese patients with Tourette syndrome using a next-generation sequencer. Psychiatr Genet (2015) 25(6):256–8. doi:10.1097/YPG.0000000000000104
48. Keen-Kim D, Mathews CA, Reus VI, Lowe TL, Herrera LD, Budman CL, et al. Overrepresentation of rare variants in a specific ethnic group may confuse interpretation of association analyses. Hum Mol Genet (2006) 15(22):3324–8. doi:10.1093/hmg/ddl408
49. Karagiannidis I, Rizzo R, Tarnok Z, Wolanczyk T, Hebebrand J, Nothen MM, et al. Replication of association between a SLITRK1 haplotype and Tourette syndrome in a large sample of families. Mol Psychiatry (2012) 17(7):665–8. doi:10.1038/mp.2011.151
50. Proenca CC, Gao KP, Shmelkov SV, Rafii S, Lee FS. Slitrks as emerging candidate genes involved in neuropsychiatric disorders. Trends Neurosci (2011) 34(3):143–53. doi:10.1016/j.tins.2011.01.001
51. Aruga J, Mikoshiba K. Identification and characterization of Slitrk, a novel neuronal transmembrane protein family controlling neurite outgrowth. Mol Cell Neurosci (2003) 24(1):117–29. doi:10.1016/S1044-7431(03)00129-5
52. Yim YS, Kwon Y, Nam J, Yoon HI, Lee K, Kim DG, et al. Slitrks control excitatory and inhibitory synapse formation with LAR receptor protein tyrosine phosphatases. Proc Natl Acad Sci U S A (2013) 110(10):4057–62. doi:10.1073/pnas.1209881110
53. Um JW, Kim KH, Park BS, Choi Y, Kim D, Kim CY, et al. Structural basis for LAR-RPTP/Slitrk complex-mediated synaptic adhesion. Nat Commun (2014) 5:5423. doi:10.1038/ncomms6423
54. Katayama K, Yamada K, Ornthanalai VG, Inoue T, Ota M, Murphy NP, et al. Slitrk1-deficient mice display elevated anxiety-like behavior and noradrenergic abnormalities. Mol Psychiatry (2010) 15(2):177–84. doi:10.1038/mp.2008.97
55. Lei J, Deng X, Zhang J, Su L, Xu H, Liang H, et al. Mutation screening of the HDC gene in Chinese Han patients with Tourette syndrome. Am J Med Genet B Neuropsychiatr Genet (2012) 159B(1):72–6. doi:10.1002/ajmg.b.32003
56. Panula P, Airaksinen MS, Pirvola U, Kotilainen E. A histamine-containing neuronal system in human brain. Neuroscience (1990) 34(1):127–32. doi:10.1016/0306-4522(90)90307-P
57. Watanabe T, Taguchi Y, Shiosaka S, Tanaka J, Kubota H, Terano Y, et al. Distribution of the histaminergic neuron system in the central nervous system of rats; a fluorescent immunohistochemical analysis with histidine decarboxylase as a marker. Brain Res (1984) 295(1):13–25. doi:10.1016/0006-8993(84)90811-4
58. Schlicker E, Fink K, Detzner M, Gothert M. Histamine inhibits dopamine release in the mouse striatum via presynaptic H3 receptors. J Neural Transm Gen Sect (1993) 93(1):1–10. doi:10.1007/BF01244933
59. Komori H, Nitta Y, Ueno H, Higuchi Y. Structural study reveals that Ser-354 determines substrate specificity on human histidine decarboxylase. J Biol Chem (2012) 287(34):29175–83. doi:10.1074/jbc.M112.381897
60. Zohar J, Insel TR. Drug treatment of obsessive-compulsive disorder. J Affect Disord (1987) 13(2):193–202. doi:10.1016/0165-0327(87)90023-1
61. Lieberman J. Evidence for a biological hypothesis of obsessive-compulsive disorder. Neuropsychobiology (1984) 11(1):14–21. doi:10.1159/000118043
62. Zohar J, Insel TR, Zohar-Kadouch RC, Hill JL, Murphy DL. Serotonergic responsivity in obsessive-compulsive disorder. Effects of chronic clomipramine treatment. Arch Gen Psychiatry (1988) 45(2):167–72. doi:10.1001/archpsyc.1988.01800260081011
63. Billett EA, Richter MA, King N, Heils A, Lesch KP, Kennedy JL. Obsessive compulsive disorder, response to serotonin reuptake inhibitors and the serotonin transporter gene. Mol Psychiatry (1997) 2(5):403–6. doi:10.1038/sj.mp.4000257
64. McDougle CJ, Epperson CN, Price LH, Gelernter J. Evidence for linkage disequilibrium between serotonin transporter protein gene (SLC6A4) and obsessive compulsive disorder. Mol Psychiatry (1998) 3(3):270–3. doi:10.1038/sj.mp.4000391
65. Bengel D, Greenberg BD, Cora-Locatelli G, Altemus M, Heils A, Li Q, et al. Association of the serotonin transporter promoter regulatory region polymorphism and obsessive-compulsive disorder. Mol Psychiatry (1999) 4(5):463–6. doi:10.1038/sj.mp.4000550
66. Di Bella D, Erzegovesi S, Cavallini MC, Bellodi L. Obsessive-compulsive disorder, 5-HTTLPR polymorphism and treatment response. Pharmacogenomics J (2002) 2(3):176–81. doi:10.1038/sj.tpj.6500090
67. Ozaki N, Goldman D, Kaye WH, Plotnicov K, Greenberg BD, Lappalainen J, et al. Serotonin transporter missense mutation associated with a complex neuropsychiatric phenotype. Mol Psychiatry (2003) 8(11):933–6. doi:10.1038/sj.mp.4001415
68. Wendland JR, Moya PR, Kruse MR, Ren-Patterson RF, Jensen CL, Timpano KR, et al. A novel, putative gain-of-function haplotype at SLC6A4 associates with obsessive-compulsive disorder. Hum Mol Genet (2008) 17(5):717–23. doi:10.1093/hmg/ddm343
69. Voyiaziakis E, Evgrafov O, Li D, Yoon HJ, Tabares P, Samuels J, et al. Association of SLC6A4 variants with obsessive-compulsive disorder in a large multicenter US family study. Mol Psychiatry (2011) 16(1):108–20. doi:10.1038/mp.2009.100
70. Hu XZ, Lipsky RH, Zhu G, Akhtar LA, Taubman J, Greenberg BD, et al. Serotonin transporter promoter gain-of-function genotypes are linked to obsessive-compulsive disorder. Am J Hum Genet (2006) 78(5):815–26. doi:10.1086/503850
71. Kilic F, Murphy DL, Rudnick G. A human serotonin transporter mutation causes constitutive activation of transport activity. Mol Pharmacol (2003) 64(2):440–6. doi:10.1124/mol.64.2.440
72. Jacobs BL, Azmitia EC. Structure and function of the brain serotonin system. Physiol Rev (1992) 72(1):165–229.
73. Blakely RD, De Felice LJ, Hartzell HC. Molecular physiology of norepinephrine and serotonin transporters. J Exp Biol (1994) 196:263–81.
74. Lesch KP, Wolozin BL, Estler HC, Murphy DL, Riederer P. Isolation of a cDNA encoding the human brain serotonin transporter. J Neural Transm Gen Sect (1993) 91(1):67–72. doi:10.1007/BF01244919
75. Kittler K, Lau T, Schloss P. Antagonists and substrates differentially regulate serotonin transporter cell surface expression in serotonergic neurons. Eur J Pharmacol (2010) 629(1–3):63–7. doi:10.1016/j.ejphar.2009.12.010
76. Esposito E, Di Matteo V, Di Giovanni G. Serotonin-dopamine interaction: an overview. Prog Brain Res (2008) 172:3–6. doi:10.1016/S0079-6123(08)00901-1
77. Daw ND, Kakade S, Dayan P. Opponent interactions between serotonin and dopamine. Neural Netw (2002) 15(4–6):603–16. doi:10.1016/S0893-6080(02)00052-7
78. Lam S, Shen Y, Nguyen T, Messier TL, Brann M, Comings D, et al. A serotonin receptor gene (5HT1A) variant found in a Tourette’s syndrome patient. Biochem Biophys Res Commun (1996) 219(3):853–8. doi:10.1006/bbrc.1996.0322
79. Guo Y, Deng X, Jankovic J, Su L, Zhang J, Le W, et al. Mutation screening of the HTR2B gene in patients with Tourette syndrome. Neurosci Lett (2012) 526(2):150–3. doi:10.1016/j.neulet.2012.08.013
80. Hannon J, Hoyer D. Molecular biology of 5-HT receptors. Behav Brain Res (2008) 195(1):198–213. doi:10.1016/j.bbr.2008.03.020
81. McCorvy JD, Roth BL. Structure and function of serotonin G protein-coupled receptors. Pharmacol Ther (2015) 150:129–42. doi:10.1016/j.pharmthera.2015.01.009
82. Ramboz S, Oosting R, Amara DA, Kung HF, Blier P, Mendelsohn M, et al. Serotonin receptor 1A knockout: an animal model of anxiety-related disorder. Proc Natl Acad Sci U S A (1998) 95(24):14476–81. doi:10.1073/pnas.95.24.14476
83. Toth M. 5-HT1A receptor knockout mouse as a genetic model of anxiety. Eur J Pharmacol (2003) 463(1–3):177–84. doi:10.1016/S0014-2999(03)01280-9
84. Popova NK, Naumenko VS. 5-HT1A receptor as a key player in the brain 5-HT system. Rev Neurosci (2013) 24(2):191–204. doi:10.1515/revneuro-2012-0082
85. Launay JM, Schneider B, Loric S, Da Prada M, Kellermann O. Serotonin transport and serotonin transporter-mediated antidepressant recognition are controlled by 5-HT2B receptor signaling in serotonergic neuronal cells. FASEB J (2006) 20(11):1843–54. doi:10.1096/fj.06-5724com
86. Doly S, Valjent E, Setola V, Callebert J, Herve D, Launay JM, et al. Serotonin 5-HT2B receptors are required for 3,4-methylenedioxymethamphetamine-induced hyperlocomotion and 5-HT release in vivo and in vitro. J Neurosci (2008) 28(11):2933–40. doi:10.1523/JNEUROSCI.5723-07.2008
87. Singer HS, Minzer K. Neurobiology of Tourette’s syndrome: concepts of neuroanatomic localization and neurochemical abnormalities. Brain Dev (2003) 25(Suppl 1):S70–84. doi:10.1016/S0387-7604(03)90012-X
88. Adamczyk A, Gause CD, Sattler R, Vidensky S, Rothstein JD, Singer H, et al. Genetic and functional studies of a missense variant in a glutamate transporter, SLC1A3, in Tourette syndrome. Psychiatr Genet (2011) 21(2):90–7. doi:10.1097/YPG.0b013e328341a307
89. Manev H, Favaron M, Guidotti A, Costa E. Delayed increase of Ca2+ influx elicited by glutamate: role in neuronal death. Mol Pharmacol (1989) 36(1):106–12.
90. O’Shea RD. Roles and regulation of glutamate transporters in the central nervous system. Clin Exp Pharmacol Physiol (2002) 29(11):1018–23. doi:10.1046/j.1440-1681.2002.03770.x
91. Furuta A, Rothstein JD, Martin LJ. Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci (1997) 17(21):8363–75.
92. Karlsson RM, Tanaka K, Saksida LM, Bussey TJ, Heilig M, Holmes A. Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology (2009) 34(6):1578–89. doi:10.1038/npp.2008.215
93. Zebardast N, Crowley MJ, Bloch MH, Mayes LC, Wyk BV, Leckman JF, et al. Brain mechanisms for prepulse inhibition in adults with Tourette syndrome: initial findings. Psychiatry Res (2013) 214(1):33–41. doi:10.1016/j.pscychresns.2013.05.009
94. Zhang C, Milunsky JM, Newton S, Ko J, Zhao G, Maher TA, et al. A neuroligin-4 missense mutation associated with autism impairs neuroligin-4 folding and endoplasmic reticulum export. J Neurosci (2009) 29(35):10843–54. doi:10.1523/JNEUROSCI.1248-09.2009
95. Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet (2008) 82(2):477–88. doi:10.1016/j.ajhg.2007.12.009
96. Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet (2004) 74(3):552–7. doi:10.1086/382137
97. Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet (2003) 34(1):27–9. doi:10.1038/ng1136
98. Kim JE, O’Sullivan ML, Sanchez CA, Hwang M, Israel MA, Brennand K, et al. Investigating synapse formation and function using human pluripotent stem cell-derived neurons. Proc Natl Acad Sci U S A (2011) 108(7):3005–10. doi:10.1073/pnas.1007753108
99. Lawson-Yuen A, Saldivar JS, Sommer S, Picker J. Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur J Hum Genet (2008) 16(5):614–8. doi:10.1038/sj.ejhg.5202006
100. Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature (2008) 455(7215):903–11. doi:10.1038/nature07456
101. Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, et al. Neuroligins determine synapse maturation and function. Neuron (2006) 51(6):741–54. doi:10.1016/j.neuron.2006.09.003
102. Chih B, Engelman H, Scheiffele P. Control of excitatory and inhibitory synapse formation by neuroligins. Science (2005) 307(5713):1324–8. doi:10.1126/science.1107470
103. Jamain S, Radyushkin K, Hammerschmidt K, Granon S, Boretius S, Varoqueaux F, et al. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc Natl Acad Sci U S A (2008) 105(5):1710–5. doi:10.1073/pnas.0711555105
104. Delattre V, La Mendola D, Meystre J, Markram H, Markram K. Nlgn4 knockout induces network hypo-excitability in juvenile mouse somatosensory cortex in vitro. Sci Rep (2013) 3:2897. doi:10.1038/srep02897
105. El-Kordi A, Winkler D, Hammerschmidt K, Kastner A, Krueger D, Ronnenberg A, et al. Development of an autism severity score for mice using Nlgn4 null mutants as a construct-valid model of heritable monogenic autism. Behav Brain Res (2013) 251:41–9. doi:10.1016/j.bbr.2012.11.016
106. Moya PR, Dodman NH, Timpano KR, Rubenstein LM, Rana Z, Fried RL, et al. Rare missense neuronal cadherin gene (CDH2) variants in specific obsessive-compulsive disorder and Tourette disorder phenotypes. Eur J Hum Genet (2013) 21(8):850–4. doi:10.1038/ejhg.2012.245
107. Hatta K, Okada TS, Takeichi M. A monoclonal antibody disrupting calcium-dependent cell-cell adhesion of brain tissues: possible role of its target antigen in animal pattern formation. Proc Natl Acad Sci U S A (1985) 82(9):2789–93. doi:10.1073/pnas.82.9.2789
108. Reichardt LFN-. Cadherin and integrins: two receptor systems that mediate neuronal process outgrowth on astrocyte surfaces. Neuron (2008) 60(3):398–9. doi:10.1016/j.neuron.2008.10.030
109. Tomaselli KJ, Neugebauer KM, Bixby JL, Lilien J, Reichardt LF. N-cadherin and integrins: two receptor systems that mediate neuronal process outgrowth on astrocyte surfaces. Neuron (1988) 1(1):33–43. doi:10.1016/0896-6273(88)90207-3
110. Arikkath J, Reichardt LF. Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci (2008) 31(9):487–94. doi:10.1016/j.tins.2008.07.001
111. Bamji SX, Shimazu K, Kimes N, Huelsken J, Birchmeier W, Lu B, et al. Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron (2003) 40(4):719–31. doi:10.1016/S0896-6273(03)00718-9
112. Okuda T, Yu LM, Cingolani LA, Kemler R, Goda Y. beta-Catenin regulates excitatory postsynaptic strength at hippocampal synapses. Proc Natl Acad Sci U S A (2007) 104(33):13479–84. doi:10.1073/pnas.0702334104
113. Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, et al. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell (2003) 114(5):635–45. doi:10.1016/j.cell.2003.08.008
114. Radice GL, Rayburn H, Matsunami H, Knudsen KA, Takeichi M, Hynes RO. Developmental defects in mouse embryos lacking N-cadherin. Dev Biol (1997) 181(1):64–78. doi:10.1006/dbio.1996.8443
115. Kadowaki M, Nakamura S, Machon O, Krauss S, Radice GL, Takeichi M. N-cadherin mediates cortical organization in the mouse brain. Dev Biol (2007) 304(1):22–33. doi:10.1016/j.ydbio.2006.12.014
116. Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet (2008) 82(1):160–4. doi:10.1016/j.ajhg.2007.09.015
117. Alarcon M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet (2008) 82(1):150–9. doi:10.1016/j.ajhg.2007.09.005
118. Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med (2006) 354(13):1370–7. doi:10.1056/NEJMoa052773
119. Penagarikano O, Geschwind DH. What does CNTNAP2 reveal about autism spectrum disorder? Trends Mol Med (2012) 18(3):156–63. doi:10.1016/j.molmed.2012.01.003
120. Bakkaloglu B, O’Roak BJ, Louvi A, Gupta AR, Abelson JF, Morgan TM, et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet (2008) 82(1):165–73. doi:10.1016/j.ajhg.2007.09.017
121. Verkerk AJ, Mathews CA, Joosse M, Eussen BH, Heutink P, Oostra BA. CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics (2003) 82(1):1–9. doi:10.1016/S0888-7543(03)00097-1
122. Belloso JM, Bache I, Guitart M, Caballin MR, Halgren C, Kirchhoff M, et al. Disruption of the CNTNAP2 gene in a t(7;15) translocation family without symptoms of Gilles de la Tourette syndrome. Eur J Hum Genet (2007) 15(6):711–3. doi:10.1038/sj.ejhg.5201824
123. Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, et al. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron (1999) 24(4):1037–47. doi:10.1016/S0896-6273(00)81049-1
124. Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St Martin M, et al. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron (2001) 30(2):369–83. doi:10.1016/S0896-6273(01)00294-X
125. Mink JW. Basal ganglia dysfunction in Tourette’s syndrome: a new hypothesis. Pediatr Neurol (2001) 25(3):190–8. doi:10.1016/S0887-8994(01)00262-4
126. Mink JW. Neurobiology of basal ganglia circuits in Tourette syndrome: faulty inhibition of unwanted motor patterns? Adv Neurol (2001) 85:113–22.
127. Penagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell (2011) 147(1):235–46. doi:10.1016/j.cell.2011.08.040
128. Prontera P, Napolioni V, Ottaviani V, Rogaia D, Fusco C, Augello B, et al. DPP6 gene disruption in a family with Gilles de la Tourette syndrome. Neurogenetics (2014) 15(4):237–42. doi:10.1007/s10048-014-0418-9
129. Egger G, Roetzer KM, Noor A, Lionel AC, Mahmood H, Schwarzbraun T, et al. Identification of risk genes for autism spectrum disorder through copy number variation analysis in Austrian families. Neurogenetics (2014) 15(2):117–27. doi:10.1007/s10048-014-0394-0
130. Tanaka S, Syu A, Ishiguro H, Inada T, Horiuchi Y, Ishikawa M, et al. DPP6 as a candidate gene for neuroleptic-induced tardive dyskinesia. Pharmacogenomics J (2013) 13(1):27–34. doi:10.1038/tpj.2011.36
131. Kin Y, Misumi Y, Ikehara Y. Biosynthesis and characterization of the brain-specific membrane protein DPPX, a dipeptidyl peptidase IV-related protein. J Biochem (2001) 129(2):289–95. doi:10.1093/oxfordjournals.jbchem.a002856
132. Nadal MS, Amarillo Y, Vega-Saenz de Miera E, Rudy B. Differential characterization of three alternative spliced isoforms of DPPX. Brain Res (2006) 1094(1):1–12. doi:10.1016/j.brainres.2006.03.106
133. Strop P, Bankovich AJ, Hansen KC, Garcia KC, Brunger AT. Structure of a human A-type potassium channel interacting protein DPPX, a member of the dipeptidyl aminopeptidase family. J Mol Biol (2004) 343(4):1055–65. doi:10.1016/j.jmb.2004.09.003
134. Clark BD, Kwon E, Maffie J, Jeong HY, Nadal M, Strop P, et al. DPP6 localization in brain supports function as a Kv4 channel associated protein. Front Mol Neurosci (2008) 1:8. doi:10.3389/neuro.02.008.2008
135. Schoppa NE, Westbrook GL. Regulation of synaptic timing in the olfactory bulb by an A-type potassium current. Nat Neurosci (1999) 2(12):1106–13. doi:10.1038/16033
136. Johnston D, Hoffman DA, Magee JC, Poolos NP, Watanabe S, Colbert CM, et al. Dendritic potassium channels in hippocampal pyramidal neurons. J Physiol (2000) 525(Pt 1):75–81. doi:10.1111/j.1469-7793.2000.00075.x
137. Liss B, Franz O, Sewing S, Bruns R, Neuhoff H, Roeper J. Tuning pacemaker frequency of individual dopaminergic neurons by Kv4.3L and KChip3.1 transcription. EMBO J (2001) 20(20):5715–24. doi:10.1093/emboj/20.20.5715
138. Nadal MS, Ozaita A, Amarillo Y, Vega-Saenz de Miera E, Ma Y, Mo W, et al. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron (2003) 37(3):449–61. doi:10.1016/S0896-6273(02)01185-6
139. Lin L, Sun W, Throesch B, Kung F, Decoster JT, Berner CJ, et al. DPP6 regulation of dendritic morphogenesis impacts hippocampal synaptic development. Nat Commun (2013) 4:2270. doi:10.1038/ncomms3270
140. Liao C, Fu F, Li R, Yang WQ, Liao HY, Yan JR, et al. Loss-of-function variation in the DPP6 gene is associated with autosomal dominant microcephaly and mental retardation. Eur J Med Genet (2013) 56(9):484–9. doi:10.1016/j.ejmg.2013.06.008
141. Boghosian-Sell L, Comings DE, Overhauser J. Tourette syndrome in a pedigree with a 7;18 translocation: identification of a YAC spanning the translocation breakpoint at 18q22.3. Am J Hum Genet (1996) 59(5):999–1005.
142. Petek E, Windpassinger C, Vincent JB, Cheung J, Boright AP, Scherer SW, et al. Disruption of a novel gene (IMMP2L) by a breakpoint in 7q31 associated with Tourette syndrome. Am J Hum Genet (2001) 68(4):848–58. doi:10.1086/319523
143. Patel C, Cooper-Charles L, McMullan DJ, Walker JM, Davison V, Morton J. Translocation breakpoint at 7q31 associated with tics: further evidence for IMMP2L as a candidate gene for Tourette syndrome. Eur J Hum Genet (2011) 19(6):634–9. doi:10.1038/ejhg.2010.238
144. Bertelsen B, Melchior L, Jensen LR, Groth C, Glenthoj B, Rizzo R, et al. Intragenic deletions affecting two alternative transcripts of the IMMP2L gene in patients with Tourette syndrome. Eur J Hum Genet (2014) 22(11):1283–9. doi:10.1038/ejhg.2014.24
145. Gakh O, Cavadini P, Isaya G. Mitochondrial processing peptidases. Biochim Biophys Acta (2002) 1592(1):63–77. doi:10.1016/S0167-4889(02)00265-3
146. Burri L, Strahm Y, Hawkins CJ, Gentle IE, Puryer MA, Verhagen A, et al. Mature DIABLO/Smac is produced by the IMP protease complex on the mitochondrial inner membrane. Mol Biol Cell (2005) 16(6):2926–33. doi:10.1091/mbc.E04-12-1086
147. Nunnari J, Fox TD, Walter P. A mitochondrial protease with two catalytic subunits of nonoverlapping specificities. Science (1993) 262(5142):1997–2004. doi:10.1126/science.8266095
148. Lu B, Poirier C, Gaspar T, Gratzke C, Harrison W, Busija D, et al. A mutation in the inner mitochondrial membrane peptidase 2-like gene (Immp2l) affects mitochondrial function and impairs fertility in mice. Biol Reprod (2008) 78(4):601–10. doi:10.1095/biolreprod.107.065987
149. Ma Y, Mehta SL, Lu B, Li PA. Deficiency in the inner mitochondrial membrane peptidase 2-like (Immp21) gene increases ischemic brain damage and impairs mitochondrial function. Neurobiol Dis (2011) 44(3):270–6. doi:10.1016/j.nbd.2011.06.019
150. Sundaram SK, Huq AM, Sun Z, Yu W, Bennett L, Wilson BJ, et al. Exome sequencing of a pedigree with Tourette syndrome or chronic tic disorder. Ann Neurol (2011) 69(5):901–4. doi:10.1002/ana.22398
151. Guo Y, Deng X, Zhang J, Su L, Xu H, Luo Z, et al. Analysis of the MRPL3, DNAJC13 and OFCC1 variants in Chinese Han patients with TS-CTD. Neurosci Lett (2012) 517(1):18–20. doi:10.1016/j.neulet.2012.03.097
152. Gruschke S, Grone K, Heublein M, Holz S, Israel L, Imhof A, et al. Proteins at the polypeptide tunnel exit of the yeast mitochondrial ribosome. J Biol Chem (2010) 285(25):19022–8. doi:10.1074/jbc.M110.113837
153. Galmiche L, Serre V, Beinat M, Assouline Z, Lebre AS, Chretien D, et al. Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum Mutat (2011) 32(11):1225–31. doi:10.1002/humu.21562
154. Vilarino-Guell C, Rajput A, Milnerwood AJ, Shah B, Szu-Tu C, Trinh J, et al. DNAJC13 mutations in Parkinson disease. Hum Mol Genet (2014) 23(7):1794–801. doi:10.1093/hmg/ddt570
155. Girard M, Poupon V, Blondeau F, McPherson PS. The DnaJ-domain protein RME-8 functions in endosomal trafficking. J Biol Chem (2005) 280(48):40135–43. doi:10.1074/jbc.M505036200
156. Fujibayashi A, Taguchi T, Misaki R, Ohtani M, Dohmae N, Takio K, et al. Human RME-8 is involved in membrane trafficking through early endosomes. Cell Struct Funct (2008) 33(1):35–50. doi:10.1247/csf.07045
157. Davies SJ, Wise C, Venkatesh B, Mirza G, Jefferson A, Volpi EV, et al. Mapping of three translocation breakpoints associated with orofacial clefting within 6p24 and identification of new transcripts within the region. Cytogenet Genome Res (2004) 105(1):47–53. doi:10.1159/000078008
158. Ohnishi T, Yamada K, Watanabe A, Ohba H, Sakaguchi T, Honma Y, et al. Ablation of Mrds1/Ofcc1 induces hyper-gamma-glutamyl transpeptidasemia without abnormal head development and schizophrenia-relevant behaviors in mice. PLoS One (2011) 6(12):e29499. doi:10.1371/journal.pone.0029499
159. Mattheisen M, Samuels JF, Wang Y, Greenberg BD, Fyer AJ, McCracken JT, et al. Genome-wide association study in obsessive-compulsive disorder: results from the OCGAS. Mol Psychiatry (2015) 20:337–44. doi:10.1038/mp.2014.43
160. Cukier HN, Dueker ND, Slifer SH, Lee JM, Whitehead PL, Lalanne E, et al. Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol Autism (2014) 5(1):1. doi:10.1186/2040-2392-5-1
161. Weimann M, Grossmann A, Woodsmith J, Ozkan Z, Birth P, Meierhofer D, et al. A Y2H-seq approach defines the human protein methyltransferase interactome. Nat Methods (2013) 10(4):339–42. doi:10.1038/nmeth.2397
162. Thompson MD, Comings DE, Abu-Ghazalah R, Jereseh Y, Lin L, Wade J, et al. Variants of the orexin2/hcrt2 receptor gene identified in patients with excessive daytime sleepiness and patients with Tourette’s syndrome comorbidity. Am J Med Genet B Neuropsychiatr Genet (2004) 129B(1):69–75. doi:10.1002/ajmg.b.30047
163. Willie JT, Chemelli RM, Sinton CM, Yanagisawa M. To eat or to sleep? Orexin in the regulation of feeding and wakefulness. Annu Rev Neurosci (2001) 24:429–58. doi:10.1146/annurev.neuro.24.1.429
164. Tsujino N, Sakurai T. Role of orexin in modulating arousal, feeding, and motivation. Front Behav Neurosci (2013) 7:28. doi:10.3389/fnbeh.2013.00028
165. Geschwind DH. Autism: many genes, common pathways? Cell (2008) 135(3):391–5. doi:10.1016/j.cell.2008.10.016
166. Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A (1992) 89(12):5547–51. doi:10.1073/pnas.89.12.5547
167. Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, et al. Human genomics. The human transcriptome across tissues and individuals. Science (2015) 348(6235):660–5. doi:10.1126/science.aaa0355
168. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (2006) 126(4):663–76. doi:10.1016/j.cell.2006.07.024
169. Young-Pearse TL, Morrow EM. Modeling developmental neuropsychiatric disorders with iPSC technology: challenges and opportunities. Curr Opin Neurobiol (2015) 36:66–73. doi:10.1016/j.conb.2015.10.006
170. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet (1999) 23(2):185–8. doi:10.1038/13810
171. Cheung AY, Horvath LM, Grafodatskaya D, Pasceri P, Weksberg R, Hotta A, et al. Isolation of MECP2-null Rett syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum Mol Genet (2011) 20(11):2103–15. doi:10.1093/hmg/ddr093
172. Griesi-Oliveira K, Acab A, Gupta AR, Sunaga DY, Chailangkarn T, Nicol X, et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry (2015) 20(11):1350–65. doi:10.1038/mp.2014.141
173. Wen Z, Nguyen HN, Guo Z, Lalli MA, Wang X, Su Y, et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature (2014) 515(7527):414–8. doi:10.1038/nature13716
174. Egawa N, Kitaoka S, Tsukita K, Naitoh M, Takahashi K, Yamamoto T, et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci Transl Med (2012) 4(145):145ra04. doi:10.1126/scitranslmed.3004052
175. Thomson PA, Malavasi EL, Grunewald E, Soares DC, Borkowska M, Millar JK. DISC1 genetics, biology and psychiatric illness. Front Biol (2013) 8(1):1–31. doi:10.1007/s11515-012-1254-7
177. Jiang H, Ren Y, Yuen EY, Zhong P, Ghaedi M, Hu Z, et al. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat Commun (2012) 3:668. doi:10.1038/ncomms1669
178. Beevers JE, Caffrey TM, Wade-Martins R. Induced pluripotent stem cell (iPSC)-derived dopaminergic models of Parkinson’s disease. Biochem Soc Trans (2013) 41(6):1503–8. doi:10.1042/BST20130194
179. Su YC, Qi X. Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum Mol Genet (2013) 22(22):4545–61. doi:10.1093/hmg/ddt301
180. Reinhardt P, Schmid B, Burbulla LF, Schondorf DC, Wagner L, Glatza M, et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell (2013) 12(3):354–67. doi:10.1016/j.stem.2013.01.008
181. Chen H, Qian K, Du Z, Cao J, Petersen A, Liu H, et al. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell (2014) 14(6):796–809. doi:10.1016/j.stem.2014.02.004
182. Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell (2011) 8(3):267–80. doi:10.1016/j.stem.2011.01.013
183. Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med (2011) 17(12):1657–62. doi:10.1038/nm.2576
184. White AK, VanInsberghe M, Petriv OI, Hamidi M, Sikorski D, Marra MA, et al. High-throughput microfluidic single-cell RT-qPCR. Proc Natl Acad Sci U S A (2011) 108(34):13999–4004. doi:10.1073/pnas.1019446108
185. White AK, Heyries KA, Doolin C, Vaninsberghe M, Hansen CL. High-throughput microfluidic single-cell digital polymerase chain reaction. Anal Chem (2013) 85(15):7182–90. doi:10.1021/ac400896j
186. Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc (2014) 9(10):2329–40. doi:10.1038/nprot.2014.158
187. Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature (2013) 501(7467):373–9. doi:10.1038/nature12517
188. Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell (2015) 162(2):375–90. doi:10.1016/j.cell.2015.06.034
189. Bobis-Wozowicz S, Osiak A, Rahman SH, Cathomen T. Targeted genome editing in pluripotent stem cells using zinc-finger nucleases. Methods (2011) 53(4):339–46. doi:10.1016/j.ymeth.2010.12.019
190. Soldner F, Laganiere J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell (2011) 146(2):318–31. doi:10.1016/j.cell.2011.06.019
191. Smith C, Abalde-Atristain L, He C, Brodsky BR, Braunstein EM, Chaudhari P, et al. Efficient and allele-specific genome editing of disease loci in human iPSCs. Mol Ther (2015) 23(3):570–7. doi:10.1038/mt.2014.226
192. Byrne SM, Mali P, Church GM. Genome editing in human stem cells. Methods Enzymol (2014) 546:119–38. doi:10.1016/B978-0-12-801185-0.00006-4
193. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol (2013) 31(9):822–6. doi:10.1038/nbt.2623
194. Costa V, Aprile M, Esposito R, Ciccodicola A. RNA-Seq and human complex diseases: recent accomplishments and future perspectives. Eur J Hum Genet (2013) 21(2):134–42. doi:10.1038/ejhg.2012.129
195. Marchetto MC, Brennand KJ, Boyer LF, Gage FH. Induced pluripotent stem cells (iPSCs) and neurological disease modeling: progress and promises. Hum Mol Genet (2011) 20(R2):R109–15. doi:10.1093/hmg/ddr336
196. Soneson C, Delorenzi M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics (2013) 14:91. doi:10.1186/1471-2105-14-91
197. Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol (2013) 31(1):46–53. doi:10.1038/nbt.2450
198. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol (2010) 28(5):511–5. doi:10.1038/nbt.1621
199. Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res (2014) 42(Database issue):D199–205. doi:10.1093/nar/gkt1076
200. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res (2000) 28(1):27–30. doi:10.1093/nar/28.1.27
201. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc (2009) 4(1):44–57. doi:10.1038/nprot.2008.211
202. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res (2009) 37(1):1–13. doi:10.1093/nar/gkn923
203. Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB – a database for integrating human functional interaction networks. Nucleic Acids Res (2009) 37(Database issue):D623–8. doi:10.1093/nar/gkn698
204. Parikshak NN, Gandal MJ, Geschwind DH. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genet (2015) 16(8):441–58. doi:10.1038/nrg3934
205. Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature (2012) 489(7416):391–9. doi:10.1038/nature11405
206. Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, et al. Spatio-temporal transcriptome of the human brain. Nature (2011) 478(7370):483–9. doi:10.1038/nature10523
207. Keen JC, Moore HM. The genotype-tissue expression (GTEx) project: linking clinical data with molecular analysis to advance personalized medicine. J Pers Med (2015) 5(1):22–9. doi:10.3390/jpm5010022
208. Cotney J, Muhle RA, Sanders SJ, Liu L, Willsey AJ, Niu W, et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat Commun (2015) 6:6404. doi:10.1038/ncomms7404
209. Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci U S A (2015) 112(23):7285–90. doi:10.1073/pnas.1507125112
210. Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res (2006) 34(Database issue):D535–9. doi:10.1093/nar/gkj109
211. Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron (2011) 70(5):898–907. doi:10.1016/j.neuron.2011.05.021
212. Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell (2013) 155(5):997–1007. doi:10.1016/j.cell.2013.10.020
213. Chang J, Gilman SR, Chiang AH, Sanders SJ, Vitkup D. Genotype to phenotype relationships in autism spectrum disorders. Nat Neurosci (2015) 18(2):191–8. doi:10.1038/nn.3907
214. Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell (2014) 159(2):440–55. doi:10.1016/j.cell.2014.09.014
215. Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell (2013) 154(6):1370–9. doi:10.1016/j.cell.2013.08.022
216. Godar SC, Mosher LJ, Di Giovanni G, Bortolato M. Animal models of tic disorders: a translational perspective. J Neurosci Methods (2014) 238:54–69. doi:10.1016/j.jneumeth.2014.09.008
217. Comings DE, Wu S, Chiu C, Ring RH, Gade R, Ahn C, et al. Polygenic inheritance of Tourette syndrome, stuttering, attention deficit hyperactivity, conduct, and oppositional defiant disorder: the additive and subtractive effect of the three dopaminergic genes – DRD2, D beta H, and DAT1. Am J Med Genet (1996) 67(3):264–88. doi:10.1002/(SICI)1096-8628(19960531)67:3<264::AID-AJMG4>3.0.CO;2-N
218. Paschou P. The genetic basis of Gilles de la Tourette syndrome. Neurosci Biobehav Rev (2013) 37(6):1026–39. doi:10.1016/j.neubiorev.2013.01.016
219. Clarke RA, Lee S, Eapen V. Pathogenetic model for Tourette syndrome delineates overlap with related neurodevelopmental disorders including autism. Transl Psychiatry (2012) 2:e158. doi:10.1038/tp.2012.75
220. Yee CM, Javitt DC, Miller GA. Replacing DSM categorical analyses with dimensional analyses in psychiatry research: the research domain criteria initiative. JAMA Psychiatry (2015) 72(12):1159–60. doi:10.1001/jamapsychiatry.2015.1900
221. Kraemer HC. Research domain criteria (RDoC) and the DSM-two methodological approaches to mental health diagnosis. JAMA Psychiatry (2015) 72(12):1163–4. doi:10.1001/jamapsychiatry.2015.2134
222. Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry (2010) 167(7):748–51. doi:10.1176/appi.ajp.2010.09091379
223. Casey BJ, Oliveri ME, Insel T. A neurodevelopmental perspective on the research domain criteria (RDoC) framework. Biol Psychiatry (2014) 76(5):350–3. doi:10.1016/j.biopsych.2014.01.006
224. Cuthbert BN. Research domain criteria: toward future psychiatric nosologies. Dialogues Clin Neurosci (2015) 17(1):89–97.
225. Heiman GA, King RA, Tischfield JA. New Jersey center for Tourette syndrome sharing repository: methods and sample description. BMC Med Genomics (2008) 1:58. doi:10.1186/1755-8794-1-58
226. Dietrich A, Fernandez TV, King RA, State MW, Tischfield JA, Hoekstra PJ, et al. The Tourette International Collaborative Genetics (TIC Genetics) study, finding the genes causing Tourette syndrome: objectives and methods. Eur Child Adolesc Psychiatry (2015) 24(2):141–51. doi:10.1007/s00787-014-0543-x
Keywords: Tourette’s disorder, functional studies, genes, neurobiology, induced pluripotent stem cells
Citation: Sun N, Tischfield JA, King RA and Heiman GA (2016) Functional Evaluations of Genes Disrupted in Patients with Tourette’s Disorder. Front. Psychiatry 7:11. doi: 10.3389/fpsyt.2016.00011
Received: 14 December 2015; Accepted: 18 January 2016;
Published: 09 February 2016
Edited by:
Peristera Paschou, Democritus University of Thrace, GreeceReviewed by:
Fotis Tsetsos, Democritus University of Thrace, GreeceMarianthi Georgitsi, Aristotle University of Thessaloniki, Greece
Copyright: © 2016 Sun, Tischfield, King and Heiman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gary A. Heiman, aGVpbWFuQGRscy5ydXRnZXJzLmVkdQ==