 Lucy M. Collins
Lucy M. Collins Caroline H. Williams-Gray
Caroline H. Williams-Gray- John Van Geest Centre for Brain Repair, University of Cambridge, Cambridge, UK
Cognitive dysfunction is a common feature of Parkinson’s disease (PD) with mild cognitive impairment affecting around a quarter of patients in the early stages of their disease, and approximately half developing dementia by 10 years from diagnosis. However, the pattern of cognitive impairments and their speed of evolution vary markedly between individuals. While some of this variability may relate to extrinsic factors and comorbidities, inherited genetic heterogeneity is also known to play an important role. A number of common genetic variants have been identified, which contribute to cognitive function in PD, including variants in catechol-O-methyltransferase, microtubule-associated protein tau, and apolipoprotein E. Furthermore, rarer mutations in glucocerebrosidase and α-synuclein and are strongly associated with dementia risk in PD. This review explores the functional impact of these variants on cognition in PD and discusses how such genotype–phenotype associations provide a window into the mechanistic basis of cognitive heterogeneity in this disorder. This has consequent implications for the development of much more targeted therapeutic strategies for cognitive symptoms in PD.
Introduction
Parkinson’s disease (PD) is typically characterized as a movement disorder with the cardinal motor features of bradykinesia, rigidity, and rest tremor reflecting dysfunction within dopaminergic nigrostriatal circuitry. However, non-motor symptoms constitute a significant part of the symptom burden in PD, reflecting more widespread pathology throughout both the brain and the peripheral and enteric nervous systems. Among the most relevant of these non-motor symptoms in terms of quality of life and planning care requirements is cognitive dysfunction. This evolves through the course of the disease and dementia ensues in nearly 50% of PD patients by 10 years into their illness (1). The factors that underlie this evolution are not fully understood, but pathologically the dementing process seems to involve Lewy body deposition and Alzheimer’s type changes in cortical brain regions (2) as well as widespread cholinergic deficits (3).
Cognitive deficits in PD are heterogeneous in terms of the domains affected including executive dysfunction, memory, and visuospatial impairments. There is also considerable variation in the speed at which cognitive deficits develop, with some patients progressing rapidly to dementia while others have a slower course (1). To some extent, this course is predictable early on in the disease: in an incident population-representative PD cohort followed up for 10 years from diagnosis, patients who were older at diagnosis with more motor impairment and early dysfunction on neuropsychological tests with a posterior cortical basis (semantic fluency and pentagon copying) progressed more rapidly to dementia. In contrast, more frontostriatally based executive deficits were not associated with earlier dementia and, in fact, even improved in some patients as the disease progressed (1) (Figure 1). Similarly, verbal fluency and silhouette perception impairments preceded onset of dementia in an independent longitudinal study (4). Further support for the concept that posterior cortical dysfunction predicts dementia comes from neuroimaging studies with longitudinal follow-up demonstrating that hypometabolism (5) and atrophy (6) in posterior cortical regions precede cognitive decline.
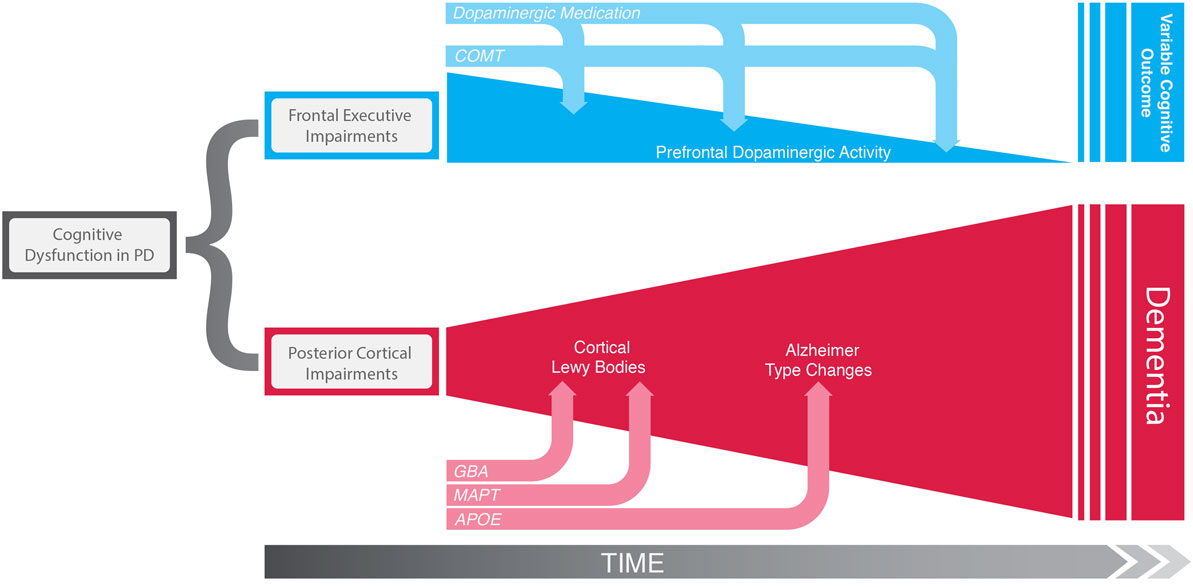
Figure 1. Schematic representation of the two distinct cognitive syndromes of Parkinson’s disease. “Frontal executive” impairments in early disease appear to be a consequence of a hyperdopaminergic state in the prefrontal cortex, which is in turn modulated by COMT genotype and dopaminergic medication. These deficits can get better or worse over time but are not associated global cognitive decline and dementia risk. In contrast, early deficits on more posterior cortically based cognitive tasks do not have a dopaminergic basis, but reflect the early stages of a dementing process due to Lewy body deposition and Alzheimer’s type changes in posterior cortical areas. This irreversible pathological process is influenced by early on by GBA mutations, MAPT H1/H2 haplotypes, and at a later disease stage by variation in APOE.
Thus, mild cognitive impairment in PD (PD-MCI) per se is not necessarily a prodrome of dementia: some individuals with PD-MCI remain cognitively stable over time and some revert back to normal cognitive function (7, 8). While cognitive phenotype is helpful in predicting prognosis in these “PD-MCI” cases as discussed above, knowledge of genetic variation may also be useful in this regard. The underlying genetic basis of PD is complex: a small proportion of PD cases are accounted for by monogenic forms of PD inherited in a Mendelian fashion (e.g., related to mutations in α-synuclein (SNCA), leucine-rich repeat kinase 2 (LRRK2), parkin, Parkinson’s disease-associated kinase-1 (PINK-1), DJ-1, ATP13A2), but numerous susceptibility genes and loci have also been identified (at least 24) (9), which contribute to the risk of idiopathic PD. In this article, we will review how this complex genetic heterogeneity maps onto phenotypic cognitive heterogeneity of PD. We will first discuss the cognitive characteristics of Mendelian forms of PD. We will then focus on two PD susceptibility genes, namely, glucocerebrosidase (GBA) and microtubule-associated protein tau (MAPT), both of which influence progression to dementia in PD. Finally, we will discuss candidate genes implicated in modulating cognition in PD but not associated with increased risk of development of PD per se. These include catechol-O-methyltransferase (COMT) and apolipoprotein E (APOE). A better understanding of these genetic influences on cognition in PD will lead to improved patient stratification and could guide novel therapeutic strategies for cognitive dysfunction in PD, better targeted at the most relevant underlying pathogenic pathways.
Insights from Monogenic Parkinson’s Disease
Although most cases of PD are idiopathic, 3–5% of cases are caused by a single gene variant (10). Genes associated with autosomal dominantly inherited PD include SNCA (11–13) and LRRK2 (14, 15), and more recently eukaryotic translation initiation factor 4 gamma-1 (EIF4G1) (16) and vacuolar protein sorting-associated protein 35 (VPS35) (17) have been identified. Autosomal recessive forms of PD are associated with mutations in Parkin (Parkin E3 ubiquitin ligase) (18), PINK-1 (19), DJ-1 (PARK7, Parkinson protein 7) (20). These different genetic forms of PD vary in their clinical and cognitive phenotypes (see Table 1).
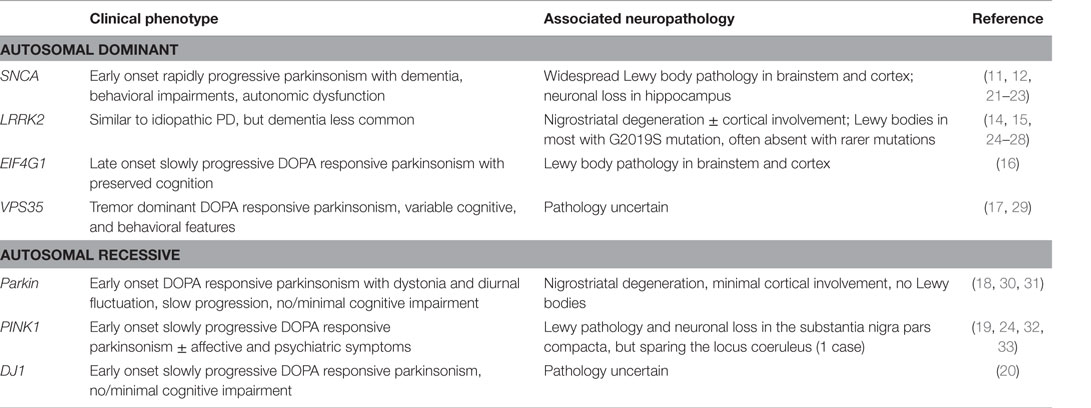
Table 1. Monogenic PD genes.
Some of these genotype–phenotype associations are very well established and provide useful insights into pathogenesis. For example, monogenic forms of PD caused by mutations within, or a change in dose of, the SNCA gene implicate α-synuclein in the development of dementia in PD. SNCA mutations, including A53T, A30P, and E46K, are often associated with an early onset severe clinical phenotype with cognitive decline and behavioral change as well as autonomic dysfunction (21–23). Similarly, individuals with SNCA triplications have early onset rapidly progressive parkinsonism and dementia (11). The pathology in these cases is characterized by widespread Lewy body not only in the brainstem but also in the cortex, as well as neuronal loss in the hippocampal CA2/3 region (11, 24). However, SNCA duplication leads to PD with a more benign phenotype, similar to idiopathic PD (34). In contrast, Parkin-associated PD is a more “pure” motor disorder with a very low incidence of dementia. A meta-analysis of available phenotypic data in Parkin mutation carriers indicated that only 7% of 58 cases carrying 1 mutant allele developed dementia over 12 years of follow-up, with incidence rates being even lower in those carrying 2 mutant alleles at 1% of 232 cases over 19 years (30). This is consistent with neuropathological findings in Parkin-associated PD, which indicate that the disease is largely restricted to the nigrostriatal tract with the absence of Lewy bodies (31). LRRK2 mutations represent the commonest cause of autosomal dominantly inherited PD (25), and hence the clinical phenotype has been well described, particularly in association with the most frequently occurring G2019S mutation. The clinical phenotype is similar to idiopathic PD (26), although the prevalence of dementia is reported to be lower (27, 28). Most LRRK2 G2019S PD cases have the typical pattern of brainstem pathology with Lewy bodies with variable cortical involvement, but Lewy bodies are not a universal finding, and are present in less than half of those with LRRK2 mutations other than G2019S (24), in keeping with the more benign phenotype when compared to SNCA-associated PD. Other observed but less well-replicated genotype–phenotype associations include a high prevalence of psychiatric symptoms in PINK-1 families (32) and learning difficulties and cognitive impairment in a Swiss family with the D620N mutation in VPS35 causing early onset tremor dominant PD (29).
Although these observations are of interest, in general there is a paucity of longitudinal data on phenotypic characteristics in Mendelian forms of PD. A meta-analysis in 2010 identified 119 studies reporting information on non-motor symptoms in 990 cases with monogenic forms of PD, but the available phenotypic data were limited, with dementia frequency being reported in only 54% of studies, and most studies did not clearly distinguish between point and lifetime prevalence of non-motor symptoms, thus making interpretation of their data difficult. With these important caveats, the meta-analysis confirmed the highest prevalence of dementia among SNCA mutation carriers (approximately 25%) and the lowest among Parkin mutation carriers (approximately 3%) (35).
Hence, although these monogenic forms provide some insight into the pathogenesis of PD dementia, a more systematic evaluation of their phenotypic characteristics is needed to clarify how they contribute to the cognitive heterogeneity of PD.
Glucocerebrosidase
Mutations in the glucocerebrosidase gene, GBA, cause the autosomal recessive storage disorder Gaucher’s disease, a lysosomal storage disorder, but the observation that parkinsonism is frequently observed in this disease and in family members of Gaucher’s patients (36), in association with Lewy body deposition in the brainstem and cortex, led to the recognition of an increased frequency of heterozygote GBA mutations in PD cases versus controls. These mutations occur even in those with seemingly sporadic disease and have now been established as the commonest genetic risk factor for PD identified to date (37). GBA mutations are estimated to account for around 3–4% of PD cases (38, 39) with much higher frequencies of around 15% in Ashkenazi Jewish populations (38). However, these mutations also occur in around 3% Ashkenazi Jewish individuals and 1% of non-Jewish individuals (38, 39), hence their classification as a susceptibility factor for PD rather than a monogenic cause of the disease.
There is now clear evidence that GBA-associated PD has a more aggressive phenotype than idiopathic PD, particularly in terms of progression to dementia. In cross-sectional studies, the prevalence of dementia in GBA-PD cases is reported to be around 50% (39, 40), compared to a dementia prevalence of 24–31% in idiopathic PD cases (41), and this difference is particularly striking given that many GBA-associated cases are young onset (39). GBA mutations have also been reported to be associated with a younger onset of Dementia with Lewy Bodies, as well as male gender (42), although this gender association has not been replicated and, in fact, an opposite gender effect has been reported for PD with GBA mutations being more common in women (40). Longitudinal studies have confirmed a faster progression to dementia in PD cases carrying GBA mutations (43, 44), with an odds ratio for dementia of 4.6 (95% CI 1.3–15.9) for GBA-PD compared to idiopathic PD in a population-representative cohort followed up for 10 years from diagnosis (43). Cognitive deficits are also described in patients with Gaucher’s disease and healthy carriers of GBA mutations (45, 46) and may be an early indicator of neurodegenerative pathology in these cases.
The mechanism underlying this association between GBA mutations and PD/PD dementia is still not entirely clear. GBA is a lysosomal enzyme responsible for the breakdown of glucosylceramide, resulting in the accumulation of glucosylceramide in cells (47). Pathologically, PD-GBA cases are characterized by limbic or neocortical Lewy body deposition (48), and colocalization of glucocerebrosidase with Lewy bodies in these GBA mutation carriers has been demonstrated (49), supporting the theory that mutant glucocerebrosidase may contribute directly to increased α-synuclein aggregation (50, 51). It has been proposed that accumulation of the substrate glucocerebroside in the lysosome, due to impaired function of the enzyme, acts as a backbone for α-synuclein aggregation. This in turn leads to compromise of GBA activity and lysosomal function and a consequent increase in α-synuclein aggregation in a positive feedback loop (51). Further understanding of these mechanisms may lead to the development of novel disease-modifying therapies targeted at GBA to slow progression to dementia in PD. However, the overall impact of GBA mutations on the incidence of dementia in PD remains low given that these mutations account for only 3–4% of PD cases (38, 39), and it is unclear how relevant such therapeutic strategies might be in idiopathic PD.
Microtubule-Associated Protein Tau
Microtubule-associated protein tau is a protein involved in microtubule assembly and stabilization, which forms pathological aggregates in a number of neurodegenerative diseases. The gene is encoded on chromosome 17q21 in the center of a 900-kb long fragment, which is inverted in a proportion of the population due to the divergence of two chromosomal lineages (H1 and H2) more than 3 million years ago with no recombination since (52). The inverted H2 haplotype accounts for approximately 25% of haplotypes in those with European ancestry. MAPT H1/H1 genotype has been firmly established not only as a risk factor for tauopathies, including progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD), but also associated with increased PD risk (53). Although significant tau pathology is not found in the majority of idiopathic PD cases at postmortem, there is clear evidence that tau interacts with α-synuclein in Lewy body formation. Tau epitopes are found within Lewy bodies on immunostaining of PD postmortem brains (54), α-synuclein fibrils have been shown to induce polymerization of tau into hyperphosphorylated aggregates in vitro (55), and transgenic mice overexpressing α-synuclein have increased levels of hyperphosphorylated tau colocalizing with α-synuclein deposits (56). Thus, MAPT variation is a good candidate to influence the development of dementia in PD given the widespread cortical Lewy body deposition which characterizes this state.
Association between the MAPT H1/H1 genotype and cognitive dysfunction in PD was first reported in 2007 in a longitudinal study of a population-representative incident PD cohort (n = 108): over a mean (SD) follow-up of 3.5 (0.7) years from diagnosis, age was strongly correlated with cognitive decline in H1 homozygotes but cognitive performance remained unchanged in H2 carriers, regardless of advancing age. In the same CamPaIGN cohort at 5 years from diagnosis, all but one of the PD cases who had developed dementia carried the H1/H1 genotype (18/65 of H1/H1 versus 1/34 of H2 carriers), with an estimated odds ratio (with adjustment for age and other confounding factors) for dementia of 12.1 (57), strikingly greater than the estimated odds ratio for PD susceptibility for MAPT of 1.4 (53), suggesting that MAPT H1 has a much more profound influence on cognitive function in early PD than on risk of disease per se. The 10-year follow-up data from CamPaIGN continued to support an association between H1/H1 genotype and progression to dementia, although the magnitude of the effect seems to lessen with increasing disease duration (age-adjusted hazard ratio 2.9) (1). While this association has been replicated by other groups, including a large case–control study reporting a much stronger association with H1 haplotype for PD dementia (OR = 3.73; P = 0.002) than for PD without dementia (OR = 1.89; P = 0.04) (58), some studies have failed to find such an association (59, 60). The discrepancy may reflect the nature of the cohorts assessed, with negative associations in prevalent cohorts with longer disease durations, suggesting that the predominant effect of the MAPT H1 variant may be to accelerate cognitive decline within the early years of the disease.
Further support for this concept comes from functional MRI studies in patients with early PD, demonstrating association between MAPT H1/H1 and reduced brain activation in medial temporal and parietal regions during memory and visuospatial tasks, respectively, but no association with frontostriatal function (61, 62). Hence, the MAPT H1/H1 effect seems to be specific to posterior cortical circuitry, and given the observations that posterior cortical deficits predict early dementia in PD (discussed above), this genetic variant may be a critical driver of this dementing pathway (see Figure 1).
Evidence from postmortem studies supports the hypothesis that the MAPT H1 variant exerts it phenotypic effects through promoting protein aggregation and Lewy body formation. In a cohort of 22 dementia with Lewy body cases, total Lewy body counts were found to be significantly higher in H1/H1 (n = 12) versus H2 (n = 10) carriers matched for demographics and clinical variables, with no between-group difference in Alzheimer’s type pathology (63). Furthermore, in a large postmortem series of cases with Alzheimer’s disease (AD), Lewy body disease or vascular pathology (n = 762), the MAPT H1 variant was associated with higher cortical Lewy body counts and reduced Alzheimer’s type changes (64). The functional mechanism by which the MAPT H1 variant might lead to changes in protein aggregation is unknown, but increased expression levels of total tau or of four repeat tau isoforms has been reported in PD as well as the tauopathies (65–67), and the H1 haplotype is associated with an increase in four repeat tau transcription in PD brain (57, 68).
Catechol-O-Methyltransferase
The COMT enzyme is a key regulator of synaptic dopamine levels, particularly in the frontal cortex where the dopamine transporter (DAT) is rarely expressed (69). The COMT gene contains a common functional polymorphism of valine (val) for methionine (met) at codon 158, which alters the thermostability of the enzyme, resulting in a reduction in activity of 40% in human frontal cortex in met versus val homozygotes (70). Healthy subjects with low activity COMT genotypes (met/met, with putatively higher prefrontal dopamine levels) show improved performance on working memory (WM) and attentional control tasks activating the prefrontal cortex (71–73). Hence, COMT val158met is a good candidate for modulating the frequently observed executive deficits in PD, which are known to be mediated by dopaminergic dysfunction in frontostriatal networks (74).
This genetic variant has indeed been shown to alter performance on such tasks in PD, although the direction of the effect is opposite to that seen in controls. Specifically, in a cohort of patients with early PD (n = 288), an increase in met alleles (i.e., lower COMT activity and putatively higher dopamine levels) was associated with impaired performance on the CANTAB one-touch Tower of London, a planning task dependent on prefrontal cortical areas, and the impairment was greatest in patients on dopaminergic medications (75). This association between COMT val158met and executive function has since been replicated in an independent early PD cohort (CamPaIGN study), although the effect size was small (57). A further study found evidence of an interactive effect of COMT met alleles and dopaminergic medication on executive performance, although it did not demonstrate a main effect of COMT possibly due to insufficient power (76). Functional MRI studies further support a role for COMT val158met in mediating executive function in PD, demonstrating that impaired performance on both planning and attentional control tasks in met homozygotes is associated with reduced BOLD activation in frontoparietal networks (77, 78), and revealing an interaction between genotype and dopaminergic medication (62), which has potential implications for planning treatment in PD patients with significant executive dysfunction.
The detrimental effect of COMT met alleles on executive performance in early PD (in contrast to the positive effect in healthy controls) seems likely to relate to an “overload” of dopamine in the prefrontal cortex, which can be further exacerbated by dopaminergic medication. This hypothesis is consistent with [18F]DOPA-PET studies indicating a hyperdopaminergic state in prefrontal areas in early PD (79, 80) (presumably compensating for nigrostriatal dopamine loss), as well as a more recent [18F]-DOPA study indicating that COMT met alleles are associated with reduced dopamine metabolism and higher synaptic dopamine levels in frontal brain regions in early PD patients (81). Indeed, it is well established in the animal literature that there is an inverted U-shaped relationship between executive function and dopamine levels in the frontal cortex, with high and low synaptic dopamine levels causing impaired performance (82). As PD progresses, dopamine levels in the prefrontal cortex decline (83) thus resulting in a shift in position on the inverted U-shaped curve, and an alteration in the relationship between COMT genotype and executive performance with time (Figure 2), which has been demonstrated when comparing subgroups of patients with early versus late PD (57). Furthermore, longitudinal analysis in the CamPaIGN incident PD cohort has shown that executive performance improves in met/met individuals over 5 years of disease progression, in contrast to other COMT genotypic groups. So, this genetic variant, while modulating executive heterogeneity in PD through its effects on frontal dopaminergic networks, is not necessarily associated with poor cognitive outcomes in PD and, in particular, does not appear to be associated with dementia risk (57).
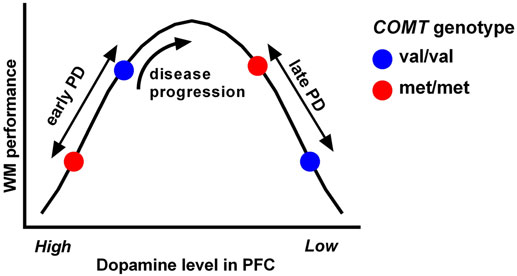
Figure 2. The relationship between working memory (WM) performance and dopamine levels in the prefrontal cortex follows an inverted U-shaped curve. Behavioral and functional imaging data from PD patients indicates that an individual’s position on the curve is dependent on their stage of disease as well as their COMT val158met genotype (which determines the activity of the COMT enzyme). Hence, the relationship between executive function and COMT genotype in PD is complex, and executive deficits may improve rather than worsen in certain genotypic groups as the disease progresses. Reproduced from Williams-Gray et al. (84).
Apolipoprotein E
Association between the APOE gene and susceptibility to AD is well established (85). There is an overlap between AD and PD both clinically and pathologically: dementia and extrapyramidal features occur in both, and neuronal loss and aberrant protein aggregation are common pathological features; and hence, a possible association of APOE with susceptibility to PD and PD dementia has been debated for some time. APOE has three major alleles: APOE-ɛ2, APOE-ɛ3, and APOE-ɛ4. APOE-ɛ4 is associated with both AD risk and lower age of onset, whereas APOE-ɛ2 is protective (85). The functional basis of these effects is thought to be mediated at least in part through altered amyloid metabolism, but APOE has also been implicated in the molecular pathway of α-synuclein-mediated neurodegeneration (86).
A large meta-analysis of over 4000 cases and 10,000 controls has suggested an over-representation of APOE-ɛ2 carriers among PD patients compared to controls, although the odds ratio was very modest at 1.16 (95% CI 1.03–1.31) (84), and association between APOE and PD risk has not been replicated in further large scale genetic association studies (87) or genome-wide association studies in PD. Further meta-analysis indicated an over-representation of APOE-ɛ4 carriers among PD dementia cases (n = 501) compared to PD non-dementia cases (n = 1145) [OR 1.74 (1.36–2.23)] (84). However, the validity of this finding is limited by the small number of PDD cases in each study, the significant heterogeneity of odds ratios between studies, and evidence of publication bias (84). Perhaps most importantly, such cross-sectional studies are probably misleading because any effect of APOE-ɛ4 is likely to be time-dependent analogous to its effect in AD. Longitudinal analyses should be more informative, but only a small number of such studies have been published to date, and again findings have been inconsistent.
The effect of APOE-ɛ4 carrier status on longitudinal cognitive decline in PD was investigated in the CamPaIGN cohort (n = 107) over a 5-year period from diagnosis, and no evidence for association with rate of change in MMSE scores, age-dependent cognitive decline, or incidence of dementia was found (84). Similarly, no associations between APOE genotype and development of dementia were found in another incident PD cohort (n = 64) followed up for a mean of 9.7 years from diagnosis (88). In contrast, a study of 212 PD patients found more rapid cognitive decline (Mattis Dementia Rating Scale) over 4 years in APOE-ɛ4 carriers compared to non-carriers, but these were more advanced cases at study entry with a mean disease duration of 7 years (60). In the same study, MAPT H1/H1 genotype was not associated with cognitive decline over time, although there was an association with memory performance (60). Hence, the disparity between studies may reflect the fact that these genetic variants have time-dependent effects on cognition which vary with disease stage: MAPT seems to have its greatest impact on cognitive decline in early PD, whereas APOE may have a more pronounced effect in later disease. This raises the question of whether association between APOE genotype and dementia in PD reflects the development of Alzheimer’s type pathology in the aging brain. Indeed, a recent study of 232 PD cases reporting association between APOE-ɛ4 and dementia status (OR 5.15) found Alzheimer’s type pathology including moderate to high frequency neuritic plaques and Braak tau stage ≥3 in 90% of APOE-ɛ4 carriers coming to postmortem (n = 10) compared to 43–53% of non-carriers (n = 19) (89). However, postmortem examination over 900 dementia cases and controls genotyped for APOE has demonstrated that APOE-ɛ4 frequency is increased not only in cases with pathologically confirmed AD (38.1%) and mixed Lewy body-AD pathology (40.6%) but also in those with “pure” DLB (31.9%) and “pure” PDD (19.1%) compared to controls (7.2%), indicating that the contribution of APOE-ɛ4 to the dementing process in PD is not mediated entirely through effects on amyloid metabolism and Alzheimer’s type changes (90).
Discussion and Conclusion
As our knowledge of the genetic basis of PD continues to expand, it is becoming increasingly apparent that genetic factors are important not only in determining an individual’s susceptibility to the disease but also how their disease evolves phenotypically (Table 2). Clinical evaluation of cases with Mendelian forms of PD has been informative to some extent, in particular, in terms of corroborating the importance of α-synuclein in the pathogenesis of dementia in PD, but longitudinal data still remains limited in many monogenic forms of PD, and more detailed follow-up data from these rare cases, ideally with postmortem evaluation, would be very valuable. GBA mutations are the most common genetic risk factor for PD, and consequently phenotypic data on GBA-PD cases are rapidly accumulating, suggesting that these cases have an aggressive form of the disease with early onset dementia and florid cortical Lewy body pathology. This knowledge has prompted a wealth of research into the mechanistic relationship between glucocerebrosidase, lysosomal function, and α-synuclein aggregation, which may lead to novel therapeutic strategies.
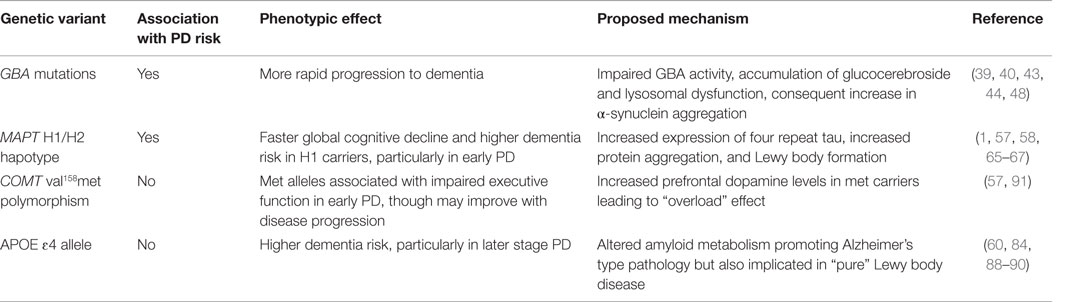
Table 2. Genes contributing to cognitive heterogeneity in PD.
The approach of investigating candidate genes within putatively relevant neurobiological pathways mediating cognitive dysfunction in PD is of course inherently biased, but nonetheless has yielded some very interesting insights into how cognitive deficits evolve in this disorder, and in particular has helped to define distinct cognitive syndromes (57). The frontal dysexecutive syndrome is related to dopaminergic dysfunction in frontostriatal networks and modulated by COMT genotype and does not necessarily indicate the onset of a dementing process. In contrast the posterior cortical syndrome is an age-dependent process modulated by MAPT and APOE genotypes, which is likely to reflect irreversible protein aggregation and neuronal dysfunction in the temporal and parietal cortices, and evolves into clinical dementia (Figure 1). Further support for this idea of distinct cognitive syndromes in PD has recently come from a study which used functional MRI to further interrogate the association between COMT, MAPT, and APOE genotypes and brain function in the ICICLE-PD cohort of newly diagnosed PD patients (n = 168). This study used tasks designed to probe executive, visuospatial, and memory domains and demonstrated that these three genes had dissociable effects on performance in the different cognitive domains associated with regionally specific changes in cortical activation. Specifically, COMT val158met was found to interact with levodopa dose to modulate activation in frontostriatal regions during the executive task, MAPT H1 versus H2 haplotype-modulated activation in parietal regions during the visuospatial task, and APOE genotype-modulated activation in temporoparietal regions during the memory encoding task (62). This concept of different cognitive syndromes in PD has practical implications in term of planning therapy in the clinic, for example, those with a predominantly dysexecutive syndrome in early PD may benefit from a reduction in dopaminergic therapy, whereas those with early posterior cortical deficits warrant close follow-up for an emerging dementia, with a low threshold for starting anticholinesterase inhibitors.
Genetic factors differ not only with respect to the cognitive domains that they influence but also in terms of their temporal relationship with cognition. The relationship between COMT val158met and executive function in PD can vary over time, both according to disease stage (and underlying frontal dopamine levels) and secondary to changes in dopaminergic medication (57). The dementing process appears to be accelerated in early disease by MAPT H1/H1 genotype, although ultimately the same process occurs at a delayed rate in H2 carriers (1); hence, studies in later disease do not necessarily detect a phenotypic effect. In contrast, there is evidence to suggest that APOE genotype is associated with the development of dementia in later disease (60), but an impact on early dementia in PD has not been demonstrated in longitudinal studies (84, 88). This highlights the importance of longitudinal rather than cross-sectional studies to gain a true picture of the relationship between genetic factors and phenotypic variation in PD.
The advent of new technologies allowing whole-genome investigations, such as genome-wide association studies (GWAS) and whole-exome sequencing of large cohorts, has led to the recent discovery of a number of new unanticipated genetic loci associated with PD. These include signal-induced proliferation-associated 1 like 2 (SIPA1L2), inositol polyphosphate-5-phosphatase F (INPP5F), microRNA 4697 (MIR4697), GTP cyclohydrolase (GCH1), vacuolar protein sorting 13 homolog C (VPS13C), and DDRGK domain containing 1 (DDRGK1) (9). However, the contribution of genetic variation at these loci to the clinical and cognitive heterogeneity of PD is yet to be explored. Given that the contribution of individual genetic variants may be small, the optimal strategy for the future will be to pool genetic datasets with longitudinal clinical data from multiple centers to provide sufficient power to adequately interrogate genotypic–phenotypic associations. Ultimately, detailed genetic profiling may provide us with a reliable tool for stratification of cognitive prognosis in PD, which will be invaluable for the selection of the most appropriate patients for future trials of therapies to prevent dementia in PD, as well as to better guide management strategies for this devastating aspect of the disease. Furthermore, as our understanding of the genetic basis of cognitive dysfunction in PD evolves, so will our knowledge of the underlying pathophysiological pathways involved, with consequent implications for the development of novel therapies.
Author Contributions
Both LC and CW-G were responsible for drafting the article, critical revision of the article, and final approval of the version to be published.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
LC’s current work is funded by The Rosetrees Trust and Alzheimers Research UK (ARUK). CW-G’s current work is funded by The Rosetrees Trust, Addenbrookes Charitable Trust (ACT), The Academy of Medical Sciences UK (AMS), and the Stevenage Bioscience Catalyst.
References
1. Williams-Gray CH, Mason SL, Evans JR, Foltynie T, Brayne C, Robbins TW, et al. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J Neurol Neurosurg Psychiatry (2013) 84:1258–64. doi:10.1136/jnnp-2013-305277
2. Compta Y, Parkkinen L, O’Sullivan SS, Vandrovcova J, Holton JL, Collins C, et al. Lewy-and Alzheimer-type pathologies in Parkinson’s disease dementia: which is more important? Brain (2011) 134(Pt 5):1493–505. doi:10.1093/brain/awr031
3. Bohnen NI, Kaufer DI, Ivanco LS, Lopresti B, Koeppe RA, Davis JG, et al. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch Neurol (2003) 60:1745–8. doi:10.1001/archneur.60.12.1745
4. Compta Y, Pereira JB, Ríos J, Ibarretxe-Bilbao N, Junqué C, Bargalló N, et al. Combined dementia-risk biomarkers in Parkinson’s disease: a prospective longitudinal study. Parkinsonism Relat Disord (2013) 19:717–24. doi:10.1016/j.parkreldis.2013.03.009
5. Bohnen NI, Koeppe RA, Minoshima S, Giordani B, Albin RL, Frey KA, et al. Cerebral glucose metabolic features of Parkinson disease and incident dementia: longitudinal study. J Nucl Med (2011) 52:848–55. doi:10.2967/jnumed.111.089946
6. Mak E, Su L, Williams GB, Firbank MJ, Lawson RA, Yarnall AJ, et al. Baseline and longitudinal grey matter changes in newly diagnosed Parkinson’s disease: ICICLE-PD study. Brain (2015) 138:2974–86. doi:10.1093/brain/awv211
7. Pedersen KF, Larsen JP, Tysnes OB, Alves G. Prognosis of mild cognitive impairment in early Parkinson disease: the Norwegian ParkWest study. Neurology (2013) 70(5):580–6. doi:10.1001/jamaneurol.2013.2110
8. Broeders M, de Bie RMA, Velseboer DC, Speelman JD, Muslimovic D, Schmand B. Evolution of mild cognitive impairment in Parkinson disease. Neurology (2013) 81:346–52. doi:10.1212/WNL.0b013e31829c5c86
9. Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet (2014) 46:989–93. doi:10.1038/ng.3043
10. Klein C, Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med (2012) 2:a008888. doi:10.1101/cshperspect.a008888
11. Farrer M, Kachergus J, Forno L, Lincoln S, Wang D-S, Hulihan M, et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol (2004) 55:174–9. doi:10.1002/ana.10846
12. Ibáñez P, Bonnet AM, Débarges B, Lohmann E, Tison F, Pollak P, et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet (2004) 364:1169–71. doi:10.1016/S0140-6736(04)17104-3
13. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science (1997) 276:2045–7. doi:10.1126/science.276.(5321)0.2045-2047
14. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron (2004) 44:601–7. doi:10.1016/j.neuron.2004.11.005
15. Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron (2004) 44:595–600. doi:10.1016/j.neuron.2004.10.023
16. Chartier-Harlin M-C, Dachsel JC, Vilariño-Güell C, Lincoln SJ, Leprêtre F, Hulihan MM, et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet (2011) 89:398–406. doi:10.1016/j.ajhg.2011.08.009
17. Vilariño-Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet (2011) 89:162–7. doi:10.1016/j.ajhg.2011.06.001
18. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature (1998) 392(6676):605–8. doi:10.1038/33416
19. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science (2004) 304:1158–60. doi:10.1126/science.1096284
20. Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science (2003) 299(5604):256–9. doi:10.1126/science.1077209
21. Somme JH, Gomez-Esteban JC, Molano A, Tijero B, Lezcano E, Zarranz JJ. Initial neuropsychological impairments in patients with the E46K mutation of the α-synuclein gene (PARK 1). J Neurol Sci (2011) 310:86–9. doi:10.1016/j.jns.2011.07.047
22. Puschmann A, Ross OA, Vilariño-Güell C. A Swedish family with de novo α-synuclein A53T mutation: evidence for early cortical dysfunction. Parkinsonism Relat Disord (2009) 15(9):627–32. doi:10.1016/j.parkreldis.2009.06.007
23. Seidel K, Schöls L, Nuber S, Petrasch-Parwez E, Gierga K, Wszolek Z, et al. First appraisal of brain pathology owing to A30P mutant alpha-synuclein. Ann Neurol (2010) 67:684–9. doi:10.1002/ana.21966
24. Poulopoulos M, Levy OA, Alcalay RN. The neuropathology of genetic Parkinson’s disease. Mov Disord (2012) 27(7):831–42. doi:10.1002/mds.24962
25. Williams-Gray CH, Goris A, Foltynie T, Brown J, Maranian M, Walton A, et al. Prevalence of the LRRK2 G2019S mutation in a UK community based idiopathic Parkinson’s disease cohort. J Neurol Neurosurg Psychiatry (2006) 77:665–7. doi:10.1136/jnnp.2005.085019
26. Munhoz RP, Rogaeva E, Langston JW, Kasten M, Meaney C, Klein C, et al. Phenotype in parkinsonian and nonparkinsonian LRRK2 G2019S mutation carriers. Neurology (2011) 77(4):325–33. doi:10.1212/WNL.0b013e318227042d
27. Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol (2008) 7(7):583–90. doi:10.1016/S1474-4422(08)70117-0
28. Srivatsal S, Cholerton B, Leverenz JB, Wszolek ZK, Uitti RJ, Dickson DW, et al. Cognitive profile of LRRK2-related Parkinson’s disease. Mov Disord (2015) 30:728–33. doi:10.1002/mds.26161
29. Wider C, Skipper L, Solida A, Brown L, Farrer M, Dickson D, et al. Autosomal dominant DOPA-responsive parkinsonism in a multigenerational Swiss family. Parkinsonism Relat Disord (2008) 14:465–70. doi:10.1016/j.parkreldis.2007.11.013
30. Grünewald A, Kasten M, Ziegler A, Klein C. Next-generation phenotyping using the parkin example: time to catch up with genetics. JAMA Neurol (2013) 70:1186–91. doi:10.1001/jamaneurol.2013.488
31. van de Warrenburg BP, Lammens M, Lücking CB, Denèfle P, Wesseling P, Booij J, et al. Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology (2001) 56:555–7. doi:10.1212/WNL.56.4.555
32. Steinlechner S, Stahlberg J, Völkel B, Djarmati A, Hagenah J, Hiller A, et al. Co-occurrence of affective and schizophrenia spectrum disorders with PINK1 mutations. J Neurol Neurosurg Psychiatry (2007) 78:532–5. doi:10.1136/jnnp.2006.105676
33. Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, et al. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet (2001) 68:895–900. doi:10.1086/319522
34. Chartier-Harlin M-C, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet (2004) 364:1167–9. doi:10.1016/S0140-6736(04)17103-1
35. Kasten M, Kertelge L, Brüggemann N, van der Vegt J, Schmidt A, Tadic V, et al. Nonmotor symptoms in genetic Parkinson disease. Arch Neurol (2010) 67:670–6. doi:10.1001/archneurol.67.6.670
36. Capablo JL, Saenz de Cabezón A, Fraile J, Alfonso P, Pocovi M, Giraldo P. Spanish group on Gaucher disease. Neurological evaluation of patients with Gaucher disease diagnosed as type 1. J Neurol Neurosurg Psychiatry (2008) 79:219–22. doi:10.1136/jnnp.2006.111518
37. Velayati A, Yu WH, Sidransky E. The role of glucocerebrosidase mutations in Parkinson disease and Lewy body disorders. Curr Neurol Neurosci Rep (2010) 10(3):190–8. doi:10.1007/s11910-010-0102-x
38. Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med (2009) 361:1651–61. doi:10.1056/NEJMoa0901281
39. Neumann J, Bras J, Deas E, O’Sullivan SS, Parkkinen L, Lachmann RH, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain (2009) 132:1783–94. doi:10.1093/brain/awp044
40. Setó-Salvia N, Pagonabarraga J, Houlden H, Pascual-Sedano B, Dols-Icardo O, Tucci A, et al. Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson’s disease course. Mov Disord (2011) 27:393–9. doi:10.1002/mds.24045
41. Aarsland D, Zaccai J, Brayne C. A systematic review of prevalence studies of dementia in Parkinson’s disease. Mov Disord (2005) 20:1255–63. doi:10.1002/mds.20527
42. Gámez-Valero A, Prada-Dacasa P, Santos C, Adame-Castillo C, Campdelacreu J, Reñé R, et al. GBA mutations are associated with earlier onset and male sex in dementia with Lewy bodies. Mov Disord (2016). doi:10.1002/mds.26593
43. Winder-Rhodes SE, Evans JR, Ban M, Mason SL, Williams-Gray CH, Foltynie T, et al. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain (2013) 136:392–9. doi:10.1093/brain/aws318
44. Brockmann K, Srulijes K, Pflederer S, Hauser A-K, Schulte C, Maetzler W, et al. GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord (2014) 30:407–11. doi:10.1002/mds.26071
45. McNeill A, Duran R, Proukakis C, Bras J, Hughes D, Mehta A, et al. Hyposmia and cognitive impairment in Gaucher disease patients and carriers. Mov Disord (2012) 27:526–32. doi:10.1002/mds.24945
46. Beavan M, McNeill A, Proukakis C, Hughes DA, Mehta A, Schapira AHV. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol (2015) 72:201–8. doi:10.1001/jamaneurol.2014.2950
47. Brady RO, Kanfer JN, Shapiro D. Metabolism of glucocerebrosides. II. Evidence of an enzymatic deficiency in Gaucher’s disease. Biochem Biophys Res Commun (1965) 18:221–5. doi:10.1016/0006-291X(65)90743-6
48. Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab (2004) 82:192–207. doi:10.1016/j.ymgme.2004.04.011
49. Goker-Alpan O, Stubblefield BK, Giasson BI, Sidransky E. Glucocerebrosidase is present in α-synuclein inclusions in Lewy body disorders. Acta Neuropathol (2010) 120:641–9. doi:10.1007/s00401-010-0741-7
50. Westbroek W, Gustafson AM, Sidransky E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol Med (2011) 17:485–93. doi:10.1016/j.molmed.2011.05.003
51. Mazzulli JR, Xu Y-H, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell (2011) 146:37–52. doi:10.1016/j.cell.2011.06.001
52. Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, et al. A common inversion under selection in Europeans. Nat Genet (2005) 37:129–37. doi:10.1038/ng1508
53. Goris A, Williams-Gray CH, Clark GR, Foltynie T, Lewis SJG, Brown J, et al. Tau and alpha-synuclein in susceptibility to, and dementia in, Parkinson’s disease. Ann Neurol (2007) 62:145–53. doi:10.1002/ana.21192
54. Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol (2003) 62:389–97. doi:10.1093/jnen/62.4.389
55. Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by α-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci (2011) 31:7604–18. doi:10.1523/JNeurosci.0297-11.2011
56. Haggerty T, Credle J, Rodriguez O, Wills J, Oaks AW, Masliah E, et al. Hyperphosphorylated Tau in an α-synuclein-overexpressing transgenic model of Parkinson’s disease. Eur J Neurosci (2011) 33:1598–610. doi:10.1111/j.1460-9568.2011.07660.x
57. Williams-Gray CH, Evans JR, Goris A, Foltynie T, Ban M, Robbins TW, et al. The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain (2009) 132(Pt 11):2958–69. doi:10.1093/brain/awp245
58. Setó-Salvia N, Clarimón J, Pagonabarraga J, Pascual-Sedano B, Campolongo A, Combarros O, et al. Dementia risk in Parkinson disease: disentangling the role of MAPT haplotypes. Arch Neurol (2011) 68:359–64. doi:10.1001/archneurol.2011.17
59. Mata IF, Leverenz JB, Weintraub D, Trojanowski JQ, Hurtig HI, Van Deerlin VM, et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol (2014) 71:1405–12. doi:10.1001/jamaneurol.2014.1455
60. Morley JF, Xie SX, Hurtig HI, Stern MB, Colcher A, Horn S, et al. Genetic influences on cognitive decline in Parkinson’s disease. Mov Disord (2012) 27:512–8. doi:10.1002/mds.24946
61. Winder-Rhodes SE, Hampshire A, Rowe JB, Peelle JE, Robbins TW, Owen AM, et al. Association between MAPT haplotype and memory function in patients with Parkinson’s disease and healthy aging individuals. Neurobiol Aging (2015) 36:1519–28. doi:10.1016/j.neurobiolaging.2014.12.006
62. Nombela C, Rowe JB, Winder-Rhodes SE, Hampshire A, Owen AM, Breen DP, et al. Genetic impact on cognition and brain function in newly diagnosed Parkinson’s disease: ICICLE-PD study. Brain (2014) 137:2743–58. doi:10.1093/brain/awu201
63. Colom-Cadena M, Gelpi E, Martí MJ, Charif S, Dols-Icardo O, Blesa R, et al. MAPT H1 haplotype is associated with enhanced α-synuclein deposition in dementia with Lewy bodies. Neurobiol Aging (2013) 34:936–42. doi:10.1016/j.neurobiolaging.2012.06.015
64. Wider C, Ross OA, Nishioka K, Heckman MG, Vilariño-Güell C, Jasinska-Myga B, et al. An evaluation of the impact of MAPT, SNCA and APOE on the burden of Alzheimer’s and Lewy body pathology. J Neurol Neurosurg Psychiatry (2012) 83:424–9. doi:10.1136/jnnp-2011-301413
65. Myers AJ, Pittman AM, Zhao AS, Rohrer K, Kaleem M, Marlowe L, et al. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis (2007) 25:561–70. doi:10.1016/j.nbd.2006.10.018
66. Kwok JBJ, Teber ET, Loy C, Hallupp M, Nicholson G, Mellick GD, et al. Tau haplotypes regulate transcription and are associated with Parkinson’s disease. Ann Neurol (2004) 55:329–34. doi:10.1002/ana.10826
67. Rademakers R, Melquist S, Cruts M, Theuns J, Del-Favero J, Poorkaj P, et al. High-density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy. Hum Mol Genet (2005) 14:3281–92. doi:10.1093/hmg/ddi361
68. Tobin JE, Latourelle JC, Lew MF, Klein C, Suchowersky O, Shill HA, et al. Haplotypes and gene expression implicate the MAPT region for Parkinson disease: the GenePD Study. Neurology (2008) 71:28–34. doi:10.1212/01.wnl.0000304051.01650.23
69. Lewis DA, Melchitzky DS, Sesack SR, Whitehead RE, Auh S, Sampson A. Dopamine transporter immunoreactivity in monkey cerebral cortex: regional, laminar, and ultrastructural localization. J Comp Neurol (2001) 432:119–36. doi:10.1002/cne.1092
70. Chen JS, Lipska BK, Halim N, Ma QD, Matsumoto M, Melhem S, et al. Functional analysis of genetic variation in catechol-o-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am J Hum Genet (2004) 75:807–21. doi:10.1086/425589
71. Barnett JH, Jones PB, Robbins TW, Müller U. Effects of the catechol-O-methyltransferase Val158Met polymorphism on executive function: a meta-analysis of the Wisconsin Card Sort Test in schizophrenia and healthy controls. Mol Psychiatry (2007) 12:502–9. doi:10.1038/sj.mp.4001973
72. Blasi G, Mattay VS, Bertolino A, Elvevåg B, Callicott JH, Das S, et al. Effect of catechol-O-methyltransferase Val158Met genotype on attentional control. J Neurosci (2005) 25:5038–45. doi:10.1523/JNeurosci.0476-05.2005
73. Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE, et al. Effect of COMT Val108/158 met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A (2001) 98:6917–22. doi:10.1073/pnas.111134598
74. Owen AM. Cognitive dysfunction in Parkinson’s disease: the role of frontostriatal circuitry. Neuroscientist (2004) 10:525–37. doi:10.1177/1073858404266776
75. Foltynie T, Goldberg TE, Lewis SGJ, Blackwell AD, Kolachana BS, Weinberger DR, et al. Planning ability in Parkinson’s disease is influenced by the COMT Val158Met polymorphism. Mov Disord (2004) 19:885–91. doi:10.1002/mds.20118
76. Hoogland J, de Bie R, Gray CW. Catechol-O-methyltransferase Val158Met and cognitive function in Parkinson’s disease. Mov Disord (2010) 25(15):2550–4. doi:10.1002/mds.23319
77. Williams-Gray CH, Hampshire A, Barker RA, Owen AM. Attentional control in Parkinson’s disease is dependent on COMT val 158 met genotype. Brain (2008) 131:397–408. doi:10.1093/brain/awm313
78. Williams-Gray CH, Hampshire A, Robbins TW, Owen AM, Barker RA. Catechol O-methyltransferase Val158Met genotype influences frontoparietal activity during planning in patients with Parkinson’s disease. J Neurosci (2007) 27:4832–8. doi:10.1523/JNeurosci.0774-07.2007
79. Kaasinen V, Nurmi E, Brück A, Eskola O, Bergman J, Solin O, et al. Increased frontal [(18)F]fluorodopa uptake in early Parkinson’s disease: sex differences in the prefrontal cortex. Brain (2001) 124:1125–30. doi:10.1093/brain/124.6.1125
80. Rakshi JS, Uema T, Ito K, Bailey DL, Morrish PK, Ashburner J, et al. Frontal, midbrain and striatal dopaminergic function in early and advanced Parkinson’s disease A 3D [(18)F]DOPA-PET study. Brain (1999) 122(Pt 9):1637–50. doi:10.1093/brain/122.9.1637
81. Wu K, O’Keeffe D, Politis M, O’Keeffe GC, Robbins TW. The catechol-O-methyltransferase Val158Met polymorphism modulates fronto-cortical dopamine turnover in early Parkinson’s disease: a PET study. Brain (2012) 135(Pt 8):2449–57. doi:10.1093/brain/aws157
82. Goldman-Rakic PS, Muly EC III, Williams GV. D 1 receptors in prefrontal cells and circuits. Brain Res Rev (2000) 31(2–3):295–301. doi:10.1016/S0165-0173(99)00045-4
83. Brooks DJ, Piccini P. Imaging in Parkinson’s disease: the role of monoamines in behavior. Biol Psychiatry (2006) 59:908–18. doi:10.1016/j.biopsych.2005.12.017
84. Williams-Gray CH, Goris A, Saiki M, Foltynie T, Compston DA, Sawcer SJ, et al. Apolipoprotein E genotype as a risk factor for susceptibility to and dementia in Parkinson’s disease. J Neurol (2009) 256(3):493–8. doi:10.1007/s00415-009-0119-8
85. Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA (1997) 278:1349–56. doi:10.1001/jama.1997.03550160069041
86. Gallardo G, Schlüter OM, Südhof TC. A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nat Neurosci (2008) 11:301–8. doi:10.1038/nn2058
87. Federoff M, Jimenez-Rolando B, Nalls MA, Singleton AB. A large study reveals no association between APOE and Parkinson’s disease. Neurobiol Dis (2012) 46:389–92. doi:10.1016/j.nbd.2012.02.002
88. Kurz MW, Dekomien G, Nilsen OB, Larsen JP, Aarsland D, Alves G. APOE alleles in Parkinson disease and their relationship to cognitive decline: a population-based, longitudinal study. J Geriatr Psychiatry Neurol (2009) 22:166–70. doi:10.1177/0891988709332945
89. Monsell SE, Besser LM, Heller KB, Checkoway H, Litvan I, Kukull WA. Clinical and pathologic presentation in Parkinson’s disease by apolipoprotein e4 allele status. Parkinsonism Relat Disord (2014) 20:503–7. doi:10.1016/j.parkreldis.2014.02.001
90. Tsuang D, Leverenz JB, Lopez OL. APOE ϵ4 increases risk for dementia in pure synucleinopathies. Neurology (2013) 70(2):223–8. doi:10.1001/jamaneurol.2013.600
Keywords: Parkinson’s disease, cognition, dementia, genetics, COMT, MAPT, APOE, GBA
Citation: Collins LM and Williams-Gray CH (2016) The Genetic Basis of Cognitive Impairment and Dementia in Parkinson’s Disease. Front. Psychiatry 7:89. doi: 10.3389/fpsyt.2016.00089
Received: 15 December 2015; Accepted: 09 May 2016;
Published: 20 May 2016
Edited by:
Angie A. Kehagia, King’s College London, UKReviewed by:
Roland Brandt, University of Osnabrück, GermanyChrystalina A. Antoniades, University of Oxford, UK
Copyright: © 2016 Collins and Williams-Gray. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucy M. Collins, bG1jNThAY2FtLmFjLnVr