 Ryoma Morigaki
Ryoma Morigaki Hideo Mure1,3
Hideo Mure1,3 Ryuji Kaji
Ryuji Kaji Shinji Nagahiro
Shinji Nagahiro Satoshi Goto
Satoshi Goto- 1Parkinson’s Disease and Dystonia Research Center, Tokushima University Hospital, Tokushima University, Tokushima, Japan
- 2Department of Neurodegenerative Disorders Research, Graduate School of Medical Sciences, Institute of Biomedical Sciences, Tokushima University, Tokushima, Japan
- 3Department of Neurosurgery, Graduate School of Medical Sciences, Institute of Biomedical Sciences, Tokushima University, Tokushima, Japan
- 4Department of Clinical Neuroscience, Graduate School of Medical Sciences, Institute of Biomedical Sciences, Tokushima University, Tokushima, Japan
Tardive syndrome (TDS) is a potentially permanent and irreversible hyperkinetic movement disorder caused by exposure to dopamine receptor blocking agents. Guidelines published by the American Academy of Neurology recommend pharmacological first-line treatment for TDS with clonazepam (level B), ginkgo biloba (level B), amantadine (level C), and tetrabenazine (level C). Recently, a class II study provided level C evidence for use of deep brain stimulation (DBS) of the globus pallidus internus (GPi) in patients with TDS. Although the precise pathogenesis of TDS remains to be elucidated, the beneficial effects of GPi-DBS in patients with TDS suggest that the disease may be a basal ganglia disorder. In addition to recent advances in understanding the pathophysiology of TDS, this article introduces the current use of DBS in the treatment of medically intractable TDS.
Introduction
The term “tardive” originates from the French “tardif,” meaning “late”; tardive syndrome (TDS) refers to delayed onset motor disturbances following treatment with psychotropic medication (1, 2). DSM-5 diagnostic criteria for TDS include a history of more than 3 months cumulative exposure to dopamine receptor blocking agents (DRBAs), except in elderly patients in whom 1 month is adequate (3). They also contain the presence of “mild” or “moderate” abnormal involuntary movements (AIMs) in one or more body areas, and the absence of other conditions that might produce AIMs (4, 5).
Tardive syndrome can manifest heterogeneous features of AIMs that comprise dystonia, chorea, athetosis, akathisia, myoclonus, stereotyped behavior, tremor, and tourettism or tics (6–8). Orofacial dyskinesia is the most common symptom in less severe cases, while generalized hyperkinetic movements with predominance of axial dystonia also occur in severe cases (9). Two-thirds of patients with TDS have cervical involvement (10). As many various types of motor symptoms can emerge, it has been suggested that TDS is a more accurate term for the condition than the traditionally used term “tardive dyskinesia (TDD)” (2, 11). TDD is now used to refer to more specific involuntary movements (e.g., lingual–facial–buccal dyskinesia) which are caused by DRBAs (8, 11).
The causative agents are usually typical or atypical antipsychotic drugs (APDs). Recent reports, however, suggest that TDS could also be caused by a wide variety of psychotropic drugs, such as antidepressants and antiparkinsonian medications (7). Systematic overview and meta-regression analyses of 52 randomized controlled trials conducted by Geddes et al. revealed that there are no differential effects between typical and atypical antipsychotics in causing extrapyramidal side effects (12). Recently, O’Brien et al. reviewed studies that investigated the prevalence or incidence of TDS in elderly patients exposed to APDs from 1957 to 2015. The inclusion criteria of this meta-analysis were prospective studies (n > 20), which used validated rating scales and research diagnostic criteria (13). According to this meta-analysis, the estimated prevalence for probable TDS—defined according to the Schooler and Kane Research Diagnostic Criteria where abnormal movements in at least one body part are labeled “moderate” and in two or more body parts are rated “mild” (4) — was higher in patients after being treated with typical APDs for 1 year (23 vs. 7%). In more than 50% of cases, TDS was irreversible even after withdrawal from the responsible neuroleptics (14).
In guidelines proposed by the American Academy of Neurology, clonazepam (level B), ginkgo biloba (level B) and amantadine (level C), and tetrabenazine (level C) are recommended for the treatment of TDS (Table 1) (5). Among them, tetrabenazine is most effective at reducing TDS, but has the risk of inducing depression or Parkinsonism (15, 16). Neuroleptic agents cannot be recommended in this guideline since they may cause TDS and mask its symptoms, instead of treating it (5). However, clozapine is the most acceptable alternative for patients with schizophrenia (6). It has the lowest risk among all APDs that cause TDS by inhibiting dopamine D1 and D2 receptors (6, 17). Although its efficacy in reducing TDS is undetermined due to conflicting class III studies, the currently used APDs treatment should be replaced with clozapine as an alternative therapy for suppressing TDS prior to attempting surgical procedures in deep brain stimulation (DBS) clinical trials (18, 19). As published in our previous report (20), accumulating evidence suggests that patients with TDS could be good candidates for undergoing DBS that targets the globus pallidus internus (GPi). Recently, Pouclet-Courtemanche et al. reported a class II evidence trial indicating that GPi-DBS significantly relieves motor symptoms in patients with medically intractable TDS. In this article, we describe recent understandings of the pathophysiology of TDS, and introduce the current use of GPi-DBS in treatment of the disease (19).
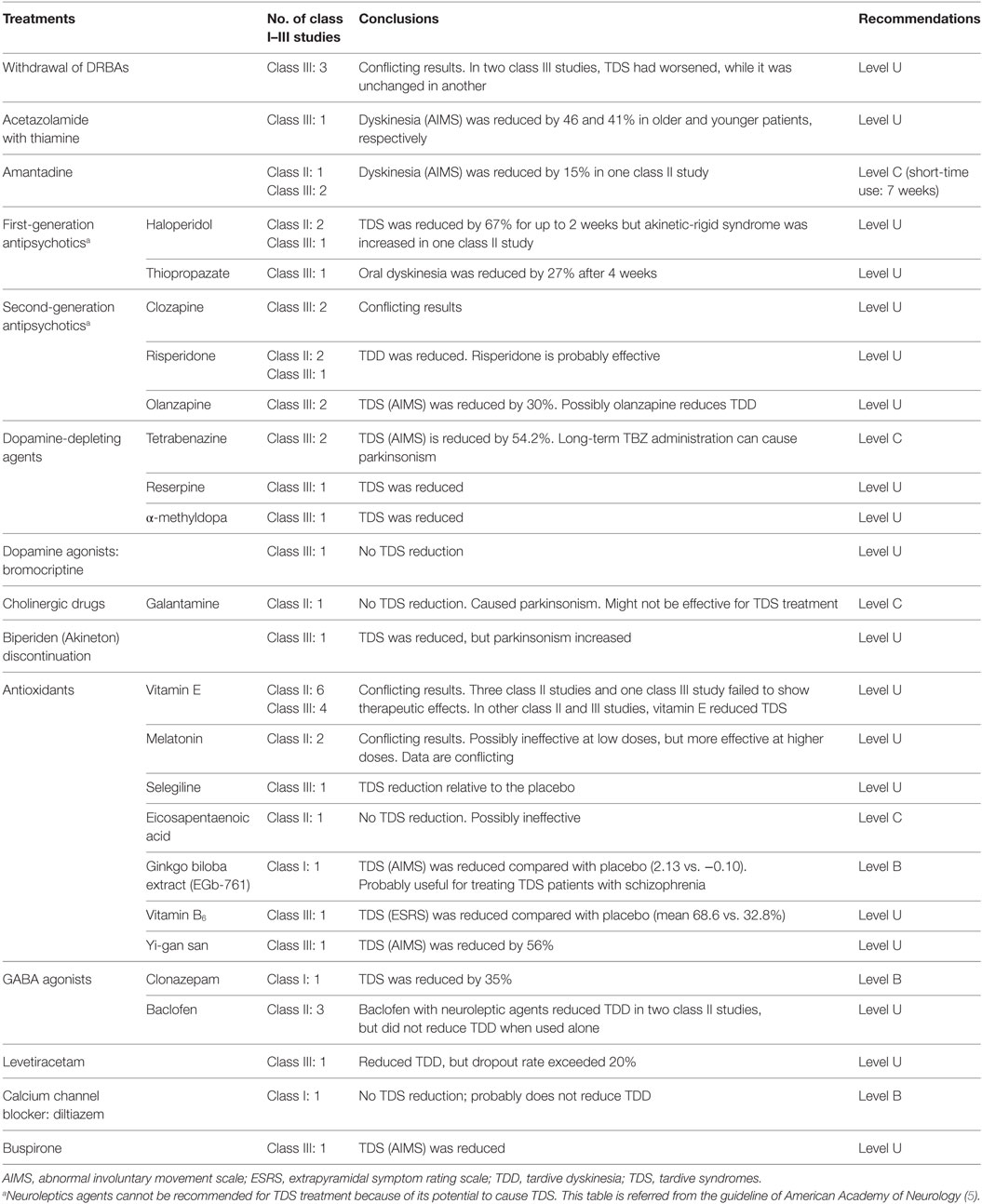
Table 1. Evidence-based medical treatments of tardive syndrome.
Pathophysiology of TDS
Dopamine Receptor Hypersensitivity
Striatal dopamine receptor supersensitivity has so far been the most plausible explanation for development of TDS. Chronic exposure to DRBAs can induce upregulation of postsynaptic dopamine receptors, particularly of the D2 subclass, in the striatum (21). Notably, medications that act on the presynaptic D2 receptors, such as reserpine and tetrabenazine, do not cause TDS (6). The proposed model of a postsynaptic dopamine hypersensitivity mechanism occurring due to upregulation of the D2 receptors is supported by findings obtained from experimental animal models (22–25) and in a human study using positron emission tomography (PET) (26). In the animal models, sub-chronic treatment with antipsychotics increased vacuous chewing movements (VCM) associated with upregulation of striatal D2 receptors (24). Teo et al. hypothesized that hypersensitivity of D2 receptors could cause maladaptive plasticity in the cortico-striatal transmission, resulting in an inability to normalize the miscoded motor program in patients with TDS (27). This notion might be supported by PET findings in patients with TDS (9). In addition to an increase in regional cerebral blood flow during the rest condition in the prefrontal and anterior cingulate cortex and the cerebellum, Thobois et al. (16) reported an excess of brain activity in the prefrontal and premotor cortical areas during motor execution, which might reflect a loss of motor selectivity leading to generation of abnormal movements (9). Trugman et al. hypothesized that the D2 receptor blockade concomitant with repetitive activation of the D1 receptors could be a fundamental cause of TDS (17). This hypothesis might be consistent with the delayed onset of TDS after exposure to neuroleptics and the persistence of TDS even after withdrawal from them (17). In addition, maladaptive changes in non-dopaminergic neurotransmitter systems, such as those involving opioids (enkephalin and dynorphin), glutamate, and acetylcholine, have also been reported in patients with TDS (28, 29) and in animal models of TDS (30–34).
Neurotoxicity Induced by Oxidative Stress
More recently, oxidative stress has been suggested as a mechanism for TDS pathogenesis. Neuroleptics can exert direct toxic effects on neurons by inhibiting the complex I of the electron transport chain. They also can increase dopamine turnover through chronic dopamine receptor blockade, thereby generating hydrogen peroxide and free radicals, leading to neurotoxicity (8, 35, 36). In animal studies, antipsychotics increase membrane lipid peroxidation, free radical activity, and glutamate transmission, but decrease antioxidant enzyme activity for glutathione (28, 37–39). Defects in the antioxidant systems might cause development of TDS (40). Several authors suggest that oxidative damage leading to neuronal degeneration may explain the irreversibility of TDS (41, 42). In support of this notion, neuroimaging studies using CT and MRI showed that among patients with schizophrenia, a significant reduction in structural volume of the caudate nucleus was found in patients with TDS when compared to non-TDS patients (43–45). Moreover, variances in the gene encoding manganese superoxide dismutase (MnSOD) and the gene for an enzyme that eliminates free radicals have also been found to correlate with presence of TDS symptoms (35, 46–49). Based on these findings, a wide variety of antioxidants has been tested in clinical trials (5). The guidelines of the American Academy of Neurology suggest that ginkgo biloba extract (EGb-761) is probably useful (Level B) in TDS therapy (5). Although data conflictingly support or oppose the use of other antioxidative agents, class I and II studies have shown that TDS could be significantly alleviated by vitamin B6, vitamin E, and melatonin (Table 1) (5, 36).
Genetic Predisposition
Genetic studies suggest that there is an intrinsic susceptibility to develop AIMs in patients with schizophrenia and that the role of antipsychotics is one of promotion or acceleration of rather than causation of symptoms (45, 50). There is solid evidence for a genetic predisposition to TDS (7). Family studies showed that occurrence of TDS was influenced by polymorphisms in the genes coding for the D2 and D3 receptors (DRD2 and DRD3), catechol-O-methyl-transferase (COMT), 5-HT2A receptors (HTR2A), manganese super-dismutase (MnSOD), and cytochrome P450 (CYP2D6) (8, 51). Mutations in genes related to GABAergic pathways (SLCA11, GABRB2, and GABRC3), N-methyl-d-aspartate (NMDA) receptor (GRIN2A), and oxidative stress related genes (GSTM1, GSTP1, NQO1, and NOS3) are also suggested to play a role in developing TDS (8, 51). Souza et al. reported that GSK-3β polymorphism might be a risk factor for TDS in patients with schizophrenia (52). A single nucleotide polymorphism marker located in the 3′-untranslated regulatory region of the Nurr77 mRNA is nominally associated with risk and severity of AIMs in TDS patients with schizophrenia (53, 54).
Animal Models of TDS
Rats, mice, and non-human primates have been commonly used as TDS models, in order to investigate disease pathogenesis and evaluate the efficacy of TDS pharmacotherapy. Since the early 1970s, rats that were exposed to dopamine receptor blocking agents for consecutive weeks manifested different patterns of purposeless, chewing activity, which is termed “vacuous chewing movements”(22–25, 30–34). VCM are also observed in mouse models of TDS (55, 56). The VCM induced by haloperidol was further exacerbated by knocking out Nur77 (57). Knocking out aquaporin-4, however, abolished VCM that were induced by chronic haloperidol treatment (58). The expression patterns of immediate early genes in the striatum, which were induced by clozapine or haloperidol, have been demonstrated using transgenic dopamine D3 receptor knockout mice (59, 60). Thus, transgenic rodent models are beneficial for addressing drug-induced neural changes. Non-human primate model of TDS appeared as early as the late 1970s. Given the marked interspecies difference in susceptibility of New World monkey species, TDS developed in proportions of 0, 45, and 71% in squirrels (Saimiri sciureus), capuchins (Cebus apella), and marmosets (Callithrix jacchus), respectively (61). In non-human primates, chronic APD exposure, typically of haloperidol, for at least 1 year, was required to model TDS (61). Abnormal stereotypical movements observed in non-human primate models of TDS include various orofacial dyskinetic movements, neck rotation, brief back extension, flexion/extension movements of the toes, and upper limb chorea, which persisted for several months following drug withdrawal (61). Since the latency of onset, individual susceptibility, phenomenological expression, and persistence of TDS is similar to humans, non-human primate models of TDS are best suited to address therapeutic issues (61).
DBS for TDS
TDS as a Basal Ganglia Circuit Disorder
Accumulating evidence suggests that TDS might result from abnormal plasticity in the motor circuit that links with the basal ganglia (9, 17, 45). Consistent with this concept, TDS was successfully treated with DBS of the GPi, which is the major basal ganglia output nucleus (see Tables 2 and 3). During GPi-DBS surgery in patients with TDS, microelectrode recordings (MERs) of GPi neurons show abnormal bursts and irregular activities (62, 63). In addition, simultaneous recording on pairs of GPi cells also showed a high degree of discharge synchronization (63). By means of a fast Fourier transform analysis, Nandi et al. reported that local field potentials in the GPi showed significant strength of correlation and coherence with the EMG data of AIMs in a patient with TDS (64). Given the evidence that in patients with TDS, GPi cells fired before onset of AIMs, Magariños-Ascone et al. suggest that the burst and irregular patterns of neuronal discharges might indicate an imperfect code that becomes arranged in a confused order at the cortical level, and that GPi-DBS could disrupt these “noisy signals” and allow the motor program to be gated with ease (63). Evidence that GPi-DBS could influence the brain CBF levels in the primary and associative motor cortices has also been reported (9, 62). It has also been noted that not only the GPi but also the STN and thalamus could be targets for DBS in the treatment of TDS (65, 66). These observations indicate that TDS might be a network disorder involving cortico-thalamo-basal ganglia motor circuitry.
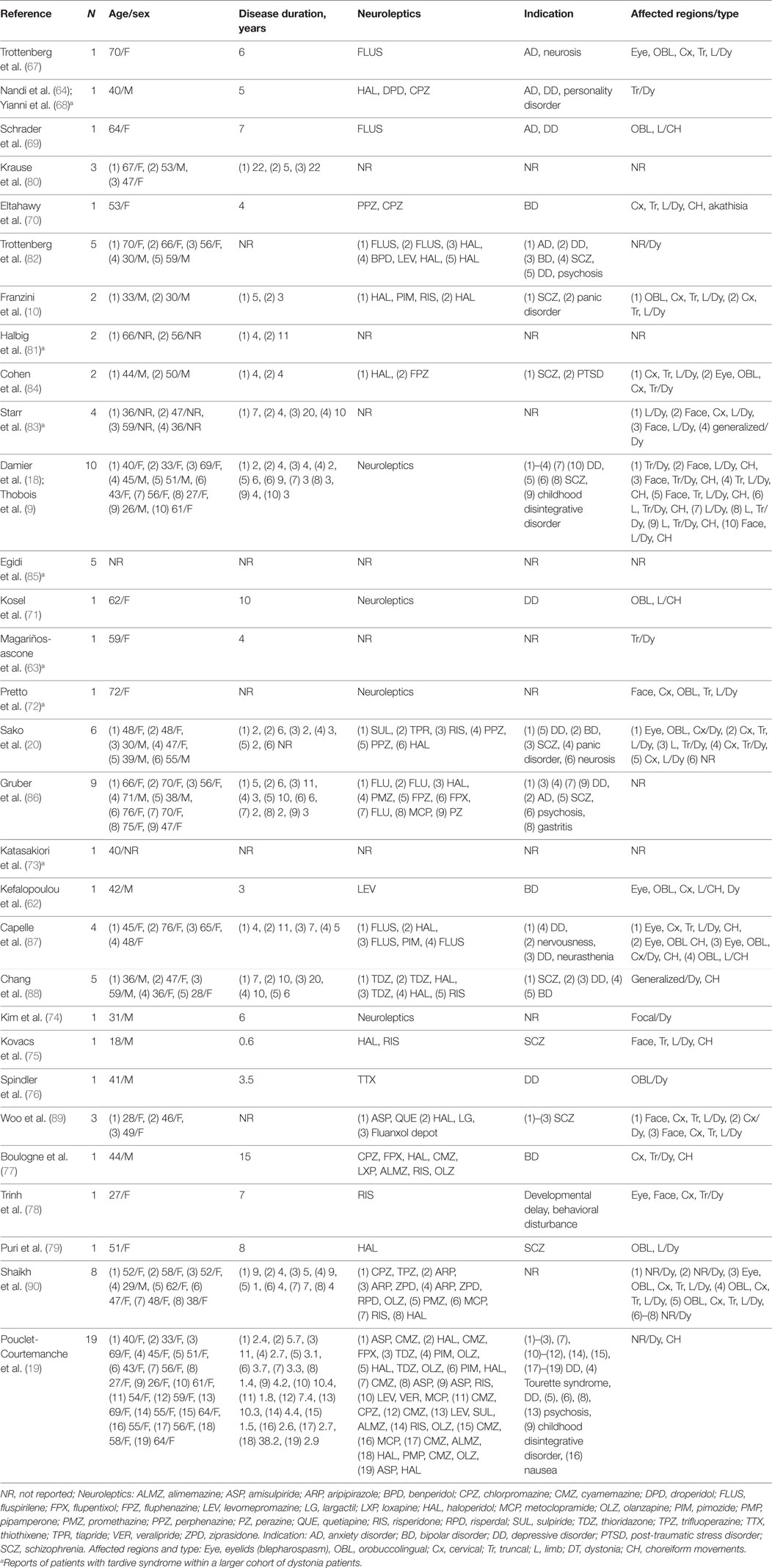
Table 2. Reported cases of GPi-DBS in patients with tardive syndrome.
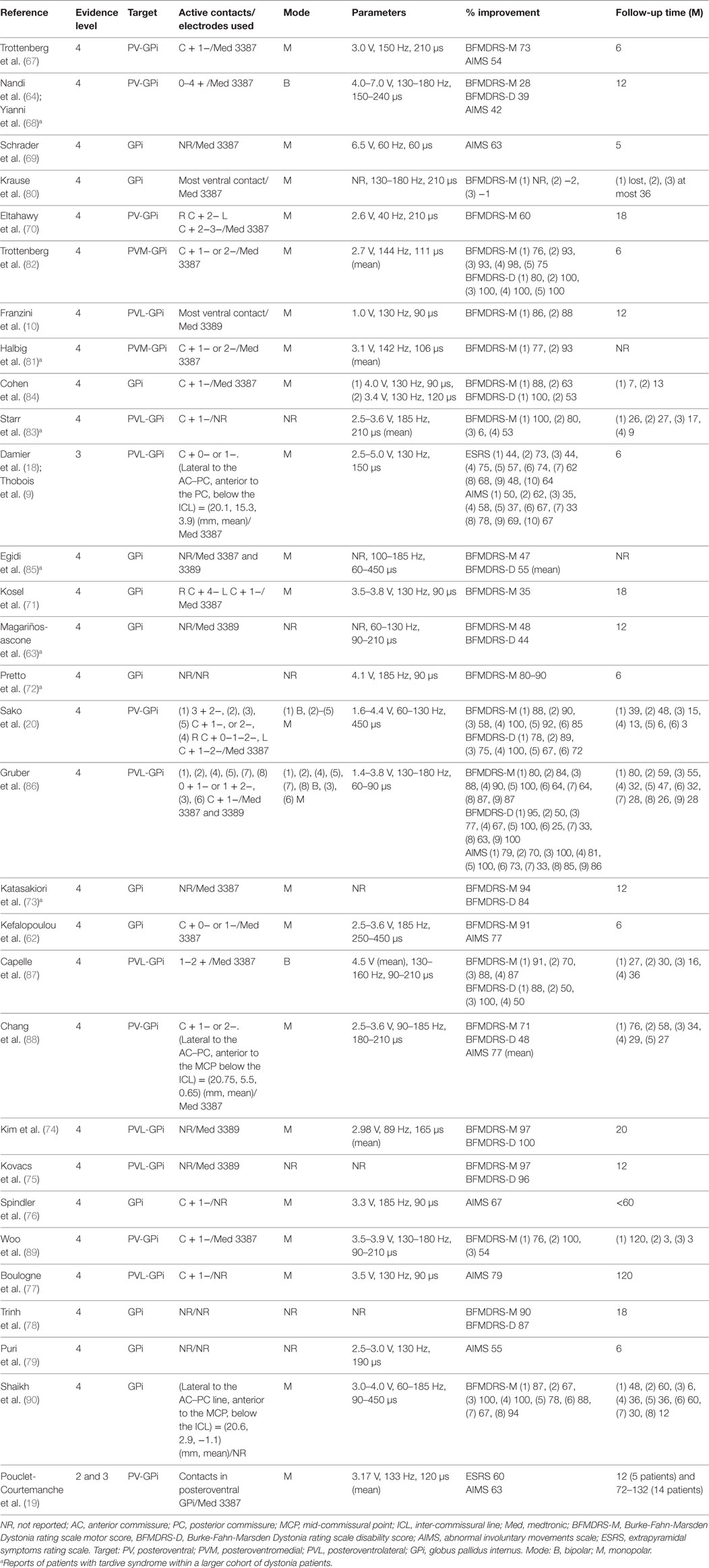
Table 3. Detailed information about GPi-DBS in patients with tardive syndrome.
Current Use of GPi-DBS in TDS
Multiple single case reports (62–64, 67–79) and open-labeled small case series (10, 20, 80–90) have shown that GPi-DBS could be highly effective in the treatment of patients with medically intractable TDS (see Tables 2 and 3). Recently, a class II study provided level C evidence for positive effects of GPi-DBS in TDS therapy (19). Here, we introduce the current state of GPi-DBS use in the treatment of patients with TDS.
Patient Selection
Selection of candidates for GPi-DBS is a critical step for obtaining good outcome results and for avoiding adverse events. The primary inclusion criterion is that patients experience medically intractable and markedly disabling motor symptoms associated with TDS. According to the criteria proposed by The French Stimulation for TDD (STARDYS), which might so far be the most rigorous and strict, DBS should only be considered for patients with persistent (>1 year) and severely disabling TDS, for whom treatment with clozapine or tetrabenazine at their maximum tolerable dosages had been attempted for at least 4 weeks (18, 19). The exclusion criteria are essentially the same as those applied to patients with primary dystonias, which include marked cognitive impairment, acute psychiatric changes, severe depression, and other coexisting medical disorders that would increase the surgical risk (86, 91). To predict the potential risks inherent to the surgical procedures, a preoperative brain MRI should be performed to check for the presence of brain atrophy and/or other organic lesions. It is also important to evaluate if the psychiatric conditions of the patient are satisfactorily stable with the current medication, for at least several months prior to the surgery and to confirm the ability to provide consent for the surgical procedure (18, 19).
GPi Target Determination
So far, bilateral DBS targeting the posteroventral part of the GPi has been used in patients with TDS (see Table 3). More specifically, the posteroventrolateral part of the GPi was chosen as the optimal target in most previous reports (9, 10, 18, 74, 75, 77, 83, 86, 87) but the posteroventromedial part of the GPi was also targeted in two reports (67, 81) (Figures 1A–C). Ventral two-thirds of the posterior GPi is the primary motor cortex-related territory that shows a somatotopic organization (Figure 1C) (92). The supplementary motor area-related territory locates more dorsal and anterior to the motor cortex-related territory (92). Dorsal one-third of the posterior GPi is the prefrontal cortex-related territory, while the most medial part of the GPi corresponds to the limbic cortex-related territory (92). Imaging with stereotactic MRI or CT-MRI fusion method is usually employed to define the anatomical targets (76). The stereotactic coordinates for the GPi are 19–22 mm lateral to the anterior commissure–posterior commissure line, 2–4 mm anterior to the mid-commissural point, and 4–6 mm inferior to the intercommissural line (10, 62, 63, 70, 73, 74, 80–82, 84, 86, 87, 89). Pouclet-Courtemanche et al. suggested that the locations of active electrodes as far as they were positioned within the posterolateral part of the ventral GPi might not be optimal in terms of clinical benefit (19). In a previous case report, a target that was 1–2 mm above, 1.5 mm rostral, and 2 mm medial to the usual target in dystonia was chosen to selectively stimulate the facial area (71). However, this single case was an exception because, as shown in Table 3, the GPi active contacts that are usually used are the same as in primary dystonia. During surgery, MERs are often used to detect neuronal discharges in the GPi. Intraoperative macrostimulation has also been used to assess the therapeutic effects of DBS and to determine thresholds for capsular stimulation and visual phosphene detection (76).
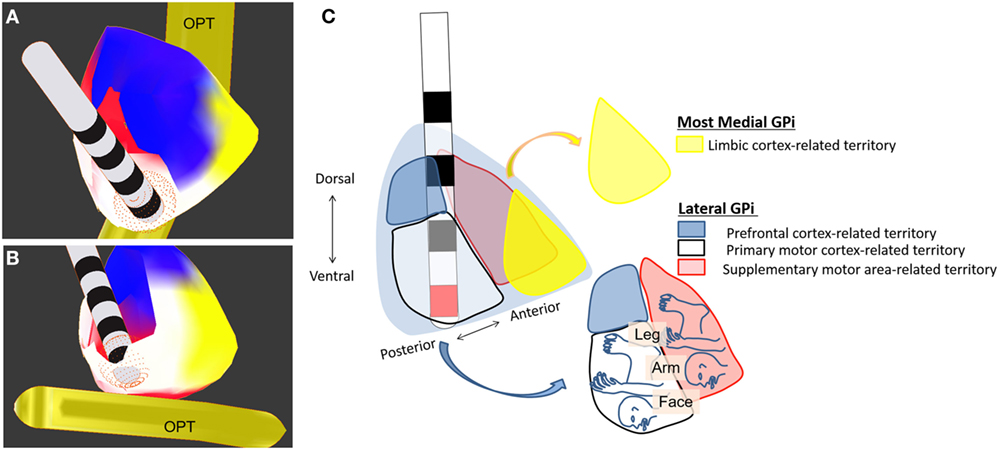
Figure 1. Deep brain stimulation of the globus pallidus internus (GPi). Electrodes are placed in the ventroposterolateral part of the GPi (the posterodorsolateral part of the GPi is partially removed). (A) Dorsoposterior view of the GPi. (B) Ventroposterior view of the GPi. (C) Schematic drawing of GPi-DBS with active contact (red) placed within the posteroventrolateral GPi. Colors indicate the territories receiving limbic- (yellow), prefrontal- (blue), motor- (white), and supplementary motor (red) cortex-related inputs. OPT, optic tract.
Stimulating Paradigms
Postoperatively, most ventral contacts of the DBS leads located within the GPi were usually used with the monopolar stimulating modes (9, 10, 16, 18–20, 62, 67, 69–71, 74, 76, 77, 80–82, 84–86, 88–90), and rarely with the bipolar modes (20, 64, 68, 86, 87) (Table 3). Stimuli were applied with amplitudes ranging from 2.7 to 4.5 V and a high frequency setting (>100 Hz) with a pulse width of 60–240 µs; alternatively, low frequency stimulation (<100 Hz) with a pulse width of 120–450 µs was also often applied (10, 18–20, 67, 68, 71, 74, 76, 77, 79, 84–90) (Table 3). The stimulation parameters used in TDS were similar to those applied in primary dystonia.
Effects on Motor Symptoms
Data from the STARDYS study group (18, 19) have verified the beneficial effects of bilateral GPi-DBS in patients with TDS. Following a prospective multicenter trial using double-blind evaluations at 6 months after surgery, reports showed that in all patients, the extrapyramidal symptoms rating scale (ESRS) scores decreased to less than 60% of the preoperative baseline, and that there was a 49% reduction of the total ESRS scores in the stimulation “on” conditions when compared to the “off” conditions. Pouclet-Coutemanche et al. showed that this therapeutic impact remained at 12 months after surgery, with a 58% (p < 0.0001) decrease of the total ESRS scores and a 50% (p < 0.0001) decrease of the total AIMS scores (19). Given the results obtained from the study with long-term (6–11 years) follow-up with the patients (n = 14), they also reported a persistent improvement of TDS patients’ conditions, with a 60 and 63% decrease from preoperative baselines in the total ESRS and AIMS scores, respectively (19).
Multiple case reports document that TDS-associated motor symptoms could be alleviated immediately or within a few days after the GPi-DBS was initiated (10, 67, 75, 77, 81, 82, 84, 86, 87). Among the TDS symptoms, choreiform dyskinesia tended to respond to DBS earlier than tonic postural dystonia, which gradually improved over weeks or months (18, 68, 70, 77, 84, 87, 88). Therapeutic efficacy of GPi-DBS seemed to be higher in the choreiform and dystonic movements than in the fixed dystonias (10, 20, 86, 90). Shaikh et al. reported that meaningful improvements in neck and truncal dystonias were most challenging to achieve, but could develop gradually over 48 months after the stimulation was initiated (90). Prospective studies with blind assessments also showed that GPi-DBS could alleviate TDS symptoms regardless of their subtypes (e.g., chorea and dystonia) or body distributions (18, 19).
The beneficial effects from GPi-DBS could produce an improvement in daily life activities in patients with disabilities due to TDS. Using the Burke-Fahn-Marsden Dystonia rating scale (BFMDRS), a systematic review showed that GPi-DBS produced a 74% improvement of disability scores (p < 0.0001) (93). Using the 36-item Short Form General Health Survey, Gruber et al. also reported a 46% improvement in total subscores for physical health (86). However, a prospective study using Lehman quality of life (QOL) Interview showed no significant change in QOL before and 6 months after surgery in seven patients with TDS (19).
Effects on Non-Motor Symptoms
Two separate case series reports (71, 86) show that GPi-DBS produced a significant improvement of mood in patients with TDS, as determined by the Hamilton rating scale for depression, the Beck Depression Inventory Score, and the Montgomery-Åsberg Depression rating scale (MADRS). However, a prospective study on 16–19 patients found that the mean scores of both the MADRS and the positive and negative syndrome scale did not change significantly up to 1 year after surgery (19).
Gruber et al. reported no significant change in cognitive functions of nine patients before and after surgery, as determined by the Mattis Dementia rating scale (MDRS), the Multiple Wording Test part B, the Rey Auditory Verbal Learning Test, and the digit span task (86). Pouclet-Courtemanche et al. published a prospective study on 16–19 patients to show the results of neuropsychological tests using Mini-Mental State Examination (MMSE), the Frontal Assessment Battery (FAB), and the MDRS (19). They found that there were no significant changes in the mean scores of both the MMSE and FAB up to 1 year after surgery, while the mean scores of the MDRS improved at 3 months and persisted for 1 year after surgery (p < 0.05). Thus, it is likely that in TDS patients, GPi-DBS might not exert a negative impact on QOL, mood, or cognition.
Adverse Events
The overall complication rate of GPi-DBS for TDS is 9%, which is almost equivalent to that of GPi-DBS for other movement disorders (76, 94). There are no reports of death related to DBS in patients with TDS (95). However, a potential risk of suicide after GPi-DBS surgery has been suggested in patients with TDS (76, 96). Complications relating to the implanted DBS devices in patients with TDS, such as displacement and misplacement of the DBS leads, have also been noted (19, 87). Pouclet-Courtemanche et al. reported other complications that include dysfunction of the active contacts, painful traction by the cable connection, and sudden stopping of the stimulator (19). Surgery-related infection (80, 88) and venous infarction (83, 88) have also been documented. Electrostimulation-dependent complications, such as paresthesia, shuffling gait, decreased sensitivity for precise and skillful movements, muscular contractions, phosphenes, scotoma, and dysarthria, have also been reported (67, 69, 80, 82, 87), although they were transient and addressed by adjustment of settings. Concerning psychiatric issues, Trottenberg et al. reported that one of five patients with schizophrenia manifested a psychotic relapse 6 months after surgery (82). Pouclet-Courtemanche et al. reported that within 1 year after surgery, 8 of 19 patients experienced adverse psychiatric events that included depression, anxiety, manic states, delirium, agitation, and aggressiveness, although mental health was successfully restored with medical treatments (19).
Could STN-DBS Be a New Target for TDS?
Two separate case series reports document that STN-DBS produced striking improvement of motor symptoms in patients with TDS, as determined by the BFMDRS (65, 66). The average score improved by 89% compared to the baseline. Sun et al. reported that STN-DBS produced immediate symptomatic improvement, using lower stimulation parameters with longer battery life (66). They proposed that STN-DBS might enable better symptomatic control over GPi-DBS. However, there is currently a lack of head-to-head comparison between GPi and STN stimulation for primary dystonia and TDS (97). Furthermore, the effects of STN-DBS on the non-motor symptoms of TDS patients are still unknown. Several recent meta-analyses comparing the effects of GPi-DBS with STN-DBS in patients with Parkinson’s disease concluded that the risk of worsening depression with GPi-stimulated patients was the same or even smaller than that with STN-stimulated patients (98–101). A selective decline in cognitive functions with STN-DBS has also been highlighted in almost all the meta-analyses (98–103). These observations could be in part attributed to the reduction in dopaminergic drugs for STN-stimulated patients (99, 100, 104). Given the extent of dopamine withdrawal in STN-stimulated patients in Parkinson’s disease, the results of studies comparing GPi and STN stimulations in these patients cannot be directly applied to primary dystonia or TDS. Therefore, well-designed randomized controlled trials will be required to select better targets for patients with dystonia, including those with TDS.
Summary
Globus pallidus internus-DBS results in promising and continuous improvement in motor function over months and possibly years, which may persist over 6–11 years in patients with TDS. There is no available evidence to demonstrate that GPi-DBS negatively impacts QOL, mood, or cognition in patients with TDS. The complication rate of GPi-DBS for TDS is almost equivalent to that of GPi-DBS for other movement disorders. To obtain a higher level of clinical evidence about the precise efficacy of GPi-DBS in reducing TDS, more well-designed double-blind trials are needed. In particular, it is important to clarify specific inclusion criteria for patient selection. One of the particular questions to be addressed in the near future is a comparison of STN-DBS and GPi-DBS efficacy in patients with TDS.
Author Contributions
The conception or design of the work: RM and SG. The acquisition, analysis, or interpretation of data for the work: RM and HM. Drafting the work: RM and SG. Revising the work critically for important intellectual content: SN, RK, and SG. Final approval of the version to be published and agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: RM, HM, SN, RK, and SG.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewers H-KM and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Funding
This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (grants-in-aid for Scientific Research no. 24390223, 26461272, 26430054, and 16k10788), and Japan Agency for Medical Research and Development (AMED) (no. 16ek0109182h0001).
References
1. Owens DGC. Tardive Dyskinesia. A Guide to the Extrapyramidal Side-Effects of Antipsychotic Drugs. Cambridge, UK: Cambridge University Press (1999). p. 166–226.
2. Lerner PP, Miodownik C, Lerner V. Tardive dyskinesia (syndrome): current concept and modern approaches to its management. Psychiatry Clin Neurosci (2015) 69:321–34. doi: 10.1111/pcn.12270
3. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association (2013). 712 p.
4. Schooler NR, Kane JM. Research diagnosis for tardive dyskinesia. Arch Gen Psychiatry (1982) 39:486–7. doi:10.1001/archpsyc.1982.04290040080012
5. Bhidayasiri R, Fahn S, Weiner WJ, Gronseth GS, Sullivan K, Zesiewicz TA, et al. Evidence-based guideline: treatment of tardive syndromes: report of the guideline development subcommittee of the American Academy of Neurology. Neurology (2013) 81:463–9. doi:10.1212/WNL.0b013e31829d86b6
6. Aia PG, Revuelta GJ, Cloud LJ, Factor SA. Tardive dyskinesia. Curr Treat Options Neurol (2011) 13:231–41. doi:10.1007/s11940-011-0117-x
7. Lerner V, Miodownik C. Motor symptoms of schizophrenia: is tardive dyskinesia a symptom or side effect? A modern treatment. Curr Psychiatry Rep (2011) 13:295–304. doi:10.1007/s11920-011-0202-6
8. Aquino CCH, Lang AE. Tardive dyskinesia syndromes: current concepts. Parkinsonism Relat Disord (2014) 20:s113–7. doi:10.1016/S1353-8020(13)70028-2
9. Thobois S, Ballanger B, Xie-Brustolin J, Damier P, Durif F, Azulay JP, et al. Globus pallidus stimulation reduces frontal hyperactivity in tardive dystonia. J Cereb Blood Flow Metab (2008) 28:1127–38. doi:10.1038/sj.jcbfm.9600610
10. Franzini A, Marras C, Ferroli P, Zorzi G, Bugiani O, Romito L, et al. Long-term high-frequency bilateral pallidal stimulation for neuroleptic-induced tardive dystonia. Report of two cases. J Neurosurg (2005) 102:721–5. doi:10.3171/jns.2005.102.4.0721
11. Bhidayasiri R, Boonyawairoj S. Spectrum of tardive syndromes: clinical recognition and management. Postgrad Med J (2011) 87:132–41. doi:10.1136/pgmj.2010.103234
12. Geddes J, Freemantle N, Harrison P, Bebbington P. Atypical antipsychotics in the treatment of schizophrenia: systematic overview and meta-regression analysis. BMJ (2000) 321:1371–6. doi:10.1136/bmj.321.7273.1371
13. O’Brien A. Comparing the risk of tardive dyskinesia in older adults with first-generation and second-generation antipsychotics: a systematic review and meta-analysis. Int J Geriatr Psychiatry (2016) 31:683–93. doi:10.1002/gps.4399
14. Najib J. Tardive dyskinesia: a review and current treatment options. Am J Ther (1999) 6:51–60. doi:10.1097/00045391-199901000-00008
15. Kenney C, Hunter C, Jankovic J. Long-term tolerability of tetrabenazine in the treatment of hyperkinetic movement disorders. Mov Disord (2007) 22:193–7. doi:10.1002/mds.21222
16. Thobois S, Poisson A, Damier P. Surgery for tardive dyskinesia. Int Rev Neurobiol (2011) 98:289–96. doi:10.1016/B978-0-12-381328-2.00012-2
17. Trugman JM, Leadbetter R, Zalis ME, Burgdorf RO, Wooten FG. Treatment of severe axial tardive dystonia with clozapine: case report and hypothesis. Mov Disord (1994) 9:441–6. doi:10.1002/mds.870090411
18. Damier P, Thobois S, Witjas T, Cuny E, Derost P, Raoul S, et al. Bilateral deep brain stimulation of the globus pallidus to treat tardive dyskinesia. Arch Gen Psychiatry (2007) 64:170–6. doi:10.1001/archpsyc.64.2.170
19. Pouclet-Courtemanche H, Rouaud T, Thobois S, Nguyen JM, Brefel-Courbon C, Chreau I, et al. Long-term efficacy and tolerability of bilateral pallidal stimulation to treat tardive dyskinesia. Neurology (2016) 86:651–9. doi:10.1212/WNL.0000000000002370
20. Sako W, Goto S, Shimazu H, Murase N, Matsuzaki K, Tamura T, et al. Bilateral deep brain stimulation of the globus pallidus internus in tardive dystonia. Mov Disord (2008) 23:1929–31. doi:10.1002/mds.22100
21. Loonen AJ, Ivanova SA. New insights into the mechanism of drug-induced dyskinesia. CNS Spectr (2013) 18:15–20. doi:10.1017/S1092852912000752
22. Clow A, Jenner P, Theodorou A, Mardsen CD. Striatal dopamine receptors become supersensitive while rats are given trifluperazine for 6 months. Nature (1979) 278:59–67. doi:10.1038/278059a0
23. Waddington JL, Cross AJ, Gambele SJ, Bourne RC. Spontaneous orofacial dyskinesia and dopaminergic function in rats after 6 months of neuroleptic treatment. Science (1983) 220:530–2. doi:10.1126/science.6132447
24. Ellison G, See RE. Rats administered chronic neuroleptics develop oral movements which are similar in form to those in humans with tardive dyskinesia. Psychopharmacology (Berl) (1989) 98:564–6. doi:10.1007/BF00441962
25. Turrone P, Remington G, Kapur S, Nobrega JN. The relationship between dopamine D2 receptor occupancy and the vacuous chewing movement syndrome in rats. Psychopharmacology (Berl) (2003) 165:166–71. doi:10.1007/s00213-002-1259-z
26. Silvestri S, Seeman MV, Negrete JC, Houle S, Shammi CM, Remington GJ, et al. Increased dopamine D2 receptor binding after long-term treatment with antipsychotics in humans: a clinical PET study. Psychopharmacology (Berl) (2000) 152:174–80. doi:10.1007/s002130000532
27. Teo JT, Edwards MJ, Bhatia K. Tardive dyskinesia is caused by maladaptive synaptic plasticity: a hypothesis. Mov Disord (2012) 27:1205–15. doi:10.1002/mds.25107
28. Tsai G, Goff DC, Chang RW, Flood J, Baer L, Coyle JT. Makers of glutamatergic neurotransmission and oxidative stress associated with tardive dyskinesia. Am J Psychiatry (1998) 155:1207–13. doi:10.1176/ajp.155.9.1207
29. Lu RB, Ko HC, Lin WL, Lin YT, Ho SL. CSF neurochemical study of tardive dyskinesia. Biol Psychiatry (1989) 25:717–24. doi:10.1016/0006-3223(89)90243-6
30. Kelley JJ, Roberts RC. Effects of haloperidol on cholinergic striatal interneurons: relationship to oral dyskinesias. J Neural Transm (Vienna) (2004) 111:1075–91. doi:10.1007/s00702-004-0131-1
31. Bordia T, McIntosh JM, Quik M. Nicotine reduces antipsychotic-induced orofacial dyskinesia in rats. J Pharmacol Exp Ther (2012) 340:612–9. doi:10.1124/jpet.111.189100
32. Egan MF, Hurd Y, Hyde TM, Weinberger DR, Wyatt RJ, Kleinman JE. Alterations in mRNA levels of D2 receptors and neuropeptides in striatonigral and striatopallidal neurons of rats with neuroleptic-induced dyskinesias. Synapse (1994) 18:178–89. doi:10.1002/syn.890180303
33. Egan MF, Hurd Y, Ferguson J, Bachus SE, Hamid EH, Hyde TM. Pharmacological and neurochemical differences between acute and tardive vacuous chewing movements induced by haloperidol. Psychopharmacology (Berl) (1996) 127:337–45. doi:10.1007/s002130050095
34. Grimm JW, Chapman MA, Zahm DS, See RE. Decreased choline acetyltransferase immunoreactivity in discrete striatal subregions following chronic haloperidol in rats. Synapse (2001) 39:51–7. doi:10.1002/1098-2396(20010101)39:1<51:AID-SYN7>3.0.CO;2-Z
35. Hori H, Ohmori O, Shinkai T, Kojima H, Okano C, Suzuki T, et al. Manganese superoxide dismutase gene polymorphism and schizophrenia: relation to tardive dyskinesia. Neuropsychopharmacology (2000) 23:170–7. doi:10.1016/S0893-133X(99)00156-6
36. Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurothetherapeutics (2014) 11:166–76. doi:10.1007/s13311-013-0222-5
37. Cadet JL, Perumal AS. Chronic treatment with prolixin causes oxidative stress in rat brain. Biol Psychiatry (1990) 28:738–40. doi:10.1016/0006-3223(90)90461-A
38. Cohen G, Spina MB. Deprenyl suppresses the oxidant stress associated with increased dopamine turnover. Ann Neurol (1989) 26:689–90. doi:10.1002/ana.410260518
39. Shivakumar BR, Ravindranath V. Oxidative stress induced by administration of the neuroleptic drug haloperidol is attenuated by higher doses of haloperidol. Brain Res (1992) 595:256–62. doi:10.1016/0006-8993(92)91058-M
40. Cho CH, Lee HJ. Oxidative stress and tardive dyskinesia: pharmacogenetic evidence. Prog Neuropsychopharmacol Biol Psychiatry (2013) 46:207–13. doi:10.1016/j.pnpbp.2012.10.018
41. Elkashef AM, Wyatt RJ. Tardive dyskinesia: possible involvement of free radicals and treatment with vitamine E. Schizophr Bull (1999) 25:731–40. doi:10.1093/oxfordjournals.schbul.a033414
42. Sachdev P, Saharov T, Cathcart S. The preventive role of antioxidants (selegiline and vitamine E) in a rat model of tardive dyskinesia. Biol Psychiatry (1999) 15:1672–81. doi:10.1016/S0006-3223(99)00091-8
43. Bartels M, Themelis J. Computerized tomography in tardive dyskinesia. Evidence of structural abnormalities in the basal ganglia system. Arch Psychiatr Nervenkr (1970) 233:371–9. doi:10.1007/BF00346087
44. Mion CC, Andreasen NC, Arndt S, Swayze VW II, Cohen GA. MRI abnormalities in tardive dyskinesia. Psychiatry Res (1991) 40:157–66. doi:10.1016/0925-4927(91)90007-D
45. Sarró S, Pomarol-Clotet E, Canales-Rodríguez EJ, Salvador R, Gomar JJ, Ortiz-Gil J, et al. Structural brain changes associated with tardive dyskinesia in schizophrenia. Br J Psychiatry (2013) 203:51–7. doi:10.1192/bjp.bp.112.114538
46. Gałecki P, Pietras T, Szemraj J. Manganese superoxide dismutase gene (MnSOD) polymorphism in schizophrenics with tardive dyskinesia from central Poland. Psychiatr Pol (2006) 40:937–48.
47. Hitzeroth A, Niehaus DJ, Koen L, Botes WC, Deleuze JF, Warnich L. Association between the MnSOD Ala-9Val polymorphism and development of schizophrenia and abnormal involuntary movements in the Xhosa population. Prog Neuropsychopharmacol Biol Psychiatry (2007) 31:664–72. doi:10.1016/j.pnpbp.2006.12.019
48. Bakker PR, van Harten PN, van Os J. Antipsychotic-induced tardive dyskinesia and polymorphic variations in COMT, DRD2, CYT1A2 and MnSOD genes: a meta-analysis of pharmacogenetic interactions. Mol Psychiatry (2008) 13:544–56. doi:10.1038/sj.mp.4002142
49. Kang SG, Choi JE, An H, Park YM, Lee HJ, Han C, et al. Manganese superoxide dismutase gene Ala-9Val polymorphism might be related to the severity of abnormal involuntary movements in Korean schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry (2008) 32:1844–7. doi:10.1016/j.pnpbp.2008.08.013
50. Whitty PF, Owoeye O, Waddington JL. Neurological signs and involuntary movements in schizophrenia: intrinsic to and informative on systems pathobiology. Schizophr Bull (2009) 35:415–24. doi:10.1093/schbul/sbn126
51. Ferentinos P, Dikeos D. Genetic correlates of medial comorbidity associated with schizophrenia and treatment with antipsychotics. Curr Opin Psychiatry (2012) 25:381–90. doi:10.1097/YCO.0b013e3283568537
52. Souza RP, Remington G, Chowdhury NI, Lau MK, Voineskos AN, Lieberman JA, et al. Association study of the GSK-3B gene with tardive dyskinesia in European Caucasians. Eur Neuropsychopharmacol (2010) 20:688–94. doi:10.1016/j.euroneuro.2010.05.002
53. Ethier I, Kagechika H, Shudo K, Rouillard C, Lévesque D. Docosahexaenoic acid reduces haloperidol-induced dyskinesias in mice: involvement of Nurr77 and retinoid receptors. Biol Psychiatry (2004) 56:522–6. doi:10.1016/j.biopsych.2004.06.036
54. Novak G, Gallo A, Zai CC, Meltzer HY, Lieberman JA, Potkin SG, et al. Association of the orphan nuclear receptor NR4A1 with tardive dyskinesia. Psychiatr Genet (2010) 20:39–43. doi:10.1097/YPG.0b013e3283351221
55. Crowley JJ, Adkins DE, Pratt AL, Quackenbush CR, van den Oord EJ, Moy SS, et al. Antipsychotic-induced vacuous chewing movements and extrapyramidal side effects are highly heritable in mice. Pharmacogenomics J (2012) 12:147–55. doi:10.1038/tpj.2010.82
56. Crowley JJ, Kim Y, Szatkiewicz JP, Pratt AL, Quackenbush CR, Adkins DE, et al. Genome-wide association mapping of loci for antipsychotic-induced extrapyramidal symptoms in mice. Mamm Genome (2012) 23:322–35. doi:10.1007/s00335-011-9385-8
57. Éthier I, Kagechika H, Shudo K, Rouillard C, Lévesque D. Docosahexaenoic acid reduced haloperidol-induced dyskinesia in mice: involvement of Nurr77 and retinoid receptors. Biol Psychiatry (2004) 56:522–6. doi:10.1016/j.biopsych.2004.06.036
58. Su CJ, Xu XQ, Fan Y, Du RH, Hu G. Aquaporin-4 knockout abolishes apomorphine-induced tardive dyskinesia following chronic treatment with neuroleptis. CNS Neurosci Ther (2012) 18:1024–6. doi:10.1111/cns.12020
59. Carta AR, Gerfen CR. Lack of a role for the D3 receptor in clozapine induction of c-fos demonstrated in D3 dopamine receptor-deficient mice. Neuroscience (1999) 90:1021–9. doi:10.1016/S0306-4522(98)00498-9
60. Robertson GS, Lee CJ, Sridhar K, Nakabeppu Y, Cheng M, Wang YM, et al. Clozapine-, but not haloperidol-, induced increases in ΔFosB-like immunoreactivity are completely blocked in the striatum of mice lacking D3 dopamine receptors. Eur J Neurosci (2004) 20:3189–94. doi:10.1111/j.1460-9568.2004.03774.x
61. Blanchet PJ, Parent MT, Rompré PH, Lévesque D. Relevance of animal models to human tardive dyskinesia. Behav Brain Funct (2012) 8:12. doi:10.1186/1744-9081-8-12
62. Kefalopoulou Z, Paschali A, Markaki E, Vassilakos P, Ellul J, Constantoyannis C. A double-blind study on a patient with tardive dyskinesia treated with pallidal deep brain stimulation. Acta Neurol Scand (2009) 119:269–73. doi:10.1111/j.1600-0404.2008.01115.x
63. Magariños-ascone CM, Regidor I, Gómez-galán M, Cabañes-martínez L, Figueiras-méndez R. Deep brain stimulation in the globus pallidus to treat dystonia: electrophysiological characteristics and 2 years’ follow-up in 10 patients. Neuroscience (2008) 152:558–71. doi:10.1016/j.neuroscience.2008.01.001
64. Nandi D, Parkin S, Scott R, Winter JL, Joint C, Gregory R, et al. Camptocormia treated with bilateral pallidal stimulation: case report. Neurosurg Focus (2002) 12:1–6. doi:10.3171/foc.2002.12.2.5
65. Zhang JG, Zhang K, Wang ZC. Deep brain stimulation in the treatment of tardive dystonia. Chin Med J (2006) 5:789–92.
66. Sun B, Chen S, Zhan S, Le W, Krahl SE. Subthalamic nucleus stimulation for primary dystonia and tardive dystonia. Acta Neurochir Suppl (2007) 97:207–14. doi:10.1007/978-3-211-33081-4_23
67. Trottenberg T, Paul G, Meissner W, Maier-Hauff K, Taschner C, Kupsch A. Pallidal and thalamic neurostimulation in severe tardive dystonia. J Neurol Neurosurg Psychiatry (2001) 70:557–9. doi:10.1136/jnnp.70.4.557
68. Yianni J, Bain P, Giladi N, Auca M, Gregory R, Joint C, et al. Globus pallidus internus deep brain stimulation for dystonic conditions: a prospective audit. Mov Disord (2003) 18:436–42. doi:10.1002/mds.10380
69. Schrader C, Peschel T, Petermeyer M, Dengler R, Hellwig D. Unilateral deep brain stimulation of the internal globus pallidus alleviates tardive dyskinesia. Mov Disord (2004) 19:583–5. doi:10.1002/mds.10705
70. Eltahawy HA, Feinstein A, Khan F, Saint-Cyr J, Lang AE, Lozano AM. Bilateral globus pallidus internus deep brain stimulation in tardive dyskinesia: a case report. Mov Disord (2004) 19:969–72. doi:10.1002/mds.20092
71. Kosel M, Sturm V, Frick C, Lenartz D, Zeidler G, Brodesser D, et al. Mood improvement after deep brain stimulation of the internal globus pallidus for tardive dyskinesia in a patient suffering from major depression. J Psychiatr Res (2007) 41:801–3. doi:10.1016/j.jpsychires.2006.07.010
72. Pretto TE, Dalvi A, Kang UJ, Penn RD. A prospective blinded evaluation of deep brain stimulation for the treatment of secondary dystonia and primary torticollis syndromes. J Neurosurg (2008) 109:405–9. doi:10.3171/JNS/2008/109/9/0405
73. Katasakiori PF, Kefalopoulou Z, Markaki E, Paschali A, Ellul J, Kagadis GC, et al. Deep brain stimulation for secondary dystonia: results in 8 patients. Acta Neurochir (Wien) (2009) 151:473–8. doi:10.1007/s00701-009-0281-x
74. Kim JP, Chang WS, Chang JW. Treatment of secondary dystonia with a combined stereotactic procedure: long-term surgical outcomes. Acta Neurochir (Wien) (2011) 153:2319–28. doi:10.1007/s00701-011-1147-6
75. Kovacs N, Balas I, Janszky J, Simon M, Fekete S, Komoly S. Status dystonicus in tardive dystonia successfully treated by bilateral deep brain stimulation. Clin Neurol Neurosurg (2011) 113:808–9. doi:10.1016/j.clineuro.2011.08.003
76. Spindler MA, Galifianakis NB, Wilkinson JR, Duda JE. Globus pallidus interna deep brain stimulation for tardive dyskinesia: case report and review of the literature. Parkinsonism Relat Disord (2013) 19:141–7. doi:10.1016/j.parkreldis.2012.09.016
77. Boulogne S, Danaila T, Polo G, Broussolle E, Thobois S. Relapse of tardive dystonia after globus pallidus deep-brain stimulation discontinuation. J Neurol (2014) 261:1636–7. doi:10.1007/s00415-014-7404-x
78. Trinh B, Ha AD, Mahant N, Kim SD, Owler B, Fung VS. Dramatic improvement of truncal tardive dystonia following globus pallidus pars interna deep brain stimulation. J Clin Neurosci (2014) 21:515–7. doi:10.1016/j.jocn.2013.03.035
79. Puri M, Albassam A, Silver B. Deep brain stimulation in the treatment of tardive dyskinesia. Psychiatr Ann (2014) 44:123–5. doi:10.3928/00485713-20140306-02
80. Krause M, Fogel W, Kloss M, Rasche D, Volkmann J, Tronnier V. Pallidal stimulation for dystonia. Neurosurgery (2004) 55:1361–70. doi:10.1227/01.NEU.0000143331.86101.5E
81. Hälbig TD, Gruber D, Kopp UA, Schneider GH, Trottenberg T, Kupsch A. Pallidal stimulation in dystonia: effects on cognition, mood, and quality of life. J Neurol Neurosurg Psychiatry (2005) 76:1713–6. doi:10.1136/jnnp.2004.057992
82. Trottenberg T, Volkmann J, Deuschl G, Kühn AA, Schneider GH, Müller J, et al. Treatment of severe tardive dystonia with pallidal deep brain stimulation. Neurology (2005) 64:344–6. doi:10.1212/01.WNL.0000149762.80932.55
83. Starr PA, Turner RS, Rau G, Lindsey N, Heath S, Volz M, et al. Microelectrode-guided implantation of deep brain stimulators into the globus pallidus internus for dystonia: techniques, electrode locations, and outcomes. J Neurosurg (2006) 104:488–501. doi:10.3171/jns.2006.104.4.488
84. Cohen OS, Hassin-Baer S, Spiegelmann R. Deep brain stimulation of the internal globus pallidus for refractory tardive dystonia. Parkinsonism Relat Disord (2007) 13:541–4. doi:10.1016/j.parkreldis.2006.11.007
85. Egidi M, Franzini A, Marras C, Cavallo M, Mondani M, Lavano A, et al. A survey of Italian cases of dystonia treated by deep brain stimulation. J Neurosurg Sci (2007) 51:153–8.
86. Gruber D, Trottenberg T, Kivi A, Schoenecker T, Kopp UA, Hoffmann KT, et al. Long-term effects of pallidal deep brain stimulation in tardive dystonia. Neurology (2009) 73:53–8. doi:10.1212/WNL.0b013e3181aaea01
87. Capelle HH, Blahak C, Schrader C, Baezner H, Kinfe TM, Herzog J, et al. Chronic deep brain stimulation in patients with tardive dystonia without a history of major psychosis. Mov Disord (2010) 25:1477–81. doi:10.1002/mds.23123
88. Chang EF, Schrock LE, Starr PA, Ostrem JL. Long-term benefit sustained after bilateral pallidal deep brain stimulation in patients with refractory tardive dystonia. Stereotact Funct Neurosurg (2010) 88:304–10. doi:10.1159/000316763
89. Woo PY, Chan DT, Zhu XL, Yeung JH, Chan AY, Au AC, et al. Pallidal deep brain stimulation: an effective treatment in Chinese patients with tardive dystonia. Hong Kong Med J (2014) 20:455–9. doi:10.12809/hkmj134082
90. Shaikh AG, Mewes K, DeLong MR, Gross RE, Triche SD, Jinnah HA, et al. Temporal profile of improvement of tardive dystonia after globus pallidus deep brain stimulation. Parkinsonism Relat Disord (2015) 21:116–9. doi:10.1016/j.parkreldis.2014.11.013
91. Kupsch A, Benecke R, Müller J, Trottenberg T, Schneider GH, Poewe W, et al. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. N Engl J Med (2006) 355:1978–90. doi:10.1056/NEJMoa063618
92. Nambu A. Somatotopic organization of the primate basal ganglia. Front Neuroanat (2011) 5:26. doi:10.3389/fnana.2011.00026
93. Mentzel CL, Tenback DE, Tijssen MA, Visser-Vandewalle VE, van Harten PN. Efficacy and safety of deep brain stimulation in patients with medication-induced tardive dyskinesia and/or dystonia: a systematic review. J Clin Psychiatry (2012) 73:1434–8. doi:10.4088/JCP.12r07643
94. Smith KM, Spindler MA. Uncommon applications of deep brain stimulation in hyperkinetic movement disorders. Tremor Other Hyperkinet Mov (N Y) (2015) 5:278. doi:10.7916/D84X56HP
95. Sobstyl M, Ząbek M. Deep brain stimulation for intractable tardive dystonia: literature overview. Neurol Neurochir Pol (2016) 50:114–22. doi:10.1016/j.pjnns.2016.01.004
96. Foncke EM, Schuurman PR, Speelman JD. Suicide after deep brain stimulation of the internal globus pallidus for dystonia. Neurology (2006) 66:142–3. doi:10.1212/01.wnl.0000191328.05752.e2
97. Hu W, Stead M. Deep brain stimulation for dystonia. Transl Neurodegener (2014) 3:2. doi:10.1186/2047-9158-3-2
98. Sako W, Miyazaki Y, Izumi Y, Kaji R. Which target is best for patients with Parkinson’s disease? A meta-analysis of pallidal and subthalamic stimulation. J Neurol Neurosurg Psychiatry (2014) 85:982–6. doi:10.1136/jnnp-2013-306090
99. Liu Y, Li W, Tan C, Liu X, Wang X, Gui Y, et al. Meta-analysis comparing deep brain stimulation of the globus pallidus and subthalamic nucleus to treat advanced Parkinson disease. J Neurosurg (2014) 121:709–18. doi:10.3171/2014.4.JNS131711
100. Tan ZG, Zhou Q, Huang T, Jiang Y. Efficacies of globus pallidus stimulation and subthalamic nucleus stimulation for advanced Parkinson’s disease: a meta-analysis of randomized controlled trials. Clin Interv Aging (2016) 11:777–86. doi:10.2147/CIA.S105505
101. Wang JW, Zhang YQ, Zhang XH, Wang YP, Li JP, Li YJ. Cognitive and psychiatric effects of STN versus GPi deep brain stimulation in Parkinson’s disease: a meta-analysis of randomized controlled trials. PLoS One (2016) 11:e0156721. doi:10.1371/journal.pone.0156721
102. Combs HL, Folley BS, Berry DT, Segerstrom SC, Han DY, Anderson-Mooney AJ, et al. Cognition and depression following deep brain stimulation of the subthalamic nucleus and globus pallidus pars internus in Parkinson’s disease: a meta-analysis. Neuropsychol Rev (2015) 25:439–54. doi:10.1007/s11065-015-9302-0
103. Rothlind JC, York MK, Carlson K, Luo P, Marks WJ Jr, Weaver FM, et al. Neuropsychological changes following deep brain stimulation surgery for Parkinson’s disease: comparisons of treatment at pallidal and subthalamic targets versus best medical therapy. J Neurol Neurosurg Psychiatry (2015) 86:622–9. doi:10.1136/jnnp-2014-308119
Keywords: deep brain stimulation, globus pallidus internus, antipsychotic agents, abnormal involuntary movements, tardive dyskinesia, tardive syndrome, secondary dystonia, pathophysiology
Citation: Morigaki R, Mure H, Kaji R, Nagahiro S and Goto S (2016) Therapeutic Perspective on Tardive Syndrome with Special Reference to Deep Brain Stimulation. Front. Psychiatry 7:207. doi: 10.3389/fpsyt.2016.00207
Received: 07 September 2016; Accepted: 15 December 2016;
Published: 26 December 2016
Edited by:
Paul Croarkin, Mayo Clinic, USAReviewed by:
Hoon-Ki Min, Mayo Clinic, USAOsama Ali Abulseoud, National Institute on Drug Abuse (NIDA), USA
Copyright: © 2016 Morigaki, Mure, Kaji, Nagahiro and Goto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Satoshi Goto, c2dvdG9AdG9rdXNoaW1hLXUuYWMuanA=