Abstract
Norepinephrine (NE) is recognized as having a key role in the pathophysiology of major depressive disorder (MDD) and schizophrenia, although its distinct actions via α-adrenergic receptors (α-ARs) are not well defined. We performed a systematic review examining the roles of NE and α-ARs in MDD and schizophrenia. PubMed and ProQuest database searches were performed to identify English language papers published between 2008 and 2015. In total, 2,427 publications (PubMed, n = 669; ProQuest, n = 1,758) were identified. Duplicates, articles deemed not relevant, case studies, reviews, meta-analyses, preclinical reports, or articles on non-target indications were excluded. To limit the review to the most recent data representative of the literature, the review further focused on publications from 2010 to 2015, which were screened independently by all authors. A total of 16 research reports were identified: six clinical trial reports, six genetic studies, two biomarker studies, and two receptor studies. Overall, the studies provided indirect evidence that α-AR activity may play an important role in aberrant regulation of cognition, arousal, and valence systems associated with MDD and schizophrenia. Characterization of the NE pathway in patients may provide clinicians with information for more personalized therapy of these heterogeneous diseases. Current clinical studies do not provide direct evidence to support the role of NE α-ARs in the pathophysiology of MDD and schizophrenia and in the treatment response of patients with these diseases, in particular with relation to specific valence systems. Clinical studies that attempt to define associations between specific receptor binding profiles of psychotropics and particular clinical outcomes are needed.
Introduction
Major depressive disorder (MDD) and schizophrenia affect 151.2 and 26.3 million people worldwide, respectively, and are associated with high rates of morbidity and mortality (1). These disorders exert a large mental health burden and socioeconomic cost across the world, accounting for 24.5 and 5.3%, respectively, of disability-adjusted life-years attributed to all mental, neurological, and substance use disorders (2). The catecholamine neurotransmitter, norepinephrine (NE), has been speculated as having a role in depressive disorders since the 1950s (3), and schizophrenia since the 1970s (4), although a full understanding of the actions of NE in the pathophysiology of these diseases remains unclear.
An overview of the synthesis, actions, and metabolism of NE is shown in Figure 1. NE exerts its effects through binding to G-protein coupled α- and β-adrenergic receptors (ARs). α-ARs are further divided into α1 and α2, and each of these has three subtypes: α1A, α1B, and α1D; α2A, α2B, and α2C. β-ARs include β1, β2, and β3 subtypes (5). Generally, α1- and β-ARs have a stimulatory effect on cell signaling, as they have been shown to increase intracellular phospholipase C or cyclic adenosine monophosphate (cAMP), respectively, whereas α2-ARs suppress intracellular cAMP and generally have an inhibitory influence on signaling (Figure 1) (6). NE has the highest affinity for α2-ARs (5) so low-level NE release may inhibit neuronal activity, whereas increased neural transmission arising from NE binding to stimulatory α1- and β-ARs only occurs at higher NE concentrations (7).
Figure 1
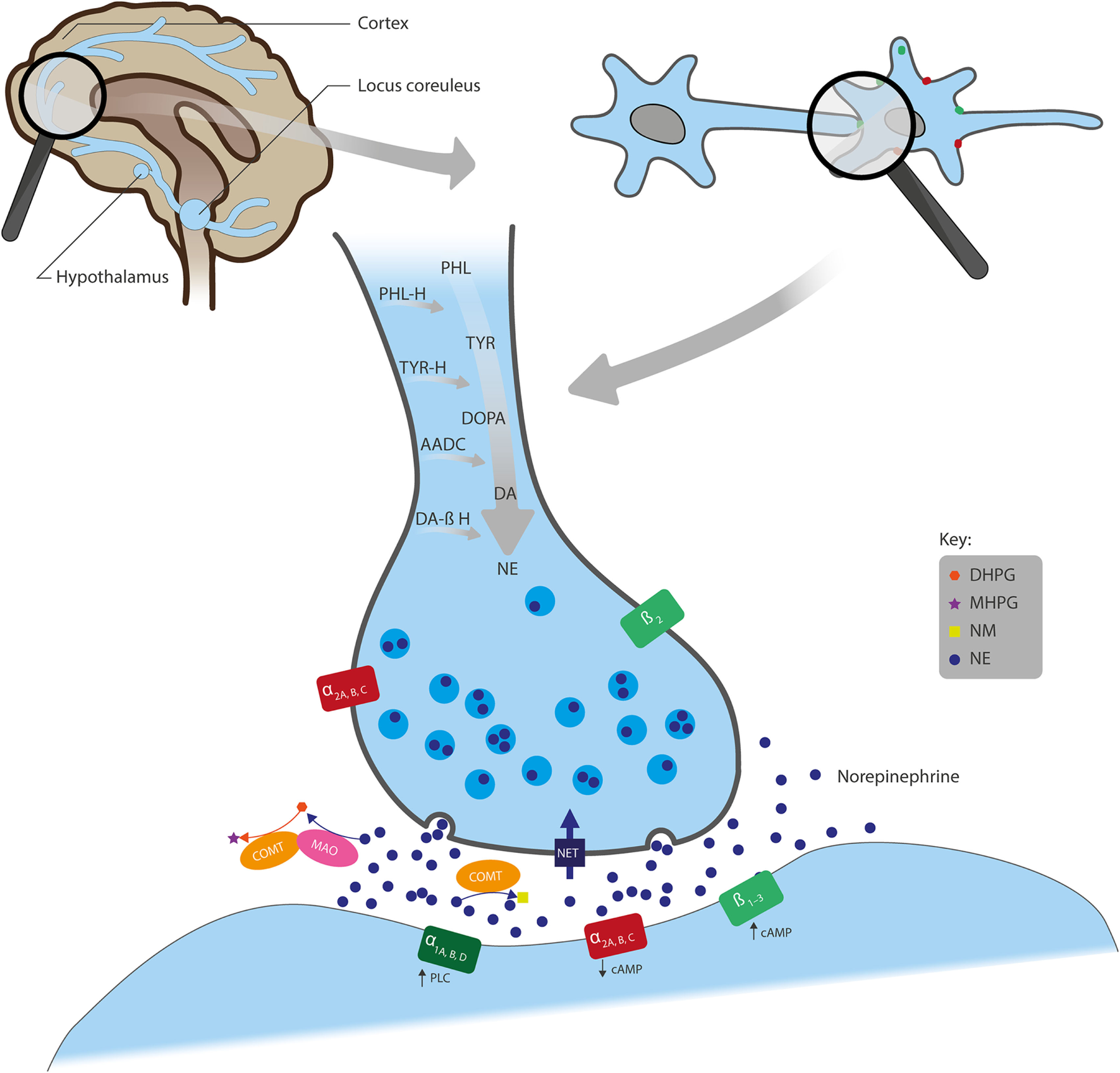
Overview of the actions of NE in neural synapses. Noradrenergic neurons originate from the locus coeruleus and project to regions of the forebrain, including the cortex and hypothalamus. NE is synthesized from the amino acids TYR and PHL and converted first to DOPA, DA, and further to NE by the enzymes TYR-H, AADC, and DA β-H, respectively, after which NE is stored in presynaptic vesicles. Following its release into the synaptic cleft, NE exerts its effects through binding to the adrenergic receptors (ARs): α1A, α1B, and α1D; α2A, α2B, and α2C; or β1, β2, and β3. α1- and β-ARs have a stimulatory effect on cell signaling, whereas α2-ARs inhibit signaling. Also, while ARs are mainly located post-synaptically, α2- and β2-AR subtypes can also be localized pre-synaptically. NE is removed from the synaptic cleft by either reuptake via NET (expressed on the presynaptic terminals of NE neurons and glial cells), inactivation through the catabolic enzyme COMT to NM, or metabolism by MAO into several transitional metabolites, including its principal brain metabolite, MHPG. AADC, l-aromatic amino acid decarboxylase; cAMP, cyclic adenosine monophosphate; COMT, catechol O-methyltransferase; DA, dopamine; DA β-H, dopamine β-hydroxylase; DHPG, dihydroxyphenylglycol; DOPA, 3,4-dihydroxyphenylalanine; MAO, monoamine oxidase; MHPG, 3-methoxy-4-hydroxyphenylglycol; NE, norepinephrine; NET, NE transporter; NM, normetanephrine; PHL, phenylalanine; PHL-H, phenylalanine hydroxylase; PLC, phospholipase C; TYR, tyrosine; TYR-H, tyrosine hydroxylase.
Noradrenergic receptors are found on nerve fibers that originate from the locus coeruleus (LC) and project to many parts of the forebrain, including the cortex, cerebellum, amygdala, hippocampus, basal ganglia, thalamus, and hypothalamus (Figure 1) (8). Noradrenergic heteroreceptors are also located on glutamate, gamma-aminobutyric acid (GABA), dopamine (DA), serotonin (5-HT), histamine, and orexin neurons, as well as in glial and immune cells. Therefore, in addition to being autoregulated by presynaptic α2A-ARs, α2C-ARs (5), and β2-ARs (9), NE signaling is also regulated by other neurotransmitters, such as inhibitory GABA and excitatory glutamate (3, 10). Taken together, this suggests that NE receptors within these pathways play a role in a broad range of brain functions, such as arousal, stress response, memory consolidation, immune response, endocrine function, sleep/wakefulness, and pain-threshold regulation (11).
The Research Domain Criteria (RDoC) matrix was created to help identify brain mechanisms that explain the pathology of psychiatric disorders, improve accuracy of diagnosis, and predict responses to treatment (12). The RDoC matrix classifies symptoms into negative valence systems (including fear and anxiety), positive valence systems (including motivation and reward-seeking behavior), cognitive systems (including attention, perception, declarative, and working memory), social processing systems (including affiliation and attachment), and arousal/regulatory systems (including circadian rhythms and sleep) (13). By including genetic and other factors that influence neurotransmission, the RDoC matrix provides a more comprehensive model of psychiatric diseases, including MDD and schizophrenia.
Major depressive disorder is mostly characterized by a depressed mood, fatigue, a diminished ability to think or concentrate, and disruptions to sleep/wakefulness, circadian rhythms, and immune responses (14). Evidence has shown that these symptoms can be influenced by NE activity in the LC via α-AR modulation. Two of the first antidepressant treatments (ADTs) were iproniazid, which inhibits monoamine oxidase (MAO), and imipramine, which blocks reuptake of serotonin and NE, leading to increased concentrations of these neurotransmitters (15). NE activity in the LC has been shown to be altered in patients with MDD compared with controls: histopathology studies have suggested patients with MDD have increased levels of tyrosine hydroxylase and a reduced density of NE transporter (NET) in the LC (16), the latter confirmed by radioligand binding studies (17). Concentrations of 3-methoxy-4-hydroxyphenylglycol (MHPG), the major NE metabolite, in the cerebrospinal fluid (CSF) have been shown to positively correlate with lifetime mood burden, a composite measure reflecting the number, duration, and intensity of depressive episodes (18). Moreover, salivary MHPG levels in men were recently shown to correlate with depressive symptom scores (19).
Desensitized α1-ARs in the brains of depressed patients have previously been identified (20). Conversely, studies have shown that both the affinity and density of inhibitory α2-ARs are increased in the LC and prefrontal cortex of patients with MDD compared with controls (21, 22), which may reflect a compensatory response related to high NE levels. However, blocking α2-ARs using yohimbine has been shown to improve memory consolidation in patients with MDD, suggesting that increased α2-AR density may also have detrimental effects in these patients (23). MDD could, therefore, be conceptualized as a highly phenotypically and biologically heterogeneous condition, whereby depressed patients may experience both under- and over-arousal that may vary regionally (24).
Like MDD, schizophrenia is a heterogeneous disease where symptomatology includes positive (e.g., paranoid delusions, auditory hallucinations, incoherent thinking), negative (e.g., affective blunting, inactivity, impoverished speech), affective, and cognitive symptoms that can vary independently in each patient (4). In general, positive symptoms are aggravated by selective, indirect NE agonists such as yohimbine, and ameliorated by functional NE antagonists such as clonidine and oxypertine (4). Furthermore, adjunctive ADTs that alter NE activity (e.g., duloxetine) have been shown to relieve negative symptoms (25), suggesting NE may have a role in the pathophysiology of schizophrenia. However, the heterogeneity of the disease is reflected in postmortem studies, which have shown that brain concentrations of NE in patients with schizophrenia vary across populations (4).
An increased understanding of the involvement of α-ARs in the pathophysiology of MDD and schizophrenia is reflected in the pharmacology of recently approved ADTs and antipsychotics (APs). Many treatments for MDD act on overall NE levels (including uptake and MAO inhibitors) or on α-ARs (15). More recent therapies include selective NE reuptake inhibitors, such as reboxetine (26); NE and DA uptake inhibitors, such as bupropion (27); mixed serotonergic and noradrenergic reuptake inhibitors, such as venlafaxine, duloxetine, milnacipran, levomilnacipran, and desvenlafaxine (15); the selective serotonin reuptake inhibitor (SSRI) and 5-HT1A partial agonist vilazodone (28); multimodal agents, such as vortioxetine, a SSRI, 5-HT1A agonist, 5-HT1B partial agonist, and 5-HT1D, 5-HT3, and 5-HT7 antagonist (29); and compounds that block α2-ARs and 5-HT2 receptors and increase activity at 5-HT1 receptors, such as mirtazapine (30). Many of these agents either directly or indirectly modulate NE.
Although most current APs are DA D2 receptor antagonists or partial agonists/antagonists, several second generation APs also act on other neurotransmitter receptors, including α-ARs (31). In schizophrenia, it is generally thought that blocking α1-ARs suppresses positive symptoms, while blocking α2-ARs relieves negative and cognitive symptoms (31). Indeed, clozapine, a prototypical second generation AP whose mechanism of action is not fully understood and has not yet been replicated by any other agent, is known to be an antagonist of α1-ARs with higher affinity for these receptors than for D2 receptors, as well as acting as an antagonist of α2-ARs (32). Administration of clozapine in rats increases α1-AR density in the cortex and thalamus (33). While clozapine results in superior efficacy in treatment-resistant schizophrenia and reduced extrapyramidal symptoms compared with standard APs and other second generation APs (34), its ability to induce potentially fatal agranulocytosis restricts its use clinically (32). Other APs such as risperidone, olanzapine, quetiapine, aripiprazole, asenapine, lurasidone, and cariprazine were subsequently developed in an attempt to improve efficacy and side effect profiles (32). Indeed, risperidone is a potent α-AR antagonist (35). Moreover, the second generation AP brexpiprazole has a distinct pharmacologic profile with purported serotonin-DA modulating activity and α-AR antagonist activity (36). In phase II and III studies, brexpiprazole was efficacious in the treatment of patients with schizophrenia (37), and when administered adjunctively in patients with MDD and an inadequate response to ADTs (38). Therefore, it is of considerable interest to understand the role that NE antagonism may play in ameliorating the symptoms of MDD and schizophrenia.
This systematic review will focus on the role of α-ARs in the pathophysiology of MDD and schizophrenia, as well as in the mechanism of action of compounds used in clinical studies of these diseases. In particular, this review aims to assess how the action of these compounds at α-ARs may be related to the RDoC negative and positive valence systems, arousal, and cognition. The review will include only those compounds exerting their action on α-ARs receptors, and not β-ARs or NET, as the hope is to elucidate the role of α-ARs in the perceived efficacy of these compounds. Moreover, this review aims to establish whether the current clinical evidence is sufficient to link α-ARs to clinical treatments or outcomes of MDD and schizophrenia, with the understanding that the nature of these heterogeneous diseases may make it difficult to identify a single common thread that definitively establishes such a link. The review will also identify gaps in the literature that future investigations need to address.
Materials and Methods
Literature Searches
Two literature searches were performed on the role of NE and α-ARs in MDD and schizophrenia. A PubMed search was carried out on December 11, 2015 and a ProQuest search was carried out on December 18, 2015. Both searches were conducted using the following Medical Subject Heading (MeSH) term search string: “norepinephrine” OR “noradrenaline” OR “alpha adrenergic receptor(s)” OR “beta adrenergic receptor(s)” AND “schizophrenia” OR “major depressive disorder” AND “translational medicine” OR “immunity” OR “microglia” OR “astrocyte(s)” OR “inflammation” OR “gene(s)” OR “brain chemistry” OR “biomarker” OR “neuronal plasticity” OR “cognition” OR “arousal” OR “sleep” OR “psychopharmacology” OR “brain development.” The original searches were limited to the period January 2008–December 2015 and publications in the English language.
Literature Review Results
The searches yielded a total of 2,427 publications (PubMed, n = 669; ProQuest, n = 1,758). After exclusion of duplicates, articles deemed not relevant, case studies, reviews and meta-analyses, preclinical reports, or articles concerning other indications, abstracts, and full-length text were independently screened for eligibility by all the authors. To focus the review on the most recent information, it was decided that only articles published during the period January 2010–December 2015 would be included in full article screening. Psychotropics used in the treatment of MDD and schizophrenia do not generally exert a direct effect on β-ARs, with these receptors only influenced by a change in overall NE transmission. While some adverse events of psychotropics may be mediated by β-ARs, in order to focus the review on the role of NE related to the pathophysiology of psychiatric disorders, the review was limited to articles discussing the actions of NE at α-ARs only and on drugs with at least a moderate affinity for α-ARs (Ki < 100 nM). A total of 16 research reports were identified as meeting these inclusion criteria (Figure 2) including six clinical trial reports, six genetic studies, two biomarker studies, and two receptor studies (Table 1).
Figure 2

Flow diagram of article selection. MDD, major depressive disorder; NE, norepinephrine; PK, pharmacokinetics.
Table 1
| Reference | Indication | Type of study |
|---|---|---|
| Chandley et al. (3) | MDD | Genetic |
| Houston et al. (45) | Genetic | |
| Egami et al. (47) | Biomarker | |
| Nyberg et al. (55) | PET study | |
| Rivero et al. (48) | Receptor density | |
| Kuffel et al. (40) | Clinical trial | |
| Nagao et al. (41) | Clinical trial | |
| Cheng et al. (49) | SCZ | Genetic |
| De Luca et al. (51) | Genetic | |
| Liu et al. (50) | Genetic | |
| Lochman et al. (53) | Genetic | |
| Evers et al. (52) | Biomarker | |
| Oranje and Glenthoj (39) | Clinical trial | |
| Oranje and Glenthoj (42) | Clinical trial | |
| Stenberg et al. (44) | Clinical trial | |
| Terevnikov et al. (43) | Clinical trial | |
Summary of articles identified.
MDD, major depressive disorder; SCZ, schizophrenia.
Results
Overall, we found that direct clinical evidence for the role of NE at α-ARs in the symptomatology of MDD and schizophrenia is limited. Six clinical trials were identified (three investigating clonidine and three investigating mirtazapine; one for each in MDD and two for each in schizophrenia), none of which were designed a priori to compare the effect of compounds with differing pharmacological profiles on outcomes of the study.
Clinical Trials
Major Depressive Disorder
Clonidine, a specific α2-AR agonist, may suppress NE output, while simultaneously stimulating postsynaptic α2-ARs (39). In a study investigating the effects of clonidine in MDD patients and healthy controls, clonidine was shown to impair memory consolidation (word list learning) but did not affect working memory or memory retrieval (40). Conversely, mirtazapine, a noradrenergic and specific serotonergic ADT, selectively blocks α2-ARs and serotonin 5-HT2 and 5-HT3 receptors as well as histaminergic H1 receptors, and indirectly stimulates serotonin 5-HT1A receptors (30). Mirtazapine was compared with the selective serotonin and norepinephrine reuptake inhibitor duloxetine and was shown to be superior in the reduction of Hamilton Rating Scale for Depression scores in patients with MDD (41). Therefore, caution may be needed when targeting α2-AR in order to balance effects on learning and memory consolidation, while effectively reducing depressive symptoms in patients with MDD.
Schizophrenia
Administration of clonidine to patients with schizophrenia increased pre-pulse inhibition of the startle reflex, a measure of sensorimotor gating known to be defective in schizophrenia, to levels similar to healthy controls, whereas inhibition of this reflex remained deficient in placebo-treated patients (39). Similarly, P50 suppression, a related measure of sensory gating, was reduced in patients with schizophrenia compared with healthy controls. However, clonidine increased P50 suppression in patients, thereby causing enhanced sensory gating and potentially improving cognitive abilities in these patients (42).
Patients treated with adjunctive mirtazapine showed an overall improvement in total Positive and Negative Syndrome Scale (PANSS) scores, predominantly driven by improvements in positive symptom scores (43), although this may have been due to its sedating properties. Further investigation into factors that may predict and mediate this response revealed that improvements were attributable to increased spatial visualization ability and motor skill as measured by the block design test. Furthermore, pathway analysis indicated that perceived perceptual/cognitive benefits, related to the improvement in attention and processing speed, may be attributable to mirtazapine’s antidepressant properties, rather than direct impact on cognition and negative symptoms of schizophrenia (44). One possible interpretation of these findings is that mirtazapine-associated modulation of α1-ARs and α2-ARs and consequent “tuning” of NE/5-HT signaling may have played a key role in its ameliorative effect on the positive symptoms of schizophrenia.
Predicting Response to Treatment
Many investigations have been carried out to determine genetic or metabolic signatures associated with MDD and schizophrenia. As well as providing insights into the pathophysiology of these disorders, these reports may assist clinicians with predicting how patients will respond to therapy and providing more personalized care. We identified six reports investigating the genetic variants of noradrenergic receptors or proteins expressed within the noradrenergic synthetic pathway, and their relationship to either symptoms of MDD and schizophrenia or the side effects of treatment with agents that have activity at α-ARs. We also identified two studies investigating NE metabolites as biomarkers of MDD and schizophrenia, and two studies investigating the binding or distribution of NE-associated receptors or transporters.
Major Depressive Disorder
Locus coeruleus NE activity is non-specifically modulated by glutamatergic input, and so changes in glutamate levels could impact the effects of NE in this brain region. Analysis of genes related to glutamatergic function showed reduced expression in LC astrocytes of patients with MDD (3). Additionally, this study provides evidence of astrocyte dysfunction in the noradrenergic LC region in patients with MDD.
In another study, expression of SLC6A2rs36024 [a single nucleotide polymorphism (SNP) of NET] was found to be associated with improvement of Montgomery–Åsberg Depression Rating Scale scores during olanzapine–fluoxetine treatment of patients (45). Olanzapine acts as an α1-AR antagonist with moderate affinity (46), which may in part explain its beneficial therapeutic response given dysfunctional reuptake by NET. Moreover, saliva levels of MHPG were shown to be higher in patients than controls, likely reflecting aberrant NE turnover and less efficient neurotransmission (47). These studies support targeting the reuptake of NE or antagonism of stimulatory α1-ARs for the treatment of patients with MDD.
Rivero et al. (2014) investigated the density of ARs in MDD (48). Using radioligand saturation binding experiments, α2- and β1-AR densities were found to be higher in the prefrontal cortex of ADT-free patients with MDD than controls, possibly as a compensation associated with altered NE turnover in MDD. In antidepressant-treated patients, α2-AR density remained increased over controls, whereas no difference was observed in β1-AR densities (48). These results suggest a resistance of α2-ARs to the downregulatory effect of ADTs, a potentially important consideration in the development of future ADTs. However, as the majority of brain tissue used in this study was collected from patients who committed suicide (48), these results also indicate that it is important to consider the role of α2-ARs in patients with MDD with respect to suicidality and treatment response.
Schizophrenia
Weight gain has been noted as a common side effect of second generation APs. We identified three genetic studies focusing on the association of genes encoding ARs and weight gain. Two reports provided evidence of a link between ADRA1A gene expression and increased weight gain, as well as body mass index during AP treatment (49, 50). Moreover, ADRA1A gene expression was positively associated with the cumulative number of metabolic syndrome components, with the presence of Arg347 allele of ADRA1A identified as a risk factor for metabolic abnormalities (49). Conversely, in nine genetic polymorphisms tested across genes coding for seven proteins related to noradrenergic receptors or NE synthesis or metabolism (ADRA1A, ADRA2A, ADRA2C, ADRB3, DBH, MAOA, and COMT), no association with AP-induced weight gain in patients with schizophrenia was identified (51).
Other identified articles provided evidence that genetic changes in NE metabolic markers, α-ARs, or other receptors that alter NE activity may influence relevant clinical characteristics of patients with schizophrenia. An investigation into metabolite biomarkers of schizophrenia revealed that levels of DA, homovanillic acid, NE, vanillylmandelic acid, and serotonin positively correlated with Full Scale Intelligence Quotient scores in patients with chromosome 22q11 deletion syndrome, which results in a higher risk of developing MDD, schizophrenia, and other psychiatric disorders (52). Another report discovered an interaction between an α2A-AR gene SNP and a specific methylenetetrahydrofolate reductase gene SNP, which encodes for an enzyme involved in DNA methylation, in patients with schizophrenia (53), suggesting a potential role for NE in the epigenetic control relevant to the pathogenesis of schizophrenia.
The overall pharmacological profile of a compound may play a role in its efficacy that may not initially be recognized. Like many APs, quetiapine binds to a broad range of receptors at varying affinities, including 5-HT1A/2A/2C, D2, histamine H1, and adrenergic α1/2 receptors (54). Quetiapine-extended release (XR) has been shown to also occupy NET in healthy participants, which may help contribute to its broad efficacy (55). Ziprasidone, another AP with affinity for NET and adrenergic α1-ARs, has also demonstrated efficacy as adjunctive treatment to SSRIs in treatment-resistant MDD (56) and anxious depression (57), although it is not approved for the treatment of MDD. Ziprasidone has also been effectively used as an adjunct in depressed patients who have had an inadequate response to escitalopram alone (58). However, it is unlikely that affinity for NET is necessary for the antidepressant effect of APs, as both olanzapine and clozapine have been shown to provide a greater relief of depressive symptoms in patients suffering from schizophrenia, compared to quetiapine (59).
Discussion
Although it is known that NE and its α-ARs play key roles in the pathophysiology of MDD and schizophrenia, direct evidence from recent clinical reports is limited. This appears to be largely due to the trial designs used in most clinical studies. Indeed, there is a lack of direct comparisons of ADTs and APs with differing pharmacologic profiles that measure their impact on specific symptoms or disease outcomes. Overall, our review provided data illustrating that NE action at α-ARs can impact the RDoC negative valence systems and cognitive systems proposed to be involved in MDD and schizophrenia. We also identified several studies that suggested that not only can NE activity be used as a biomarker of MDD and schizophrenia, but that it may also be a useful tool for predicting patient response and adverse reactions related to pharmacological intervention.
NE Effects on Negative Valence Systems
The RDoC negative valence system domain includes fear and anxiety (13), and can, along with disturbances in cognitive domain, be used to categorize some of the positive symptoms of schizophrenia, such as hallucinations (60). A biomarker study showed that elevated NE may be associated with cognitive disorganization, poor impulse control, suspiciousness, hostility, and hallucinations (61). Our findings also suggest that NE manipulation can potentially affect the positive symptoms associated with schizophrenia (43, 44), with the long-term study of adjunctive mirtazapine in patients with schizophrenia showing it improved PANSS positive scores (43), although, this may also be related to modulation of several 5-HT receptors (62). The effectiveness of mirtazapine in schizophrenia is supported by a preclinical study that showed that mirtazapine enhanced the AP activity of haloperidol in an apomorphine-induced climbing test in mice (63). Mirtazapine acts as an antagonist of α2-ARs, as well as serotonin 5-HT2 and 5-HT3 receptors (30), so that when used in combination with a first generation AP such as the D2 receptor antagonist haloperidol, the overall pharmacological profile resembles that of clozapine (43), which was reported to be superior to haloperidol for treating positive symptoms of schizophrenia (64). These findings are also supported by the pharmacological properties of brexpiprazole, a partial agonist at serotonin 5-HT1A and DA D2 receptors, and a potent antagonist at serotonin 5-HT2A and NE α1B/2C receptors, all at similar potencies (36). Studies with brexpiprazole, recently approved for the treatment of schizophrenia, have demonstrated AP activity in rat models of the positive psychotic symptoms of schizophrenia (65), as well as significantly improved PANSS positive scores in patients with schizophrenia (37).
NE Effects on Cognitive Systems
Compromised ability to think or concentrate is one of the diagnostic features of MDD. An imaging study showed that hyperactivity of the frontal and prefrontal cortices, areas of the brain innervated by noradrenergic neurons, is required for depressed patients to complete a working memory task at a level similar to healthy controls (66). Several cognitive processes, including attention and working memory, have been shown to be linked to NE activity (5). Indeed, cognitive defects in chromosome 22q11 deletion syndrome are associated with the abnormal function of several neurotransmitters, including NE (52). Furthermore, clonidine may exert dual activity by engaging inhibitory presynaptic α2-ARs, and therefore suppressing NE activity, while at the same time directly stimulating prefrontal cortex postsynaptic α2-ARs (39). Our review identified a study in which clonidine suppressed memory consolidation in patients with MDD and healthy controls (40). This role of α2-ARs in learning and memory consolidation is supported by preclinical studies in rats (67, 68), although Kuffel et al. (40) found no effect of noradrenergic blockade on working memory in patients with MDD or healthy participants. This is in contrast to an infusion study in which α2-AR agonists applied directly to the prefrontal cortex of monkeys and rats resulted in improved working memory performance (69). In addition, it is thought that the α2A-AR subtype may be the most important for cognitive improvement (5), as the α2A selective agonist guanfacine is more effective at improving working memory without side effects than clonidine, which also acts at α2B- and α2C-ARs (70). High concentrations of NE have also been shown to reduce working memory function through actions at α1-ARs; indeed, stress-induced cognitive deficits have been shown to be blocked by infusion of an α1-AR antagonist into the prefrontal cortex of rats (71). Therefore, the balance between the activation of α2A-ARs and reducing the activity of α1-ARs may be a factor for consideration when designing pharmacological agents for the improvement of cognitive functions in MDD.
Recent studies have indicated a role for α1- and α2-ARs in regulation of cognitive processes, where α2-AR stimulation may ameliorate working memory deficits and α1-AR agonism may enhance sustained attention (72). Preclinical studies point to a differential role for subtypes of α1-ARs in cognition. While activation of α1A-ARs may have a positive impact on cognition and neurogenesis, α1B-mediated signaling may be detrimental to cognition and promote apoptosis (73, 74). Furthermore, while α2A-AR activation mediates improvement in working memory following guanfacine treatment (75), activity at α2C-ARs may also mediate stress-related psychiatric diseases such as MDD and anxiety disorders (76). Furthermore, α2C-AR antagonism may be associated with a pro-cognitive effect, in addition to ameliorating anxiety and depression (77). Thus, selective modulation of α-AR subtypes may be advantageous in therapy of MDD. This is indirectly supported by the pharmacological profile of brexpiprazole, which has antagonist activity at both α1B- and α2C-ARs (36), and has been shown to be an efficacious adjunctive treatment for patients with MDD (38).
In schizophrenia, defects in the early sensory filter mechanisms are thought to result in cognitive fragmentation, and ultimately hallucinations and delusions (78). Reduced filtering of sensory and sensorimotor information, demonstrated by both a reduced P50 suppression or reduced pre-pulse inhibition of the startle reflex, was corrected by activating α2-ARs with clonidine (39, 42). Other evidence from our search showed that mirtazapine increased mental speed and attention control in schizophrenia (44), which may be related to its activation of α2-ARs.
Glutamate has been shown to be crucial for cognitive processing, and pharmacological agents that modulate glutamate transmission, such as ketamine, have demonstrated ADT-like properties (14). Our search results suggested that glutamate transporter gene expression was reduced in LC astrocytes in postmortem brain tissue of patients with MDD (3). LC–NE activity is modulated by glutamatergic input, and so changes in glutamate levels could also indirectly impact the NE signaling. Elsewhere, increased glutamate levels have been reported in the occipital cortex, prefrontal cortex, and plasma of patients with MDD (79–81). Therefore, the benefit of glutamate modulators may in part be due to indirect actions on NE activity.
Effects of NE on Other RDoC Systems
There was little direct evidence from our review to support a role of NE and α-ARs in the positive valence systems, systems for social processes, or arousal/regulation systems associated with MDD and schizophrenia. However, other studies outside the selection criteria for this review provided some data. Negative symptoms of schizophrenia, such as flat affect and poverty of speech, can be categorized into the social process RDoC system (13). Clinical studies have shown that mirtazapine enhanced the effects of haloperidol, risperidone, and clozapine on negative symptoms (82–84), which support the identified study that showed adjunctive mirtazapine improved PANSS negative scores (43).
Disruptions in sleep cycles and circadian rhythms are common in patients with MDD (85). Moreover, genetic studies have suggested that patients with MDD have both a phase shift of rhythms and a disrupted regulation of circadian genes (86). Since NE neurons project to the hypothalamus, which contains the suprachiasmatic nucleus—the master pacemaker, which coordinates rhythms throughout the brain and body (85), it is possible that NE has a role in modulating these effects. As reviewed elsewhere, imaging studies provide evidence of sleep disruption due to excessive arousal in MDD, while biochemical studies point to an association between elevated NE levels and decreased sleep efficiency (87). Thirty-hour sampling of CSF samples from depressed patients has found elevated NE relative to healthy controls around the clock (88).
Predicting Response to Treatment
When considering individual patient needs, biomarkers which aid in the diagnosis of patients or predict responses to treatment are valuable for successful therapeutic intervention. MDD and schizophrenia were originally thought to be diseases located entirely in the CNS, although recent hypotheses and findings suggest that the pathophysiology of these diseases can be shaped by peripheral endocrine, autonomic, immune, and gut-brain events (24, 89, 90). Several metabolic studies have attempted to identify potential biomarkers for MDD and schizophrenia by assessing changes in enzymatic activity in plasma and saliva. Baseline levels of MHPG in saliva were shown to positively correlate with a high Beck Depression Inventory score in men (19), which provides support to the findings that MHPG concentrations in CSF were correlated with a lifetime burden of mood swings (18). In addition, plasma MHPG has been shown to correlate with negative symptoms and cognitive impairment during early-stage schizophrenia (91).
A patient’s NE metabolites or genetic profile may also help to predict how well they will respond to different treatments, not only with regards to improving symptoms, but also the emergence of adverse events. A study found that patients with MDD that responded to mirtazapine or SSRI treatment had higher baseline saliva MHPG concentrations, although this did not reach statistical significance in patients receiving mirtazapine alone, or when it was used as adjunctive therapy with SSRIs (47). Similarly, risperidone, which acts as an antagonist of α1- and α2-ARs (as well as other receptors), was shown not to affect NE, vanillylmandelic acid, or MHPG in the plasma or urine of patients with schizophrenia (35).
Genetic studies identified here suggested that differences in the gene which codes for α1A-AR are associated with weight gain (50) and metabolic abnormalities (49) during AP treatment of schizophrenia. However, there are many factors that affect weight gain in patients with schizophrenia, and attribution of this adverse reaction to a single receptor type is inappropriate. For example, it has been shown that antagonism of the DA D2 receptor has both an effect on eating, mediated by 5-HT2C receptors, and disinhibition of prolactin secretion, which has an impact on lipid and glucose metabolism (92).
The atypical AP olanzapine and the SSRI fluoxetine are often used in combination in patients with treatment-resistant MDD. Previously, olanzapine and fluoxetine have been shown to have a synergistic effect on elevation of NE and DA levels in the rat prefrontal cortex (93), which may be responsible for some of the efficacy of this combination therapy. Therefore, genetic variants in the NET identified in patients with MDD who were not initially responsive to fluoxetine alone may be a marker to predict improvements in these patients after switching to olanzapine–fluoxetine combination (45). Quetiapine was also shown to occupy NET in healthy patients at doses that produce antidepressive effects. This may explain the unique broad spectrum efficacy of quetiapine therapy in psychiatric disorders (55).
The higher density of α2-ARs in both ADT-free and antidepressant-treated patients with depression compared with matched controls suggests that these receptors are resistant to the observed downregulation of β1-ARs by ADTs (48). In a preclinical study in rats, treatment with the SSRI citalopram did not result in any adaptive changes in LC neurons, but did indirectly enhance the inhibitory effect of NE on neurons in the LC (94). The activation of inhibitory α2-ARs resulting from elevation of NE concentrations in the synaptic cleft may underlie this effect (94). Although, desensitization of these receptors may also have occurred, as previously observed in animal studies (95, 96). Furthermore, preclinical evidence also suggests that intact α1-AR-mediated signaling may be necessary for SSRI induced 5-HT elevation (97).
Both MDD and schizophrenia have been associated with immune abnormalities and dysfunctional inflammatory signaling (98, 99). Elevated peripheral inflammation may precipitate aberrant CNS inflammatory signaling with subsequent disruption of monoamine and glutamate transmission, in part by inappropriately activating brain microglia (100). Both α- and β-ARs have a key role in regulating macrophage and microglia activity (101, 102). Thus, compromised NE transmission may promote a pro-inflammatory phenotype consistent with depressive psychopathology. Intriguingly, a recent study reported a preferential response to an SSRI (escitalopram) in patients with lower levels of an inflammatory marker [C-reactive protein (CRP) <1 mg/mL], compared with depressed patients with higher inflammation (CRP > 3 mg/mL) who had a robust response to a predominantly noradrenergic ADT (nortriptyline) (103). Therefore, one may speculate that noradrenergic ADTs exert some of their therapeutic effect by attenuating excessive inflammatory signaling.
Limitations
The objective of our systematic review was to determine the role of NE and α-ARs in the pathophysiology of MDD and schizophrenia using evidence from clinical reports. In order to achieve this, a number of specific search terms were implemented, including the use of “alpha adrenergic receptor(s)” as a limiter in the MeSH search-term string. Therefore, any reports that did not contain this term were excluded, even though they may indirectly support the role of NE activity at α-ARs in MDD or schizophrenia. It should also be noted that in order to focus this literature review on recent data, the scope was restricted to publications between 2010 and 2015, which will limit our findings. Finally, it is a daunting challenge to attempt to link receptor activity to specific therapeutic effect, especially as many of the drugs discussed in this review have clinically relevant binding affinities for a number of different receptors, and additionally, these diseases are phenotypically, genetically, and biologically heterogeneous. The lack of a common pathophysiology or homogenous symptom presentation between all patients diagnosed with MDD or schizophrenia will impact the summary of effects linked to α-AR modulation. Moreover, it has been reported that females have a larger LC than males, comprising nearly 3,000 more neurons, which could impact their capacity for NE production and release (104). Given these neurobiological differences, we suggest that gender should be considered as a factor in future clinical studies.
Conclusion
Preclinical evidence suggests that targeting NE α-ARs is beneficial for the treatment of both MDD and schizophrenia. However, the complex and heterogeneous nature of both of these diseases means that interpretation of clinical findings is not always straightforward. Using a systematic approach, we have provided indirect evidence from recent clinical studies and other reports in humans that support the concept that, in general, NE and α-ARs, and drugs that act through these receptors, have an important role in the negative valence systems and cognitive systems related to MDD and schizophrenia. However, there is no direct clinical evidence for the specific effects of α-ARs, likely due to a lack of agents that have proven differential subtype affinity. Moreover, no direct clinical comparisons have been made between therapies that have α-AR activity. Therefore, advantages of the interactions at these receptors are theoretical in the clinical situation, supported only by evidence from preclinical experiments. In addition, due to both the fact that clinical trials have not been designed to determine if there are differences in efficacy between different symptom domains, and the added complexity of the heterogeneity of these diseases across populations of patients, the effects of targeting distinct α-ARs on specific symptom domains are yet to be determined. These points should be considered when studying future medications.
It is worth noting that genetic studies may suggest that candidate genes could have predictive properties enabling their use as putative biomarkers for disease in target populations. However, there are multiple influences on MDD and schizophrenia, and blanket SNP studies conducted to date have not revealed any specific relationships with NE. The limited number of studies identified here suggests that more up-to-date clinical investigations, designed to tease apart the contribution of α-ARs and their subtypes to the pathophysiology of MDD and schizophrenia, are required.
Statements
Author contributions
VM provided substantial contributions to the manuscript in leading the design of the literature search, defining the inclusion and exclusion criteria, screening of the articles, providing critical reviews and revisions for each of the drafts, and approving the final version for submission. AE, KG, SO, and RD provided substantial contributions to the manuscript in closely participating in designing the literature search, defining the inclusion and exclusion criteria, screening of the articles, providing critical reviews and revisions for each of the drafts, and approving the final version for submission. VM, AE, KG, SO, and RD agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy and integrity of the work are appropriately investigated and resolved.
Acknowledgments
Rebecca Hurst, Ph.D., QXV Communications (an Ashfield Business, part of UDG Healthcare plc), Macclesfield, UK, provided medical writing support, which was funded by Lundbeck LLC, Deerfield, IL, USA and Otsuka Pharmaceutical Development and Commercialization, Inc., Princeton, NJ, USA.
Conflict of interest
AE and KG are employees of Lundbeck LLC. SO and RD are employees of Otsuka Pharmaceutical Development and Commercialization, Inc.
References
1
World Health Organization. The Global Burden of Disease 2004 Update. (2008). Available from: http://www.who.int/healthinfo/global_burden_disease/GBD_report_2004update_full.pdf
2
WhitefordHAFerrariAJDegenhardtLFeiginVVosT. The global burden of mental, neurological and substance use disorders: an analysis from the global burden of disease study 2010. PLoS One (2015) 10(2):e0116820.10.1371/journal.pone.0116820
3
ChandleyMJSzebeniKSzebeniACrawfordJStockmeierCATureckiGet alGene expression deficits in pontine locus coeruleus astrocytes in men with major depressive disorder. J Psychiatry Neurosci (2013) 38(4):276–84.10.1503/jpn.120110
4
YamamotoKHornykiewiczO. Proposal for a noradrenaline hypothesis of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2004) 28(5):913–22.10.1016/j.pnpbp.2004.05.033
5
RamosBPArnstenAF. Adrenergic pharmacology and cognition: focus on the prefrontal cortex. Pharmacol Ther (2007) 113(3):523–36.10.1016/j.pharmthera.2006.11.006
6
DragoACrisafulliCSidotiASerrettiA. The molecular interaction between the glutamatergic, noradrenergic, dopaminergic and serotoninergic systems informs a detailed genetic perspective on depressive phenotypes. Prog Neurobiol (2011) 94(4):418–60.10.1016/j.pneurobio.2011.05.009
7
ArnstenAFWangMJPaspalasCD. Neuromodulation of thought: flexibilities and vulnerabilities in prefrontal cortical network synapses. Neuron (2012) 76(1):223–39.10.1016/j.neuron.2012.08.038
8
ChamberlainSRRobbinsTW. Noradrenergic modulation of cognition: therapeutic implications. J Psychopharmacol (2013) 27(8):694–718.10.1177/0269881113480988
9
HeinL. Adrenoceptors and signal transduction in neurons. Cell Tissue Res (2006) 326(2):541–51.10.1007/s00441-006-0285-2
10
JinXLiSBondyBZhongWOginskyMFWuYet alIdentification of a group of GABAergic neurons in the dorsomedial area of the locus coeruleus. PLoS One (2016) 11(1):e0146470.10.1371/journal.pone.0146470
11
AtzoriMCuevas-OlguinREsquivel-RendonEGarcia-OscosFSalgado-DelgadoRCSaderiNet alLocus coeruleus norepinephrine release: a central regulator of CNS spatio-temporal activation?Front Synaptic Neurosci (2016) 8:25.10.3389/fnsyn.2016.00025
12
InselTCuthbertBGarveyMHeinssenRPineDSQuinnKet alResearch domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry (2010) 167(7):748–51.10.1176/appi.ajp.2010.09091379
13
SanislowCAQuinnKJSypherI. NIMH research domain criteria (RDoC). In: CautinRLLilienfeldSO, editors. The Encyclopedia of Clinical Psychology. Hoboken, NJ: Wiley-Blackwell (2015).
14
SaltielPFSilversheinDI. Major depressive disorder: mechanism-based prescribing for personalized medicine. Neuropsychiatr Dis Treat (2015) 11:875–88.10.2147/NDT.S73261
15
MontoyaABruinsRKatzmanMABlierP. The noradrenergic paradox: implications in the management of depression and anxiety. Neuropsychiatr Dis Treat (2016) 12:541–57.10.2147/NDT.S91311
16
RajkowskaG. Histopathology of the prefrontal cortex in major depression: what does it tell us about dysfunctional monoaminergic circuits?Prog Brain Res (2000) 126:397–412.10.1016/S0079-6123(00)26026-3
17
KlimekVStockmeierCOverholserJMeltzerHYKalkaSDilleyGet alReduced levels of norepinephrine transporters in the locus coeruleus in major depression. J Neurosci (1997) 17(21):8451–8.
18
EhnvallASjogrenMZachrissonOCAgrenH. Lifetime burden of mood swings and activation of brain norepinephrine turnover in patients with treatment-refractory depressive illness. J Affect Disord (2003) 74(2):185–9.10.1016/S0165-0327(02)00011-3
19
WatanabeILiGYImamuraYNabetaHKunitakeYIshiiHet alAssociation of saliva 3-methoxy-4-hydroxyphenylglycol levels and a later depressive state in older subjects living in a rural community: 3-year follow-up study. Int J Geriatr Psychiatry (2012) 27(3):321–6.10.1002/gps.2729
20
AsnisGMSandersonWCvan PraagHM. Cortisol response to intramuscular desipramine in patients with major depression and normal control subjects: a replication study. Psychiatry Res (1992) 44(3):237–50.10.1016/0165-1781(92)90027-Z
21
OrdwayGASchenkJStockmeierCAMayWKlimekV. Elevated agonist binding to alpha2-adrenoceptors in the locus coeruleus in major depression. Biol Psychiatry (2003) 53(4):315–23.10.1016/S0006-3223(02)01728-6
22
Garcia-SevillaJAEscribaPVOzaitaALa HarpeRWalzerCEytanAet alUp-regulation of immunolabeled alpha2A-adrenoceptors, Gi coupling proteins, and regulatory receptor kinases in the prefrontal cortex of depressed suicides. J Neurochem (1999) 72(1):282–91.10.1046/j.1471-4159.1999.0720282.x
23
WingenfeldKKuffelAUhlmannCTerfehrKSchreinerJKuehlLKet alEffects of noradrenergic stimulation on memory in patients with major depressive disorder. Stress (2013) 16(2):191–201.10.3109/10253890.2012.708951
24
GoldPW. The organization of the stress system and its dysregulation in depressive illness. Mol Psychiatry (2015) 20(1):32–47.10.1038/mp.2014.163
25
TerevnikovVJoffeGStenbergJH. Randomized controlled trials of add-on antidepressants in schizophrenia. Int J Neuropsychopharmacol (2015) 18(9):yv049.10.1093/ijnp/pyv049
26
EydingDLelgemannMGrouvenUHarterMKrompMKaiserTet alReboxetine for acute treatment of major depression: systematic review and meta-analysis of published and unpublished placebo and selective serotonin reuptake inhibitor controlled trials. BMJ (2010) 341:c4737.10.1136/bmj.c4737
27
ManeetonNManeetonBEurviriyanukulKSrisurapanontM. Efficacy, tolerability, and acceptability of bupropion for major depressive disorder: a meta-analysis of randomized-controlled trials comparison with venlafaxine. Drug Des Devel Ther (2013) 7:1053–62.10.2147/DDDT.S46849
28
WangSMHanCLeeSJPatkarAAMasandPSPaeCU. Vilazodone for the treatment of depression: an update. Chonnam Med J (2016) 52(2):91–100.10.4068/cmj.2016.52.2.91
29
MeekerASHerinkMCHaxbyDGHartungDM. The safety and efficacy of vortioxetine for acute treatment of major depressive disorder: a systematic review and meta-analysis. Syst Rev (2015) 4:21.10.1186/s13643-015-0001-y
30
FawcettJBarkinRL. Review of the results from clinical studies on the efficacy, safety and tolerability of mirtazapine for the treatment of patients with major depression. J Affect Disord (1998) 51(3):267–85.10.1016/S0165-0327(98)00224-9
31
SvenssonTH. Alpha-adrenoceptor modulation hypothesis of antipsychotic atypicality. Prog Neuropsychopharmacol Biol Psychiatry (2003) 27(7):1145–58.10.1016/j.pnpbp.2003.09.009
32
MiyamotoSDuncanGEMarxCELiebermanJA. Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry (2005) 10(1):79–104.10.1038/sj.mp.4001556
33
CahirMMawhinneyTKingDJ. Differential region-specific regulation of central alpha 1-adrenoceptor binding following chronic haloperidol and clozapine administration in the rat. Psychopharmacology (Berl) (2004) 172(2):196–201.10.1007/s00213-003-1639-z
34
KaneJHonigfeldGSingerJMeltzerH. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry (1988) 45(9):789–96.10.1001/archpsyc.1988.01800330013001
35
CaiHLLiHDYanXZSunBZhangQYanMet alMetabolomic analysis of biochemical changes in the plasma and urine of first-episode neuroleptic-naive schizophrenia patients after treatment with risperidone. J Proteome Res (2012) 11(8):4338–50.10.1021/pr300459d
36
MaedaKSuginoHAkazawaHAmadaNShimadaJFutamuraTet alBrexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther (2014) 350(3):589–604.10.1124/jpet.114.213793
37
CorrellCUSkubanAHobartMOuyangJWeillerEWeissCet alEfficacy of brexpiprazole in patients with acute schizophrenia: review of three randomized, double-blind, placebo-controlled studies. Schizophr Res (2016) 174(1–3):82–92.10.1016/j.schres.2016.04.012
38
ThaseMEYouakimJMSkubanAHobartMAugustineCZhangPet alEfficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry (2015) 76(9):1224–31.10.4088/JCP.14m09688
39
OranjeBGlenthojBY. Clonidine normalizes sensorimotor gating deficits in patients with schizophrenia on stable medication. Schizophr Bull (2013) 39(3):684–91.10.1093/schbul/sbs071
40
KuffelAEikelmannSTerfehrKMauGKuehlLKOtteCet alNoradrenergic blockade and memory in patients with major depression and healthy participants. Psychoneuroendocrinology (2014) 40:86–90.10.1016/j.psyneuen.2013.11.001
41
NagaoKKishiTMoriwakiMFujitaKHiranoSYamanouchiYet alComparative clinical profile of mirtazapine and duloxetine in practical clinical settings in Japan: a 4-week open-label, parallel-group study of major depressive disorder. Neuropsychiatr Dis Treat (2013) 9:781–6.10.2147/NDT.S43600
42
OranjeBGlenthojBY. Clonidine normalizes levels of P50 gating in patients with schizophrenia on stable medication. Schizophr Bull (2014) 40(5):1022–9.10.1093/schbul/sbt144
43
TerevnikovVStenbergJHJoffeMTiihonenJBurkinMTchoukhineEet alMore evidence on additive antipsychotic effect of adjunctive mirtazapine in schizophrenia: an extension phase of a randomized controlled trial. Hum Psychopharmacol (2010) 25(6):431–8.10.1002/hup.1137
44
StenbergJHTerevnikovVJoffeMTiihonenJChukhinEBurkinMet alPredictors and mediators of add-on mirtazapine-induced cognitive enhancement in schizophrenia – a path model investigation. Neuropharmacology (2013) 64:248–53.10.1016/j.neuropharm.2012.06.028
45
HoustonJPLauKArisVLiuWFijalBAHeinlothANet alAssociation of common variations in the norepinephrine transporter gene with response to olanzapine-fluoxetine combination versus continued-fluoxetine treatment in patients with treatment-resistant depression: a candidate gene analysis. J Clin Psychiatry (2012) 73(6):878–85.10.4088/JCP.10m06744
46
BymasterFPHemrick-LueckeSKPerryKWFullerRW. Neurochemical evidence for antagonism by olanzapine of dopamine, serotonin, alpha 1-adrenergic and muscarinic receptors in vivo in rats. Psychopharmacology (Berl) (1996) 124(1–2):87–94.10.1007/BF02245608
47
EgamiMImamuraYNabetaHMizoguchiYYamadaS. Saliva levels of 3-methoxy-4-hydroxyphenylglycol and clinical efficacy of mirtazapine or selective serotonin reuptake inhibitors in patients with major depression. Hum Psychopharmacol (2013) 28(1):7–14.10.1002/hup.2273
48
RiveroGGabilondoAMGarcia-SevillaJALa HarpeRCalladoLFMeanaJJ. Increased alpha2- and beta1-adrenoceptor densities in postmortem brain of subjects with depression: differential effect of antidepressant treatment. J Affect Disord (2014) 167:343–50.10.1016/j.jad.2014.06.016
49
ChengCChiuHJLoh elWChanCHHwuTMLiuYRet alAssociation of the ADRA1A gene and the severity of metabolic abnormalities in patients with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2012) 36(1):205–10.10.1016/j.pnpbp.2011.10.011
50
LiuYRLohEWLanTHChenSFYuYHChangYHet alADRA1A gene is associated with BMI in chronic schizophrenia patients exposed to antipsychotics. Pharmacogenomics J (2010) 10(1):30–9.10.1038/tpj.2009.55
51
De LucaVSouzaRPViggianoESickertLTeoCZaiCet alGenetic interactions in the adrenergic system genes: analysis of antipsychotic-induced weight gain. Hum Psychopharmacol (2011) 26(6):386–91.10.1002/hup.1219
52
EversLJCurfsLMBakkerJABootEda Silva AlvesFAbelingNet alSerotonergic, noradrenergic and dopaminergic markers are related to cognitive function in adults with 22q11 deletion syndrome. Int J Neuropsychopharmacol (2014) 17(8):1159–65.10.1017/S1461145714000376
53
LochmanJPlesnikJJanoutVPovovaJMisekIDvorakovaDet alInteractive effect of MTHFR and ADRA2A gene polymorphisms on pathogenesis of schizophrenia. Neuro Endocrinol Lett (2013) 34(8):792–7.
54
NemeroffCBKinkeadBGoldsteinJ. Quetiapine: preclinical studies, pharmacokinetics, drug interactions, and dosing. J Clin Psychiatry (2002) 63(Suppl 13):5–11.
55
NybergSJucaiteATakanoAKagedalMCselenyiZHalldinCet alNorepinephrine transporter occupancy in the human brain after oral administration of quetiapine XR. Int J Neuropsychopharmacol (2013) 16(10):2235–44.10.1017/S1461145713000680
56
PapakostasGIPetersenTJNierenbergAAMurakamiJLAlpertJERosenbaumJFet alZiprasidone augmentation of selective serotonin reuptake inhibitors (SSRIs) for SSRI-resistant major depressive disorder. J Clin Psychiatry (2004) 65(2):217–21.10.4088/JCP.v65n0212
57
IonescuDFSheltonRCBaerLMeadeKHSweeMBFavaMet alZiprasidone augmentation for anxious depression. Int Clin Psychopharmacol (2016) 31(6):341–6.10.1097/yic.0000000000000133
58
PapakostasGIFavaMBaerLSweeMBJaegerABoboWVet alZiprasidone augmentation of escitalopram for major depressive disorder: efficacy results from a randomized, double-blind, placebo-controlled study. Am J Psychiatry (2015) 172(12):1251–8.10.1176/appi.ajp.2015.14101251
59
NakajimaSTakeuchiHFervahaGPlitmanEChungJKCaravaggioFet alComparative efficacy between clozapine and other atypical antipsychotics on depressive symptoms in patients with schizophrenia: analysis of the CATIE phase 2E data. Schizophr Res (2015) 161(2–3):429–33.10.1016/j.schres.2014.12.024
60
FordJMMorrisSEHoffmanRESommerIWatersFMcCarthy-JonesSet alStudying hallucinations within the NIMH RDoC framework. Schizophr Bull (2014) 40(Suppl 4):S295–304.10.1093/schbul/sbu011
61
Fryar-WilliamsSStrobelJE. Biomarkers of a five-domain translational substrate for schizophrenia and schizoaffective psychosis. Biomark Res (2015) 3:3.10.1186/s40364-015-0028-1
62
HaddjeriNBlierPde MontignyC. Acute and long-term actions of the antidepressant drug mirtazapine on central 5-HT neurotransmission. J Affect Disord (1998) 51(3):255–66.10.1016/S0165-0327(98)00223-7
63
BerendsenHHBroekkampCLPinderRM. Mirtazapine enhances the effect of haloperidol on apomorphine-induced climbing behaviour in mice and attenuates haloperidol-induced catalepsy in rats. Psychopharmacology (Berl) (1998) 135(3):284–9.10.1007/s002130050511
64
BreierABuchananRWKirkpatrickBDavisORIrishDSummerfeltAet alEffects of clozapine on positive and negative symptoms in outpatients with schizophrenia. Am J Psychiatry (1994) 151(1):20–6.10.1176/ajp.151.1.20
65
MaedaKLerdrupLSuginoHAkazawaHAmadaNMcQuadeRDet alBrexpiprazole II: antipsychotic-like and procognitive effects of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther (2014) 350(3):605–14.10.1124/jpet.114.213819
66
HarveyPOFossatiPPochonJBLevyRLebastardGLehericySet alCognitive control and brain resources in major depression: an fMRI study using the n-back task. Neuroimage (2005) 26(3):860–9.10.1016/j.neuroimage.2005.02.048
67
GazariniLSternCACarobrezAPBertoglioLJ. Enhanced noradrenergic activity potentiates fear memory consolidation and reconsolidation by differentially recruiting alpha1- and beta-adrenergic receptors. Learn Mem (2013) 20(4):210–9.10.1101/lm.030007.112
68
SirvioJRiekkinenPJrEkonsaloTLammintaustaRRiekkinenPJ. The effects of dexmedetomidine, an alpha 2 agonist, on learning and memory, assessed using passive avoidance and water maze tasks in rats. Neuropharmacology (1992) 31(2):163–8.10.1016/0028-3908(92)90027-M
69
RamosBPStarkDVerduzcoLvan DyckCHArnstenAF. Alpha2A-adrenoceptor stimulation improves prefrontal cortical regulation of behavior through inhibition of cAMP signaling in aging animals. Learn Mem (2006) 13(6):770–6.10.1101/lm.298006
70
ArnstenAFCaiJXGoldman-RakicPS. The alpha-2 adrenergic agonist guanfacine improves memory in aged monkeys without sedative or hypotensive side effects: evidence for alpha-2 receptor subtypes. J Neurosci (1988) 8(11):4287–98.
71
BirnbaumSGobeskeKTAuerbachJTaylorJRArnstenAF. A role for norepinephrine in stress-induced cognitive deficits: alpha-1-adrenoceptor mediation in the prefrontal cortex. Biol Psychiatry (1999) 46(9):1266–74.10.1016/S0006-3223(99)00138-9
72
BerridgeCWSpencerRC. Differential cognitive actions of norepinephrine a2 and a1 receptor signaling in the prefrontal cortex. Brain Res (2016) 1641(Pt B):189–96.10.1016/j.brainres.2015.11.024
73
PerezDMDozeVA. Cardiac and neuroprotection regulated by alpha(1)-adrenergic receptor subtypes. J Recept Signal Transduct Res (2011) 31(2):98–110.10.3109/10799893.2010.550008
74
NalepaIKreinerGBielawskiARafa-ZablockaKRomanA. alpha1-Adrenergic receptor subtypes in the central nervous system: insights from genetically engineered mouse models. Pharmacol Rep (2013) 65(6):1489–97.10.1016/S1734-1140(13)71509-3
75
FranowiczJSArnstenAF. The alpha-2a noradrenergic agonist, guanfacine, improves delayed response performance in young adult rhesus monkeys. Psychopharmacology (Berl) (1998) 136(1):8–14.10.1007/s002130050533
76
SallinenJHaapalinnaAMacDonaldEViitamaaTLahdesmakiJRybnikovaEet alGenetic alteration of the alpha2-adrenoceptor subtype c in mice affects the development of behavioral despair and stress-induced increases in plasma corticosterone levels. Mol Psychiatry (1999) 4(5):443–52.10.1038/sj.mp.4000543
77
KalkmanHOSubramanianNHoyerD. Extended radioligand binding profile of iloperidone: a broad spectrum dopamine/serotonin/norepinephrine receptor antagonist for the management of psychotic disorders. Neuropsychopharmacology (2001) 25(6):904–14.10.1016/S0893-133X(01)00285-8
78
PerryWGeyerMABraffDL. Sensorimotor gating and thought disturbance measured in close temporal proximity in schizophrenic patients. Arch Gen Psychiatry (1999) 56(3):277–81.10.1001/archpsyc.56.3.277
79
HashimotoKSawaAIyoM. Increased levels of glutamate in brains from patients with mood disorders. Biol Psychiatry (2007) 62(11):1310–6.10.1016/j.biopsych.2007.03.017
80
KimJSSchmid-BurgkWClausDKornhuberHH. Increased serum glutamate in depressed patients. Arch Psychiatr Nervenkr (1970) (1982) 232(4):299–304.10.1007/BF00345492
81
MitaniHShirayamaYYamadaTMaedaKAshbyCRJrKawaharaR. Correlation between plasma levels of glutamate, alanine and serine with severity of depression. Prog Neuropsychopharmacol Biol Psychiatry (2006) 30(6):1155–8.10.1016/j.pnpbp.2006.03.036
82
ZoccaliRMuscatelloMRCedroCNeriPLa TorreDSpinaEet alThe effect of mirtazapine augmentation of clozapine in the treatment of negative symptoms of schizophrenia: a double-blind, placebo-controlled study. Int Clin Psychopharmacol (2004) 19(2):71–6.10.1097/00004850-200403000-00003
83
BerkMIchimCBrookS. Efficacy of mirtazapine add on therapy to haloperidol in the treatment of the negative symptoms of schizophrenia: a double-blind randomized placebo-controlled study. Int Clin Psychopharmacol (2001) 16(2):87–92.10.1097/00004850-200103000-00003
84
AbbasiSHBehpourniaHGhoreshiASalehiBRaznahanMRezazadehSAet alThe effect of mirtazapine add on therapy to risperidone in the treatment of schizophrenia: a double-blind randomized placebo-controlled trial. Schizophr Res (2010) 116(2–3):101–6.10.1016/j.schres.2009.11.008
85
EdgarNMcClungCA. Major depressive disorder: a loss of circadian synchrony?Bioessays (2013) 35(11):940–4.10.1002/bies.201300086
86
LiJZBunneyBGMengFHagenauerMHWalshDMVawterMPet alCircadian patterns of gene expression in the human brain and disruption in major depressive disorder. Proc Natl Acad Sci U S A (2013) 110(24):9950–5.10.1073/pnas.1305814110
87
NuttDWilsonSPatersonL. Sleep disorders as core symptoms of depression. Dialogues Clin Neurosci (2008) 10(3):329–36.
88
WongMLKlingMAMunsonPJListwakSLicinioJProloPet alPronounced and sustained central hypernoradrenergic function in major depression with melancholic features: relation to hypercortisolism and corticotropin-releasing hormone. Proc Natl Acad Sci U S A (2000) 97(1):325–30.10.1073/pnas.97.1.325
89
Pyndt JorgensenBKrychLPedersenTBPlathNRedrobeJPHansenAKet alInvestigating the long-term effect of subchronic phencyclidine-treatment on novel object recognition and the association between the gut microbiota and behavior in the animal model of schizophrenia. Physiol Behav (2015) 141:32–9.10.1016/j.physbeh.2014.12.042
90
RaisonCLLowryCARookGA. Inflammation, sanitation, and consternation: loss of contact with coevolved, tolerogenic microorganisms and the pathophysiology and treatment of major depression. Arch Gen Psychiatry (2010) 67(12):1211–24.10.1001/archgenpsychiatry.2010.161
91
GotoNYoshimuraRKakedaSMoriyaJHayashiKIkenouchi-SugitaAet alAssociations between plasma levels of 3-methoxy-4-hydroxyphenylglycol (MHPG) and negative symptoms or cognitive impairments in early-stage schizophrenia. Hum Psychopharmacol (2009) 24(8):639–45.10.1002/hup.1070
92
ReynoldsGPKirkSL. Metabolic side effects of antipsychotic drug treatment – pharmacological mechanisms. Pharmacol Ther (2010) 125(1):169–79.10.1016/j.pharmthera.2009.10.010
93
ZhangWPerryKWWongDTPottsBDBaoJTollefsonGDet alSynergistic effects of olanzapine and other antipsychotic agents in combination with fluoxetine on norepinephrine and dopamine release in rat prefrontal cortex. Neuropsychopharmacology (2000) 23(3):250–62.10.1016/S0893-133X(00)00119-6
94
GrandosoLTorrecillaMPinedaJUgedoL. alpha(2)-Adrenoceptor involvement in the in vitro inhibitory effect of citalopram on a subpopulation of rat locus coeruleus neurons. Eur J Pharmacol (2005) 517(1–2):51–8.10.1016/j.ejphar.2005.05.033
95
MongeauRde MontignyCBlierP. Electrophysiologic evidence for desensitization of alpha 2-adrenoceptors on serotonin terminals following long-term treatment with drugs increasing norepinephrine synaptic concentration. Neuropsychopharmacology (1994) 10(1):41–51.10.1038/npp.1994.6
96
EstebanSLladoJSastre-CollAGarcia-SevillaJA. Activation and desensitization by cyclic antidepressant drugs of alpha2-autoreceptors, alpha2-heteroreceptors and 5-HT1A-autoreceptors regulating monamine synthesis in the rat brain in vivo. Naunyn Schmiedebergs Arch Pharmacol (1999) 360(2):135–43.10.1007/s002109900045
97
ReaKFolgeringJWesterinkBHCremersTI. Alpha1-adrenoceptors modulate citalopram-induced serotonin release. Neuropharmacology (2010) 58(7):962–71.10.1016/j.neuropharm.2009.12.018
98
MillerAHMaleticVRaisonCL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry (2009) 65(9):732–41.10.1016/j.biopsych.2008.11.029
99
NaKSJungHYKimYK. The role of pro-inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2014) 48:277–86.10.1016/j.pnpbp.2012.10.022
100
MillerAHRaisonCL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol (2016) 16(1):22–34.10.1038/nri.2015.5
101
KatoTAYamauchiYHorikawaHMonjiAMizoguchiYSekiYet alNeurotransmitters, psychotropic drugs and microglia: clinical implications for psychiatry. Curr Med Chem (2013) 20(3):331–44.10.2174/092986713804870800
102
WeinsteinLIRevueltaAPandoRH. Catecholamines and acetylcholine are key regulators of the interaction between microbes and the immune system. Ann N Y Acad Sci (2015) 1351:39–51.10.1111/nyas.12792
103
UherRTanseyKEDewTMaierWMorsOHauserJet alAn inflammatory biomarker as a differential predictor of outcome of depression treatment with escitalopram and nortriptyline. Am J Psychiatry (2014) 171(12):1278–86.10.1176/appi.ajp.2014.14010094
104
BangasserDAWiersielisKRKhantsisS. Sex differences in the locus coeruleus-norepinephrine system and its regulation by stress. Brain Res (2016) 1641(Pt B):177–88.10.1016/j.brainres.2015.11.021
Summary
Keywords
norepinephrine, α-adrenergic receptors, major depressive disorder, schizophrenia, valence systems, pathophysiology, antidepressants, antipsychotics
Citation
Maletic V, Eramo A, Gwin K, Offord SJ and Duffy RA (2017) The Role of Norepinephrine and Its α-Adrenergic Receptors in the Pathophysiology and Treatment of Major Depressive Disorder and Schizophrenia: A Systematic Review. Front. Psychiatry 8:42. doi: 10.3389/fpsyt.2017.00042
Received
20 December 2016
Accepted
01 March 2017
Published
17 March 2017
Volume
8 - 2017
Edited by
Luis F. Callado, University of the Basque Country, Spain
Reviewed by
Andrzej Pilc, Polish Academy of Sciences, Poland; Wladyslaw Lason, Polish Academy of Sciences, Poland; Jesús A. García-Sevilla, University of the Balearic Islands, Spain
Updates
Copyright
© 2017 Maletic, Eramo, Gwin, Offord and Duffy.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vladimir Maletic, vmaletic@bellsouth.net
Specialty section: This article was submitted to Neuropharmacology, a section of the journal Frontiers in Psychiatry
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.