 Alma Y. Galvez-Contreras1,2*
Alma Y. Galvez-Contreras1,2* Tania Campos-Ordonez3,4
Tania Campos-Ordonez3,4 Rocio E. Gonzalez-Castaneda1
Rocio E. Gonzalez-Castaneda1 Oscar Gonzalez-Perez3,5
Oscar Gonzalez-Perez3,5
- 1Department of Neuroscience, Institute of Translational Neuroscience, Centro Universitario de Ciencias de la Salud, University of Guadalajara, Guadalajara, Mexico
- 2Unidad de Atencion en Neurosciencias, Department of Neuroscience, Centro Universitario de Ciencias de la Salud, University of Guadalajara, Guadalajara, Mexico
- 3Laboratory of Neuroscience, School of Psychology, University of Colima, Colima, Mexico
- 4Medical Science PhD Program, School of Medicine, University of Colima, Colima, Mexico
- 5El Colegio de Colima, Colima, Mexico
Growth factors (GFs) are cytokines that regulate the neural development. Recent evidence indicates that alterations in the expression level of GFs during embryogenesis are linked to the pathophysiology and clinical manifestations of attention-deficit/hyperactivity disorder (ADHD) and autism spectrum disorders (ASD). In this concise review, we summarize the current evidence that supports the role of brain-derived neurotrophic factor, insulin-like growth factor 2, hepatocyte growth factor (HGF), glial-derived neurotrophic factor, nerve growth factor, neurotrophins 3 and 4, and epidermal growth factor in the pathogenesis of ADHD and ASD. We also highlight the potential use of these GFs as clinical markers for diagnosis and prognosis of these neurodevelopmental disorders.
Introduction
During neural development, a myriad of biological events occurs simultaneously, i.e., neurogenesis, gliogenesis, cellular migration, cell differentiation, synapse formation, etc. These neurobiological processes are orchestrated by several growth factors (GFs) and help shape the postnatal brain (1). In the postnatal brain, GFs have been extensively studied and most of them share similar cell functions to those reported in the neurodevelopment (2–4). Increasing evidence indicates that GFs modulate motor, emotional, and cognitive functions, which may explain several clinical manifestations of psychiatric disorders (5).
Neurodevelopmental disorders comprise a group of neurological conditions that are considered a public health problem with strong socioeconomic impact (6). These disorders have a very complex etiology and are characterized by early-onset during childhood. The initial pathological change appears to involve abnormal growth-factor expression during embryogenesis, which persist in the adulthood and may contribute to some clinical manifestations (6).
Some of the most common neurodevelopment disorders are autism spectrum disorder (ASD) and attention-deficit/hyperactivity disorder (ADHD). These disorders generate a poor global performance throughout life. ADHD and ASD have a complex etiology and their pathophysiology remains unclear. ADHD and ASD are often comorbid disorders (7) that share some morphological, molecular, and functional characteristics, such as: abnormal growth of neural tissue (8), cognitive impairment (9), male preponderance, epigenetic components (8, 10), and abnormal expression levels of GFs in serum and brain. Remarkably, the expression level of some growth-factor correlates with the clinical manifestations of ADHD and ASD (11). This evidence suggests the role of GFs in the pathophysiology of these disorders. Herein, we summarize the current evidence obtained in humans and animal models that associate GFs levels with ADHD and ASD (Table 1). This correlation unveils the attractive possibility to use GFs as serological biomarkers to establish diagnosis and prognosis for these disorders.
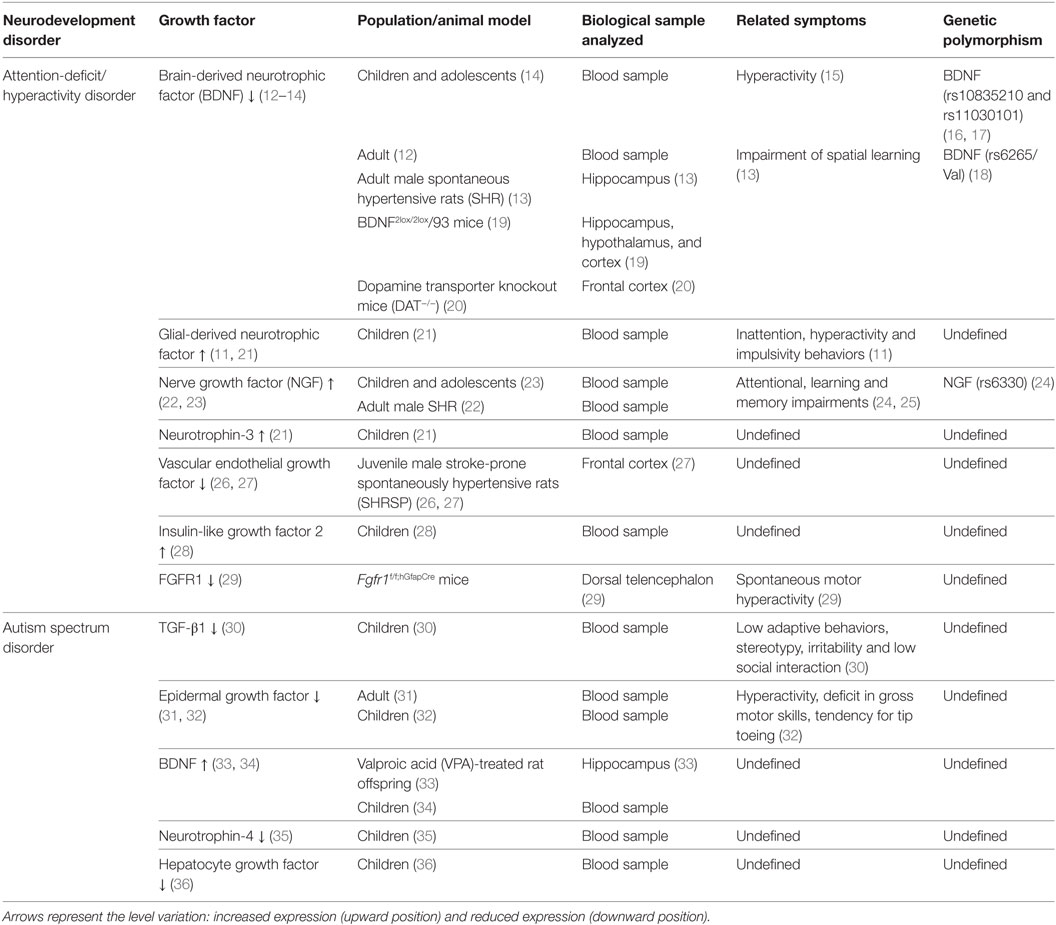
Table 1. Relationship between growth-factor levels and clinical symptoms in neurodevelopmental disorders.
Attention-Deficit/Hyperactivity Disorder
The ADHD has the highest incidence rate among all neurodevelopmental disorders (37). ADHD is characterized by inappropriate levels of inattention, hyperactivity, and impulsivity (15). These patients have evident social and academic problems that affect their global performance (38, 39). During the childhood, the main manifestation of ADHD is hyperactivity, which is commonly identified at the preschool, whereas inattention becomes more evident at elementary school (7). Children with ADHD also present negative emotionality, high emotional lability, and poor emotion management (39). During adolescence, patients with ADHD have high risk to suffer motor vehicle accidents, spontaneous sexual encounters, sexual diseases, unwanted pregnancies, drug abuse, poor social relationship, legal problems, etc. (37). Some of these ADHD symptoms persist until adulthood (37). In adults, ADHD induces a predisposition toward deficient relationships, substandard job performance, low socioeconomic status, and poor quality of life (37). ADHD is highly comorbid with other psychiatric or neurodevelopment disorders, such as oppositional defiant disorder, major depressive disorder, and anxiety disorders (37, 40).
Catecholamine dysfunction is the main hypothesis to explain ADHD pathophysiology; specifically, the dysfunction in dopamine receptors D4, D5, and in dopamine transporter proteins (15, 41) in prefrontal cortex, nucleus accumbens, striatum, substantial nigra, ventral tegmentum, and frontal cortex (6). The homeostasis of dopamine system requires the brain-derived neurotrophic factor (BDNF), a widely expressed neurotrophin in brain cortex and hippocampus (42). BDNF is critical in the synthesis, release, and uptake of dopamine in nigro-striatal dopaminergic neurons (43, 44) and plays a fundamental role in neuronal survival, plasticity, and proliferation (12). During development, BDNF and its receptors TrkB not only promote survival and differentiation of neurons but also are involved in neural plasticity in adulthood (13). Alteration in BDNF/TrkB activity is implicated in midbrain dopaminergic dysfunction reported in ADHD, which may explain the development of the main symptoms (45). Low serum levels of BDNF in ADHD can persist until adulthood (12). This indicates that BDNF signaling alteration occurs across life spam in patients with ADHD.
Polymorphisms rs11030101 and rs10835210 have been associated with a high risk to develop ADHD (16, 17). Some BDNF polymorphism are related to gender. The BDNF polymorphism rs6265/Val is more frequent in women with ADHD (18). This polymorphism has also been associated with susceptibility to neuroticism and anxiety (46), which may explain some psychiatry comorbid disorders. Conditional BDNF mice (BDNF2lox/2lox/93), characterized by low BDNF expression in the hippocampus, hypothalamus, and cortex, develop hyperactivity and aggression after stress (19), which supports the role of BDNF in the processing of motor control or in the worsening of hyperactivity symptoms of ADHD.
The role of BDNF in ADHD pathophysiology is not fully understood, but the evidence in animal research provides clues to understand the biochemical mechanism that underlie this condition. These patients show several alterations in cognitive process and memory performance (47) that may be due to hypoactivation of prefrontal cortex (48). In the spontaneous hypertensive rats, an animal model for ADHD, have been found low levels of BDNF and TrkB in the hippocampus that were related to memory impairment (13). These findings suggest that cognitive manifestations of ADHD might be associated with alterations in BDNF signaling. In dopamine transporter knockout mice (DAT−/−), another animal model for ADHD, low expression level of BDNF mRNA and TrkB receptors were found in frontal cortex (20). BDNF regulates two crucial circuits, the fronto-striatal-cerebellar and the ventral striatal-limbic circuits in normal brain (45). Neural circuits in prefrontal cortex and cerebellum, which modulate the attentional process, thoughts, emotions, social behavior, and motor control, are implicated in ADHD symptomatology (37).
Patients with ADHD show ~5% reduction in brain volume (15) in several regions, such as corpus callosum, orbitofrontal cortex, hippocampus, amygdala, basal ganglia, temporal lobe, prefrontal cortex, caudate, and cerebellum (49). Low levels in BDNF expression may explain this volume reduction, as demonstrated in late-onset forebrain-specific BDNF knockout (CaMK-BDNFKO) mice (50). On the other hand, patients with the pure form of ADHD lack microstructural changes in white matter tracts (40). In this study, the authors associated these microstructural changes with the clinical manifestations of ADHD and reported that the brain volume in gray and white matter correlates to poor cognitive processing, attention and motor planning (37). Interestingly, BDNF-deficient (bdnf−/−) mice show a significant reduction in myelin proteolipid protein and myelin basic protein in the hippocampus and cortex, with a subsequent deficit in myelination (15). Altogether, these data support the hypothesis that the BDNF signaling pathway is associated with changes of cognitive performance and brain structure in ADHD. Several pharmacological treatments that modulate the symptoms of ADHD can also modify GFs levels. Tricyclic antidepressants and the selective serotonin reuptake inhibitors increase the levels of BDNF (15). Methylphenidate, the main drug prescribed for ADHD, increases the BDNF expression in the prefrontal cortex (51, 52). A 6-week administration of methylphenidate recuperates the plasma levels of BDNF in children with ADHD (42).
Glial-derived neurotrophic factor (GDNF) is another GF-related to the pathophysiology of ADHD. GDNF is widely involved in the survival of serotonergic and dopaminergic neurons because it has neuroprotective effects against neuroinflammation and oxidative damage (21). Untreated children with ADHD show high plasma levels of GDNF (21). These high GDNF levels have a positive correlation with inattention, hyperactivity, and impulsivity behaviors (11), which are the main clinical manifestations of ADHD. Remarkably, psychostimulants, such as MPH, increase levels of GDNF mRNA in the hippocampus and prefrontal cortex (51). Furthermore, nerve growth factor (NGF) is involved in neuronal development and brain plasticity of cholinergic neurons that are important in attentional processing. Therefore, dysregulation of NGF levels have been associated with the pathophysiology of ADHD (23). At genetic levels, the single nucleotide polymorphism (rs6330) is associated with the risk of ADHD (24). High NGF levels are found in an animal model of ADHD (22). Interestingly, children and adolescents with ADHD show high NGF serum levels (23). These alterations in the pro-NGF and/or NGF levels are related to attentional, learning, and memory impairments shown by ADHD patients (24, 25). Neurotrophins also play a role in the pathophysiology of ADHD (21). Alterations in neurotrophin-3 (NTF3) expression are considered a risk factor for ADHD in childhood (2). Serum NTF3 levels are increased in untreated patients (21).
Vascular endothelial growth factor (VEGF) has an important role during brain development and repair (27). In stroke-prone spontaneously hypertensive rats were found downregulation of VEGF (26, 27), VEGFR-1 (Flt-1), and VEGFR-2 (Flk-1) receptors, endothelial nitric oxide synthase and the phosphorylated Akt in frontal cortex (26, 27). Since alterations on VEGF signaling have been associated with degeneration of cerebral cortex, it is possible that these alterations are implicated in cerebral abnormalities of patients with ADHD (26, 27).
Insulin-like growth factor 2 (IGF2) regulates normal development of cerebellum and hippocampus (50, 53), both of which are affected in ADHD (43, 44). Recently, IGF2 DNA methylation may be as predisposing factor to develop ADHD (33, 54, 55). In fact, prenatal exposure to high-fat and -sugar diet promotes IGF2 DNA methylation at birth that, in turn, has been positively associated with higher ADHD symptoms (28). Another GF that has also been implicated in hyperactive behavior include the fibroblast growth factor. In rodents, disruption of the Fgfr1 gene in dorsal telencephalon causes spontaneous motor hyperactivity and significant reductions in specific types of cortical inhibitory neurons (29). In humans, the role of FGFR in the etiology of ADHD has been suggested by pathway analysis of FGFR1b and FGFR2b activation, but further study is needed to support this assertion (56). In summary, the development of ADHD may be influenced by the interaction of multiple molecules and concomitant epigenetic factors, but confirmatory studies are required to reveal more definitive associations.
Autism Spectrum Disorder
Autism spectrum disorder is a neurodevelopmental disorder (35, 57) that is typically diagnosed between 2 and 6 years of age (8). ASD is characterized by impaired social communication, repetitive, or stereotyped behaviors and low interest for environment stimuli (7, 58). Patients with ASD have maladjustment in emotional response, anxiety, impaired emotional learning, limited interest in surrounding environment, and deficit in communication and social interactions (32, 59). ASD pathology has several associated symptoms that are generated by comorbid disorders. These comorbid symptoms often include: seizures, anxiety, intellectual impairment, hyperactivity, hyper or hypo- responsiveness to stimulus, sleep disruption, aggressive behavior, etc. (8). Consequently, ASD is considered a complex disorder with important epigenetic components (8, 60, 61).
Acetylation is a common feature of the neurotrophic proteins encoded by at least 18 genes dysregulated in patients with ASD (62, 63). While there is some evidence of the role of immune system dysregulation in the etiology of autism (64), it is possible that acetylation, lysine methylation/demethylation of histones, and inflammatory mediators affect mutual signaling pathways in both the nervous system and the immune system (65). Increasing evidence suggests that the abnormal increase of brain cortex and minicolumnar abnormalities observed in autism are driven by excess neuronal production (66–68). This hypothesis has been supported by neuroimaging studies (69–71) and three-dimensional neural cultures (a cerebral organoid model) with induced pluripotent stem cells (72). Since GFs regulate different aspects of neural development, including brain growth, stem cell proliferation, and cell survival, this evidence supports their role in the development of ASD (32) and may explain the enlargement of prefrontal and temporal cortex that persists until adulthood (73). The hyper-functioning in certain neural circuits observed in autism may be due to this uncontrolled growth of neural connections (35, 57, 74).
The brain volume is considered as clinical indicator of certain psychiatry disorder (75). In patients with ASD, the brain enlargement and abnormal neuronal migration have been observed in regions, such as the subependymal cell layer, the granule layer in the dentate gyrus, the cornu ammonis subfield, and the amygdala (76). In contrast to this abnormal migration and brain overgrowth, some authors have reported a low rate of growth in other brain areas (77). In the post-mortem brain of patients with ASD has been observed a significant reduction of neuronal density in layer III, the total number of neuron in layer III, V and VI, and a decrease in the volume of neurons in layers V and VI at the fusiform gyrus (77). Another study reported a decrease in pyramidal neuron size in the inferior frontal cortex, specifically in the Brodmann’s areas 44 and 45, which are brain regions involved in language processing, imitation function, and sociality processing (78).
Children with autism show low expression levels of Neurotrophin-4 (NTF4) in blood, which have been correlated to impairments in neuroplasticity (35). TGF-β1 plasma levels are also reduced in autism and have a significant correlation with the low scores obtained in adaptive behaviors, stereotypy, irritability, and low social interaction (30). Similar findings have been reported in juvenile mice in which the treatment with TGF-β1 impairs social interaction and promotes repetitive and stereotyped behaviors. Intriguingly, TGF-β1 overexpression has the opposite effects in adult stages (79).
Brain-derived neurotrophic factor serum levels are significantly increased in autism (34). High expression of BDNF is also found in a model of autism [valproic acid (VPA)-treated rat offspring] (54). In addition to the high BDNF expression in hippocampus, the VPA-treated rats show low expression of p-Akt, Bcl-2, p-CaMKII, as well as a significant increase in Bax and caspase-3 expression (33). This evidence is consistent with that found in BTBR T+tf/J mice (another autistic mouse strain), which present a significant upregulation of BDNF expression and myelin protein, and low expression of glial fibrillary acidic protein (55).
Another GF involved in the pathophysiology of ASD is the epidermal growth factor (EGF). EGF strongly promotes cell proliferation and differentiation via MAPK, PKC, and Akt pathways in neural progenitor cells (4). Recently, EGF and its receptor protein (EGFR) have been proposed as biomarkers of schizophrenia, depression, and bipolar disorder [reviewed in Ref. (5)]. Interestingly, adult patients with high-functioning ASD show a significant reduction in serum levels of EGF as compared with controls (31). Similar findings are observed in children with ASD (32), who present persistent low plasma levels of EGF until adulthood (80). Remarkably, these low plasma levels negatively correlates with the severity of hyperactivity, the deficit in gross motor skills, and the tendency for tip toeing (32). In addition, patients with ASD show a reduction in Akt phosphorylation, EGFR overexpression and low levels of gamma-aminobutyric acid that correlate to the severity of alterations in several language components (81). Elevated Akt phosphorylation has also been found in prenatal exposure to valproate, a well-known animal model of autism (82). These animals show a significant growth of several brain regions, a common alteration that is also observed in autistic patients. Taken together, these data strongly support the notion that EGFR/AKT pathway may play an important role in the pathophysiology of ASD.
Hepatocyte growth factor (HGF) is another molecule that has been involved in the development of ASD (36, 83). Although low levels of HGF have been reported in patients with ASD, these findings could not be correlated to the severity of symptoms (36). HGF promotes morphogenesis and cell proliferation after binding to the c-Met receptor, a product of the MET gene (84). Interestingly, polymorphisms in the MET gene appear to confer an increased susceptibility to autism and this gene is included in the chromosome 7q31 that has been linked to autism susceptibility (85, 86). This evidence suggests that HGF may represent an important factor in the pathogenesis of autism.
Future Approaches in Neurodevelopmental Disorders
Attention-deficit/hyperactivity disorder and ASD are neurodevelopmental disorders with significant comorbidity. Increasing evidence indicates that they share some pathological features and some etiological factors. Alterations in the expression level of BDNF, GDNF, NGF, NTF3, NTF4, or EGF are common features between ADHD and ASD (Figure 1). Therefore, identifying all these aspects will allow to establish the clinical use of these GFs as biomarkers in ADHD and ASD.
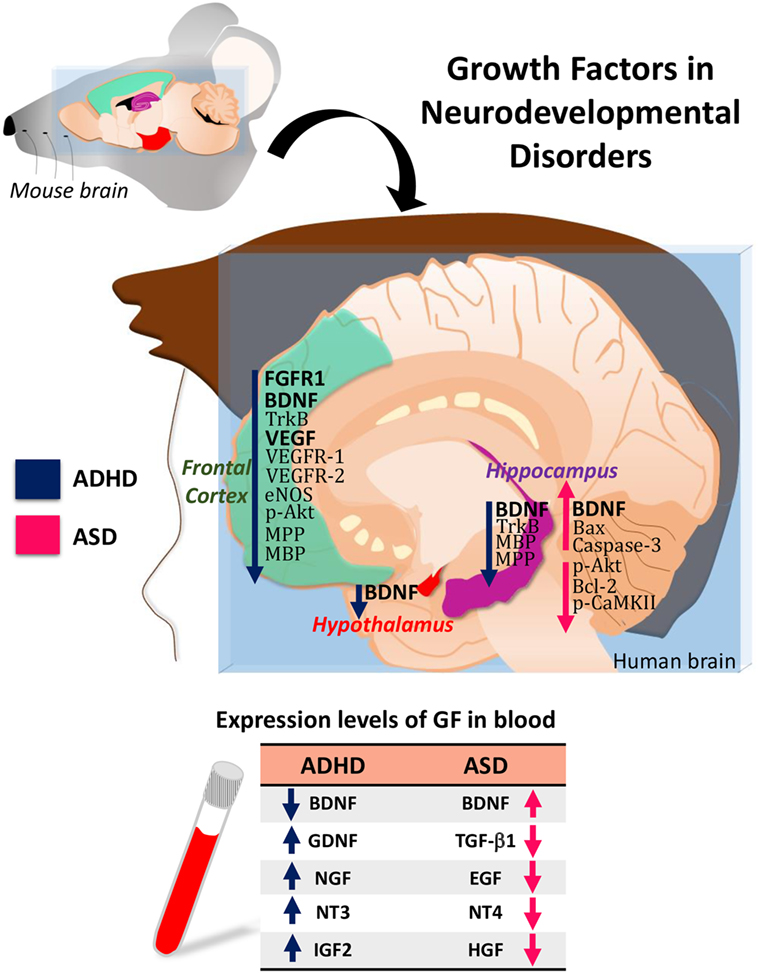
Figure 1. Hypothetical model of growth-factor expression in the human brain based on data obtained in attention-deficit/hyperactivity disorder (ADHD)/autism spectrum disorder (ASD) animal models. The table shows the blood levels of growth factors (GF) found in patients with ADHD or ASD.
Author Contributions
AG-C contributed in the conception idea for the article, in the manuscript writing, and in the table design. TC-O participated in the manuscript writing and in the table and the figure design. RG-C participated in the manuscript writing and in the table and figure design. OG-P contributed to the conception of the idea for the article and in the manuscript writing type.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was kindly supported by grants from Consejo Nacional de Ciencia y Tecnologia (CONACyT No. PN 2015-01-465 and INFR-280414), the Red Temática Células Troncales y Medicina Regenerativa (No. 271609), and the CONACyT’s Fellowship grant (No. 736339).
References
1. Costales J, Kolevzon A. The therapeutic potential of insulin-like growth factor-1 in central nervous system disorders. Neurosci Biobehav Rev (2016) 63:207–22. doi:10.1016/j.neubiorev.2016.01.001
2. Ribases M, Ramos-Quiroga JA, Hervas A, Bosch R, Bielsa A, Gastaminza X, et al. Exploration of 19 serotoninergic candidate genes in adults and children with attention-deficit/hyperactivity disorder identifies association for 5HT2A, DDC and MAOB. Mol Psychiatry (2009) 14(1):71–85. doi:10.1038/sj.mp.4002100
3. Galvez-Contreras AY, Gonzalez-Castaneda RE, Luquin S, Gonzalez-Perez O. Role of fibroblast growth factor receptors in astrocytic stem cells. Curr Signal Transduct Ther (2012) 7(1):81–6. doi:10.2174/157436212799278205
4. Galvez-Contreras AY, Quinones-Hinojosa A, Gonzalez-Perez O. The role of EGFR and ErbB family related proteins in the oligodendrocyte specification in germinal niches of the adult mammalian brain. Front Cell Neurosci (2013) 7:258. doi:10.3389/fncel.2013.00258
5. Galvez-Contreras AY, Campos-Ordonez T, Lopez-Virgen V, Gomez-Plascencia J, Ramos-Zuniga R, Gonzalez-Perez O. Growth factors as clinical biomarkers of prognosis and diagnosis in psychiatric disorders. Cytokine Growth Factor Rev (2016) 32:85–96. doi:10.1016/j.cytogfr.2016.08.004
6. Homberg JR, Kyzar EJ, Nguyen M, Norton WH, Pittman J, Poudel MK, et al. Understanding autism and other neurodevelopmental disorders through experimental translational neurobehavioral models. Neurosci Biobehav Rev (2016) 65:292–312. doi:10.1016/j.neubiorev.2016.03.013
7. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders DSM-V. Washington, DC: American Psychiatric Association (2013).
8. Pasciuto E, Borrie SC, Kanellopoulos AK, Santos AR, Cappuyns E, D’Andrea L, et al. Autism spectrum disorders: translating human deficits into mouse behavior. Neurobiol Learn Mem (2015) 124:71–87. doi:10.1016/j.nlm.2015.07.013
9. Craig F, Margari F, Legrottaglie AR, Palumbi R, de Giambattista C, Margari L. A review of executive function deficits in autism spectrum disorder and attention-deficit/hyperactivity disorder. Neuropsychiatr Dis Treat (2016) 12:1191–202. doi:10.2147/NDT.S104620
10. Visser JC, Rommelse NN, Greven CU, Buitelaar JK. Autism spectrum disorder and attention-deficit/hyperactivity disorder in early childhood: a review of unique and shared characteristics and developmental antecedents. Neurosci Biobehav Rev (2016) 65:229–63. doi:10.1016/j.neubiorev.2016.03.019
11. Shim SH, Hwangbo Y, Yoon HJ, Kwon YJ, Lee HY, Hwang JA, et al. Increased levels of plasma glial-derived neurotrophic factor in children with attention deficit hyperactivity disorder. Nord J Psychiatry (2015) 69(7):546–51. doi:10.3109/08039488.2015.1014834
12. Corominas-Roso M, Ramos-Quiroga JA, Ribases M, Sanchez-Mora C, Palomar G, Valero S, et al. Decreased serum levels of brain-derived neurotrophic factor in adults with attention-deficit hyperactivity disorder. Int J Neuropsychopharmacol (2013) 16(6):1267–75. doi:10.1017/s1461145712001629
13. Jeong HI, Ji ES, Kim SH, Kim TW, Baek SB, Choi SW. Treadmill exercise improves spatial learning ability by enhancing brain-derived neurotrophic factor expression in the attention-deficit/hyperactivity disorder rats. J Exerc Rehabil (2014) 10(3):162–7. doi:10.12965/jer.140111
14. Zeni CP, Tramontina S, Aguiar BW, Salatino-Oliveira A, Pheula GF, Sharma A, et al. BDNF Val66Met polymorphism and peripheral protein levels in pediatric bipolar disorder and attention-deficit/hyperactivity disorder. Acta Psychiatr Scand (2016) 134(3):268–74. doi:10.1111/acps.12587
15. Tsai SJ. Attention-deficit hyperactivity disorder and brain-derived neurotrophic factor: a speculative hypothesis. Med Hypotheses (2003) 60(6):849–51. doi:10.1016/S0306-9877(03)00052-5
16. Cho S-C, Kim H-W, Kim B-N, Kim J-W, Shin M-S, Chung S, et al. Gender-specific association of the brain-derived neurotrophic factor gene with attention-deficit/hyperactivity disorder. Psychiatry Investig (2010) 7(4):285–90. doi:10.4306/pi.2010.7.4.285
17. Kwon HJ, Ha M, Jin HJ, Hyun JK, Shim SH, Paik KC, et al. Association between BDNF gene polymorphisms and attention deficit hyperactivity disorder in Korean children. Genet Test Mol Biomarkers (2015) 19(7):366–71. doi:10.1089/gtmb.2015.0029
18. Li H, Liu L, Tang Y, Ji N, Yang L, Qian Q, et al. Sex-specific association of brain-derived neurotrophic factor (BDNF) Val66Met polymorphism and plasma BDNF with attention-deficit/hyperactivity disorder in a drug-naïve Han Chinese sample. Psychiatry Res (2014) 217(3):191–7. doi:10.1016/j.psychres.2014.03.011
19. Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, et al. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol (2001) 15(10):1748–57. doi:10.1210/mend.15.10.0706
20. Fumagalli F, Racagni G, Colombo E, Riva MA. BDNF gene expression is reduced in the frontal cortex of dopamine transporter knockout mice. Mol Psychiatry (2003) 8(11):898–9. doi:10.1038/sj.mp.4001370
21. Bilgiç A, Toker A, Işık Ü, Kılınç İ. Serum brain-derived neurotrophic factor, glial-derived neurotrophic factor, nerve growth factor, and neurotrophin-3 levels in children with attention-deficit/hyperactivity disorder. Eur Child Adolesc Psychiatry (2017) 26(3):355–63. doi:10.1007/s00787-016-0898-2
22. Clemow DB, Steers WD, Tuttle JB. Stretch-activated signaling of nerve growth factor secretion in bladder and vascular smooth muscle cells from hypertensive and hyperactive rats. J Cell Physiol (2000) 183(3):289–300. doi:10.1002/(SICI)1097-4652(200006)183:3<289:AID-JCP1>3.0.CO;2-6
23. Guney E, Ceylan MF, Kara M, Tekin N, Goker Z, Senses Dinc G, et al. Serum nerve growth factor (NGF) levels in children with attention deficit/hyperactivity disorder (ADHD). Neurosci Lett (2014) 560:107–11. doi:10.1016/j.neulet.2013.12.026
24. Syed Z, Dudbridge F, Kent L. An investigation of the neurotrophic factor genes GDNF, NGF, and NT3 in susceptibility to ADHD. Am J Med Genet B Neuropsychiatr Genet (2007) 144B(3):375–8. doi:10.1002/ajmg.b.30459
25. Tiveron C, Fasulo L, Capsoni S, Malerba F, Marinelli S, Paoletti F, et al. ProNGF\NGF imbalance triggers learning and memory deficits, neurodegeneration and spontaneous epileptic-like discharges in transgenic mice. Cell Death Differ (2013) 20(8):1017–30. doi:10.1038/cdd.2013.22
26. Jesmin S, Togashi H, Mowa CN, Ueno K, Yamaguchi T, Shibayama A, et al. Characterization of regional cerebral blood flow and expression of angiogenic growth factors in the frontal cortex of juvenile male SHRSP and SHR. Brain Res (2004) 1030(2):172–82. doi:10.1016/j.brainres.2004.10.004
27. Jesmin S, Togashi H, Sakuma I, Mowa CN, Ueno K, Yamaguchi T, et al. Gonadal hormones and frontocortical expression of vascular endothelial growth factor in male stroke-prone, spontaneously hypertensive rats, a model for attention-deficit/hyperactivity disorder. Endocrinology (2004) 145(9):4330–43. doi:10.1210/en.2004-0487
28. Rijlaarsdam J, Cecil CAM, Walton E, Mesirow MSC, Relton CL, Gaunt TR, et al. Prenatal unhealthy diet, insulin-like growth factor 2 gene (IGF2) methylation, and attention deficit hyperactivity disorder symptoms in youth with early-onset conduct problems. J Child Psychol Psychiatry (2017) 58(1):19–27. doi:10.1111/jcpp.12589
29. Smith KM, Fagel DM, Stevens HE, Rabenstein RL, Maragnoli ME, Ohkubo Y, et al. Deficiency in inhibitory cortical interneurons associates with hyperactivity in fibroblast growth factor receptor 1 mutant mice. Biol Psychiatry (2008) 63(10):953–62. doi:10.1016/j.biopsych.2007.09.020
30. Ashwood P, Enstrom A, Krakowiak P, Hertz-Picciotto I, Hansen R, Croen LA, et al. Decreased transforming growth factor beta1 in autism: a potential link between immune dysregulation and impairment in clinical behavioral outcomes. J Neuroimmunol (2008) 204(1–2):149–53. doi:10.1016/j.jneuroim.2008.07.006
31. Suzuki K, Hashimoto K, Iwata Y, Nakamura K, Tsujii M, Tsuchiya KJ, et al. Decreased serum levels of epidermal growth factor in adult subjects with high-functioning autism. Biol Psychiatry (2007) 62(3):267–9. doi:10.1016/j.biopsych.2006.08.001
32. Russo AJ. Decreased epidermal growth factor (EGF) associated with HMGB1 and increased hyperactivity in children with autism. Biomark Insights (2013) 8:35–41. doi:10.4137/BMI.S11270
33. van Mil NH, Steegers-Theunissen RPM, Bouwland-Both MI, Verbiest MMPJ, Rijlaarsdam J, Hofman A, et al. DNA methylation profiles at birth and child ADHD symptoms. J Psychiatr Res (2014) 49:51–9. doi:10.1016/j.jpsychires.2013.10.017
34. Qin X, Feng J, Cao C, Wu H, Loh Y, Cheng Y. Association of peripheral blood levels of brain-derived neurotrophic factor with autism spectrum disorder in children: a systematic review and meta-analysis. JAMA Pediatr (2016) 170(11):1079–86. doi:10.1001/jamapediatrics.2016.1626
35. Abdallah MW, Mortensen EL, Greaves-Lord K, Larsen N, Bonefeld-Jorgensen EC, Norgaard-Pedersen B, et al. Neonatal levels of neurotrophic factors and risk of autism spectrum disorders. Acta Psychiatr Scand (2013) 128(1):61–9. doi:10.1111/acps.12020
36. Russo AJ. Correlation between hepatocyte growth factor (HGF) and gamma-aminobutyric acid (GABA) plasma levels in autistic children. Biomark Insights (2013) 8:69–75. doi:10.4137/bmi.s11448
37. Sharma A, Couture J. A review of the pathophysiology, etiology, and treatment of attention-deficit hyperactivity disorder (ADHD). Ann Pharmacother (2014) 48(2):209–25. doi:10.1177/1060028013510699
38. Loe IM, Feldman HM. Academic and educational outcomes of children with ADHD. J Pediatr Psychol (2007) 32(6):643–54. doi:10.1093/jpepsy/jsl054
39. McQuade JD, Breaux RP. Are elevations in ADHD symptoms associated with physiological reactivity and emotion dysregulation in children? J Abnorm Child Psychol (2016):1–13. doi:10.1007/s10802-016-0227-8
40. Adisetiyo V, Tabesh A, Di Martino A, Falangola MF, Castellanos FX, Jensen JH, et al. Attention-deficit/hyperactivity disorder without comorbidity is associated with distinct atypical patterns of cerebral microstructural development. Hum Brain Mapp (2014) 35(5):2148–62. doi:10.1002/hbm.22317
41. Banaschewski T, Becker K, Scherag S, Franke B, Coghill D. Molecular genetics of attention-deficit/hyperactivity disorder: an overview. Eur Child Adolesc Psychiatry (2010) 19(3):237–57. doi:10.1007/s00787-010-0090-z
42. Amiri A, Torabi Parizi G, Kousha M, Saadat F, Modabbernia MJ, Najafi K, et al. Changes in plasma brain-derived neurotrophic factor (BDNF) levels induced by methylphenidate in children with attention deficit-hyperactivity disorder (ADHD). Prog Neuropsychopharmacol Biol Psychiatry (2013) 47:20–4. doi:10.1016/j.pnpbp.2013.07.018
43. Castellanos F, Lee PP, Sharp W, Jeffries NO, Greenstein DK, Clasen LS, et al. Developmental trajectories of brain volume abnormalities in children and adolescents with attention-deficit/hyperactivity disorder. JAMA (2002) 288(14):1740–8. doi:10.1001/jama.288.14.1740
44. Plessen KJ, Bansal R, Zhu H, Whiteman R, Amat J, Quackenbush GA, et al. Hippocampus and amygdala morphology in attention-deficit/hyperactivity disorder. Arch Gen Psychiatry (2006) 63(7):795–807. doi:10.1001/archpsyc.63.7.795
45. Liu DY, Shen XM, Yuan FF, Guo OY, Zhong Y, Chen JG, et al. The physiology of BDNF and its relationship with ADHD. Mol Neurobiol (2015) 52(3):1467–76. doi:10.1007/s12035-014-8956-6
46. Gadow KD, Roohi J, DeVincent CJ, Kirsch S, Hatchwell E. Association of COMT (Val158Met) and BDNF (Val66Met) gene polymorphisms with anxiety, ADHD and tics in children with autism spectrum disorder. J Autism Dev Disord (2009) 39(11):1542–51. doi:10.1007/s10803-009-0794-4
47. Mattfeld AT, Whitfield-Gabrieli S, Biederman J, Spencer T, Brown A, Fried R, et al. Dissociation of working memory impairments and attention-deficit/hyperactivity disorder in the brain. Neuroimage Clin (2016) 10:274–82. doi:10.1016/j.nicl.2015.12.003
48. Bollmann S, Ghisleni C, Poil SS, Martin E, Ball J, Eich-Höchli D, et al. Age-dependent and -independent changes in attention-deficit/hyperactivity disorder (ADHD) during spatial working memory performance. World J Biol Psychiatry (2017) 18(4):279–90. doi:10.3109/15622975.2015.1112034
49. Amico F, Stauber J, Koutsouleris N, Frodl T. Anterior cingulate cortex gray matter abnormalities in adults with attention deficit hyperactivity disorder: a voxel-based morphometry study. Psychiatry Res (2011) 191(1):31–5. doi:10.1016/j.pscychresns.2010.08.011
50. Ouchi Y, Banno Y, Shimizu Y, Ando S, Hasegawa H, Adachi K, et al. Reduced adult hippocampal neurogenesis and working memory deficits in the Dgcr8-deficient mouse model of 22q11.2 deletion-associated schizophrenia can be rescued by IGF2. J Neurosci (2013) 33(22):9408–19. doi:10.1523/jneurosci.2700-12.2013
51. Simchon-Tenenbaum Y, Weizman A, Rehavi M. Alterations in brain neurotrophic and glial factors following early age chronic methylphenidate and cocaine administration. Behav Brain Res (2015) 282:125–32. doi:10.1016/j.bbr.2014.12.058
52. Lukkes JL, Freund N, Thompson BS, Meda S, Andersen SL. Preventative treatment in an animal model of ADHD: behavioral and biochemical effects of methylphenidate and its interactions with ovarian hormones in female rats. Eur Neuropsychopharmacol (2016) 26(9):1496–506. doi:10.1016/j.euroneuro.2016.06.003
53. Pidsley R, Dempster E, Troakes C, Al-Sarraj S, Mill J. Epigenetic and genetic variation at the IGF2/H19 imprinting control region on 11p15.5 is associated with cerebellum weight. Epigenetics (2012) 7(2):155–63. doi:10.4161/epi.7.2.18910
54. Schuch V, Utsumi DA, Costa TVMM, Kulikowski LD, Muszkat M. Attention deficit hyperactivity disorder in the light of the epigenetic paradigm. Front Psychiatry (2015) 6(126):1–7. doi:10.3389/fpsyt.2015.00126
55. Walton E, Pingault JB, Cecil CAM, Gaunt TR, Relton CL, Mill J, et al. Epigenetic profiling of ADHD symptoms trajectories: a prospective, methylome-wide study. Mol Psychiatry (2017) 22(2):250–6. doi:10.1038/mp.2016.85
56. Mooney MA, McWeeney SK, Faraone SV, Hinney A, Hebebrand J, Consortium I, et al. Pathway analysis in attention deficit hyperactivity disorder: an ensemble approach. Am J Med Genet B Neuropsychiatr Genet (2016) 171(6):815–26. doi:10.1002/ajmg.b.32446
57. Onore C, Van de Water J, Ashwood P. Decreased levels of EGF in plasma of children with autism spectrum disorder. Autism Res Treat (2012) 2012:205362. doi:10.1155/2012/205362
58. Bambini-Junior V, Rodrigues L, Behr GA, Moreira JC, Riesgo R, Gottfried C. Animal model of autism induced by prenatal exposure to valproate: behavioral changes and liver parameters. Brain Res (2011) 1408:8–16. doi:10.1016/j.brainres.2011.06.015
59. Banerjee A, Engineer CT, Sauls BL, Morales AA, Kilgard MP, Ploski JE. Abnormal emotional learning in a rat model of autism exposed to valproic acid in utero. Front Behav Neurosci (2014) 8:387. doi:10.3389/fnbeh.2014.00387
60. Hill DS, Cabrera R, Wallis Schultz D, Zhu H, Lu W, Finnell RH, et al. Autism-like behavior and epigenetic changes associated with autism as consequences of in utero exposure to environmental pollutants in a mouse model. Behav Neurol (2015) 2015:426263. doi:10.1155/2015/426263
61. Castro K, Baronio D, Perry IS, Riesgo RDS, Gottfried C. The effect of ketogenic diet in an animal model of autism induced by prenatal exposure to valproic acid. Nutr Neurosci (2017) 20(6):343–50. doi:10.1080/1028415X.2015.1133029
62. Cohen OS, Varlinskaya EI, Wilson CA, Glatt SJ, Mooney SM. Acute prenatal exposure to a moderate dose of valproic acid increases social behavior and alters gene expression in rats. Int J Dev Neurosci (2013) 31(8):740–50. doi:10.1016/j.ijdevneu.2013.09.002
63. De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature (2014) 515(7526):209–15. doi:10.1038/nature13772
64. Meltzer A, Van de Water J. The role of the immune system in autism spectrum disorder. Neuropsychopharmacology (2017) 42(1):284–98. doi:10.1038/npp.2016.158
65. Gonzalez-Perez O, Gutierrez-Fernandez F, Lopez-Virgen V, Collas-Aguilar J, Quinones-Hinojosa A, Garcia-Verdugo JM. Immunological regulation of neurogenic niches in the adult brain. Neuroscience (2012) 226:270–81. doi:10.1016/j.neuroscience.2012.08.053
66. Casanova MF, Buxhoeveden DP, Switala AE, Roy E. Minicolumnar pathology in autism. Neurology (2002) 58(3):428–32. doi:10.1212/WNL.58.3.428
67. Casanova MF, van Kooten IAJ, Switala AE, van Engeland H, Heinsen H, Steinbusch HWM, et al. Minicolumnar abnormalities in autism. Acta Neuropathol (2006) 112(3):287. doi:10.1007/s00401-006-0085-5
68. Kaushik G, Zarbalis KS. Prenatal neurogenesis in autism spectrum disorders. Front Chem (2016) 4(12):1–7. doi:10.3389/fchem.2016.00012
69. Hazlett H, Poe M, Gerig G. Magnetic resonance imaging and head circumference study of brain size in autism: birth through age 2 years. Arch Gen Psychiatry (2005) 62(12):1366–76. doi:10.1001/archpsyc.62.12.1366
70. Courchesne E, Campbell K, Solso S. Brain growth across the life span in autism: age-specific changes in anatomical pathology. Brain Res (2011) 1380:138–45. doi:10.1016/j.brainres.2010.09.101
71. Lange N, Travers BG, Bigler ED, Prigge MBD, Froehlich AL, Nielsen JA, et al. Longitudinal volumetric brain changes in autism spectrum disorder ages 6–35 years. Autism Res (2015) 8(1):82–93. doi:10.1002/aur.1427
72. Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell (2015) 162(2):375–90. doi:10.1016/j.cell.2015.06.034
73. Ha S, Sohn I-J, Kim N, Sim HJ, Cheon K-A. Characteristics of brains in autism spectrum disorder: structure, function and connectivity across the lifespan. Exp Neurobiol (2015) 24(4):273–84. doi:10.5607/en.2015.24.4.273
74. Chomiak T, Turner N, Hu B. What we have learned about autism spectrum disorder from valproic acid. Patholog Res Int (2013) 2013:712758. doi:10.1155/2013/712758
75. Stevens MC, Haney-Caron E. Comparison of brain volume abnormalities between ADHD and conduct disorder in adolescence. J Psychiatry Neurosci (2012) 37(6):389–98. doi:10.1503/jpn.110148
76. Wegiel J, Kuchna I, Nowicki K, Imaki H, Marchi E, Ma SY, et al. The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol (2010) 119(6):755–70. doi:10.1007/s00401-010-0655-4
77. van Kooten IAJ, Palmen SJMC, von Cappeln P, Steinbusch HWM, Korr H, Heinsen H, et al. Neurons in the fusiform gyrus are fewer and smaller in autism. Brain (2008) 131(4):987–99. doi:10.1093/brain/awn033
78. Jacot-Descombes S, Uppal N, Wicinski B, Santos M, Schmeidler J, Giannakopoulos P, et al. Decreased pyramidal neuron size in Brodmann areas 44 and 45 in patients with autism. Acta Neuropathol (2012) 124(1):67–79. doi:10.1007/s00401-012-0976-6
79. Depino AM, Lucchina L, Pitossi F. Early and adult hippocampal TGF-beta1 overexpression have opposite effects on behavior. Brain Behav Immun (2011) 25(8):1582–91. doi:10.1016/j.bbi.2011.05.007
80. Toyoda T, Nakamura K, Yamada K, Thanseem I, Anitha A, Suda S, et al. SNP analyses of growth factor genes EGF, TGFbeta-1, and HGF reveal haplotypic association of EGF with autism. Biochem Biophys Res Commun (2007) 360(4):715–20. doi:10.1016/j.bbrc.2007.06.051
81. Russo AJ. Decreased phosphorylated protein kinase B (Akt) in individuals with autism associated with high epidermal growth factor receptor (EGFR) and low gamma-aminobutyric acid (GABA). Biomark Insights (2015) 10:89–94. doi:10.4137/BMI.S21946
82. Yang E-J, Ahn S, Lee K, Mahmood U, Kim H-S. Early behavioral abnormalities and perinatal alterations of PTEN/AKT pathway in valproic acid autism model mice. PLoS One (2016) 11(4):e0153298. doi:10.1371/journal.pone.0153298
83. Russo AJ, Krigsman A, Jepson B, Wakefield A. Decreased serum hepatocyte growth factor (HGF) in autistic children with severe gastrointestinal disease. Biomark Insights (2009) 4:181–90.
84. Naldini L, Weidner KM, Vigna E, Gaudino G, Bardelli A, Ponzetto C, et al. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J (1991) 10(10):2867–78.
85. Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S, et al. A genetic variant that disrupts MET transcription is associated with autism. Proc Natl Acad Sci U S A (2006) 103(45):16834–9. doi:10.1073/pnas.0605296103
Keywords: biomarker, developmental disorders, autism spectrum disorder, attention-deficit/hyperactivity disorder, growth factors, cytokines, cerebral cortex, cognitive impairment
Citation: Galvez-Contreras AY, Campos-Ordonez T, Gonzalez-Castaneda RE and Gonzalez-Perez O (2017) Alterations of Growth Factors in Autism and Attention-Deficit/Hyperactivity Disorder. Front. Psychiatry 8:126. doi: 10.3389/fpsyt.2017.00126
Received: 02 May 2017; Accepted: 29 June 2017;
Published: 13 July 2017
Edited by:
Hanna E. Stevens, University of Iowa, United StatesReviewed by:
Didem Oztop, Erciyes University, TurkeyKaren M. Smith, University of Louisiana at Lafayette, United States
Copyright: © 2017 Galvez-Contreras, Campos-Ordonez, Gonzalez-Castaneda and Gonzalez-Perez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alma Y. Galvez-Contreras, YWxtYS5nYWx2ZXpAYWNhZGVtaWNvcy51ZGcubXg=