 Zhewu Wang
Zhewu Wang Blaine Caughron3
Blaine Caughron3 M. Rita I. Young
M. Rita I. Young- 1Mental Health Service, Ralph H. Johnson VA Medical Center, Charleston, SC, United States
- 2Department of Psychiatry, Medical University of South Carolina, Charleston, SC, United States
- 3Research Service, Ralph H. Johnson VA Medical Center, Charleston, SC, United States
- 4Department of Otolaryngology – Head and Neck Surgery, Medical University of South Carolina, Charleston, SC, United States
Patients with posttraumatic stress disorder (PTSD) exhibit an increased state of inflammation. Various animal models for PTSD have shown some of the same immune imbalances as have been shown in human subjects with PTSD, and some of these studies are discussed in this review. However, animal studies can only indirectly implicate immune involvement in PTSD in humans. This review of mainly studies with human subjects focuses on dissecting the immunological role in the pathogenesis of PTSD following initial trauma exposure. It addresses both the inflammatory state associated with PTSD and the immune imbalance between stimulatory and inhibitory immune mediators, as well as variables that can lead to discrepancies between analyses. The concept of immunological treatment approaches is proposed for PTSD, as new treatments are needed for this devastating disorder that is affecting unprecedented numbers of Veterans from the long-standing wars in the Middle East and which affects civilians following severe trauma.
Immune Imbalances in Posttraumatic Stress Disorder (PTSD)
Posttraumatic stress disorder is a debilitating psychiatric disorder that follows trauma exposure. There are four symptom clusters that characterize PTSD: reliving the traumatic event, avoidance of situations reminiscent of the traumatic event, negative thoughts and mood, and hyperarousal. These symptoms are debilitating to function. Trauma exposure is a required risk factor for developing PTSD, but is not sufficient as not all who are exposed to trauma develop PTSD (1, 2). The complex phenotype of PTSD emerges from interactions among genetic, environmental, and other biological risk factors. Dissecting the causes of PTSD could identify individuals who would be at increased risk of developing PTSD following trauma exposure.
A number of studies assessing cytokine levels and, in a few instances, blood immune cell functions have provided support for immunological involvement in PTSD following an initial trauma event. Although somewhat inconsistent, the compilation of these studies points to immune alterations in PTSD that indicate the immunological balance is skewed toward a pro-inflammatory state (Table 1). This is supported by increased levels of pro-inflammatory cytokines such as IFN-γ, IL-6, TNF-α, and IL-17 in the plasma, and increased levels of immune stimulatory Th1 and inflammatory Th17 cells in the blood (3–8). The increase in levels of the pro-inflammatory mediators IL-12 and IFN-γ in plasma of PTSD subjects is associated with multiple genetic and epigenetic modifications in peripheral blood mononuclear cells (9).
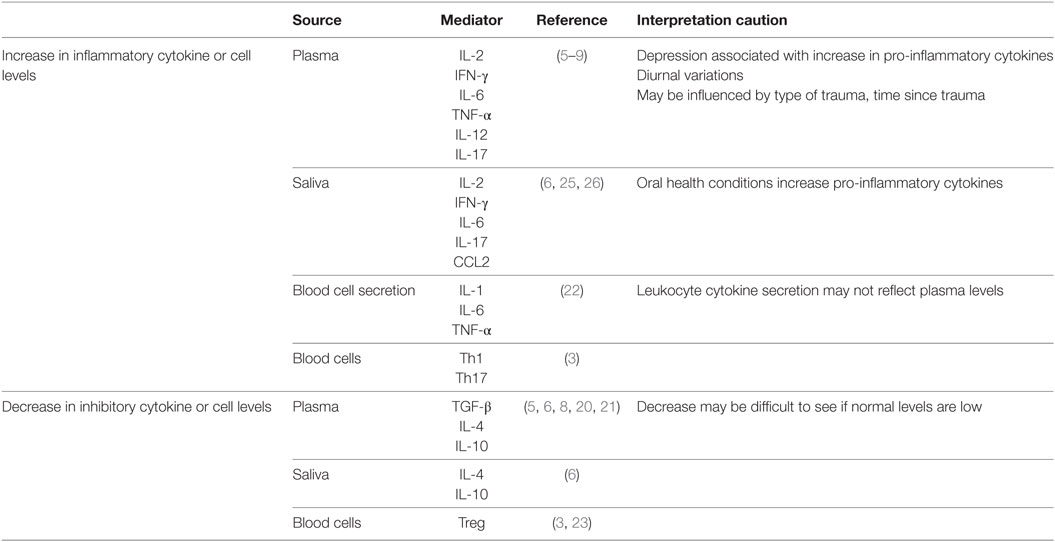
Table 1. Correlative association between posttraumatic stress disorder and immune imbalance in humans.
Multiple molecular genetics studies that have sought to pinpoint genetic variations associated with the risk for PTSD after trauma exposure (10). Some of these include genes encoding mediators involved in immune regulation. The best studied is the FK506-binding protein 5 gene, FKBP5, which encodes an immune regulatory immunophilin (11). FKBP5 gene variants moderate the effect of trauma exposure on the risk of PTSD. The minor allele of FKBP5 SNPs rs1360780 was associated with increased hurricane-associated PTSD (12). In subjects with chronic pain, heterozygous T-allele carriers of the FKBP5 SNP rs9470080 were associated with an increased risk for PTSD (13). A study of mainly African-American subjects who had experienced severe childhood trauma showed that those who carried minor alleles of FKBP5 SNPs rs9296158, rs3800373, rs1360780, or rs9470080 were more likely to exhibit PTSD (14).
In addition to FKBP5 polymorphisms, there are associations between variants in the pro-inflammatory C-reactive protein (CRP) and PTSD. A study of mainly African-American inner-city individuals who had been exposed to trauma showed a link between a CRP SNP, rs1130864, and the risk of PTSD, with the most prominent symptom of being overly alert (15). This risk SNP was also associated with increased serum CRP levels, which indicates an inflammatory state.
Multiple studies have indicated that immune hyperactivation could be a predictor of PTSD risk. For example, high levels of CRP in Marines before their deployment were predictive of PTSD following deployment; this study suggested inflammation to be a contributor to PTSD (16). Blood levels of inflammatory cytokines were increased in hospitalized patients with traumatic orthopedic injuries who subsequently developed PTSD (8). In a pre- and post-deployment analysis that used whole-transcriptome RNA-Seq gene expression of blood from U.S. Marines, both pairs of samples from subjects who developed PTSD over-expressed genes enriched for immune responses (17). This supports the concept that immune hyperactivation before trauma exposure could predict PTSD.
Some studies have shown that increases in inflammatory mediators correlate with PTSD severity (3), although others have not (18). A polymorphism in the gene of the inflammatory marker, TNF-α (rs1800629) was associated with PTSD in Vietnam war combat Veterans, and correlated with PTSD severity (19). While rs1800629 was a risk genotype for PTSD severity, serum levels of TNF-α were associated with symptom severity, but only trended to significance when controlling for covariates.
Although studies have shown increases in pro-inflammatory mediators in subjects with PTSD, fewer have measured inhibitory cytokines (5, 8, 20). In such studies, PTSD subjects have generally lower inhibitory cell levels such as Treg and reduced levels of the inhibitory mediators TGF-β and IL-4 (3, 6) in the blood. A comparison of individuals who were exposed to urban violence-associated trauma showed that those with PTSD had lower blood levels of the inhibitory mediator IL-10 than those who were resilient to the trauma (21). The importance of these deficiencies in immune inhibitory regulators is that a healthy immune status is composed of a balance of stimulatory and inhibitory cells and mediators. In studies where balances in stimulatory and inhibitory mediators were assessed, immune skewing was toward the pro-inflammatory direction in Veterans and civilians with PTSD (6, 8).
Most studies examining the changes in cytokine levels in PTSD have measured cytokine levels in the blood. However, several studies have also examined the sources of the pro-inflammatory cytokines. Blood leukocytes of war-exposed refugees with PTSD spontaneously produced increased levels of IL-1, IL-6, and TNF-α than did leukocytes of controls, although plasma cytokine levels were similar among the two groups of subjects (22). Also shown in this study was a direct correlation between PTSD severity and spontaneous secretion of IL-6 and TNF-α by the leukocytes. Stimulation of the PTSD subjects’ blood leukocytes with LPS further increased IL-6 production to higher levels than those produced by leukocytes of controls. A separate analysis of peripheral blood of combat Veterans with PTSD showed increases in blood pro-inflammatory Th1 and Th17 subsets and a reduction in the number of inhibitory Treg (3). Reduced levels of Treg cells were similarly seen in war-exposed civilians with PTSD as compared to exposed civilians without PTSD or controls (23). In contrast, a separate study comparing war Veterans to age-matched healthy controls showed reduced T-cell production of pro-inflammatory cytokines IL-2 and IFN-γ (24). Possible reasons for the discrepancies among some of these studies are discussed below.
There have been a few studies that also examined levels of immune mediators in saliva. Similar to the blood observations, inflammatory mediator levels in saliva were increased following stress exposure (25, 26). Veterans with PTSD had higher levels of pro-inflammatory mediators IL-2, IFN-γ, IL-6, and IL-17 and reduced levels of the inhibitory mediators IL-4 and IL-10 in saliva compared to Veterans without combat-related PTSD (6). In this latter study, it was shown that the immune cytokine imbalances in PTSD patients are more prominently expressed in saliva than in blood (6). A study of hurricane survivors with PTSD showed that the increased saliva levels of the inflammatory mediator CCL2 (MCP-1) correlated with PTSD severity (25). The origin of cytokines in saliva and whether salivary cytokine levels are a reflection of blood levels has been questioned, but increases in inflammatory cytokines have appeared rapidly in saliva following acute stress and could reflect mental health status (26–28).
Comorbidities need to be considered when studying immune imbalances in saliva, plasma, or blood cells of PTSD subjects. For example, oral health increases salivary cytokine levels in individuals with periodontitis, gingivitis, premalignant oral lesions, and oral cancer (29, 30). Depression, which is common in subjects with PTSD, also increases levels of inflammatory cytokines (31). However, several studies that examined the contribution of depression to the increases in inflammatory cytokines in subjects with PTSD showed that such increases were independent of a depression diagnosis (6, 32). Other variables that can impact on cytokine or immune cell analyses could include treatments and recovery from PTSD. Traumatized women with PTSD had increased plasma levels of IL-6, but those who had recovered from PTSD had the same lower levels as did healthy controls (33). The reduced levels of Treg in war-exposed PTSD subjects were restored to levels of healthy controls following narrative exposure therapy (23). Cytokine levels can also be influenced by technical complications. For example, cytokines such as IL-1 and IL-6 exhibit diurnal variations, which can contribute to differing results among studies (34, 35). The type of trauma can also impact on cytokine measurements. This was highlighted by an analysis showing that interpersonal-related traumas had distinct gene expression signatures from combat-related traumas, but there was convergence on immune cascades between the different trauma categories (36).
More difficult to control in studies with human subjects is the impact of the duration between the PTSD-associated trauma and the time of immunological analysis, in particular if the immunological skewing is a predisposing factor for PTSD (37). However, this can be controlled in animal models of PTSD. Using the stress-enhanced fear learning model, IL-1 expression was shown to rapidly increase (within 6 h) in the dorsal hippocampus and remain increased for the 72-h duration of assessments (38). Brain levels of IL-α, IL-6, and TNF-α were also increased within 1 day of stress re-exposure in a rat predator stress model for PTSD (39). Also using the predator model, a separate study showed increased brain levels of the inflammatory mediator IL-1 and reduced levels of the inhibitory mediators IL-4 and IL-10 after 7 days following re-exposure (40). While these studies in animal models of PTSD demonstrate dysregulation of cytokines within the brain within a short period of time following stress exposure, there is a deficiency in studies to demonstrate the duration of this immune imbalance. This leaves a gap in understanding the kinetics of the immune imbalances associated with PTSD as diagnosis, immune analysis, and treatment for humans with PTSD typically occur considerably later after exposure to the traumatic event. However, these animal models are in a unique position to determine whether immunological dysregulation is a consequence or a cause of PTSD; this is further discussed below.
Clinical Impacts of Immune Imbalances in PTSD
Associated with PTSD is not only inflammation but its consequence on poorer health outcomes. For example, health-related quality of life was lower in military persons with PTSD who had higher plasma levels of IL-6 (41). The pro-inflammatory milieu of subjects with PTSD may predispose them to autoimmune diseases since combat Veterans with PTSD have an increased incidence of thyroiditis, inflammatory bowel disease, multiple sclerosis, rheumatoid arthritis, and systemic lupus erythematosus (42). Patients exhibiting trauma-related symptoms had a slower neutrophil recovery time following stem cell transplantation than those without PTSD (43). Other comorbidities of subjects with PTSD include an increased risk of coronary heart disease (25, 44). A study of a Vietnam Era Twin Registry showed that the rate of coronary heart disease in twins with PTSD was twice that of twins without PTSD (44). The increased risk of coronary heart disease in PTSD subjects could be due to the increased levels of immune chemokines such as CCL-2, which recruits monocytes toward the inflamed endothelium (25). Monocytes, whose role has not been extensively examined in PTSD, have upregulation of target genes of the pro-inflammatory NF-κB/Rel family of transcription factors, which further contributes to the inflammatory state of PTD subjects (45).
It could also be argued that the increase in autoimmune diseases and other immune-associated comorbidities of PTSD are not the result of PTSD but are predisposing factors for developing PTSD following a traumatic event. Although the immune skewing in PTSD subjects toward an inflammatory state is becoming established, it is difficult to design studies with human subjects to test causality between immune dysregulation and PTSD. However, triggering of CNS neuroinflammation by peripheral inflammation has been shown in animal models (38, 46–55). Activation of the peripheral immune system by endotoxin injection triggered neuroinflammation in the brain (49, 50). Studies with animal models have also shown the requirement for functional immune competence for anxiety behavior to be evident following stress sensitization. For example, neutralization of the inflammatory cytokine IL-1 lessened the maladaptation to acute psychological stress (38). Also shown was monocyte recruitment from the periphery to the brain in stress-sensitized mice, but splenectomy before stress sensitization prevented monocyte migration to the brain and, in turn, prevented anxiety is stress-sensitized mice (38, 46–55).
Studies with human subjects have been less frequent and less definitive, but nevertheless suggest the contribution of peripheral inflammation to neuroinflammation and behavior (Table 2). Traumatic brain injury resulting from military deployment is associated with inflammation and, more recently, this has been associated with a higher extent of PTSD comorbidity (56). This study speculated that the chronic inflammatory state associated with the traumatic brain injury results in elevated cytokine levels in the central nervous system and, in turn, microglial over-activation. In a clinical trial, patients with acute respiratory distress who were treated with GM-CSF had more severe PTSD symptoms than those treated with placebo (57). Since GM-CSF stimulates proliferation and differentiation of hematopoietic cells and can cross the blood–brain barrier, this study suggested the possibility of GM-CSF stimulating either brain microglia or production of inflammatory cytokines within the brain to result in more severe PTSD.
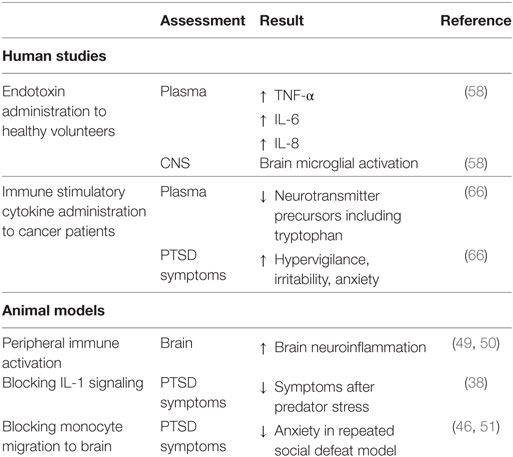
Table 2. Suggestions of causal associations between immune imbalances and posttraumatic stress disorder (PTSD).
While not directly studied, it could be expected that increased persistent infection could stimulate a chronic inflammatory state to, in turn, increase vulnerability to PTSD following trauma. This concept could be in part supported by studies with human subjects in which healthy volunteers who systemically received endotoxin exhibited stimulated peripheral levels of inflammatory mediators TNF-α, IL-6, and IL-8, and microglial activation in the brain (58). Once activated, brain microglia release mediators such as nitric oxide, IL-1, IL-6, TNF-α, and glutamate, which impact neurotransmission, neuronal apoptosis, neuroendocrine function, neural plasticity, and behavior (59–64). Induction of peripheral inflammation in healthy human volunteers via typhoid vaccination resulted in functional impairments in the form of reduced spatial memory performance (65). The impact of peripheral immune activation on behavior was inadvertently made evident by immune-activating treatment of cancer patients who resulted in symptoms characteristic of PTSD such as hypervigilance, irritability, anxiety, and decreased concentrations of tryptophan, a precursor to the neurotransmitter serotonin (66). The serotonergic axis influences mood, aggression, arousal, anxiety, sleep, learning, nociception, fear, and appetite (67).
PTSD Treatments in the Context of Immune Imbalances
A few studies have examined the impact of therapies for PTSD on the inflammatory status of patients with PTSD, although more of these studies have been conducted in mouse PTSD models. A study with PTSD patients showed that narrative exposure psychotherapy improved PTSD symptoms and restored the levels of immune inhibitory Treg cells, which were reduced before treatment (23). However, the immune imbalances were not fully corrected as the proportion of naïve T-cell remained low relative to memory T-cells, suggesting premature immune senescence. PTSD subjects who received pharmacotherapy with selective serotonin reuptake inhibitors (SSRIs) improved clinically and showed reductions in levels of the pro-inflammatory mediator IL-1 (68). In a mouse model of PTSD, treatment with the SSRI inhibitor fluoxetine prevented stress-induced inflammatory gene expression and improved PTSD-like symptoms (69). The results of this study indicated the role of inflammation in PTSD pathology and suggested using anti-inflammatory agents to treat PTSD. A rat study of PTSD showed that treatment with ibuprofen to directly target inflammation reduced both levels of inflammatory cytokines and PTSD-like symptoms (70). Similarly, treating mice with intraperitoneal injections of COX-2 inhibitors to diminish inflammation attenuated their PTSD-like symptoms and reduced neuronal excitability in the basolateral amygdala (71).
While studies showing immune restoration with PTSD treatment suggest a psycho-neuro-immune relationship, it is not possible to determine if treatment impacted the PTSD disorder to, in turn, lessen the extent of inflammation, or if the restoration of immune balance led to PTSD psychological improvement. SSRIs, by modulating serotonin levels, may be influencing immune function since serotonin has been shown to be an immune modulator (72–74). There remains a need for studies to determine the associations between improvements in PTSD clinical status and the immune rebalancing.
Despite the availability of psychological and pharmacological treatments for PTSD, approximately half of combat Veterans do not respond favorably to treatments (75). Therefore, studies need to be expanded to assess if PTSD treatment responsiveness or resistance is associated with a respective immune rebalancing or resistance to rebalancing. If the pro-inflammatory state contributes to PTSD, and if inflammation-skewed leukocytes of PTSD subjects still retain plasticity, then there is the opportunity for immune redirection as an additional PTSD treatment approach. Such immune rebalancing approaches have become more common in treating inflammation-associated diseases, such as rheumatoid arthritis or type 1 diabetes, and as treatments for cancer (76–79).
The plasticity of immune T-cells and monocytes provides optimism that the inflammation-skewed immune state in patients with PTSD can be rebalanced. This rebalancing could be driven by the composition of the cytokine milieu (80, 81). Inflammatory Th17 cells share a common lineage with inhibitory Treg cells, and their plasticity is evident by examples of one phenotype differentiating from the other (82). Although cytokines have been widely used to define immune plasticity, a pharmaco-immunological approach may be a more practical means by which to attain immune rebalancing. For example, the STAT3 inhibitor STA-21 was effective in restoring immune balance in a mouse model of inflammatory arthritis (80). Studies in both mouse models and in humans have shown vitamin D metabolites can restore immune balance in several different clinical conditions (83–86). These alternative agents could be used in future clinical studies to restore immune balance in Veterans and civilians, with the goal of tempering the clinical course of their PTSD.
Conclusion
The interconnections between immune imbalances and PTSD are becoming better defined, but much is left to be resolved. It is clear that PTSD is associated with a pro-inflammatory state but whether this contributes to the symptoms of PTSD or whether it is a consequence of disease has yet to be clarified. There is, however, increasing evidence that hyperinflammation is not only a biomarker for PTSD but increases the risk of PTSD following trauma. This is important to enable identification of those at risk for PTSD that could, for example, result from military combat exposure. It could also expose new opportunities for prevention and treatment of PTSD. Immune modulating approaches are accepted means of treating various disorders such as autoimmune diseases. Unraveling the psycho-neuro-immunological interplay in PTSD is an ongoing challenge that could results in effective means to prevent and to treat PTSD.
Author Contributions
This manuscript was written by MY with close collaboration and contribution by ZW and BC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Clinical Sciences Research and Development Program of the Department of Veterans Affairs (Merit Review grants I01 CX000851 to MY and 1 I01 CX000487 to ZW).
References
1. Dursa EK, Reinhard MJ, Barth SK, Schneiderman AI. Prevalence of a positive screen for PTSD among OEF/OIF and OEF/OIF-era veterans in a large population-based cohort. J Trauma Stress (2014) 27:542–9. doi:10.1002/jts.21956
2. Creamer M, Burgess P, McFarlane AC. Post-traumatic stress disorder: findings from the Australian National Survey of Mental Health and Well-being. Psychol Med (2001) 31:1237–47. doi:10.1017/S0033291701004287
3. Zhou J, Nagarkatti P, Zhong Y, Ginsberg JP, Singh NP, Zhang J, et al. Dysregulation in microRNA expression is associated with alterations in immune functions in combat veterans with post-traumatic stress disorder. PLoS One (2014) 9:e94075. doi:10.1371/journal.pone.0094075
4. Sommershof A, Aichinger H, Engler H, Adenauer H, Catani C, Boneberg EM, et al. Substantial reduction of naive and regulatory T cells following traumatic stress. Brain Behav Immun (2009) 23:1117–24. doi:10.1016/j.bbi.2009.07.003
5. von Kanel R, Hepp U, Kraemer B, Traber R, Keel M, Mica L, et al. Evidence for low-grade systemic proinflammatory activity in patients with posttraumatic stress disorder. J Psychiatr Res (2007) 41:744–52. doi:10.1016/j.jpsychires.2006.06.009
6. Wang Z, Mandel H, Levingston CA, Young MRI. An exploratory approach demonstrating immune skewing and a loss of coordination among cytokines in plasma and saliva of veterans with combat-related PTSD. Hum Immunol (2016) 77:652–7. doi:10.1016/j.humimm.2016.05.018
7. Guo M, Liu T, Guo JC, Jiang XL, Chen F, Gao YS. Study on serum cytokine levels in posttraumatic stress disorder patients. Asian Pac J Trop Med (2012) 5:323–5. doi:10.1016/s1995-7645(12)60048-0
8. Cohen M, Meir T, Klein E, Volpin G, Assaf M, Pollack S. Cytokine levels as potential biomarkers for predicting the development of posttraumatic stress symptoms in casualties of accidents. Int J Psychiatry Med (2011) 42:117–31. doi:10.2190/PM.42.2.b
9. Bam M, Yang X, Zhou J, Ginsberg JP, Leyden Q, Nagarkatti PS, et al. Evidence for epigenetic regulation of pro-inflammatory cytokines, interleukin-12 and interferon-γ, in peripheral blood mononuclear cells from PTSD patients. J Neuroimmune Pharmacol (2016) 11:168–81. doi:10.1007/s11481-015-9643-8
10. Voisey J, Young RM, Lawford BR, Morris CP. Progress towards understanding the genetics of posttraumatic stress disorder. J Anxiety Disord (2014) 28:873–83. doi:10.1016/j.janxdis.2014.09.014
11. Romano S, D’Angelillo A, Staibano S, Simeone E, D’Arrigo P, Ascierto PA, et al. Immunomodulatory pathways regulate expression of a spliced FKBP51 isoform in lymphocytes of melanoma patients. Pigment Cell Melanoma Res (2015) 28:442–52. doi:10.1111/pcmr.12378
12. Dunn EC, Solovieff N, Lowe SR, Gallagher PJ, Chaponis J, Rosand J, et al. Interaction between genetic variants and exposure to Hurricane Katrina on post-traumatic stress and post-traumatic growth: a prospective analysis of low income adults. J Affect Disord (2014) 152-154:243–9. doi:10.1016/j.jad.2013.09.018
13. Boscarino JA, Erlich PM, Hoffman SN, Zhang X. Higher FKBP5, COMT, CHRNA5, and CRHR1 allele burdens are associated with PTSD and interact with trauma exposure: implications for neuropsychiatric research and treatment. Neuropsychiatr Dis Treat (2012) 8:131–9. doi:10.2147/ndt.s29508
14. Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA (2008) 299:1291–305. doi:10.1001/jama.299.11.1291
15. Michopoulos V, Rothbaum AO, Jovanovic T, Almli LM, Bradley B, Rothbaum BO, et al. Association of CRP genetic variation and CRP level with elevated PTSD symptoms and physiological responses in a civilian population with high levels of trauma. Am J Psychiatry (2015) 172:353–62. doi:10.1176/appi.ajp.2014.14020263
16. Eraly SA, Nievergelt CM, Maihofer AX, Barkauskas DA, Biswas N, Agorastos A, et al. Assessment of plasma C-reactive protein as a biomarker of posttraumatic stress disorder risk. JAMA Psychiatry (2014) 71:423–31. doi:10.1001/jamapsychiatry.2013.4374
17. Breen MS, Maihofer AX, Glatt SJ, Tylee DS, Chandler SD, Tsuang MT, et al. Gene networks specific for innate immunity define post-traumatic stress disorder. Mol Psychiatry (2015) 20:1538–45. doi:10.1038/mp.2015.9
18. Lindqvist D, Dhabhar FS, Mellon SH, Yehuda R, Grenon SM, Flory JD, et al. Increased pro-inflammatory milieu in combat related PTSD – a new cohort replication study. Brain Behav Immun (2017) 59:260–4. doi:10.1016/j.bbi.2016.09.012
19. Bruenig D, Mehta D, Morris CP, Harvey W, Lawford B, Young RM, et al. Genetic and serum biomarker evidence for a relationship between TNFα and PTSD in Vietnam war combat veterans. Compr Psychiatry (2017) 74:125–33. doi:10.1016/j.comppsych.2017.01.015
20. Newton TL, Fernandez-Botran R, Miller JJ, Burns VE. Interleukin-6 and soluble interleukin-6 receptor levels in posttraumatic stress disorder: associations with lifetime diagnostic status and psychological context. Biol Psychol (2014) 99:150–9. doi:10.1016/j.biopsycho.2014.03.009
21. Teche SP, Rovaris DL, Aguiar BW, Hauck S, Vitola ES, Bau CHD, et al. Resilience to traumatic events related to urban violence and increased IL10 serum levels. Psychiatry Res (2017) 250:136–40. doi:10.1016/j.psychres.2017.01.072
22. Gola H, Engler H, Sommershof A, Adenauer H, Kolassa S, Schedlowski M, et al. Posttraumatic stress disorder is associated with an enhanced spontaneous production of pro-inflammatory cytokines by peripheral blood mononuclear cells. BMC Psychiatry (2013) 13:40. doi:10.1186/1471-244x-13-40
23. Morath J, Gola H, Sommershof A, Hamuni G, Kolassa S, Catani C, et al. The effect of trauma-focused therapy on the altered T cell distribution in individuals with PTSD: evidence from a randomized controlled trial. J Psychiatr Res (2014) 54:1–10. doi:10.1016/j.jpsychires.2014.03.016
24. Jergovic M, Tomicevic M, Vidovic A, Bendelja K, Savic A, Vojvoda V, et al. Telomere shortening and immune activity in war veterans with posttraumatic stress disorder. Prog Neuropsychopharmacol Biol Psychiatry (2014) 54:275–83. doi:10.1016/j.pnpbp.2014.06.010
25. An K, Salyer J, Kao HF. Psychological strains, salivary biomarkers, and risks for coronary heart disease among hurricane survivors. Biol Res Nurs (2015) 17:311–20. doi:10.1177/1099800414551164
26. Slavish DC, Graham-Engeland JE, Smyth JM, Engeland CG. Salivary markers of inflammation in response to acute stress. Brain Behav Immun (2015) 44:253–69. doi:10.1016/j.bbi.2014.08.008
27. Minetto MA, Gazzoni M, Lanfranco F, Baldi M, Saba L, Pedrola R, et al. Influence of the sample collection method on salivary interleukin-6 levels in resting and post-exercise conditions. Eur J Appl Physiol (2007) 101:249–56. doi:10.1007/s00421-007-0484-x
28. Izawa S, Sugaya N, Kimura K, Ogawa N, Yamada KC, Shirotsuki K, et al. An increase in salivary interleukin-6 level following acute psychosocial stress and its biological correlates in healthy young adults. Biol Psychol (2013) 94:249–54. doi:10.1016/j.biopsycho.2013.06.006
29. Ebersole JL, Nagarajan R, Akers D, Miller CS. Targeted salivary biomarkers for discrimination of periodontal health and disease(s). Front Cell Infect Microbiol (2015) 5:62. doi:10.3389/fcimb.2015.00062
30. Juretic M, Cerovic R, Belusic-Gobic M, Brekalo Prso I, Kqiku L, Spalj S, et al. Salivary levels of TNF-α and IL-6 in patients with oral premalignant and malignant lesions. Folia Biol (2013) 59:99–102.
31. van Dooren FE, Schram MT, Schalkwijk CG, Stehouwer CD, Henry RM, Dagnelie PC, et al. Associations of low grade inflammation and endothelial dysfunction with depression – the Maastricht Study. Brain Behav Immun (2016) 56:390–6. doi:10.1016/j.bbi.2016.03.004
32. Lindqvist D, Wolkowitz OM, Mellon S, Yehuda R, Flory JD, Henn-Haase C, et al. Proinflammatory milieu in combat-related PTSD is independent of depression and early life stress. Brain Behav Immun (2014) 42:81–8. doi:10.1016/j.bbi.2014.06.003
33. Gill JM, Saligan L, Lee H, Rotolo S, Szanton S. Women in recovery from PTSD have similar inflammation and quality of life as non-traumatized controls. J Psychosom Res (2013) 74:301–6. doi:10.1016/j.jpsychores.2012.10.013
34. Ghazali NB, Steele M, Koh D, Idris A. The diurnal pattern of salivary IL-1β in healthy young adults. Int J Adolesc Med Health (2017). doi:10.1515/ijamh-2017-0058
35. Nilsonne G, Lekander M, Akerstedt T, Axelsson J, Ingre M. Diurnal variation of circulating interleukin-6 in humans: a meta-analysis. PLoS One (2016) 11:e0165799. doi:10.1371/journal.pone.0165799
36. Breen MS, Tylee DS, Maihofer AX, Neylan TC, Mehta D, Binder E, et al. PTSD blood transcriptome mega-analysis: shared inflammatory pathways across biological sex and modes of trauma. Neuropsychopharmacology (2017). doi:10.1038/npp.2017.220
37. Olff M, van Zuiden M. Neuroendocrine and neuroimmune markers in PTSD: pre-, peri- and post-trauma glucocorticoid and inflammatory dysregulation. Curr Opin Psychol (2017) 14:132–7. doi:10.1016/j.copsyc.2017.01.001
38. Jones ME, Lebonville CL, Barrus D, Lysle DT. The role of brain interleukin-1 in stress-enhanced fear learning. Neuropsychopharmacology (2015) 40:1289–96. doi:10.1038/npp.2014.317
39. Levkovitz Y, Fenchel D, Kaplan Z, Zohar J, Cohen H. Early post-stressor intervention with minocycline, a second-generation tetracycline, attenuates post-traumatic stress response in an animal model of PTSD. Eur Neuropsychopharmacol (2015) 25:124–32. doi:10.1016/j.euroneuro.2014.11.012
40. Wilson CB, McLaughlin LD, Ebenezer PJ, Nair AR, Dange R, Harre JG, et al. Differential effects of sertraline in a predator exposure animal model of post-traumatic stress disorder. Front Behav Neurosci (2014) 8:256. doi:10.3389/fnbeh.2014.00256
41. Gill J, Lee H, Barr T, Baxter T, Heinzelmann M, Rak H, et al. Lower health related quality of life in U.S. Military personnel is associated with service-related disorders and inflammation. Psychiatry Res (2014) 216:116–22. doi:10.1016/j.psychres.2014.01.046
42. O’Donovan A, Cohen BE, Seal KH, Bertenthal D, Margaretten M, Nishimi K, et al. Elevated risk for autoimmune disorders in Iraq and Afghanistan veterans with posttraumatic stress disorder. Biol Psychiatry (2015) 77:365–74. doi:10.1016/j.biopsych.2014.06.015
43. Hobfoll SE, Gerhart JI, Zalta AK, Wells K, Maciejewski J, Fung H. Posttraumatic stress symptoms predict impaired neutrophil recovery in stem cell transplant recipients. Psychooncology (2015) 24:1529–35. doi:10.1002/pon.3761
44. Vaccarino V, Goldberg J, Rooks C, Shah AJ, Veledar E, Faber TL, et al. Post-traumatic stress disorder and incidence of coronary heart disease: a twin study. J Am Coll Cardiol (2013) 62:970–8. doi:10.1016/j.jacc.2013.04.085
45. O’Donovan A, Sun B, Cole S, Rempel H, Lenoci M, Pulliam L, et al. Transcriptional control of monocyte gene expression in post-traumatic stress disorder. Dis Markers (2011) 30:123–32. doi:10.3233/dma-2011-0768
46. Wohleb ES, McKim DB, Shea DT, Powell ND, Tarr AJ, Sheridan JF, et al. Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry (2014) 75:970–81. doi:10.1016/j.biopsych.2013.11.029
47. Banks WA, Kastin AJ, Broadwell RD. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation (1995) 2:241–8. doi:10.1159/000097202
48. Vargas-Caraveo A, Perez-Ishiwara DG, Martinez-Martinez A. Chronic psychological distress as an inducer of microglial activation and leukocyte recruitment into the area postrema. Neuroimmunomodulation (2015) 22:311–21. doi:10.1159/000369350
49. Ho YH, Lin YT, Wu CW, Chao YM, Chang AY, Chan JY. Peripheral inflammation increases seizure susceptibility via the induction of neuroinflammation and oxidative stress in the hippocampus. J Biomed Sci (2015) 22:46. doi:10.1186/s12929-015-0157-8
50. Cazareth J, Guyon A, Heurteaux C, Chabry J, Petit-Paitel A. Molecular and cellular neuroinflammatory status of mouse brain after systemic lipopolysaccharide challenge: importance of CCR2/CCL2 signaling. J Neuroinflammation (2014) 11:132. doi:10.1186/1742-2094-11-132
51. McKim DB, Patterson JM, Wohleb ES, Jarrett BL, Reader BF, Godbout JP, et al. Sympathetic release of splenic monocytes promotes recurring anxiety following repeated social defeat. Biol Psychiatry (2015) 79:803–13. doi:10.1016/j.biopsych.2015.07.010
52. Cohen H, Ziv Y, Cardon M, Kaplan Z, Matar MA, Gidron Y, et al. Maladaptation to mental stress mitigated by the adaptive immune system via depletion of naturally occurring regulatory CD4+CD25+ cells. J Neurobiol (2006) 66:552–63. doi:10.1002/neu.20249
53. Muhie S, Gautam A, Meyerhoff J, Chakraborty N, Hammamieh R, Jett M. Brain transcriptome profiles in mouse model simulating features of post-traumatic stress disorder. Mol Brain (2015) 8:14. doi:10.1186/s13041-015-0104-3
54. Gautam A, D’Arpa P, Donohue DE, Muhie S, Chakraborty N, Luke BT, et al. Acute and chronic plasma metabolomic and liver transcriptomic stress effects in a mouse model with features of post-traumatic stress disorder. PLoS One (2015) 10:e0117092. doi:10.1371/journal.pone.0117092
55. Liu X, Nemeth DP, Tarr AJ, Belevych N, Syed ZW, Wang Y, et al. Euflammation attenuates peripheral inflammation-induced neuroinflammation and mitigates immune-to-brain signaling. Brain Behav Immun (2016) 54:140–8. doi:10.1016/j.bbi.2016.01.018
56. Devoto C, Arcurio L, Fetta J, Ley M, Rodney T, Kanefsky R, et al. Inflammation relates to chronic behavioral and neurological symptoms in military with traumatic brain injuries. Cell Transplant (2016) 26:1169–77. doi:10.3727/096368916x693455
57. Spencer-Segal JL, Hyzy RC, Iwashyna TJ, Standiford TJ. Psychiatric symptoms in survivors of the acute respiratory distress syndrome: effects of age, sex, and immune modulation. Ann Am Thorac Soc (2017) 14:960–7. doi:10.1513/AnnalsATS.201606-468OC
58. Sandiego CM, Gallezot JD, Pittman B, Nabulsi N, Lim K, Lin SF, et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc Natl Acad Sci U S A (2015) 112:12468–73. doi:10.1073/pnas.1511003112
59. Ye L, Huang Y, Zhao L, Li Y, Sun L, Zhou Y, et al. IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J Neurochem (2013) 125:897–908. doi:10.1111/jnc.12263
60. Kaur C, Sivakumar V, Zou Z, Ling EA. Microglia-derived proinflammatory cytokines tumor necrosis factor-α and interleukin-1β induce Purkinje neuronal apoptosis via their receptors in hypoxic neonatal rat brain. Brain Struct Funct (2014) 219:151–70. doi:10.1007/s00429-012-0491-5
61. Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, et al. Tumor necrosis factor-α induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem (2006) 281:21362–8. doi:10.1074/jbc.M600504200
62. Lee YY, Lee EJ, Park JS, Jang SE, Kim DH, Kim HS. Anti-inflammatory and antioxidant mechanism of tangeretin in activated microglia. J Neuroimmune Pharmacol (2016) 11:294–305. doi:10.1007/s11481-016-9657-x
63. Reshef R, Kreisel T, Beroukhim Kay D, Yirmiya R. Microglia and their CX3CR1 signaling are involved in hippocampal- but not olfactory bulb-related memory and neurogenesis. Brain Behav Immun (2014) 41:239–50. doi:10.1016/j.bbi.2014.04.009
64. Qian J, Zhu L, Li Q, Belevych N, Chen Q, Zhao F, et al. Interleukin-1R3 mediates interleukin-1-induced potassium current increase through fast activation of Akt kinase. Proc Natl Acad Sci U S A (2012) 109:12189–94. doi:10.1073/pnas.1205207109
65. Harrison NA, Doeller CF, Voon V, Burgess N, Critchley HD. Peripheral inflammation acutely impairs human spatial memory via actions on medial temporal lobe glucose metabolism. Biol Psychiatry (2014) 76:585–93. doi:10.1016/j.biopsych.2014.01.005
66. Capuron L, Ravaud A, Neveu PJ, Miller AH, Maes M, Dantzer R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol Psychiatry (2002) 7:468–73. doi:10.1038/sj.mp.4000995
67. Davis LL, Suris A, Lambert MT, Heimberg C, Petty F. Post-traumatic stress disorder and serotonin: new directions for research and treatment. J Psychiatry Neurosci (1997) 22:318–26.
68. Tucker P, Ruwe WD, Masters B, Parker DE, Hossain A, Trautman RP, et al. Neuroimmune and cortisol changes in selective serotonin reuptake inhibitor and placebo treatment of chronic posttraumatic stress disorder. Biol Psychiatry (2004) 56:121–8. doi:10.1016/j.biopsych.2004.03.009
69. Kao CY, He Z, Zannas AS, Hahn O, Kuhne C, Reichel JM, et al. Fluoxetine treatment prevents the inflammatory response in a mouse model of posttraumatic stress disorder. J Psychiatr Res (2016) 76:74–83. doi:10.1016/j.jpsychires.2016.02.003
70. Lee B, Sur B, Yeom M, Shim I, Lee H, Hahm DH. Effects of systemic administration of ibuprofen on stress response in a rat model of post-traumatic stress disorder. Korean J Physiol Pharmacol (2016) 20:357–66. doi:10.4196/kjpp.2016.20.4.357
71. Gamble-George JC, Baldi R, Halladay L, Kocharian A, Hartley N, Silva CG, et al. Cyclooxygenase-2 inhibition reduces stress-induced affective pathology. Elife (2016) 5:e14137. doi:10.7554/eLife.14137
72. Kut JL, Young MRI, Crayton JW, Wright MA, Young ME. Regulation of murine T-lymphocyte function by spleen cell-derived and exogenous serotonin. Immunopharmacol Immunotoxicol (1992) 14:783–96. doi:10.3109/08923979209009235
73. Young MR, Kut JL, Coogan MP, Wright MA, Young ME, Matthews J. Stimulation of splenic T-lymphocyte function by endogenous serotonin and by low dose exogenous serotonin. Immunology (1993) 80:395–400.
74. Young MRI, Matthews JP. Serotonin regulation of T-cell subpopulations and of macrophage accessory function. Immunology (1995) 84:148–52.
75. Rodriguez P, Holowka DW, Marx BP. Assessment of posttraumatic stress disorder-related functional impairment: a review. J Rehabil Res Dev (2012) 49:649–65. doi:10.1682/JRRD.2011.09.0162
76. George S, Motzer RJ, Hammers HJ, Redman BG, Kuzel TM, Tykodi SS, et al. Safety and efficacy of nivolumab in patients with metastatic renal cell carcinoma treated beyond progression: a subgroup analysis of a randomized clinical trial. JAMA Oncol (2016) 2:1179–86. doi:10.1001/jamaoncol.2016.0775
77. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Transl Med (2015) 7:290ra87. doi:10.1126/scitranslmed.aaa9301
78. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7:315ra189. doi:10.1126/scitranslmed.aad4134
79. Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med (2012) 4:134ra62. doi:10.1126/scitranslmed.3003330
80. Park JS, Kwok SK, Lim MA, Kim EK, Ryu JG, Kim SM, et al. STA-21, a promising STAT3 inhibitor that reciprocally regulates Th17 and Treg, inhibits osteoclastogenesis and alleviates autoimmune inflammation. Arthritis Rheum (2014) 66:918–29. doi:10.1002/art.38305
81. Astry B, Venkatesha SH, Moudgil KD. Involvement of the IL-23/IL-17 axis and the Th17/Treg balance in the pathogenesis and control of autoimmune arthritis. Cytokine (2015) 74:54–61. doi:10.1016/j.cyto.2014.11.020
82. Valmori D, Raffin C, Raimbaud I, Ayyoub M. Human RORγt+ TH17 cells preferentially differentiate from naive FOXP3+ Treg in the presence of lineage-specific polarizing factors. Proc Natl Acad Sci U S A (2010) 107:19402–7. doi:10.1073/pnas.1008247107
83. Meeker S, Seamons A, Paik J, Treuting PM, Brabb T, Grady WM, et al. Increased dietary vitamin D suppresses MAPK signaling, colitis and colon cancer. Cancer Res (2014) 74:4398–408. doi:10.1158/0008-5472.can-13-2820
84. Joshi S, Pantalena LC, Liu XK, Gaffen SL, Liu H, Rohowsky-Kochan C, et al. 1,25-Dihydroxyvitamin D3 ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol Cell Biol (2011) 31:3653–69. doi:10.1128/MCB.05020-11
85. Dyring-Andersen B, Bonefeld CM, Bzorek M, Lovendorf MB, Lauritsen JP, Skov L, et al. The vitamin D analogue calcipotriol reduces the frequency of CD8+ IL-17+ T cells in psoriasis lesions. Scand J Immunol (2015) 82:84–91. doi:10.1111/sji.12304
Keywords: Immune, Inflammation, PTSD, Stress, Disorder
Citation: Wang Z, Caughron B and Young MRI (2017) Posttraumatic Stress Disorder: An Immunological Disorder? Front. Psychiatry 8:222. doi: 10.3389/fpsyt.2017.00222
Received: 28 August 2017; Accepted: 23 October 2017;
Published: 06 November 2017
Edited by:
Dorota Frydecka, Wroclaw Medical University, PolandReviewed by:
Kelly M. Standifer, University of Oklahoma Health Sciences Center, United StatesTamas Kozicz, Radboud University Nijmegen, Netherlands
Anđelko Vidović, Klinika za psihijatriju Vrapče, Croatia
Copyright: © 2017 Wang, Caughron and Young. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Rita I. Young, eW91bmdtckBtdXNjLmVkdQ==