 Chieh-Hsin Lin
Chieh-Hsin Lin Eugene Lin
Eugene Lin Hsien-Yuan Lane
Hsien-Yuan Lane- 1Graduate Institute of Biomedical Sciences, China Medical University, Taichung, Taiwan
- 2Department of Psychiatry, Kaohsiung Chang Gung Memorial Hospital, Chang Gung University College of Medicine, Kaohsiung, Taiwan
- 3Center for General Education, Cheng Shiu University, Kaohsiung, Taiwan
- 4Department of Electrical Engineering, University of Washington, Seattle, WA, United States
- 5TickleFish Systems Corporation, Seattle, WA, United States
- 6Department of Psychiatry, Brain Disease Research Center, China Medical University Hospital, Taichung, Taiwan
With ever-increasing elder populations, age-related cognitive decline, which is characterized as a gradual decline in cognitive capacity in the aging process, has turned out to be a mammoth public health concern. Since genetic information has become increasingly important to explore the biological mechanisms of cognitive decline, the search for genetic biomarkers of cognitive aging has received much attention. There is growing evidence that single-nucleotide polymorphisms (SNPs) within the ADAMTS9, BDNF, CASS4, COMT, CR1, DNMT3A, DTNBP1, REST, SRR, TOMM40, circadian clock, and Alzheimer’s diseases-associated genes may contribute to susceptibility to cognitive aging. In this review, we first illustrated evidence of the genetic contribution to disease susceptibility to age-related cognitive decline in recent studies ranging from approaches of candidate genes to genome-wide association studies. We then surveyed a variety of association studies regarding age-related cognitive decline with consideration of gene–gene and gene–environment interactions. Finally, we highlighted their limitations and future directions. In light of advances in precision medicine and multi-omics technologies, future research in genomic medicine promises to lead to innovative ideas that are relevant to disease prevention and novel drugs for cognitive aging.
Introduction
Generally speaking, age-related cognitive decline, or cognitive aging, is recognized as a gradual and enduring process of transition in cognitive capacity with increasing age (1). Although cognitive decline is common in old age, the relationship between aging and neurodegenerative disease such as dementia remains unclear. Whereas aging is a well-known risk factor for dementia, dementia is not an inevitable consequence of the process of aging. The concept and underlying mechanisms of normal aging and pathological aging might be different. It should be noted that cognitive aging may raise the likelihood of many age-associated diseases and neurodegenerative disorders, such as mild cognitive impairment (MCI), Alzheimer’s diseases (AD), Parkinson’s disease, and other dementias, due to the fact that prior research work has projected that rates of age-associated diseases and neurodegenerative disorders gain rapidly with advancing age (2). While ever-increasing elder populations exist in both developed and developing countries, the pervasiveness of age-associated diseases and neurodegenerative disorders has become a huge public health concern owing to high social and economic burdens (3). What is more, cognitive aging processes implicate multiple complex pathogeneses including genetic and environmental factors (4). In this light, the identification of genetic biomarkers has become an important area of research that aims to preclude the advancement of cognitive aging and to grasp the biology of cognitive aging in an increasingly aging society (5). It is noteworthy that untangling genetic biomarkers for cognitive aging has been at the center of major investigations in the field of precision medicine, and the relevant biomarkers for AD are generally utilized in cognitive aging research as well because of the increased risk for AD in the elderly individuals (6).
More recent research in genome-wide association studies (GWAS) has implicated that single-nucleotide polymorphisms (SNPs) among 11 genes appear to affect the risk of AD, encompassing the PICALM, MS4A6E, MS4A4E, MS4A4A, EPHA1, CR1, CLU, CD33, CD2AP, BIN1, APOE, and ABCA7 gene (7–11). Consequently, a meta-analysis of GWAS studies (n = 74,046) identified 14 risk genes in AD, encompassing the ZCWPW1, SORL1, SLC24A4, RIN3, PTK2B, NME8, MEF2C, INPP5D, HLA-DRB4, HLA-DRB1, FERMT2, DSG2, CELF1, and CASS4 genes (12). The succeeding whole-exome sequencing analysis further tracked down the PLD3 gene to likely be a risk gene for AD (13). Moreover, it has been found that cognitive decline was linked with the CR1 rs6656401 SNP by using the established AD-associated genes (14). Additionally, recent epistasis studies suggested that the CLU-MS4A4E (15, 16) and CD33-MS4A4E (16) gene–gene interactions might have a considerable influence on the susceptibility of AD. By utilizing the known AD-associated genes, it has also been demonstrated that cognitive decline was related with the ABCA7 rs3764650 and CD33 rs3865444 SNPs in the elderly Caucasian women (17).
In this review, we first surveyed some genetic biomarkers that were linked with age-related cognitive decline in several recent association studies (Table 1). Furthermore, we assessed some potential gene–gene and gene–environment interactions on age-related cognitive decline. This review does not intend to comprehensively survey all literature. We mainly focused on the most recent developments for biomarker research in cognitive aging. Finally, the limitations and future perspectives associated with cognitive aging in terms of genetic biomarkers were summarized. Future replication studies in larger samples with longitudinal follow-up are required to confirm the findings of the biomarkers for cognitive aging discovered in the association studies.
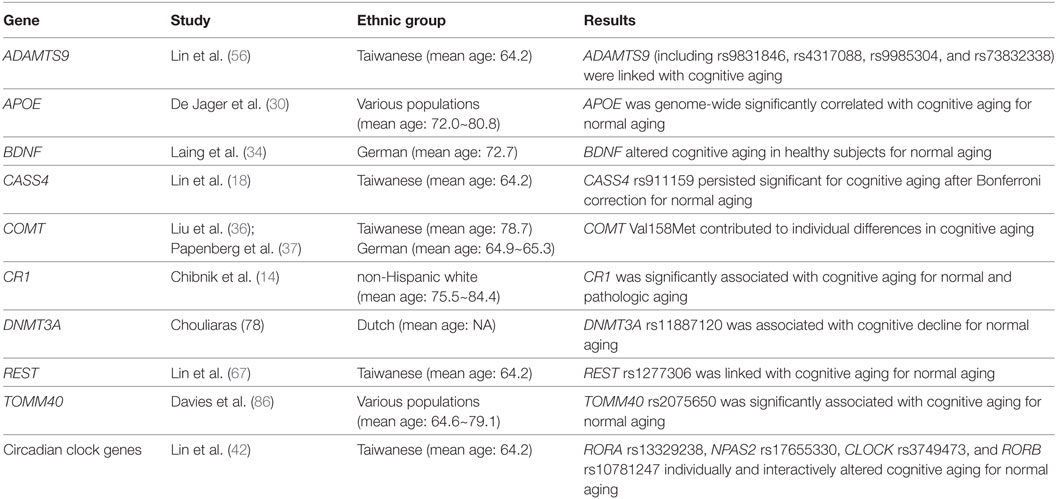
Table 1. Relevant studies in genetic biomarkers on age-related cognitive decline.
Recent Association Studies
AD-Associated Genes
As mentioned previously in the Section “Introduction,” it has been revealed that AD risk is linked with ZCWPW1, SORL1, SLC24A4, RIN3, PTK2B, PLD3, PICALM, NME8, MS4A6E, MS4A4E, MS4A4A, MEF2C, INPP5D, HLA-DRB4, HLA-DRB1, FERMT2, EPHA1, DSG2, CR1, CLU, CELF1, CD33, CD2AP, CASS4, BIN1, APOE, and ABCA7 in GWAS and meta-analyses (7–12). To recognize probable genes implicated in the regulation of age-related cognitive decline, a recent association study has analyzed whether SNPs within these 27 AD-associated genes are linked with cognitive aging as well as via complex gene–gene and gene–environment interactions in a cohort of older Taiwanese adults (n = 634) aged over 60 years (mean age: 64.2 years) from the Taiwan Biobank (18). In order to weigh cognitive functions, the mini-mental state examination (MMSE) method was administered for all participants (18). Lin et al. tested 588 SNPs, but only the CASS4-rs911159 SNP persisted significant for cognitive aging after Bonferroni correction (18). In addition, their analysis results suggested an association with 6 more SNPs in the AD-related genes, encompassing the SLC24A4-rs67063100, RIN3-rs1885747, PLD3-rs11672825, MEF2C-rs9293506, FERMT2-rs4901317, and EPHA1-rs10952552 SNPs (18). Lin et al. also displayed the gene–gene interactions among the SLC24A4-rs67063100, MEF2C-rs9293506, FERMT2-rs4901317, EPHA-rs10952552, and CASS4-rs911159 SNPs on cognitive aging by using the generalized multifactor dimensionality reduction (GMDR) approach (18). Furthermore, they disclosed the gene–environment interactions of the MEF2C-rs9293506 and SLC24A4-rs67063100 SNP with environmental factors including social support, physical activity, smoking status, and alcohol consumption on cognitive aging by using the GMDR approach (18).
Consistent with the findings by Lin et al. (18), two preceding GWAS studies (8, 10) have reported that the EPHA1 rs11767557 SNP may affect the vulnerability to AD. In addition, the FERMT2 rs17125944, MEF2C rs190982, and SLC24A4 rs10498633 SNPs were prone to AD in a meta-analysis study (12). Moreover, a whole-exome sequencing study pinpointed a rare rs145999145 (Val232Met) variant in the PLD3 gene was liable to AD (13). The PLD3 gene may play a central role in dealing with amyloid-beta precursor protein (13).
The CASS4 protein is proposed to better characterize the functions of cell growing, spreading, adhesion, and other activities (19). Moreover, it was speculated that CASS4 plays a vital part in the hallmarks of AD such as the amyloid precursor protein (APP) and Tau protein (20). More recent studies also indicated an association between AD and the CASS4 rs7274581, rs6024870, and rs16979934 SNPs (12, 21–23). In contrast, a replication study indicated that the CASS4 rs7274581 SNP did not affect the risk of AD in a Spanish population (24).
Gene–gene interaction study inferred that SLC24A4, MEF2C, FERMT2, EPHA1, and CASS4 synergistically raised the propensity of cognitive aging by using the GMDR approach (18). It was further speculated that these five genes are comprised in the relevant pathology and pathways (18). EPHA1 is implicated in regulating neurodevelopment (20). FERMT2 contributes to Tau neurotoxicity and cell adhesion (25). MEF2C may influence hippocampal synaptic connectivity and thereby mediate hippocampal-dependent memory and learning (26). SLC24A4, in the vicinity of the RIN3 gene, is linked with neurodevelopment (27) as well as Tau pathology and APP (28).
APOE
The APOE gene, located on chromosome 19q13.32, encodes a major protein which is crucial for the regular catabolism of triglyceride-rich lipoprotein constituents (29). Conducting a GWAS study with the longitudinal cognitive testing data such as memory and perceptual speed, De Jager et al. discovered that APOE was genome-wide significantly correlated with age-related cognitive decline (mean age: 72.0~80.8 years) (30). A meta-analysis of 77 studies (n = 40,942) also suggested that carriers of the APOE ɛ4 allele, which were linked with late-onset AD, performed worse on several domains of cognitive functions including overall global cognitive ability, episodic memory, and executive functioning (31). Further, the APOE rs405509 and APOE rs440446 SNPs were more likely to develop non-pathological cognitive aging, independent of APOE major isoforms, in a Finnish population (32). In contrast, a replication study indicated that the APOE gene did not affect the risk of age-related cognitive decline in older Taiwanese adults (18).
BDNF
Another potential candidate gene such as BDNF was reported to be implicated in age-related cognitive decline among the elderly (33). The BDNF gene, encoding proteins of the nerve growth factor family, was demonstrated to alter cognitive deficits in healthy subjects (mean age: 72.7 years) for normal aging by using the mean Z-scores based on three cognitive domains including motor function, memory, and perceptual speed (34). Ward et al. also disclosed an interaction between APOE and BDNF that predicted a cognitive effect in healthy older adults (35).
COMT
The COMT gene, located on chromosome 22q11.21, is essential in the metabolic degradation of prefrontal dopamine (36). A functional COMT Val158Met polymorphism has been shown to contribute to individual differences in cognitive aging in Taiwanese (mean age: 78.7 years) and German (mean age: 64.9~65.3 years) populations using MMSE scores (36, 37). Based on a 4-year longitudinal study, Dixon et al. also revealed that the APOE and COMT genes are complementary biomarkers in normal cognitive aging and early MCI for older adults (38).
CR1
The CR1 gene encodes a protein that plays a role in cellular binding and immune complexes (14). A replication study reported that the rs6656401 SNP in the CR1 gene, which is one of the susceptibility loci for AD, was significantly associated with cognitive deficits (mean age: 75.5~84.4 years) for normal aging (in terms of global cognitive decline) as well as for pathological aging (in terms of global AD pathology) using the cognitive testing data such as memory and global cognition (14). A subsequent replication study also indicated that the rs4844609 SNP in the CR1 gene modulates episodic memory decline and an interaction between APOE and CR1 influences cognitive decline for normal aging and for pathological aging (39). In contrast, a replication study indicated that the CR1 gene did not affect the risk of age-related cognitive decline in older Taiwanese adults (18).
Circadian Clock Genes
Circadian rhythms are instinctively recurring cycles that determine the timing of biological events such as energy metabolism, hormone release, and sleep–wake cycles (40). Additionally, dysregulation of circadian rhythms is characteristic of the natural process of ongoing aging and cognitive decline (40). The circadian rhythms are maintained and triggered by a composition of core circadian clock genes, which can be classified as vital genes on provoking circadian rhythms in specific cells (41).
Circadian clock genes, encompassing RORB, RORA, PER3, PER2, PER1, NR1D1, NPAS2, CRY2, CRY1, CLOCK, and ARNT, may be also involved in cognitive impairment (40, 41). To find out potential genes implicated in the regulation of age-related cognitive decline, a recent study analyzed whether the 11 aforementioned core circadian clock genes as well as complex gene–gene and gene–environment interactions contributed to cognitive aging in more than 634 elderly individuals (mean age: 64.2 years) in Taiwan (42). As a result, four SNPs, including the RORA-rs13329238, NPAS2-rs17655330, CLOCK-rs3749473, and RORB-rs10781247 SNPs individually and interactively alters the hazard of cognitive deficits in terms of MMSE scores for normal aging (42). Finally, environmental factors such as smoking status, alcohol consumption, social support, and physical activity also interacted with these SNPs in regulating the liability of age-related cognitive decline (42).
CLOCK encodes a protein which creates the ARNTL/CLOCK heterodimeric protein with ARNTL (43). In addition, rs1554483, 3111, and rs4580704 CLOCK SNPs influenced the tendency to AD in Chinese subjects (44–46). However, these results were not confirmed in different populations (42).
The RORA and RORB proteins create a family of nuclear hormone receptors (47). These two genes were reported to have a key part in a wide variety of regulations; for instance, cellular metabolism, circadian rhythm, embryonic development, immunity, and inflammatory responses (48, 49).
It should be noted that NPAS2 encodes transcription factors which are a component belonging to the basic helix-loop-helix family, and the ARNTL/NPAS2 and ARNTL/CLOCK heterodimeric proteins can bind to chromatin, bringing about the upregulation in gene expression for PER1, PER2, PER3, CRY1, and CRY2 (50). In this manner, products of these CRY1, CRY2, PER1, PER2, and PER3 genes build a complex that inhibits the action of the ARNTL-containing complexes and in turn forms a negative feedback loop (50).
Insulin Resistance-Associated Genes
To identify possible genes implicated in the process of age-related cognitive decline, a recent association study of the Taiwan Biobank has hypothesized that SNPs in insulin resistance-associated genes, including the PPARG, GCKR, and ADAMTS9 genes, might be associated with cognitive aging individually or collectively in a cohort of Taiwanese individuals (n = 547; age > 60 years; mean age: 64.1 years) (51). The results indicated that 4 SNPs (including rs9831846, rs4317088, rs9985304, and rs73832338) within ADAMTS9 were linked with MMSE scores (that is, cognitive aging) after performing Bonferroni correction (51). In addition, Lin et al. identified a SNP–SNP interaction between ADAMTS9-rs76346246 and ADAMTS9-rs9985304 that may influence cognitive aging by using the GMDR approach (51).
ADAMTS9 is demonstrated in the regulation of a wide variety of processes, such as the control of organ shape during growth, the inhibition of angiogenesis, and the cleavage of proteoglycans (51). Since one of the risk factors for cognitive aging and AD is insulin resistance, insulin resistance-related genes might be linked with cognitive aging (52, 53). Insulin abnormalities raise the uncertainty for neurodegenerative disorders including MCI, AD, and cognitive aging (54, 55). Insulin is also a fundamental factor for normal brain functioning. It should be noted that ADAMTS9 is associated with insulin resistance, insulin sensitivity, and type 2 diabetes (56, 57). There is growing evidence that the ADAMTS-9 protein might participate in the processes of brain disease states including spinal cord injury, ischemic stroke, and transient middle cerebral artery occlusion in animal studies (58–60). In accordance, a GWAS study has identified the ADAMTS9 rs6795735 SNP as a candidate biomarker for age-related macular degeneration, which commonly occurs in elder adults (61).
RE1-Silencing Transcription Factor (REST) Gene
A growing body of evidence suggests that REST may be involved with AD and cognitive aging (62). A recent replication study has assessed whether REST SNPs are associated with cognitive aging as well as via SNP–SNP interactions for normal aging in elder Taiwanese subjects (n = 634; mean age: 64.2 years) (62). Their analysis results demonstrated that REST-rs1277306 was linked with cognitive aging, which was measured by MMSE scores (62). This prediction is further supported by evidence that the association remained significant for individuals without APOE ε4 allele after Bonferroni correction (62). On the other hand, the REST rs1277306 SNP was not a predicting factor for cognitive aging among individuals with at least one APOE ε4 allele (62). In addition, Lin et al. tracked down an SNP–SNP interaction between the REST rs1713985 and REST rs1277306 SNPs on cognitive aging by using the GMDR. The REST protein is indicated in the modulation of synaptic plasticity, ion channels, vesicular transport, axonal growth, and neuronal differentiation (63, 64). In addition, REST is associated with amyloid β-protein toxicity, protection from oxidative stress, AD pathology, MCI, brain aging, and slow hippocampal loss (65–67). Additionally, REST might act as both a primary protector against neurodegeneration and an essential repressor for normal neurogenesis (65–67). Although APOE is well-established regarding its major role in cognitive decline in elder adults (68, 69), the biologically synergistic effects between the APOE and REST genes on cognitive aging are still unknown. It was speculated that APOE and REST might involve in a comparable pathway relevant to cognitive aging (70). Consistent with the findings by Lin et al. (62), several other studies pinpointed an interaction of APOE with PSEN2 (71), PSEN1 (72), PICALM (70), and APP (71, 73) by using patient stratification based on APOE ε4 status.
DNA Methylation
Recent studies indicate that DNA methylation, one of main epigenetic mechanisms, plays a crucial role in cognitive aging (74, 75). DNA methylation involves the inclusion of a methyl group to the DNA molecule, especially when a cytosine is followed by a guanine (76). DNA methylation is regularly associated with reduced transcriptional activity and is triggered by a family of DNA methyl-transferase proteins (77). Using repeated measures of composite scores for annual cognitive testing, Chouliaras et al. investigated associations among common SNPs in genes modulating DNA methylation and cognitive aging. They found that the rs11887120 SNP in the DNMT3A gene was associated with annual decline in cognitive deficits for normal aging in a Dutch population (78). In contrast, this finding was not replicated in German subjects (79). DNMT3A, encoding a DNA methyltransferase, is located in the cytoplasm and nucleus in de novo methylation (80).
TOMM40
With longitudinal cognitive ability data, a GWAS study reported that the rs2075650 SNP in the TOMM40 gene, which is adjacent to the APOE gene, was significantly associated with age-related cognitive decline (mean age: 64.6~79.1 years) (81). After fine SNP mapping of the TOMM40/APOE region, both APOE rs429358 and TOMM40 rs11556505 were correlated with cognitive aging (81). Furthermore, SNPs within the TOMM40/APOE zone possessed a non-protein-coding regulatory and functional effect in a functional genomic analysis, indicating that the TOMM40/APOE zone may be linked with nonpathological cognitive aging (81). The TOMM40 gene, located on chromosome 19q13.32, encodes the mitochondrial outer membrane complex relevant to the channel-forming subunit of the translocase, which is indispensable for construction of protein precursors to mitochondria (82).
Other Potential Genes
The DTNBP1 gene encodes a protein that plays a role in the biogenesis of organelle linked with lysosomes, platelet dense granules, and melanosomes (83). It has been suggested that the DTNBP1 gene modulates general cognitive abilities both in schizophrenia patients and in healthy subjects in Japanese (mean age: 34.1~39.2) and German (mean age: 24.8) populations (84–87). In addition, Burdick et al. found that the CTCTAC risk haplotype of 6 SNPs including rs909706, rs1018381, rs2619522, rs760761, rs2619528, and rs1011313 in the DTNBP1 gene was associated with general cognitive ability and cognitive decline in schizophrenia patients (84).
In a recent systems genetics study applying a genetically diverse population of mice, Neuner et al. pinpointed Hp1bp3 gene to be a novel modulator of cognitive aging (88). Their findings also confirmed that as compared to cognitively healthy individuals, levels of HP1BP3 protein were significantly decreased in the hippocampi of elderly subjects with cognitive impairment, suggesting that reduced expression of Hp1bp3 may contribute to cognitive aging in both mice and humans (88). The HP1BP3 gene is located on chromosome 1p36.12 and encodes a histone H1 related protein with non-redundant and specific roles vital for viability and gain in humans (89).
It is worth mentioning a potential gene called SRR although, to our knowledge, there are only animal studies but no population studies for this gene in cognitive aging. SRR encodes the serine racemase enzyme which converts l-serine to d-serine. d-serine is an endogenous co-agonist for N-methyl-D-aspartate receptors (90). The SRR rs408067 SNP, located in the promoter region, may affect the transcription activity of the SRR gene (91). Reduced SRR expression impaired hippocampal age-related cognitive function in an animal study, suggesting that the SRR-dependent pathway might be one possible target of the hippocampus-related cognitive decline in aging (92). Another expression profiling study reported that various genes that are associated with cognitive ability were influenced by the SRR mutation (93).
Limitations in Current Studies
Notwithstanding, there were several limitations with respect to the aforementioned studies. First, there is certainly room for development of much further research and comprehensive evaluation to reassess whether the current results remain in other ethnic populations for the investigated genetic variants with cognitive aging in terms of the association and interactions (62).
Second, given the relatively young mean age of the sample in several studies mentioned previously, the current results are unable to be extrapolated to much older populations that have higher risk for developing age-associated diseases and neurodegenerative disorders, such as MCI, AD, and other dementias (18).
Because of logistical and ethical matters, it is challenging to assess homogeneous genetic backgrounds and recruit a large enough cohort of participants at the same time (94). Furthermore, some findings were not replicated, and the discordant results found among these studies may be due to issues in the sample size, ethnicities, study design, and phenotype definitions. Moreover, confounding factors may not be fully handled, and thereby considerable bias may not be excluded.
In order to reinforce the statistical findings, it is appealing to seek more supplementary biologically relevant evidences owing to the fact that the investigated SNPs might be greatly enhanced in association studies (95).
Besides, the aforementioned studies utilized various methods to assess cognitive function. A major challenge is to ensure a proper approach for evaluating cognitive function. The well-established MMSE approach is chosen to evaluate cognitive function in several aforementioned studies because it is the most widely used screening test of cognition (96). Nevertheless, the ceiling effect of MMSE in healthy young subjects as well as its floor effect in the oldest subjects diminishes the variability (96). Similar to MMSE, another strategy is the General Practitioner Assessment of Cognition (GPCOG) with psychometric properties. However, using the GPCOG is required to further examine for its possible language or cultural tendency (97, 98). Another more recent language independent method is the CANTAB, a visual and cognitive assessment tool used on computers (99). Nonetheless, because the correlations between CANTAB and commonly used cognitive tools such as GPCOG and MMSE are only modest, the application of CANTAB should be justified in future studies (100). Moreover, an alternative is the ADAS-Cog which achieves higher sensitivity with a change of four-point (101). Although the ADAS-Cog is a well-validated scale in cognitive performance, the drawback is that about 40 min is needed to complete the task and this fact causes it unacceptable in most large-scale studies (96).
Cross-sectional design for cognitive aging studies has been adopted by many researchers because it is less feasible to examine aging trajectories for individual participants with longitudinal studies due to the high cost and long follow-up time (102). Nevertheless, it is always important to recognize the limitations of transversal studies on aging. Most importantly, we are unable to make longitudinal or causal inferences about changes in cognitive function by using cross-sectional data.
Future Outlook
Over the past decade, advances in genome science have spawned numerous lines of research into precision medicine and multi-omics (103). In spite of spectacular progress in precision medicine and multi-omics technology, which can assemble a mammoth amount of multi-omics data, there are no established approaches to take advantage of that data in a predictive fashion (103). Therefore, we face a challenge of developing a fundamental, personalizable, mathematical model, which is calibrated on a broad range of multi-omics and clinical data (103). To conquer this challenge, a key component of future projects is to be able to advance the aforementioned predictive capability by facilitating machine learning and predictive approaches (103).
Building up a set of genetic biomarkers which are immensely dependable as a benchmark of disease status or drug response for cognitive aging will be considerably indispensable in the future (104). At this juncture, no genetic biomarkers found in the aforementioned studies would be unquestionably qualified to be incorporated in the panel owing to the aforementioned limitations (104).
Moreover, machine learning and predictive techniques such as Bayesian networks may present a conceivable approach to forecast novel drug efficacy and establish statistical models for predicting disease status (103). In future research, we will be able to help physicians in the prescription by creating predictive models which forecast the likelihood of diseases or treatment response (103, 104). In addition, predictive and machine learning approaches such as Bayesian networks might be important in weighing correlations in RNA–RNA molecule, correlations between miRNA and mRNA, as well as interactions between gene and environment (105). Moreover, the statistical modeling such as meta-analysis, pathway analysis, and gene–gene expression correlations is intrinsic to eliminate the false positive biomarkers observed during the association analyses of current precision medicine studies (105).
Essentially, evidence shows that multi-omics data and biomarkers such as genetic, epigenetic, metabolomic, transcriptomic, and proteomic profiles are important in assorted pathophysiology for a certain disease and novel drug treatment (103, 105). Subsequently, the systematic and integrative analyses of different profiles with apparently cooperative functions might have a big impact on the disclosing for the mysterious pathogenic processes of a certain disease and novel drug treatment (103, 105–107). Finally, in order to unquestionably carry out disease pathogenesis as well as novel drug therapy, future studies will have to accomplish an integrative and systematic way of using clinical information, biomarkers, and multi-omics data (103, 105).
Summary
In this review, we have focused primarily on recent findings and relevant studies for age-related cognitive decline. The current review also highlighted the merit of association studies with relatively large sample size to incorporate a wide variety of populations for cognitive aging. In order to advance personalized treatment and prevention strategies worldwide, a main challenge is how best to integrate these findings with other pieces until the picture of cognitive aging is adequately apparent. Similarly, these findings have indicated that machine learning and predictive tools might be beneficial for clinical decision making by integrating multi-omics data and biomarkers.
In light of recent developments, novel machine learning and predictive algorithms will be the new frontier in the decades to come for establishing prognostic and diagnostic assessments by using huge data technologies for precision medicine (103, 105). Future research using machine learning and predictive approaches is warranted in the matter of managing the interactions of biomarkers and foretelling the relationship between biomarkers and drug response in precision medicine studies (103, 105). In our opinion, yet a number of challenges remain and a host of deeply key and crucial research issues must be ironed out. As we enter a period of the new envisioned science of precision medicine, personalized therapy for individuals would undoubtedly become a reality.
Author Contributions
EL, H-YL, and C-HL involved in conception and design; EL and C-HL involved in literature review and interpretation, and manuscript writing; EL, H-YL, and C-HL involved in financial support and final approval of manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors extend their sincere thanks to Vita Genomics, Inc. for funding this research. This work was supported by the Ministry of Economic Affairs in Taiwan (SBIR Grant S099000280249-154), Ministry of Science and Technology in Taiwan (MOST 105-2314-B-182A-059-), Taiwan Ministry of Health and Welfare Clinical Trial and Research Center of Excellence (MOHW105-TDU-B-212-133019), and China Medical University Hospital, Taiwan (DMR-101-091 and DMR-102-069). Role of Funding Source: The aforementioned institutes had no further role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
References
1. Blazer DG, Wallace RB. Cognitive aging: what every geriatric psychiatrist should know. Am J Geriatr Psychiatry (2016) 24(9):776–81. doi:10.1016/j.jagp.2016.06.013
2. Katz MJ, Lipton RB, Hall CB, Zimmerman ME, Sanders AE, Verghese J, et al. Age-specific and sex-specific prevalence and incidence of mild cognitive impairment, dementia, and Alzheimer dementia in blacks and whites: a report from the Einstein Aging Study. Alzheimer Dis Assoc Disord (2012) 26(4):335–43. doi:10.1097/WAD.0b013e31823dbcfc
3. Barnes LL, Bennett DA. Alzheimer’s disease in African Americans: risk factors and challenges for the future. Health Aff (Millwood) (2014) 33(4):580–6. doi:10.1377/hlthaff.2013.1353
4. Konar A, Singh P, Thakur MK. Age-associated cognitive decline: insights into molecular switches and recovery avenues. Aging Dis (2016) 7(2):121–9. doi:10.14336/AD.2015.1004
5. Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol (2014) 13(6):614–29. doi:10.1016/S1474-4422(14)70090-0
6. Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology (2016) 87(5):539–47. doi:10.1212/WNL.0000000000002923
7. Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet (2009) 41(10):1088–93. doi:10.1038/ng.440
8. Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet (2011) 43(5):429–35. doi:10.1038/ng.803
9. Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet (2009) 41(10):1094–9. doi:10.1038/ng.439
10. Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet (2011) 43(5):436–41. doi:10.1038/ng.801
11. Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA (2010) 303(18):1832–40. doi:10.1001/jama.2010.574
12. Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet (2013) 45(12):1452–8. doi:10.1038/ng.2802
13. Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature (2014) 505(7484):550–4. doi:10.1038/nature12825
14. Chibnik LB, Shulman JM, Leurgans SE, Schneider JA, Wilson RS, Tran D, et al. CR1 is associated with amyloid plaque burden and age-related cognitive decline. Ann Neurol (2011) 69(3):560–9. doi:10.1002/ana.22277
15. Ebbert MT, Boehme KL, Wadsworth ME, Staley LA; Alzheimer’s Disease Neuroimaging Initiative, Alzheimer’s Disease Genetics Consortium, et al. Interaction between variants in CLU and MS4A4E modulates Alzheimer’s disease risk. Alzheimers Dement (2016) 12(2):121–9. doi:10.1016/j.jalz.2015.08.163
16. Ebbert MT, Ridge PG, Wilson AR, Sharp AR, Bailey M, Norton MC, et al. Population-based analysis of Alzheimer’s disease risk alleles implicates genetic interactions. Biol Psychiatry (2014) 75(9):732–7. doi:10.1016/j.biopsych.2013.07.008
17. Nettiksimmons J, Tranah G, Evans DS, Yokoyama JS, Yaffe K. Gene-based aggregate SNP associations between candidate AD genes and cognitive decline. Age (Dordr) (2016) 38(2):41. doi:10.1007/s11357-016-9885-2
18. Lin E, Tsai SJ, Kuo PH, Liu YL, Yang AC, Kao CF. Association and interaction effects of Alzheimer’s disease-associated genes and lifestyle on cognitive aging in older adults in a Taiwanese population. Oncotarget (2017) 8(15):24077–87. doi:10.18632/oncotarget.15269
19. Singh MK, Dadke D, Nicolas E, Serebriiskii IG, Apostolou S, Canutescu A, et al. A novel Cas family member, HEPL, regulates FAK and cell spreading. Mol Biol Cell (2008) 19(4):1627–36. doi:10.1091/mbc.E07-09-0953
20. Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry (2015) 77(1):43–51. doi:10.1016/j.biopsych.2014.05.006
21. Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, Myers AJ, et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet (2014) 10(9):e1004606. doi:10.1371/journal.pgen.1004606
22. Rosenthal SL, Barmada MM, Wang X, Demirci FY, Kamboh MI. Connecting the dots: potential of data integration to identify regulatory SNPs in late-onset Alzheimer’s disease GWAS findings. PLoS One (2014) 9(4):e95152. doi:10.1371/journal.pone.0095152
23. Wang X, Lopez OL, Sweet RA, Becker JT, DeKosky ST, Barmada MM, et al. Genetic determinants of disease progression in Alzheimer’s disease. J Alzheimers Dis (2015) 43(2):649–55. doi:10.3233/JAD-140729
24. Ruiz A, Heilmann S, Becker T, Hernandez I, Wagner H, Thelen M, et al. Follow-up of loci from the International Genomics of Alzheimer’s Disease Project identifies TRIP4 as a novel susceptibility gene. Transl Psychiatry (2014) 4:e358. doi:10.1038/tp.2014.2
25. Shulman JM, Imboywa S, Giagtzoglou N, Powers MP, Hu Y, Devenport D, et al. Functional screening in Drosophila identifies Alzheimer’s disease susceptibility genes and implicates Tau-mediated mechanisms. Hum Mol Genet (2014) 23(4):870–7. doi:10.1093/hmg/ddt478
26. Akhtar MW, Kim MS, Adachi M, Morris MJ, Qi X, Richardson JA, et al. In vivo analysis of MEF2 transcription factors in synapse regulation and neuronal survival. PLoS One (2012) 7(4):e34863. doi:10.1371/journal.pone.0034863
27. Larsson M, Duffy DL, Zhu G, Liu JZ, Macgregor S, McRae AF, et al. GWAS findings for human iris patterns: associations with variants in genes that influence normal neuronal pattern development. Am J Hum Genet (2011) 89(2):334–43. doi:10.1016/j.ajhg.2011.07.011
28. Chapuis J, Hansmannel F, Gistelinck M, Mounier A, Van Cauwenberghe C, Kolen KV, et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry (2013) 18(11):1225–34. doi:10.1038/mp.2013.1
29. Zhao N, Liu CC, Qiao W, Bu G. Apolipoprotein E, receptors, and modulation of Alzheimer’s disease. Biol Psychiatry (2017). doi:10.1016/j.biopsych.2017.03.003
30. De Jager PL, Shulman JM, Chibnik LB, Keenan BT, Raj T, Wilson RS, et al. A genome-wide scan for common variants affecting the rate of age-related cognitive decline. Neurobiol Aging (2012) 33(5):1017.e1–15. doi:10.1016/j.neurobiolaging.2011.09.033
31. Wisdom NM, Callahan JL, Hawkins KA. The effects of apolipoprotein E on non-impaired cognitive functioning: a meta-analysis. Neurobiol Aging (2011) 32(1):63–74. doi:10.1016/j.neurobiolaging.2009.02.003
32. Rantalainen V, Lahti J, Henriksson M, Kajantie E, Tienari P, Eriksson JG, et al. APOE and aging-related cognitive change in a longitudinal cohort of men. Neurobiol Aging (2016) 44:151–8. doi:10.1016/j.neurobiolaging.2016.04.024
33. Harris SE, Deary IJ. The genetics of cognitive ability and cognitive ageing in healthy older people. Trends Cogn Sci (2011) 15(9):388–94. doi:10.1016/j.tics.2011.07.004
34. Laing KR, Mitchell D, Wersching H, Czira ME, Berger K, Baune BT. Brain-derived neurotrophic factor (BDNF) gene: a gender-specific role in cognitive function during normal cognitive aging of the MEMO-Study? Age (Dordr) (2012) 34(4):1011–22. doi:10.1007/s11357-011-9275-8
35. Ward DD, Summers MJ, Saunders NL, Janssen P, Stuart KE, Vickers JC. APOE and BDNF Val66Met polymorphisms combine to influence episodic memory function in older adults. Behav Brain Res (2014) 271:309–15. doi:10.1016/j.bbr.2014.06.022
36. Liu ME, Hong CJ, Liou YJ, Tsai YL, Hsieh CH, Tsai SJ. Association study of a functional catechol-O-methyltransferase polymorphism and executive function in elderly males without dementia. Neurosci Lett (2008) 436(2):193–5. doi:10.1016/j.neulet.2008.03.018
37. Papenberg G, Backman L, Nagel IE, Nietfeld W, Schroder J, Bertram L, et al. COMT polymorphism and memory dedifferentiation in old age. Psychol Aging (2014) 29(2):374–83. doi:10.1037/a0033225
38. Dixon RA, DeCarlo CA, MacDonald SW, Vergote D, Jhamandas J, Westaway D. APOE and COMT polymorphisms are complementary biomarkers of status, stability, and transitions in normal aging and early mild cognitive impairment. Front Aging Neurosci (2014) 6:236. doi:10.3389/fnagi.2014.00236
39. Keenan BT, Shulman JM, Chibnik LB, Raj T, Tran D, Sabuncu MR, et al. A coding variant in CR1 interacts with APOE-epsilon4 to influence cognitive decline. Hum Mol Genet (2012) 21(10):2377–88. doi:10.1093/hmg/dds054
40. Ribas-Latre A, Eckel-Mahan K. Interdependence of nutrient metabolism and the circadian clock system: importance for metabolic health. Mol Metab (2016) 5(3):133–52. doi:10.1016/j.molmet.2015.12.006
41. Buhr ED, Takahashi JS. Molecular components of the mammalian circadian clock. Handb Exp Pharmacol (2013) 217:3–27. doi:10.1007/978-3-642-25950-0_1
42. Lin E, Kuo PH, Liu YL, Yang AC, Kao CF, Tsai SJ. Effects of circadian clock genes and environmental factors on cognitive aging in old adults in a Taiwanese population. Oncotarget (2017) 8(15):24088–98. doi:10.18632/oncotarget.15493
43. Woon PY, Kaisaki PJ, Braganca J, Bihoreau MT, Levy JC, Farrall M, et al. Aryl hydrocarbon receptor nuclear translocator-like (BMAL1) is associated with susceptibility to hypertension and type 2 diabetes. Proc Natl Acad Sci U S A (2007) 104(36):14412–7. doi:10.1073/pnas.0703247104
44. Chen HF, Huang CQ, You C, Wang ZR, Si-qing H. Polymorphism of CLOCK gene rs 4580704 C > G is associated with susceptibility of Alzheimer’s disease in a Chinese population. Arch Med Res (2013) 44(3):203–7. doi:10.1016/j.arcmed.2013.01.002
45. Chen Q, Huang CQ, Hu XY, Li SB, Zhang XM. Functional CLOCK gene rs1554483 G/C polymorphism is associated with susceptibility to Alzheimer’s disease in the Chinese population. J Int Med Res (2013) 41(2):340–6. doi:10.1177/0300060513476430
46. Yang YK, Peng XD, Li YH, Wang ZR, Chang-quan H, Hui W, et al. The polymorphism of CLOCK gene 3111T/C C>T is associated with susceptibility of Alzheimer disease in Chinese population. J Investig Med (2013) 61(7):1084–7. doi:10.2310/JIM.0b013e31829f91c0
47. Duez H, Staels B. Rev-erb-alpha: an integrator of circadian rhythms and metabolism. J Appl Physiol (1985) (2009) 107(6):1972–80. doi:10.1152/japplphysiol.00570.2009
48. Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal (2009) 7:e003. doi:10.1621/nrs.07003
49. Kang HS, Okamoto K, Takeda Y, Beak JY, Gerrish K, Bortner CD, et al. Transcriptional profiling reveals a role for RORalpha in regulating gene expression in obesity-associated inflammation and hepatic steatosis. Physiol Genomics (2011) 43(13):818–28. doi:10.1152/physiolgenomics.00206.2010
50. Kondratova AA, Kondratov RV. The circadian clock and pathology of the ageing brain. Nat Rev Neurosci (2012) 13(5):325–35. doi:10.1038/nrn3208
51. Lin E, Tsai SJ, Kuo PH, Liu YL, Yang AC, Kao CF, et al. The ADAMTS9 gene is associated with cognitive aging in the elderly in a Taiwanese population. PLoS One (2017) 12(2):e0172440. doi:10.1371/journal.pone.0172440
52. Gao L, Cui Z, Shen L, Ji HF. Shared genetic etiology between type 2 diabetes and Alzheimer’s disease identified by bioinformatics analysis. J Alzheimers Dis (2016) 50(1):13–7. doi:10.3233/JAD-150580
53. Hao K, Di Narzo AF, Ho L, Luo W, Li S, Chen R, et al. Shared genetic etiology underlying Alzheimer’s disease and type 2 diabetes. Mol Aspects Med (2015) 4(3–44):66–76. doi:10.1016/j.mam.2015.06.006
54. Craft S. Insulin resistance and Alzheimer’s disease pathogenesis: potential mechanisms and implications for treatment. Curr Alzheimer Res (2007) 4(2):147–52. doi:10.2174/156720507780362137
55. Watson GS, Craft S. Insulin resistance, inflammation, and cognition in Alzheimer’s disease: lessons for multiple sclerosis. J Neurol Sci (2006) 245(1–2):21–33. doi:10.1016/j.jns.2005.08.017
56. Boesgaard TW, Gjesing AP, Grarup N, Rutanen J, Jansson PA, Hribal ML, et al. Variant near ADAMTS9 known to associate with type 2 diabetes is related to insulin resistance in offspring of type 2 diabetes patients – EUGENE2 study. PLoS One (2009) 4(9):e7236. doi:10.1371/journal.pone.0007236
57. Trombetta M, Bonetti S, Boselli ML, Miccoli R, Trabetti E, Malerba G, et al. PPARG2 Pro12Ala and ADAMTS9 rs4607103 as “insulin resistance loci” and “insulin secretion loci” in Italian individuals. The GENFIEV study and the Verona Newly Diagnosed Type 2 Diabetes Study (VNDS) 4. Acta Diabetol (2013) 50(3):401–8. doi:10.1007/s00592-012-0443-9
58. Demircan K, Yonezawa T, Takigawa T, Topcu V, Erdogan S, Ucar F, et al. ADAMTS1, ADAMTS5, ADAMTS9 and aggrecanase-generated proteoglycan fragments are induced following spinal cord injury in mouse. Neurosci Lett (2013) 544:25–30. doi:10.1016/j.neulet.2013.02.064
59. Lemarchant S, Pruvost M, Montaner J, Emery E, Vivien D, Kanninen K, et al. ADAMTS proteoglycanases in the physiological and pathological central nervous system. J Neuroinflammation (2013) 10:133. doi:10.1186/1742-2094-10-133
60. Reid MJ, Cross AK, Haddock G, Allan SM, Stock CJ, Woodroofe MN, et al. ADAMTS-9 expression is up-regulated following transient middle cerebral artery occlusion (tMCAo) in the rat. Neurosci Lett (2009) 452(3):252–7. doi:10.1016/j.neulet.2009.01.058
61. Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, et al. Seven new loci associated with age-related macular degeneration. Nat Genet (2013) 45(4):433–9, 439e1–2. doi:10.1038/ng.2578
62. Lin E, Tsai SJ, Kuo PH, Liu YL, Yang AC, Kao CF, et al. The rs1277306 variant of the REST gene confers susceptibility to cognitive aging in an elderly Taiwanese population. Dement Geriatr Cogn Disord (2017) 43(3–4):119–27. doi:10.1159/000455833
63. Bahn S, Mimmack M, Ryan M, Caldwell MA, Jauniaux E, Starkey M, et al. Neuronal target genes of the neuron-restrictive silencer factor in neurospheres derived from fetuses with Down’s syndrome: a gene expression study. Lancet (2002) 359(9303):310–5. doi:10.1016/S0140-6736(02)07497-4
64. Baldelli P, Meldolesi J. The transcription repressor REST in adult neurons: physiology, pathology, and diseases (1,2,3). eNeuro (2015) 2(4). doi:10.1523/ENEURO.0010-15.2015
65. Dallagnol KM, Remor AP, da Silva RA, Prediger RD, Latini A, Aguiar AS Jr. Running for REST: physical activity attenuates neuroinflammation in the hippocampus of aged mice. Brain Behav Immun (2017) 61:31–5. doi:10.1016/j.bbi.2016.07.159
66. Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y, et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature (2014) 507(7493):448–54. doi:10.1038/nature13163
67. Nho K, Kim S, Risacher SL, Shen L, Corneveaux JJ, Swaminathan S, et al. Protective variant for hippocampal atrophy identified by whole exome sequencing. Ann Neurol (2015) 77(3):547–52. doi:10.1002/ana.24349
68. Cheng D, Noble J, Tang MX, Schupf N, Mayeux R, Luchsinger JA. Type 2 diabetes and late-onset Alzheimer’s disease. Dement Geriatr Cogn Disord (2011) 31(6):424–30. doi:10.1159/000324134
69. Ronnemaa E, Zethelius B, Lannfelt L, Kilander L. Vascular risk factors and dementia: 40-year follow-up of a population-based cohort. Dement Geriatr Cogn Disord (2011) 31(6):460–6. doi:10.1159/000330020
70. Jun G, Naj AC, Beecham GW, Wang LS, Buros J, Gallins PJ, et al. Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol (2010) 67(12):1473–84. doi:10.1001/archneurol.2010.201
71. Wijsman EM, Daw EW, Yu X, Steinbart EJ, Nochlin D, Bird TD, et al. APOE and other loci affect age-at-onset in Alzheimer’s disease families with PS2 mutation. Am J Med Genet B Neuropsychiatr Genet (2005) 132B(1):14–20. doi:10.1002/ajmg.b.30087
72. Pastor P, Roe CM, Villegas A, Bedoya G, Chakraverty S, Garcia G, et al. Apolipoprotein Eepsilon4 modifies Alzheimer’s disease onset in an E280A PS1 kindred. Ann Neurol (2003) 54(2):163–9. doi:10.1002/ana.10636
73. St George-Hyslop P, McLachlan DC, Tsuda T, Rogaev E, Karlinsky H, Lippa CF, et al. Alzheimer’s disease and possible gene interaction. Science (1994) 263(5146):537. doi:10.1126/science.8290965
74. Xu X. DNA methylation and cognitive aging. Oncotarget (2015) 6(16):13922–32. doi:10.18632/oncotarget.4215
75. Mather KA, Kwok JB, Armstrong N, Sachdev PS. The role of epigenetics in cognitive ageing. Int J Geriatr Psychiatry (2014) 29(11):1162–71. doi:10.1002/gps.4183
76. Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci (2006) 31(2):89–97. doi:10.1016/j.tibs.2005.12.008
77. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet (2012) 13(7):484–92. doi:10.1038/nrg3230
78. Chouliaras L, Kenis G, Visser PJ, Scheltens P, Tsolaki M, Jones RW, et al. DNMT3A moderates cognitive decline in subjects with mild cognitive impairment: replicated evidence from two mild cognitive impairment cohorts. Epigenomics (2015) 7(4):533–7. doi:10.2217/epi.15.22
79. Bey K, Wolfsgruber S, Karaca I, Wagner H, Lardenoije R, Becker J, et al. No association of the variant rs11887120 in DNMT3A with cognitive decline in individuals with mild cognitive impairment. Epigenomics (2016) 8(5):593–8. doi:10.2217/epi-2015-0014
80. Chen BF, Chan WY. The de novo DNA methyltransferase DNMT3A in development and cancer. Epigenetics (2014) 9(5):669–77. doi:10.4161/epi.28324
81. Davies G, Harris SE, Reynolds CA, Payton A, Knight HM, Liewald DC, et al. A genome-wide association study implicates the APOE locus in nonpathological cognitive ageing. Mol Psychiatry (2014) 19(1):76–87. doi:10.1038/mp.2012.159
82. Gottschalk WK, Lutz MW, He YT, Saunders AM, Burns DK, Roses AD, et al. The broad impact of TOM40 on neurodegenerative diseases in aging. J Parkinsons Dis Alzheimers Dis (2014) 1(1). doi:10.13188/2376-922X.1000003
83. Guo AY, Sun J, Riley BP, Thiselton DL, Kendler KS, Zhao Z. The dystrobrevin-binding protein 1 gene: features and networks. Mol Psychiatry (2009) 14(1):18–29. doi:10.1038/mp.2008.88
84. Burdick KE, Goldberg TE, Funke B, Bates JA, Lencz T, Kucherlapati R, et al. DTNBP1 genotype influences cognitive decline in schizophrenia. Schizophr Res (2007) 89(1–3):169–72. doi:10.1016/j.schres.2006.09.008
85. Hashimoto R, Noguchi H, Hori H, Nakabayashi T, Suzuki T, Iwata N, et al. A genetic variation in the dysbindin gene (DTNBP1) is associated with memory performance in healthy controls. World J Biol Psychiatry (2010) 11(2 Pt 2):431–8. doi:10.1080/15622970902736503
86. Hashimoto R, Noguchi H, Hori H, Ohi K, Yasuda Y, Takeda M, et al. Association between the dysbindin gene (DTNBP1) and cognitive functions in Japanese subjects. Psychiatry Clin Neurosci (2009) 63(4):550–6. doi:10.1111/j.1440-1819.2009.01985.x
87. Kircher T, Markov V, Krug A, Eggermann T, Zerres K, Nothen MM, et al. Association of the DTNBP1 genotype with cognition and personality traits in healthy subjects. Psychol Med (2009) 39(10):1657–65. doi:10.1017/S0033291709005388
88. Neuner SM, Garfinkel BP, Wilmott LA, Ignatowska-Jankowska BM, Citri A, Orly J, et al. Systems genetics identifies Hp1bp3 as a novel modulator of cognitive aging. Neurobiol Aging (2016) 46:58–67. doi:10.1016/j.neurobiolaging.2016.06.008
89. Garfinkel BP, Melamed-Book N, Anuka E, Bustin M, Orly J. HP1BP3 is a novel histone H1 related protein with essential roles in viability and growth. Nucleic Acids Res (2015) 43(4):2074–90. doi:10.1093/nar/gkv089
90. Wolosker H, Blackshaw S, Snyder SH. Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc Natl Acad Sci U S A (1999) 96(23):13409–14. doi:10.1073/pnas.96.23.13409
91. Morita Y, Ujike H, Tanaka Y, Otani K, Kishimoto M, Morio A, et al. A genetic variant of the serine racemase gene is associated with schizophrenia. Biol Psychiatry (2007) 61(10):1200–3. doi:10.1016/j.biopsych.2006.07.025
92. Turpin FR, Potier B, Dulong JR, Sinet PM, Alliot J, Oliet SH, et al. Reduced serine racemase expression contributes to age-related deficits in hippocampal cognitive function. Neurobiol Aging (2011) 32(8):1495–504. doi:10.1016/j.neurobiolaging.2009.09.001
93. Labrie V, Fukumura R, Rastogi A, Fick LJ, Wang W, Boutros PC, et al. Serine racemase is associated with schizophrenia susceptibility in humans and in a mouse model. Hum Mol Genet (2009) 18(17):3227–43. doi:10.1093/hmg/ddp261
94. Chen CH, Yang JH, Chiang CWK, Hsiung CN, Wu PE, Chang LC, et al. Population structure of Han Chinese in the modern Taiwanese population based on 10,000 participants in the Taiwan Biobank project. Hum Mol Genet (2016) 25(24):5321–31. doi:10.1093/hmg/ddw346
95. Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res (2012) 22(9):1748–59. doi:10.1101/gr.136127.111
96. Sheehan B. Assessment scales in dementia. Ther Adv Neurol Disord (2012) 5(6):349–58. doi:10.1177/1756285612455733
97. Brodaty H, Low LF, Gibson L, Burns K. What is the best dementia screening instrument for general practitioners to use? Am J Geriatr Psychiatry (2006) 14(5):391–400. doi:10.1097/01.JGP.0000216181.20416.b2
98. Milne A, Culverwell A, Guss R, Tuppen J, Whelton R. Screening for dementia in primary care: a review of the use, efficacy and quality of measures. Int Psychogeriatr (2008) 20(5):911–26. doi:10.1017/S1041610208007394
99. Wild K, Howieson D, Webbe F, Seelye A, Kaye J. Status of computerized cognitive testing in aging: a systematic review. Alzheimers Dement (2008) 4(6):428–37. doi:10.1016/j.jalz.2008.07.003
100. Smith PJ, Need AC, Cirulli ET, Chiba-Falek O, Attix DK. A comparison of the Cambridge Automated Neuropsychological Test Battery (CANTAB) with “traditional” neuropsychological testing instruments. J Clin Exp Neuropsychol (2013) 35(3):319–28. doi:10.1080/13803395.2013.771618
101. Rockwood K, Fay S, Gorman M, Carver D, Graham JE. The clinical meaningfulness of ADAS-Cog changes in Alzheimer’s disease patients treated with donepezil in an open-label trial. BMC Neurol (2007) 7:26. doi:10.1186/1471-2377-7-26
102. Thomas ML, Kaufmann CN, Palmer BW, Depp CA, Martin AS, Glorioso DK, et al. Paradoxical trend for improvement in mental health with aging: a community-based study of 1,546 adults aged 21–100 years. J Clin Psychiatry (2016) 77(8):e1019–25. doi:10.4088/JCP.16m10671
103. Lin E, Lane HY. Machine learning and systems genomics approaches for multi-omics data. Biomark Res (2017) 5:2. doi:10.1186/s40364-017-0082-y
104. Lin E, Lane HY. Genome-wide association studies in pharmacogenomics of antidepressants. Pharmacogenomics (2015) 16(5):555–66. doi:10.2217/pgs.15.5
105. Lin E, Tsai SJ. Genome-wide microarray analysis of gene expression profiling in major depression and antidepressant therapy. Prog Neuropsychopharmacol Biol Psychiatry (2016) 64:334–40. doi:10.1016/j.pnpbp.2015.02.008
106. Lin CH, Chen PK, Chang YC, Chuo LJ, Chen YS, Tsai GE, et al. Benzoate, a D-amino acid oxidase inhibitor, for the treatment of early-phase Alzheimer disease: a randomized, double-blind, placebo-controlled trial. Biol Psychiatry (2014) 75(9):678–85. doi:10.1016/j.biopsych.2013.08.010
Keywords: Alzheimer’s diseases, biomarker, age-related cognitive decline, cognitive aging, gene–gene interactions, neurodegeneration, single-nucleotide polymorphisms, SNP–SNP interactions
Citation: Lin C-H, Lin E and Lane H-Y (2017) Genetic Biomarkers on Age-Related Cognitive Decline. Front. Psychiatry 8:247. doi: 10.3389/fpsyt.2017.00247
Received: 29 August 2017; Accepted: 07 November 2017;
Published: 21 November 2017
Edited by:
Kenji Hashimoto, Chiba University, JapanReviewed by:
Po See Chen, National Cheng Kung University, TaiwanRogerio Panizzutti, Universidade Federal do Rio de Janeiro, Brazil
Copyright: © 2017 Lin, Lin and Lane. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hsien-Yuan Lane, aHlsYW5lQGdtYWlsLmNvbQ==