 Aliya M. Lucatch
Aliya M. Lucatch Darby J. E. Lowe
Darby J. E. Lowe Rachel C. Clark1
Rachel C. Clark1- 1Addictions Division, Centre for Addiction and Mental Health (CAMH), Toronto, ON, Canada
- 2Institute of Medical Sciences, University of Toronto, Toronto, ON, Canada
- 3Division and Brain and Therapeutics, Department of Psychiatry, University of Toronto, Toronto, ON, Canada
Purpose of review: To provide an overview of the underlying neurobiology of tobacco smoking in schizophrenia, and implications for treatment of this comorbidity.
Recent findings: Explanations for heavy tobacco smoking in schizophrenia include pro-cognitive effects of nicotine, and remediation of the underlying pathophysiology of schizophrenia. Nicotine may ameliorate neurochemical deficits through nicotine acetylcholine receptors (nAChRs) located on the dopamine, glutamate, and GABA neurons. Neurophysiological indices including electroencephalography, electromyography, and smooth pursuit eye movement (SPEM) paradigms may be biomarkers for underlying neuronal imbalances that contribute to the specific risk of tobacco smoking initiation, maintenance, and difficulty quitting within schizophrenia. Moreover, several social factors including socioeconomic factors and permissive smoking culture in mental health facilities, may contribute to the smoking behaviors (initiation, maintenance, and inability to quit smoking) within this disorder.
Summary: Tobacco smoking may alleviate specific symptoms associated with schizophrenia. Understanding the neurobiological underpinnings and psychosocial determinants of this comorbidity may better explain these potential beneficial effects, while also providing important insights into effective treatments for smoking cessation.
Introduction
The high rates of tobacco use in the schizophrenia (SZ) population are widely recognized, but the underlying neurobiological factors contributing to this comorbidity are not fully understood. Rates of tobacco smoking are between 45 and 88% in SZ compared to <16% of the general population (1, 2). In this review, we aim to highlight the recent literature on the latter category of neurobiological determinants and discuss some potential treatment targets.
The high prevalence of smoking in SZ is maintained in large part by resistance to quitting (3); quit rates from an American nationally representative sample range from 10 to 27.2% for those with psychotic disorders compared to 42.5% in the general population (4). Additionally, relapse rates pose a common challenge in delivering cessation treatments, but there is some indication that longer courses of pharmacological treatments could reduce the possibility of relapse (2). Unfortunately, these high smoking rates come with a cost, and smokers with SZ are at higher risk for tobacco-related morbidity and mortality; people with SZ have ~25 years of shortened lifespan, with 53% of this being related to tobacco-smoking conditions (5–7). One population-based study in the United States (U.S.) found that among individuals with SZ, cardiovascular disease, lung cancer, and respiratory diseases such as chronic obstructive pulmonary disease and pneumonia contributed to the most deaths (8). It is clear from the evidence that reducing smoking rates has the potential to drastically change mortality rates and improve outcomes for these patients.
Etiology of Tobacco Smoking in SZ
Many explanations have been proposed for the higher prevalence of smoking in persons with SZ. In this section, we briefly review some of these important factors before highlighting current findings on the neurobiology of this comorbidity. These major factors include increased craving in SZ, modulating negative symptoms, pro-cognitive effects of nicotine, and genetic factors (3). In addition, we compare the self-medication hypothesis with the addiction vulnerability hypothesis for tobacco use in SZ.
It has been proposed that due to the pathophysiology of SZ, these individuals may have an enhanced experience of the reinforcing effects of nicotine (3, 9). This idea has been corroborated with a study that compared smokers with SZ to non-psychiatric control smokers in an abstinence condition; they found that the SZ group reported stronger cravings and withdrawal symptoms and had a shorter time to smoking lapse compared to the control group (10). This effect was moderated by negative affect and withdrawal symptom severity (10). Another study using an animal model produced lesions in the ventral hippocampus, a region associated with SZ, and found increased reinforcing effects and drug-seeking behavior for nicotine (11).
There appears to be a link between the enhanced reinforcing effects of nicotine and the role that negative affect has on increasing the smoking rate in SZ. This may be due to the deficits in reward processing and alterations in reward-related brain circuitry that is characteristic of negative symptoms in SZ (12, 13). In an fMRI study of smokers with SZ compared to control smokers, researchers found that both groups had brain reactivity to smoking cues, but SZ group had reduced brain reactivity to neutral cues, and that this effect was related to negative symptoms (14). This finding indicates that the enhanced addictive properties of nicotine in SZ is not due to a stronger reactivity to nicotine-related cues, but rather may be related to the underlying negative symptomatology (14). A recent study using a cognitive assessment of reward learning in smokers found a negative correlation between general reward responsiveness and intensity of nicotine craving (15). This finding suggests that individuals with negative affect and a dysfunctional reward circuit, such as those with SZ, may be more susceptible to nicotine addiction (15). Together, these findings indicate that increased negative symptomatology may play a role in the enhanced susceptibility to smoking in SZ, yet does not reveal the full picture.
Pro-cognitive Effects of Nicotine in SZ
Another key etiological factor to consider is the potential pro-cognitive effects that nicotine has on SZ; nonetheless there have been mixed findings in this field. For instance, much of the epidemiological research surrounding this comorbidity has found no effect or worsened cognition within chronic smokers with SZ (16–19). However, lack of control for time since last cigarette may result in nicotine withdrawal-related cognitive impairment (20–22) and may explain some of these negative findings as the participants were likely to be experiencing significant smoking deprivation. Table 1 has been included below to illustrate the variety of study methodologies examining the pro-cognitive effects of smoking and how each study accounted for the duration since last cigarette. However, studies that carefully control for time since last cigarette have found that smoking produces cognitive deficits in SZ, particularly in working memory, visual learning, and attention (3, 23, 24, 31). In laboratory studies where nicotine is acutely administered or acute overnight abstinence and reinstatement paradigms are used (thereby avoiding any confounding effects of nicotine withdrawal), smoking groups have shown marked improvement for attention, working memory, pre-pulse inhibition, visuospatial working memory, processing speed, and verbal learning and memory(23, 24, 26, 28–37).
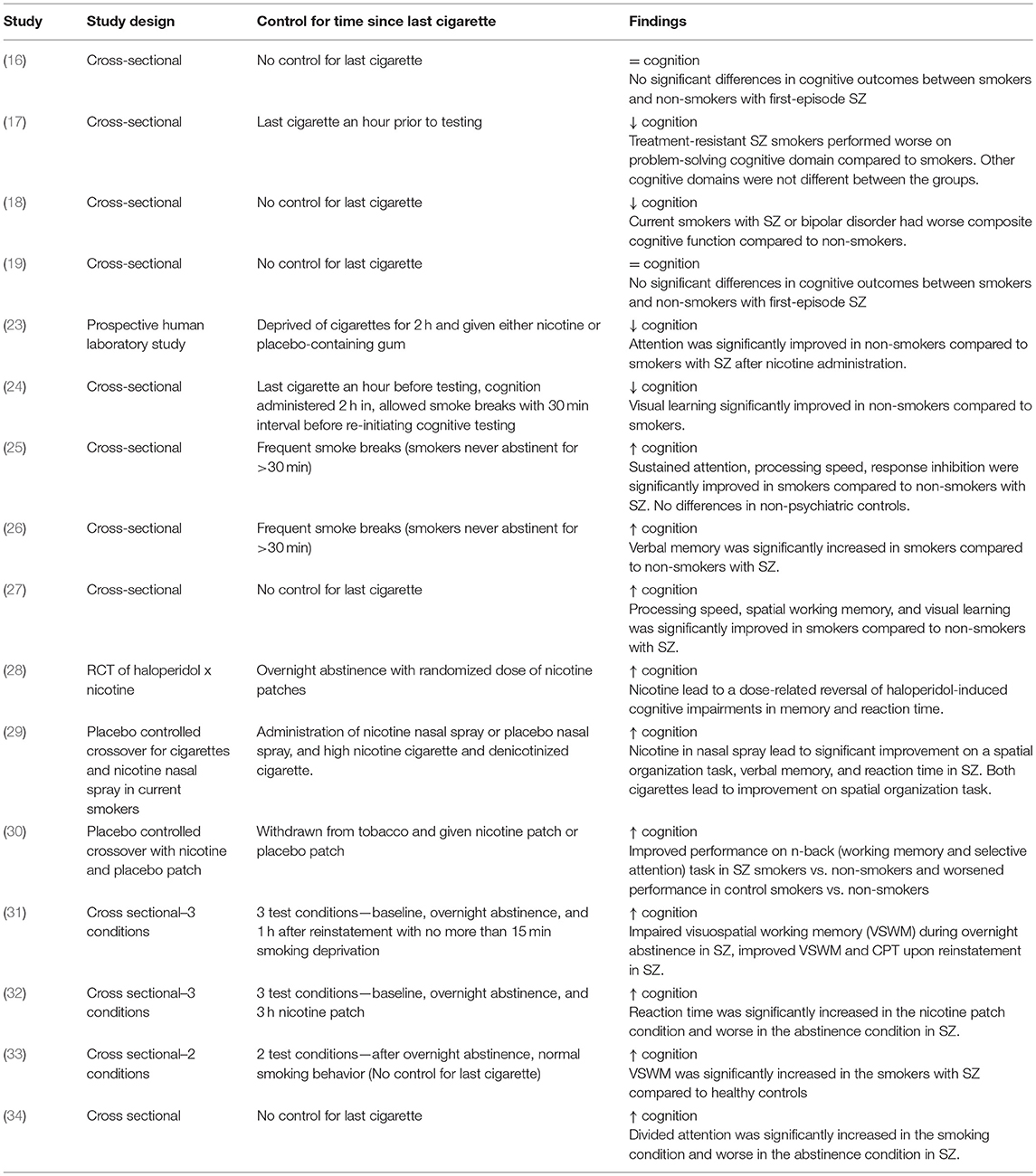
Table 1. Cognitive Effects in Acute vs. Chronic Smokers with SZ.
Studies have also compared cognitive performance between non-smoking and smoking patients with SZ. These studies account for the level of nicotine in the participant's system by allowing frequent smoke breaks so as to avoid inducing a state of withdrawal (25, 26). Non-smokers revealed significantly worse cognitive deficits, particularly in verbal memory (25, 26).
Interestingly, this effect is specific to those with schizophrenia, as no such finding was observed in patients with major depression, bipolar disorder or non-psychiatric controls (25, 26). Furthermore, individuals categorized as ultra-high-risk (UHR) for developing psychosis may also demonstrate this effect (27).
Other studies employed cognitive testing in both satiated and abstinent states and demonstrated that smokers with SZ show improvements in various cognitive domains (28–30, 33, 38). These studies are shown in Table 1. A satiated state was produced by administering nicotine throughout the study session with a patch, gum, or nasal spray, providing the benefit of acute nicotine exposure (28–30, 32); however, these were regular smokers. There are few studies examining acute nicotine administration in non-smokers, due to the nature of tobacco use disorder and the all-or-nothing tendency for people to be regular smokers or non-smokers. The few studies that have examined nicotine administration in non-smokers found an overall improvement in attention following nicotine administration and a specific effect at improving cognitive outcomes in the SZ participants (23, 39). Nonetheless, nicotine administration improves cognitive outcomes in SZ individuals, this may account for the increased frequency and severity of tobacco use in SZ and also the perseverance of tobacco use disorder in this population (40).
There are two primary theories proposed to explain the pro-cognitive effects of nicotine in SZ, and the relationship to the increase prevalence of smoking in SZ. First, the self-medication hypothesis proposes that individuals with SZ choose to smoke to alleviate the clinical symptoms and cognitive deficits that are characteristic of the illness as well as the side effects of antipsychotic medications (41). Many of the studies examining the procognitive effects of nicotine lend support to the theory, but others have refuted this theory. For example, one group found that it was a stronger tendency for those with SZ to experience withdrawal when abstinent that led to cognitive deficits and that blood nicotine concentration did not affect performance when compared to healthy controls (42). In response to these challenges identified with the self-medication hypothesis, researchers have developed an alternate theory to explain the heightened prevalence of smoking in SZ which is termed the addiction vulnerability hypothesis (43). This theory proposes that it is due to genetic, neurobiological, and environmental factors that are inherent to the SZ diagnosis that make these patients susceptible to smoking (43). Understanding the unique factors contributing this vulnerability can provide us with novel treatments targeting smoking cessation in this specific population, in particular, building our knowledge about the underlying neurotransmitter systems and brain circuitry is essential (44).
Neurobiological Determinants of Tobacco Smoking in Schizophrenia
Nicotine
Nicotine, the addictive component in tobacco cigarettes, binds to nicotinic acetylcholine receptors (nAChRs), which are endogenously expressed in the human brain and influenced by the native agonist, acetylcholine (45). nAChRs are a heterogeneous family of ion channels, that are expressed on various cellular regions of both excitatory and inhibitory neurons, allowing for modulation of neurotransmitters (45, 46). The composition of the nAChRs is a variety of subunits which define the receptors' actions and properties (47). The most common high-affinity nAChRs include receptors consisting of two α4 subunits, two β2 subunits, and an undefined fifth subunit (48). Single nucleotide polymorphisms (SNPs) found on the CHRNA4 gene coding for the α4 subunit has been associated with nicotine dependence (49–51). Another important nAChR type to consider is that of the α7 receptor, which consists entirely of α7 subunits. SNPs located on the receptor coding gene CHRNA7, has been associated with both SZ diagnoses (52, 53) and nicotine dependence (54). Notably, there are two nicotinic acetylcholine receptors (nAChR) subtypes linked to cognition: high-affinity α4β2 and low-affinity α7 nAChRs (45, 55). High-affinity nAChRs are sensitive to nicotinic antagonists such as mecamylamine (56), and mediate nicotine reinforcement and cognition (36, 57), whereas low-affinity nAChRs are less nicotine-sensitive.
Dopamine, norepinephrine, serotonin, glutamate, aminobutyric acid (GABA) and opioid peptides are neurotransmitter systems influenced by nAChRs (58). The neurobiological phenotype of SZ involves dysfunction of similar neurotransmitters, such as the dopaminergic, glutamatergic and GABAergic systems, as well as overarching dysfunction of cortical and subcortical communicative circuitry. Developmental and genetically predisposed abnormalities observed in the prefrontal and hippocampal regions in individuals with SZ may facilitate neural circuitry dysfunction, promoting a vulnerability toward addiction, such as tobacco use disorder (59). The neurobiological abnormalities of SZ will be discussed after which the corresponding effects of nicotine will be supplemented in order to provide insight into this prevalent comorbidity.
Pathophysiology of SZ and Nicotine Effects
The following section describes the role of each neurotransmitter system on both nicotine addiction and schizophrenia. An overall summary and simplification of these effects is illustrated with Figure 1.
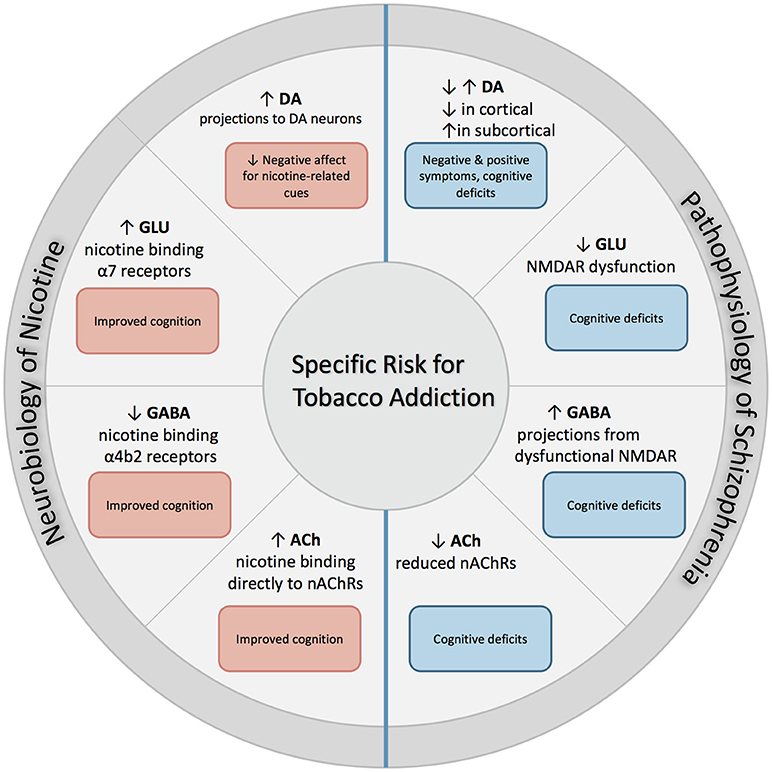
Figure 1. Excessive dopaminergic activity has been proposed within subcortical regions in individuals with SZ, and is associated with positive symptoms of SZ. Conversely, a hypo-dopaminergic state has been postulated in the cortical regions, and is associated with cognitive deficits and increased negative symptoms. NMDAR abnormalities found in SZ contribute to both hypo-glutamatergic activity and hyper-GABAergic activity, and leads to cognitive dysfunction. Individuals with SZ have reduced expression of nAChRs which leads to altered nicotinic cholinergic transmission, which may contribute to cognitive dysfunction. When nicotine is administered through tobacco smoking, these deficits may be partially attenuated. First, nicotine binds directly to nAChRs that are located in mesolimbic dopaminergic pathways, which increases its expression and contributes to reduction in negative affect in response to smoking-related cues. In addition, nicotine binds to α7 and α4β2 receptors on glutamatergic and GABAergic neurons in the prefrontal cortex, attenuating deficits found in SZ and enhancing cognition.
Dopaminergic Dysfunction
The dopamine (DA) hypothesis for SZ features the imbalances in dopaminergic neurotransmission throughout the brain, such as presynaptic abnormalities of DA neurons that are described in both SZ and high-risk populations (60, 61). DA dysfunction influences both cortical and subcortical circuitry, facilitating symptomatology of SZ differentially. Subcortical regions in the brain have been associated with increased dopaminergic activity, leading to over-stimulation of D2 receptors (62, 63). Hyper-dopaminergic activity in subcortical regions, specifically in the associative striatum, has been associated with positive symptoms of SZ, including psychosis (61, 64–66). Cortical regions, however, have been linked with dampened dopaminergic activity. This has been investigated through various functional imaging studies, demonstrating an under-stimulation of D1 receptors in the frontal regions of individuals with SZ (67). Additionally, in vivo findings of decreased dopaminergic activity in the dorsolateral prefrontal cortex has been associated with cognitive impairment severity, such as worsened working memory, as well as negative symptom severity (68–71). Treatment that targets dopamine dysfunction, the most pervasive form of medication for SZ (targeting dopamine D2 receptors), has proven to aid with positive symptoms; however, in individuals who lack substantial dopaminergic dysfunction, this treatment does not robustly align with symptom improvement (61, 72).
Glutamatergic and GABAergic Dysfunction
A more recent hypothesis for SZ pathology involves glutamatergic dysfunction, involving the hypofunction of N-methyl-D-aspartate (NMDA) receptors of which glutamate, the major excitatory transmitter in the brain, binds (73, 74). Supported by genetic convergence, brain tissue analysis and brain imaging studies (74–76), the glutamatergic theory offers a unique conceptualization that encompasses the widespread deficits observed in SZ (73). For example, NMDAR antagonists, such as phencyclidine and ketamine, correlate with the emergence of both negative and positive symptoms as well as general neuropsychological and sensory deficits associated with SZ in contrast to amphetamine, a dopamine receptor agonist, which is mostly associated with inducing the positive symptoms of the disorder (73). NMDA receptors influence the majority of input, output and interneuronal cortical projections and, therefore, have a diffuse influence on brain function (77). In SZ, glutamatergic dysfunction due to NMDAR abnormalities has been noted in regions within the limbic system, the hippocampus and the dorsolateral prefrontal cortex (78–81). NMDAR activity is also important in considering the functioning and maintenance of the brain's main inhibitory transmitter -aminobutyric acid (GABA) (82). Deficits in GABA synthesis (deficits in glutamic acid decarboxylase (GAD)-67, which aids GABA synthesis) have been denoted throughout the cortex of individuals with SZ (83). Decreased functioning of GABAergic interneurons has been posed to contribute to cognitive impairment in SZ by means of decreased synchronization in neuronal cortical activity (84). Specifically, the hypofunctionality of NMDARs in SZ has been proposed to lead to dysfunctional GABAergic transmission (85). Dampened activity of NMDARs at GABAergic interneurons disrupts and reduces inhibitory control over cortical activity as well as the overall synchrony of gamma oscillations, leading to clinical disruptions in SZ (86).
Additionally, NMDARs, which influence both glutamate and GABA, highly influence dopamine synthesis and transmission (75, 87). Glutamate, stimulated by NMDARs, regulates dopamine neurons that project from the ventral tegmentum area (VTA) toward the nucleus accumbens (NAc) or the prefrontal cortex (PFC), as well as GABA neurons that also play a role in regulating dopamine neurons (88). Dysfunction within the glutamatergic system in SZ has been proposed to facilitate the dopaminergic dysfunction linked to cognitive disruptions of the illness (87, 89). Additionally, presynaptic dopamine released both subcortically and within the frontal regions of the brain are influenced by inhibitory GABA interneurons, therefore disruption in GABA signaling via NMDAR abnormalities has also been linked to abnormalities in dopamine signaling (73, 90, 91).
DA, GLU, GABA, and Nicotine
Nicotine modulates dopaminergic transmission in both subcortical and cortical regions. Primary dopaminergic projections involve transmission from the VTA toward the NAc; major components of reward circuitry in addiction of which nicotine enhances through this pathway (92, 93). Nicotine is able to influence dopamine's activity by directly binding nAChRs on dopaminergic projections sourced in the VTA as well as by regulating glutamate and GABA activity, which excites and inhibits dopaminergic activity, respectively (94). Specifically, nicotine binds to the α7 receptors along glutamatergic neurons, stimulating their activity and enhancing NMDAR function, which together enhances dopaminergic neuronal activity (94). Nicotine also binds α4β2, a high-affinity receptor along GABA neurons, which, with chronic nicotine use, become desensitized, therefore dampening the inhibition on dopaminergic transmission from the VTA to the NAc, while α7, low-affinity nAChRs on glutamatergic neurons are less prone to desensitization, therefore continue to enhance dopamine transmission (94, 95). Nicotine directly binds nAChRs along dopamine neuronal cells, facilitating burst firing and increased dopamine activity directly (96), which, combined with enhanced glutamatergic tone, leads to an overall increased level of dopamine transmission and release in the NAc that supports the reinforcing effects of nicotine (97).
It has been posed that nicotine leads to increases in dopamine levels within prefrontal regions through direct and indirect (GABA and glutamate influences) enhancement of dopaminergic activity that makes up for the lowered dopamine D1 stimulation and ensuing cognitive deficits observed in SZ (44, 67, 98). Nicotine facilitates increased dopamine in the cortex similarly to observations in subcortical regions in that the drug binds high-affinity nAChRs on dopamine neurons that project from the VTA, but in this case toward the cortex, and binds low-affinity receptors on excitatory glutamatergic neurons projecting toward the prefrontal cortex (99, 100). The reported beneficial effects that nicotine influences in the frontal cortex have been proposed to be largely due to its effects on the α7 nAChR subunit, although there is some evidence surrounding α4b2 receptor subunit activity leading to improved higher cognition (98). Because GABA contains many α7 as well as α4b2 nAChRs, nicotine could counteract the deficits observed in GABAergic transmission in SZ and the coinciding prefrontal dysfunction by stabilizing cortical inhibition through enhancing interneuronal activity and frontal gamma oscillations (3, 83, 101–103).
naChRs and Nicotine
Additionally, SZ involves the dysregulation of both high- and low-affinity nAChRs (98, 104). Studies have shown that individuals with SZ have a reduction in nAChRs expression throughout brain regions that are central to higher cognitive functioning (105). Additionally, it has been found that chronic nicotine use leads to nAChR receptor desensitization and inactivation during stages of withdrawal, which are reactivated upon overnight abstinence (3, 106, 107). Clinically, this pattern of receptor desensitization may explain the phenomenon where smokers prefer the first cigarette in the morning, and why cognitive deficits are present during periods of withdrawal (106). However, in the SZ population, this pattern of desensitization and resensitization may have a different presentation due to the decreased expression of nicotinic receptors, which may account for their increased severity of tobacco addiction (108).
SZ has also been linked genetically to the CHRNA7 gene, a potential site of genetic heritability, which codes for the α7 subunit of nAChRs (109). Individuals with SZ who smoke have exhibited improvements in their cognition, highlighting the potential benefits of stimulating this receptor in this population (26, 29). For example, nAChR agonists and antagonists, such as varenicline and mecamylamine, have been used in various smoking cessation and treatment trials in which the results further support the cognitive improvements observed in smokers with SZ (31, 36, 110). Additionally, levels of CHRNA7 protein and mRNA became comparable to non-psychiatric smokers following smoking in SZ (53). Overall, nAChR dysfunction may influence the aberrant signaling of glutamate, GABA, and dopamine of which nicotine use may partially alleviate (3).
Circuitry Dysfunction
It is posed that each transmitter system, including dopamine, glutamate, GABA and cholinergic neuronal transmission, incorporates a circuit, supported by genetic risk, that facilitates a risk for SZ presentation (61, 111). The dysfunction within SZ and nicotine's influence on these abnormalities do not exist in isolation. The combination of abnormal dopamine neurotransmission and nAChR signaling, along with imbalances in glutamate and GABA transmission, which influences the former dysfunctions, may lead to the widespread deficits and symptoms observed in SZ (3). Nicotine stimulates nAChRs, which are situated along glutamatergic and GABAergic neurons. Nicotine, therefore, may modulate glutamate-GABA interactions and normalize excitation-inhibition influences over dopamine signaling and communication within the brain through improving baseline nAChR-stimulation dysregulated in SZ. The influence nicotine has on the transmission and general circuitry in SZ has been shown to alleviate certain symptomatic characteristics and cognitive deficits, as described above, which may place this population at an enhanced risk to developing tobacco use disorder.
Tobacco Use and Antipsychotic Medication
Cigarette smoking has been found to increase activity of the liver enzyme, cytochrome P450 1A2 (CYP 1A2), which in turn break down drugs in the body, including antipsychotic medications such as olanzapine and clozapine (112). As a result, there is reduced concentration of these antipsychotics in medicated SZ smokers, which predictably leads to a reduction in side effects, motivating further use (113) (supporting the self-medication hypothesis). An important implication of the reduction in antipsychotic medication is the potential for worsened symptoms of psychosis (114), to account for this, researchers of a meta-analysis study have indicated that smokers with SZ should be prescribed antipsychotics at a dose double that of non-smokers (115).
Biomarkers of Vulnerability
P50 Suppression and Mismatch Negativity
P50 suppression is an electroencephalographic measure of cortical inhibition that follows a second tone that is presented 500 ms after an initial tone (116). SZ, as well as the pathological and heritable characteristics of the disorder, is associated with the sensory gating deficit of diminished suppression (117–119). This deficit has been genetically linked to polymorphisms found on the promoter region of the CHRNA7 coding gene for the α7 nAChR subunit, as well as decreased function of the α7 nAChR (120–122). It is thought that GABAergic neurotransmission within the hippocampal region, which is dysfunctional in SZ, mediates the production of P50 suppression, therefore may contribute to this population's sensory deficit (123). Nicotine, through its influence on nAChRs and the downstream effects, has been shown to remediate the deficits in P50 suppression for this population (35, 124, 125). Moreover, nAChR agonists also show improvements in cognitive functioning, including P50 suppression (126).
MMN is a neurophysiological test that quantitatively and temporally measures central auditory functioning or, more specifically, the neuronal processing in response to an auditory “oddball paradigm,” which involves the intervention of a deviant stimulus within sequential auditory tones (127, 128). From clinically high-risk to chronic classifications of SZ, this population exhibits reductions in MMN amplitudes based on frontocentral electroencephalographic recordings that are shown across all dimensions of auditory deviance (127–130). This deficit has been linked to NMDAR dysfunction, which correlates to the glutamatergic hypothesis of SZ pathology (119, 131, 132). Nicotine seems to enhance the duration of MMN amplitude, facilitating improvement in this neurophysiological deficit (119, 133, 134).
Pre-pulse Inhibition (PPI)
PPI is an electromyography measure of eye blink responses (i.e., one's eye muscle movement) to a startling auditory tone. If a “prepulse” tone occurs before the main auditory stimulus, one's blinking response is attenuated; however, individuals with SZ exhibit a deficit in this gating response (135, 136). This deficit has been associated with CHRNA3 polymorphisms, relating to nAChR dysfunction that is characteristic of SZ, and observed as heritable within this population (137–140). Nicotine has been noted to improve this deficit in smokers with and without SZ (137, 141, 142). Furthermore, abstinence related deficits in SZ (vs. non-psychiatric controls) were ameliorated by smoking reinstatement and blocked by nAChR antagonist, mecamylamine (143), suggesting that nAChR stimulation may remediate PPI deficits in SZ.
Smooth Pursuit Eye Movement
Smooth pursuit eye movement (SPEM) tasks involve the measurement of saccades, which are eye movements toward a target stimulus, as well as anti-saccades, which involves the movement away from a stimulus. Individuals with SZ have more intruding saccades to the extent of being described as a heritable characteristic of the diagnosis (144–147). Nicotine has shown to influence this measurement by improving the reliability of saccadic measures in individuals with SZ, but not in non-psychiatric controls, by potentially lowering the hyperactivation in regions facilitating this response and improving cortical inhibitory control (148–152).
Psychosocial Determinants of Tobacco Addiction in Schizophrenia
There are a variety of psychosocial factors that increase the vulnerability of individuals with SZ to develop and sustain tobacco addictions. Individuals with SZ tend to be of lower socioeconomic status compared to general population, which is associated with an increased likelihood of smoking initiation and a decreased likelihood of smoking cessation (153, 154). These individuals often have fixed, government-assisted income and may spend close to 30% of this income on cigarettes (155).
Elements of the mental health system itself may make individuals afflicted with SZ more vulnerable to smoking. There is a longstanding and pervasive smoking culture in mental health institutions that tolerates and even encourages tobacco consumption (156). Although, the smoking culture impacts all people with mental illness, SZ patients are particularly likely to be exposed, as they receive treatment largely in institutions and mental health settings (5). In fact, 80% of light smokers and 57% of moderate smokers have actually been found to increase their cigarette consumption following psychiatric admission (157). Despite the fact that programs to treat tobacco addiction in inpatient settings have been shown to be effective, mental health staff are reluctant to treat nicotine dependence in psychiatric patients and counseling for smoking cessation is rarely provided (156, 158, 159). Moreover, they are hesitant to ban cigarette smoking in institutions because of concern over patient resistance, infringing on patients' right to smoke and potential negative effects of smoking cessation on treatment outcomes (160, 161). Despite this common concern, inpatient psychiatric facilities that have implemented smoking bans, have demonstrated positive outcomes and had far fewer problems than anticipated (162, 163).
Treatment Implications
The advantage of broadening our understanding about the underlying neurobiology of this comorbidity is that it may lead to more novel, targeted treatments to be developed. In this section, we will briefly discuss some new developments in the area of treating smokers with SZ. Some major advancements in this field have been drugs targeting nAChRs and the potential for neuromodulation.
Currently, the most commonly studied and accepted treatments for this population in the order of effectiveness have been varenicline, bupropion and nicotine replacement therapy (NRT) (164). Varenicline acts as a partial agonist at the α4β2 nAChR, while buproprion acts at several targets including at the norepinephrine-dopamine receptors and at the nicotinic receptor (165, 166). Pharmacological treatments for smoking are more effective than any behavioral treatments in this population, and maintenance treatment is also an important way to prevent relapse in SZ (164). However, although varenicline has been found effective at reducing overall smoking in SZ, it has not been found to compensate as a cognitive enhancer (167). This is consistent with the findings of a recent meta analysis that determined α7-nAChRs as ineffective treatments for improving cognitive and negative symptom outcomes in SZ based on 8 RCTs (168). Additionally, cessation rates in patients with SZ remain significantly lower than those of the general population, and it is evident that a more holistic treatment strategy is required (164).
Neuromodulation is a promising new treatment modality which may have considerable promise for smokers with SZ (169). For example, repetitive transcranial magnetic stimulation (rTMS) delivers high-frequency magnetic fields to a targeted area of the brain (e.g., dorsolateral prefrontal cortex), stimulating neurons in that region and altering the brain circuitry. One study conducted in our lab found that a short course of rTMS was not effective at reducing craving in an overnight abstinence condition in patients with SZ (170). However, three other studies examined rTMS delivered over a longer course (10, 21, and 28 days, respectively) and found significant improvements on cigarette consumption and craving, but these effects dissipated over time (3, 171, 172). While the mechanism behind the impact of rTMS on nicotine addiction has not be fully elucidated (173), one hypothesis is that rTMS directed to the dorsolateral prefrontal cortex (169) reduces drug craving experienced by the user (174–176) (3. SZ Res.). Another neuromodulation method is transcranial direct current stimulation (tDCS), which provides a weaker electrical current over a longer duration to the brain, modulating neural firing without producing stimulation of neurons (177). A recent RCT of tDCS delivered in 5 sessions over 21 days in smokers with SZ found significant improvements on several cognitive deficits, but no improvements on cigarette use and craving outcomes (178). The findings within the field of neuromodulation are promising, but further studies are needed to corroborate these techniques as an effective treatment for smoking in SZ.
Conclusions
In summary, it is evident that the comorbidity of SZ and cigarette smoking is widespread, and that the underlying neurobiological factors are complex. Research on these factors is contributing to the development of treatment strategies that may help to reduce smoking and in turn the high mortality rates that arise due to the high smoking prevalence in this population. It is also important to consider a holistic approach because although neurobiology plays a large role in this comorbidity, etiological factors for smoking are multifaceted and all things must be considered. Further research and discussion should continue, and it is important that clinicians work against stigma and toward promoting education about high smoking rates as a specific vulnerability for individuals with SZ.
Author Contributions
AL: literature review, drafting manuscript, and final submission. DL and RC: literature review and contributing to content. TG: oversight, editor, and finalizing manuscript. KK: literature review for revision recommendations, contributing to content of revisions and final editing.
Funding
This work was supported in part by research support from the Astra H. Flaska Funds/CAMH Foundation and NIDA grant R21-DA-043949 to TG.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Morisano D, Bacher I, Audrain-McGovern J, George TP. Mechanisms underlying the comorbidity of tobacco use in mental health and addictive disorders. Can J Psychiatry (2009) 54:356–67. doi: 10.1177/070674370905400603
2. Tidey JW, Miller ME. Smoking cessation and reduction in people with chronic mental illness. BMJ (2015) 351:h4065. doi: 10.1136/bmj.h4065
3. Wing VC, Wass CE, Soh DW, George T. A review of neurobiological vulnerability factors and treatment implications for comorbid tobacco dependence in schizophrenia. Ann N Y Acad Sci. (2012) 1248:89–106. doi: 10.1111/j.1749-6632.2011.06261.x
4. Lasser K, Boyd JW, Woolhandler S, Himmelstein DU, McCormick D, Bor DH. Smoking and mental illness: a population-based prevalence study. JAMA (2000) 284:2606–10. doi: 10.1001/jama.284.20.2606
5. Ziedonis D, Hitsman B, Beckham JC, Zvolensky M, Adler LE, Audrain-McGovern J, et al. Tobacco use and cessation in psychiatric disorders: National Institute of Mental Health report. Nicotine Tob Res. (2008) 10:1691–715. doi: 10.1080/14622200802443569
6. George TP, Ziedonis DM. Addressing tobacco dependence in psychiatric practice: promises and pitfalls. Can J Psychiatry (2009) 54:353–5. doi: 10.1177/070674370905400602
7. Callaghan RC, Veldhuizen S, Jeysingh T, Orlan C, Graham C, Kakouris G, et al. Patterns of tobacco-related mortality among individuals diagnosed with schizophrenia, bipolar disorder, or depression. J Psychiatr Res. (2014) 48:102–10. doi: 10.1016/j.jpsychires.2013.09.014
8. Olfson M, Gerhard T, Huang C, Crystal S, Stroup TS. Premature mortality among adults with schizophrenia in the United States. JAMA Psychiatry (2015) 72:1172–81. doi: 10.1001/jamapsychiatry.2015.1737
9. Brunzell DH, McIntosh JM. Alpha7 nicotinic acetylcholine receptors modulate motivation to self-administer nicotine: implications for smoking and schizophrenia. Neuropsychopharmacology (2012) 37:1134–43. doi: 10.1038/npp.2011.299
10. Tidey JW, Colby SM, Xavier EM. Effects of smoking abstinence on cigarette craving, nicotine withdrawal, and nicotine reinforcement in smokers with and without schizophrenia. Nicotine Tob Res. (2014) 16:326–34. doi: 10.1093/ntr/ntt152
11. Berg SA, Chambers RA. Accentuated behavioral sensitization to nicotine in the neonatal ventral hippocampal lesion model of schizophrenia. Neuropharmacology (2008) 54:1201–7. doi: 10.1016/j.neuropharm.2008.03.011
12. Gold JM, Waltz JA, Prentice KJ, Morris S, Heerey EA. Reward processing in schizophrenia: a deficit in the representation of value. Schizophr Bull. (2008) 34:835–47. doi: 10.1093/schbul/sbn068
13. Whitton AE, Treadway MT, Pizzagalli DA. Reward processing dysfunction in major depression, bipolar disorder and schizophrenia. Curr Opin Psychiatry (2015) 28:7–12. doi: 10.1097/YCO.0000000000000122
14. Moran LV, Betts JM, Ongur D, Janes AC. Neural responses to smoking cues in schizophrenia. Schizophr Bull. (2017) 44:525–34. doi: 10.1093/schbul/sbx085
15. Peechatka AL, Whitton AE, Farmer SL, Pizzagalli D, Janes AC. Cigarette craving is associated with blunted reward processing in nicotine-dependent smokers. Drug Alcohol Depend. (2015) 155:202–7. doi: 10.1016/j.drugalcdep.2015.07.015
16. Zhang XY, Chen DC, Xiu MH, Haile CN, Sun H, Lu L, et al. Cigarette smoking and cognitive function in Chinese male schizophrenia: a case-control study. PLoS ONE (2012) 7:e36563. doi: 10.1371/journal.pone.0036563
17. Iasevoli F, Balletta R, Gilardi V, Giordano S, de Bartolomeis A. Tobacco smoking in treatment-resistant schizophrenia patients is associated with impaired cognitive functioning, more severe negative symptoms, and poorer social adjustment. Neuropsychiatr Dis Treat. (2013) 9:1113–20. doi: 10.2147/NDT.S47571
18. Depp CA, Bowie CR, Mausbach BT, Wolyniec P, Thornquist MH, Luke JR, et al. Current smoking is associated with worse cognitive and adaptive functioning in serious mental illness. Acta Psychiatr Scand. (2015) 131:333–41. doi: 10.1111/acps.12380
19. Hickling LM, Perez-Iglesias R, Ortiz-García de la Foz V, Balanzá-Martínez V, McGuire P, Crespo-Facorro B, et al. Tobacco smoking and its association with cognition in first episode psychosis patients. Schizophr Res. (2018) 192:269–73. doi: 10.1016/j.schres.2017.04.018
20. Hatsukami D, Fletcher L, Morgan S, Keenan R, Amble P. The effects of varying cigarette deprivation duration on cognitive and performance tasks. J Subst Abuse (1989) 1:407–16.
21. Snyder FR, Davis FC, Henningfield JE. The tobacco withdrawal syndrome: performance decrements assessed on a computerized test battery. Drug Alcohol Depend. (1989) 23:259–66. doi: 10.1016/0376-8716(89)90090-2
22. Foulds J, Stapleton JA, Bell N, Swettenham J, Jarvis M, Russell MA. Mood and physiological effects of subcutaneous nicotine in smokers and never-smokers. Drug Alcohol Depend. (1997) 44:105–15. doi: 10.1016/S0376-8716(96)01327-0
23. Harris JG, Kongs S, Allensworth D, Martin L, Tregellas J, Sullivan B, et al. Effects of nicotine on cognitive deficits in schizophrenia. Neuropsychopharmacology (2004) 29:1378–85. doi: 10.1038/sj.npp.1300450
24. Reed AC, Harris JG, Olincy A. Schizophrenia, smoking status, and performance on the matrics cognitive consensus battery. Psychiatry Res. (2016) 246:1–8. doi: 10.1016/j.psychres.2016.08.062
25. Wing VC, Bacher I, Sacco KA, George TP. Neuropsychological performance in patients with schizophrenia and controls as a function of cigarette smoking status. Psychiatry Res. (2011) 188:320–6. doi: 10.1016/j.psychres.2011.05.037
26. Morisano D, Wing VC, Sacco KA, Arenovich T, George TP. Effects of tobacco smoking on neuropsychological function in schizophrenia in comparison to other psychiatric disorders and non-psychiatric controls. Am J Addict. (2013) 22:46–53. doi: 10.1111/j.1521-0391.2013.00313.x
27. Gupta T, Mittal VA. Nicotine usage is associated with elevated processing speed, spatial working memory, and visual learning performance in youth at ultra high-risk for psychosis. Psychiatry Res. (2014) 220:687–90. doi: 10.1016/j.psychres.2014.07.085
28. Levin ED, Wilson W, Rose JE, McEvoy J. Nicotine-haloperidol interactions and cognitive performance in schizophrenics. Neuropsychopharmacology (1996) 15:429–36. doi: 10.1016/S0893-133X(96)00018-8
29. Smith RC, Singh A, Infante M, Khandat A, Kloos A. Effects of cigarette smoking and nicotine nasal spray on psychiatric symptoms and cognition in schizophrenia. Neuropsychopharmacology (2002) 27:479–97. doi: 10.1016/S0893-133X(02)00324-X
30. Jacobsen LK, D'Souza DC, Mencl WE, Pugh K, Skudlarski P, Krystal JH. Nicotine effects on brain function and functional connectivity in schizophrenia. Biol Psychiatry (2004) 55:850–8. doi: 10.1016/j.biopsych.2003.12.023
31. Sacco KA, Termine A, Seyal A, Dudas MM, Vessicchio J, Krishnan-Sarin S, et al. Effects of cigarette smoking on spatial working memory and attentional deficits in schizophrenia: involvement of nicotinic receptor mechanisms. Arch Gen Psychiatry (2005) 62:649–59. doi: 10.1001/archpsyc.62.6.649
32. AhnAllen CG, Nestor PG, Shenton ME, McCarley R, Niznikiewicz MA. Early nicotine withdrawal and transdermal nicotine effects on neurocognitive performance in schizophrenia. Schizophr Res. (2008) 100:261–9. doi: 10.1016/j.schres.2007.07.030
33. Beck AK, Baker AL, Todd J. Smoking in schizophrenia: cognitive impact of nicotine and relationship to smoking motivators. Schizophr Res Cogn. (2015) 2:26–32. doi: 10.1016/j.scog.2014.12.001
34. Ahlers E, Hahn E, Ta TM, Goudarzi E, Dettling M, Neuhaus AH. Smoking improves divided attention in schizophrenia. Psychopharmacology (2014) 231:3871–7. doi: 10.1007/s00213-014-3525-2
35. Adler LE, Hoffer LD, Wiser A, Freedman R. Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am J Psychiatry (1993) 150:1856. doi: 10.1176/ajp.150.12.1856
36. George TP, Vessicchio JC, Termine A, Sahady DM, Head CA, Pepper WT, et al. Effects of smoking abstinence on visuospatial working memory function in schizophrenia. Neuropsychopharmacology (2002) 26:75–85. doi: 10.1016/S0893-133X(01)00296-2
37. Boggs DL, Carlson J, Cortes-Briones J, Krystal JH, D'Souza D. Going up in smoke? A review of nAChRs-based treatment strategies for improving cognition in schizophrenia. Curr Pharm Des. (2014) 20:5077–92. doi: 10.2174/1381612819666131216121019
38. AhnAllen CG, Bidwell LC, Tidey JW. Cognitive effects of very low nicotine content cigarettes, with and without nicotine replacement, in smokers with schizophrenia and controls. Nicotine Tob Res. (2015) 17:510–4. doi: 10.1093/ntr/ntu163
39. Barr RS, Culhane MA, Jubelt LE, Mufti R, Dyer MA, Weiss AP, et al. The effects of transdermal nicotine on cognition in nonsmokers with schizophrenia and nonpsychiatric controls. Neuropsychopharmacology (2008) 33:480–90. doi: 10.1038/sj.npp.1301423
40. de Leon J, Diaz FJ. A meta-analysis of worldwide studies demonstrates an association between schizophrenia and tobacco smoking behaviors. Schizophr Res. (2005) 76:135–57. doi: 10.1016/j.schres.2005.02.010
41. Winterer G. Why do patients with schizophrenia smoke? Curr Opin Psychiatry (2010) 23:112–9. doi: 10.1097/YCO.0b013e3283366643
42. Hahn B, Harvey AN, Concheiro-Guisan M, Huestis MA, Holcomb HH, Gold JM. A test of the cognitive self-medication hypothesis of tobacco smoking in schizophrenia. Biol Psychiatry (2013) 74:436–43. doi: 10.1016/j.biopsych.2013.03.017
43. Chambers RA. A nicotine challenge to the self-medication hypothesis in a neurodevelopmental animal model of schizophrenia. J Dual Diagn. (2009) 5:139–48. doi: 10.1080/15504260902869808
44. George TP. Neurobiological links between nicotine addiction and schizophrenia. J Dual Diagn. (2007) 3:27–42. doi: 10.1300/J374v03n03_04
45. Picciotto MR, Caldarone BJ, King SL, Zachariou V. Nicotinic receptors in the brain. Links between molecular biology and behavior. Neuropsychopharmacology (2000) 22:451–65. doi: 10.1016/S0893-133X(99)00146-3
46. Changeux JP. Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat Rev Neurosci. (2010) 11:389. doi: 10.1038/nrn2849
47. Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. (2006) 27:482–91. doi: 10.1016/j.tips.2006.07.004
48. Zoli M, Pistillo F, Gotti C. Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology (2015) 96:302–11. doi: 10.1016/j.neuropharm.2014.11.003
49. Hutchison KE, Allen DL, Filbey FM, Jepson C, Lerman C, Benowitz NL, et al. CHRNA4 and tobacco dependence: from gene regulation to treatment outcome. Arch Gen Psychiatry (2007) 64:1078–86. doi: 10.1001/archpsyc.64.9.1078
50. Breitling LP, Dahmen N, Mittelstrass K, Rujescu D, Gallinat J, Fehr C, et al. Association of nicotinic acetylcholine receptor subunit alpha 4 polymorphisms with nicotine dependence in 5500 Germans. Pharmacogenomics J. (2009) 9:219–24. doi: 10.1038/tpj.2009.6
51. Greenwood PM, Parasuraman R, Espeseth T. A cognitive phenotype for a polymorphism in the nicotinic receptor gene CHRNA4. Neurosci Biobehav Rev. (2012) 36:1331–41. doi: 10.1016/j.neubiorev.2012.02.010
52. Stephens SH, Logel J, Barton A, Franks A, Schultz J, Short M, et al. Association of the 5'-upstream regulatory region of the alpha7 nicotinic acetylcholine receptor subunit gene (CHRNA7) with schizophrenia. Schizophr Res. (2009) 109:102–12. doi: 10.1016/j.schres.2008.12.017
53. Mexal S, Berger R, Logel J, Ross RG, Freedman R, Leonard S. (2010). Differential regulation of α7 nicotinic receptor gene (CHRNA7) expression in schizophrenic smokers. J Mol Neurosci. 40:185–95. doi: 10.1007/s12031-009-9233-4
54. De Luca V, Wong AH, Muller DJ, Wong G, Tyndale RF, Kennedy JL. Evidence of association between smoking and alpha7 nicotinic receptor subunit gene in schizophrenia patients. Neuropsychopharmacology (2004) 29:1522–6. doi: 10.1038/sj.npp.1300466
55. Bencherif M, Schmitt JD. Targeting neuronal nicotinic receptors: a path to new therapies. Curr Drug Targets CNS Neurol Disord. (2002) 1:349–57. doi: 10.2174/1568007023339094
56. Wooltorton JR, Pidoplichko VI, Broide RS, Dani J. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci. (2003) 23:3176–85. doi: 10.1523/JNEUROSCI.23-08-03176.2003
57. Dani JA, De Biasi M. Cellular mechanisms of nicotine addiction. Pharmacol Biochem Behav. (2001) 70:439–46. doi: 10.1016/S0091-3057(01)00652-9
58. Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors, from structure to function. Physiol Rev. (2009) 89:73–120. doi: 10.1152/physrev.00015.2008
59. Kozak K, Barr MS, George TP. Traits and biomarkers for addiction risk in schizophrenia. Curr Addict Rep. (2017) 4:14–24. doi: 10.1007/s40429-017-0130-6
60. Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment: meta-analysis of imaging studies. Arch Gen Psychiatry (2012) 69:776–86. doi: 10.1001/archgenpsychiatry.2012.169
61. Kesby J, Eyles D, McGrath J, Scott J. Dopamine, psychosis and schizophrenia: the widening gap between basic and clinical neuroscience. Transl Psychiatry (2018) 8:30. doi: 10.1038/s41398-017-0071-9
62. Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl J, Bowers M, et al. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry (1998) 155:761–7.
63. Howes OD, Williams M, Ibrahim K, Leung G, Egerton A, McGuire P, et al. Midbrain dopamine function in schizophrenia and depression: a post-mortem and positron emission tomographic imaging study. Brain (2013) 136:3242–51. doi: 10.1093/brain/awt264
64. Laruelle M, Abi-Dargham A. Dopamine as the wind of the psychotic fire: new evidence from brain imaging studies. J Psychopharmacol. (1999) 13:358–71. doi: 10.1177/026988119901300405
65. Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles L, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci USA. (2000) 97:8104–9. doi: 10.1073/pnas.97.14.8104
66. Artiges E, Leroy C, Dubol M, Prat M, Pepin A, Mabondo A, et al. Striatal and extrastriatal dopamine transporter availability in schizophrenia and its clinical correlates: a voxel-based and high-resolution PET study. Schizophr Bull. (2017) 43:1134–42. doi: 10.1093/schbul/sbw192
67. Abi-Dargham A, Moore H. Prefrontal DA transmission at D1 receptors and the pathology of schizophrenia. Neuroscientist (2003) 9:404–16. doi: 10.1177/1073858403252674
68. Davis KL, Kahn RS. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry (1991) 148:1474. doi: 10.1176/ajp.148.11.1474
69. Okubo Y, Suhara T, Suzuki K, Kobayashi K, Inoue O, Terasaki O, et al. Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature (1997) 385:634. doi: 10.1038/385634a0
70. Goldman-Rakic PS, Castner SA, Svensson TH, Siever L, Siever LJ, Williams GV. Targeting the dopamine D 1 receptor in schizophrenia: insights for cognitive dysfunction. Psychopharmacology (2004) 174:3–16. doi: 10.1007/s00213-004-1793-y
71. Slifstein ME, van de Giessen E, Van Snellenberg J, Thompson JL, Narendran R, Gil R, et al. Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry (2015) 72:316–24. doi: 10.1001/jamapsychiatry.2014.2414
72. Miyamoto S, Duncan G, Marx C, Lieberman J. Treatments for schizophrenia, a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry (2005) 10:79. doi: 10.1038/sj.mp.4001556
73. Kantrowitz JT, Javitt DC. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. (2010). 83:108–21. doi: 10.1016/j.brainresbull.2010.04.006
74. Poels E, Kegeles L, Kantrowitz J, Slifstein M, Javitt D, Lieberman J, et al. Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Mol Psychiatry (2014) 19:20. doi: 10.1038/mp.2013.136
75. Gao WJ, Snyder MA. NMDA hypofunction as a convergence point for progression and symptoms of schizophrenia. Front Cell Neurosci. (2013) 7:31. doi: 10.3389/fncel.2013.00031
76. Hu W, MacDonald ML, Elswick DE, Sweet R. The glutamate hypothesis of schizophrenia: evidence from human brain tissue studies. Ann N Y Acad Sci. (2015) 1338:38–57. doi: 10.1111/nyas.12547
77. Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology (2012) 37:4. doi: 10.1038/npp.2011.181
78. Gao X-M, Sakai K, Roberts RC, Conley RR, Dean B, Tamminga CA. Ionotropic glutamate receptors and expression of N-methyl-D-aspartate receptor subunits in subregions of human hippocampus: effects of schizophrenia. Am J Psychiatry (2000) 157:1141–9. doi: 10.1176/appi.ajp.157.7.1141
79. Ghose S, Gleason KA, Potts BW, Lewis-Amezcua K, Tamminga CA. Differential expression of metabotropic glutamate receptors 2 and 3 in schizophrenia: a mechanism for antipsychotic drug action? Am J Psychiatry (2009) 166:812–20. doi: 10.1176/appi.ajp.2009.08091445
80. Stan A, Ghose S, Zhao C, Hulsey K, Mihalakos P, Yanagi M, et al. Magnetic resonance spectroscopy and tissue protein concentrations together suggest lower glutamate signaling in dentate gyrus in schizophrenia. Mol Psychiatry (2015) 20:433. doi: 10.1038/mp.2014.54
81. Coyle JT, Konopaske G. Glutamatergic dysfunction in schizophrenia evaluated with magnetic resonance spectroscopy. JAMA Psychiatry (2016) 73:649–50. doi: 10.1001/jamapsychiatry.2016.0575
82. De Marco García NV, Karayannis T, Fishell G. Neuronal activity is required for the development of specific cortical interneuron subtypes. Nature (2011) 472:351. doi: 10.1038/nature09865
83. Gonzalez-Burgos G, Hashimoto T, Lewis DA. Alterations of cortical GABA neurons and network oscillations in schizophrenia. Curr Psychiatry Rep. (2010) 12:335–44. doi: 10.1007/s11920-010-0124-8
84. Lewis DA. Inhibitory neurons in human cortical circuits: substrate for cognitive dysfunction in schizophrenia. Curr Opin Neurobiol. (2014) 26:22–6. doi: 10.1016/j.conb.2013.11.003
85. Cohen SM, Tsien RW, Goff DC, Halassa M. The impact of NMDA receptor hypofunction on GABAergic neurons in the pathophysiology of schizophrenia. Schizophr Res. (2015) 167:98–107. doi: 10.1016/j.schres.2014.12.026
86. Nakazawa K, Zsiros V, Jiang Z, Nakao K, Kolata S, Zhang S, et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology (2012) 62:1574–83. doi: 10.1016/j.neuropharm.2011.01.022
87. Tost H, Meyer-Lindenberg A. Dopamine-glutamate interactions: a neural convergence mechanism of common schizophrenia risk variants. Biol Psychiatry (2011) 69:912–3. doi: 10.1016/j.biopsych.2011.03.013
88. Sesack SR, Carr DB. Selective prefrontal cortex inputs to dopamine cells: implications for schizophrenia. Physiol Behav. (2002) 77:513–7. doi: 10.1016/S0031-9384(02)00931-9
89. Lewis DA, González-Burgos G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology (2008) 33:141. doi: 10.1038/sj.npp.1301563
90. Kegeles LS, Abi-Dargham A, Zea-Ponce Y, Rodenhiser-Hill J, Mann JJ, Van Heertum RL, et al. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: implications for schizophrenia. Biol Psychiatry (2000) 48:627–40. doi: 10.1016/S0006-3223(00)00976-8
91. Adell A, Jiménez-Sánchez L, López-Gil X, Romón T. Is the acute NMDA receptor hypofunction a valid model of schizophrenia? Schizophr Bull. (2011) 38:9–14. doi: 10.1093/schbul/sbr133
92. Laviolette SR, Van Der Kooy D. The neurobiology of nicotine addiction: bridging the gap from molecules to behaviour. Nat Rev Neurosci. (2004) 5:55. doi: 10.1038/nrn1298
93. Jasinska AJ, Zorick T, Brody AL, Stein EA. Dual role of nicotine in addiction and cognition: a review of neuroimaging studies in humans. Neuropharmacology (2014) 84:111–22. doi: 10.1016/j.neuropharm.2013.02.015
94. Pistillo F, Clementi F, Zoli M, Gotti C. Nicotinic, glutamatergic and dopaminergic synaptic transmission and plasticity in the mesocorticolimbic system: focus on nicotine effects. Prog Neurobiol. (2015) 124:1–27. doi: 10.1016/j.pneurobio.2014.10.002
95. Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron (2002) 33:905–19. doi: 10.1016/S0896-6273(02)00625-6
96. Mameli-Engvall M, Evrard A, Pons S, Maskos U, Svensson TH, Changeux JP, et al. Hierarchical control of dopamine neuron-firing patterns by nicotinic receptors. Neuron (2006) 50:911–21. doi: 10.1016/j.neuron.2006.05.007
97. D'Souza MS, Markou A. The stop and go of nicotine dependence: role of GABA and glutamate. Cold Spring Harb Perspect Med. (2013) 3:a012146. doi: 10.1101/cshperspect.a012146
98. Parikh V, Kutlu MG, Gould TJ. nAChR dysfunction as a common substrate for schizophrenia and comorbid nicotine addiction: current trends and perspectives. Schizophr Res. (2016) 171:1–15. doi: 10.1016/j.schres.2016.01.020
99. George TP, Verrico CD, Picciotto MR, Roth R. Nicotinic modulation of mesoprefrontal dopamine neurons: pharmacologic and neuroanatomic characterization. J Pharmacol Exp Ther. (2000) 295:58–66.
100. Livingstone PD, Srinivasan J, Kew JN, Dawson LA, Gotti C, Moretti M, et al. α7 and non-α7 nicotinic acetylcholine receptors modulate dopamine release in vitro and in vivo in the rat prefrontal cortex. Eur J Neurosci. (2009) 29:539–50. doi: 10.1111/j.1460-9568.2009.06613.x
101. Song C, Murray TA, Kimura R, Wakui M, Ellsworth K, Javedan SP, et al. Role of α7-nicotinic acetylcholine receptors in tetanic stimulation-induced γ oscillations in rat hippocampal slices. Neuropharmacology (2005) 48:869–80. doi: 10.1016/j.neuropharm.2005.01.003
102. Wanaverbecq N, Semyanov A, Pavlov I, Walker MC, Kullmann D. Cholinergic axons modulate GABAergic signaling among hippocampal interneurons via postsynaptic α7 nicotinic receptors. J Neurosci. (2007) 27:5683–93. doi: 10.1523/JNEUROSCI.1732-07.2007
103. Maloku E, Kadriu B, Zhubi A, Dong E, Pibiri F, Satta R, et al. Selective α 4 β 2 nicotinic acetylcholine receptor agonists target epigenetic mechanisms in cortical GABAergic neurons. Neuropsychopharmacology (2011) 36:1366. doi: 10.1038/npp.2011.21
104. Martin LF, Freedman R. Schizophrenia and the α7 nicotinic acetylcholine receptor. Int Rev Neurobiol. (2007) 78:225–46. doi: 10.1016/S0074-7742(06)78008-4
105. D'Souza DC, Esterlis I, Carbuto M, Krasenics M, Seibyl J, Bois F, et al. Lower β2*-nicotinic acetylcholine receptor availability in smokers with schizophrenia. Am J Psychiatry (2012) 169:326–34. doi: 10.1176/appi.ajp.2011.11020189
106. Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature (1997) 390:401. doi: 10.1038/37120
107. Sacco KA, Bannon KL, George TP. Nicotinic receptor mechanisms and cognition in normal states and neuropsychiatric disorders. J Psychopharmacol. (2004) 18:457–74. doi: 10.1177/0269881104047273
108. Leonard S, Adler LE, Benhammou K, Berger R, Breese C, Drebing C, et al. Smoking and mental illness. Pharmacol Biochem Behav. (2001) 70:561–70. doi: 10.1016/S0091-3057(01)00677-3
109. Farrell M, Werge T, Sklar P, Owen MJ, Ophoff R, O'Donovan MC, et al. Evaluating historical candidate genes for schizophrenia. Mol Psychiatry (2015) 20:555. doi: 10.1038/mp.2015.16
110. Wing VC, Wass CE, Bacher I, Rabin RA, George TP. Varenicline modulates spatial working memory deficits in smokers with schizophrenia. Schizophr Res. (2013) 149:190–1. doi: 10.1016/j.schres.2013.06.032
111. Lisman JE, Coyle JT, Green RW, Javitt D, Benes FM, Heckers S, et al. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. (2008) 31:234–42. doi: 10.1016/j.tins.2008.02.005
112. Bozikas VP, Papakosta M, Niopas I, Karavatos A, Mirtsou-Fidani V. Smoking impact on CYP1A2 activity in a group of patients with schizophrenia. Eur Neuropsychopharmacol. (2004) 14:39–44. doi: 10.1016/S0924-977X(03)00061-0
113. Sagud M, Mihaljevic-Peles A, Muck-Seler D, Pivac N, Vuksan-Cusa B, Brataljenovic T, et al. Smoking and schizophrenia. Psychiatr Danub (2009) 21:371–5.
114. Nozawa M, Ohnuma T, Matsubara Y, Sakai Y, Hatano T, Hanzawa R, et al. The relationship between the response of clinical symptoms and plasma olanzapine concentration, based on pharmacogenetics: Juntendo University Schizophrenia Projects (JUSP). Ther Drug Monit. (2008) 30:35–40. doi: 10.1097/FTD.0b013e31816336fd
115. Tsuda Y, Saruwatari J, Yasui-Furukori N. Meta-analysis: the effects of smoking on the disposition of two commonly used antipsychotic agents, olanzapine and clozapine. BMJ Open (2014) 4:e004216. doi: 10.1136/bmjopen-2013-004216
116. Nagamoto HT, Adler LE, Waldo MC, Griffith J, Freedman R. Gating of auditory response in schizophrenics and normal controls: effects of recording site and stimulation interval on the P50 wave. Schizophr Res. (1991) 4:31–40. doi: 10.1016/0920-9964(91)90007-E
117. Adler LE, Pachtman E, Franks R, Pecevich M, Waldo M, Freedman R. Neurophysiological evidence for a defect in neuronal mechanisms involved in sensory gating in schizophrenia. Biol Psychiatry (1982) 17:639–54.
118. Olincy A, Braff DL, Adler LE, Cadenhead K, Calkins ME, Dobie DJ, et al. Inhibition of the P50 cerebral evoked response to repeated auditory stimuli: results from the Consortium on Genetics of Schizophrenia. Schizophr Res. (2010) 119:175–82. doi: 10.1016/j.schres.2010.03.004
119. Hamilton H, Kort N, Ford J, Roach B, Gunduz-Bruce H, Krystal J, et al. 540. Effects of N-methyl-D-aspartate glutamate receptor disruption and nicotinic acetylcholine enhancement on mismatch negativity. Biol Psychiatry (2017) 81:S219. doi: 10.1016/j.biopsych.2017.02.1148
120. Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, et al. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci. USA. (1997) 94:587–92. doi: 10.1073/pnas.94.2.587
121. Leonard S, Gault J, Hopkins J, Logel J, Vianzon R, Short M, et al. Association of promoter variants in the α7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Arch Gen Psychiatry (2002) 59:1085–96. doi: 10.1001/archpsyc.59.12.1085
122. Houy E, Raux G, Thibaut F, Belmont A, Demily C, Allio G, et al. The promoter– 194 C polymorphism of the nicotinic alpha 7 receptor gene has a protective effect against the P50 sensory gating deficit. Mol Psychiatry (2004) 9:320. doi: 10.1038/sj.mp.4001443
123. Freedman R, Adams CE, Leonard S. The α7-nicotinic acetylcholine receptor and the pathology of hippocampal interneurons in schizophrenia. J Chem Neuroanat. (2000) 20:299–306. doi: 10.1016/S0891-0618(00)00109-5
124. Chen XS, Li CB, Smith RC, Xiao ZP, Wang JJ. Differential sensory gating functions between smokers and non-smokers among drug-naive first episode schizophrenic patients. Psychiatry Res. (2011) 188:327–33. doi: 10.1016/j.psychres.2010.12.009
125. Ishikawa M, Hashimoto K. α7 nicotinic acetylcholine receptor as a potential therapeutic target for schizophrenia. Curr Pharm Design (2011) 17:121–9. doi: 10.2174/138161211795049561
126. Freedman R. α7-nicotinic acetylcholine receptor agonists for cognitive enhancement in schizophrenia. Ann Rev Med. (2014) 65:245–61. doi: 10.1146/annurev-med-092112-142937
127. Light GA, Näätänen R. Mismatch negativity is a breakthrough biomarker for understanding and treating psychotic disorders. Proc Natl Acad Sci USA. (2013) 110:15175–6. doi: 10.1073/pnas.1313287110
128. Erickson MA, Ruffle A, Gold JM. A meta-analysis of mismatch negativity in schizophrenia: from clinical risk to disease specificity and progression. Biol Psychiatry (2016) 79:980–7. doi: 10.1016/j.biopsych.2015.08.025
129. Murphy JR, Rawdon C, Kelleher I, Twomey D, Markey PS, Cannon M, et al. Reduced duration mismatch negativity in adolescents with psychotic symptoms: further evidence for mismatch negativity as a possible biomarker for vulnerability to psychosis. BMC Psychiatry (2013) 13:45. doi: 10.1186/1471-244X-13-45
130. Hay RA, Roach BJ, Srihari VH, Woods S, Ford JM, Mathalon DH. Equivalent mismatch negativity deficits across deviant types in early illness schizophrenia-spectrum patients. Biol Psychol. (2015) 105:130–7. doi: 10.1016/j.biopsycho.2015.01.004
131. Umbricht D, Koller R, Vollenweider FX, Schmid L. Mismatch negativity predicts psychotic experiences induced by NMDA receptor antagonist in healthy volunteers. Biol Psychiatry (2002) 51:400–6. doi: 10.1016/S0006-3223(01)01242-2
132. Lee M, Sehatpour P, Hoptman MJ, Lakatos P, Dias E, Kantrowitz JT, et al. Neural mechanisms of mismatch negativity (MMN) dysfunction in schizophrenia. Mol Psychiatry (2017). 22:1585–93. doi: 10.1038/mp.2017.3
133. Dulude L, Labelle A, Knott VJ. Acute nicotine alteration of sensory memory impairment in smokers with schizophrenia. J Clin Psychopharmacol. (2010) 30:541–8. doi: 10.1097/JCP.0b013e3181f0c9c6
134. Hamilton HK, D'Souza DC, Ford JM, Roach B, Kort NS, Ahn KH, et al. Interactive effects of an N-methyl-d-aspartate receptor antagonist and a nicotinic acetylcholine receptor agonist on mismatch negativity: Implications for schizophrenia. Schizophr Res. (2018) 191:87–94. doi: 10.1016/j.schres.2017.06.040
135. Cadenhead KS, Swerdlow NR, Shafer KM, Diaz M, Braff DL. Modulation of the startle response and startle laterality in relatives of schizophrenic patients and in subjects with schizotypal personality disorder: evidence of inhibitory deficits. Am J Psychiatry (2000) 157:1660–8. doi: 10.1176/appi.ajp.157.10.1660
136. Hasenkamp W, Epstein MP, Green A, Wilcox L, Boshoven W, Lewison B, et al. Heritability of acoustic startle magnitude, prepulse inhibition, and startle latency in schizophrenia and control families. Psychiatry Res. (2010) 178:236–43. doi: 10.1016/j.psychres.2009.11.012
137. Hong LE, Wonodi I, Lewis J, Thaker GK. Nicotine effect on prepulse inhibition and prepulse facilitation in schizophrenia patients. Neuropsychopharmacology (2008) 33:2167. doi: 10.1038/sj.npp.1301601
138. Petrovsky N, Quednow BB, Ettinger U, Schmechtig A, Mössner R, Collier DA, et al. Sensorimotor gating is associated with CHRNA3 polymorphisms in schizophrenia and healthy volunteers. Neuropsychopharmacology (2010) 35:1429. doi: 10.1038/npp.2010.12
139. Petrovsky N, Ettinger U, Kessler H, Mössner R, Wolfsgruber S, Dahmen N, et al. The effect of nicotine on sensorimotor gating is modulated by a CHRNA3 polymorphism. Psychopharmacology (2013) 229:31–40. doi: 10.1007/s00213-013-3081-1
140. Greenwood TA, Light GA, Swerdlow NR, Calkins M, Green MF, Gur RE, et al. Gating deficit heritability and correlation with increased clinical severity in schizophrenia patients with positive family history. Am J Psychiatry (2015) 173:385–91. doi: 10.1176/appi.ajp.2015.15050605
141. Postma P, Gray JA, Sharma T, Geyer M, Mehrotra R, Das M, et al. A behavioural and functional neuroimaging investigation into the effects of nicotine on sensorimotor gating in healthy subjects and persons with schizophrenia. Psychopharmacology (2006). 184:589–99. doi: 10.1007/s00213-006-0307-5
142. Woznica AA, Sacco KA, George TP. Prepulse inhibition deficits in schizophrenia are modified by smoking status. Schizophr Res. (2009) 112:86–90. doi: 10.1016/j.schres.2009.04.016
143. George TP, Termine A, Sacco KA, Allen TM, Reutenauer E, Vessicchio JC, et al. A preliminary study of the effects of cigarette smoking on prepulse inhibition in schizophrenia: involvement of nicotinic receptor mechanisms. Schizophr Res. (2006) 87:307–15. doi: 10.1016/j.schres.2006.05.022
144. Hommer DW, Clem T, Litman R, Pickar D. Maladaptive anticipatory saccades in schizophrenia. Biol Psychiatry (1991) 30:779–94. doi: 10.1016/0006-3223(91)90234-D
145. Ross RG, Harris JG, Olincy A, Radant A, Adler L, Freedman R. Familial transmission of two independent saccadic abnormalities in schizophrenia. Schizophr Res. (1998) 30:59–70. doi: 10.1016/S0920-9964(97)00133-3
146. Karoumi BB, Saoud MT, d'Amato T, Rosenfeld F, Denise P, Gutknecht C, et al. Poor performance in smooth pursuit and antisaccadic eye-movement tasks in healthy siblings of patients with schizophrenia. Psychiatry Res. (2001) 101:209–19. doi: 10.1016/S0165-1781(01)00227-X
147. Krebs MO, Gut-Fayand A, Amado I, Daban C, Bourdel MC, Poirier MF, et al. Impairment of predictive saccades in schizophrenia. Neuroreport (2001) 12:465–9. doi: 10.1097/00001756-200103050-00009
148. Avila MT, Sherr JD, Hong E, Myers CS, Thaker GK. Effects of nicotine on leading saccades during smooth pursuit eye movements in smokers and nonsmokers with schizophrenia. Neuropsychopharmacology (2003) 28:2184. doi: 10.1038/sj.npp.1300265
149. Larrison-Faucher AL, Matorin AA, Sereno AB. Nicotine reduces antisaccade errors in task impaired schizophrenic subjects. Prog Neuro Psychopharm Biol Psychiatry (2004) 28:505–16. doi: 10.1016/j.pnpbp.2004.01.002
150. Tregellas JR, Tanabe JL, Martin LF, Freedman R. FMRI of response to nicotine during a smooth pursuit eye movement task in schizophrenia. Am J Psychiatry (2005) 162:391–3. doi: 10.1176/appi.ajp.162.2.391
151. Tanabe J, Tregellas JR, Martin LF, Freedman R. Effects of nicotine on hippocampal and cingulate activity during smooth pursuit eye movement in schizophrenia. Biol Psychiatry (2006) 59:754–61. doi: 10.1016/j.biopsych.2005.09.004
152. Petrovsky N, Ettinger U, Quednow BB, Walter H, Schnell K, Kessler H, et al. Nicotine differentially modulates antisaccade performance in healthy male non-smoking volunteers stratified for low and high accuracy. Psychopharmacology (2012) 221:27–38. doi: 10.1007/s00213-011-2540-9
153. Hiscock R, Bauld L, Amos A, Fidler JA, Munafo M. Socioeconomic status and smoking: a review. Ann N Y Acad Sci. (2012) 1248:107–23. doi: 10.1111/j.1749-6632.2011.06202.x
154. Jiang J, See YM, Subramaniam M, Lee J. Investigation of cigarette smoking among male schizophrenia patients. PLoS ONE (2013) 8:e71343. doi: 10.1371/journal.pone.0071343
155. Steinberg ML, Williams JM, Ziedonis DM. Financial implications of cigarette smoking among individuals with schizophrenia. Tobacco Control (2004) 13:206.
156. Olivier D, Lubman DI, Fraser R. Tobacco smoking within psychiatric inpatient settings: biopsychosocial perspective. Aust N Z J Psychiatry (2007) 41:572–80. doi: 10.1080/00048670701392809
157. Keizer I, Eytan A. Variations in smoking during hospitalization in psychiatric in-patient units and smoking prevalence in patients and health-care staff. Int J Soc Psychiatry (2005) 51:317–28. doi: 10.1177/0020764005057377
158. Prochaska JJ, Gill P, Hall SM. Treatment of tobacco use in an inpatient psychiatric setting. Psychiatr Serv. (2004) 55:1265–70. doi: 10.1176/appi.ps.55.11.1265
159. Prochaska JJ, Fletcher L, Hall SE, Hall SM. Return to smoking following a smoke-free psychiatric hospitalization. Am J Addict. (2006) 15:15–22. doi: 10.1080/10550490500419011
160. Willemsen MC, Gorts CA, Van Soelen P, Jonkers R, Hilberink S. Exposure to environmental tobacco smoke (ETS) and determinants of support for complete smoking bans in psychiatric settings. Tob Control (2004) 13:180–5. doi: 10.1136/tc.2003.004804
161. Hitsman B, Moss TG, Montoya ID, George T. Treatment of tobacco dependence in mental health and addictive disorders. Can J Psychiatry (2009) 54:368–78. doi: 10.1177/070674370905400604
162. Lawn S, Pols R. Smoking bans in psychiatric inpatient settings? A review of the research. Aust N Z J Psychiatry (2005) 39:866–85. doi: 10.1080/j.1440-1614.2005.01697.x
163. Riad-Allen L, Dermody SS, Herman Y, Bellissimo K, Selby P, George TP. Becoming tobacco-free: changes in staff and patient attitudes and incident reports in a large academic mental health and addictions hospital. Am J Addict. (2017) 26:183–91. doi: 10.1111/ajad.12513
164. Cather C, Pachas GN, Cieslak KM, Evins A. Achieving smoking cessation in individuals with schizophrenia: special considerations. CNS Drugs (2017) 31:471–81. doi: 10.1007/s40263-017-0438-8
165. Dwoskin LP. Emerging Targets and Therapeutics in the Treatment of Psychostimulant Abuse. Elsevier Science (2014).
166. Tashkin DP. Smoking cessation in chronic obstructive pulmonary disease. Semin Respir Crit Care Med. (2015) 36:491–507. doi: 10.1055/s-0035-1555610
167. Smith RC, Amiaz R, Si TM, Maayan L, Jin H, Boules S, et al. Varenicline effects on smoking, cognition, and psychiatric symptoms in schizophrenia: a double-blind randomized trial. PLoS ONE (2016) 11:e0143490. doi: 10.1371/journal.pone.0143490
168. Jin Y, Wang Q, Wang Y, Liu M, Sun A, Geng Z, et al. Alpha7 nAChR agonists for cognitive deficit and negative symptoms in schizophrenia: a meta-analysis of randomized double-blind controlled trials. Shanghai Arch Psychiatry (2017) 29:191–9. doi: 10.11919/j.issn.1002-0829.217044
169. Coles AS, Kozak K, George TP. A review of brain stimulation for treatment of substance use disorders. Am J Addict. (2018) 27:71–91. doi: 10.1111/ajad.12674
170. Kozak K, Sharif-Razi M, Morozova M, Gaudette EV, Barr M, Daskalakis ZJ, et al. Effects of short-term, high-frequency repetitive transcranial magnetic stimulation to bilateral dorsolateral prefrontal cortex on smoking behavior and cognition in patients with schizophrenia and non-psychiatric controls. Schizophr Res. (2018). 197:441–3. doi: 10.1016/j.schres.2018.02.015
171. Amiaz R, Levy D, Vainiger D, Grunhaus L, Zangen A. Repeated high-frequency transcranial magnetic stimulation over the dorsolateral prefrontal cortex reduces cigarette craving and consumption. Addiction (2009) 104:653–60. doi: 10.1111/j.1360-0443.2008.02448.x
172. Prikryl R, Ustohal L, Kucerova HP, Kasparek T, Jarkovsky J, Hublova V, et al. Repetitive transcranial magnetic stimulation reduces cigarette consumption in schizophrenia patients. Prog Neuropsychopharmacol Biol Psychiatry (2014) 49:30–5. doi: 10.1016/j.pnpbp.2013.10.019
173. Barr MS, Farzan F, Wing VC, George TP, Fitzgerald PB, Daskalakis ZJ. Repetitive transcranial magnetic stimulation and drug addiction. Int Rev Psychiatry (2011) 23:454–66. doi: 10.3109/09540261.2011.618827
174. de la Fuente-Fernandez R, Schulzer M, Stoessl AJ. The placebo effect in neurological disorders. Lancet Neurol. (2002) 1:85–91. doi: 10.1016/S1474-4422(02)00038-8
175. Perkins K, Sayette M, Conklin C, Caggiula A. Placebo effects of tobacco smoking and other nicotine intake. Nicotine Tob Res. (2003) 5:695–709. doi: 10.1080/1462220031000158636
176. Ernst E. Placebo: new insights into an old enigma. Drug Discov Today (2007) 12:413–18. doi: 10.1016/j.drudis.2007.03.007
177. Wu AD, Fregni F, Simon DK, Deblieck C, Pascual-Leone A. Noninvasive brain stimulation for Parkinson's disease and dystonia. Neurotherapeutics (2008) 5:345–61. doi: 10.1016/j.nurt.2008.02.002
Keywords: schizophrenia, nicotine, tobacco, neurobiology, nicotinic acetylcholine receptor
Citation: Lucatch AM, Lowe DJE, Clark RC, Kozak K and George TP (2018) Neurobiological Determinants of Tobacco Smoking in Schizophrenia. Front. Psychiatry 9:672. doi: 10.3389/fpsyt.2018.00672
Received: 31 July 2018; Accepted: 21 November 2018;
Published: 06 December 2018.
Edited by:
Amanda L. Baker, University of Newcastle, AustraliaReviewed by:
Robert C. Smith, New York University, United StatesJose Antonio Apud, National Institute of Mental Health (NIMH), United States
Vanessa Clark, University of Newcastle, Australia
Copyright © 2018 Lucatch, Lowe, Clark, Kozak and George. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aliya M. Lucatch, YWxpeWEubHVjYXRjaEBtYWlsLnV0b3JvbnRvLmNh
Tony P. George, dG9ueS5nZW9yZ2VAY2FtaC5jYQ==