Abstract
Early life stressors display a high universal prevalence and constitute a major public health problem. Prolonged psychoneurobiological alterations as sequelae of early life stress (ELS) could represent a developmental risk factor and mediate risk for disease, leading to higher physical and mental morbidity rates in later life. ELS could exert a programming effect on sensitive neuronal brain networks related to the stress response during critical periods of development and thus lead to enduring hyper- or hypo-activation of the stress system and altered glucocorticoid signaling. In addition, alterations in emotional and autonomic reactivity, circadian rhythm disruption, functional and structural changes in the brain, as well as immune and metabolic dysregulation have been lately identified as important risk factors for a chronically impaired homeostatic balance after ELS. Furthermore, human genetic background and epigenetic modifications through stress-related gene expression could interact with these alterations and explain inter-individual variation in vulnerability or resilience to stress. This narrative review presents relevant evidence from mainly human research on the ten most acknowledged neurobiological allostatic pathways exerting enduring adverse effects of ELS even decades later (hypothalamic-pituitary-adrenal axis, autonomic nervous system, immune system and inflammation, oxidative stress, cardiovascular system, gut microbiome, sleep and circadian system, genetics, epigenetics, structural, and functional brain correlates). Although most findings back a causal relation between ELS and psychobiological maladjustment in later life, the precise developmental trajectories and their temporal coincidence has not been elucidated as yet. Future studies should prospectively investigate putative mediators and their temporal sequence, while considering the potentially delayed time-frame for their phenotypical expression. Better screening strategies for ELS are needed for a better individual prevention and treatment.
Introduction
Stress is defined as the state of threatened homeodynamic balance of the organism (1, 2). Inadequate, excessive or prolonged stress reactions may exceed the organism's natural adaptive capacity and permanently affect stress responses (2, 3). Excessive stress exposure, especially in stress-sensitive developmental stages of higher brain plasticity (e.g., early childhood), may over- or under-sensitize neuroendocrine responses to stress, leading to an altered homeodynamic state (i.e., allostasis/cacostasis) with profound and debilitating effects on physiological development and close association to chronic physical and mental morbidity (2, 4–10).
The term Early Life Stress (ELS) describes a broad spectrum of adverse and stressful experiences (e.g., maltreatment, neglect, separation, parental loss, extreme poverty, starvation, domestic/community/school violence) during the first months of life, early and late childhood and adolescence, while the term has been recently extended by some authors and includes also prenatal life events (11). Childhood Trauma (CT) represents a more specific form of ELS and is defined as “a traumatic event that threatens injury, death, or the physical integrity of self or others and also causes horror, terror, or helplessness at the time it occurs and overwhelms a person's ability to cope” (e.g., physical/sexual abuse, medical trauma, motor vehicle accident, acts of terrorism, war experiences, natural and human-made disasters, witnessed homicides/suicides) (12). ELS/CT constitute a major public health issue, as they occur at ominously high rates, with over 30–40% of the general adult population having experienced some form of disrupting early life adversities (13–16).
In addition, many studies report a negative association of ELS/CT with general adult mental and physical health-related quality of life (17–21). Especially an increased risk for mental disorders (e.g., depression, post-traumatic stress disorder, schizophrenia) and their unfavorable outcomes after ELS/CT experience has been repeatedly reported in several retrospective (5, 22–25) but also prospective studies (26–29). Similarly, history of ELS/CT has been associated with risk behavior patterns, such as substance abuse and suicide attempts in later life (30–35). Furthermore, several larger-scale studies and meta-analyses also suggest a close association of ELS/CT with adverse physical health and in particular with cardiovascular, gastrointestinal, neuromusculoskeletal, pulmonary, inflammatory, and metabolic diseases, chronic pain syndromes, frequency of medical consultations, as well as number of medical diagnoses (24, 36–42).
ELS/CT rarely occurs as a single event but frequently consists of continued maltreatment involving one or more malicious acts. In addition, in most cases, several negative risk factors may co-exist (e.g., poverty, parental absence and parental mental disease, drug addiction) leading to a multifaceted context of multiple chronic stressors. The severity of physical and psychological consequences may be also associated with the number of experienced ELS/CT events (13, 17, 43, 44). More recent studies confirmed that increasing number of ELS/CT may result in higher adult risk for psychopathological complexity and severity, mental comorbidities, prescribed psychotropic medication, poor mental and physical quality of life, as well as several physical conditions (e.g., chronic pain syndromes, cephalgias, heart disease, asthma, diabetes mellitus, and arthritis) (23, 24, 45–50). Apart from number of ELS/CT experiences, the specific nature of ELS/CT and particularly its exact timing could greatly influence downstream biological pathways. Furthermore, genetic factors, presence of caregivers and psychological support, family history of major psychiatric disorders, as well as additional traumatic stress experiences in adulthood may all further influence the individual vulnerability for later disease (51). The continuum of trauma-provoked aftermath reaches from healthy adaptation with high resilience, to severe maladjustment with co-occurring psychiatric and physical pathologies in children, adolescents, and adults. Despite the resilience of many abused individuals in their early years, ELS/CT significantly increases the risk for impaired physical and psychological well-being and adaptive functioning in adulthood.
All these findings suggest that ELS/CT may trigger a health-related risk cascade and be conceptualized as a common developmental risk factor and cumulative health risk mediator, associated with an increased physical and mental morbidity and all-cause mortality in later life (13, 15, 36, 52–59). Although prospective findings support the causal relation between ELS/CT and its long-term adverse health-related effects, so far, little is known about the exact pathways through which ELS/CT is translated into health risk. Observational human studies and experimental animal models suggest that ELS/CT is related to remarkable functional and structural changes even decades later in adulthood. The current hypothetical model suggests that ELS/CT may trigger enduring systemic alterations of fundamental, mainly brain-related plasticity mechanisms and so enhance the biological embedment of distinct “biological memories” of ELS/CT during the sensitive period of early organism development, thus enhancing disease vulnerability in later life (60). To date, most studies assessing the link between ELS/CT and adult disease risk suggest stress system related neuroendocrine alterations as the main pathway of disease development. However, many other related, but distinct biological systems may also play a role and have lately emerged as important pathophysiological pathways.
This current review discusses additionally further potential pathophysiological mechanisms exerting the enduring adverse effects of ELS/CT and mediating the cumulative long-term risk for disease vulnerability in later life, a topic that cannot yet be approached via systematic reviews. Therefore, the literature is presented as a narrative review, providing an overview on the most relevant and acknowledged neurobiological findings from mainly human research. Literature searches were undertaken using PubMed/Medline, PsychINFO, Scopus, and Google Scholar from inception to March 2018 to identify publications (reviews, discussion papers, clinical, observational, and preclinical studies, etc.) addressing neurobiological aspects of ELS/CT and relevant information was extracted. Additionally, the search was complemented through manual review of related terms and citations from article reference lists. The ten most important neurobiological concepts, as backed from current evidence, were synthesized under the headings reported in this narrative review.
The Human Stress System
ELS/CT can irreversibly disrupt vital central neurobiological systems during vulnerable human development periods and lead to sustainable alterations in stress regulation and psychophysiological reactivity (13, 15, 36, 52–59, 61–63). Because of their pivotal role in the regulation of the dynamic stress response and perhaps also due to a historical focus on these two systems, the hypothalamic-pituitary-adrenal (HPA) axis and the locus ceruleus/autonomic nervous system (LC/ANS) have been more investigated and are considered more crucially affected by ELS/CT than other biological systems (64–66).
Hypothalamic-Pituitary-Adrenal (HPA) Axis
The chronic dysregulation of the HPA axis is of vital importance in the pathophysiology of stress-related disorders. However, our understanding is hampered by complex and often conflicting relations between HPA axis markers and history of ELS/CT (i.e., findings on both increased and decreased HPA axis activity) (2, 64), as well as the broad definition of ELS/CT (i.e., broad time window of 0–18 y.o.a.). For example, positive ELS/CT history has been repeatedly associated with HPA axis hyperactivity in adults patients with depression and anxiety, but also in healthy individuals [e.g., higher circulating cortisol levels, enhanced cortisol awakening response (CAR), increased adrenocorticotropic hormone (ACTH) and cortisol responses to psychosocial stress or endocrine challenges] (67–75). Chronic hyperactivation of the stress system is related to hypersecretion of corticotropin releasing hormone and arginine-vasopressin (CRH, AVP) by the hypothalamus and ACTH hypersecretion by the pituitary (76), resulting in higher circulating cortisol levels due also to an “insensitive” negative glucocorticoid (GC) feedback of the HPA axis loop (77). The typical example of chronic HPA axis hyper-activation is depression (2, 78), while other conditions such as anorexia nervosa, obsessive-compulsive disorder, panic disorder, alcohol withdrawal, excessive exercising, poorly controlled diabetes mellitus, and hyperthyroidism amongst others, are also associated with increased cortisol levels and HPA axis hyper-activation (79).
On the other hand, several ELS/CT studies have reported HPA axis hypo-activity (e.g., lower circulating cortisol levels, blunted cortisol stress responses) in similar populations and study designs (80–84). This diminished activity could represent a compensatory physiologic adaptation possibly related to a negative feedback hypersensitivity of GC by an up-regulated leukocyte GC-receptor (GR) number and sensitivity (5, 63, 85–87), downregulated secretion of CRH/AVP to the pituitary (76) or a long-lasting GC catabolism drop leading to higher active cortisol persistence in liver and kidney without elevation in the periphery (88). This seems to be the case for patients with post-traumatic stress disorder (PTSD), atypical depression, chronic fatigue syndrome, fibromyalgia, and hypothyroidism.
These results suggest a particularly vital role of GC-signaling in the pathophysiology of ELS/CT (89–91). Insufficient multilevel GC-signaling (resulting from either hyper- or hypo-activation of the HPA axis), may have comparable deleterious effects on the organisms' physiology, as for example seen in the development and long-term effects of both PTSD (i.e., HPA axis hypo-activation) and melancholic depression (i.e., HPA axis hyper-activation) (91–93). These effects appear even greater in individuals with ELS history, suggesting a developmental programming through GC signaling.
Thereby, several factors may have influenced study findings, such as the exact subtype and nature of trauma, sex, the timing and duration of exposure and the assessment of phasic (e.g., diurnal saliva cortisol, cortisol reactivity to challenge) vs. time-integrated cortisol values (e.g., hair cortisol) (94, 95). However, probably the most significant factor modulating the ELS/CT impact on future HPA axis activity may be its exact timing, suggesting a degree of developmental programming through GC signaling.
Timing of ELS/CT and Developmental Programming of HPA-Axis
The HPA axis activity trajectory develops from infancy to early adulthood and beyond. Together with the HPA axis, however, the amygdala and the hippocampus also develop during the same period following non-linear patterns until early adulthood (96–100). Thereby, periods of greater HPA axis plasticity may represent specific periods of greater vulnerability (96, 100, 101), while mounting evidence suggests a differential impact of ELS/CT on HPA axis activity according to the specific developmental age of exposure (102).
Infancy and early childhood (age 0–5 y.o.a.) represent one of the most vulnerable periods in brain development (6, 101, 103, 104). After an initial hyper-responsive period, the HPA axis may later transition into a stress hypo-responsive period (SHRP) with lower basal cortisol levels and blunted stress-induced cortisol reactivity (101, 104–106). Some longitudinal studies suggest that stress responsivity in early childhood decreases with age throughout the preschool period (101, 105–107), suggesting a potential social buffering of the HPA axis by a nurturing caregiver, who may operate as a safety signal (108–110). This could partly rely on important interactions of GC-signaling with oxytocin pathways, as recently reviewed by Struber et al. (111). Accordingly, this shift from a hyper- to a hypo-responsive stress axis in the first 5 years in life may be represent an particularly crucial stress-sensitive period, especially in the absence of a nurturing caregiver (104). ELS/CT together with higher cortisol during this SHRP could possibly lead to GR insensitivity through greater exposure to GC over time, thus altering the physiological of HPA axis development (101, 112). Studies from Kuhlman et al. (94, 113) confirmed that ELS/CT exposure in the first 2 years in life is associated with prolonged cortisol reactivity to acute social stressors among adolescents.
The later developmental stage of puberty/adolescence represents the second particularly sensitive and vulnerable period with a new major change in HPA axis activity. In this phase, the HPA axis transitions from hypo-responsivity into a period of increased activity (101, 114–116) with progressive higher basal (106, 110, 117, 118) and reactive (106, 118–121) cortisol levels. Interestingly, parental support no longer buffers HPA axis reactivity during this developmental stage (110). On the other hand, it is rather sexual maturation, in interaction with sex and environmental cues, which represents a new major confounder of HPA axis reprogramming (113). The onset of gonadal hormone production plays a vital role in stress and HPA axis reactivity, since estrogen secretion influences GC hyperactivity (122). Some studies on ELS/CT during adolescence reported lower baseline cortisol (123) and blunted cortisol responses to psychosocial stress (124), accordingly suggesting an opposite effect of ELS/CT on HPA axis basal activity and reactivity than in infancy and early childhood.
Summarizing, ELS/CT during the first hypo-sensitive 2 years of life may lead to a hyper-activity and -responsiveness of HPA axis, while ELS/CT during the hyper-active phase of adolescence to a hypo-active and hypo-responsive HPA axis (101). Bosch et al. (125) confirmed this hypothesis showing a relation between ELS/CT in the first year of life, but not late childhood or adolescence, and higher cortisol reactivity in adult life. They could also show higher adult cortisol levels after ELS/CT experience during childhood, but lower cortisol output after experience of ELS/CT in adolescence. These age-dependent differences in HPA axis plasticity could be also reflected on the specific risk for a mental disorder in adulthood. Taken together, ELS/CT exposure in early childhood leads to a similarly higher risk for developing major depressive disorder or PTSD in adulthood, while after ELS/CT exposure in adolescence, the risk for PTSD is greater than for depression (22).
Locus Ceruleus/Autonomic Nervous System (LC/ANS)
The LC/ANS is also vitally implicated in the stress-related pathophysiological trajectories of trauma (126). LC/ANS and HPA axis are closely interconnected at several neuroendocrine levels throughout the brain and body and their activity normally shows a certain degree of analogy and complementarity. The appropriate regulation of the HPA axis depends at least in part on LC/ANS, especially on vagal influences (127). HPA axis and LC/ANS are both integrated components of an internal neural regulation system (central autonomic network, CAN) (128). Dysregulation of the CAN (129–131) may affect downstream autonomic core centers (i.e., PFC, amygdala, hypothalamus, brain stem nuclei), and alter peripheral ANS activity and overall stress responsivity (130, 132, 133). The significant overlap of the fear/arousal circuitry with the CAN (134) could be, at least partly, responsible for ELS/CT-related autonomic dysregulation. The very high comorbidity of stress- and trauma-related disorders and cardiovascular disease (135–140) confirms a central pathophysiological link between the stress axis and ANS (141–143).
With respect to ELS/CT in particular, a limited number of studies have reported altered autonomic activity in adults with ELS/CT exposure. For example, Otte et al. (144) reported higher catecholamine responses to psychological stress in police recruits, while O'Hare et al. (145) found higher rates of syncope frequency in adulthood in individuals with ELS/CT experience. Heleniak et al. (146) reported blunted cardiac output reactivity and increased vascular resistance associated during a social stress task in ELS/CT-exposed adolescents. However, most studies assessing ANS activity in adult population after trauma included PTSD patients with adult exposure to traumatic stress, repeatedly suggesting an increased sympathetic and/or decreased vagal activity in sequel of a trauma (147).
Some pediatric studies have also lately tried to better investigate the interplay of HPA axis and ANS after ELS/CT. For example, De Bellis et al. (148) reported significantly higher 24 h urinary concentrations of catecholamines in sexually abused girls in comparison to matched controls. Another pediatric study by Gordis et al. (149) reported an asymmetry between the HPA axis and ANS reactivity to a social stressor with absent associations between the peripheral biomarkers of HPA axis (cortisol) and sympathetic activity (salivary alpha-amylase, sAA) only in the maltreated group. In a study longitudinally assessing children after trauma exposure to a motor vehicle accident, Pervanidou et al. (150) could show a successive normalization of cortisol levels but continuously higher catecholamine levels 6 months after trauma exposure, suggesting a lifted cortisol-mediated restraint on catecholamine responses leading to a mid- and long-term enhanced ANS activity. Lower cortisol levels and higher ANS activity found in adult PTSD patients and after ELS/CT exposure may, thus, represent a resulting state of a progredient stress-axes divergence in trauma-related disorders (151). Accordingly, Pervanidou et al. (152) proposed that such a progredient divergence of the two limbs of the stress system following ELS/CT, may represent a vital pathophysiological pathway leading to the long-term impact of ELS/CT on health and the chronic preservation of related symptoms.
Immune System and Inflammation
Inflammation is a natural immune response to pathogens and injury, an integral part of the stress response and, thus, crucial to tissue healing, adaptation and survival (4, 153, 154). Acute stress activates the secretion of pro-inflammatory cytokines, presumably by adrenergic and CRH-peptidergic stimulation, which help orchestrate the further immune response (e.g., stimulation of systemic acute-phase proteins, such as C-reactive protein, CRP) (4, 155). Pro-inflammatory cytokines, however, unfold systemic effects far beyond the canonical immune response and also stimulate the secretion of CGs, while CGs, in turn, among their numerous pleiotropic effects, help terminate the inflammatory response (153, 154, 156, 157). This is part of a very complex, two-way neuroimmunoendocrine interaction between the central and peripheral limbs of the stress system and the immune axis (156, 158, 159). Growing evidence, accordingly, implicates the immune system in stress resilience and coping through peripheral and central immune mechanisms of action, affecting the brain and all stress-related neurobiological and neuroendocrine responses (160). Vice versa, a dysregulated stress system could allow a disinhibition or excessive inhibition of inflammatory processes, promoting biological aging, inflammatory-related or immunosuppressed medical conditions and compromised overall health (63, 89, 161–163). There is growing evidence suggesting that positive ELS/CT history is an independent risk factor for peripheral immune dysregulation and long-term, low-grade inflammatory excess (i.e., a pro-inflammatory phenotype) in adulthood (101, 164–172).
Given this, the dysfunctional neuroendocrine interface following ELS/CT may be closely correlated to immunological alterations and related long-term health consequences (4, 36, 101, 153, 154, 172, 173), while adults with ELS/CT experience could be at increased risk of disease with potentially immune origin (36, 53). Most human research has been concentrating on pro-inflammatory cytokines and CRP for the immune status characterization. Among all cytokines assessed, interleukin-6 (IL-6) findings are the most robust.
Interleukin-6 and CRP
IL-6 is a pleiotropic cytokine and simultaneously one of the most suitable inflammatory markers for the characterization of inflammatory status in humans (174), but also an applicable stress biomarker (155), as IL-6 may have a reciprocal modulatory effect on the stress system (175). Indeed, animal and human research confirms that IL-6 stimulates the HPA axis at hypothalamic, pituitary and adrenal level (176–183). Basal IL-6, through activation of the JAK/STAT3 signaling cascade, is required for the sustained cortisol response to chronic stress and is therefore a possible mediator of HPA axis plasticity, in particular in chronic stress states (184). Conversely, cortisol exerts a mild inhibitory effect on the peripheral production of IL-6 (185) and is a major moderator of circadian IL-6 changes (186, 187), while prednisone administration flattens the diurnal rise of IL-6 in the early morning (188). Norepinephrine and epinephrine, on the other hand, lead to an increase of plasma IL-6 in both humans and rats (189–191), in part via beta-adrenergic receptor mechanisms regulating hepatic and splenic IL-6 production (192–194). A recent animal finding also suggested that basal IL-6 signaling in the hypothalamus is a potential determinant of plasticity in the HPA axis response, specifically during chronic stress exposure (184), suggesting that both central and peripheral IL-6 play crucials role on the development of stress susceptibility and related behaviors (175, 195). Several studies have reported dysregulated IL-6 levels in individuals with ELS/CT experience. Carpenter et al. (169) reported higher IL-6 baseline concentrations and a higher inflammatory IL-6 response to acute psychosocial stress challenge in healthy adults with a history of ELS/CT. Using the same paradigm (Trier Social Stress Test; TSST), Pace et al. (196) have shown the same exaggerated IL-6 response to an acute psychosocial stressor in depressed male patients with positive ELS/CT history, compared to depressed patients without ELS/CT history. Interestingly, Kunz-Ebrecht et al. (197) reported an inverse relation between IL-6 and cortisol release to mild mental stress challenges, while Pervanidou et al. (150) provided evidence that IL-6 was involved in the initial biological alterations in the aftermath of trauma, and predictive of PTSD development 6 months later in a longitudinal study design following motor vehicle accidents in children. Finally, in one of the few large (over 3,500 children) prospective studies, Slopen et al. (198) reported ELS/CT being associated with increased levels of IL-6 years later.
With respect to CRP, there are a large number of studies reporting on the association of ELS/CT with increased circulating CRP levels. Most findings, but not all, suggest a robust correlation between ELS/CT and adult CRP levels (165, 166, 170, 199–202). In their seminal study of a birth cohort followed to age 32 years, Danese et al. (165) reported an independent effect of ELS/CT on adult inflammation and suggested that more than 10% of the low-grade inflammation cases in the population may be attributable to ELS/CT. In their prospective study, Slopen et al. (198) found that ELS/CT is a significant independent predictor of persisting inflammation almost 10 years after ELS/CT exposure. Finally, a recent meta-analysis, including over 20,000 samples, confirmed that individuals exposed to ELS/CT show significantly elevated baseline peripheral levels of CRP, IL-6 and TNF-α (203). This study also suggested that specific types of ELS/CT may have differential impacts on single inflammatory markers.
Neuroimmune Pathways
Although numerous neurobiological links between ELS/CT and inflammation have been put forth, the underlying mechanisms are still not completely understood (159). On the one hand, ELS/CT-related autonomic imbalance with reduced vagal activity may further directly augment inflammation through a direct vagal efferent effect of autonomic brain regions (204–206). On the other hand, HPA axis dysregulation in ELS/CT affects GR-mediated transcriptional and post-transcriptional responses of immune-related genes with lower recovery ability (89, 207). Preclinical research has shown GC resistance in immune cells following repeated acute stress (208, 209), while in humans, prolonged or chronic stress leads also to GR insensitivity of immune cells and, respectively, altered GC inhibitory signal (112, 210). Respectively, several recent human gene expression studies show accumulating evidence for innate immune dysregulation after trauma and a particular and specific (i.e., comorbidity-independent) role of cytokines (211–215). Smid et al. (175) have recently reported both higher mitogen-stimulated T-cell cytokine and innate cytokine production with increasing PTSD symptoms, suggesting a direct effect of cytokine production in stress sensitization. Further human PTSD research suggested that elevated expression of pro-inflammatory cytokines after traumatic stress exposure is probably regulated by multiple epigenetic mechanisms, including dysregulation of microRNA expression (216–218). Interestingly, animal findings suggest that pro-inflammatory cytokines also mediate chronic, stress-induced impairments in hippocampal neurogenesis (167), suggesting that ELS/CT-related subsequent pro-inflammatory diathesis could impair neurogenesis in vital central nervous system (CNS) areas during critical developmental periods and result in a reduced hippocampal volume (see below) and a related malfunction of the fear response circuit in context-dependent situations in adulthood.
Human Microbiome and the gut-Brain-Axis
During the last decade, the human microbiome and the microbiota-gut-brain (MGB)-axis have become a novel epicenter in mental health and specifically stress-related research and have been already acknowledged as a potentially vital new determinant in the field of neuroimmunoregulation, brain development and behavior (219–223). The MGB-axis represents a bidirectional, key communication pathway between the immune system and the CNS, thus partly mediating the regulation of stress response and early life programming of the neuroimmune system (221, 224). The gut microbiota is a complex ecosystem with a great organism variety and refined genomic structure that resides in the intestinal tract and has a central position in human health and disease (225).
The microbiome produces directly and indirectly significant amounts of antimicrobial peptides, hormones, short chain fatty acids, vitamins, and several neurotransmitters (e.g., 5-HT, catecholamines) and strongly influences our metabolic, endocrine, immune, and CNS (219). In addition, a special role of macrobiota wall constituents on CNS function and development has been suggested recently. For example, peptidoglycans and lipopolysaccharides have been shown to cross the intestinal epithelial barrier and to bind to specific pattern recognition receptors and lead to an activation of the central and peripheral immune system and HPA axis (226, 227). Furthermore, gut microbiota may modulate CNS microglia maturation and functioning and thus also affect neural circuitry of the developing brain (228, 229).
The other way around, the CNS can also modulate the composition and balance of the intestinal microbial community (and mostly Gram-negative bacteria) through the stress system (ANS, HPA axis), (230). For example, PTSD patients show differences in the total abundance of specific bacterial taxa in comparison to trauma-exposed controls (231), while chronic social defeat stress animals models have also lead to shifts in intestinal microbiota composition (232, 233). A chronic bacterial dysbiosis weakens the intestinal mucosal barrier and affects intestinal permeability (“leaky gut”) (234), which possibly results in a microbiota-driven proinflammatory state (235). Thus, a major candidate source of systemic stress-related inflammation could be the disordered gut barrier function (236). A stress-driven microbiome imbalance could then feedback and affect brain functioning by reprogramming the HPA axis through cytokines-related CRH release in the hypothalamus and elsewhere (224, 237–240).
The human microbiome follows a dynamic trajectory development throughout the lifespan and establishes a symbiotic relationship with the organism early in life. Thereby, the development of the intestinal microbiota occurs in parallel with the CNS, having similar critical windows with rapid and profound developmental changes during infancy, childhood, and adolescence (241). Stress-related disruption of the dynamic host-microbe interaction at these critical periods can lead to alterations of the bacterial colonization of the gut in early life and vice versa (242, 243). As the microbiome plays an important role in the programming of the HPA axis and stress reactivity (244), ELS/CT may affect the signaling of the MGB axis in a major fashion and alter not only immune, but also CNS and stress system functioning with lifelong emotional and behavioral consequences (i.e., higher risk of neurodevelopmental disorders) (223, 239, 241, 245, 246).
Taken together, the imbalanced human microbiome might be another vital pathway linking ELS/CT with altered neuroimmune reactions and neurodevelopment, as well as long-lasting effects on general health, behavior, emotions, and cognition (247). Risk and resilience to stress- and immune-related disorders may, thus, depend on the diversity and complexity of gastrointestinal microbiota (229), which could play a pivotal role in the etiology of psychiatric illness and make individuals more susceptible to develop psychopathology after ELS/CT (241, 248, 249).
Oxidative Stress and Cardiovascular System
Redox State and Antioxidant Defenses
Oxidative stress (OXS), defined as a disequilibrium between oxidant generation and antioxidant defenses (i.e., an altered redox state), has been proposed recently to link ELS/CT to a higher risk of developing psychiatric but also physical morbidity in general (250). Animal findings confirmed that ELS (e.g., maternal separation) has a significant impact on parameters of OXS in mitochondrial function and has shown an association with reactive oxygen species, mitochondrial glutathione, ATP and cytochrome c release in cardiac tissue (251). Furthermore, decreased levels of superoxide dismutase and catalase activity, as well as higher levels of protein carbonylation have been reported in the brain of adult animals exposed to ELS (252). Human research been successfully replicated similar findings. For example, increased OXS markers (i.e., reduced glutathione peroxidase levels, increased protein carbonylation and total reactive antioxidant potential kinetics, etc.) have been reported recently in otherwise healthy ELS/CT-exposed adolescents (253). ELS/CT may so lead to long-term molecular consequences in the basal antioxidant defenses with elevated systemic levels of OXS, stimulating inflammation and driving oxidative damage and accelerated cellular aging in both the CNS and the periphery of the organism (254, 255).
Telomere Length
Telomeres are DNA-protein complexes located at the ends of linear chromosomes capping and protecting the genome from damage, while inflammation and OXS have been suggested to reduce telomere length. Telomere length is an emerging marker of biological age and OXS, with shorter length being associated with accelerated biological aging, premature cell death and increased morbidity and mortality from age-related diseases (256). Not only has PTSD been associated with shorter telomere length, but also the experience of ELS/CT (257–260). For example, Tyrka et al. (261) investigated healthy adults with absent Axis-I disorders and reported shorter whole-blood telomere length in association with ELS/CT. In a longitudinal study, Shalev et al. (262) showed higher telomere erosion in children 5–10 years old exposed to more than 2 violent events. Chen et al. (263) reported that greater ELS/CT exposure was associated with reduced telomere length and normal telomerase activity in healthy volunteers. A recent study by Mitchell et al. (264) also found a significant association between father loss and children's telomere length, with the death of father showing the greatest effect, and a 90% greater effect in the children with the most reactive alleles of the 5-HTTLPR gene. Finally, two current meta-analytic studies, confirmed the significant association between ELS/CT and accelerated telomere erosion in adulthood (265, 266). ELS/CT could, thus, possibly partly mediate their long-term biological impact also through shorter telomere length, representing another biomarker of increased cacostatic load (51, 256).
Oxidative Stress and Endothelial Dysfunction
Emerging epidemiologic evidence strongly supports that ELS/CT is an independent albeit silent risk factor of future chronic cardiovascular risk through various systemic and molecular mechanisms (267–272) and that its effect is particularly heightened among women (273). The recent American Heart Association scientific statement offers a comprehensive review of the literature on the influence of ELS/CT on cardiovascular outcomes (274). Besides genetic, metabolic, autonomic, circadian and inflammatory pathways reviewed elsewhere in this article, OXS-related endothelial dysfunction plays a similarly major role in total cardiovascular risk. Animal findings suggest that ELS/CT-related significant endothelial dysfunction is linked to increased superoxide production (275) and reduced endothelial nitrous oxide system buffering capacity with dysfunctional endothelial Angiotensin II-mediated signaling and sensitization to Angiotensin II-induced vasoconstriction (276).
Metabolism
The stress system is closely interconnected with metabolism. GCs, as the end-effectors of the HPA axis, stimulate appetite (277), alter insulin and leptin secretion and target tissue effects by increasing body weight through the orexigenic and food reward effect of the hypothalamic feeding signal NPY (278, 279) [an effect inhibited by leptin and insulin (280)]. Consequently, in individuals with ELS/CT history, the disrupted biological background described above promotes a tendency toward a dysmetabolic syndrome (281, 282). Accordingly, in the obese population, rates of ELS/CT exposure are reported to be almost twice as high as in the non-obese population (69 vs. 39%) (283). Furthermore, ELS/CT has been repeatedly found to be independently associated with increased overall metabolic risk (284, 285), obesity and increased visceral fat deposition (286–288), decreased HDL, increased LDL levels and lower HDL/LDL ratio (289, 290), higher triglyceride levels (285), an overall prediabetic state (e.g., impaired insulin sensitivity) (291), reduced T3 levels and abnormal metabolism of thyroid hormones (292), enhanced risk for emotional eating as a self-regulatory coping strategy (293) and higher prevalence of metabolic syndrome (290, 294, 295) in later life, while some studies have suggested a dose-dependent relation in these associations (288, 296).
ELS/CT-induced metabolic derangements, such as hyperinsulinemia and altered insulin sensitivity on exposure to a high energy diet later in life, can be a result of altered peripheral gene expression. For example, the interaction between HPA axis activity and liver 11-beta hydroxysteroid dehydrogenase (11β-HSD1) could modulate both tissue and circulating GC availability, with adverse metabolic consequences (297). In addition, genetic interactions with ELS/CT could influence risk for dysmetabolic consequences. HPA axis related FKBP5 polymorphisms, in combination with ELS/CT exposure predict higher insulin and glucose values in midlife (298). Animal findings suggest that ELS/CT is associated with increased food intake, weight gain, increased deposition of abdominal fat, higher plasma triglycerides levels, n-3 PUFA deficiency, etc. (299).
On the other hand, there is also evidence that ELS/CT can exert a programming effect on the adipose tissue and alter the highly sensitive process of adipogenesis (282), leading for example to alterations in adipokine regulation and higher fat accumulations in mice (300). Leptin is an important, circadially secreted adipokine and a vital regulator of energy homeostasis and metabolism, reward processing, brain development and neuroendocrine and immune function (301). Leptin directly interacts with the HPA axis (302), showing an inverse relation to circulating corticotropin and cortisol in healthy men and exerts an anorexigenic effect in conjunction with inhibition of orexigenic pathways via leptin-responsive hypothalamic neurons (303). The adipose tissue–derived protein adiponectin, is another adipokine that may also play a central role in the metabolic dysregulation after ELS/CT. Adiponectin is decreased in obesity (304), whereas hypoadiponectinemia is related to adverse metabolic and cardiovascular outcomes in humans (305). Prospective pediatric studies of physical injury (i.e., burn, MVA) have shown a persistently elevated insulin resistance index up to 3 years (306) and decreased adiponectin levels up to 6 months after physical stress exposure (152).
Taken together, mounting evidence suggests that stress during critical periods of growth and development disrupts the interplay between the stress, circadian and metabolic system and has permanent adverse effects on body size and composition and is often accompanied by associated lifestyle and nutritional risk behaviors (i.e., physical inactivity, emotional eating, disrupted sleep) (282).
Sleep and Circadian System
The human circadian system (CS) enables the nyctohemeral organization and coordination of many physiological processes and promotes homeostasis and environmental adaptation (307). The HPA axis activity is closely linked to the CS and displays circadian rhythmicity (308–311). Through various pathways, the central circadian system synchronizes hypothalamic neuroendocrine neurons secreting CRH and AVP, modulates adrenal ACTH sensitivity, stimulates GC secretion and defines the peripheral circadian changes in target tissue GC sensitivity (308, 312–314). Circadian acetylation and deacetylation of the GR, modulated by melatonin, allows for these changes in tissue sensitivity (308, 312, 315, 316). In addition, animal studies demonstrated a circadian regulation of peripheral clock gene oscillation in the adrenal gland (317, 318) confirming a nyctohemeral change in its responsiveness to ACTH. Central and peripheral circadian rhythmicity also modulates ANS control through projections to pre-autonomic neurons of the hypothalamus and is essential for the physiologic diurnal fluctuations seen in humans (319–321). Finally, animal and human studies demonstrate responsiveness of cognitive performance to the CS (322, 323). Memory processing, formation and consolidation are directly influenced by the circadian clock and stress (322, 324, 325). Besides light, an important regulator of CS activity is sleep. Sleep acts synergistically and bidirectionally with the central CS, but also independently to reinstate the internal temporal synchrony (326). Specific sleep stages are associated with CLOCK gene expression in the suprachiasmatic nuclei and are tightly ruled by the CS (326–328).
A critical loss of this timed order across several organizational levels of the organism is defined as chronodisruption and promotes a dysharmony of internal biological systems and appropriate biobehavioral adaptations to external stimuli (329) with short- and long-term pathophysiologic and epigenetic impact (330, 331). Chronodisruption may progressively alter the fundamental properties of brain systems regulating neuroendocrine, immune and autonomic function, similar to ELS/CT-related stress axis dysregulation, and may play a central role in the development of stress-related disorders (328).
Direct and indirect human and animal stress research supports the important supraordinate role of CS on stress system and GCs, linking circadian misalignment in ELS/CT-related pathophysiology and potentially resulting in the extensive co-morbidities of ELS/CT through an impaired homeostatic balance. Some animal (332), but—most importantly—numerous human studies including large cohorts, have repeatedly confirmed that ELS/CT is independently associated with enduring adult sleep disruption including global sleep pathology (i.e., insomnia), as well as specific types of sleep problems, such as shortened total sleep time, prolonged sleep onset latency, decreased sleep efficiency, increased number of awakenings, nightmare related distress, sleep apnea and higher nocturnal activity in a probably dose-response manner (333–344).
Sleep deprivation, which is tightly associated with chronodisruption (326–328), has been recurrently related to HPA axis dysregulation findings, such as a flattened cortisol amplitude, decreased CAR and cortisol reactivity, increased but also decreased diurnal cortisol concentrations and increased CRH levels in humans (345–347). Both animal and human studies show that sleep deprivation is associated with increased sympathoadrenal activity and blunted cardiovascular autonomic rhythmicity and responsiveness, thus representing a key cardiovascular risk factor (347–349). Human and animal sleep deprivation studies have reported hypo-responsive medial-frontal cortical regions, hyper-responsive amygdala, and a smaller hippocampal volume (350–352), as shown in adults with ELS/CT history (see above). Sleep disturbances have been associated with altered CLOCK gene expression in humans, which vitally affects neurobiological response to stress (353, 354). Chronodisruption may, thus, sensitize individuals to stress and increase their vulnerability to stress-related disorders (347, 355).
Numerous human and animal studies suggest that acute and chronic physical and/or psychological stress affects the sleep centers of the brain (356–363). Stress, thus, influences sleep physiology and dream patterns and may cause both immediate and long-lasting sleep disruption (364–366), which may, in turn, enhance maladaptive stress regulation (367). For example, REM sleep disruption immediately after trauma exposure has been associated with higher REM-related sympathoadrenal activity, and represents an important predictive factor for the development of trauma-related disorders in humans (368–370). As sleep promotes memory consolidation, in particular for emotional content, sleep deprivation after stress exposure can affect amygdala-cortical connectivity and disrupt this process (371–373).
Such findings suggest that sleep disruption occurring after trauma exposure may represent a core, rather than a secondary pathway that mediates the enduring neurobiological correlates of ELS/CT (364, 368–370, 374, 375) and that chronodisruption may be the common underlying neurobiologic link (370, 374, 376).
Genetics and Epigenetics
Genome-wide association studies (GWAS) have identified several disease-associated candidate genes, which, however, explain only a minor part of heritability in such complex disorders. In recent few years, the interest has shifted to the central role of the interaction of specific candidate genes with environmental factors, as well as to gene programming through epigenetic regulation (e.g., DNA methylation, histone modification of chromatin, aberrant expression of miRNA) (377, 378). The combination of specific genetic polymorphism profiles and density or activity of functional sites controlling the human stress axis may increase or decrease the risk of psychobiological maladjustment after exposure to ELS/CT. A thorough understanding of the interaction between genes, environment, DNA methylation patterns (methylome) and subsequent gene expression profiles (transcriptome) is integral to our understanding and treatment of stress-related disorders (378).
Gene × Environment Interactions
Two of the first ground-breaking human studies investigating the interaction between ELS/CT and gene polymorphisms were conducted by Caspi and collaborators. In the first study, abused children with a monoamine oxidase A (MAOA) genotype associated with low levels of MAOA expression, were more likely to show antisocial-personality disorder and commit violent crimes in adulthood (379). In the second prospective-longitudinal study of a representative birth cohort, functional polymorphisms in the promoter region of the serotonin transporter (5-HTT) gene (5-HTTLPR) was found to moderate the influence of ELS/CT on depression, with the presence of the short allele being associated with more depressive symptoms, diagnosable depression, and suicidality (380). These findings were later confirmed by Karg et al. (381) and are consistent with the assumption that 5-HTTLPR moderates emotional responsivity to stress in interaction with ELS/CT (382).
More recent findings suggest a vital role of genes involved with HPA axis function and GC sensitivity, in conjunction with exposure to child maltreatment or abuse (383). To date, findings mainly implicate two key genes: the GC response element (GRE) and the CRH-releasing hormone receptor 1 (CRHR1) of the FKBP5 gene (383, 384). The co-chaperone FKBP5 regulates steroid receptors such as the GR, resulting in a resistance (reduced sensitivity) against GCs. As first shown by Binder et al. (385), specific single-nucleotide poly-morphisms (SNPs) of the FKBP5 gene interacting with ELS/CT predict the level of adult PTSD symptoms. An allele-specific demethylation in the GREs of FKBP5 may result in a dysregulated expression of GRs (386). Further clinical studies confirm minor alleles of FKBP5 being particularly sensitive and interact with ELS/CT to increase aggressive behavior (387), suicide attempts (388), and depression (389). The CRHR1 acts as a mediator in initiating the stress response, possibly leading to a hypersensitive negative feedback loop of cortisol. Bradley et al. reported in two separate cohorts, independently, that specific CRHR1 polymorphisms interact with ELS/CT to increase the risk of adult depression (390), similar to Heim et al. (391), while Ben-Efraim et al. (392) reported comparable findings with respect to suicide attempts.
Taken together, gene × environment interactions of gene polymorphisms may affect the acute biological response to ELS/CT and mediate long-term risk of disease to some extent, most probably through their effects on stress responsiveness.
Epigenetic Regulation
Epigenetic modifications are dynamic—and to some extend reversible—changes, that mediate the interaction between genetic predisposition and environmental factors through regulating functional expression of genes by decreasing, silencing or increasing gene expression (393, 394). The installment of such epigenetic marks by ELS/CT exposure and its genetic moderation by related factors represents a critical factor for vulnerability or resilience to stress-related disorders and may explain inter-individual variation. The interpretation of epigenetic findings is critical due to the complexity of the epigenetic mechanisms and the large number of involved genes.
ELS/CT exposure has been repeatedly related to epigenetic changes and altered gene expression profiles, particularly in the CNS (e.g., hippocampus, amygdala), thus affecting stress responses and memory consolidation (395–398). There is accumulating evidence for gene programming and epigenetic regulation of specific genes in the pathophysiology of PTSD in humans (399–402). Especially, several GC-signaling-related genes (e.g., GCR gene promoter 1F) are sensitive to traumatic-stress-related epigenetic regulation across the lifespan and may represent useful biomarkers related to the development, symptomology and prognosis of PTSD (403, 404). For example, in a recent human brain autopsy material study, history of childhood abuse was associated with changes in DNA methylation related to the neuron-specific GR (NR3C1) promoter in the hippocampus, suggesting distinct effects of ELS/CT on the epigenetic regulation of hippocampal GR expression (405). With respect to the promoter and exon 1F of the human GR gene Nr3c1, Oberlander et al. (406) showed specific epigenetic effects (gene hypermethylation) and elevated cortisol stress reactivity in the offspring due to maternal depression even during late pregnancy. Other animal findings also suggested ELS/CT-related epigenetic changes in the CNS growth and differentiation-related BDNF gene expression (407), while in a genome-wide blood DNA methylation analysis study by Houtepen et al. (408), a locus in the Kit ligand gene (KITLG; cg27512205) was shown to strongly modulate the relation between ELS/CT and cortisol stress reactivity.
Lately, various studies have investigated large-scale methylation patterns with respect to ELS/CT in cross-sectional settings. Bick et al. (409) reported significant differences in methylation in 72 of investigated 173 genes (responsible for HPA and immune system regulation) in children with and without foster care experience. Yang et al. (410) reported significant differences in methylation in 2,868 CpG sites on genes of all 23 chromosomes with respect to presence of ELS/CT, while Essex et al. (411) described similar transgenerational results in more than 150 of 28,000 CpG sites in a prospective study assessing parental stress and its consequences in their offspring. Interestingly, Mehta et al. (412) found that gene expression profiles of PTSD patients with and without ELS/CT are 98% non-overlapping. Moreover, these changes were mostly mediated by DNA methylation changes to a much larger proportion in the childhood abuse group, suggesting that changes in DNA methylation may exert a much greater impact during early life and possibly reflect differences in PTSD pathophysiology, depending on preceding exposure to ELS/CT.
Taken together, enduring changes in the transcriptome may facilitate the response to early developmental challenges and thus play a central role in the long-term (and sometimes transgenerational) biological trajectories of stress-related disease through programming effects for stress reactivity after ELS/CT exposure (104, 413, 414).
Structural and Functional Imaging Findings
ELS/CT during critical periods of brain development crucially affects the interaction between developing brain regions and neural circuits, exerts epigenetic influences and alters the functions of the HPA axis and GCs; indeed, it has been associated with remarkable structural and functional brain changes even decades later, in adulthood, defining both vulnerability and resilience (383, 415, 416) [for an in-depth review see (417)]. Studies in animals have shown that elevated levels of GCs and catecholamines may lead to alterations in brain development through accelerated loss of neurons (418), delays in myelination (419), or abnormalities in developmentally appropriate synapse pruning (420). ELS/CT-related remodeling of structure, responsiveness and connectivity of specific brain areas and circuits can accordingly alter behavioral, cognitive, emotional, and physiologic responses (51, 421). For example, as cognitive function is heavily dependent on HPA axis and CG activity, childhood adversity associated with HPA axis dysfunction and GC excess or deficiency can result in diminished cognitive functioning and maladaptive emotional behavior (422). Accordingly, in a human resting activity neuroimaging PET study by Insana et al. (423), ELS/CT was associated with altered frontolimbic adult neural activity in the left orbital frontal cortex and left hippocampus, regions involved in executive functioning and emotional autoregulation, socioemotional processes, autonomic function, and sleep/wake regulation. ELS/CT has been also associated with several altered cognitive function findings, such as poor processing speed, defective executive functioning, and memory deficits (e.g., impaired spatial working memory performance, pattern recognition memory) in adulthood, which in turn might pose risks for the development of psychopathology (424–426).
There have been several additional studies assessing structural and functional brain correlates of ELS/CT, but the results have to be explored with caution, given the complexity of brain function, the simplicity of most study paradigms, the age of ELS/CT and assessment, the specific morbid population (i.e., type of psychopathology) and a number of other parameters not taken into account (427, 428). With respect to structural correlates, ELS/CT is associated with disruptive development and reduced volume of corpus callosum, insula, dorsolateral prefrontal cortex (PFC), orbitofrontal cortex (OFC), anterior cingulate gyrus, and caudate, as well as decreased cortical thickness of medial and lateral prefrontal and temporal lobe regions, and reduced overall brain volume in humans (416, 417, 425, 426, 428–434). A study of Teicher et al. (435), utilizing high-resolution T1-weighted MRI scans to assess network connectivity, also reported substantial changes in the cortical network architecture in these areas in young adults with ELS/CT history. Interestingly, the distinct neural plasticity during development can lead to cortical adaptation with very specific regionally altered cortical representation fields (436, 437) and be potentially protecting from the specific sensory processing of different ELS/CT (417). Thus, experience of sexual abuse has been associated with cortical thinning specifically in the genital representation field of the primary somatosensory cortex, while emotional abuse specifically in regions relevant to self-awareness and self-evaluation (438). Such plastic reorganization may be initially protective under abusive conditions, but may underlie later behavioral problems in the same areas (e.g., sexual dysfunction) and be selectively associated with increased vulnerability to internalizing and externalizing psychopathology (434).
The amygdala and the hippocampus are the two brain structures so far mostly reported to be impaired in adult victims of ELS/CT, suggesting most vital effects of ELS/CT on prefrontal-limbic gray matter. The hippocampus is of particular importance because of its role in cognition, but also its rich density of GR, while the amygdala because of its pivotal role in stress responsivity and the extensive related research in mood and anxiety disorders. There are numerous reports and meta-analytic studies confirming the association of ELS/CT with reduced hippocampal volume in adulthood (416, 417, 428, 430, 431, 433, 439). Interestingly, several studies assessing the effects of ELS/CT on hippocampal volume in patients with MDD, suggested that it is rather the history of ELS/CT than depression which is associated with hippocampal atrophy (440–442). However, hippocampal volume seems to be unaffected in children but not in adults with maltreatment-related PTSD, suggesting an initially volumetrically normal hippocampus with subsequent abnormal disrupted development (443). With respect to amygdala, the results from human studies regarding the volumetric effect of ELS/CT are inconclusive, with some studies reporting reduced volume (416, 428, 430, 444), some differential effects according to specific type of ELS/CT (432, 445), and some even greater amygdala volume (in non-human primates) (446). However, findings are conclusive concerning amygdala responsiveness, as ELS/CT has been repeatedly associated with facial threat- or negative-emotion-related amygdala hyper-responsiveness (416, 417, 447, 448). In addition, some studies even suggested that the relation between ELS/CT and risk for adult depression is actually mediated by this preceding amygdala hyperactivity (448, 449).
Finally, imaging studies have investigated the potential influence of genetics (i.e., specific polymorphisms in candidate genes) on the ELS/CT effects described above (417). For example, van Velzen et al. (444) showed that the magnitude of amygdala atrophy in maltreated individuals was significantly associated with the BDNF Val66Met genotype, while Booij et al. (450) demonstrated that greater peripheral serotonin transporter methylation in smaller hippocampal volume in adults with ELS/CT experience. More importantly, there have been a number of studies suggesting a moderating effect of FKBP5 (451–453) and mineralocorticoid receptor genotypes (454) on amygdala volume, reactivity and connectivity of ELS/CT exposed adults, thus implicating HPA axis-related genes in brain development. Genetic susceptibility may, thus, represent a crucial factor leading to related structural and functional trajectories of ELS/CT on brain development (455).
Taken together, altered amygdala-PFC connectivity with reduced top-down regulation of the amygdala by the PFC, reduced contextual input to the amygdala from the hippocampus, and increased connectivity of the amygdala with the LC (leading to increased limbic activity and PFC dysfunction), all suggest that ELS/CT plays a seminal role in functional and structural changes in the brain that may persist along the lifespan (51, 417, 430). Developmental differences in sensitivity to specific forms of childhood maltreatment may lead to different susceptibility of various brain regions and pathways to maltreatment at different ages (417). These results suggest that previously reported structural and functional findings in adolescent or adult psychiatric disease should be re-evaluated addressing ELS/CT as a potential confounder (417).
Explanatory Models
The developmental origin hypothesis of evolutionary biology suggests that the origins of adult disease are often found among early-life disruptions of physiological developmental processes, ranging from direct causal associations to complex, interacting environmental effects (58, 456–460). The previous sections confirm that ELS/CT during critical phases of perinatal and juvenile brain development is associated with increased cacostatic load and reduced stress adaptability in adulthood, leading to enhanced vulnerability to several chronic diseases. Consequently, various explanatory models have been suggested during the past decades.
According to the cumulative stress model (diathesis-stress model) put forth by McEven et al. (4), when the accumulation of stressors along the life span exceeds a certain threshold, disease development is enhanced in individuals with higher stress exposure. Gluckman et al. (458, 459) suggested a pivotal role of ELS/CT that could prompt developmental (epigenetic) changes underlying predictive adaptive responses leading to a mismatch between the phenotypic outcome of adaptive plasticity and the ability to cope with current stressors increasing risk for disease (match/mismatch hypothesis). In contrast to the cumulative stress model, the mismatch hypothesis explicitly assumes that ELS/CT may also have advantageous effects by representing a possible source of adaptation, potentially even promoting active coping (stress inoculation) to moderate stressors and, thus, resilience. Similarly, the for-better-and-for-worse model suggested by Belsky and Beaver (461) assumes that genetic susceptibility should be contextually interpreted and, according to the specific environment, could be beneficial or not. Nederhof and colleagues have proposed an integrated model based on programming effects of ELS/CT interacting with individual genetic vulnerability (462, 463). Recently, Daskalakis et al. (104) have expanded this model suggesting a three-hit concept for vulnerability and resilience. Accordingly, vulnerability in a given context is enhanced when failure to cope with adversity accumulates. The interaction of the individual genetic background (hit-1) with ELS/CT exposure (hit-2) results in an evolving phenotype with altered stress axis regulation and sensitivity due to early developmental programming, which, in turn, interacts with later-life challenges (hit-3) to result in a higher or lower vulnerability risk according to the type of challenge experienced. This model underlines the extraordinary plasticity of the brain and suggests that “nothing is written in stone” (464).
Discussion
Coordination of the stress, immune and circadian systems is essential to individual development, adaptation, survival, and well-being (1, 2, 153). ELS/CT, in interaction with genetic factors, disrupts developmental programming of the related neural circuitry and leads to alterations in neuroendocrine, immune, circadian, emotional, and autonomic (re-)activity, with related structural, functional, and epigenetic modifications both in the brain and peripheral tissues. These persistent structural and functional neuropsychobiological changes as sequelae of ELS/CT could mediate risk for chronic disease in adulthood, and lead to cumulative disadvantages and increased adult physical and mental health morbidity (15, 55, 58, 62). Nevertheless, although most studies support a causal relation between ELS/CT and psychobiological maladjustment in later life, the developmental course of such changes and its temporal coincidence has not been elucidated as yet. Thereby, non-linear patterns in neurodevelopment lead to specific periods of greater stress system plasticity, which represent important vulnerability periods (96, 100, 101). Thus, ELS/CT experience is probably associated with a differential impact on stress system activity according to the specific developmental period of exposure (102). ELS/CT exposure during the first hypo-sensitive 2 years of life may lead to a hyper-activity and -responsiveness of HPA axis and accordingly higher risk for developing depression than PTSD, while ELS/CT during the hyper-active phase of adolescence may lead to a hypo-active and hypo-responsive HPA axis and accordingly higher risk for developing PTSD than depression in adulthood (22, 101).
Figure 1 summarizes the above developmental approaches and provides an integrative schematic model of moderating factors and allostatic neurobiological trajectory networks involved in the enduring biopsychological effects of ELS. However, further biological pathways (i.e., gonadal steroids, amyloid beta, mitochondrial function, leptin/ghrelin system), psychiatric states (i.e., depression, PTSD), and behavioral patterns (i.e., substance abuse, physical exercise, nutrition) could also play an important role in the mediation of the overall biological risk after ELS/CT and should be better investigated.
Figure 1
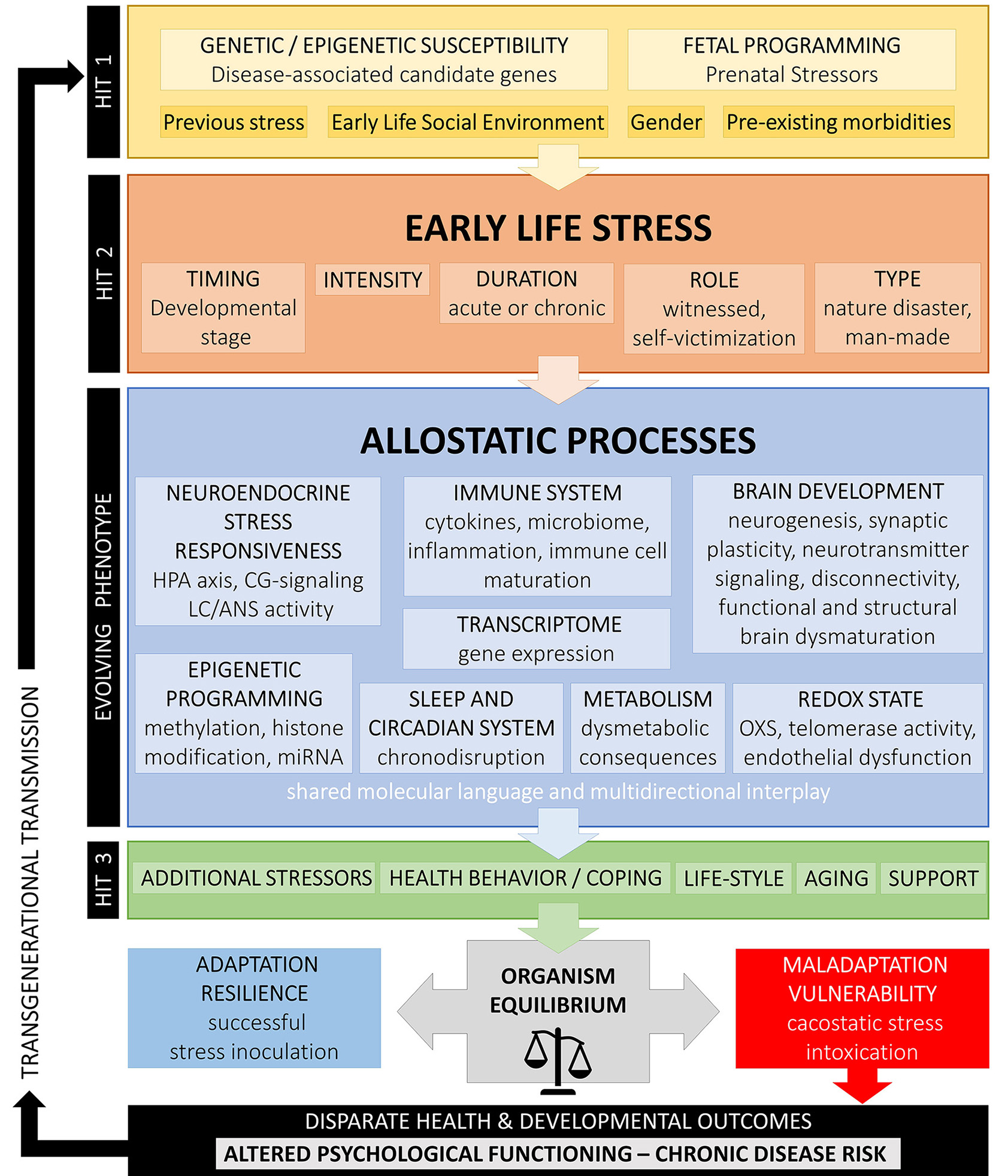
Conceptual model on developmental trajectories of early life stress. Schematic model of moderating factors and allostatic neurobiological trajectory networks involved in the enduring biopsychological effects of ELS/CT. Exposure to ELS/CT can lead to disruption in critical phases of perinatal and juvenile brain development and an evolving programmed phenotype with altered allostatic processes and reduced adaptability to stress. The individual effects on ELS/CT on the organism depend on the specific genetic background and fetal programming (hit-1), the timing, duration, intensity and type of ELS/CT (hit-2) and other later-life challenges, such as additional stressors, coping strategies, support existence, life style, and aging (hit-3). Depending on their interaction, these factors explain inter-individual variation in resilience or vulnerability to altered biopsychological functioning and disparate health outcomes.
Conclusions
The identification of factors related to risk and resilience in the wake of child abuse is a matter of central importance for public health interventions (465). Understanding the pathways susceptible to disruption following ELS/CT exposure and the effects of a dysregulated interconnection between all neural systems involved could provide new insights into the pathophysiologic trajectories that link toxic stress during developmental stages of childhood and adolescence to adult maladjustment and psychopathology. Future studies should prospectively investigate potential confounders, their temporal sequence and combined effects at the epidemiological, biological, and epigenetic level (466, 467), while considering the potentially delayed time-frame for the expression of their effects. Finally, screening strategies for ELS/CT and trauma need to be improved. Information about ELS/CT history and the number of adverse experiences could help to better identify the individual risk for disease development, predict individual treatment response and design prevention strategies to reduce the negative effects of ELS/CT (468). Detecting and healing of the “hidden wounds” left by ELS/CT should thus be a public health priority.
Statements
Author contributions
AA managed all literature searches. AA and PP and wrote the first draft of the paper. GC and DB contributed with significant text passages and revised the draft for important intellectual content. All authors have contributed to, read, and approved the final version of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
ChrousosGPGoldPW. The concepts of stress and stress system disorders. Overview Phys Behav Homeost. (1992) 267:1244–52. 10.1001/jama.1992.03480090092034
2.
ChrousosGP. Stress and disorders of the stress system. Nat Rev Endocrinol. (2009) 5:374–81. 10.1038/nrendo.2009.106
3.
DhabharFSMcEwenBSSpencerRL. Adaptation to prolonged or repeated stress–comparison between rat strains showing intrinsic differences in reactivity to acute stress. Neuroendocrinology. (1997) 65:360–8. 10.1159/000127196
4.
McEwenBS. Protective and damaging effects of stress mediators. N Engl J Med. (1998) 338:171–9. 10.1056/NEJM199801153380307
5.
HeimCNemeroffCB. The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry. (2001) 49:1023–39. 10.1016/S0006-3223(01)01157-X
6.
LupienSJMcEwenBSGunnarMRHeimC. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. (2009) 10:434–45. 10.1038/nrn2639
7.
KoolhaasJMBartolomucciABuwaldaBde BoerSFFluggeGKorteSMet al. Stress revisited: a critical evaluation of the stress concept. Neurosci Biobehav Rev. (2011) 35:1291–301. 10.1016/j.neubiorev.2011.02.003
8.
FrodlTO'KeaneV. How does the brain deal with cumulative stress? A review with focus on developmental stress, HPA axis function and hippocampal structure in humans. Neurobiol Dis. (2013) 52:24–37. 10.1016/j.nbd.2012.03.012
9.
SeoDTsouKAAnsellEBPotenzaMNSinhaR. Cumulative adversity sensitizes neural response to acute stress: association with health symptoms. Neuropsychopharmacology. (2014) 39:670–80. 10.1038/npp.2013.250
10.
Stults-KolehmainenMATuitKSinhaR. Lower cumulative stress is associated with better health for physically active adults in the community. Stress. (2014) 17:157–68. 10.3109/10253890.2013.878329
11.
ReynoldsRMLabadJBussCGhaemmaghamiPRaikkonenK. Transmitting biological effects of stress in utero: implications for mother and offspring. Psychoneuroendocrinology. (2013) 38:1843–9. 10.1016/j.psyneuen.2013.05.018
12.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th Edition Washington, DC: American Psychiatric Association (2013).
13.
EdwardsVJHoldenGWFelittiVJAndaRF. Relationship between multiple forms of childhood maltreatment and adult mental health in community respondents: results from the adverse childhood experiences study. Am J Psychiatry. (2003) 160:1453–60. 10.1176/appi.ajp.160.8.1453
14.
ScherCDFordeDRMcQuaidJRSteinMB. Prevalence and demographic correlates of childhood maltreatment in an adult community sample. Child Abuse Negl. (2004) 28:167–80. 10.1016/j.chiabu.2003.09.012
15.
GilbertRWidomCSBrowneKFergussonDWebbEJansonS. Burden and consequences of child maltreatment in high-income countries. Lancet. (2009) 373:68–81. 10.1016/S0140-6736(08)61706-7
16.
GreenJGMcLaughlinKABerglundPAGruberMJSampsonNAZaslavskyAMet al. Childhood adversities and adult psychiatric disorders in the national comorbidity survey replication I: associations with first onset of DSM-IV disorders. Arch Gen Psychiatry. (2010) 67:113–23. 10.1001/archgenpsychiatry.2009.186
17.
WalkerEAGelfandAKatonWJKossMPVon KorffMBernsteinDet al. Adult health status of women with histories of childhood abuse and neglect. Am J Med. (1999) 107:332–9. 10.1016/S0002-9343(99)00235-1
18.
SpertusILYehudaRWongCMHalliganSSeremetisSV. Childhood emotional abuse and neglect as predictors of psychological and physical symptoms in women presenting to a primary care practice. Child Abuse Negl. (2003) 27:1247–58. 10.1016/j.chiabu.2003.05.001
19.
ElstadJI. Childhood adversities and health variations among middle-aged men: a retrospective lifecourse study. Eur J Public Health. (2005) 15:51–8. 10.1093/eurpub/cki114
20.
DraperBPfaffJJPirkisJSnowdonJLautenschlagerNTWilsonIet al. Long-term effects of childhood abuse on the quality of life and health of older people: results from the depression and early prevention of suicide in general practice project. J Am Geriatr Soc. (2008) 56:262–71. 10.1111/j.1532-5415.2007.01537.x
21.
DubeSRCookMLEdwardsVJ. Health-related outcomes of adverse childhood experiences in Texas, 2002. Prev Chronic Dis. (2010) 7:A52.
22.
MaerckerAMichaelTFehmLBeckerESMargrafJ. Age of traumatisation as a predictor of post-traumatic stress disorder or major depression in young women. Br J Psychiatry. (2004) 184:482–7. 10.1192/bjp.184.6.482
23.
PirkolaSIsometsaEAroHKestilaLHamalainenJVeijolaJet al. Childhood adversities as risk factors for adult mental disorders: results from the Health 2000 study. Soc Psychiatry Psychiatr Epidemiol. (2005) 40:769–77. 10.1007/s00127-005-0950-x
24.
ScottKMVon KorffMAngermeyerMCBenjetCBruffaertsRde GirolamoGet al. Association of childhood adversities and early-onset mental disorders with adult-onset chronic physical conditions. Arch Gen Psychiatry. (2011) 68:838–44. 10.1001/archgenpsychiatry.2011.77
25.
NanniVUherRDaneseA. Childhood maltreatment predicts unfavorable course of illness and treatment outcome in depression: a meta-analysis. Am J Psychiatry. (2012) 169:141–51. 10.1176/appi.ajp.2011.11020335
26.
KoenenKCMoffittTEPoultonRMartinJCaspiA. Early childhood factors associated with the development of post-traumatic stress disorder: results from a longitudinal birth cohort. Psychol Med. (2007) 37:181–92. 10.1017/S0033291706009019
27.
WangZInslichtSSMetzlerTJHenn-HaaseCMcCaslinSETongHet al. A prospective study of predictors of depression symptoms in police. Psychiatry Res. (2010) 175:211–6. 10.1016/j.psychres.2008.11.010
28.
BerntsenDJohannessenKBThomsenYDBertelsenMHoyleRHRubinDC. Peace and war: trajectories of posttraumatic stress disorder symptoms before, during, and after military deployment in afghanistan. Psychol Sci. (2012) 23:1557–65. 10.1177/0956797612457389
29.
HovensJGGiltayEJWiersmaJESpinhovenPPenninxBWZitmanFG. Impact of childhood life events and trauma on the course of depressive and anxiety disorders. Acta Psychiatr Scand. (2012) 126:198–207. 10.1111/j.1600-0447.2011.01828.x
30.
SprattEGBackSEYeattsSDSimpsonANMcRae-ClarkAMoran-Santa MariaMMet al. Relationship between child abuse and adult smoking. Int J Psychiatry Med. (2009) 39:417–26. 10.2190/PM.39.4.f
31.
KhouryLTangYLBradleyBCubellsJFResslerKJ. Substance use, childhood traumatic experience, and posttraumatic stress disorder in an urban civilian population. Depress Anxiety. (2010) 27:1077–86. 10.1002/da.20751
32.
StrineTWDubeSREdwardsVJPrehnAWRasmussenSWagenfeldMet al. Associations between adverse childhood experiences, psychological distress, and adult alcohol problems. Am J Health Behav. (2012) 36:408–23. 10.5993/AJHB.36.3.11
33.
FentonMCGeierTKeyesKSkodolAEGrantBFHasinDS. Combined role of childhood maltreatment, family history, and gender in the risk for alcohol dependence. Psychol Med. (2013) 43:1045–57. 10.1017/S0033291712001729
34.
Fuller-ThomsonEFilippelliJLue-CrisostomoCA. Gender-specific association between childhood adversities and smoking in adulthood: findings from a population-based study. Public Health. (2013) 127:449–60. 10.1016/j.puhe.2013.01.006
35.
ZattiCRosaVBarrosAValdiviaLCalegaroVCFreitasLHet al. Childhood trauma and suicide attempt: a meta-analysis of longitudinal studies from the last decade. Psychiatry Res. (2017) 256:353–8. 10.1016/j.psychres.2017.06.082
36.
DongMGilesWHFelittiVJDubeSRWilliamsJEChapmanDPet al. Insights into causal pathways for ischemic heart disease: adverse childhood experiences study. Circulation. (2004) 110:1761–6. 10.1161/01.CIR.0000143074.54995.7F
37.
SpringerKWSheridanJKuoDCarnesM. Long-term physical and mental health consequences of childhood physical abuse: results from a large population-based sample of men and women. Child Abuse Negl. (2007) 31:517–30. 10.1016/j.chiabu.2007.01.003
38.
ParasMLMuradMHChenLPGoransonENSattlerALColbensonKMet al. Sexual abuse and lifetime diagnosis of somatic disorders: a systematic review and meta-analysis. JAMA. (2009) 302:550–61. 10.1001/jama.2009.1091
39.
WegmanHLStetlerC. A meta-analytic review of the effects of childhood abuse on medical outcomes in adulthood. Psychosom Med. (2009) 71:805–12. 10.1097/PSY.0b013e3181bb2b46
40.
KorkeilaJVahteraJKorkeilaKKivimakiMSumanenMKoskenvuoKet al. Childhood adversities as predictors of incident coronary heart disease and cerebrovascular disease. Heart. (2010) 96:298–303. 10.1136/hrt.2009.188250
41.
SteinDJScottKHaro AbadJMAguilar-GaxiolaSAlonsoJAngermeyerMet al. Early childhood adversity and later hypertension: data from the World Mental Health Survey. Ann Clin Psychiatry. (2010) 22:19–28.
42.
TamayoTChristianHRathmannW. Impact of early psychosocial factors (childhood socioeconomic factors and adversities) on future risk of type 2 diabetes, metabolic disturbances and obesity: a systematic review. BMC Public Health. (2010) 10:525. 10.1186/1471-2458-10-525
43.
HuangMCSchwandtMLRamchandaniVAGeorgeDTHeiligM. Impact of multiple types of childhood trauma exposure on risk of psychiatric comorbidity among alcoholic inpatients. Alcohol Clin Exp Res. (2012) 36:1099–107. 10.1111/j.1530-0277.2011.01695.x
44.
AgorastosAPittmanJOAngkawACNievergeltCMHansenCJAversaLHet al. The cumulative effect of different childhood trauma types on self-reported symptoms of adult male depression and PTSD, substance abuse and health-related quality of life in a large active-duty military cohort. J Psychiatr Res. (2014) 58:46–54. 10.1016/j.jpsychires.2014.07.014
45.
AndaRFFelittiVJBremnerJDWalkerJDWhitfieldCPerryBDet al. The enduring effects of abuse and related adverse experiences in childhood. A convergence of evidence from neurobiology and epidemiology. Eur Arch Psychiatry Clin Neurosci. (2006) 256:174–86. 10.1007/s00406-005-0624-4
46.
AfifiTOEnnsMWCoxBJde GraafRten HaveMSareenJ. Child abuse and health-related quality of life in adulthood. J Nerv Ment Dis. (2007) 195:797–804. 10.1097/NMD.0b013e3181567fdd
47.
AndaRFBrownDWFelittiVJBremnerJDDubeSRGilesWH. Adverse childhood experiences and prescribed psychotropic medications in adults. Am J Prev Med. (2007) 32:389–94. 10.1016/j.amepre.2007.01.005
48.
BriereJKaltmanSGreenBL. Accumulated childhood trauma and symptom complexity. J Trauma Stress. (2008) 21:223–6. 10.1002/jts.20317
49.
LangAJAaronsGAGearityJLaffayeCSatzLDresselhausTRet al. Direct and indirect links between childhood maltreatment, posttraumatic stress disorder, and women's health. Behav Med. (2008) 33:125–35. 10.3200/BMED.33.4.125-136
50.
SulimanSMkabileSGFinchamDSAhmedRSteinDJSeedatS. Cumulative effect of multiple trauma on symptoms of posttraumatic stress disorder, anxiety, and depression in adolescents. Compr Psychiatry. (2009) 50:121–7. 10.1016/j.comppsych.2008.06.006
51.
NemeroffCB. Paradise lost: the neurobiological and clinical consequences of child abuse and neglect. Neuron. (2016) 89:892–909. 10.1016/j.neuron.2016.01.019
52.
MullenPEMartinJLAndersonJCRomansSEHerbisonGP. The long-term impact of the physical, emotional, and sexual abuse of children: a community study. Child Abuse Negl. (1996) 20:7–21. 10.1016/0145-2134(95)00112-3
53.
FelittiVJAndaRFNordenbergDWilliamsonDFSpitzAMEdwardsVet al. Relationship of childhood abuse and household dysfunction to many of the leading causes of death in adults. The Adverse Childhood Experiences (ACE) study. Am J Prev Med. (1998) 14:245–58. 10.1016/S0749-3797(98)00017-8
54.
HeimCNemeroffCB. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry. (1999) 46:1509–22. 10.1016/S0006-3223(99)00224-3
55.
KaufmanJPlotskyPMNemeroffCBCharneyDS. Effects of early adverse experiences on brain structure and function: clinical implications. Biol Psychiatry. (2000) 48:778–90. 10.1016/S0006-3223(00)00998-7
56.
GoodwinRDSteinMB. Association between childhood trauma and physical disorders among adults in the United States. Psychol Med. (2004) 34:509–20. 10.1017/S003329170300134X
57.
PoleNNeylanTCOtteCMetzlerTJBestSRHenn-HaaseCet al. Associations between childhood trauma and emotion-modulated psychophysiological responses to startling sounds: a study of police cadets. J Abnorm Psychol. (2007) 116:352–61. 10.1037/0021-843X.116.2.352
58.
ShonkoffJPBoyceWTMcEwenBS. Neuroscience, molecular biology, and the childhood roots of health disparities: building a new framework for health promotion and disease prevention. JAMA. (2009) 301:2252–9. 10.1001/jama.2009.754
59.
MockSEAraiSM. Childhood trauma and chronic illness in adulthood: mental health and socioeconomic status as explanatory factors and buffers. Front Psychol. (2010) 1:246.
60.
GlaserD. Child abuse and neglect and the brain–a review. J Child Psychol Psychiatry. (2000) 41:97–116. 10.1017/S0021963099004990
61.
TeicherMHAndersenSLPolcariAAndersonCMNavaltaCP. Developmental neurobiology of childhood stress and trauma (2002). Psychiatr Clin North Am.25:397–426. 10.1016/S0193-953X(01)00003-X
62.
NemeroffCB. Neurobiological consequences of childhood trauma. J Clin Psychiatry. (2004) 65(Suppl 1.):18–28.
63.
HeimCNewportDJMletzkoTMillerAHNemeroffCB. The link between childhood trauma and depression: insights from HPA axis studies in humans. Psychoneuroendocrinology. (2008) 33:693–710. 10.1016/j.psyneuen.2008.03.008
64.
HeimCNewportDJMillerAHNemeroffCB. Long-term neuroendocrine effects of childhood maltreatment. JAMA. (2000) 284:2321. 10.1001/jama.284.18.2317
65.
YehudaR. Biology of posttraumatic stress disorder. J Clin Psychiatry. (2000) 61 (Suppl. 7):14–21
66.
NemeroffCBBremnerJDFoaEBMaybergHSNorthCSSteinMB. Posttraumatic stress disorder: a state-of-the-science review. J Psychiatr Res. (2006) 40:1–21. 10.1016/j.jpsychires.2005.07.005
67.
HeimCNewportDJHeitSGrahamYPWilcoxMBonsallRet al. Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA. (2000) 284:592–7. 10.1001/jama.284.5.592
68.
HeimCNewportDJBonsallRMillerAHNemeroffCB. Altered pituitary-adrenal axis responses to provocative challenge tests in adult survivors of childhood abuse. Am J Psychiatry. (2001) 158:575–81. 10.1176/appi.ajp.158.4.575
69.
HeimCMletzkoTPurselleDMusselmanDLNemeroffCB. The dexamethasone/corticotropin-releasing factor test in men with major depression: role of childhood trauma. Biol Psychiatry. (2008) 63:398–405. 10.1016/j.biopsych.2007.07.002
70.
MuhtzCWesterMYassouridisAWiedemannKKellnerM. A combined dexamethasone/corticotropin-releasing hormone test in patients with chronic PTSD–first preliminary results. J Psychiatr Res. (2008) 42:689–93. 10.1016/j.jpsychires.2007.08.006
71.
TyrkaARWierLPriceLHRossNAndersonGMWilkinsonCWet al. Childhood parental loss and adult hypothalamic-pituitary-adrenal function. Biol Psychiatry. (2008) 63:1147–54. 10.1016/j.biopsych.2008.01.011
72.
PesonenAKRaikkonenKFeldtKHeinonenKOsmondCPhillipsDIet al. Childhood separation experience predicts HPA axis hormonal responses in late adulthood: a natural experiment of World War II. Psychoneuroendocrinology. (2010) 35:758–67. 10.1016/j.psyneuen.2009.10.017
73.
KumariMHeadJBartleyMStansfeldSKivimakiM. Maternal separation in childhood and diurnal cortisol patterns in mid-life: findings from the Whitehall II study. Psychol Med. (2013) 43:633–43. 10.1017/S0033291712001353
74.
LuSGaoWHuangMLiLXuY. In search of the HPA axis activity in unipolar depression patients with childhood trauma: Combined cortisol awakening response and dexamethasone suppression test. J Psychiatr Res. (2016) 78:24–30. 10.1016/j.jpsychires.2016.03.009
75.
ButlerKKlausKEdwardsLPenningtonK. Elevated cortisol awakening response associated with early life stress and impaired executive function in healthy adult males. Horm Behav. (2017) 95:13–21. 10.1016/j.yhbeh.2017.07.013
76.
AguileraGLiuY. The molecular physiology of CRH neurons. Front Neuroendocrinol. (2012) 33:67–84. 10.1016/j.yfrne.2011.08.002
77.
StetlerCMillerGE. Depression and hypothalamic-pituitary-adrenal activation: a quantitative summary of four decades of research. Psychosom Med. (2011) 73:114–26. 10.1097/PSY.0b013e31820ad12b
78.
CharmandariETsigosCChrousosG. Endocrinology of the stress response. Annu Rev Physiol. (2005) 67:259–84. 10.1146/annurev.physiol.67.040403.120816
79.
PervanidouPChrousosGP. Neuroendocrinology of post-traumatic stress disorder. Prog Brain Res. (2010) 182:149–60. 10.1016/S0079-6123(10)82005-9
80.
CarpenterLLTyrkaARRossNSKhouryLAndersonGMPriceLH. Effect of childhood emotional abuse and age on cortisol responsivity in adulthood. Biol Psychiatry. (2009) 66:69–75. 10.1016/j.biopsych.2009.02.030
81.
CarpenterLLShattuckTTTyrkaARGeraciotiTDPriceLH. Effect of childhood physical abuse on cortisol stress response. Psychopharmacology. (2011) 214:367–75. 10.1007/s00213-010-2007-4
82.
HinkelmannKMuhtzCDettenbornLAgorastosAWingenfeldKSpitzerCet al. Association between childhood trauma and low hair cortisol in depressed patients and healthy control subjects. Biol Psychiatry. (2013) 74:e15–7. 10.1016/j.biopsych.2013.04.021
83.
SuzukiAPoonLPapadopoulosASKumariVCleareAJ. Long term effects of childhood trauma on cortisol stress reactivity in adulthood and relationship to the occurrence of depression. Psychoneuroendocrinology. (2014) 50:289–99. 10.1016/j.psyneuen.2014.09.007
84.
VoellminAWinzelerKHugEWilhelmFHSchaeferVGaabJet al. Blunted endocrine and cardiovascular reactivity in young healthy women reporting a history of childhood adversity. Psychoneuroendocrinology. (2015) 51:58–67. 10.1016/j.psyneuen.2014.09.008
85.
HeimCEhlertUHellhammerDH. The potential role of hypocortisolism in the pathophysiology of stress-related bodily disorders. Psychoneuroendocrinology. (2000) 25:1–35. 10.1016/S0306-4530(99)00035-9
86.
de KloetERJoelsMHolsboerF. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. (2005) 6:463–75. 10.1038/nrn1683
87.
de KloetCSVermettenEBikkerAMeulmanEGeuzeEKavelaarsAet al. Leukocyte glucocorticoid receptor expression and immunoregulation in veterans with and without post-traumatic stress disorder. Mol Psychiatry. (2007) 12:443–53. 10.1038/sj.mp.4001934
88.
YehudaRSecklJ. Minireview: Stress-related psychiatric disorders with low cortisol levels: a metabolic hypothesis. Endocrinology. (2011) 152:4496–503. 10.1210/en.2011-1218
89.
RaisonCLMillerAH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. (2003) 160:1554–65. 10.1176/appi.ajp.160.9.1554
90.
PervanidouP. Biology of post-traumatic stress disorder in childhood and adolescence. J Neuroendocrinol. (2008) 20:632–8. 10.1111/j.1365-2826.2008.01701.x
91.
RohlederNWolfJMWolfOT. Glucocorticoid sensitivity of cognitive and inflammatory processes in depression and posttraumatic stress disorder. Neurosci Biobehav Rev. (2010) 35:104–14. 10.1016/j.neubiorev.2009.12.003
92.
SartorCEGrantJDLynskeyMTMcCutcheonVVWaldronMStathamDJet al. Common heritable contributions to low-risk trauma, high-risk trauma, posttraumatic stress disorder, and major depression. Arch Gen Psychiatry. (2012) 69:293–9. 10.1001/archgenpsychiatry.2011.1385
93.
VaccarinoV. An inflammatory phenotype for posttraumatic stress disorder and depression?Brain Behav Immun. (2018)
94.
KuhlmanKRGeissEGVargasILopez-DuranNL. Differential associations between childhood trauma subtypes and adolescent HPA-axis functioning. Psychoneuroendocrinology. (2015) 54:103–14. 10.1016/j.psyneuen.2015.01.020
95.
SchalinskiIElbertTSteudte-SchmiedgenSKirschbaumC. The cortisol paradox of trauma-related disorders: lower phasic responses but higher tonic levels of cortisol are associated with sexual abuse in childhood. PLoS ONE. (2015) 10:e0136921. 10.1371/journal.pone.0136921
96.
ThompsonJVSullivanRMWilsonDA. Developmental emergence of fear learning corresponds with changes in amygdala synaptic plasticity. Brain Res. (2008) 1200:58–65. 10.1016/j.brainres.2008.01.057
97.
GieddJN. The digital revolution and adolescent brain evolution. J Adolesc Health. (2012) 51:101–5. 10.1016/j.jadohealth.2012.06.002
98.
UematsuAMatsuiMTanakaCTakahashiTNoguchiKSuzukiMet al. Developmental trajectories of amygdala and hippocampus from infancy to early adulthood in healthy individuals. PLoS ONE. (2012) 7:e46970. 10.1371/journal.pone.0046970
99.
QiuAMoriSMillerMI. Diffusion tensor imaging for understanding brain development in early life. Annu Rev Psychol. (2015) 66:853–76. 10.1146/annurev-psych-010814-015340
100.
TallotLDoyereVSullivanRM. Developmental emergence of fear/threat learning: neurobiology, associations and timing. Genes Brain Behav. (2016) 15:144–54. 10.1111/gbb.12261
101.
KuhlmanKRChiangJJHornSBowerJE. Developmental psychoneuroendocrine and psychoneuroimmune pathways from childhood adversity to disease. Neurosci Biobehav Rev. (2017) 80:166–84. 10.1016/j.neubiorev.2017.05.020
102.
SullivanRM. The neurobiology of attachment to nurturing and abusive caregivers. Hastings Law J. (2012) 63:1553–70.
103.
GunnarMR. Quality of early care and buffering of neuroendocrine stress reactions: potential effects on the developing human brain. Prev Med. (1998) 27:208–11. 10.1006/pmed.1998.0276
104.
DaskalakisNPBagotRCParkerKJVinkersCHde KloetER. The three-hit concept of vulnerability and resilience: toward understanding adaptation to early-life adversity outcome. Psychoneuroendocrinology. (2013) 38:1858–73. 10.1016/j.psyneuen.2013.06.008
105.
GunnarMQuevedoK. The neurobiology of stress and development. Annu Rev Psychol. (2007) 58:145–73. 10.1146/annurev.psych.58.110405.085605
106.
GunnarMRWewerkaSFrennKLongJDGriggsC. Developmental changes in hypothalamus-pituitary-adrenal activity over the transition to adolescence: normative changes and associations with puberty. Dev Psychopathol. (2009) 21:69–85. 10.1017/S0954579409000054
107.
DavisEPGrangerDA. Developmental differences in infant salivary alpha-amylase and cortisol responses to stress. Psychoneuroendocrinology. (2009) 34:795–804. 10.1016/j.psyneuen.2009.02.001
108.
HostinarCEGunnarMR. Future directions in the study of social relationships as regulators of the HPA axis across development. J Clin Child Adolesc Psychol. (2013) 42:564–75. 10.1080/15374416.2013.804387
109.
HostinarCESullivanRMGunnarMR. Psychobiological mechanisms underlying the social buffering of the hypothalamic-pituitary-adrenocortical axis: a review of animal models and human studies across development. Psychol Bull. (2014) 140:256–82. 10.1037/a0032671
110.
HostinarCEJohnsonAEGunnarMR. Early social deprivation and the social buffering of cortisol stress responses in late childhood: an experimental study. Dev Psychol. (2015) 51:1597–608. 10.1037/dev0000029
111.
StruberNStruberDRothG. Impact of early adversity on glucocorticoid regulation and later mental disorders. Neurosci Biobehav Rev. (2014) 38:17–37. 10.1016/j.neubiorev.2013.10.015
112.
CohenSJanicki-DevertsDDoyleWJMillerGEFrankERabinBSet al. Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc Natl Acad Sci USA. (2012) 109:5995–9. 10.1073/pnas.1118355109
113.
KuhlmanKRVargasIGeissEGLopez-DuranNL. Age of trauma onset and HPA axis dysregulation among trauma-exposed youth. J Trauma Stress. (2015) 28:572–9. 10.1002/jts.22054
114.
SomervilleLHJonesRMCaseyBJ. A time of change: behavioral and neural correlates of adolescent sensitivity to appetitive and aversive environmental cues. Brain Cogn. (2010) 72:124–33. 10.1016/j.bandc.2009.07.003
115.
GalvanAMcGlennenKM. Enhanced striatal sensitivity to aversive reinforcement in adolescents versus adults. J Cogn Neurosci. (2013) 25:284–96. 10.1162/jocn_a_00326
116.
FuhrmannDKnollLJBlakemoreSJ. Adolescence as a sensitive period of brain development. Trends Cogn Sci. (2015) 19:558–66. 10.1016/j.tics.2015.07.008
117.
TornhageCJ. Reference values for morning salivary cortisol concentrations in healthy school-aged children. J Pediatr Endocrinol Metab. (2002) 15:197–204. 10.1515/JPEM.2002.15.2.197
118.
StroudLRFosterEPapandonatosGDHandwergerKGrangerDAKivlighanKTet al. Stress response and the adolescent transition: performance versus peer rejection stressors. Dev Psychopathol. (2009) 21:47–68. 10.1017/S0954579409000042
119.
SumterSRBokhorstCLMiersACVan PeltJWestenbergPM. Age and puberty differences in stress responses during a public speaking task: do adolescents grow more sensitive to social evaluation?Psychoneuroendocrinology. (2010) 35:1510–6. 10.1016/j.psyneuen.2010.05.004
120.
BlumenthalHLeen-FeldnerEWBadourCLTrainorCDBabsonKA. Pubertal maturation and cortisol level in response to a novel social environment among female adolescents. J Adolesc. (2014) 37:893–900. 10.1016/j.adolescence.2014.06.005
121.
van den BosEde RooijMMiersACBokhorstCLWestenbergPM. Adolescents' increasing stress response to social evaluation: pubertal effects on cortisol and alpha-amylase during public speaking. Child Dev. (2014) 85:220–36. 10.1111/cdev.12118
122.
SolomonMBHermanJP. Sex differences in psychopathology: of gonads, adrenals and mental illness. Physiol Behav. (2009) 97:250–8. 10.1016/j.physbeh.2009.02.033
123.
VaillancourtTDukuEDecatanzaroDMacmillanHMuirCSchmidtLA. Variation in hypothalamic-pituitary-adrenal axis activity among bullied and non-bullied children. Aggress Behav. (2008) 34:294–305. 10.1002/ab.20240
124.
TrickettPKGordisEPeckinsMKSusmanEJ. Stress reactivity in maltreated and comparison male and female young adolescents. Child Maltreat. (2014) 19:27–37. 10.1177/1077559513520466
125.
BoschNMRieseHReijneveldSABakkerMPVerhulstFCOrmelJet al. Timing matters: long term effects of adversities from prenatal period up to adolescence on adolescents' cortisol stress response. The TRAILS study. Psychoneuroendocrinology. (2012) 37:1439–47. 10.1016/j.psyneuen.2012.01.013
126.
ThayerJFLaneRD. A model of neurovisceral integration in emotion regulation and dysregulation. J Affect Disord. (2000) 61:201–16. 10.1016/S0165-0327(00)00338-4
127.
ThayerJFSternbergE. Beyond heart rate variability: vagal regulation of allostatic systems. Ann NY Acad Sci. (2006) 1088:361–72. 10.1196/annals.1366.014
128.
LichtCMVreeburgSAvan Reedt DortlandAKGiltayEJHoogendijkWJDeRijkRHet al. Increased sympathetic and decreased parasympathetic activity rather than changes in hypothalamic-pituitary-adrenal axis activity is associated with metabolic abnormalities. J Clin Endocrinol Metab. (2010) 95:2458–66. 10.1210/jc.2009-2801
129.
LoewyADSpyerKM. Central Regulation of Autonomic Functions. Oxford: Oxford University Press (1990).
130.
DavisAMNatelsonBH. Brain-heart interactions. The neurocardiology of arrhythmia and sudden cardiac death. Tex Heart Inst J. (1993) 20:158–69.
131.
SaperCB. Central autonomic system. In: Paxinos G, editor. The Rat Nervous System. 3rd ed. Amsterdam: Elsevier (2004). p. 761–96. 10.1016/B978-012547638-6/50025-0
132.
ThayerJFLaneRD. Claude Bernard and the heart-brain connection: further elaboration of a model of neurovisceral integration. Neurosci Biobehav Rev. (2009) 33:81–8. 10.1016/j.neubiorev.2008.08.004
133.
StiedlOYounJJansenRF. Cardiovascular conditioning: neural substrates. In: KoobGFLeMoalMThompsonRF editors. Encyclopedia of Behavioral Neuroscience.Amsterdam: Elsevier (2010). p. 226–35. 10.1016/B978-0-08-045396-5.00130-5
134.
Ter HorstGJHautvastRWDe JongsteMJKorfJ. Neuroanatomy of cardiac activity-regulating circuitry: a transneuronal retrograde viral labelling study in the rat. Eur J Neurosci. (1996) 8:2029–41. 10.1111/j.1460-9568.1996.tb00723.x
135.
GormanJMSloanRP. Heart rate variability in depressive and anxiety disorders. Am Heart J. (2000) 140:77–83. 10.1067/mhj.2000.109981
136.
BoscarinoJA. Posttraumatic stress disorder and physical illness: results from clinical and epidemiologic studies. Ann NY Acad Sci. (2004) 1032:141–53. 10.1196/annals.1314.011
137.
BediUSAroraR. Cardiovascular manifestations of posttraumatic stress disorder. J Natl Med Assoc. (2007) 99:642–9.
138.
BoscarinoJA. A prospective study of PTSD and early-age heart disease mortality among Vietnam veterans: implications for surveillance and prevention. Psychosom Med. (2008) 70:668–76. 10.1097/PSY.0b013e31817bccaf
139.
KempAHQuintanaDSGrayMAFelminghamKLBrownKGattJM. Impact of depression and antidepressant treatment on heart rate variability: a review and meta-analysis. Biol Psychiatry. (2010) 67:1067–74. 10.1016/j.biopsych.2009.12.012
140.
KempAHQuintanaDS. The relationship between mental and physical health: insights from the study of heart rate variability. Int J Psychophysiol. (2013) 89:288–96. 10.1016/j.ijpsycho.2013.06.018
141.
CarneyRMFreedlandKEVeithRC. Depression, the autonomic nervous system, and coronary heart disease. Psychosom Med. (2005) 67(Suppl 1.):S29–33. 10.1097/01.psy.0000162254.61556.d5
142.
KamphuisMHGeerlingsMIDekkerJMGiampaoliSNissinenAGrobbeeDEet al. Autonomic dysfunction: a link between depression and cardiovascular mortality? The FINE Study. Eur J Cardiovasc Prev Rehabil. (2007) 14:796–802. 10.1097/HJR.0b013e32829c7d0c
143.
HarrisonNACooperEVoonVMilesKCritchleyHD. Central autonomic network mediates cardiovascular responses to acute inflammation: relevance to increased cardiovascular risk in depression?Brain Behav Immun. (2013) 31:189–96. 10.1016/j.bbi.2013.02.001
144.
OtteCNeylanTCPoleNMetzlerTBestSHenn-HaaseCet al. Association between childhood trauma and catecholamine response to psychological stress in police academy recruits. Biol Psychiatry. (2005) 57:27–32. 10.1016/j.biopsych.2004.10.009
145.
O'HareCMcCroryCO'LearyNO'BrienHKennyRA. Childhood trauma and lifetime syncope burden among older adults. J Psychosom Res. (2017) 97:63–9. 10.1016/j.jpsychores.2017.03.019
146.
HeleniakCMcLaughlinKAOrmelJRieseH. Cardiovascular reactivity as a mechanism linking child trauma to adolescent psychopathology. Biol Psychol. (2016) 120:108–19. 10.1016/j.biopsycho.2016.08.007
147.
AgorastosAKellnerMBakerDGStiedlO. Diminished vagal and/or increased sympathetic activity in post-traumatic stress disorder. In: MartinCPreedyVRPatelVB. The Comprehensive Guide to Post-Traumatic Stress Disorders. BerlinSpringer. (2015). 1–1510.1007/978-3-319-08613-2_30-1
148.
De BellisMDChrousosGPDornLDBurkeLHelmersKKlingMAet al. Hypothalamic-pituitary-adrenal axis dysregulation in sexually abused girls. J Clin Endocrinol Metab. (1994) 78:249–55.
149.
GordisEBGrangerDASusmanEJTrickettPK. Salivary alpha amylase-cortisol asymmetry in maltreated youth. Horm Behav. (2008) 53:96–103. 10.1016/j.yhbeh.2007.09.002
150.
PervanidouPKolaitisGCharitakiSMargeliAFerentinosSBakoulaCet al. Elevated morning serum interleukin (IL)-6 or evening salivary cortisol concentrations predict posttraumatic stress disorder in children and adolescents six months after a motor vehicle accident. Psychoneuroendocrinology. (2007) 32:991–9. 10.1016/j.psyneuen.2007.07.001
151.
PervanidouPKolaitisGCharitakiSLazaropoulouCPapassotiriouIHindmarshPet al. The natural history of neuroendocrine changes in pediatric posttraumatic stress disorder (PTSD) after motor vehicle accidents: progressive divergence of noradrenaline and cortisol concentrations over time. Biol Psychiatry. (2007) 62:1095–102. 10.1016/j.biopsych.2007.02.008
152.
PervanidouPMargeliALazaropoulouCPapassotiriouIChrousosGP. The immediate and long-term impact of physical and/or emotional stress from motor vehicle accidents on circulating stress hormones and adipo-cytokines in children and adolescents. Stress. (2008) 11:438–47. 10.1080/10253890801890622
153.
ChrousosGP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. (1995) 332:1351–62. 10.1056/NEJM199505183322008
154.
GlaserRKiecolt-GlaserJK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol. (2005) 5:243–51. 10.1038/nri1571
155.
SteptoeAHamerMChidaY. The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav Immun. (2007) 21:901–12. 10.1016/j.bbi.2007.03.011
156.
CainDWCidlowskiJA. Immune regulation by glucocorticoids. Nat Rev Immunol. (2017) 17:233–47. 10.1038/nri.2017.1
157.
SternbergEM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol. (2006) 6:318–28. 10.1038/nri1810
158.
PaceTWHuFMillerAH. Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun. (2007) 21:9–19. 10.1016/j.bbi.2006.08.009
159.
PaceTWHeimCM. A short review on the psychoneuroimmunology of posttraumatic stress disorder: from risk factors to medical comorbidities. Brain Behav Immun. (2011) 25:6–13. 10.1016/j.bbi.2010.10.003
160.
MenardCPfauMLHodesGERussoSJ. Immune and neuroendocrine mechanisms of stress vulnerability and resilience. Neuropsychopharmacology. (2017) 42:62–80. 10.1038/npp.2016.90
161.
RohlederNKarlA. Role of endocrine and inflammatory alterations in comorbid somatic diseases of post-traumatic stress disorder. Minerva Endocrinol. (2006) 31:273–88.
162.
GillJMSaliganLWoodsSPageG. PTSD is associated with an excess of inflammatory immune activities. Perspect Psychiatr Care. (2009) 45:262–77. 10.1111/j.1744-6163.2009.00229.x
163.
HogeEABrandstetterKMoshierSPollackMHWongKKSimonNM. Broad spectrum of cytokine abnormalities in panic disorder and posttraumatic stress disorder. Depress Anxiety. (2009) 26:447–55. 10.1002/da.20564
164.
ChidaYSudoNSonodaJHiramotoTKuboC. Early-life psychological stress exacerbates adult mouse asthma via the hypothalamus-pituitary-adrenal axis. Am J Respir Crit Care Med. (2007) 175:316–22. 10.1164/rccm.200607-898OC
165.
DaneseAParianteCMCaspiATaylorAPoultonR. Childhood maltreatment predicts adult inflammation in a life-course study. Proc Natl Acad Sci USA. (2007) 104:1319–24. 10.1073/pnas.0610362104
166.
DaneseAMoffittTEParianteCMAmblerAPoultonRCaspiA. Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch Gen Psychiatry. (2008) 65:409–15. 10.1001/archpsyc.65.4.409
167.
GoshenIKreiselTBen-Menachem-ZidonOLichtTWeidenfeldJBen-HurTet al. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry. (2008) 13:717–28. 10.1038/sj.mp.4002055
168.
PowellNDBaileyMTMaysJWStiner-JonesLMHankeMLPadgettDAet al. Repeated social defeat activates dendritic cells and enhances Toll-like receptor dependent cytokine secretion. Brain Behav Immun. (2009) 23:225–31. 10.1016/j.bbi.2008.09.010
169.
CarpenterLLGawugaCETyrkaARLeeJKAndersonGMPriceLH. Association between plasma IL-6 response to acute stress and early-life adversity in healthy adults. Neuropsychopharmacology. (2010) 35:2617–23. 10.1038/npp.2010.159
170.
RooksCVeledarEGoldbergJBremnerJDVaccarinoV. Early trauma and inflammation: role of familial factors in a study of twins. Psychosom Med. (2012) 74:146–52. 10.1097/PSY.0b013e318240a7d8
171.
WeiLSimenAManeSKaffmanA. Early life stress inhibits expression of a novel innate immune pathway in the developing hippocampus. Neuropsychopharmacology. (2012) 37:567–80. 10.1038/npp.2011.239
172.
DaneseAJ LewisS. Psychoneuroimmunology of early-life stress: the hidden wounds of childhood trauma?Neuropsychopharmacology. (2017) 42:99–114. 10.1038/npp.2016.198
173.
ElenkovIJ. Neurohormonal-cytokine interactions: implications for inflammation, common human diseases and well-being. Neurochem Int. (2008) 52:40–51. 10.1016/j.neuint.2007.06.037
174.
BrowningLMKrebsJDJebbSA. Discrimination ratio analysis of inflammatory markers: implications for the study of inflammation in chronic disease. Metabolism. (2004) 53:899–903. 10.1016/j.metabol.2004.01.013
175.
SmidGEvan ZuidenMGeuzeEKavelaarsAHeijnenCJVermettenE. Cytokine production as a putative biological mechanism underlying stress sensitization in high combat exposed soldiers. Psychoneuroendocrinology. (2015) 51:534–46. 10.1016/j.psyneuen.2014.07.010
176.
NaitohYFukataJTominagaTNakaiYTamaiSMoriKet al. Interleukin-6 stimulates the secretion of adrenocorticotropic hormone in conscious, freely-moving rats. Biochem Biophys Res Commun. (1988) 155:1459–63. 10.1016/S0006-291X(88)81305-6
177.
VankelecomHCarmelietPVan DammeJBilliauADenefC. Production of interleukin-6 by folliculo-stellate cells of the anterior pituitary gland in a histiotypic cell aggregate culture system. Neuroendocrinology. (1989) 49:102–6. 10.1159/000125097
178.
SalasMAEvansSWLevellMJWhicherJT. Interleukin-6 and ACTH act synergistically to stimulate the release of corticosterone from adrenal gland cells. Clin Exp Immunol. (1990) 79:470–3. 10.1111/j.1365-2249.1990.tb08114.x
179.
TominagaTFukataJNaitoYUsuiTMurakamiNFukushimaMet al. Prostaglandin-dependent in vitro stimulation of adrenocortical steroidogenesis by interleukins. Endocrinology. (1991) 128:526–31. 10.1210/endo-128-1-526
180.
MastorakosGChrousosGPWeberJS. Recombinant interleukin-6 activates the hypothalamic-pituitary-adrenal axis in humans. J Clin Endocrinol Metab. (1993) 77:1690–4.
181.
StouthardJMRomijnJAVan der PollTEndertEKleinSBakkerPJet al. Endocrinologic and metabolic effects of interleukin-6 in humans. Am J Physiol. (1995) 268(5 Pt 1), E813–9. 10.1152/ajpendo.1995.268.5.E813
182.
WilderRL. Neuroendocrine-immune system interactions and autoimmunity. Annu Rev Immunol. (1995) 13:307–38. 10.1146/annurev.iy.13.040195.001515
183.
CroffordLJKalogerasKTMastorakosGMagiakouMAWellsJKanikKSet al. Circadian relationships between interleukin (IL)-6 and hypothalamic-pituitary-adrenal axis hormones: failure of IL-6 to cause sustained hypercortisolism in patients with early untreated rheumatoid arthritis. J Clin Endocrinol Metab. (1997) 82:1279–83. 10.1210/jcem.82.4.3852
184.
GirottiMDoneganJJMorilakDA. Influence of hypothalamic IL-6/gp130 receptor signaling on the HPA axis response to chronic stress. Psychoneuroendocrinology. (2013) 38:1158–69. 10.1016/j.psyneuen.2012.11.004
185.
PapanicolaouDATsigosCOldfieldEHChrousosGP. Acute glucocorticoid deficiency is associated with plasma elevations of interleukin-6: does the latter participate in the symptomatology of the steroid withdrawal syndrome and adrenal insufficiency?J Clin Endocrinol Metab. (1996) 81:2303–6. 10.1210/jcem.81.6.8964868
186.
FantidisPPerez De PradaTFernandez-OrtizACarcia-TouchardAAlfonsoFSabateMet al. Morning cortisol production in coronary heart disease patients. Eur J Clin Invest. (2002) 32:304–8. 10.1046/j.1365-2362.2002.00988.x
187.
AlesciSMartinezPEKelkarSIliasIRonsavilleDSListwakSJet al. Major depression is associated with significant diurnal elevations in plasma interleukin-6 levels, a shift of its circadian rhythm, and loss of physiological complexity in its secretion: clinical implications. J Clin Endocrinol Metab. (2005) 90:2522–30. 10.1210/jc.2004-1667
188.
GalboHKallL. Circadian variations in clinical symptoms and concentrations of inflammatory cytokines, melatonin, and cortisol in polymyalgia rheumatica before and during prednisolone treatment: a controlled, observational, clinical experimental study. Arthritis Res Ther. (2016) 18:174. 10.1186/s13075-016-1072-4
189.
van GoolJvan VugtHHelleMAardenLA. The relation among stress, adrenalin, interleukin 6 and acute phase proteins in the rat. Clin Immunol Immunopathol. (1990) 57:200–10. 10.1016/0090-1229(90)90034-N
190.
DeRijkRHBoelenATildersFJBerkenboschF. Induction of plasma interleukin-6 by circulating adrenaline in the rat. Psychoneuroendocrinology. (1994) 19:155–63. 10.1016/0306-4530(94)90005-1
191.
SondergaardSROstrowskiKUllumHPedersenBK. Changes in plasma concentrations of interleukin-6 and interleukin-1 receptor antagonists in response to adrenaline infusion in humans. Eur J Appl Physiol. (2000) 83:95–8. 10.1007/s004210000257
192.
JuddAM. Cytokine expression in the rat adrenal cortex. Horm Metab Res. (1998) 30:404–10. 10.1055/s-2007-978905
193.
EnglerHDoenlenRRietherCEnglerABesedovskyHODel ReyAet al. Chemical destruction of brain noradrenergic neurons affects splenic cytokine production. J Neuroimmunol. (2010) 219:75–80. 10.1016/j.jneuroim.2009.12.001
194.
HankeMLPowellNDStinerLMBaileyMTSheridanJF. Beta adrenergic blockade decreases the immunomodulatory effects of social disruption stress. Brain Behav Immun. (2012) 26:1150–9. 10.1016/j.bbi.2012.07.011
195.
HodesGEMenardCRussoSJ. Integrating Interleukin-6 into depression diagnosis and treatment. Neurobiol Stress. (2016) 4:15–22. 10.1016/j.ynstr.2016.03.003
196.
PaceTWMletzkoTCAlagbeOMusselmanDLNemeroffCBMillerAHet al. Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry. (2006) 163:1630–3. 10.1176/ajp.2006.163.9.1630
197.
Kunz-EbrechtSRMohamed-AliVFeldmanPJKirschbaumCSteptoeA. Cortisol responses to mild psychological stress are inversely associated with proinflammatory cytokines. Brain Behav Immun. (2003) 17:373–83. 10.1016/S0889-1591(03)00029-1
198.
SlopenNKubzanskyLDMcLaughlinKAKoenenKC. Childhood adversity and inflammatory processes in youth: a prospective study. Psychoneuroendocrinology. (2013) 38:188–200. 10.1016/j.psyneuen.2012.05.013
199.
TaylorSELehmanBJKiefeCISeemanTE. Relationship of early life stress and psychological functioning to adult C-reactive protein in the coronary artery risk development in young adults study. Biol Psychiatry. (2006) 60:819–24. 10.1016/j.biopsych.2006.03.016
200.
DaneseACaspiAWilliamsBAmblerASugdenKMikaJet al. Biological embedding of stress through inflammation processes in childhood. Mol Psychiatry. (2011) 16:244–6. 10.1038/mp.2010.5
201.
HepgulNParianteCMDipasqualeSDifortiMTaylorHMarquesTRet al. Childhood maltreatment is associated with increased body mass index and increased C-reactive protein levels in first-episode psychosis patients. Psychol Med. (2012) 42:1893–901. 10.1017/S0033291711002947
202.
MillerGEColeSW. Clustering of depression and inflammation in adolescents previously exposed to childhood adversity. Biol Psychiatry. (2012) 72:34–40. 10.1016/j.biopsych.2012.02.034
203.
BaumeisterDAkhtarRCiufoliniSParianteCMMondelliV. Childhood trauma and adulthood inflammation: a meta-analysis of peripheral C-reactive protein, interleukin-6 and tumour necrosis factor-alpha. Mol Psychiatry. (2016) 21:642–9. 10.1038/mp.2015.67
204.
MatteoliGBoeckxstaensGE. The vagal innervation of the gut and immune homeostasis. Gut. (2013) 62:1214–22. 10.1136/gutjnl-2012-302550
205.
OhiraHMatsunagaMOsumiTFukuyamaSShinodaJYamadaJet al. Vagal nerve activity as a moderator of brain-immune relationships. J Neuroimmunol. (2013) 260:28–36. 10.1016/j.jneuroim.2013.04.011
206.
MichopoulosVPowersAGillespieCFResslerKJJovanovicT. Inflammation in fear- and anxiety-based disorders: PTSD, GAD, and beyond. Neuropsychopharmacology. (2017) 42:254–70. 10.1038/npp.2016.146
207.
DaskalakisNPCohenHNievergeltCMBakerDGBuxbaumJDRussoSJet al. New translational perspectives for blood-based biomarkers of PTSD: From glucocorticoid to immune mediators of stress susceptibility. Exp Neurol. (2016) 284(Pt B):133–40. 10.1016/j.expneurol.2016.07.024
208.
EnglerHBaileyMTEnglerAStiner-JonesLMQuanNSheridanJF. Interleukin-1 receptor type 1-deficient mice fail to develop social stress-associated glucocorticoid resistance in the spleen. Psychoneuroendocrinology. (2008) 33:108–17. 10.1016/j.psyneuen.2007.10.007
209.
AvitsurRPowellNPadgettDASheridanJF. Social interactions, stress, and immunity. Immunol Allergy Clin North Am. (2009) 29:285–93. 10.1016/j.iac.2009.02.006
210.
MillerGECohenSRitcheyAK. Chronic psychological stress and the regulation of pro-inflammatory cytokines: a glucocorticoid-resistance model. Health Psychol. (2002) 21:531–41. 10.1037/0278-6133.21.6.531
211.
LindqvistDJanelidzeSHagellPErhardtSSamuelssonMMinthonLet al. Interleukin-6 is elevated in the cerebrospinal fluid of suicide attempters and related to symptom severity. Biol Psychiatry. (2009) 66:287–92. 10.1016/j.biopsych.2009.01.030
212.
GlattSJTyleeDSChandlerSDPazolJNievergeltCMWoelkCHet al. Blood-based gene-expression predictors of PTSD risk and resilience among deployed marines: a pilot study. Am J Med Genet B Neuropsychiatr Genet. (2013) 162B:313–26. 10.1002/ajmg.b.32167
213.
TursichMNeufeldRWFrewenPAHarricharanSKiblerJLRhindSGet al. Association of trauma exposure with proinflammatory activity: a transdiagnostic meta-analysis. Transl Psychiatry. (2014) 4:e413. 10.1038/tp.2014.56
214.
TyleeDSChandlerSDNievergeltCMLiuXPazolJWoelkCHet al. Blood-based gene-expression biomarkers of post-traumatic stress disorder among deployed marines: a pilot study. Psychoneuroendocrinology. (2015) 51:472–94. 10.1016/j.psyneuen.2014.09.024
215.
GuardadoPOliveraARuschHLRoyMMartinCLejbmanNet al. Altered gene expression of the innate immune, neuroendocrine, and nuclear factor-kappa B (NF-kappaB) systems is associated with posttraumatic stress disorder in military personnel. J Anxiety Disord. (2016) 38:9–20. 10.1016/j.janxdis.2015.12.004
216.
SmithAKConneelyKNKilaruVMercerKBWeissTEBradleyBet al. Differential immune system DNA methylation and cytokine regulation in post-traumatic stress disorder. Am J Med Genet B Neuropsychiatr Genet. (2011) 156B:700–8. 10.1002/ajmg.b.31212
217.
ZhouJNagarkattiPZhongYGinsbergJPSinghNPZhangJet al. Dysregulation in microRNA expression is associated with alterations in immune functions in combat veterans with post-traumatic stress disorder. PLoS ONE. (2014) 9:e94075. 10.1371/journal.pone.0094075
218.
BamMYangXZhouJGinsbergJPLeydenQNagarkattiPSet al. Evidence for epigenetic regulation of pro-inflammatory cytokines, interleukin-12 and interferon gamma, in peripheral blood mononuclear cells from PTSD patients. J Neuroimmune Pharmacol. (2016) 11:168–81. 10.1007/s11481-015-9643-8
219.
CryanJFDinanTG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. (2012) 13:701–12. 10.1038/nrn3346
220.
RookGARaisonCLLowryCA. Microbiota, immunoregulatory old friends and psychiatric disorders. Adv Exp Med Biol. (2014) 817:319–56. 10.1007/978-1-4939-0897-4_15
221.
CarabottiMSciroccoAMaselliMASeveriC. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. (2015) 28:203–9.
222.
FlowersSAEllingrodVL. The microbiome in mental health: potential contribution of gut microbiota in disease and pharmacotherapy management. Pharmacotherapy. (2015) 35:910–6. 10.1002/phar.1640
223.
Malan-MullerSValles-ColomerMRaesJLowryCASeedatSHemmingsSMJ. The gut microbiome and mental health: implications for anxiety- and trauma-related disorders. OMICS. (2018) 22:90–107. 10.1089/omi.2017.0077
224.
DinanTGCryanJF. Regulation of the stress response by the gut microbiota: implications for psychoneuroendocrinology. Psychoneuroendocrinology. (2012) 37:1369–78. 10.1016/j.psyneuen.2012.03.007
225.
SekirovIRussellSLAntunesLCFinlayBB. Gut microbiota in health and disease. Physiol Rev. (2010) 90:859–904. 10.1152/physrev.00045.2009
226.
ClarkeTBDavisKMLysenkoESZhouAYYuYWeiserJN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med. (2010) 16:228–31. 10.1038/nm.2087
227.
ArentsenTQianYGkotzisSFemeniaTWangTUdekwuKet al. The bacterial peptidoglycan-sensing molecule Pglyrp2 modulates brain development and behavior. Mol Psychiatry. (2017) 22:257–66. 10.1038/mp.2016.182
228.
SchaferDPStevensB. Microglia function in central nervous system development and plasticity. Cold Spring Harb Perspect Biol. (2015) 7:a020545. 10.1101/cshperspect.a020545
229.
ReaKDinanTGCryanJF. The microbiome: a key regulator of stress and neuroinflammation. Neurobiol Stress. (2016) 4:23–33. 10.1016/j.ynstr.2016.03.001
230.
GalleyJDBaileyMT. Impact of stressor exposure on the interplay between commensal microbiota and host inflammation. Gut Microbes. (2014) 5:390–6. 10.4161/gmic.28683
231.
HemmingsSMJMalan-MullerSvan den HeuvelLLDemmittBAStanislawskiMASmithDGet al. The microbiome in posttraumatic stress disorder and trauma-exposed controls: an exploratory study. Psychosom Med. (2017) 79:936–46. 10.1097/PSY.0000000000000512
232.
Aoki-YoshidaAAokiRMoriyaNGotoTKubotaYToyodaAet al. Omics studies of the murine intestinal ecosystem exposed to subchronic and mild social defeat stress. J Proteome Res. (2016) 15:3126–38. 10.1021/acs.jproteome.6b00262
233.
BharwaniAMianMFFosterJASuretteMGBienenstockJForsytheP. Structural & functional consequences of chronic psychosocial stress on the microbiome & host. Psychoneuroendocrinology. (2016) 63:217–27. 10.1016/j.psyneuen.2015.10.001
234.
SoderholmJDPerdueMH. Stress and gastrointestinal tract. II Stress and intestinal barrier function. Am J Physiol Gastrointest Liver Physiol. (2001) 280:G7–13. 10.1152/ajpgi.2001.280.1.G7
235.
KellyJRKennedyPJCryanJFDinanTGClarkeGHylandNP. Breaking down the barriers: the gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front Cell Neurosci. (2015) 9:392. 10.3389/fncel.2015.00392
236.
MaesMKuberaMLeunisJCBerkM. Increased IgA and IgM responses against gut commensals in chronic depression: further evidence for increased bacterial translocation or leaky gut. J Affect Disord. (2012) 141:55–62. 10.1016/j.jad.2012.02.023
237.
CardingSVerbekeKVipondDTCorfeBMOwenLJ. Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis. (2015) 26:26191. 10.3402/mehd.v26.26191
238.
DinanTGCryanJF. The impact of gut microbiota on brain and behaviour: implications for psychiatry. Curr Opin Clin Nutr Metab Care. (2015) 18:552–8. 10.1097/MCO.0000000000000221
239.
DinanTGCryanJF. Microbes, immunity, and behavior: psychoneuroimmunology meets the microbiome. Neuropsychopharmacology. (2017) 42:178–92. 10.1038/npp.2016.103
240.
MoussaouiNJacobsJPLaraucheMBiraudMMillionMMayerEet al. Chronic early-life stress in rat pups alters basal corticosterone, intestinal permeability, and fecal microbiota at weaning: influence of sex. J Neurogastroenterol Motil. (2017) 23:135–43. 10.5056/jnm16105
241.
BorreYEO'KeeffeGWClarkeGStantonCDinanTGCryanJF. Microbiota and neurodevelopmental windows: implications for brain disorders. Trends Mol Med. (2014) 20:509–18. 10.1016/j.molmed.2014.05.002
242.
BaileyMTLubachGRCoeCL. Prenatal stress alters bacterial colonization of the gut in infant monkeys. J Pediatr Gastroenterol Nutr. (2004) 38:414–21. 10.1097/00005176-200404000-00009
243.
O'MahonySMMarchesiJRScullyPCodlingCCeolhoAMQuigleyEMet al. Early life stress alters behavior, immunity, and microbiota in rats: implications for irritable bowel syndrome and psychiatric illnesses. Biol Psychiatry. (2009) 65:263–7. 10.1016/j.biopsych.2008.06.026
244.
FarziAFrohlichEEHolzerP. Gut microbiota and the neuroendocrine system. Neurotherapeutics. (2018) 15:5–22. 10.1007/s13311-017-0600-5
245.
Diaz HeijtzRWangSAnuarFQianYBjorkholmBSamuelssonAet al. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci USA. (2011) 108:3047–52. 10.1073/pnas.1010529108
246.
DohertyFDO'MahonySMPetersonVLO'SullivanOCrispieFCotterPDet al. Post-weaning social isolation of rats leads to long-term disruption of the gut microbiota-immune-brain axis. Brain Behav Immun. (2018) 68:261–73. 10.1016/j.bbi.2017.10.024
247.
FosterJARinamanLCryanJF. Stress & the gut-brain axis: regulation by the microbiome. Neurobiol Stress. (2017) 7:124–36. 10.1016/j.ynstr.2017.03.001
248.
PetraAIPanagiotidouSHatziagelakiEStewartJMContiPTheoharidesTC. Gut-microbiota-brain axis and its effect on neuropsychiatric disorders with suspected immune dysregulation. Clin Ther. (2015) 37:984–95. 10.1016/j.clinthera.2015.04.002
249.
LeclercqSForsythePBienenstockJ. Posttraumatic stress disorder: does the gut microbiome hold the key?Can J Psychiatry. (2016) 61:204–13. 10.1177/0706743716635535
250.
SchiavoneSColaiannaMCurtisL. Impact of early life stress on the pathogenesis of mental disorders: relation to brain oxidative stress. Curr Pharm Des. (2015) 21:1404–12. 10.2174/1381612821666150105143358
251.
SahafiEPeeriMHosseiniMJAzarbyjaniMA. Cardiac oxidative stress following maternal separation stress was mitigated following adolescent voluntary exercise in adult male rat. Physiol Behav. (2018) 183:39–45. 10.1016/j.physbeh.2017.10.022
252.
ReusGZFernandesGCde MouraABSilvaRHDarabasACde SouzaTGet al. Early life experience contributes to the developmental programming of depressive-like behaviour, neuroinflammation and oxidative stress. J Psychiatr Res. (2017) 95:196–207. 10.1016/j.jpsychires.2017.08.020
253.
do PradoCHGrassi-OliveiraRWieckAZaparteAFilhoLDda Silva MorroneMet al. The impact of childhood maltreatment on redox state: Relationship with oxidative damage and antioxidant defenses in adolescents with no psychiatric disorder. Neurosci Lett. (2016). 617:173–7. 10.1016/j.neulet.2016.01.062
254.
MarascoVSpencerKARobinsonJHerzykPCostantiniD. Developmental post-natal stress can alter the effects of pre-natal stress on the adult redox balance. Gen Comp Endocrinol. (2013) 191:239–46. 10.1016/j.ygcen.2013.07.003
255.
MillerMWLinAPWolfEJMillerDR. Oxidative Stress, Inflammation, and Neuroprogression in Chronic PTSD. Harv Rev Psychiatry. (2018) 26:57–69. 10.1097/HRP.0000000000000167
256.
ShalevIEntringerSWadhwaPDWolkowitzOMPutermanELinJet al. Stress and telomere biology: a lifespan perspective. Psychoneuroendocrinology. (2013) 38:1835–42. 10.1016/j.psyneuen.2013.03.010
257.
O'DonovanAEpelELinJWolkowitzOCohenBMaguenSet al. Childhood trauma associated with short leukocyte telomere length in posttraumatic stress disorder. Biol Psychiatry. (2011) 70:465–71. 10.1016/j.biopsych.2011.01.035
258.
PriceLHKaoHTBurgersDECarpenterLLTyrkaAR. Telomeres and early-life stress: an overview. Biol Psychiatry. (2013) 73:15–23. 10.1016/j.biopsych.2012.06.025
259.
JergovicMTomicevicMVidovicABendeljaKSavicAVojvodaVet al. Telomere shortening and immune activity in war veterans with posttraumatic stress disorder. Prog Neuropsychopharmacol Biol Psychiatry. (2014) 54:275–83. 10.1016/j.pnpbp.2014.06.010
260.
ZhangLHuXZBenedekDMFullertonCSForstenRDNaifehJAet al. The interaction between stressful life events and leukocyte telomere length is associated with PTSD. Mol Psychiatry. (2014) 19:855–6. 10.1038/mp.2013.141
261.
TyrkaARPriceLHKaoHTPortonBMarsellaSACarpenterLL. Childhood maltreatment and telomere shortening: preliminary support for an effect of early stress on cellular aging. Biol Psychiatry. (2010) 67:531–4. 10.1016/j.biopsych.2009.08.014
262.
ShalevIMoffittTESugdenKWilliamsBHoutsRMDaneseAet al. Exposure to violence during childhood is associated with telomere erosion from 5 to 10 years of age: a longitudinal study. Mol Psychiatry. (2013) 18:576–81. 10.1038/mp.2012.32
263.
ChenSHEpelESMellonSHLinJReusVIRosserRet al. Adverse childhood experiences and leukocyte telomere maintenance in depressed and healthy adults. J Affect Disord. (2014) 169:86–90. 10.1016/j.jad.2014.07.035
264.
MitchellCMcLanahanSSchneperLGarfinkelIBrooks-GunnJNottermanD. Father loss and child telomere length. Pediatrics. (2017) 140:e20163245. 10.1542/peds.2016-3245
265.
HanssenLMSchutteNSMalouffJMEpelES. The relationship between childhood psychosocial stressor level and telomere length: a meta-analysis. Health Psychol Res. (2017) 5:6378. 10.4081/hpr.2017.6378
266.
LiZHeYWangDTangJChenX. Association between childhood trauma and accelerated telomere erosion in adulthood: a meta-analytic study. J Psychiatr Res. (2017) 93:64–71. 10.1016/j.jpsychires.2017.06.002
267.
AlastaloHRaikkonenKPesonenAKOsmondCBarkerDJHeinonenKet al. Cardiovascular morbidity and mortality in Finnish men and women separated temporarily from their parents in childhood–a life course study. Psychosom Med. (2012) 74:583–7. 10.1097/PSY.0b013e31825b3d76
268.
WilsonRSBoylePALevineSRYuLAnagnosSEBuchmanASet al. Emotional neglect in childhood and cerebral infarction in older age. Neurology. (2012) 79:1534–9. 10.1212/WNL.0b013e31826e25bd
269.
LoriaASHoDHPollockJS. A mechanistic look at the effects of adversity early in life on cardiovascular disease risk during adulthood. Acta Physiol. (2014) 210:277–87. 10.1111/apha.12189
270.
McCroryCDooleyCLayteRKennyRA. The lasting legacy of childhood adversity for disease risk in later life. Health Psychol. (2015) 34:687–96. 10.1037/hea0000147
271.
SuSJimenezMPRobertsCTLoucksEB. The role of adverse childhood experiences in cardiovascular disease risk: a review with emphasis on plausible mechanisms. Curr Cardiol Rep. (2015) 17:88. 10.1007/s11886-015-0645-1
272.
MurphyMOCohnDMLoriaAS. (2017). Developmental origins of cardiovascular disease: Impact of early life stress in humans and rodents. Neurosci Biobehav Rev.74(Pt B):453–65. 10.1016/j.neubiorev.2016.07.018
273.
GaradYMaximovaKMacKinnonNMcGrathJJKozyrskyjALColmanI. Sex-specific differences in the association between childhood adversity and cardiovascular disease in adulthood: evidence from a national cohort study. Can J Cardiol. (2017) 33:1013–9. 10.1016/j.cjca.2017.05.008
274.
SugliaSFKoenenKCBoynton-JarrettRChanPSClarkCJDaneseAet al. Childhood and adolescent adversity and cardiometabolic outcomes: a scientific statement from the American heart association. Circulation. (2018) 137:e15–28. 10.1161/CIR.0000000000000536
275.
HoDHBurchMLMusallBMusallJBHyndmanKAPollockJS. Early life stress in male mice induces superoxide production and endothelial dysfunction in adulthood. Am J Physiol Heart Circ Physiol. (2016) 310:H1267–74. 10.1152/ajpheart.00016.2016
276.
LoriaASKangKTPollockDMPollockJS. Early life stress enhances angiotensin II-mediated vasoconstriction by reduced endothelial nitric oxide buffering capacity. Hypertension. (2011) 58:619–26. 10.1161/HYPERTENSIONAHA.110.168674
277.
DallmanMFStrackAMAkanaSFBradburyMJHansonESScribnerKAet al. Feast and famine: critical role of glucocorticoids with insulin in daily energy flow. Front Neuroendocrinol. (1993) 14:303–47. 10.1006/frne.1993.1010
278.
StephensTWBasinskiMBristowPKBue-ValleskeyJMBurgettSGCraftLet al. The role of neuropeptide Y in the antiobesity action of the obese gene product. Nature. (1995) 377:530–2. 10.1038/377530a0
279.
FiglewiczDP. Adiposity signals and food reward: expanding the CNS roles of insulin and leptin. Am J Physiol Regul Integr Comp Physiol. (2003) 284:R882–92. 10.1152/ajpregu.00602.2002
280.
AskariHLiuJDagogo-JackS. Energy adaptation to glucocorticoid-induced hyperleptinemia in human beings. Metabolism. (2005) 54:876–80. 10.1016/j.metabol.2005.01.035
281.
PervanidouPChrousosGP. Post-traumatic stress disorder in children and adolescents: from Sigmund Freud's “trauma” to psychopathology and the (Dys)metabolic syndrome. Horm Metab Res. (2007) 39:413–9. 10.1055/s-2007-981461
282.
PervanidouPChrousosGP. Metabolic consequences of stress during childhood and adolescence. Metabolism. (2012) 61:611–9. 10.1016/j.metabol.2011.10.005
283.
MutluHBilgicVErtenSArasSTayfurM. Evaluation of the relationship between childhood traumas and adulthood obesity development. Ecol Food Nutr. (2016) 55:390–401. 10.1080/03670244.2016.1198791
284.
van Reedt DortlandAKGiltayEJvan VeenTZitmanFGPenninxBW. Metabolic syndrome abnormalities are associated with severity of anxiety and depression and with tricyclic antidepressant use. Acta Psychiatr Scand. (2010) 122:30–9. 10.1111/j.1600-0447.2010.01565.x
285.
van Reedt DortlandAKGiltayEJvan VeenTZitmanFGPenninxBW. Personality traits and childhood trauma as correlates of metabolic risk factors: the Netherlands Study of Depression and Anxiety (NESDA). Prog Neuropsychopharmacol Biol Psychiatry. (2012) 36:85–91. 10.1016/j.pnpbp.2011.10.001
286.
HollingsworthKCallawayLDuhigMMathesonSScottJ. The association between maltreatment in childhood and pre-pregnancy obesity in women attending an antenatal clinic in Australia. PLoS ONE. (2012) 7:e51868. 10.1371/journal.pone.0051868
287.
LiLChassanRABruerEHGowerBASheltonRC. Childhood maltreatment increases the risk for visceral obesity. Obesity. (2015) 23:1625–32. 10.1002/oby.21143
288.
WangYWuBYangHSongX. The effect of childhood abuse on the risk of adult obesity. Ann Clin Psychiatry. (2015) 27:175–84.
289.
SpannSJGillespieCFDavisJSBrownASchwartzAWingoAet al. The association between childhood trauma and lipid levels in an adult low-income, minority population. Gen Hosp Psychiatry. (2014) 36:150–5. 10.1016/j.genhosppsych.2013.10.004
290.
MisiakBKiejnaAFrydeckaD. The history of childhood trauma is associated with lipid disturbances and blood pressure in adult first-episode schizophrenia patients. Gen Hosp Psychiatry. (2015) 37:365–7. 10.1016/j.genhosppsych.2015.03.017
291.
LiLGarveyWTGowerBA. Childhood maltreatment is an independent risk factor for prediabetic disturbances in glucose regulation. Front Endocrinol. (2017) 8:151. 10.3389/fendo.2017.00151
292.
MachadoTDSalumGABosaVLGoldaniMZMeaneyMJAgranonikMet al. Early life trauma is associated with decreased peripheral levels of thyroid-hormone T3 in adolescents. Int J Dev Neurosci. (2015) 47:304–8. 10.1016/j.ijdevneu.2015.10.005
293.
MichopoulosVPowersAMooreCVillarrealSResslerKJBradleyB. The mediating role of emotion dysregulation and depression on the relationship between childhood trauma exposure and emotional eating. Appetite. (2015) 91:129–36. 10.1016/j.appet.2015.03.036
294.
MideiAJMatthewsKABrombergerJT. Childhood abuse is associated with adiposity in midlife women: possible pathways through trait anger and reproductive hormones. Psychosom Med. (2010) 72:215–23. 10.1097/PSY.0b013e3181cb5c24
295.
LeeCTsenkovaVCarrD. Childhood trauma and metabolic syndrome in men and women. Soc Sci Med. (2014) 105:122–30. 10.1016/j.socscimed.2014.01.017
296.
DelpierreCFantinRBarboza-SolisCLepageBDarnauderyMKelly-IrvingM. The early life nutritional environment and early life stress as potential pathways towards the metabolic syndrome in mid-life? A lifecourse analysis using the 1958 British birth cohort. BMC Public Health. (2016) 16:815. 10.1186/s12889-016-3484-0
297.
ManiamJAntoniadisCMorrisMJ. Early-life stress, HPA axis adaptation, and mechanisms contributing to later health outcomes. Front Endocrinol. (2014) 5:73. 10.3389/fendo.2014.00073
298.
SuarezALahtiJKajantieEErikssonJGRaikkonenK. Early life stress, FKBP5 polymorphisms, and quantitative glycemic traits. Psychosom Med. (2017) 79:524–32. 10.1097/PSY.0000000000000439
299.
BernardiJRFerreiraCFSenterGKrolowRde AguiarBWPortellaAKet al. Early life stress interacts with the diet deficiency of omega-3 fatty acids during the life course increasing the metabolic vulnerability in adult rats. PLoS ONE. (2013) 8:e62031. 10.1371/journal.pone.0062031
300.
YamKYNaninckEFGAbbinkMRla FleurSESchipperLvan den BeukelJCet al. Exposure to chronic early-life stress lastingly alters the adipose tissue, the leptin system and changes the vulnerability to western-style diet later in life in mice. Psychoneuroendocrinology. (2017) 77:186–95. 10.1016/j.psyneuen.2016.12.012
301.
MantzorosCSMagkosFBrinkoetterMSienkiewiczEDardenoTAKimSYet al. Leptin in human physiology and pathophysiology. Am J Physiol Endocrinol Metab. (2011) 301:E567–84. 10.1152/ajpendo.00315.2011
302.
LicinioJMantzorosCNegraoABCizzaGWongMLBongiornoPBet al. Human leptin levels are pulsatile and inversely related to pituitary-adrenal function. Nat Med. (1997) 3:575–9. 10.1038/nm0597-575
303.
AhimaRSQiYSinghalNS. Adipokines that link obesity and diabetes to the hypothalamus. Prog Brain Res. (2006) 153:155–74. 10.1016/S0079-6123(06)53009-2
304.
AritaYKiharaSOuchiNTakahashiMMaedaKMiyagawaJet al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. (1999) 257:79–83. 10.1006/bbrc.1999.0255
305.
KyndeIHeitmannBLBygbjergICAndersenLBHelgeJW. Hypoadiponectinemia in overweight children contributes to a negative metabolic risk profile 6 years later. Metabolism. (2009) 58:1817–24. 10.1016/j.metabol.2009.06.014
306.
GauglitzGGHerndonDNKulpGAMeyerWJ 3rdJeschkeMG. Abnormal insulin sensitivity persists up to three years in pediatric patients post-burn. J Clin Endocrinol Metab. (2009) 94:1656–64. 10.1210/jc.2008-1947
307.
SaperCB. The central circadian timing system. Curr Opin Neurobiol. (2013) 23:747–51. 10.1016/j.conb.2013.04.004
308.
DickmeisT. Glucocorticoids and the circadian clock. J Endocrinol. (2009) 200:3–22. 10.1677/JOE-08-0415
309.
GanEHQuintonR. Physiological significance of the rhythmic secretion of hypothalamic and pituitary hormones. Prog Brain Res. (2010) 181:111–26. 10.1016/S0079-6123(08)81007-2
310.
NaderNChrousosGPKinoT. Interactions of the circadian CLOCK system and the HPA axis. Trends Endocrinol Metab. (2010) 21:277–86. 10.1016/j.tem.2009.12.011
311.
QianXDrosteSKLightmanSLReulJMHMLinthorstACE. Circadian and ultradian rhythms of free glucocorticoid hormone are highly synchronized between the blood, the subcutaneous tissue, and the brain. Endocrinology. (2012) 153:4346–53. 10.1210/en.2012-1484
312.
OsterHDamerowSKiesslingSJakubcakovaVAbrahamDTianJet al. The circadian rhythm of glucocorticoids is regulated by a gating mechanism residing in the adrenal cortical clock. Cell Metab. (2006) 4:163–73. 10.1016/j.cmet.2006.07.002
313.
WuYHZhouJNBalesarRUnmehopaUBaoAJockersRet al. Distribution of MT1 melatonin receptor immunoreactivity in the human hypothalamus and pituitary gland: colocalization of MT1 with vasopressin, oxytocin, and corticotropin-releasing hormone. J Comp Neurol. (2006) 499:897–910. 10.1002/cne.21152
314.
ClowAHucklebridgeFStalderTEvansPThornL. The cortisol awakening response: more than a measure of HPA axis function. Neurosci Biobehav Rev. (2010) 35:97–103. 10.1016/j.neubiorev.2009.12.011
315.
ChrousosGPKinoT. Glucocorticoid signaling in the cell. Expanding clinical implications to complex human behavioral and somatic disorders. Ann NY Acad Sci. (2009) 1179:153–66. 10.1111/j.1749-6632.2009.04988.x
316.
CharmandariEChrousosGPLambrouGIPavlakiAKoideHNgSSet al. Peripheral CLOCK regulates target-tissue glucocorticoid receptor transcriptional activity in a circadian fashion in man. PLoS ONE. (2011) 6:e25612. 10.1371/journal.pone.0025612
317.
ValenzuelaFJTorres-FarfanCRichterHGMendezNCampinoCTorrealbaFet al. Clock gene expression in adult primate suprachiasmatic nuclei and adrenal: is the adrenal a peripheral clock responsive to melatonin?Endocrinology. (2008) 149:1454–61. 10.1210/en.2007-1518
318.
Torres-FarfanCMendezNAbarzua-CatalanLVilchesNValenzuelaGJSeron-FerreM. A circadian clock entrained by melatonin is ticking in the rat fetal adrenal. Endocrinology. (2011) 152:1891–900. 10.1210/en.2010-1260
319.
VandewalleGMiddletonBRajaratnamSMStoneBMThorleifsdottirBArendtJet al. Robust circadian rhythm in heart rate and its variability: influence of exogenous melatonin and photoperiod. J Sleep Res. (2007) 16:148–55. 10.1111/j.1365-2869.2007.00581.x
320.
KalsbeekAYiCXla FleurSEBuijsRMFliersE. Suprachiasmatic nucleus and autonomic nervous system influences on awakening from sleep. Int Rev Neurobiol. (2010) 93:91–107. 10.1016/S0074-7742(10)93004-3
321.
PortaluppiFTiseoRSmolenskyMHHermidaRCAyalaDEFabbianF. Circadian rhythms and cardiovascular health. Sleep Med Rev. (2012) 16:151–66. 10.1016/j.smrv.2011.04.003
322.
GerstnerJRLyonsLCWrightKPJrLohDHRawashdehOEckel-MahanKLet al. Cycling behavior and memory formation. J Neurosci. (2009) 29:12824–30. 10.1523/JNEUROSCI.3353-09.2009
323.
GerstnerJRYinJC. Circadian rhythms and memory formation. Nat Rev Neurosci. (2010) 11:577–88. 10.1038/nrn2881
324.
RubyNFHwangCEWessellsCFernandezFZhangPSapolskyRet al. Hippocampal-dependent learning requires a functional circadian system. Proc Natl Acad Sci USA. (2008) 105:15593–8. 10.1073/pnas.0808259105
325.
AlbrechtU. The circadian clock, reward, and memory. Front Mol Neurosci. (2011) 4:41. 10.3389/fnmol.2011.00041
326.
ZisapelN. Sleep and sleep disturbances: biological basis and clinical implications. Cell Mol Life Sci. (2007) 64:1174–86. 10.1007/s00018-007-6529-9
327.
RichardsonGS. The human circadian system in normal and disordered sleep. J Clin Psychiatry. (2005) 66(Suppl 9):3–9; quiz 42–43.
328.
MorrisCJAeschbachDScheerFAJL. Circadian system, sleep and endocrinology. Mol Cell Endocrinol. (2012) 349:91–104. 10.1016/j.mce.2011.09.003
329.
ErrenTCReiterRJ. Defining chronodisruption. J Pineal Res. (2009) 46:245–7. 10.1111/j.1600-079X.2009.00665.x
330.
ErrenTCReiterRJ. Revisiting chronodisruption: when the physiological nexus between internal and external times splits in humans. Naturwissenschaften. (2013) 100:291–8. 10.1007/s00114-013-1026-5
331.
ZelinskiELDeibelSHMcDonaldRJ. The trouble with circadian clock dysfunction: Multiple deleterious effects on the brain and body. Neurosci Biobehav Rev. (2014) 24:80–101. 10.1016/j.neubiorev.2014.01.007
332.
MrdaljJPallesenSMildeAMJellestadFKMurisonRUrsinRet al. Early and later life stress alter brain activity and sleep in rats. PLoS ONE. (2013) 8:e69923. 10.1371/journal.pone.0069923
333.
BaderKSchaferVSchenkelMNissenLSchwanderJ. Adverse childhood experiences associated with sleep in primary insomnia. J Sleep Res. (2007) 16:285–96. 10.1111/j.1365-2869.2007.00608.x
334.
KoskenvuoKHublinCPartinenMPaunioTKoskenvuoM. Childhood adversities and quality of sleep in adulthood: a population-based study of 26,000 Finns. Sleep Med. (2010) 11:17–22. 10.1016/j.sleep.2009.03.010
335.
GreenfieldEALeeCFriedmanELSpringerKW. Childhood abuse as a risk factor for sleep problems in adulthood: evidence from a U.S. national study. Ann Behav Med. (2011) 42:245–56. 10.1007/s12160-011-9285-x
336.
SchaferVBaderK. Relationship between early-life stress load and sleep in psychiatric outpatients: a sleep diary and actigraphy study. Stress Health. (2013) 29:177–89. 10.1002/smi.2438
337.
BaidenPFallonBden DunnenWBoatengGO. The enduring effects of early-childhood adversities and troubled sleep among Canadian adults: a population-based study. Sleep Med. (2015) 16:760–7. 10.1016/j.sleep.2015.02.527
338.
KajeepetaSGelayeBJacksonCLWilliamsMA. Adverse childhood experiences are associated with adult sleep disorders: a systematic review. Sleep Med. (2015) 16:320–30. 10.1016/j.sleep.2014.12.013
339.
LindMJAggenSHKendlerKSYorkTPAmstadterAB. An epidemiologic study of childhood sexual abuse and adult sleep disturbances. Psychol Trauma. (2016) 8:198–205. 10.1037/tra0000080
340.
PetrovMEDavisMCBelyeaMJZautraAJ. Linking childhood abuse and hypertension: sleep disturbance and inflammation as mediators. J Behav Med. (2016) 39:716–26. 10.1007/s10865-016-9742-x
341.
TaylorDJPruiksmaKEHaleWJKellyKMaurerDPetersonALet al. Prevalence, correlates, and predictors of insomnia in the US army prior to deployment. Sleep. (2016) 39:1795–806. 10.5665/sleep.6156
342.
ThordardottirEBHansdottirIValdimarsdottirUAShipherdJCResnickHGudmundsdottirB. The manifestations of sleep disturbances 16 years post-trauma. Sleep. (2016) 39:1551–4. 10.5665/sleep.6018
343.
WangYRaffeldMRSlopenNHaleLDunnEC. Childhood adversity and insomnia in adolescence. Sleep Med. (2016) 21:12–8. 10.1016/j.sleep.2016.01.011
344.
SteineIMWinjeDKrystalJHBjorvatnBMildeAMGronliJet al. Cumulative childhood maltreatment and its dose-response relation with adult symptomatology: findings in a sample of adult survivors of sexual abuse. Child Abuse Negl. (2017) 65:99–111. 10.1016/j.chiabu.2017.01.008
345.
VgontzasANBixlerEOLinHMProloPMastorakosGVela-BuenoAet al. Chronic insomnia is associated with nyctohemeral activation of the hypothalamic-pituitary-adrenal axis: clinical implications. J Clin Endocrinol Metab. (2001) 86:3787–94. 10.1210/jcem.86.8.7778
346.
BackhausJJunghannsKHohagenF. Sleep disturbances are correlated with decreased morning awakening salivary cortisol. Psychoneuroendocrinology. (2004) 29:1184–91. 10.1016/j.psyneuen.2004.01.010
347.
MeerloPSgoifoASucheckiD. Restricted and disrupted sleep: effects on autonomic function, neuroendocrine stress systems and stress responsivity. Sleep Med Rev. (2008) 12:197–210. 10.1016/j.smrv.2007.07.007
348.
MullingtonJMHaackMTothMSerradorJMMeier-EwertHK. Cardiovascular, inflammatory, and metabolic consequences of sleep deprivation. Prog Cardiovasc Dis. (2009) 51:294–302. 10.1016/j.pcad.2008.10.003
349.
RugerMScheerFA. Effects of circadian disruption on the cardiometabolic system. Rev Endocr Metab Disord. (2009) 10:245–60. 10.1007/s11154-009-9122-8
350.
TakiYHashizumeHThyreauBSassaYTakeuchiHWuKet al. Sleep duration during weekdays affects hippocampal gray matter volume in healthy children. Neuroimage. (2012) 60:471–5. 10.1016/j.neuroimage.2011.11.072
351.
KillgoreWD. Self-reported sleep correlates with prefrontal-amygdala functional connectivity and emotional functioning. Sleep. (2013) 36:1597–608. 10.5665/sleep.3106
352.
MotomuraYKitamuraSObaKTerasawaYEnomotoMKatayoseYet al. Sleep debt elicits negative emotional reaction through diminished amygdala-anterior cingulate functional connectivity. PLoS ONE. (2013) 8:e56578. 10.1371/journal.pone.0056578
353.
AckermannKPlompRLaoOMiddletonBRevellVLSkeneDJet al. Effect of sleep deprivation on rhythms of clock gene expression and melatonin in humans. Chronobiol Int. (2013) 30:901–9. 10.3109/07420528.2013.784773
354.
Moller-LevetCSArcherSNBuccaGLaingEESlakAKabiljoRet al. Effects of insufficient sleep on circadian rhythmicity and expression amplitude of the human blood transcriptome. Proc Natl Acad Sci USA. (2013) 110:E1132–41. 10.1073/pnas.1217154110
355.
WeibelLMaccariSVan ReethO. Circadian clock functioning is linked to acute stress reactivity in rats. J Biol Rhythms. (2002) 17:438–46. 10.1177/074873002237138
356.
GogenurIOcakUAltunpinarOMiddletonBSkeneDJRosenbergJ. Disturbances in melatonin, cortisol and core body temperature rhythms after major surgery. World J Surg. (2007) 31:290–8. 10.1007/s00268-006-0256-5
357.
ParedesSDSanchezSParvezHRodriguezABBarrigaC. Altered circadian rhythms of corticosterone, melatonin, and phagocytic activity in response to stress in rats. Neuro Endocrinol Lett. (2007) 28:489–95.
358.
Couto-MoraesRPalermo-NetoJMarkusRP. The immune-pineal axis: stress as a modulator of pineal gland function. Ann NY Acad Sci. (2009) 1153:193–202. 10.1111/j.1749-6632.2008.03978.x
359.
PinaGBrunJTissotSClaustratB. Long-term alteration of daily melatonin, 6-sulfatoxymelatonin, cortisol, and temperature profiles in burn patients: a preliminary report. Chronobiol Int. (2010) 27:378–92. 10.3109/07420520903502234
360.
ChristiansenSBouzinovaEVPalmeRWiborgO. Circadian activity of the hypothalamic-pituitary-adrenal axis is differentially affected in the rat chronic mild stress model of depressionStress. (2012) 15:647–57. 10.3109/10253890.2011.654370
361.
KoreshOKozlovskyNKaplanZZoharJMatarMACohenH. The long-term abnormalities in circadian expression of Period 1 and Period 2 genes in response to stress is normalized by agomelatine administered immediately after exposure. Eur Neuropsychopharmacol. (2012) 22:205–21. 10.1016/j.euroneuro.2011.07.012
362.
TakahashiKYamadaTTsukitaSKanekoKShiraiYMunakataYet al. Chronic mild stress alters circadian expressions of molecular clock genes in the liver. Am J Physiol Endocrinol Metab. (2013) 304:E301–9. 10.1152/ajpendo.00388.2012
363.
WeberGFJohnsonBNYamamotoBKGudelskyGA. Effects of stress and MDMA on hippocampal gene expression. Biomed Res Int. (2014) 2014:141396. 10.1155/2014/141396
364.
LavieP. Sleep disturbances in the wake of traumatic events. N Engl J Med. (2001) 345:1825–32. 10.1056/NEJMra012893
365.
ToumaCFenzlTRuschelJPalmeRHolsboerFKimuraMet al. Rhythmicity in mice selected for extremes in stress reactivity: behavioural, endocrine and sleep changes resembling endophenotypes of major depression. PLoS ONE. (2009) 4:e4325. 10.1371/journal.pone.0004325
366.
PhilbertJPichatPBeeskeSDecobertMBelzungCGriebelG. Acute inescapable stress exposure induces long-term sleep disturbances and avoidance behavior: a mouse model of post-traumatic stress disorder (PTSD). Behav Brain Res. (2011) 221:149–54. 10.1016/j.bbr.2011.02.039
367.
GermainA. Sleep disturbances as the hallmark of PTSD: where are we now?Am J Psychiatry. (2013) 170:372–82. 10.1176/appi.ajp.2012.12040432
368.
MellmanTABustamanteVFinsAIPigeonWRNolanB. REM sleep and the early development of posttraumatic stress disorder. Am J Psychiatry. (2002) 159:1696–701. 10.1176/appi.ajp.159.10.1696
369.
MellmanTAKnorrBRPigeonWRLeiterJCAkayM. Heart rate variability during sleep and the early development of posttraumatic stress disorder. Biol Psychiatry. (2004) 55:953–6. 10.1016/j.biopsych.2003.12.018
370.
MellmanTAHipolitoMM. Sleep disturbances in the aftermath of trauma and posttraumatic stress disorder. CNS Spectr. (2006) 11:611–5. 10.1017/S1092852900013663
371.
YooSSGujarNHuPJoleszFAWalkerMP. The human emotional brain without sleep–a prefrontal amygdala disconnect. Curr Biol. (2007) 17:R877–8. 10.1016/j.cub.2007.08.007
372.
HagewoudRWhitcombSNHeeringaANHavekesRKoolhaasJMMeerloP. A time for learning and a time for sleep: the effect of sleep deprivation on contextual fear conditioning at different times of the day. Sleep. (2010) 33:1315–22. 10.1093/sleep/33.10.1315
373.
MenzMMRihmJSSalariNBornJKalischRPapeHCet al. The role of sleep and sleep deprivation in consolidating fear memories. Neuroimage. (2013) 75:87–96. 10.1016/j.neuroimage.2013.03.001
374.
GermainABuysseDJNofzingerE. Sleep-specific mechanisms underlying posttraumatic stress disorder: integrative review and neurobiological hypotheses. Sleep Med Rev. (2008) 12:185–95. 10.1016/j.smrv.2007.09.003
375.
SpoormakerVIMontgomeryP. Disturbed sleep in post-traumatic stress disorder: secondary symptom or core feature?Sleep Med Rev. (2008) 12:169–84. 10.1016/j.smrv.2007.08.008
376.
AgorastosAKellnerMBakerDGOtteC. When time stands still. An integrative review on the role of chronodisruption in PTSD. Curr Opin Psychiatry. (2014) 27:385–92. 10.1097/YCO.0000000000000079
377.
SkeltonKResslerKJNorrholmSDJovanovicTBradley-DavinoB. PTSD and gene variants: new pathways and new thinking. Neuropharmacology. (2012) 62:628–37. 10.1016/j.neuropharm.2011.02.013
378.
Malan-MullerSSeedatSHemmingsSM. Understanding posttraumatic stress disorder: insights from the methylome. Genes Brain Behav. (2014) 13:52–68. 10.1111/gbb.12102
379.
CaspiAMcClayJMoffittTEMillJMartinJCraigIWet al. Role of genotype in the cycle of violence in maltreated children. Science. (2002) 297:851–4. 10.1126/science.1072290
380.
CaspiASugdenKMoffittTETaylorACraigIWHarringtonHet al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. (2003) 301:386–9. 10.1126/science.1083968
381.
KargKBurmeisterMSheddenKSenS. The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch Gen Psychiatry. (2011) 68:444–54. 10.1001/archgenpsychiatry.2010.189
382.
SteinMBSchorkNJGelernterJ. Gene-by-environment (serotonin transporter and childhood maltreatment) interaction for anxiety sensitivity, an intermediate phenotype for anxiety disorders. Neuropsychopharmacology. (2008) 33:312–9. 10.1038/sj.npp.1301422
383.
GillespieCFPhiferJBradleyBResslerKJ. Risk and resilience: genetic and environmental influences on development of the stress response. Depress Anxiety. (2009) 26:984–92. 10.1002/da.20605
384.
HaugerRLOlivares-ReyesJADautzenbergFMLohrJBBraunSOakleyRH. Molecular and cell signaling targets for PTSD pathophysiology and pharmacotherapy. Neuropharmacology. (2012) 62:705–14. 10.1016/j.neuropharm.2011.11.007
385.
BinderEBBradleyRGLiuWEpsteinMPDeveauTCMercerKBet al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. (2008) 299:1291–305. 10.1001/jama.299.11.1291
386.
KlengelTMehtaDAnackerCRex-HaffnerMPruessnerJCParianteCMet al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci. (2013) 16:33–41. 10.1038/nn.3275
387.
BevilacquaLCarliVSarchiaponeMGeorgeDKGoldmanDRoyAet al. Interaction between FKBP5 and childhood trauma and risk of aggressive behavior. Arch Gen Psychiatry. (2012) 69:62–70. 10.1001/archgenpsychiatry.2011.152
388.
RoyAGorodetskyEYuanQGoldmanDEnochMA. Interaction of FKBP5, a stress-related gene, with childhood trauma increases the risk for attempting suicide. Neuropsychopharmacology. (2010) 35:1674–83. 10.1038/npp.2009.236
389.
ZimmermannPBrucklTNoconAPfisterHBinderEBUhrMet al. Interaction of FKBP5 gene variants and adverse life events in predicting depression onset: results from a 10-year prospective community study. Am J Psychiatry. (2011) 168:1107–16. 10.1176/appi.ajp.2011.10111577
390.
BradleyRGBinderEBEpsteinMPTangYNairHPLiuWet al. Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone receptor gene. Arch Gen Psychiatry. (2008) 65:190–200. 10.1001/archgenpsychiatry.2007.26
391.
HeimCBradleyBMletzkoTCDeveauTCMusselmanDLNemeroffCBet al. Effect of childhood trauma on adult depression and neuroendocrine function: sex-specific moderation by CRH receptor 1 gene. Front Behav Neurosci. (2009) 3:41. 10.3389/neuro.08.041.2009
392.
Ben-EfraimYJWassermanDWassermanJSokolowskiM. Gene-environment interactions between CRHR1 variants and physical assault in suicide attempts. Genes Brain Behav. (2011) 10:663–72. 10.1111/j.1601-183X.2011.00703.x
393.
JaenischRBirdA. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. (2003) 33(Suppl):245–54. 10.1038/ng1089
394.
BjornssonHTFallinMDFeinbergAP. An integrated epigenetic and genetic approach to common human disease. Trends Genet. (2004) 20:350–8. 10.1016/j.tig.2004.06.009
395.
TrollopeAFGutierrez-MecinasMMifsudKRCollinsASaundersonEAReulJMHM. Stress, epigenetic control of gene expression and memory formation. Exp Neurol. (2012) 233:3–11. 10.1016/j.expneurol.2011.03.022
396.
McGowanPO. Epigenomic mechanisms of early adversity and HPA dysfunction: considerations for PTSD research. Front Psychiatry. (2013) 4:110. 10.3389/fpsyt.2013.00110
397.
StankiewiczAMSwiergielAHLisowskiP. Epigenetics of stress adaptations in the brain. Brain Res Bull. (2013) 98:76–92. 10.1016/j.brainresbull.2013.07.003
398.
ReulJMHM. Making memories of stressful events: a journey along epigenetic, gene transcription, and signaling pathways. Front Psychiatry. (2014) 5:5. 10.3389/fpsyt.2014.00005
399.
SchmidtUHolsboerFReinT. Epigenetic aspects of posttraumatic stress disorder. Dis Markers. (2011) 30:77–87. 10.1155/2011/343616
400.
WingoAPAlmliLMStevensJJKlengelTUddinMLiYet al. DICER1 and microRNA regulation in post-traumatic stress disorder with comorbid depression. Nat Commun. (2015) 6:10106. 10.1038/ncomms10106
401.
ZannasASProvencalNBinderEB. Epigenetics of posttraumatic stress disorder: current evidence, challenges, and future directions. Biol Psychiatry. (2015) 78:327–35. 10.1016/j.biopsych.2015.04.003
402.
AlmliLMStevensJSSmithAKKilaruVMengQFloryJet al. A genome-wide identified risk variant for PTSD is a methylation quantitative trait locus and confers decreased cortical activation to fearful faces. Am J Med Genet B Neuropsychiatr Genet. (2015). 168B:327–36. 10.1002/ajmg.b.32315
403.
YehudaRDaskalakisNPDesarnaudFMakotkineILehrnerALKochEet al. Epigenetic biomarkers as predictors and correlates of symptom improvement following psychotherapy in combat veterans with PTSD. Front Psychiatry. (2013) 4:118. 10.3389/fpsyt.2013.00118
404.
YehudaRFloryJDBiererLMHenn-HaaseCLehrnerADesarnaudFet al. Lower methylation of glucocorticoid receptor gene promoter 1F in peripheral blood of veterans with posttraumatic stress disorder. Biol Psychiatry. (2015) 77:356–64. 10.1016/j.biopsych.2014.02.006
405.
McGowanPOSasakiAD'AlessioACDymovSLabonteBSzyfMet al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. (2009) 12:342–8. 10.1038/nn.2270
406.
OberlanderTFWeinbergJPapsdorfMGrunauRMisriSDevlinAM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. (2008) 3:97–106. 10.4161/epi.3.2.6034
407.
RothTLLubinFDFunkAJSweattJD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry. (2009) 65:760–9. 10.1016/j.biopsych.2008.11.028
408.
HoutepenLCVinkersCHCarrillo-RoaTHiemstraMvan LierPAMeeusWet al. Genome-wide DNA methylation levels and altered cortisol stress reactivity following childhood trauma in humans. Nat Commun. (2016) 7:10967. 10.1038/ncomms10967
409.
BickJNaumovaOHunterSBarbotBLeeMLutharSSet al. Childhood adversity and DNA methylation of genes involved in the hypothalamus-pituitary-adrenal axis and immune system: whole-genome and candidate-gene associations. Dev Psychopathol. (2012) 24:1417–25. 10.1017/S0954579412000806
410.
YangBZZhangHGeWWederNDouglas-PalumberiHPerepletchikovaFet al. Child abuse and epigenetic mechanisms of disease risk. Am J Prev Med. (2013) 44:101–7. 10.1016/j.amepre.2012.10.012
411.
EssexMJBoyceWTHertzmanCLamLLArmstrongJMNeumannSMet al. Epigenetic vestiges of early developmental adversity: childhood stress exposure and DNA methylation in adolescence. Child Dev. (2013) 84:58–75. 10.1111/j.1467-8624.2011.01641.x
412.
MehtaDKlengelTConneelyKNSmithAKAltmannAPaceTWet al. Childhood maltreatment is associated with distinct genomic and epigenetic profiles in posttraumatic stress disorder. Proc Natl Acad Sci USA. (2013) 110:8302–7. 10.1073/pnas.1217750110
413.
YehudaRBiererLM. The relevance of epigenetics to PTSD: implications for the DSM-V. J Trauma Stress. (2009) 22:427–34. 10.1002/jts.20448
414.
KlengelTPapeJBinderEBMehtaD. The role of DNA methylation in stress-related psychiatric disorders. Neuropharmacology. (2014). 80:115–32. 10.1016/j.neuropharm.2014.01.013
415.
TaylorSE. Mechanisms linking early life stress to adult health outcomes. Proc Natl Acad Sci USA. (2010) 107:8507–12. 10.1073/pnas.1003890107
416.
DannlowskiUStuhrmannABeutelmannVZwanzgerPLenzenTGrotegerdDet al. Limbic scars: long-term consequences of childhood maltreatment revealed by functional and structural magnetic resonance imaging. Biol Psychiatry. (2012) 71:286–93. 10.1016/j.biopsych.2011.10.021
417.
TeicherMHSamsonJAAndersonCMOhashiK. The effects of childhood maltreatment on brain structure, function and connectivity. Nat Rev Neurosci. (2016) 17:652–66. 10.1038/nrn.2016.111
418.
SapolskyRMUnoHRebertCSFinchCE. Hippocampal damage associated with prolonged glucocorticoid exposure in primates. J Neurosci. (1990) 10:2897–902. 10.1523/JNEUROSCI.10-09-02897.1990
419.
DunlopSAArcherMAQuinlivanJABeazleyLDNewnhamJP. Repeated prenatal corticosteroids delay myelination in the ovine central nervous system. J Matern Fetal Med. (1997) 6:309–13. 10.1002/(SICI)1520-6661(199711/12)6:6<309::AID-MFM1>3.0.CO;2-S
420.
LauderJM. Neurotransmitters as morphogens. Prog Brain Res. (1988) 73:365–87. 10.1016/S0079-6123(08)60516-6
421.
ChenYBaramTZ. Toward understanding how early-life stress reprograms cognitive and emotional brain networks. Neuropsychopharmacology. (2016) 41:197–206. 10.1038/npp.2015.181
422.
EhlertU. Enduring psychobiological effects of childhood adversity. Psychoneuroendocrinology. (2013) 38:1850–7. 10.1016/j.psyneuen.2013.06.007
423.
InsanaSPBanihashemiLHerringaRJKolkoDJGermainA. Childhood maltreatment is associated with altered frontolimbic neurobiological activity during wakefulness in adulthood. Dev Psychopathol. (2016) 28:551–64. 10.1017/S0954579415000589
424.
MajerMNaterUMLinJMCapuronLReevesWC. Association of childhood trauma with cognitive function in healthy adults: a pilot study. BMC Neurol. (2010) 10:61. 10.1186/1471-2377-10-61
425.
HansonJLChungMKAvantsBBRudolphKDShirtcliffEAGeeJCet al. Structural variations in prefrontal cortex mediate the relationship between early childhood stress and spatial working memory. J Neurosci. (2012) 32:7917–25. 10.1523/JNEUROSCI.0307-12.2012
426.
SalehAPotterGGMcQuoidDRBoydBTurnerRMacFallJRet al. Effects of early life stress on depression, cognitive performance and brain morphology. Psychol Med. (2017) 47:171–81. 10.1017/S0033291716002403
427.
DanielsJKLamkeJPGaeblerMWalterHScheelM. White matter integrity and its relationship to PTSD and childhood trauma–a systematic review and meta-analysis. Depress Anxiety. (2013) 30:207–16. 10.1002/da.22044
428.
PaquolaCBennettMRLagopoulosJ. Understanding heterogeneity in grey matter research of adults with childhood maltreatment-A meta-analysis and review. Neurosci Biobehav Rev. (2016) 69:299–312. 10.1016/j.neubiorev.2016.08.011
429.
De BellisMDKeshavanMSClarkDBCaseyBJGieddJNBoringAMet al. A.E. Bennett Research Award. Developmental traumatology. Part II: Brain development. Biol Psychiatry. (1999) 45:1271–84. 10.1016/S0006-3223(99)00045-1
430.
Grassi-OliveiraRAshyMSteinLM. Psychobiology of childhood maltreatment: effects of allostatic load?Rev Bras Psiquiatr. (2008) 30:60–8. 10.1590/S1516-44462008000100012
431.
ThomaesKDorrepaalEDraijerNde RuiterMBvan BalkomAJSmitJHet al. Reduced anterior cingulate and orbitofrontal volumes in child abuse-related complex PTSD. J Clin Psychiatry. (2010) 71:1636–44. 10.4088/JCP.08m04754blu
432.
EdmistonEEWangFMazureCMGuineyJSinhaRMayesLCet al. Corticostriatal-limbic gray matter morphology in adolescents with self-reported exposure to childhood maltreatment. Arch Pediatr Adolesc Med. (2011) 165:1069–77. 10.1001/archpediatrics.2011.565
433.
LimLRaduaJRubiaK. Gray matter abnormalities in childhood maltreatment: a voxel-wise meta-analysis. Am J Psychiatry. (2014) 171:854–63. 10.1176/appi.ajp.2014.13101427
434.
BussoDSMcLaughlinKABrueckSPeverillMGoldALSheridanMA. Child abuse, neural structure, and adolescent psychopathology: a longitudinal study. J Am Acad Child Adolesc Psychiatry. (2017) 56:321–328e321. 10.1016/j.jaac.2017.01.013
435.
TeicherMHAndersonCMOhashiKPolcariA. Childhood maltreatment: altered network centrality of cingulate, precuneus, temporal pole and insula. Biol Psychiatry. (2014) 76:297–305. 10.1016/j.biopsych.2013.09.016
436.
McEwenBSMorrisonJH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. (2013) 79:16–29. 10.1016/j.neuron.2013.06.028
437.
SempleBDBlomgrenKGimlinKFerrieroDMNoble-HaeussleinLJ. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol. (2013) 106–7:1–16. 10.1016/j.pneurobio.2013.04.001
438.
HeimCMMaybergHSMletzkoTNemeroffCBPruessnerJC. Decreased cortical representation of genital somatosensory field after childhood sexual abuse. Am J Psychiatry. (2013) 170:616–23. 10.1176/appi.ajp.2013.12070950
439.
Van DamNTRandoKPotenzaMNTuitKSinhaR. Childhood maltreatment, altered limbic neurobiology, and substance use relapse severity via trauma-specific reductions in limbic gray matter volume. JAMA Psychiatry. (2014) 71:917–25. 10.1001/jamapsychiatry.2014.680
440.
VythilingamMHeimCNewportJMillerAHAndersonEBronenRet al. Childhood trauma associated with smaller hippocampal volume in women with major depression. Am J Psychiatry .(2002) 159:2072–80. 10.1176/appi.ajp.159.12.2072
441.
FrodlTReinholdEKoutsoulerisNReiserMMeisenzahlEM. Interaction of childhood stress with hippocampus and prefrontal cortex volume reduction in major depression. J Psychiatr Res. (2010) 44:799–807. 10.1016/j.jpsychires.2010.01.006
442.
OpelNRedlichRZwanzgerPGrotegerdDAroltVHeindelWet al. Hippocampal atrophy in major depression: a function of childhood maltreatment rather than diagnosis?Neuropsychopharmacology. (2014) 39:2723–31. 10.1038/npp.2014.145
443.
WoonFLHedgesDW. Hippocampal and amygdala volumes in children and adults with childhood maltreatment-related posttraumatic stress disorder: a meta-analysis. Hippocampus. (2008) 18:729–36. 10.1002/hipo.20437
444.
van VelzenLSSchmaalLJansenRMilaneschiYOpmeerEMElzingaBMet al. Effect of childhood maltreatment and brain-derived neurotrophic factor on brain morphology. Soc Cogn Affect Neurosci. (2016) 11:1841–52. 10.1093/scan/nsw086
445.
AustSStaschJJentschkeSAlkan HartwigEKoelschSHeuserIet al. Differential effects of early life stress on hippocampus and amygdala volume as a function of emotional abilities. Hippocampus. (2014) 24:1094–101. 10.1002/hipo.22293
446.
CoplanJDFathyHMJackowskiAPTangCYPereraTDMathewSJet al. Early life stress and macaque amygdala hypertrophy: preliminary evidence for a role for the serotonin transporter gene. Front Behav Neurosci. (2014) 8:342. 10.3389/fnbeh.2014.00342
447.
DannlowskiUKugelHHuberFStuhrmannARedlichRGrotegerdDet al. Childhood maltreatment is associated with an automatic negative emotion processing bias in the amygdala. Hum Brain Mapp. (2013) 34:2899–909. 10.1002/hbm.22112
448.
SwartzJRWilliamsonDEHaririAR. Developmental change in amygdala reactivity during adolescence: effects of family history of depression and stressful life events. Am J Psychiatry. (2015) 172:276–83. 10.1176/appi.ajp.2014.14020195
449.
GrantMMCannistraciCHollonSDGoreJSheltonR. Childhood trauma history differentiates amygdala response to sad faces within MDD. J Psychiatr Res. (2011) 45:886–95. 10.1016/j.jpsychires.2010.12.004
450.
BooijLSzyfMCarballedoAFreyEMMorrisDDymovSet al. DNA methylation of the serotonin transporter gene in peripheral cells and stress-related changes in hippocampal volume: a study in depressed patients and healthy controls. PLoS ONE. (2015) 10:e0119061. 10.1371/journal.pone.0119061
451.
WhiteMGBogdanRFisherPMMunozKEWilliamsonDEHaririAR. FKBP5 and emotional neglect interact to predict individual differences in amygdala reactivity. Genes Brain Behav. (2012) 11:869–78. 10.1111/j.1601-183X.2012.00837.x
452.
HolzNEBuchmannAFBoeckerRBlomeyerDBaumeisterSWolfIet al. Role of FKBP5 in emotion processing: results on amygdala activity, connectivity and volume. Brain Struct Funct. (2015) 220:1355–68. 10.1007/s00429-014-0729-5
453.
GrabeHJWittfeldKVan der AuweraSJanowitzDHegenscheidKHabesMet al. Effect of the interaction between childhood abuse and rs1360780 of the FKBP5 gene on gray matter volume in a general population sample. Hum Brain Mapp. (2016) 37:1602–13. 10.1002/hbm.23123
454.
BogdanRWilliamsonDEHaririAR. Mineralocorticoid receptor Iso/Val (rs5522) genotype moderates the association between previous childhood emotional neglect and amygdala reactivity. Am J Psychiatry. (2012) 169:515–22. 10.1176/appi.ajp.2011.11060855
455.
HymanSE. How adversity gets under the skin. Nat Neurosci. (2009) 12:241–3. 10.1038/nn0309-241
456.
GluckmanPDCutfieldWHofmanPHansonMA. The fetal, neonatal, and infant environments-the long-term consequences for disease risk. Early Hum Dev. (2005) 81:51–9. 10.1016/j.earlhumdev.2004.10.003
457.
GluckmanPDHansonMAMortonSMPinalCS. Life-long echoes–a critical analysis of the developmental origins of adult disease model. Biol Neonate. (2005) 87:127–39. 10.1159/000082311
458.
GluckmanPDHansonMAPinalC. The developmental origins of adult disease. Matern Child Nutr. (2005) 1:130–41. 10.1111/j.1740-8709.2005.00020.x
459.
GluckmanPDHansonMASpencerHG. Predictive adaptive responses and human evolution. Trends Ecol Evol. (2005) 20:527–33. 10.1016/j.tree.2005.08.001
460.
GluckmanPDHansonMASpencerHGBatesonP. Environmental influences during development and their later consequences for health and disease: implications for the interpretation of empirical studies. Proc Biol Sci. (2005) 272:671–7. 10.1098/rspb.2004.3001
461.
BelskyJBeaverKM. Cumulative-genetic plasticity, parenting and adolescent self-regulation. J Child Psychol Psychiatry. (2011) 52:619–26. 10.1111/j.1469-7610.2010.02327.x
462.
NederhofESchmidtMV. Mismatch or cumulative stress: Toward an integrated hypothesis of programming effects. Physiol Behav. (2012) 106:691–700. 10.1016/j.physbeh.2011.12.008
463.
NederhofE. The mismatch hypothesis of psychiatric disease. Physiol Behav. (2012) 106:689–90. 10.1016/j.physbeh.2012.02.014
464.
LevineS. Developmental determinants of sensitivity and resistance to stress. Psychoneuroendocrinology. (2005) 30:939–46. 10.1016/j.psyneuen.2005.03.013
465.
CuijpersPSmitFUngerFStikkelbroekYTen HaveMde GraafR. The disease burden of childhood adversities in adults: a population-based study. Child Abuse Negl. (2011) 35:937–45. 10.1016/j.chiabu.2011.06.005
466.
NelsonECAgrawalAPergadiaMLLynskeyMTTodorovAAWangJCet al. Association of childhood trauma exposure and GABRA2 polymorphisms with risk of posttraumatic stress disorder in adults. Mol Psychiatry. (2009) 14:234–5. 10.1038/mp.2008.81
467.
DinanTGCryanJShanahanFKeelingPWQuigleyEM. IBS: An epigenetic perspective. Nat Rev Gastroenterol Hepatol. (2010) 7:465–71. 10.1038/nrgastro.2010.99
468.
WiersmaJEHovensJGvan OppenPGiltayEJvan SchaikDJBeekmanATet al. The importance of childhood trauma and childhood life events for chronicity of depression in adults. J Clin Psychiatry. (2009) 70:983–9. 10.4088/JCP.08m04521
Summary
Keywords
early life stress, trauma, childhood adversity, stress, stress-related-disorders, neurobiology, gene × environment interaction, epigenetics
Citation
Agorastos A, Pervanidou P, Chrousos GP and Baker DG (2019) Developmental Trajectories of Early Life Stress and Trauma: A Narrative Review on Neurobiological Aspects Beyond Stress System Dysregulation. Front. Psychiatry 10:118. doi: 10.3389/fpsyt.2019.00118
Received
22 March 2018
Accepted
15 February 2019
Published
11 March 2019
Volume
10 - 2019
Edited by
Meichun Mohler-Kuo, University of Applied Sciences and Arts of Western Switzerland, Switzerland
Reviewed by
Erika Comasco, Uppsala University, Sweden; Michael Arthur Van Der Kooij, Johannes Gutenberg University Mainz, Germany
Updates
Copyright
© 2019 Agorastos, Pervanidou, Chrousos and Baker.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agorastos Agorastos aagorast@auth.gr
This article was submitted to Public Mental Health, a section of the journal Frontiers in Psychiatry
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.