Abstract
Thiamine (vitamin B1) is an essential nutrient that serves as a cofactor for a number of enzymes, mostly with mitochondrial localization. Some thiamine-dependent enzymes are involved in energy metabolism and biosynthesis of nucleic acids whereas others are part of the antioxidant machinery. The brain is highly vulnerable to thiamine deficiency due to its heavy reliance on mitochondrial ATP production. This is more evident during rapid growth (i.e., perinatal periods and children) in which thiamine deficiency is commonly associated with either malnutrition or genetic defects. Thiamine deficiency contributes to a number of conditions spanning from mild neurological and psychiatric symptoms (confusion, reduced memory, and sleep disturbances) to severe encephalopathy, ataxia, congestive heart failure, muscle atrophy, and even death. This review discusses the current knowledge on thiamine deficiency and associated morbidity of neurological and psychiatric disorders, with special emphasis on the pediatric population, as well as the putative beneficial effect of thiamine supplementation in autism spectrum disorder (ASD) and other neurological conditions.
Introduction
The essential nutrient thiamine (vitamin B1) is a water-soluble, sulfur-containing vitamin belonging to the vitamin B complex family (Figure 1A). Not being endogenously synthesized, the only available source of thiamine is dietary (beef, poultry, cereals, nuts, and beans). The body does not store thiamine in levels >30 mg, and the half-life for thiamine is only 9–18 days. For an average of ∼2,000 kcal consumed daily, the minimum thiamine requirement is calculated at 0.66 mg (1), although the recommended daily intake for adult men and women is 1.2 and 1.1 mg, respectively (1, 2). During pregnancy or breast-feeding, this requirement increases to 1.4 mg/day (1). In children, the recommended dietary allowance (RDA) is age-dependent and spanning from 0.2 mg (from birth up to 6 months old) to 0.6 mg (from 6 months old to 8 years old) (3). In the human body, thiamine-rich tissues are skeletal muscles, heart, liver, kidney, and brain (4).
Figure 1
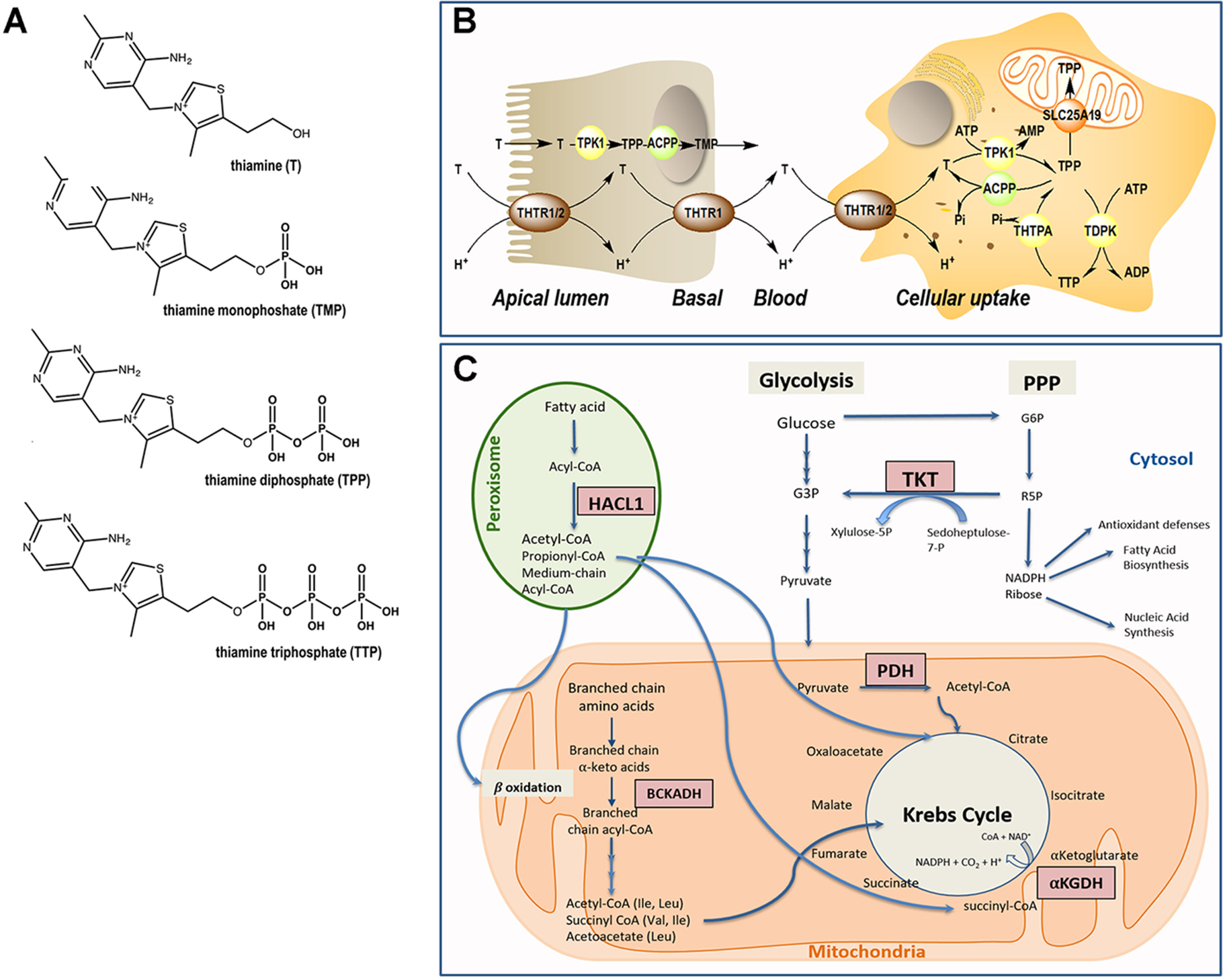
Thiamine metabolism. (A) Chemical structure of thiamine and phosphorylated derivatives. (B) Absorption and cellular uptake of thiamine. Dietary thiamine is mainly in the form of phosphate derivatives and, before absorption, these are converted to free thiamine by intestinal phosphatases. Mammalians utilize THTR1 and THTR2 to transport thiamine. The THTR1 appears to function in the micromolar range, whereas the THTR-2 appears to function in the nanomolar range. Both transporters are expressed in the human small and large intestine, and the expression of THTR1 is higher than that of THTR2, with THTR1 found at both the apical and basolateral membranes of enterocytes (somewhat higher expression at the latter compared to the former domain). Expression of the THTR2 protein was found to be restricted to the apical membrane domain only [see (5) and references therein]. Free thiamine is converted to TPP by the action of TPK1 in the cytosol and dephosphorylation of TPP to TMP and thiamine by ACPP. At higher concentrations of thiamine, simple passive diffusion takes place [see (6) and references therein]. From blood, thiamine is taken up by either transporter (THTR1 or THTR2). Both carriers are widely distributed in the body, but the levels differ in different tissues. Once inside the target cells, thiamine is phosphorylated to TPP by TPK1 and to TTP by TDPK. Both TPP and TTP can be dephosphorylated by ACPP and THTPA, respectively. TPP in the cytosol is utilized as a cofactor for TKT, but it is also required as a cofactor in the mitochondrial compartment; hence, the mitochondrial SLC25A19 exists at the inner mitochondrial membrane. TPP must be bound to HACL1 and HACL2 for its transport to the peroxisomes. Abbreviations: ACPP, prostatic acid phosphatase; ADP, adenosine diphosphate; AMP, adenosine monophosphate; ATP, adenosine triphosphate; T, thiamine; TDPK, thiamine diphosphokinase; THTPA, thiamine triphosphatase; THTR, thiamine transporter; TMP, thiamine monophosphate; TPP, thiamine pyrophosphate. (C) Pathways involving TPP-dependent enzymes. In the cytosol, TPP is the cofactor of transketolase (TKT), an enzyme involved in the pentose phosphate pathway (PPP). The PPP produces nicotinamide adenine dinucleotide phosphate (NADPH) and ribose 5-phosphate (R5P), essential for antioxidant defense and fatty acid synthesis, and precursor in the biosynthesis of nucleotides, respectively. In mitochondria, TPP acts as a cofactor for branched-chain α-ketoacid dehydrogenase (BCKDH) complex, pyruvate dehydrogenase (PDH) complex, and α-ketoglutarate dehydrogenase. In peroxisomes, TPP is a cofactor for 2-hydroxyacyl-CoA lyase 1 (HACL1) as part of the fatty acid α-oxidation pathway.
Despite the availability of dietary thiamine in wealthy countries, thiamine deficiency represents an important and usually overlooked issue. In developed countries, the predominant use of industrial food processing often depletes thiamine content along with other vitamins and nutrients. An increased consumption of processed food in the form of simple carbohydrates, not supplemented with adequate levels of thiamine, has been named “high calorie malnutrition” (7, 8). Thus, despite the caloric density, the diet is often of poor nutrition quality and does not meet recommended dietary guidelines for micronutrient intake, making this an at-risk population for micronutrient malnutrition (8). For instance, at least 29% of obese subjects that will undergo bariatric surgery have been reported as thiamine deficient (9). This condition highlights the fine balance between adequate caloric intake and balanced nutritional diet. As thiamine is a key factor in the metabolism of glucose, an increased carbohydrate intake will proportionally increase thiamine’s dietary demand (a minimum of 0.33 mg per 1,000 kcal) (1). Thus, rather than focusing on thiamine’s RDA, it is critical to match its intake with carbohydrate consumption as well as total caloric intake.
In developing countries, thiamine deficiency remains a widespread concern due to high rates of white rice consumption (3). As home-pounding techniques are replaced with industrial rice milling and processing, essential nutrients (such as thiamine) within the bran are stripped away (10). Asian countries consume about 90% of the rice produced worldwide, fulfilling an estimated 60% of the population’s daily dietary energy intake requirement, and consequently, thiamine deficiency has become prevalent in the 15% of the adolescent population (using the most conservative approximation) (11). Thiamine deficiency may develop by ingesting diets either contaminated with thiamine-metabolizing enzymes (e.g., thiaminase) (12) or that underwent thiamine inactivation by heat and/or sulfur dioxide (13). Heavy consumption of tannin-containing or food rich in caffeine, theobromine, and theophylline (such as those present in coffee, chocolate, and tea, respectively) can inactivate thiamine, thereby compromising the thiamine status (7, 14, 15).
Other risk factors that increase the likelihood of insufficient thiamine intake include aging, economic status, eating disorders, medical conditions affecting the gastrointestinal tract, subjects receiving parental nutrition, bariatric surgery, diabetes, and alcohol abuse (9, 16–23). Unmet needs for increasing the nutritional intake of thiamine are reported during lactation, pregnancy, and increased physical activity (11, 24). During lactation, infants have increased risks of developing beriberi from newly deficient but asymptomatic mothers (11). For instance, 27% of women of childbearing age are considered thiamine deficient in Cambodia, with 38% of infants being diagnosed as severely deficient in thiamine, a critical issue that contributes significantly to the mortality of 3-month-old babies (11).
However, even in the presence of an adequate thiamine intake, its deficiency can result from genetic factors, i.e., pathogenic gene mutations in key regulators of the thiamine pathway, including thiamine pyrophosphokinase 1 (TPK1), thiamine diphosphate kinase (TDPK), thiamine triphosphatase (THTPA), and thiamine transporters (SLC25A19, SLC19A2/THTR1, and SLC19A3/THTR2; Figure 1B). More recently, the organic cation transporter 1 (OCT1) has been claimed to act as a hepatic thiamine transporter (25).
Regardless of the underlying cause, thiamine deficits may have severe detrimental effects, with most of the symptoms manifesting at the neurological level (24, 26). Thiamine deficiency might cause brain tissue injury by inhibiting brain energy utilization given the critical role of thiamine-dependent enzymes associated within glucose utilization (27). This is supported by the significant rate of thiamine uptake by the blood–brain barrier emphasizing the high brain demand for thiamine and the need for its supply to sustain adequate brain functions (28, 29), especially in those brain areas with both high metabolic demands and high thiamine turnover (30–32).
This review will discuss the physiological and biochemical bases of thiamine metabolism and explore the pathophysiological implications of thiamine deficiency. In particular, we will highlight the effect of thiamine supplementation as a therapeutic strategy in the management of neurological disorders, with special emphasis on the pediatric population.
Biochemical Basis of Thiamine’s Cellular Role
Dietary Availability, Absorption, and Cellular Uptake
As with most hydrophilic micronutrients, thiamine absorption occurs mainly in the jejunum (33). Throughout the digestive tract, dietary proteins get hydrolyzed, releasing thiamine. In the intestinal lumen, alkaline phosphatases catalyze the hydrolysis of thiamine-phosphorylated derivatives into free thiamine (34). Unphosphorylated, free thiamine at concentrations higher than 1 µM enters the enterocyte by passive diffusion, whereas at lower levels, it is transported via the saturable thiamine/H+ antiport system (thiamine transporter 1 or THTR1) through an energy-dependent process (33). Under conditions of thiamine deficiency, an upregulation of the expression of the thiamine transporter 2 (THTR2) was observed in Caco2 cells in culture, suggesting that diet can modulate the expression of this transporter (35). Within the enterocyte, thiamine is phosphorylated to thiamine pyrophosphate (TPP) by TPK1. Then, most TPP is dephosphorylated to thiamine monophosphate (TMP) to cross the basal membrane of the enterocyte. The TMP is released into the bloodstream through an ATPase-dependent transport system (36) (Figure 1B). Free thiamine can also reach the bloodstream via the thiamine transporter 2 (THTR2) located mainly at the basolateral membrane of the enterocyte. Once in blood, while very low levels of TMP and thiamine circulate free in plasma or serum, more than 90% of the phosphorylated thiamine (in the form of TPP) is present in erythrocytes and leukocytes (37). Notably, the isoform 3 of the carrier SLC44A4 has recently been described as a TPP carrier in the colon. Originally, SLC44A4 was described as a choline transporter linked to the non-neuronal synthesis of choline (38) and required to the efferent innervation of hair cells in the olivocochlear bundle for the maintenance of physiological function of outer hair cells and the protection of hair cells from acoustic injury (39). Recent evidence indicates that this carrier may mediate the absorption of microbiota-generated TPP (especially in infants) and contribute to host thiamine homeostasis (40).
The cellular uptake of thiamine from the bloodstream can be mediated by any of the two high-affinity carriers: THTR1 [encoded by SLC19A2 (41)] and THTR2 [encoded by SLC19A3 (42, 43)]. These transporters are ubiquitously expressed (42–44), but THTR1 is most abundant in the intestine, skeletal muscle, nervous system, and eye followed by the placenta, liver, and kidney, whereas THTR2 is located mostly in adipose tissue, breast tissue, liver, lymphocytes, spleen, gallbladder, placenta, pancreas, and brain (information collected from GeneCards). Once transported intracellularly, free thiamine is rapidly phosphorylated to TPP by thiamine pyrophosphokinase (TPK1). A second kinase, TDPK, adds a phosphate group to TPP to generate thiamine triphosphate (TTP). TPP and TTP can be dephosphorylated to, respectively, TMP and TPP by phosphatases [Prostatic Acid Phosphatase (ACPP) and THTPA, respectively; Figure 1B].
Biochemical Roles of Phosphorylated Forms of Thiamine
Up to 90% of the total thiamine in the body remains in its diphosphate, metabolically active form (TPP), whereas the rest is found as TMP and TTP (45). TPP is a cofactor of several thiamine-dependent enzymes involved in carbohydrate and fatty acid metabolism, namely, cytosolic transketolase (TKT), peroxisomal 2-hydroxyacyl-CoA lyase 1, and three mitochondrial enzymes (pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and branched-chain α-ketoacid dehydrogenase complexes; Figure 1C). As the biochemical role of TPP is well understood, the biological significance and contribution of TTP is not entirely clear. It was previously considered to be a specific neuroactive form of thiamine, but more recently, it has been reported that TTP (which is ∼10% of the total brain thiamine pool) is involved in membrane excitability and nerve conduction by acting as a modulator of the permeability of sodium chloride channels (29, 32, 46).
Intracellular Location of TPP-Dependent Enzymes
Cytosol
In the cytosol, TPP acts as a cofactor for TKT, a key enzyme of the non-oxidative branch of the pentose phosphate pathway (PPP). This metabolic pathway generates nicotinamide adenine dinucleotide phosphate (NADPH) and ribose 5-phosphate (R5P) (47). NADPH is a key reducing agent in biosynthetic reactions and is a co-substrate of biosynthetic enzymes (fatty acid synthesis) and antioxidant enzymes such as the glutathione peroxidase–reductase system and thioredoxin peroxidases, among others. The crucial involvement of R5P in the biosynthesis of DNA and RNA highlights the critical role of thiamine in high-proliferating tissues.
Based on its role in the abovementioned biochemical pathways, it is expected that thiamine deficiency will result in increased oxidative stress and lower cell proliferation, as well as decreased fatty acid synthesis (including myelin) with severe consequences especially during brain development. Consistent with this premise, thiamine deficiency decreases TKT activity and leads to PPP impairment and reduced neurogenesis in murine cortex and hippocampus during neurodevelopment (48).
Peroxisomes
Peroxisomes play an important role in the catabolism of hydrogen peroxide, as well as in the shortening of very long fatty acids (which cannot undergo a direct mitochondrial β-oxidation catabolism) and α-oxidation (49). In the latter process, the TPP-dependent enzyme 2-hydroxyacyl-CoA lyase 1 (HACL1) catalyzes the cleavage of 3-methyl-branched and straight chain 2-hydroxy long-chain fatty acids (50). Phytanic acid (a 3-methyl-substituted, 20-carbon branched-chain fatty acid), unlike most fatty acids, is unable to undergo β-oxidation because of an existing methyl group in the 3-position (51). As such, it is broken down by HACL1 by an initial α-oxidation (52, 53). This branched-chain fatty acid is obtained through the diet, specifically from dairy products and red meat. The disruption of phytanic acid catabolism, due to inadequate levels of TPP, leads to triglyceride accumulation, which may cause deleterious effects such as cerebellar ataxia, peripheral polyneuropathy, vision and hearing loss, anosmia, and, in some instances, cardiac dysfunction and epiphyseal dysplasia (54). The symptoms caused by thiamine deficiency are shared by Refsum’s disease, which is caused by pathogenic mutations in HACL1 (55). Some of the symptoms are also observed in the autosomal recessive systemic disorder Zellweger syndrome and other peroxisomal-related diseases including the neonatal adrenoleukodystrophy. Zellweger syndrome is caused by pathogenic mutations in the pexin genes, which encode for proteins essential for the assembly of functional peroxisomes. It is characterized by deficits in the peroxisomal fatty acid oxidation pathway causing severe neurological and liver dysfunction as well as craniofacial abnormalities.
Mitochondria
Most (∼90%) of the cytosolic TPP is transported into mitochondria via the mitochondrial thiamine pyrophosphate transporter [MTPPT, product of the SLC25A19 gene (56)]. This transporter mediates the exchange of cytosolic TPP for the mitochondrial TMP; once in the cytosol, TMP is metabolized and converted back to TPP (56). In mitochondria, TPP is a critical cofactor for three enzymes, namely, pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and branched-chain α-ketoacid dehydrogenase (PDH, αKGDH, and BCKDH, respectively).
Studies in SLC25A19 knockout mice and in carriers of the SLC25A19 mutation, responsible for the Amish lethal microcephaly (MCPHA, see below) show, respectively, virtually undetectable and markedly reduced mitochondrial TPP levels in mouse embryonic fibroblasts and human lymphoblasts (56), which were completely restored upon addition of TPP to the PDH and KGDH complex assay mixtures (56).
Pyruvate dehydrogenase complex
This multisubunit complex catalyzes the TPP-dependent decarboxylation of pyruvate, generating acetyl-CoA, which then enters the Krebs cycle. The regulation of PDH activity constitutes a key metabolic switch influencing fuel choice, i.e., between fatty acid oxidation and glycolytic flux (57). The inability to adjust the choice of fuel for metabolic energy production has been proposed to underlie the “metabolic inflexibility” leading to the morbidity of metabolic disorders (58). Hence, thiamine-deficiency-mediated inhibition of the PDH complex will lock the system into the oxidation of glucose to pyruvate, leading to increases in lactate and decreases in cellular ATP production (59). As expected, in severe cases, the metabolic deficit presents as a fatal lactic acidosis in newborns, whereas in milder cases, neurological conditions may lead to structural abnormalities in the central nervous system (CNS), seizures, mental disability, and spasticity (60, 61).
α-Ketoglutarate dehydrogenase complex
This TPP-dependent enzyme catalyzes the formation of succinyl-CoA from α-ketoglutarate in the Krebs cycle and the consequent production of reduced nicotinamide adenine dinucleotide (NADH). Low TPP levels notably decrease αKGDH activity, thereby decreasing energy production and causing glutamate to accumulate (62), hampering oxidative metabolism and leading to neurodegeneration (63).
Branched-chain α-keto dehydrogenase complex
The TPP-dependent BCKDH enzyme, localized at the inner mitochondrial membrane, catalyzes one of the steps of the essential branched-chain amino acid (BCAAs; leucine, isoleucine, and valine) catabolic pathway. These amino acids are required for protein synthesis, and some of their catabolic products can be used for energy production via the citric acid cycle. In addition, their carbon skeletons (except Leu) can be used as precursors for gluconeogenesis (64). In a thiamine deficiency experimental model, medial thalamus was the brain region that shows the lowest levels of BCKDH with fivefold accumulation of Leu. This study suggested that the lower activity of this enzyme may be responsible for thalamic neuronal cell death observed in human cases of thiamine deficiency (65).
Similarly, a homozygous mutation in the BCKDH E1 α subunit results in the so-called maple syrup urine disease (MSUD, due to the distinct maple syrup smell to the urine as a result of keto acid buildup from unprocessed BCAA), characterized by physical and mental disabilities. A mild form of MSUD, called thiamine-responsive MSUD, is currently the only recognized variant that can be successfully treated with thiamine supplementation (OMIM 248600).
Hierarchical Order of Thiamine-Dependent Enzymatic Deficiencies: Effect of Species, Tissue, and Subcellular Compartment
While it could be assumed that a deficiency in a given micronutrient affects its dependent enzymes equally, experimental evidence indicates otherwise. For instance, copper deficiency severely reduces cytochrome c oxidase activity affecting copper/zinc-superoxide dismutase to a lesser extent (66). Moreover, this hierarchical order is also tissue-specific as different brain areas (66). Similarly, in the case of thiamine deficiency, the most affected brain regions seem to be cerebellum, mammillary bodies, thalamus, hypothalamus, and brainstem in adults (1, 67). In children, MRI studies showed primary involvement of mammillary bodies as well as basal ganglia (68, 69) [preferentially the striatum and, to a lesser extent, the globus pallidus (68)], in addition to frontal lobes (69).
With regard to thiamine deficiency, Zhao et al. showed that, in mice, thiamine deprivation for 14 days led to different degrees of enzymatic deficiencies when testing for TKT, PDH, and αKGDH activities in the cortex and hippocampus (48). TKT activity was significantly more reduced in both brain regions compared to both mitochondrial enzymes PDH and αKGDH (48). Two studies reporting on a genetic mutation in SLC19A3.1 resulted in thiamine-deficient Alaskan huskies (26, 70). A greater reduction in TKT versus PDH activity was observed in the cortex. In the thalamus, however, PDH activity was affected twice as much as TKT activity (26). These studies support the concept of tissue-specific deficits of thiamine deficiency and also point to the species-specific differences that should be taken into account when making extrapolations to human studies.
In cases of severe thiamine deficiency, however, a broad enzymatic deficiency encompassing all three mitochondrial TPP-dependent enzymes would be expected to result in symptoms similar to those of the genetic disorder dihydrolipoamide dehydrogenase (DLDD) deficiency (OMIM 246900). This pathogenic mutation affects the E3 subunit, common to all three TPP-dependent dehydrogenases. Patients clinically present with lactic acidosis and neurodegeneration from inefficient oxidative metabolism, as well as increased BCAA concentrations and α-keto acids in the urine, with many of the same symptoms as MSUD, but with a more severe phenotype (71).
While BCKDH and HACL1 are (as indicated above) TPP-dependent enzymes, their activity is rarely reported in the context of thiamine deficiency, with the majority of studies focusing on TKT and PDH or αKGDH as indicators of oxidative stress (TKT) and mitochondrial dysfunction (PDH or αKGDH). However, a study by Navarro et al. noted significantly reduced BCKDH activity in the medial thalamus (compared to frontal cortex) accompanied by a fivefold increase in Leu levels following administration of a thiamine antagonist (65).
The consequences of thiamine deficiency on HACL1, however, remain mostly obscure. Recent work in HACL1-deficient mice revealed the presence of an additional TPP-dependent enzyme with lyase activity, localized in the endoplasmic reticulum, able to cleave 2-hydroxyphytanoyl-CoA (72). This process is not exactly like the one catalyzed by the peroxisomal HACL1 and involves a hydroxylation at position C2 carried out by the fatty acid 2-hydroxylase (FA2H), followed by the cleavage catalyzed by HACL1 (73). More recently, an orphan enzyme (HACL2) located in the endoplasmic reticulum known as bacterial acetolactate synthase-like was reported to cleave straight chain 2-hydroxyacyl-CoAs (74), and it is likely to be identical to the HACL1-like enzyme. A strong inverse association of C17:0 with diabetes and cardiovascular risk has been observed, suggesting a metabolic link between α-oxidation and these diseases (75, 76). More research is needed to elucidate the role of HADCL1 and HADCL2 in the context of thiamine deficiency.
Defects in Genes Associated With Thiamine Metabolism
Pathogenic mutations in genes encoding for enzymes and transporters involved in thiamine metabolism result in symptoms similar to those found in nutrition-based thiamine deficiency and overlaps with disorders of mitochondrial dysfunction (Table 1). Those mutations affecting genes responsible for thiamine transporters 1 (SLC19A2; OMIM 249270) and 2 (SLC19A3; OMIM 607483) constitute the main cause of suboptimal intestinal thiamine absorption and, as a result, insufficient cellular distribution of thiamine through the body.
Table 1
| Name of disease | Mutation | Protein | Age at onset | Clinical symptoms | Management (dose) (reference) |
|---|---|---|---|---|---|
| TRMA/Rogers syndrome | SLC19A2 | THTR1 | Birth to adolescence | Megaloblastic anemia, diabetes mellitus, sensorineural deafness, optic atrophy, congenital heart defects, short stature | Thiamine (50–100 mg/day) (77) |
| Biotin–thiamine-responsive basal ganglia disease | SLC19A3 | THTR2 | Birth to adolescence | Episodic encephalopathy associated with febrile illness, seizures, external ophthalmoplegia, dysphagia, gait ataxia, bilateral lesions of the basal ganglia | – Biotin (5–10 mg/kg/day), thiamine (300–900 mg) (78) – Biotin (5–10 mg/kg/day), thiamine (100–200 mg) (79) |
| Amish lethal microcephaly/THMD3 | SLC25A19 | Mitochondrial TPP carrier | Birth | Episodic encephalopathy associated with lactic acidosis and alpha-ketoglutaric aciduria, microcephaly, delayed psychomotor development, seizures, increased urinary lactate | – Phenobarbital (for seizures) and physical therapy (80) – High-fat diet (81) |
| Thiamine metabolism dysfunction syndrome 4/THMD4 (progressive polyneuropathy type) | SLC25A19 | MTPC | Adolescence | Episodic encephalopathy associated with febrile illness, transient neurologic dysfunction, residual weakness, progressive axonal polyneuropathy, bilateral striatal degeneration | High-dose thiamine administered at the time of the study. Outcome not available (82) |
| Thiamine metabolism dysfunction syndrome 5 (episodic encephalopathy type) THMD5 | TPK1 | Thiamine phosphokinase 1 | Early childhood | Episodic encephalopathy (Leigh-like) associated with high serum and CSF lactate with progressive neurologic and motor dysfunctions (gait disturbances, ataxia, dystonia, and spasticity, which, in some cases, may result in loss of ability to walk) triggered by infections. Cognitive function usually preserved; some developmental delay. Some patients may recover from some neurologic deficits; in others, the outcome is fatal. | Oral thiamine (100–200; 500 mg/day) (83, 84) |
Genetic mutations affecting thiamine metabolism.
Carriers of homozygous or compound heterozygous loss-of-function or missense mutations in SLC19A2 develop a rare condition known as thiamine-responsive megaloblastic anemia (TRMA) or Rogers syndrome (OMIM 249270). Most cases are currently found in isolated populations with consanguineous partners (85). This early-onset, autosomal recessive disorder is characterized by megaloblastic anemia, macrocytosis, diabetes mellitus, and sensorineural deafness. Other, more variable features include optic atrophy, congenital heart defects, short stature, and significant neurological deficits such as stroke and epilepsy during infancy (77, 86). The term “thiamine-responsive” refers to the observation of clinical, symptomatic improvement after administration of high doses of thiamine (86, 87).
The biotin–thiamine-responsive basal ganglia disease (BTBGD; OMIM: 607483) is a neurometabolic autosomal recessive disorder caused by a mutation in the SLC19A3 gene, characterized by subacute encephalopathy with confusion, convulsions, muscle rigidity, ataxia, dysarthria, and dystonia, which can be fatal if left untreated. However, the disorder is completely reversible within by early treatment with biotin (5–10 mg/kg/day) in combination with thiamine [from 100–200 mg (79) to 300–900 mg (78); Table 1], underscoring the critical nature of a timely diagnosis. Signs and symptoms usually appear during childhood (between the ages of 3 and 10 years), although onset of the disease can vary between newborn period and adulthood (78, 88).
The Amish lethal microcephaly (MCPHA; OMIM 607196), also known as thiamine metabolism dysfunction syndrome-3 (THMD3; Table 1) (56), is caused by homozygous mutation in the SLC25A19 gene. Amish type microcephaly is a severe autosomal recessive metabolic disorder characterized by severe microcephaly apparent at birth, profoundly delayed psychomotor development, brain malformations, and episodic encephalopathy associated with lactic acidosis and α-ketoglutaric aciduria (89). The clinical management is achieved by administering a high-fat diet (81), which sustains the production of ATP by mitochondria primarily through fatty acid β-oxidation, bypassing PDH to directly enter the Krebs cycle. Importantly, the metabolic similarity of MCPHA to PDH deficiency (OMIM 312070) indicates the relevance of administering a low-carbohydrate diet to patients with MCPHA to avoid a risk of exacerbating the lactic acidemia.
A less severe variant of the disease, thiamine metabolism dysfunction syndrome 4 (THMD4; OMIM 613710), is similarly caused by a homozygous mutation in the SLC25A19 gene (Table 1). In THMD4, mitochondrial TPP uptake is reduced but not completely abolished. The disorder is characterized by childhood onset of episodic encephalopathy, causing transient neurologic dysfunction and a slight increase in cerebrospinal fluid (CSF) lactate levels during the acute phase of the disease. Most patients fully recover without therapeutic intervention; however, in some instances, mild residual weakness may persist (82).
Genetic defects in TPK1 (also known as THMD5) prevent the phosphorylation of thiamine to TPP, the active cofactor. It usually presents during childhood with acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive motor dysfunction (e.g., gait defects, ataxia, dystonia, and spasticity) associated with striatal, basal ganglial, and cerebellar regions of the brain, but cognition appeared to remain intact. Given that the phenotype is highly variable, it is likely that residual TPK1 activity and/or other cellular kinases (albeit at a lower extent and favored by the mass action of high thiamine doses) may also phosphorylate thiamine, thereby partly circumventing the metabolic block. This is based on the fact that i) some affected subjects improved with thiamine treatment [100–200 mg/day or 200 mg/twice daily (90, 91)] or with supplementation with thiamine and niacin, biotin, α-lipoic acid, and ketogenic diet (90), whereas others did not (83); and ii) deficiencies in other TPP-dependent enzymes, TKT and BCKADH, have not been observed in subjects with pathogenic mutations in TPK1 (83).
Pathology of Thiamine Deficiency
As indicated above, the pathology of thiamine deficiency entails impaired energy-production from mitochondria in the form of ATP when using pyruvate-generating substrates (e.g., glucose) as well as increased oxidative stress (27). Under these conditions, glucose via glycolysis generates pyruvate, which cannot enter the Krebs cycle as acetyl-CoA due to the low activity of PDH. As such, pyruvate is transaminated to Ala or reduced to lactate via lactate dehydrogenase. This is consistent with the elevated lactate and organic acids observed in CSF, urine, and blood during thiamine deficiency (92–94).
Thiamine deficiency has been show to stabilize HIF-1α under normoxic conditions, thereby upregulating the expression of the glucose transporter GLUT1, VEGF, aldolase A, LDH, and THTR2 (95). This stabilization might be achieved by the inhibition of prolyl hydroxylase by increased oxidative stress conditions originated from the lower TKT activity within the PPP. These changes may be accompanied by increased glycolysis and fatty acid oxidation to sustain the cellular energy needs. The latter process may ensue with the ultimate formation of acetyl-CoA, skipping the PDH-catalyzed step. The increased flux of acetyl-CoA will then build up the levels of Krebs’ intermediates including fumarate and succinate (also inhibitors of prolyl hydroxylase), especially if the biotin-dependent pyruvate carboxylase cannot sustain the production of oxaloacetate, the most limiting substrate of the cycle. This also provides a biochemical explanation for the efficacy of the biotin and thiamine supplementation in the treatment of the biotin–thiamine-responsive basal ganglia disease. Although lower thiamine deficiency also affects α-KGDH activity, which is essential to maintain the levels of Glu, Asp, and GABA, its activity seems to be less affected than PDC, thus providing an operational Krebs cycle albeit at a lower efficiency. The lower activity of BCKADH can explain the higher levels of BCAA (Leu, Ile, and Val) in human fluids.
The human central nervous system has a high-energy demand, with 2% of the body mass overseeing about 20% of the total metabolic expenditure, the majority of which is spent on firing action potentials, on neuron communication, through chemical synapses, axon growth, and myelination (96). With glucose being the primary fuel for energy production in the brain, it is not surprising that mitochondrial dysfunction and the consequent impaired glucose metabolism have been associated with several neurological and neurodevelopmental conditions (97) and major psychiatric illnesses, such as depression (98) and schizophrenia (99). The neurological symptoms in thiamine deficiency are similar to defects of PDH, which most frequently present as Leigh-like syndrome with basal ganglia involvement. Therefore, the nervous system, which is highly specialized in the use of glucose for energy generation, seems to be most vulnerable to PDHC deficiency due to TPP depletion. In the brain, the lower mitochondrial ATP production will limit the maintenance of membrane potential via the action of the Na+,K+-ATPase, thereby compromising nerve conduction and chemical synapses. Moreover, the increased oxidative stress due to the lower TKT activity will damage critical biomolecules, initiating lipid peroxidation and oxidative damage to proteins resulting in fragmentation, posttranslational modifications, and cross-linkings. The modification of epitopes on normal, endogenous molecules may result in the activation of the microglia and immune cells, compounding oxidative stress-mediated damage. The lower TKT has additional detrimental actions: by providing a suboptimal NADPH supply, biosynthetic reactions (nucleic acids and lipids) are undermined. This will limit not only cell proliferation but also the synthesis of fatty acids critical for the myelin sheath of axons. Thus, the combination of increased lipid peroxidation and the decreased fatty acid synthesis can result in demyelination, explaining some of the neurological issues caused by thiamine deficiency.
Altered thiamine metabolism has also been linked to growth of both neuronal soma and dendritic arbors in murine hippocampal neurons in culture. The individual silencing of TPK1 or each of the TPP carriers Slc25a19 and Slc19a3 in neurons significantly decreased dendrite arborization and soma size compared to controls (100). Conversely, overexpression of TPK1, Slc25a19, and Slc19a3 significantly reversed these changes, leading to increased dendritic branch length and number as well as soma size. Notably, these pathways are independent and nonredundant, as the overexpression of any of the proteins did not improve the dendritic damage caused by suppression of the other two. These findings are indicative of the crucial role that thiamine metabolism plays in soma and dendrite growth (100), and they are consistent with the microcephaly and neuronal damage reported in cases of thiamine deficiency.
In Adults
Symptoms and Consequences of Thiamine Deficiency
Thiamine deficiency can present with a broad range of neurological signs in children, such as anorexia, irritability, agitation, muscle pain, diminished or abolished deep tendon reflexes, ataxia, paralysis, and a progressively altered level of consciousness. Lactic acidosis may explain some of the generalized symptoms, including lethargy, irritability, anorexia, tachycardia, and tachypnea. These clinical manifestations are probably secondary to mitochondrial dysfunction in the heart and smooth muscle (particularly the gastrointestinal tract) and an autonomic nervous system insult (neurotransmitters). Others such as mood changes (agitation, confusion, and generalized malaise) may result from brain energy deficits as well as from a compromised synthesis of neurotransmitters (glutamate and GABA). Given the above-described varied clinical presentations, especially in children, any unexplained severe neurological signs or symptoms should raise the suspicion of thiamine deficiency [see Hiffler et al. (3) and references therein].
Generally, in blood from healthy adults, TPP levels <70 nmol/L are suggestive of thiamine deficiency [reference values: 70–180 nmol/L; https://www.mayocliniclabs.com (101, 102)]. Thiamine levels have also been assessed in CSF, in the context of Alzheimer’s disease and in instances of SLC19A3 mutations (103, 104). Although no universally accepted reference values exist for CSF, thiamine levels of 5–7 nM are considered normal in adults (105).
Blood and CSF thiamine levels provide limited information when assessing the thiamine status of a subject, as they not necessarily reflect thiamine metabolic function (102) or a direct association to its levels in the tissues. As such, assessments of erythrocytes’ TKT and, if available, that of other tissue-specific TPP-dependent enzymes (PDH, αKGDH) are considered gold standards (24, 26). Basic TKT activity is usually expressed as units per gram of hemoglobin (g Hb), but more importantly, the percentage of activation of TKT to TPP supplementation are calculated (with 0–15% considered normal). One study reported that the HPLC detection of TPP in whole blood or red blood cells is equally effective to the TKT activation ratio at determining thiamine deficiency (37).
The TKT activation ratio (red blood cells) and/or the activities of TPP-dependent enzymes (leukocytes, skin fibroblasts, and muscle biopsies) are usually accompanied by testing the levels of serum lactate and pyruvate, BCAAs, organic acids, as well as brain imaging. The only cases in which the evaluation of free thiamine in plasma/serum and CSF seems to provide a valuable diagnostic tool is when dealing with pathogenic mutations in SLC19A3 (104). Similarly, urinary excretion of thiamine is also not a reliable method for the assessment of its bodily levels as it is dependent on its intake and absorption. Generally, it is expressed per unit of creatinine to account for renal function, and age should be taken into account as normal values differ in children [120 nmol/mmol creatinine in 1–13 years old (105)] and adults [220 nmol/mmol creatinine in >18 years old (105)].
Unfortunately, early symptoms of thiamine deficiency are not pronounced or distinctive enough to warrant a direct diagnosis. They include loss of appetite, nausea, weakness, apathy, fatigue, irritation, sleep disturbances, anorexia, and abdominal discomfort (106). Furthermore, the identification of specific clinical symptoms of thiamine deficiency is problematic because it is obscured by the contribution of other confounding conditions (comorbidities) such as infections and/or multiple nutritional deficiencies.
The clinical classification of thiamine deficiency falls usually into dry (or neuritic, characterized by polyneuropathy, reduced knee jerk and other tendon reflexes, and progressive severe weakness of muscles) and wet (or cardiac, characterized by edema of the legs, trunk, and face; high cardiac output; ventricular failure; and pulmonary congestion). This classification is based on the amount of fluid that accumulates in the body due to factors like cardiac function and kidney lesions, among others (107). However, many cases of thiamine deficiency are amply systemic and encompass the nervous and cardiovascular system, as well as the gastrointestinal tract (with symptoms spanning from anorexia, abdominal pain, and constipation) (108). Long-term consequences of severe thiamine deficiency are manifested by the appearance of hemorrhagic and/or necrotic lesions with neuronal and dendritic spine loss in the thalamus, hypothalamus, and cerebellum (109–111), as well as polyneuritis, peripheral edema, cardiac enlargement, and ophthalmoplegia (27).
When early, generalized thiamine deficiency is suspected, prompt administration of thiamine is advised and typically an effective treatment. A wide range of therapeutic approaches and thiamine doses are reported in the literature, spanning from 1.5 to 600 mg/day (112), with 10–20 mg/day as divided doses for several weeks for mild polyneuropathy and 20–30 mg/day for moderate to severe, usually until after disappearance of symptoms (106). Generally, thiamine deficiency is approached with doses of 5–30 mg/day intravenously (IV) or intramuscularly (IM), three times daily, followed by 5–30 mg/day orally until after disappearance of symptoms (112). However, this approach is notably less effective for individuals with chronic forms of thiamine deficiency-related disorders involving encephalopathies (see the section Wernicke Encephalopathy) (113) or TPK1 deficiencies. In the latter case, it would be worth to explore a treatment directly with TPP; however, it is not clear whether this phosphorylated thiamine form would cross the blood–brain barrier and/or reach subcellular targets such as PDH.
Wernicke Encephalopathy
Wernicke encephalopathy (WE) is a thiamine-deficiency-related acute neuropsychiatric condition, often associated with alcohol abuse (114). It is characterized by ataxia, loss of muscle coordination, memory loss, confusion, and ocular abnormalities (ophthalmoplegia). Failure to timely diagnose the disease and the lack of an appropriate therapy result in death in 17% of the cases, while 84% will undergo permanent brain damage involving severe short-term memory loss and hallucinations [or Korsakoff’s syndrome (KS)], residual syndrome in patients previously affected with WE (115). Because of the close association between these two conditions, they often get referred to as Wernicke–Korsakoff syndrome (WKS). Recently, it has been suggested that WE may be underdiagnosed (116), with similar incidence in both adults and children (117), with ratification of the diagnosis in 0.4–2.8% of postmortem cases, but recognized only in 32% and 6% of patients with and without alcoholism, respectively. In particular, it would be critical to consider WE in any patient showing two of the following characteristics: nutritional deficiency, cerebellar or oculomotor abnormalities, or impaired mental state or memory (116). Only 16% of the WE cases are expected to fully recover after thiamine supplementation. As oral absorption is highly variable and patient-dependent (116), parenteral treatment is highly recommended with daily doses of 100–200 mg thiamine (IV) for non-alcoholics and 500 mg (up to three administrations daily) for alcoholics (114).
A substantial body of literature has been published in the past >50 years regarding WE. A detailed description of the clinical features, pathophysiology, and treatment of this syndrome is beyond the scope of this review.
Depression
A number of studies have shown an inverse association between thiamine levels and symptoms of depression in adults. A cross-sectional study on >1,500 elderly Chinese (age 50–70 years) showed that subjects with lower levels of erythrocyte thiamine (likely representing TPP bound to TKT) displayed more severe symptoms of depression (118). In this report, although no apparent differences were noted between individuals with and without depressive symptoms in dietary thiamine intake, the authors could not exclude errors in dietary assessment related to differences in food processing (118). In addition, other factors like diabetes, altered hormone profiles, and increased alcohol consumption observed in some individuals with depressive symptoms could play a critical role in the bioavailability of thiamine (118).
A direct correlation between thiamine deficiency and symptoms of major depressive disorder (MDD) had been previously found in a population of 74 individuals with history of malnutrition (119). These findings were confirmed by another study involving 118 geriatric patients (120), although in this particular instance, no information about dietary thiamine intake was provided.
Additionally, Ghaleiha and colleagues explored the interventional effect of thiamine supplementation in thiamine-deficient subjects with MDD in a 12-week, randomized, double-blind clinical trial (121). The study concluded that symptoms of depression improved significantly in subjects with MDD following 6 weeks of thiamine supplementation compared to placebo.
Finally, positive mood changes (clearheadedness, increased energy and appetite, improved sleep patterns, and decreased fatigue) have been observed following thiamine supplementation in healthy elderly (16) as well as younger (122) women, compared to the same population assigned to the placebo group. Interestingly, while in the elderly population, a marginal deficiency of thiamine was reportedly due to the lack of a national thiamine enrichment policy for grains and cereals at the time of the study, in the case of younger women, a normal thiamine intake was recorded for all participants at baseline.
In Children
In children, thiamine deficiency presents with acute symptoms and very rapid onset, and in the vast majority of cases, it affect infants who are breast-fed by thiamine-deficient mothers or with thiamine-deficient formulas.
A pivotal study that highlighted the crucial role of thiamine as an essential nutrient for infant development and discussed how TD phenotype may be confused or overlooked with other diseases was based on an epidemiological investigation on 24- to 39-month-old children presenting repeated vomiting, irritability, and lethargy, without major neurological abnormalities. All children who had consumed the same soy-based, thiamine-deficient formula (<0.5 mg/g formula) suffered from language and motor development deficits compared to sex- and age-matched children fed with other milk-based diets (123). Another study on the acute and long-term effects of thiamine-deficient formula-fed children who were not promptly diagnosed reported that a subset of patients (36%) died of cardiac and respiratory complications (124), and the remaining children developed intellectual disabilities, neurodevelopmental delay, and major motor dysfunction by the age of 9 (124). Recently, in another study by Harel et al. (125), more than 50% of a preschool children cohort, who were exposed to a thiamine-deficient diet for more than 1 month during the first 24 months of life, developed movement and motor skills difficulties compared to less than 10% among children with appropriate thiamine intake.
Infantile Thiamine Deficiency
Infantile thiamine deficiency (ITD) manifests after 2–3 weeks of thiamine deprivation with initially milder symptoms (refusal to eat, vomiting, and irritability), followed by rapid deterioration, with a high fatality rate (106). Newborns who are severely thiamine deprived may develop infantile beriberi. The disorder is rare in developed countries, as it can be readily treated with thiamine supplementation (126). However, it remains a concern in developing countries among infants breast-fed by thiamine-deficient mothers or fed with thiamine-deficient formula. Without proper thiamine supplementation, the deficiency is characterized by a rapid onset of symptoms involving vomiting, diarrhea, tachypnea, convulsions, ataxia, paralysis, cardiac dysfunction, and heart failure. In many cases, fatality occurs quickly (127). Those children who survive thiamine-deficiency-related WE [see the section Wernicke Encephalopathy and Vasconcelos et al. (128)] continue to live with developmental disabilities including severe epilepsy, language impairment, loss of motor function (to varying degrees), and mental retardation (mild to profound) (124).
In two separate case reports, one newborn (2 months old) with early infantile Leigh-like SLC19A3 gene defect (129) and one (30 days old) with THTR2 deficiency (130) responded dramatically differently to thiamine supplementation. In the first case, the 2-month-old infant initially presenting with fever, feeding difficulties, and complex partial seizures (129) subsequently developed a severe encephalopathy. MRI showed abnormalities in both cerebral hemispheres as well as in brainstem and cerebellum, basal ganglia, and thalamus. A diffuse volume loss was also noted in both hemispheres. At the biochemical level, metabolic acidosis was diagnosed. Despite a prompt therapeutic regimen consisting of thiamine (37.5 mg/kg) and biotin (10 mg/kg) twice daily, the patient’s conditions never improved and he died at 4 months of age. The lack of response to treatment in this instance seems to suggest that its effectiveness is completely halted in the presence of a null mutation leading to a complete loss of function of the thiamine transporter in the early-onset form [see Table 1 in Alfadhel (129)].
In the second case report, the infant showed acute mitochondrial encephalopathy, accompanied by increased level of lactate in the blood and cerebrospinal fluid as well as abnormal levels of α-ketoglutarate in the urine. Cortico-subcortical lesions were visualized by MRI, some of which seemed to have appeared in the perinatal period (130). Within 48 h after administration of thiamine (100 mg/day), biotin (10 mg/day), and carnitine (300 mg/day), a remarkable improvement was observed in both clinical and biochemical outcomes (130). After discontinuing biotin, the patient remained stable for 6 months on thiamine supplementation (20 mg/kg/day) (130).
It is now widely established that less severe cases of ITD can be treated (to varying degrees) with early thiamine supplementation. In general, therapeutic recommended doses are 10–25 mg/daily IV or IM for 2 weeks, or 10–50 mg/daily orally, followed by 5–10 mg/daily for 1 month (112).
Autism Spectrum Disorders
More recently, thiamine-based treatments have gained a great deal of attention as a therapeutic strategy for other developmental and neurological disorders linked to mitochondrial dysfunction. One such disorder is autism spectrum disorder (ASD), a group of complex neurological and developmental disabilities, with a prevalence of 1 in 54 and 1 in 252, respectively, for males and females age 8 years in the United States (131). “Spectrum” refers to the wide heterogeneity and range of symptoms within three characteristic patterns: atypical cognitive profile, executive dysfunction, and unusually restricted or repetitive behavior (131). Individuals with ASD show severe cognitive and verbal impairments and experience challenges in the development of critical social communication skills.
Limited findings are available on the link between thiamine bioavailability and the development of autism. However, studies show that the consumption of herbal supplements rich in thiaminase during pregnancy (132) or alcohol exposure in utero (133) may predispose a fetus to thiamine deficiency during pregnancy and increased risk for ASD. A study conducted on rat pups nursed by thiamine-deficient dams displayed comparable findings, in which the offspring exhibited behavioral abnormalities consistent with ASD symptoms (134).
A number of hypotheses have been put forward on the pathophysiological mechanisms linking thiamine deficiency and ASD (135), including i) increased apoptosis, due to the link of thiamine with p53 (136, 137), Bcl-2 (138, 139), and caspases (140); ii) deregulation of the serotoninergic system (141, 142); and iii) increased oxidative stress, due to the putative role of thiamine in prostaglandin expression, decreased lipid peroxidation as well as expression of nitric oxide synthase (143, 144), and its involvement as a cofactor for TKT in the PPP, master regulator of NADPH homeostasis.
More interestingly, a subset of children with ASD has been characterized by PDH deficiency and mitochondrial dysfunction (145, 146), suggesting that thiamine deficiency may also ensue in PDH deficits or that a borderline thiamine intake would compound the preexistent PDH deficit, precipitating energy metabolism and leading to ASD in genetically predisposed individuals. Furthermore, a more recent study has shown decreased TPP levels in plasma from ASD children compared to healthy controls (147), suggesting either lower consumption or an impaired absorption from the gastrointestinal tract, providing a link between thiamine metabolism and the gut microbiome, which has been considered in the context of ASD (148). While there is no evidence to suggest that a sole deficiency in thiamine may result in ASD, it could certainly be a contributing element in concert with other epigenetic factors in the presence of an already predisposed genetic background.
Based on the finding that children with ASD seem to have an impairment in the catabolism and excretion of thiol-containing heavy metals, Lonsdale et al. proposed that thiamine supplementation may be indicated for all children with ASD (149), with the idea that heavy metal toxicity can hamper TKT activity, increase oxidative stress, and, in turn, disrupt thiamine homeostasis. Hence, thiamine supplementation may, to some extent, improve carbohydrate metabolism, mitochondrial function, and brain energy production (150). In that study, children with ASD were administered a thiamine derivative, bi-daily for 2 months and noted improved communication skills, sociability, sensory/cognitive awareness, and behavior in 8 out of the 10 children studied as well as reduction in urine arsenic and mercury levels (149). In a separate, double-blind, placebo-controlled study, researchers noted decreased oxidative stress, increased ATP in plasma and NADH and NADPH production in red blood cells, and improved PGI-R (Parent Global Impressions—Revised) scores in hyperactivity, tantrum, and receptive language categories in autistic children administered vitamin/mineral supplementation, which included thiamine, over a 3-month period (151).
These pilot studies provide some support for a thiamine-based approach as a therapeutic intervention for ASD. However, further studies are warranted to clarify the role and specificity of thiamine supplementation for the management of the diverse and extensive range of ASD features.
Depression in Children and Child-Bearing Women
To our knowledge, to date, only one study has investigated the contribution of thiamine levels to pediatric depression (152). Surprisingly, and opposite to findings in adults, this study found a direct correlation between thiamine intake and depressive symptoms in Spanish schoolchildren (7–9 years old).
Although a fairly common condition among teenagers, little is known about depression and its treatment in children 12 years old and younger (153). Depression in children is often overlooked and oftentimes mistaken by teachers and parents and labeled as laziness, learning disabilities, or belligerence (154). Similarly, the “acting out” behavior observed in pediatric depression is often labeled as bullying, rebellion, or even psychotic behavior (154).
The effects of thiamine supplementation might be significant as palliative treatment in postpartum depression (PPD) and have a critical role on the infant’s subsequent cognitive development. PPD has been associated with an increased risk of developing learning disabilities, Attention-Deficit/Hyperactivity Disorder (ADHD), and anxiety disorders in toddlers, which makes PPD a critical concern for both mother and infant (155). Nikseresht et al. investigated the therapeutic effects of Zn2+, Mg2+, and TPP supplementation in a PPD mouse model. Improvements in depressive symptoms and anxiety-like behaviors (tested by forced swimming test and elevated plus-maze), as well as total antioxidant capacity, were noted in the dams when the nutrients were administered on postpartum day 3 (156). Taken together, these findings indicate that thiamine supplementation may be a promising therapeutic strategy for PPD. However, future studies should address the role of thiamine in PPD independently and whether supplementation in instances of PPD positively impacts infant development.
Conclusions and Future Perspectives
Thiamine deficiency, regardless of the underlying mechanism (genetic or epigenetic), impairs critical TPP-dependent enzymes involved in intermediary metabolism and antioxidant defenses. The consequent decrease in ATP production and increased oxidative stress result in neurological deficits, which become especially detrimental during critical windows of neurodevelopment. In the recent years, thiamine supplementation has been contemplated, with some success, as a therapeutic approach for neurodevelopmental disorders, including ASD and major psychiatric disorders (i.e., depression). However, the vast heterogeneity of the pathogenic mechanisms underlying these conditions, along with the difficulty of diagnosis and the assumption that diets from developed countries supply enough thiamine to match the caloric intake (including perinatal periods), has not prompted enough research to address the role and effects of thiamine supplementation as a therapeutic strategy in the context of neurodevelopment/neurodegeneration. Of note, and an often overlooked issue, is the lack of incorporation of antioxidants into supplemental treatments that are deemed critical to minimize the thiamine-deficiency-dependent oxidative-stress-derived damage. This will also allow to shed light on the contribution of oxidative stress versus mitochondrial dysfunction to the morbidity of neurodegenerative disorders.
Statements
Author contributions
SD and MT equally contributed to the writing of the manuscript. EN contributed to writing and edited the text. CG conceptualized the paper, contributed to writing, and edited the text.
Acknowledgments
We would like to thank the technical assistance of Dr. E. Penna.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
MartinPRSingletonCKHiller-SturmhofelS. The role of thiamine deficiency in alcoholic brain disease. Alcohol Res Health (2003) 27:134–42.
2
MartelJLFranklinDS. Vitamin B1 (thiamine). In: StatPearls. Treasure Island (FL), (2018).
3
HifflerLRakotoambininaBLaffertyNMartinez GarciaD. Thiamine deficiency in tropical pediatrics: new insights into a neglected but vital metabolic challenge. Front Nutr (2016) 3:16. doi: 10.3389/fnut.2016.00016
4
SingletonCKMartinPR. Molecular mechanisms of thiamine utilization. Curr Mol Med (2001) 1:197–207. doi: 10.2174/1566524013363870
5
SaidHMNexoE. Chapter 64—Mechanisms and regulation of intestinal absorption of water-soluble vitamins: cellular and molecular aspects. In: JohnsonLRGhishanFKKaunitzJDMerchantJLSaidHMWoodJD, editors. Physiology of the gastrointestinal tract, 5th ed. Boston: Academic Press (2012). p. 1711–56. doi: 10.1016/B978-0-12-382026-6.00064-6
6
CalhauCFariaAKeatingEMartelF. Chapter 39—Interaction of polyphenols with the intestinal and placental absorption of some nutrients and other compounds. In: WatsonRRPreedyVRZibadiS, editors. Polyphenols in human health and disease. San Diego: Academic Press (2014). p. 523–36. doi: 10.1016/B978-0-12-398456-2.00039-6
7
LonsdaleD. A review of the biochemistry, metabolism and clinical benefits of thiamin(e) and its derivatives. Evid Based Complement Alternat Med (2006) 3:49–59. doi: 10.1093/ecam/nek009
8
LonsdaleDMarrsC. High-calorie malnutrition and its impact on health. In: LonsdaleDMarrsC, editors. Thiamine deficiency disease, dysautonomia, and high calorie malnutrition. Cambridge, MA: Academic Press (2017). p. 213–61. doi: 10.1016/B978-0-12-810387-6.00006-X
9
RoustLRDiBaiseJK. Nutrient deficiencies prior to bariatric surgery. Curr Opin Clin Nutr Metab Care (2017) 20:138–44. doi: 10.1097/MCO.0000000000000352
10
ScrimshawNSBeharM. Malnutrition in underdeveloped countries. N Engl J Med (1965) 272:193–98 CONCL. doi: 10.1056/NEJM196501282720406
11
WhitfieldKCSmithGChamnanCKarakochukCDSophonnearyPKuongKet al. High prevalence of thiamine (vitamin B1) deficiency in early childhood among a nationally representative sample of Cambodian women of childbearing age and their children. PLoS Negl Trop Dis (2017) 11:e0005814. doi: 10.1371/journal.pntd.0005814
12
FujitaA. Thiaminase. Adv Enzymol Relat Subj Biochem (1954) 15:389–421.
13
LeichterJJoslynMA. Kinetics of thiamin cleavage by sulphite. Biochem J (1969) 113:611–15. doi: 10.1042/bj1130611
14
HilkerDMSomogyiJC. Antithiamins of plant origin: their chemical nature and mode of action. Ann N Y Acad Sci (1982) 378:137–45. doi: 10.1111/j.1749-6632.1982.tb31192.x
15
VimokesantSKunjaraSRungruangsakKNakornchaiSPanijpanB. Beriberi caused by antithiamin factors in food and its prevention. Ann N Y Acad Sci (1982) 378:123–36. doi: 10.1111/j.1749-6632.1982.tb31191.x
16
SmidtLJCreminFMGrivettiLECliffordAJ. Influence of thiamin supplementation on the health and general well-being of an elderly Irish population with marginal thiamin deficiency. J Gerontol (1991) 46:M16–22. doi: 10.1093/geronj/46.1.M16
17
ThornalleyPJBabaei-JadidiRAl AliHRabbaniNAntonysunilALarkinJet al. High prevalence of low plasma thiamine concentration in diabetes linked to a marker of vascular disease. Diabetologia (2007) 50:2164–70. doi: 10.1007/s00125-007-0771-4
18
Sequeira Lopes da SilvaJTAlmaraz VelardeROlgado FerreroFRobles MarcosMPerez CivantosDRamirez MorenoJMet al. Wernicke’s encephalopathy induced by total parental nutrition. Nutr Hosp (2010) 25:1034–36.
19
JuniorEVCesarCLFisbergRMMarchioniDM. Socio-economic variables influence the prevalence of inadequate nutrient intake in Brazilian adolescents: results from a population-based survey. Public Health Nutr (2011) 14:1533–8. doi: 10.1017/S1368980011000760
20
RenthalWMarin-ValenciaIEvansPA. Thiamine deficiency secondary to anorexia nervosa: an uncommon cause of peripheral neuropathy and Wernicke encephalopathy in adolescence. Pediatr Neurol (2014) 51:100–3. doi: 10.1016/j.pediatrneurol.2014.03.025
21
KernsJCArundelCChawlaLS. Thiamin deficiency in people with obesity. Adv Nutr (2015) 6:147–53. doi: 10.3945/an.114.007526
22
ChandrakumarABhardwajAJongGW. Review of thiamine deficiency disorders: Wernicke encephalopathy and Korsakoff psychosis. J Basic Clin Physiol Pharmacol (2018) 30:153–62. doi: 10.1515/jbcpp-2018-0075
23
KamasakTKulSTusatMOzgunNCansuA. A case of Wernicke encephalopathy developing after ileal bypass surgery. Pediatr Emerg Care (2018) 34:e223–e225. doi: 10.1097/PEC.0000000000001472
24
CroftLNapoliEHungCKSt LegerJGearhartSHeymKet al. Clinical evaluation and biochemical analyses of thiamine deficiency in Pacific harbor seals (Phoca vitulina) maintained at a zoological facility. J Am Vet Med Assoc (2013) 243:1179–89. doi: 10.2460/javma.243.8.1179
25
ChenLYeeSWGiacominiKM. OCT1 in hepatic steatosis and thiamine disposition. Cell Cycle (2015) 14:283–4. doi: 10.1080/15384101.2015.1006532
26
VernauKNapoliEWongSRoss-IntaCCameronJBannaschDet al. Thiamine deficiency-mediated brain mitochondrial pathology in Alaskan Huskies with mutation in SLC19A3.1. Brain Pathol (2015) 25:441–53. doi: 10.1111/bpa.12188
27
ButterworthRF. Thiamin deficiency and brain disorders. Nutr Res Rev (2003) 16:277–84. doi: 10.1079/NRR200367
28
GreenwoodJLoveERPrattOE. Kinetics of thiamine transport across the blood–brain barrier in the rat. J Physiol (1982) 327:95–103. doi: 10.1113/jphysiol.1982.sp014222
29
RindiGComincioliVReggianiCPatriniC. Nervous tissue thiamine metabolism in vivo. II. Brain Res (1984) 293:329–42. doi: 10.1016/0006-8993(84)91240-X
30
ButterworthRFGiguereJFBesnardAM. Activities of thiamine-dependent enzymes in two experimental models of thiamine-deficiency encephalopathy. 2. Neurochem Res (1986) 11:567–77. doi: 10.1007/BF00965326
31
BettendorffLWinsPLesourdM. Subcellular localization and compartmentation of thiamine derivatives in rat brain. Biochim Biophys Acta (1994) 1222:1–6. doi: 10.1016/0167-4889(94)90018-3
32
BettendorffLMastrogiacomoFKishSJGrisarT. Thiamine, thiamine phosphates, and their metabolizing enzymes in human brain. J Neurochem (1996) 66:250–8. doi: 10.1046/j.1471-4159.1996.66010250.x
33
RindiGLaforenzaU. Thiamine intestinal transport and related issues: recent aspects. Proc Soc Exp Biol Med (2000) 224:246–55. doi: 10.1046/j.1525-1373.2000.22428.x
34
SchallerKHollerH. Thiamine absorption in the rat. II. Intestinal alkaline phosphatase activity and thiamine absorption from rat small intestine in-vitro and in-vivo. Int J Vitam Nutr Res (1975) 45:30–8.
35
NabokinaSMSubramanianVSValleJESaidHM. Adaptive regulation of human intestinal thiamine uptake by extracellular substrate level: a role for THTR-2 transcriptional regulation. Am J Physiol Gastrointest Liver Physiol (2013) 305:G593–599. doi: 10.1152/ajpgi.00237.2013
36
LaforenzaUGastaldiGRindiG. Thiamine outflow from the enterocyte: a study using basolateral membrane vesicles from rat small intestine. J Physiol (1993) 468:401–12. doi: 10.1113/jphysiol.1993.sp019778
37
TalwarDDavidsonHCooneyJStJRD. Vitamin B(1) status assessed by direct measurement of thiamin pyrophosphate in erythrocytes or whole blood by HPLC: comparison with erythrocyte transketolase activation assay. Clin Chem (2000) 46:704–10.
38
SongPRekowSSSingletonCASekhonHSDissenGAZhouMet al. Choline transporter-like protein 4 (CTL4) links to non-neuronal acetylcholine synthesis. J Neurochem (2013) 126:451–61. doi: 10.1111/jnc.12298
39
MaZXiaWLiuFMaJSunSZhangJet al. SLC44A4 mutation causes autosomal dominant hereditary postlingual non-syndromic mid-frequency hearing loss. Hum Mol Genet (2017) 26:3234. doi: 10.1093/hmg/ddx232
40
NabokinaSMInoueKSubramanianVSValleJEYuasaHSaidHM. Molecular identification and functional characterization of the human colonic thiamine pyrophosphate transporter. J Biol Chem (2017) 292:16526. doi: 10.1074/jbc.A113.528257
41
FlemingJCTartagliniESteinkampMPSchorderetDFCohenNNeufeldEJ. The gene mutated in thiamine-responsive anaemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat Genet (1999) 22:305–8. doi: 10.1038/10379
42
SubramanianVSMarchantJSParkerISaidHM. Cell biology of the human thiamine transporter-1 (hTHTR1). J Biol Chem (2003) 278:3976–84. doi: 10.1074/jbc.M210717200
43
SubramanianVSMarchantJSSaidHM. Targeting and trafficking of the human thiamine transporter-2 in epithelial cells. J Biol Chem (2006) 281:5233–45. doi: 10.1074/jbc.M512765200
44
GanapathyVSmithSBPrasadPD. SLC19: the folate/thiamine transporter family. Pflugers Arch (2004) 447:641–6. doi: 10.1007/s00424-003-1068-1
45
LuJFrankEL. Rapid HPLC measurement of thiamine and its phosphate esters in whole blood. Clin Chem (2008) 54:901–6. doi: 10.1373/clinchem.2007.099077
46
GangolfMCzernieckiJRadermeckerMDetryONisolleMJouanCet al. Thiamine status in humans and content of phosphorylated thiamine derivatives in biopsies and cultured cells. PLoS One (2010) 5:e13616. doi: 10.1371/journal.pone.0013616
47
ButterworthRFKrilJJHarperCG. Thiamine-dependent enzyme changes in the brains of alcoholics: relationship to the Wernicke–Korsakoff syndrome. Alcohol Clin Exp Res (1993) 17:1084–88. doi: 10.1111/j.1530-0277.1993.tb05668.x
48
ZhaoYPanXZhaoJWangYPengYZhongC. Decreased transketolase activity contributes to impaired hippocampal neurogenesis induced by thiamine deficiency. J Neurochem (2009) 111:537–46. doi: 10.1111/j.1471-4159.2009.06341.x
49
FraccasciaPCasteelsMDe SchryverEVan VeldhovenPP. Role of thiamine pyrophosphate in oligomerisation, functioning and import of peroxisomal 2-hydroxyacyl-CoA lyase. Biochim Biophys Acta (2011) 1814:1226–33. doi: 10.1016/j.bbapap.2011.06.007
50
FraccasciaPSniekersMCasteelsMVan VeldhovenPP. Presence of thiamine pyrophosphate in mammalian peroxisomes. BMC Biochem (2007) 8:10. doi: 10.1186/1471-2091-8-10
51
FoulonVAntonenkovVDCroesKWaelkensEMannaertsGPVan VeldhovenPPet al. Purification, molecular cloning, and expression of 2-hydroxyphytanoyl-CoA lyase, a peroxisomal thiamine pyrophosphate-dependent enzyme that catalyzes the carbon-carbon bond cleavage during alpha-oxidation of 3-methyl-branched fatty acids. Proc Natl Acad Sci U S A (1999) 96:10039–44. doi: 10.1073/pnas.96.18.10039
52
WatkinsPAHowardAEMihalikSJ. Phytanic acid must be activated to phytanoyl-CoA prior to its alpha-oxidation in rat liver peroxisomes. Biochim Biophys Acta (1994) 1214:288–94. doi: 10.1016/0005-2760(94)90075-2
53
van den BrinkDMWandersRJ. Phytanic acid: production from phytol, its breakdown and role in human disease. Cell Mol Life Sci (2006) 63:1752–65. doi: 10.1007/s00018-005-5463-y
54
SkjeldalOHStokkeORefsumSNorsethJPetitH. Clinical and biochemical heterogeneity in conditions with phytanic acid accumulation. J Neurol Sci (1987) 77:87–96. doi: 10.1016/0022-510X(87)90209-7
55
WierzbickiASLloydMDSchofieldCJFeherMDGibberdFB. Refsum’s disease: a peroxisomal disorder affecting phytanic acid alpha-oxidation. J Neurochem (2002) 80:727–35. doi: 10.1046/j.0022-3042.2002.00766.x
56
LindhurstMJFiermonteGSongSStruysEDe LeonardisFSchwartzbergPLet al. Knockout of Slc25a19 causes mitochondrial thiamine pyrophosphate depletion, embryonic lethality, CNS malformations, and anemia. Proc Natl Acad Sci U S A (2006) 103:15927–32. doi: 10.1073/pnas.0607661103
57
RandlePJGarlandPBHalesCNNewsholmeEA. The glucose fatty-acid cycle. Lancet (1963) 1:785–89. doi: 10.1016/S0140-6736(63)91500-9
58
MuoioDM. Metabolic inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell (2014) 159:1253–62. doi: 10.1016/j.cell.2014.11.034
59
Jankowska-KulawyABielarczykHPawelczykTWroblewskaMSzutowiczA. Acetyl-CoA deficit in brain mitochondria in experimental thiamine deficiency encephalopathy. Neurochem Int (2010) 57:851–56. doi: 10.1016/j.neuint.2010.09.003
60
BrownGKBrownRMScholemRDKirbyDMDahlHH. The clinical and biochemical spectrum of human pyruvate dehydrogenase complex deficiency. Ann N Y Acad Sci (1989) 573:360–8. doi: 10.1111/j.1749-6632.1989.tb15011.x
61
De MeirleirL. Disorders of pyruvate metabolism. Handb Clin Neurol (2013) 113:1667–73. doi: 10.1016/B978-0-444-59565-2.00034-4
62
GibsonGEBlassJPBealMFBunikV. The alpha-ketoglutarate-dehydrogenase complex: a mediator between mitochondria and oxidative stress in neurodegeneration. Mol Neurobiol (2005) 31:43–63. doi: 10.1385/MN:31:1-3:043
63
ShiQKaruppagounderSSXuHPechmanDChenHGibsonGE. Responses of the mitochondrial alpha-ketoglutarate dehydrogenase complex to thiamine deficiency may contribute to regional selective vulnerability. Neurochem Int (2007) 50:921–31. doi: 10.1016/j.neuint.2007.03.010
64
SperringerJEAddingtonAHutsonSM. Branched-chain amino acids and brain metabolism. Neurochem Res (2017) 42:1697–709. doi: 10.1007/s11064-017-2261-5
65
NavarroDZwingmannCButterworthRF. Impaired oxidation of branched-chain amino acids in the medial thalamus of thiamine-deficient rats. Metab Brain Dis (2008) 23:445–55. doi: 10.1007/s11011-008-9105-6
66
ProhaskaJRBaileyWR. Persistent regional changes in brain copper, cuproenzymes and catecholamines following perinatal copper deficiency in mice. J Nutr (1993) 123:1226–34. doi: 10.1093/jn/123.7.1226
67
KrilJJHarperCG. Neuroanatomy and neuropathology associated with Korsakoff’s syndrome. Neuropsychol Rev (2012) 22:72–80. doi: 10.1007/s11065-012-9195-0
68
OpdenakkerGGelinGDe SurgelooseDPalmersY. Wernicke encephalopathy: MR findings in two patients. Eur Radiol (1999) 9:1620–4. doi: 10.1007/s003300050896
69
KornreichLBron-HarlevEHoffmannCSchwarzMKonenOSchoenfeldTet al. Thiamine deficiency in infants: MR findings in the brain. AJNR Am J Neuroradiol (2005) 26:1668–74.
70
VernauKMRunstadlerJABrownEACameronJMHusonHJHigginsRJet al. Genome-wide association analysis identifies a mutation in the thiamine transporter 2 (SLC19A3) gene associated with Alaskan Husky encephalopathy. PLoS One (2013) 8:e57195. doi: 10.1371/journal.pone.0057195
71
RobinsonBH. Prenatal diagnosis of disorders of energy metabolism. Semin Neurol (2001) 21:269–73. doi: 10.1055/s-2001-17944
72
MezzarSDe SchryverEAsselberghsSMeyhiEMorvayPLBaesMet al. Phytol-induced pathology in 2-hydroxyacyl-CoA lyase (HACL1) deficient mice. Biochim Biophys Acta Mol Cell Biol Lipids (2017) 1862:972–90. doi: 10.1016/j.bbalip.2017.06.004
73
FoulonVSniekersMHuysmansEAsselberghsSMahieuVMannaertsGPet al. Breakdown of 2-hydroxylated straight chain fatty acids via peroxisomal 2-hydroxyphytanoyl-CoA lyase: a revised pathway for the alpha-oxidation of straight chain fatty acids. J Biol Chem (2005) 280:9802–12. doi: 10.1074/jbc.M413362200
74
KitamuraTSekiNKiharaA. Phytosphingosine degradation pathway includes fatty acid alpha-oxidation reactions in the endoplasmic reticulum. Proc Natl Acad Sci U S A (2017) 114:E2616–E2623. doi: 10.1073/pnas.1700138114
75
KhawKTFriesenMDRiboliELubenRWarehamN. Plasma phospholipid fatty acid concentration and incident coronary heart disease in men and women: the EPIC-Norfolk prospective study. PLoS Med (2012) 9. doi: 10.1371/journal.pmed.1001255
76
ForouhiNGKoulmanASharpSJImamuraFKrogerJSchulzeMBet al. Differences in the prospective association between individual plasma phospholipid saturated fatty acids and incident type 2 diabetes: the EPIC-InterAct case-cohort study. Lancet Diab Endocrin (2014) 2:810–18. doi: 10.1016/S2213-8587(14)70146-9
77
OishiKDiazGA. Thiamine-responsive megaloblastic anemia syndrome. In: AdamMPArdingerHHPagonRAWallaceSEBeanLJHStephensKAmemiyaA, editors. GeneReviews((R)). Seattle (WA): University of Washington, Seattle (1993).
78
TabarkiBAl-HashemAAlfadhelM. Biotin-thiamine-responsive basal ganglia disease. In: AdamMPArdingerHHPagonRAWallaceSEBeanLJHStephensKAmemiyaA, editors. GeneReviews((R)). Seattle (WA): University of Washington, Seattle (1993). p. 1993–2019.
79
AlfadhelMAlmuntashriMJadahRHBashiriFAAl RifaiMTAl ShalaanHet al. Biotin-responsive basal ganglia disease should be renamed biotin-thiamine-responsive basal ganglia disease: a retrospective review of the clinical, radiological and molecular findings of 18 new cases. Orphanet J Rare Dis (2013) 8:83. doi: 10.1186/1750-1172-8-83
80
BieseckerLG. Amish lethal microcephaly. In: AdamMPArdingerHHPagonRAWallaceSEBeanLJHStephensKAmemiyaA, editors. GeneReviews((R)). Seattle (WA): University of Washington, Seattle (1993).
81
SiuVMRatkoSPrasadANPrasadCRuparCA. Amish microcephaly: long-term survival and biochemical characterization. Am J Med Genet A (2010) 152A:1747–51. doi: 10.1002/ajmg.a.33373
82
SpiegelRShaagAEdvardsonSMandelHStepenskyPShalevSAet al. SLC25A19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann Neurol (2009) 66:419–24. doi: 10.1002/ana.21752
83
MayrJAFreisingerPSchlachterKRolinskiBZimmermannFAScheffnerTet al. Thiamine pyrophosphokinase deficiency in encephalopathic children with defects in the pyruvate oxidation pathway. Am J Hum Genet (2011) 89:806–12. doi: 10.1016/j.ajhg.2011.11.007
84
BankaSde GoedeCYueWWMorrisAAvon BremenBChandlerKEet al. Expanding the clinical and molecular spectrum of thiamine pyrophosphokinase deficiency: a treatable neurological disorder caused by TPK1 mutations. Mol Genet Metab (2014) 113:301–6. doi: 10.1016/j.ymgme.2014.09.010
85
AlfadhelMBabikerA. Inborn errors of metabolism associated with hyperglycaemic ketoacidosis and diabetes mellitus: narrative review. Sudan J Paediatr (2018) 18:10–23. doi: 10.24911/SJP.2018.1.3
86
LorberAGazitAZKhouryASchwartzYMandelH. Cardiac manifestations in thiamine-responsive megaloblastic anemia syndrome. Pediatr Cardiol (2003) 24:476–81. doi: 10.1007/s00246-002-0215-3
87
GanieMAAliIAhangarAGWaniMMAhmedSBhatMAet al. Thiamine responsive megaloblastic anemia syndrome associated with patent ductus arteriosus: first case report from Kashmir Valley of the Indian subcontinent. Indian J Endocrinol Metab (2012) 16:646–50. doi: 10.4103/2230-8210.98033
88
AljabriMFKamalNMArifMAlQaediAMSantaliEY. A case report of biotin-thiamine-responsive basal ganglia disease in a Saudi child: is extended genetic family study recommended? Medicine (Baltimore) (2016) 95:e4819. doi: 10.1097/MD.0000000000004819
89
KelleyRIRobinsonDPuffenbergerEGStraussKAMortonDH. Amish lethal microcephaly: a new metabolic disorder with severe congenital microcephaly and 2-ketoglutaric aciduria. Am J Med Genet (2002) 112:318–26. doi: 10.1002/ajmg.10529
90
FraserJLVanderverAYangSChangTCrampLVezinaGet al. Thiamine pyrophosphokinase deficiency causes a Leigh disease like phenotype in a sibling pair: identification through whole exome sequencing and management strategies. Mol Genet Metab Rep (2014) 1:66–70. doi: 10.1016/j.ymgmr.2013.12.007
91
MahajanASidiropoulosC. TPK1 mutation induced childhood onset idiopathic generalized dystonia: report of a rare mutation and effect of deep brain stimulation. J Neurol Sci (2017) 376:42–3. doi: 10.1016/j.jns.2017.02.063
92
MurataT. Influence of thiamine on the amount of lactic acid in urine. J Vitaminol (Kyoto) (1958) 4:109–13. doi: 10.5925/jnsv1954.4.109
93
WendelOW. A study of urinary lactic acid levels in humans. J Vitaminol (Kyoto) (1960) 6:16–23. doi: 10.5925/jnsv1954.6.16
94
IsraelsSHaworthJCDunnHGApplegarthDA. Lactic acidosis in childhood. Adv Pediatr (1976) 22:267–303.
95
SweetRLZastreJA. HIF1-alpha-mediated gene expression induced by vitamin B1 deficiency. Int J Vitam Nutr Res (2013) 83:188–97. doi: 10.1024/0300-9831/a000159
96
AttwellDLaughlinSB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab (2001) 21:1133–45. doi: 10.1097/00004647-200110000-00001
97
MergenthalerPLindauerUDienelGAMeiselA. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci (2013) 36:587–97. doi: 10.1016/j.tins.2013.07.001
98
KarabatsiakisABockCSalinas-ManriqueJKolassaSCalziaEDietrichDEet al. Mitochondrial respiration in peripheral blood mononuclear cells correlates with depressive subsymptoms and severity of major depression. Transl Psychiatry (2014) 4:e397. doi: 10.1038/tp.2014.44
99
PrabakaranSSwattonJERyanMMHuffakerSJHuangJTGriffinJLet al. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psych (2004) 9:684-97:643. doi: 10.1038/sj.mp.4001511
100
LiuHSangSLuYWangZYuXZhongC. Thiamine metabolism is critical for regulating correlated growth of dendrite arbors and neuronal somata. Sci Rep (2017) 7:5342. doi: 10.1038/s41598-017-05476-w
101
GoldMChenMFJohnsonK. Plasma and red blood cell thiamine deficiency in patients with dementia of the Alzheimer’s type. Arch Neurol (1995) 52:1081–86. doi: 10.1001/archneur.1995.00540350075019
102
WhitfieldKCBourassaMWAdamolekunBBergeronGBettendorffLBrownKHet al. Thiamine deficiency disorders: diagnosis, prevalence, and a roadmap for global control programs. Ann N Y Acad Sci (2018) 1430:3–43. doi: 10.1111/nyas.13919
103
MolinaJAJimenez-JimenezFJHernanzAFernandez-VivancosEMedinaSde BustosFet al. Cerebrospinal fluid levels of thiamine in patients with Alzheimer’s disease. J Neural Transm (Vienna) (2002) 109:1035–44. doi: 10.1007/s007020200087
104
Ortigoza-EscobarJDMolero-LuisMAriasAOyarzabalADarinNSerranoMet al. Free-thiamine is a potential biomarker of thiamine transporter-2 deficiency: a treatable cause of Leigh syndrome. Brain (2016) 139:31–8. doi: 10.1093/brain/awv342
105
MagosL. Geigy scientific tables. In: LentnerC, editor. Units of measurement. Body fluids. Composition of the body. Nutrition, 8th ed. West Cadwell, N.J. (1987). p. 165–77.
106
Fattal-ValevskiA. Thiamine (vitamin B1). J Evidence-Based Compl Alt Med (2011) 16:12–20. doi: 10.1177/1533210110392941
107
WHO. International nomenclature of diseases. In: Metabolic, nutritional, and endocrine disorders, (1991). p. 278.
108
WilliamsRR. The classical deficiency diseases. Fed Proc (1961) 20(Suppl 7):323–27.
109
MulhollandPJ. Susceptibility of the cerebellum to thiamine deficiency. Cerebellum (2006) 5:55–63. doi: 10.1080/14734220600551707
110
NardoneRHollerYStortiMChristovaMTezzonFGolaszewskiSet al. Thiamine deficiency induced neurochemical, neuroanatomical, and neuropsychological alterations: a reappraisal. Sci World J (2013) 2013:309143. doi: 10.1155/2013/309143
111
InabaHKishimotoTOishiSNagataKHasegawaSWatanabeTet al. Vitamin B1-deficient mice show impairment of hippocampus-dependent memory formation and loss of hippocampal neurons and dendritic spines: potential microendophenotypes of Wernicke–Korsakoff syndrome. Biosci Biotechnol Biochem (2016) 80:2425–36. doi: 10.1080/09168451.2016.1224639
112
StargroveMBTreasureJMcKeeDL. Herb, nutrient, and drug interactions: clinical implications and therapeutic strategies. St. Louis (MO): Mosby/Elsevier (2008). p. 255.
113
ThomsonADMarshallEJ. The treatment of patients at risk of developing Wernicke’s encephalopathy in the community. Alcohol (2006) 41:159–67. doi: 10.1093/alcalc/agh250
114
GalvinRBrathenGIvashynkaAHillbomMTanasescuRLeoneMA. EFNS guidelines for diagnosis, therapy and prevention of Wernicke encephalopathy. Eur J Neurol (2010) 17:1408–18. doi: 10.1111/j.1468-1331.2010.03153.x
115
ArtsNJWalvoortSJKesselsRP. Korsakoff’s syndrome: a critical review. Neuropsychiatr Dis Treat (2017) 13:2875–90. doi: 10.2147/NDT.S130078
116
DayGSDel CampoCM. Wernicke encephalopathy: a medical emergency. CMAJ (2014) 186:E295. doi: 10.1503/cmaj.130091
117
ZuccoliGSiddiquiNBaileyABartolettiSC. Neuroimaging findings in pediatric Wernicke encephalopathy: a review. Neuroradiol (2010) 52:523–29. doi: 10.1007/s00234-009-0604-x
118
ZhangGDingHChenHYeXLiHLinXet al. Thiamine nutritional status and depressive symptoms are inversely associated among older Chinese adults. J Nutr (2013) 143:53–8. doi: 10.3945/jn.112.167007
119
CarneyMWWilliamsDGSheffieldBF. Thiamine and pyridoxine lack newly-admitted psychiatric patients. Br J Psychiatry (1979) 135:249–54. doi: 10.1192/bjp.135.3.249
120
PepersackTGarbusinskiJRobberechtJBeyerIWillemsDFussM. Clinical relevance of thiamine status amongst hospitalized elderly patients. Gerontology (1999) 45:96–101. doi: 10.1159/000022070
121
GhaleihaADavariHJahangardLHaghighiMAhmadpanahMSeifrabieMAet al. Adjuvant thiamine improved standard treatment in patients with major depressive disorder: results from a randomized, double-blind, and placebo-controlled clinical trial. Eur Arch Psychiatry Clin Neurosci (2016) 266:695–702. doi: 10.1007/s00406-016-0685-6
122
BentonDGriffithsRHallerJ. Thiamine supplementation mood and cognitive functioning. Psychopharmacology (1997) 129:66–71. doi: 10.1007/s002130050163
123
Fattal-ValevskiAAzouri-FattalIGreensteinYJGuindyMBlauAZelnikN. Delayed language development due to infantile thiamine deficiency. Dev Med Child Neurol (2009) 51:629–34. doi: 10.1111/j.1469-8749.2008.03161.x
124
Mimouni-BlochAGoldberg-SternHStrausbergRBreznerAHeymanEInbarDet al. Thiamine deficiency in infancy: long-term follow-up. Pediatr Neurol (2014) 51:311–16. doi: 10.1016/j.pediatrneurol.2014.05.010
125
HarelYZukLGuindyMNakarOLotanDFattal-ValevskiA. The effect of subclinical infantile thiamine deficiency on motor function in preschool children. Matern Child Nutr (2017) 13. doi: 10.1111/mcn.12397
126
LuxemburgerCWhiteNJKuileFSinghHMAllier-FrachonIOhnMet al. Beri-beri: the major cause of infant mortality in Karen refugees. Trans R Soc Trop Med Hyg (2003) 97:251–5. doi: 10.1016/S0035-9203(03)90134-9
127
BarennesHSengkhamyongKReneJPPhimmasaneM. Beriberi (thiamine deficiency) and high infant mortality in northern Laos. PLoS Negl Trop Dis (2015) 9:e0003581. doi: 10.1371/journal.pntd.0003581
128
VasconcelosMMSilvaKPVidalGSilvaAFDominguesRCBerditchevskyCR. Early diagnosis of pediatric Wernicke’s encephalopathy. Pediatr Neurol (1999) 20:289–94. doi: 10.1016/S0887-8994(98)00153-2
129
AlfadhelM. Early infantile Leigh-like SLC19A3 gene defects have a poor prognosis: report and review. J Cent Nerv Syst Dis (2017) 9:1179573517737521. doi: 10.1177/1179573517737521
130
Perez-DuenasBSerranoMRebolloMMuchartJGargalloEDupuitsCet al. Reversible lactic acidosis in a newborn with thiamine transporter-2 deficiency. Pediatrics (2013) 131:e1670–1675. doi: 10.1542/peds.2012-2988
131
BaioJWigginsLChristensenDLMaennerMJDanielsJWarrenZet al. Prevalence of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill Summ (2018) 67:1–23. doi: 10.15585/mmwr.ss6706a1
132
Ortega GarciaJAAnguloMGSobrino-NajulEJSoldinOPMiraAPMartinez-SalcedoEet al. Prenatal exposure of a girl with autism spectrum disorder to ‘horsetail’ (Equisetum arvense) herbal remedy and alcohol: a case report. J Med Case Rep (2011) 5:129. doi: 10.1186/1752-1947-5-129
133
AronsonMHagbergBGillbergC. Attention deficits and autistic spectrum problems in children exposed to alcohol during gestation: a follow-up study. Dev Med Child Neurol (1997) 39:583–7. doi: 10.1111/j.1469-8749.1997.tb07493.x
134
MahmoodSDaniHMMahmoodA. Effect of dietary thiamin deficiency on intestinal functions in rats. Am J Clin Nutr (1984) 40:226–34. doi: 10.1093/ajcn/40.2.226
135
LươngKVQNguyễnLTH. The role of thiamine in autism. Am J Psych Neurosci (2013) 1:22–37. doi: 10.11648/j.ajpn.20130102.11
136
McLureKGTakagiMKastanMB. NAD+ modulates p53 DNA binding specificity and function. Mol Cell Biol (2004) 24:9958–67. doi: 10.1128/MCB.24.22.9958-9967.2004
137
YangZGeJYinWShenHLiuHGuoY. The expression of p53, MDM2 and Ref1 gene in cultured retina neurons of SD rats treated with vitamin B1 and/or elevated pressure. Yan Ke Xue Bao (2004) 20:259–63.
138
IshaqueAAl-RubeaiM. Role of vitamins in determining apoptosis and extent of suppression by bcl-2 during hybridoma cell culture. Apoptosis (2002) 7:231–9. doi: 10.1023/A:1015343616059
139
RabieTMuhlhoferWBrucknerTSchwabABauerATZimmermannMet al. Transient protective effect of B-vitamins in experimental epilepsy in the mouse brain. J Mol Neurosci (2010) 41:74–9. doi: 10.1007/s12031-009-9286-4
140
ChornyySParkhomenkoJChornaN. Thiamine deficiency caused by thiamine antagonists triggers upregulation of apoptosis inducing factor gene expression and leads to caspase 3-mediated apoptosis in neuronally differentiated rat PC-12 cells. Acta Biochim Pol (2007) 54:315–22.
141
PlaitakisANicklasWJBerlS. Thiamine deficiency: selective impairment of the cerebellar serotonergic system. Neurology (1978) 28:691–8. doi: 10.1212/WNL.28.7.691
142
ChuganiDCMuzikORothermelRBehenMChakrabortyPMangnerTet al. Altered serotonin synthesis in the dentatothalamocortical pathway in autistic boys. Ann Neurol (1997) 42:666–9. doi: 10.1002/ana.410420420
143
LukienkoPIMel’nichenkoNGZverinskiiIVZabrodskayaSV. Antioxidant properties of thiamine. Bull Exp Biol Med (2000) 130:874–6. doi: 10.1007/BF02682257
144
FrustaciANeriMCesarioAAdamsJBDomeniciEDalla BernardinaBet al. Oxidative stress-related biomarkers in autism: systematic review and meta-analyses. Free Radic Biol Med (2012) 52:2128–41. doi: 10.1016/j.freeradbiomed.2012.03.011
145
GiuliviCZhangYFOmanska-KlusekARoss-IntaCWongSHertz-PicciottoIet al. Mitochondrial dysfunction in autism. JAMA (2010) 304:2389–96. doi: 10.1001/jama.2010.1706
146
GuFChauhanVKaurKBrownWTLaFauciGWegielJet al. Alterations in mitochondrial DNA copy number and the activities of electron transport chain complexes and pyruvate dehydrogenase in the frontal cortex from subjects with autism. Transl Psychiatry (2013) 3:e299. doi: 10.1038/tp.2013.68
147
AnwarAMariniMAbruzzoPMBolottaAGhezzoAViscontiPet al. Quantitation of plasma thiamine, related metabolites and plasma protein oxidative damage markers in children with autism spectrum disorder and healthy controls. Free Radic Res (2016) 50:S85–S90. doi: 10.1080/10715762.2016.1239821
148
MulleJGSharpWGCubellsJF. The gut microbiome: a new frontier in autism research. Curr Psychiatry Rep (2013) 15:337. doi: 10.1007/s11920-012-0337-0
149
LonsdaleDShambergerRJAudhyaT. Treatment of autism spectrum children with thiamine tetrahydrofurfuryl disulfide: a pilot study. Neuro Endocrinol Lett (2002) 23:303–8.
150
ObrenovichMEShambergerRJLonsdaleD. Altered heavy metals and transketolase found in autistic spectrum disorder. Biol Trace Elem Res (2011) 144:475–86. doi: 10.1007/s12011-011-9146-2
151
AdamsJBAudhyaTDonough-MeansSRubinRAQuigDGeisEet al. Effect of a vitamin/mineral supplement on children and adults with autism. BMC Pediatr (2011) 11:111. doi: 10.1186/1471-2431-11-111
152
Rubio-LopezNMorales-Suarez-VarelaMPicoYLivianos-AldanaLLlopis-GonzalezA. Nutrient intake and depression symptoms in Spanish children: the ANIVA study. Int J Environ Res Public Health (2016) 13. doi: 10.3390/ijerph13030352
153
CheungAHKozloffNSacksD. Pediatric depression: an evidence-based update on treatment interventions. Curr Psychiatry Rep (2013) 15:381. doi: 10.1007/s11920-013-0381-4
154
ColbertPNewmanBNeyPYoungJ. Learning disabilities as a symptom of depression in children. J Learn Disabil (1982) 15:333–6. doi: 10.1177/002221948201500605
155
SocietyCP. Maternal depression and child development. Paediatr Child Health (2004) 9:575–98. doi: 10.1093/pch/9.8.575
156
NiksereshtSEtebarySKarimianMNabavizadehFZarrindastMRSadeghipourHR. Acute administration of Zn, Mg, and thiamine improves postpartum depression conditions in mice. Arch Iran Med (2012) 15:306–11. doi: 012155/AIM.0012
Summary
Keywords
autism spectrum disorders, brain, depressive disorders, encephalomyopathies, Krebs cycle, pentose phosphate pathway, thiamine transporter
Citation
Dhir S, Tarasenko M, Napoli E and Giulivi C (2019) Neurological, Psychiatric, and Biochemical Aspects of Thiamine Deficiency in Children and Adults. Front. Psychiatry 10:207. doi: 10.3389/fpsyt.2019.00207
Received
08 January 2019
Accepted
22 March 2019
Published
04 April 2019
Volume
10 - 2019
Edited by
Shannon Rose, University of Arkansas for Medical Sciences, United States
Reviewed by
Brahim Tabarki, University of Sousse, Tunisia; Majid Alfadhel, King Saud bin Abdulaziz University for Health Sciences, Saudi Arabia
Updates
Copyright
© 2019 Dhir, Tarasenko, Napoli and Giulivi.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cecilia Giulivi, cgiulivi@ucdavis.edu
†These authors have contributed equally to this work.
This article was submitted to Molecular Psychiatry, a section of the journal Frontiers in Psychiatry
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.