 Eva M. Lüngen1,2†Viktoria Maier1,2†
Eva M. Lüngen1,2†Viktoria Maier1,2† Nils Venhoff3
Nils Venhoff3 Ulrich Salzer3
Ulrich Salzer3 Rick Dersch4Benjamin Berger4Anne N. Riering2
Rick Dersch4Benjamin Berger4Anne N. Riering2 Kathrin Nickel1,2
Kathrin Nickel1,2 Bernd L. Fiebich2
Bernd L. Fiebich2 Patrick Süß1,2
Patrick Süß1,2 Simon J. Maier1,2
Simon J. Maier1,2 Karl Egger5
Karl Egger5 Ludger Tebartz van Elst1,2
Ludger Tebartz van Elst1,2 Dominique Endres1,2*
Dominique Endres1,2*- 1Section for Experimental Neuropsychiatry, Department of Psychiatry and Psychotherapy, Medical Center–University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 2Department of Psychiatry and Psychotherapy, Medical Center–University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 3Department of Rheumatology and Clinical Immunology, Medical Center–University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 4Department of Neurology, Medical Center–University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 5Department of Neuroradiology, Medical Center–University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
Background: Organic psychiatric disorders can be caused by immunological disorders, such as autoimmune encephalitis or systemic lupus erythematosus (SLE). SLE can affect most organs, as well as the central nervous system (CNS). In this paper, we describe a patient with an isolated psychiatric syndrome in the context of SLE and discuss the role of antibody detection in the cerebrospinal fluid (CSF).
Case presentation: The 22-year-old German male high school graduate presented with obsessive–compulsive and schizophreniform symptoms. He first experienced obsessive–compulsive symptoms at the age of 14. At the age of 19, his obsessive thoughts, hallucinations, diffuse anxiety, depressed mood, severe dizziness, and suicidal ideation became severe and did not respond to neuroleptic or antidepressant treatment. Due to increased antinuclear antibodies (ANAs) with anti-nucleosome specificity in serum and CSF, complement activation, multiple bilateral white matter lesions, and inflammatory CSF alterations, we classified the complex syndrome as an isolated psychiatric variant of SLE. Immunosuppressive treatment with two times high-dose steroids, methotrexate, and hydroxychloroquine led to a slow but convincing improvement.
Conclusion: Some patients with psychiatric syndromes and increased ANA titers may suffer from psychiatric variants of SLE, even if the American College of Rheumatology criteria for SLE are not met. Whether the psychiatric symptoms in our patient represent a prodromal stage with the later manifestation of full-blown SLE or a subtype of SLE with isolated CNS involvement remains unclear. Regardless, early diagnosis and initiation of immunosuppressive treatment are essential steps in preventing further disease progression and organ damage. Intrathecal ANAs with extractable nuclear antigen differentiation may be a more sensitive marker of CNS involvement compared with serum analyses alone.
Background
Organic psychiatric disorders might be of immunological, infectious, epileptic, neurodegenerative, traumatic, metabolic, or vascular origins (1, 2). In recent years, limbic or nonlimbic autoimmune encephalitis received increased interest because each can mimic primary psychiatric and neuropsychiatric disorders (2–4). Most of these disorders are associated with autoantibodies (abs) directed against antigens on the cell surface or in the intracellular compartment (5). Hashimoto thyroiditis and rheumatic disorders, such as systemic lupus erythematosus (SLE), can also be associated with psychiatric involvement that allows for successful immunomodulatory treatment approaches (6–9).
SLE is a prototypic systemic autoimmune disease that can affect the central nervous system (CNS) as well as the rest of the body, including joints, skin, kidneys, heart, lungs, blood vessels, or the hematopoietic system. The typical age for mostly women to become ill is between 16 and 55 years (10); the peak age in females is between 45 and 69 years, while that in males is between 40 and 89 years (11). SLE is characterized by the presence of antinuclear abs (ANAs); these abs can affect different cell types, which explains the diversity of symptoms. The reference standard for the diagnosis of this extremely heterogeneous multisystemic disease is still clinical diagnosis by an SLE expert. The main reason for the use of SLE classification criteria is to ensure a consistent definition of SLE, especially for clinical trials and surveillance. The most commonly used criteria are those established by the American College of Rheumatology (ACR) in 1982 and revised in 1997 (12, 13; https://www.rheumatology.org/Practice-Quality/Clinical-Support/Criteria/ACR-Endorsed-Criteria; see Box 1A). If 4 or more of the 11 criteria, including at least 1 immunological and 1 clinical criterion, are present simultaneously or serially at any point in time, SLE should be highly suspected. Although the ACR’s 1997 criteria have generally performed well, concerns have been raised regarding the limited sensitivity of these criteria. Neuropsychiatric conditions, for example, might be underrepresented in the criteria. Up to 75% develop neuropsychiatric symptoms, such as cognitive dysfunction, seizures, and other psychiatric syndromes, such as mood disorders, anxiety, or psychosis (14). A subgroup of SLE patients mainly presents with manifestations involving the central, peripheral, or autonomic nervous system. These are referred to as neuropsychiatric SLE (NPSLE) (15). In 1999, the ACR published 19 case definitions of NPSLE to facilitate diagnosis and ensure comparability across clinical studies. Twelve definitions described syndromes associated with CNS involvement (15) (Box 1B).
Box 1. Criteria for (neuropsychiatric) systemic lupus erythematosus
A: Criteria for systemic lupus erythematosus of the American College of Rheumatology (12,13; https://www.rheumatology.org/Practice-Quality/Clinical-Support/Criteria/ACR-Endorsed-Criteria)
1. Malar rash; 2. discoid rash; 3. photosensitivity; 4. oral ulcers; 5. arthritis; 6. serositis; 7. renal involvement; 8. neurological involvement (seizures or psychosis); 9. hematological involvement (hemolytic anemia, leucopenia, lymphopenia, thrombocytopenia); 10. immunological changes (anti-dsDNA antibodies, anti-Sm antibodies, antiphospholipid antibodies, and 11. antinuclear antibodies.
B: Case definitions of neuropsychiatric systemic lupus erythematosus (15):
1. Aseptic meningitis; 2. cerebrovascular disease; 3. demyelinating syndrome; 4. headaches (migraines and benign intracranial hypertension); 5. movement disorders (chorea); 6. myelopathy; 7. seizure disorders; 8. acute confusional states; 9. anxiety disorders; 10. cognitive dysfunction; 11. mood disorders; and 12. psychosis.
Two distinct pathogenic mechanisms, tissue inflammation and thrombotic–ischemic events, have been recognized thus far. Increased ANA serum titers were detected in >95% of patients (16); however, older studies showed even lower ANA positivity (17). Positive dsDNA abs were found at a rate of 37–80% (16), and they were interpreted as a predictor of disease exacerbation (18). The metanalytic data showed that anti-nucleosome abs have comparable specificity but higher sensitivity than anti-dsDNA abs do for the diagnosis of SLE (19). Cerebrospinal fluid (CSF) pleocytosis was found in 30%, and oligoclonal bands (OCBs) were detected in 25–42% of cases (20). In the cerebral magnetic resonance imaging (cMRI), small focal hyperintensities, mainly subcortical frontoparietal or periventricular, can be detected in 15–60% of the patients. Electroencephalography (EEG) generally shows unspecific slowing (21). NPSLE treatment can be implemented with corticosteroids alone or in combination with other immunosuppressive drugs, including cyclophosphamide for remission induction or azathioprine for maintenance therapy (22). Antimalarial drugs (e.g., hydroxychloroquine) have been suggested for the prevention of NPSLE in SLE patients (23).
Case Presentation
We present the case of a 22-year-old German male high school graduate with a complex psychiatric syndrome including obsessive–compulsive, schizophreniform, and derealization phenotypes. In May 2016, at age 19, there was a sudden exacerbation of these syndromes. At age 14, he first experienced obsessive–compulsive symptoms (i.e., obsessive aggressive thoughts and compulsive avoidance acts). However, he recognized that the obsessional thoughts were a product of his own mind, and these symptoms were well compensated for at the time, enabling him to live a mostly normal life. After his final examinations at school, he consumed cannabis five times. He then experienced an exacerbation of his obsessive–compulsive symptoms and suffered from more severe obsessional thoughts, including the idea that he could injure other people and himself. Furthermore, he experienced involuntary obscene thoughts. At the time, he fought such thoughts and continued to recognize that the obsessional thoughts and impulses were a product of his own mind. He also suffered from hallucinatory symptoms, such as auditory hallucinations (i.e., hearing voices) and optical distortions (i.e., the shape of leaves on the ground appearing distorted). He developed diffuse anxiety and agitation and described extreme dizziness, as if he had drunk “five beers.” Because of his depressed mood and obsessive–compulsive symptoms, he experienced suicidal ideation and complained of difficulty falling asleep and reduced energy levels, especially in the morning.
Due to the severity of these symptoms, he was first hospitalized at age 19. He received pharmacological treatment with selective serotonin reuptake inhibitors for the obsessive–compulsive symptoms (citalopram up to 40 mg/day), neuroleptics for the schizophreniform symptoms (olanzapine up to 20 mg/day, risperidone up to 5 mg/day, and aripiprazole up to 7.5 mg/day; higher doses led to an increase in inner restlessness), carbamazepine for neuronal network stabilization up to 500 mg/day, and an anticholinergic agent because of extrapyramidal side effects (biperiden up to 4 mg/day) over a period of 7 months without significant improvement. Lorazepam (up to 2 mg/day) led to a transient reduction in anxiety. Seven months after symptom onset (December 2016, under treatment with aripiprazole 7.5 mg/day and carbamazepine 500 mg/day), his mental state still revealed obsessive thoughts, depressed mood, and diffuse anxiety. Moreover, he suffered from attention and concentration deficits (Figure 1, t0), inner restlessness, signs of derealization (with altered, slowed, and delayed perception of his environment), and severe dizziness (still comparable with having drunk five beers). The obsessive–compulsive thoughts were still present and very difficult to manage.
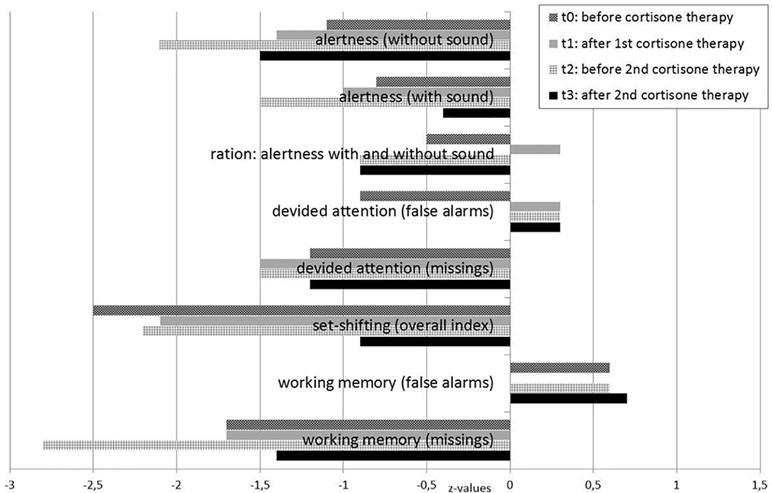
Figure 1 Test for attentional performance during the course of the disease.
Developmental, Somatic, and Family History
There were no in utero or birth complications, febrile convulsions, seizures, inflammatory brain diseases, or cerebral contusions in the patient’s history. When entering primary school, he showed subsyndromal symptoms of inattention and motor hyperactivity. Nevertheless, he finished high school successfully and his further somatic history was unremarkable. He occasionally consumed alcohol and illegal drugs (nitrous oxide three times and cannabis five times), but there was no history of severe substance abuse. The family history showed that his grandmother suffered from depression, and his mother was diagnosed with insulin-dependent diabetes mellitus. There were no known rheumatic diseases in the family history.
Investigations
The neurological examination was normal throughout the course of the disease. Initially, the CSF analyses (3 months after exacerbation, August 2016) showed positive CSF-specific OCBs. Five months after the first steroid pulse treatment (December 2016), the patient’s state deteriorated (May 2017). At that time, CSF analysis showed a mild pleocytosis (white blood cell count = 14/µl; reference <5/µl). The initial immunological screening 6 months after exacerbation in November 2016 revealed only a weak positive ANA in the indirect immunofluorescence assay. Another 6 months later (1 year after exacerbation, May 2017), we found clearly increased ANA titers in both serum and CSF (serum: titer = 800 IU; CSF: titer <100 IU) with anti-nucleosome specificity, which was also detectable in serum and CSF. At that time, we also detected decreased levels of complement component C4 and slightly increased C3d serum concentrations as indicators for increased complement activation.
Testing for rheumatoid factors, antiphospholipid abs, lupus anticoagulant, antineutrophil cytoplasmic abs, and a broad set of antineuronal and anti-thyroid abs was negative. In the cMRI, multiple diffuse periventricular white matter lesions were apparent in repeated examinations throughout the course (Figure 2). The lesions were stable. Furthermore, there was a slightly enlarged adenohypophysis not yet affecting the chiasma opticum. The hormone screening did not detect any pathological hormone activity. The fluorodeoxyglucose positron emission tomography was normal. Repeated EEGs exhibited intermittent slowing (Table 1). The neuropsychological test of attentional performances showed severe deficits in alertness, divided attention, set shifting, and working memory (Figure 1, t0). There were no further clinical, systemic SLE signs such as skin or inner organ involvement.
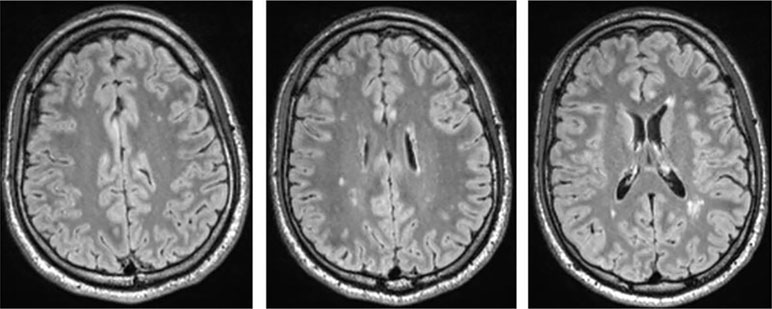
Figure 2 T2w fluid-attenuated inversion recovery (FLAIR) cerebral magnetic resonance imaging (cMRI) shows multiple disseminated dotted bilateral periventricular to subcortical white matter lesions. Shown are images of the first cMRI performed 6 months after symptom exacerbation in November 2016.

Table 1 Overview of diagnostic findings.
Differential Diagnostic Considerations
Focusing on the symptoms our patient developed during his youth, a diagnosis of obsessive–compulsive disorder with obsessive thoughts could be considered. The fact that disease symptoms exacerbated shortly after the consumption of cannabis could point to an acute episode of drug-induced psychosis. If the auditory and optical hallucinations together with signs of derealization persisted over time, paranoid hallucinatory schizophrenia could be a plausible classification, though only if all organic signs were considered pathogenetically irrelevant. The initial organic findings (positive CSF-specific OCBs, disseminated white matter lesions, no antineuronal abs, unspecific rheumatological findings) were interpreted as post-inflammatory changes. Finally, the increased ANAs with anti-nucleosome specificity in CSF and serum, serum complement activation, CSF pleocytosis, OCBs, and cognitive and psychiatric disturbances cumulated to the final diagnosis of NPSLE. Although the patient did not completely fulfill the ACR criteria for SLE, the diagnosis was established. Other supporting biomarkers, such as anti-ribosomal P protein and anti-C1q abs, as well as the direct Coombs test, were unremarkable. Autoimmune encephalitis would also be conceivable for differential diagnosis (4). However, the established antineuronal abs were negative in serum and CSF.
Immunosuppressive Treatment and Course of the Disease
Following initial assessment and based on the therapy resistance and inflammatory CSF changes with CSF-specific OCBs, we performed a high-dose corticosteroid pulse therapy with a daily dose of 1,000 mg methylprednisolone administered intravenously for five consecutive days in the context of continued treatment with aripiprazole and carbamazepine in December 2016, which was 7 months after symptom exacerbation. Following the steroid treatment, the patient’s mood, cognitive deficits, motivation, and dizziness improved. Moreover, his obsessive–compulsive symptoms were reduced. Methylprednisolone was subsequently continued orally and tapered over approximately 5 weeks (37 days). Except for steroid acne, no relevant adverse events became evident. After this immunosuppressive treatment, the patient was able to begin a volunteer social year, and treatment with aripiprazole and carbamazepine was continued. However, he still had obsessive–compulsive symptoms and cognitive deficits and felt dizzy (comparable with having drunk two or three beers). Clinically, we did not believe that these symptoms were side effects of carbamazepine because the symptoms were already reported prior to the treatment with carbamazepine, and blood concentrations were within the reference level. Neuropsychological testing showed no relevant improvement (Figure 1, t1). Over the next 4 months, his mental condition worsened again.
In May 2017, 1 year after his initial symptoms exacerbated, we repeated the diagnostic workup. The patient showed mild pleocytosis in the CSF and laboratory findings compatible with SLE (Table 1). We performed a second steroid pulse therapy with 500 mg/day methylprednisolone for five consecutive days with oral tapering over 8 weeks. At the same time, carbamazepine was stopped, and aripiprazole was reduced to 5 mg/day. As maintenance treatment, the patient received methotrexate (up to 17.5 mg/week) orally in combination with folic acid. Due to the threat of azoospermia, cyclophosphamide, which normally shows good results for NPSLE, was not initially used. The CSF white blood cell count normalized, and the cMRI showed that the multiple diffuse periventricular to subcortical white matter lesions remained stable. Neuropsychological testing revealed an improvement in alertness, mental flexibility, and working memory [Figure 1, t2 (before) versus t3 (after) the second steroid pulse treatment]. Two months later (August 2017, 15 months after his symptoms first exacerbated), we added the antimalarial drug hydroxychloroquine (200 mg/day). The maintenance therapy consisting of methotrexate (17.5 mg/week), hydroxychloroquine (200 mg/day), and aripiprazole (5 mg/day) resulted in a slow but substantial improvement of inner restlessness, cognitive deficits, derealization symptoms, and dizziness. Obsessive–compulsive symptoms disappeared completely. Over several months, he still felt dizzy, like “having drunk one beer.” Sixteen months later (December 2018), the patient was with without dizziness (“zero beers”), aripiprazole treatment was stopped in the meantime, and he enrolled in a vocational training program. Here, he was able to attend vocational school, but on long working days, he cognitively reached his limits.
Discussion
In this paper, we present the case of a male patient with a severe psychiatric variant of NPSLE suffering from obsessive–compulsive and schizophreniform symptoms who responded slowly but very well to immunological treatment.
Diagnostic Considerations
Initially, the cMRI and CSF showed signs of inflammatory CNS involvement (i.e., disseminated periventricular white matter lesions and isolated OCBs in the CSF). At that time, the ANA immunofluorescence test was only weakly positive. In view of the organic findings, the other differential diagnostic considerations mentioned above (e.g., drug-induced disorder, obsessive–compulsive disorder, or schizophreniform syndrome) were rejected. Six months after the first assessment, the ANA titers had clearly increased, with anti-nucleosome specificity found in the CSF and serum. Along with the complement alterations, the inflammatory changes in the CSF, the cMRI and EEG alterations, as well as the clinical manifestation with psychosis, we diagnosed NPSLE. Although the ACR 1997 classification criteria for SLE were not completely fulfilled, we deemed it justified to diagnose NPSLE in this patient. The strongest indicators were as follows: A) the presence of ANAs in a young male patient, which is unusual and raises suspicion for SLE; B) the detection of anti-nucleosome abs, which have the same specificity and even higher sensitivity than anti-dsDNA abs for SLE (19); C) complement activation as an indicator for disease activity; and D) the clinical presentation with psychiatric symptoms and corresponding alterations in CSF diagnostics, cMRI, and EEG.
Neuropsychiatric manifestations usually occur early in the course of SLE, and in some patients, neuropsychiatric symptoms remain the only clinical manifestations of SLE. This case demonstrates the difference between classification and diagnosis. Classification defines a homogenous group of patients for a research purpose with suboptimal sensitivity, whereas diagnosis focuses on the individual patient’s therapy and prognosis and diagnosis should also lead to the treatment of patients who do not meet the classification due to non-100% sensitivity but who should still receive treatment (24). There are limitations of the most widely used ACR 1997 classification (but not diagnostic) criteria for SLE. While photosensitivity and skin and mucous membrane involvement seems to be overrepresented, other conditions, such as neuropsychiatric involvement, might be underrepresented. Furthermore, important immunological tests, such as complement fractions, anti-b2-glycoprotein, or anti-nucleosome abs, have not been considered thus far. While our patient fulfilled only 2 (i.e., psychosis and the presence of ANA) of the possible 11 ACR criteria, he would have fulfilled 3 (i.e., psychosis, the presence of ANA, and lowered C4 levels) of the recently proposed Systemic Lupus International Collaborating Clinics (SLICC) 2012 SLE classification criteria set (25). According to the SLICC 2012 criteria, the classification of SLE also requires at least four criteria with at least one clinical and one laboratory criteria, but this case is a good example of the relevance of the composition of the applied classification criteria set.
The European League Against Rheumatism and ACR are aware of this situation, and therefore, new SLE classification standards based on weighted criteria and a continuous probability scale are currently being developed. Recent studies investigated whether the detection of certain abs (e.g., increased ANA titers) in combination with symptoms manifested in one organ system (i.e., incomplete clinical pictures of SLE) may point to a prodromal stage of SLE (26, 27). Several papers described cases with initially insufficient numbers of diagnostic findings to fulfill the ACR criteria for SLE, for example, cases with initial manifestations of the gastrointestinal tract that later developed into full-blown SLE (28, 29). There are also many cases of SLE with the presence of ANA and isolated kidney involvement with glomerulonephritis. Despite not fulfilling the ACR classification criteria, the diagnosis can be made in cases of biopsy-proven lupus nephritis. Mack et al. published a case report of a woman who suffered from a wide spectrum of psychiatric symptoms that had relapsed several times over the 25-year disease duration. Throughout the entire period, several findings indicated an immunological process as a cause of her illness, but it took approximately 23 years after first suspecting an autoimmune disorder before the diagnosis of (NP)SLE was made (9). Given this background, we conclude that an isolated psychiatric variant of SLE might well be plausible in our patient. Whether the psychiatric symptoms in our patient represent a prodromal stage with the later manifestation of full-blown SLE or a subtype of SLE with isolated CNS involvement remains unclear. Because of the autoimmune pathogenesis of this systemic disease, the continuation of the immunosuppressive maintenance therapy should prevent further disease progression and organ damage. Nevertheless, even if the patient does not currently fulfill the criteria for SLE, early diagnosis is possible, and initiating immunosuppressive treatment is essential. Therefore, it is important to consider that isolated psychiatric variants of SLE may occur without other organ manifestations.
Role of Cerebrospinal Fluid Antibody Detection
In our case, we detected ANAs with anti-nucleosome specificity not only in the serum but also in the CSF, which supported the hypothesis that the CNS is affected. Even if the CSF examinations are not established, one could hypothesize that the short-term cannabis use may have led to a temporary blood–brain barrier dysfunction and allowed the ANAs’ passage to the CNS. This could explain the rapid deterioration after cannabis consumption. Besides direct ab effects, increased interferon signaling might lead to reactive microglia and therefore could lead to neuronal damage and loss of synapses (30). Nevertheless, the precise pathophysiology of immunological mechanisms in affected brain tissue and thereby the resulting neuropsychiatric symptoms have not yet been fully clarified (31).
However, if the detection of ANAs in the CSF is a marker of CNS involvement, this would be of immense help in clinical practice. ANAs detected in serum alone are too unspecific to function as a marker for CNS involvement in clinical practice. They were found with similar prevalence rates in patients with schizophrenia and in controls in earlier studies (32). Higher rates of serum ANAs might also be due to drug-induced ANA titers, which are often observed in psychiatric patients (33). Therefore, the serum findings’ significance remains unclear in individual patients with psychiatric disorders. CSF analyses could help diagnose an autoimmune connective tissue disease with CNS involvement when detecting increased intrathecal ANA titers and other inflammatory alterations, such as increased white blood cell counts and CSF-specific OCBs. A metanalysis showed a higher rate of positive titers for antineuronal abs in the CSF of patients with NPSLE compared with SLE patients (34). In classical patients with NPSLE, an intrathecal ab synthesis was previously shown (35). Anti-SSA abs were found in CSF of patients with SLE (36) and Sjögren’s syndrome earlier (37). Future studies should investigate the sensitivity and specificity of intrathecal ANA synthesis with ENA specification in psychiatric variants of SLE.
Conclusion
Our case clearly illustrates the relevance of this issue for clinical psychiatry. Had we rejected the SLE diagnosis based on the existing classification criteria, we would not have chosen the immune-modulatory treatment approach, which proved to be extremely successful in the long run in our patient. Intrathecal ANAs with extractable nuclear antigen differentiation may be a more sensitive marker of CNS involvement than serological testing alone is.
Ethics Statement
The patient has given his signed written informed consent for this case report, including the presented images, to be published.
Author Contributions
DE, LT, NV, and RD treated the patient. EL, VM, and DE performed the data research: EL and DE summarized the case report, and VM performed a literature search. EL, VM, and DE wrote the paper. KE performed the cMRI analyses. RD and BB performed the EEG and CSF analyses and neurological interpretation. NV and US performed the rheumatological analyses, clinical interpretation and therapy suggestion. AR performed the neuropsychological testing. BF, SM, PS, and KN supported the interpretation of diagnostic findings. All authors were critically involved in the theoretical discussion and composition of the manuscript. All authors read and approved the final version of the manuscript.
Funding
The article processing charge was funded by the German Research Foundation (DFG) and the University of Freiburg in the funding program Open Access Publishing.
Conflict of Interest Statement
NV: Advisory boards, lectures, research or travel grants within the last three years: Janssen-Cilag, Roche, Novartis, AbbVie, GSK, Medac, Pfizer. BB: Travel grants and/or training expenses from Bayer Vital GmbH, Ipsen Pharma GmbH, Biogen GmbH, Norvartis, and Genzyme, as well as lecture fees from Ipsen Pharma GmbH, Alexion Pharma GmbH, Sanofi Aventis GmbH, Roche Pharma AG and Merck Serono GmbH. LT: Advisory boards, lectures, or travel grants within the last three years: Eli Lilly, Janssen-Cilag, Novartis, Shire, UCB, GSK, Servier, Janssen, and Cyberonics. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Lishman A. Lishman’s organic psychiatry: a textbook of neuropsychiatry. 4th ed. Hoboken, New Jersey: Wiley-Blackwell Pub. (2009).
2. Tebartz van Elst L. Vom Anfang und Ende der Schizophrenie: Eine neuropsychiatrische Perspektive auf das Schizophrenie-Konzept: Stuttgart: Kohlhammer. Stuttgart: Kohlhammer (2017). ISBN: 978-3-17-031258-6.
3. Tebartz van Elst L, Stich O, Endres D. Depressionen und Psychosen bei immunologischen Enzephalopathien. PSYCH up2date (2015) 9:265–80. doi: 10.1055/s-0041-102941
4. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol (2016) 15:391–404. doi: 10.1016/S1474-4422(15)00401-9
5. Stich O, Rauer S. Paraneoplastische neurologische Syndrome und Autoimmunenzephalitiden [Paraneoplastic neurological syndromes and autoimmune encephalitis]. Nervenarzt (2014) 85:485–98. doi: 10.1007/s00115-014-4030-x
6. Endres D, Perlov E, Stich O, van Tebartz Elst L. Steroid responsive encephalopathy associated with autoimmune thyroiditis (SREAT) presenting as major depression. BMC Psychiatry (2016) 16:184 doi: 10.1186/s12888-016-0897-3
7. Endres D, Perlov E, Riering AN, Maier V, Stich O, Dersch R, et al. Steroid-responsive chronic schizophreniform syndrome in the context of mildly increased antithyroid peroxidase antibodies. Front Psychiatry (2017) 8:64. doi: 10.3389/fpsyt.2017.00064
8. Endres D, Vry MS, Dykierek P, Riering AN, Lüngen E, Stich O, et al. Plasmapheresis responsive rapid onset dementia with predominantly frontal dysfunction in the context of Hashimoto’s encephalopathy. Front Psychiatry (2017) 8:212. doi: 10.3389/fpsyt.2017.00212
9. Mack A, Pfeiffer C, Schneider EM, Bechter K. Schizophrenia or atypical lupus erythematosus with predominant psychiatric manifestations over 25 years: case analysis and review. Front Psychiatry (2017) 8:131. doi: 10.3389/fpsyt.2017.00131
10. Tamirou F, Arnaud L, Talarico R, Scirè CA, Alexander T, Amoura Z, et al. Systemic lupus erythematosus: state of the art on clinical practice guidelines. RMD Open (2018) 4(2):e000793. doi: 10.1136/rmdopen-2018-000793
11. Rees F, Doherty M, Grainge MJ, Lanyon P, Zhang W. The worldwide incidence and prevalence of systemic lupus erythematosus: a systematic review of epidemiological studies. Rheumatology (Oxford) (2017) 56(11):1945–61. doi: 10.1093/rheumatology/kex260
12. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum (1982) 25(11):1271–7. doi: 10.1002/art.1780251101
13. Hochberg MC. Updating the American college of rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum (1997) 40:1725. doi: 10.1002/art.1780400928
14. Jeltsch-David H, Muller S. Neuropsychiatric systemic lupus erythematosus: pathogenesis and biomarkers. Nat Rev Neurol (2014) 10(10):579–96. doi: 10.1038/nrneurol.2014.148.
15. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum (1999) 42:599–608. doi: 10.1002/1529-0131(199904)42:4<599::AID-ANR2>3.0.CO;2-F
16. Wichainun R, Kasitanon N, Wangkaew S, Hongsongkiat S, Sukitawut W, Louthrenoo W. Sensitivity and specificity of ANA and anti-dsDNA in the diagnosis of systemic lupus erythematosus: a comparison using control sera obtained from healthy individuals and patients with multiple medical problems. Asian Pac J Allergy Immunol (2013) 31(4):292–8. doi: 10.12932/AP0272.31.4.2013
17. Sjöwall C, Sturm M, Dahle C, Bengtsson AA, Jönsen A, Sturfelt G, et al. Abnormal antinuclear antibody titers are less common than generally assumed in established cases of systemic lupus erythematosus. J Rheumatol (2008) 35(10):1994–2000.
18. ter Borg EJ, Horst G, Hummel EJ, Limburg PC, Kallenberg CG. Measurement of increases in anti-double-stranded DNA antibody levels as a predictor of disease exacerbation in systemic lupus erythematosus. Arthritis Rheum (1990) 33(5):634–43. doi: 10.1002/art.1780330505
19. Bizzaro N, Villalta D, Giavarina D, Tozzoli R. Are anti-nucleosome antibodies a better diagnostic marker than anti-dsDNA antibodies for systemic lupus erythematosus? A systematic review and a study of metanalysis. Autoimmun Rev (2012) 12(2):97–106. doi: 10.1016/j.autrev.2012.07.002
20. Weiner SM, Otte A, Uhl M, Brink I, Schumacher M, Peter HH. Neuropsychiatric involvement in systemic lupus erythematosus. Part 2: diagnostic and therapy. Med Klin (Munich) (2003) 98(2):79–90. doi: 10.1007/s00063-003-1230-8
21. Hufschmidt A, Lücking CH, Rauer S. Neurologie compact. Stuttgart: Georg Thieme Verlag (2013). ISBN: 9783131171962.
22. Bertsias GK, Ioannidis JPA, Aringer M, Bollen E, Bombardieri S, Bruce IN, et al. EULAR recommendations for the management of systemic lupus erythematosus with neuropsychiatric manifestations: report of a task force of the EULAR standing committee for clinical affairs. Ann Rheum Dis (2010) 69:2074–82. doi: 10.1136/ard.2010.130476
23. Petri M. Use of hydroxychloroquine to prevent thrombosis in systemic lupus erythematosus and in antiphospholipid antibody-positive patients. Curr Rheumatol Rep (2011) 13(1):77–80. doi: 10.1007/s11926-010-0141-y
24. Aringer M, Dörner T, Leuchten N, Johnson SR. Toward new criteria for systemic lupus erythematosus—a standpoint. Lupus (2016) 25(8):805–11. doi: 10.1177/0961203316644338
25. Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum (2012) 64(8):2677–86. doi: 10.1002/art.34473
26. Swaak AJG. Incomplete lupus erythematosus: results of a multicentre study under the supervision of the EULAR Standing Committee on International Clinical Studies Including Therapeutic Trials (ESCISIT). Rheumatology (2001) 40:89–94. doi: 10.1093/rheumatology/40.1.89
27. Chen Z, Li M-T, Xu D, Leng X-M, Wang Q, Tian X-P, et al. Organ damage in patients with incomplete lupus syndromes: from a Chinese academic center. Clin Rheumatol (2015) 34:1383–9. doi: 10.1007/s10067-015-2884-3
28. Fabio G, Carrabba M, Hu C, Floriani M, Besana C. Dramatic development of severe SLE in a patient with an incomplete disease. Rheumatol Int (2005) 25:543–7. doi: 10.1007/s00296-004-0550-1
29. Lee HA, Shim HG, Seo YH, Choi SJ, Lee BJ, Lee YH, et al. Panenteritis as an initial presentation of systemic lupus erythematosus. Korean J Gastroenterol (2016) 67:107–11. doi: 10.4166/kjg.2016.67.2.107
30. Bialas AR, Presumey J, Das A, van der Poel CE, Lapchak PH, Mesin L, et al. Microglia-dependent synapse loss in type I interferon-mediated lupus. Nature (2017) 546(7659):539–43. doi: 10.1038/nature22821
31. Faria R, Gonçalves J, Dias R. Neuropsychiatric systemic lupus erythematosus involvement: towards a tailored approach to our patients? Rambam Maimonides Med J (2017) 8(1). doi: 10.5041/RMMJ.10276
32. van Mierlo HC, Witte L de, Derksen RH, Otten HG. The prevalence of antinuclear antibodies in patients with schizophrenia spectrum disorders: results from a large cohort study. NPJ Schizophr (2015) 1:15013. doi: 10.1038/npjschz.2015.13
33. Audemard-Verger A, Comby E, Nathou C, Sultan A, Frémont M, Baldolli A, et al. Is it relevant to screen young women hospitalized in psychiatric department for neuropsychiatric systemic lupus erythematosus (NPSLE)?: a prospective study of 100 psychiatric inpatients. Medicine (Baltimore) (2016) 95:e5288. doi: 10.1097/MD.0000000000005288
34. Ho RC, Thiaghu C, Ong H, Lu Y, Ho CS, Tam WW, et al. A meta-analysis of serum and cerebrospinal fluid autoantibodies in neuropsychiatric systemic lupus erythematosus. Autoimmun Rev (2016) 15(2):124–38. doi: 10.1016/j.autrev.2015.10.003
35. Lu XY, Ye S, Wang Y, Guo L, Chen XX, Fan W, et al. [Clinical significance of neuro-reactive autoantibodies in neuro-psychiatric systemic lupus erythematosus]. Zhonghua Yi Xue Za Zhi (2006) 86(35):2462–6. doi: 10.3760/j:issn:0376-2491.2006.35.005
36. Appelgren D, Dahle C, Knopf J, Bilyy R, Vovk V, Sundgren PC, et al. Active NET formation in Libman–Sacks endocarditis without antiphospholipid antibodies: a dramatic onset of systemic lupus erythematosus. Autoimmunity (2018) 51(6):310–8. doi: 10.1080/08916934.2018.1514496
Keywords: systemic lupus erythematosus, neuropsychiatric systemic lupus erythematosus, schizophrenia, obsessive-compulsive disorder (OCD), psychosis
Citation: Lüngen EM, Maier V, Venhoff N, Salzer U, Dersch R, Berger B, Riering AN, Nickel K, Fiebich BL, Süß P, Maier SJ, Egger K, Tebartz van Elst L and Endres D (2019) Systemic Lupus Erythematosus With Isolated Psychiatric Symptoms and Antinuclear Antibody Detection in the Cerebrospinal Fluid. Front. Psychiatry 10:226. doi: 10.3389/fpsyt.2019.00226
Received: 07 January 2019; Accepted: 26 March 2019;
Published: 25 April 2019.
Edited by:
Thomas Skripuletz, Hannover Medical School, GermanyReviewed by:
Kunihiro Ichinose, Nagasaki University, JapanChristopher Sjöwall, Linköping University, Sweden
Copyright © 2019 Lüngen, Maier, Venhoff, Salzer, Dersch, Berger, Riering, Nickel, Fiebich, Süß, Maier, Egger, Tebartz van Elst and Endres. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dominique Endres ZG9taW5pcXVlLmVuZHJlc0B1bmlrbGluaWstZnJlaWJ1cmcuZGU=
†These authors have contributed equally to this work and share first authorship.