 Alec L. W. Dick
Alec L. W. Dick Khen Khermesh2
Khen Khermesh2 Fabian Stamp
Fabian Stamp Erez Y. Levanon
Erez Y. Levanon Alon Chen
Alon Chen- 1Department of Stress Neurobiology and Neurogenetics, Max Planck Institute of Psychiatry, Munich, Germany
- 2CytoReason, Tel-Aviv, Israel
- 3The Mina and Everard Goodman Faculty of LifeSciences, Bar-Ilan University, Ramat-Gan, Israel
- 4Department of Neurobiology, Weizmann Institute of Science, Rehovot, Israel
Adenosine-to-inosine (A-to-I) RNA editing is a co-/posttranscriptional modification of double-stranded RNA, catalyzed by the adenosine deaminase acting on RNA (ADAR) family of enzymes, which results in recognition of inosine as guanosine by translational and splicing machinery causing potential recoding events in amino acid sequences. A-to-I editing is prominent within brain-specific transcripts, and dysregulation of editing at several well-studied loci (e.g., Gria2, Htr2c) has been implicated in acute and chronic stress in rodents as well as neurological (e.g., Alzheimer’s) and psychopathological disorders such as schizophrenia and major depressive disorder. However, only a small fraction of recoding sites has been investigated within the brain following stress, and our understanding of the role of RNA editing in transcriptome regulation following environmental stimuli remains poorly understood. Thus, we aimed to investigate A-to-I editing at hundreds of loci following chronic social defeat stress (CSDS) in mice within corticolimbic regions responsive to chronic stress regulation. Adult male mice were subjected to CSDS or control conditions for 21 days and dynamic regulation of A-to-I editing was investigated 2 and 8 days following the final defeat within both the medial prefrontal cortex (mPFC) and basolateral amygdala (BLA). Employing a targeted resequencing approach, which utilizes microfluidics-based multiplex polymerase chain reaction (PCR) coupled with next-generation sequencing, we analyzed A-to-I editing at ∼100 high-confidence editing sites within the mouse brain. CSDS resulted in acute regulation of transcripts encoding several ADAR enzymes, which normalized 8 days following the final defeat and was specific for susceptible mice. In contrast, sequencing analysis revealed modest and dynamic regulation of A-to-I editing within numerous transcripts in both the mPFC and BLA of resilient and susceptible mice at both 2 and 8 days following CSDS with minimal overlap between regions and time points. Editing within the Htr2c transcript and relative abundance of Htr2c messenger RNA (mRNA)variants were also observed within the BLA of susceptible mice 2 days following CSDS. These results indicate dynamic RNA editing within discrete brain regions following CSDS in mice, further implicating A-to-I editing as a stress-sensitive molecular mechanism within the brain of potential relevance to resiliency and susceptibility to CSDS.
Introduction
Adenosine-to-inosine (A-to-I) RNA editing is a co-/posttranscriptional modification of double-stranded RNA (dsRNA), which is catalyzed by the adenosine deaminases acting on RNA (ADAR) family of enzymes and is the most abundant form of RNA editing in higher eukaryotes (1). ADAR enzymes deaminate adenosine bases to inosine, which is recognized as guanosine by ribosomes and splicing machinery. As such, RNA editing can induce nonsynonymous amino acid changes resulting in differential protein isoform expression and thus is considered a key mechanism of transcriptome and proteome diversification in metazoans (2).
Three members of the ADAR family are encoded in the mammalian genome including the catalytically active ADAR (ADAR1) and ADARB1 (ADAR2), as well as ADARB2 (ADAR3), which lacks a catalytic domain and is primarily restricted to low-level expression within the brain (3). Furthermore, ADAR has two distinct splicing isoforms including the constitutive 110-kDa isoform ADAR1 (p110) and the interferon-inducible isoform ADAR (p150) (4). ADARs mediate RNA editing at millions of sites in the mammalian transcriptome in both coding and noncoding RNA. Recent evidence suggests that ADAR primarily mediates A-to-I editing at repetitive elements, such as Alu repeats in primates, where ADARB1 is primarily responsible for editing at coding sites, although a degree of overlap exists between targeted sites of the two enzymes within mammalian tissues (5). Although several functions are known for editing events in noncoding sites [e.g., alteration of microRNA (miRNA) binding to 3′untranslated regions (UTRs) and alternative splicing regulation] (6–8), much interest has been focused on editing sites within coding regions capable of inducing nonsynonymous recoding events. These events are appreciated as a common form of proteome diversification in both basal and pathological states (9). Interestingly, such recoding events are enriched within the brain and also reside more commonly in transcripts associated with brain function, such as those encoding ion channels and neuromodulator receptors (10). For example, well-established ADARB1-dependent editing of the Gria2 transcript encoding the alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptor subunit Glutamate Ionotropic Receptor AMPA Type Subunit 2 (GRIA2) is essential for normal development as Adarb1 knockout mice die within 3 weeks of birth. This can be rescued upon transgenic coexpression of the fully edited Gria2 isoform in these mice (11). Another well-established role of RNA editing within the mammalian brain is regulation of the 5-hydroxytryptamine2C (5-HT2C) receptor as multiple recoding sites in the Htr2c transcript generates multiple 5-HT2C receptor isoforms with varying G protein affinities and thus receptor function (12).
RNA editing within the brain is also sensitive to environmental and pharmacological stimuli as acute and chronic stress as well as antidepressant treatment in rodents dynamically regulates Htr2c editing and thus serotonergic signaling within discrete brain regions (13, 14). Moreover, editing of the Htr2c transcript is observed within both the dorsolateral prefrontal cortex (PFC) (BA9) and anterior cingulate cortex (BA24) of patients with major depressive disorder (MDD), indicative of the translational relevance of investigating stress-induced regulation of well-conserved editing sites in rodent models of acute and chronic stress (15, 16). Despite this, our understanding of the stress-induced changes in the RNA editome remain restricted to well-studied candidate loci. Broader high-throughput approaches are necessary to identify novel stress-sensitive editing sites within the brain of potential relevance to stress-related psychiatric disorders.
Recent advancements in next-generation sequencing (NGS) technologies have significantly enhanced our ability to accurately quantify A-to-I editing throughout the transcriptome and broadened our understanding of aberrant A-to-I editing in several neurological diseases (e.g., Alzheimer’s disease, epilepsy, and amyotrophic lateral sclerosis) (17–19) and psychiatric disorders (schizophrenia, bipolar disorder, and autism spectrum disorder) (20). However, application of NGS-based techniques in rodent models of acute and chronic stress is lacking such that our understanding of the role of RNA editing in acute and long-term adaptations to stress remains poorly understood.
Thus, this study aimed to investigate aberrant RNA editing within corticolimbic brain regions following chronic social defeat stress (CSDS) in adult mice. CSDS is a well-characterized model of depression-like behavior with significant etiological, predictive, discriminative, and face validity. CSDS induces diverse transcriptome changes within corticolimbic circuits such as the medial prefrontal cortex (mPFC) and basolateral amygdala (BLA) thought to subserve stable behavioral deficits in this model (21, 22). We therefore hypothesized that CSDS would induce dynamic changes within the RNA editome within the mPFC and BLA. To identify novel stress-sensitive editing sites within these brain regions, we employed an established microfluidics-based multiplex polymerase chain reaction and deep sequencing (mmPCR-seq) approach (19, 23) to accurately quantify RNA editing at hundreds of loci within the brain following CSDS.
Methods
Animals
Male C57BL/6 mice at 10 to 12 weeks old were employed for all experiments (Charles River, Sulzfeld, Germany). Mice were single housed in the animal facilities of the Max Planck Institute of Psychiatry in Munich, Germany, for 1 week prior to experimentation and were maintained under standard conditions (12L:12D light cycle, lights on at 07:00 AM, temperature 23 ± 2°C) with food and water available ad libitum. All experiments were approved by and conducted in accordance with the regulations of the local Animal Care and Use Committee (Government of Upper Bavaria, Munich, Germany).
Chronic Social Defeat Stress
Mice were randomly assigned to stress (n = 25) or control conditions (n = 16) and were subjected to 21 days of social defeat or daily handling, respectively. CSDS was conducted essentially as previously described (24). Briefly, male CD1 mice (16 to 18 weeks of age) were employed as resident mice and were habituated to the social defeat cage for 1 week prior to experimentation. On each defeat day, experimental mice were introduced into the home cage (45 cm × 25 cm) of a dominant resident mouse, and mice were left until a social defeat was achieved with minimal injury to experimental mice (generally >2 min). Once mice were defeated, the animals were separated by a wire mesh to prevent physical contact while enabling sensory contact for 24 h. Experimental mice were defeated daily by an unfamiliar, resident mouse for 21 days between 9:00 and 16:00 h to minimize habituation to the CSDS procedure. Control mice (Con) were singly housed in their home cage and handled daily throughout the CSDS procedure. Immediately following the final defeat, experimental mice were single housed. One day following the final defeat, all mice were subjected to the social interaction (SI) test to enable classification of susceptible (SUS) [social interaction (SI) ratio <1] and resilient mice (RES) (SI ratio >1) as previously described (22, 24). Following the SI test, all mice remained in their home cage undisturbed until tissue collection either 2 days (control, n = 8; susceptible, n = 10; resilient, n = 4) or 8 days (control, n = 8; susceptible, n = 8; resilient, n = 3) following the final defeat.
Tissue Collection and Corticosterone Analysis
Either 2 or 8 days following the final defeat, mice were sacrificed (9:00–10:00) and brains were rapidly dissected, snap frozen, and stored at −80°C. Bilateral adrenal glands were dissected, cleaned of excess fat, and weighed. Whole blood was collected in Ethylenediaminetetraacetic acid tubes and centrifuged at 8,000 × g for 10 min at 4°C. Plasma was then aliquoted and stored at −20°C until corticosterone analysis, employing a commercially available radioimmunoassay kit (MP Biomedicals Inc).
Brain Region Microdissection, RNA Extraction, and Reverse Transcription
Frozen brains were serially sectioned at 250 µm on a cryostat, and the mPFC (including prelimbic, infralimbic, and cingulate cortex) and BLA were microdissected and stored at −80°C. Total RNA was extracted from mPFC and BLA tissue from each animal utilizing the miRNeasy mini kit (QIAGEN). Briefly, tissue punches were lysed in 700 µl of Qiazol, and total RNA was extracted as per manufacturer’s instructions (QIAGEN). RNA integrity was assessed on an Agilent Tapestation 2200 with the RNA screentape kit (Thermo Fisher), and all RNA samples were confirmed to have RNA integrity number (RIN) values > 8. Total RNA was DNase treated with the TURBO DNase free kit as per manufacturer’s instructions (Ambion). DNase-treated RNA was quantified with the Qubit 3.0 (Thermo Fisher), and 200 ng of RNA was reverse transcribed to cDNA in 20-µL reactions employing the iScript™ cDNA Synthesis Kit as per manufacturer’s instructions (Bio-Rad). Complementary DNA (cDNA) was stored at −20°C prior to further analysis. Due to failure of cDNA synthesis for multiple samples, final group sizes analyzed for RNA editing and quantitative PCR (qPCR) analysis were as follows: control 2 days, n = 6–8; susceptible 2 days, n = 10; resilient 2 days, n = 4; control 8 days, n = 5–8; susceptible 8 days, n = 7–8; resilient 8 days, n = 3.
Quantitative Real-Time Polymerase Chain Reaction
Determination of relative transcript expression was conducted using the 2−ΔΔCT method (25, 26). Exon spanning primers for candidate transcripts Adar, Adar variant 2, Adar variant 3, Adarb1, Adarb2, Nova1, Commd2, Gria4, and Htr2c and endogenous controls Gapdh, Rpl13a, and Sdha were designed using Primer 3 (http://frodo.wi.mit.edu/). Primer efficiencies were confirmed to be 90–110%. qPCR was conducted on a Quantistudio Flex7 PCR system (Applied Biosystems, USA) using Quantifast SYBR® Green (QIAGEN) as per manufacturer’s instructions. All data are expressed as fold change relative to control mice at each time point.
Targeted Resequencing of RNA Editing Sites in RNA Samples Using the Fluidigm Access Array Coupled With Illumina HiSeq 2500 Sequencing
To precisely detect and measure the levels of A-to-I RNA editing at candidate editing sites in mouse brain tissue, we employed a targeted resequencing approach utilizing mmPCR-seq essentially as previously described (19). Candidate editing sites were selected from the mouse RADAR database (v.2; http://rnaedit.com/) based on the following criteria: i) location within protein coding genes, ii) induction of nonsynonymous amino acid changes, iii) species conservation, and iv) location within genes associated with neuronal function. Applying these criteria, we generated a candidate list of 551 editing sites for which targeted gene and editing-site-specific exon spanning primers were designed using Primer 3.0 (http://frodo.wi.mit.edu/). Selected primers were tested for specificity and sensitivity by PCR prior to their inclusion to the finalized primer set (Supplemental Table 1). Amplification of target regions containing targeting editing loci was conducted with the Access Array™ System for Illumina Sequencing Systems as per manufacturer’s instructions. Briefly, 4 µL of single primer pair (4 μM per primer in 1× AA loading buffer) was loaded into the primer inlets of the 48.48 Access Array integrated fluidic circuits (IFC) (Fluidigm). To prepare the cDNA templates, 2.25 μL of each cDNA sample was added to 2.75 μL of presample mix containing the following enzyme and reagents from the Roche FastStart High Fidelity PCR System: 0.5 μL of 10× FastStart High Fidelity Reaction Buffer wo/Mg, 0.5 μL of dimethyl sulfoxide (DMSO) (5%), 0.1 μL of 10 mM PCR Grade Nucleotide Mix (200 μM), 0.9 μL of 25 mM MgCl2 (4.5 mM), 0.25 μL of 20× Access Array Loading Reagent (Fluidigm), 0.05 μL of FastStart High Fidelity Enzyme Blend, and 0.7 μL of PCR grade water. Four microliters of this mix was loaded into the sample inlets of the 48.48 Access Array IFC (Fluidigm). After the loading of both samples and primers via IFC Controller AX (Fluidigm) loading script, the IFC was subject to thermal cycling using the FC1 Cycler (Fluidigm) with the following program for 40 cycles: 50°C for 2 min, 70°C for 20 min, 95°C 10 min; 10 cycles of 95°C for 15 s, 59.5°C for 30 s, 72°C for 1 min; 4 cycles of 95°C for 15 s, 80°C for 30 s, 59.5°C for 30 s, 72°C for 1 min; 10 cycles of 95°C for 15 s, 59.5°C for 30 s, 72°C for 1 min; 4 cycles of 95°C for 15 s, 80°C for 30 s, 60°C for 30 s, 72°C for 1 min; 8 cycles of 95°C for 15 s, 59.5°C for 30 s, 72°C for 1 min; 4 cycles of 95°C for 15 s, 80°C for 30 s, 60°C for 30 s; 72°C for 1 min; finalizing with 72°C for 3 min. Once PCR has terminated, the IFC was transferred to another IFC Controller AX (Fluidigm) and mini-libraries were harvested by the controller harvest script. Thus, mini-libraries from each sample were obtained for further barcoding and sequencing.
Sequencing Adaptor and Barcode Addition
For each sample, 1.0 μL of the PCR products harvested from the IFC was 1:110 diluted and added to 15 μL of presample mix containing the following enzyme and reagents from the Roche FastStart High Fidelity PCR System: 2 μL of 10× FastStart High Fidelity Reaction Buffer wo/Mg, 1 μL of DMSO (5%), 0.4 μL of 10 mM PCR Grade Nucleotide Mix (200 μM), 3.6 μL of 25 mM MgCl2 (4.5 mM), 0.2 μL of FastStart High Fidelity Enzyme Blend, and 7.8 μL of PCR-grade water. To that samples mix, 4 μL of primer mix from the 2 μM Access Array Barcode Library for Illumina Sequencer—384 (Fluidigm, PN 100-3771), utilizing the B-set; PE2_BC_CS2 and PE1–CS1 barcode primer combination. We used the following PCR program: 95°C for 10 min; 10 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 1 min; and 72°C for 5 min. Amplified libraries were purified with AMPure XP beads (Beckman Coulter) and subjected to paired-end 100-bp sequencing on an Illumina Hiseq2500.
Bioinformatics Sequence Analysis
Bioinformatics analysis was conducted essentially as previously described (19). Briefly, the University of California, Santa Cruz (UCSC) mouse genome browser (NCBI37/mm9) assembly is used to identify any A/G mismatches within the target cDNA sequences. Such mismatches were summed and calculated for their signal strength according to the overall number of reads coverage and the percentage of A-to-G levels.
Prealignment Processing
Fastq files were deindexed into 48 samples according to the individual barcodes used by an in-house script. All reads were trimmed of the universal CS1 and CS2 sequences, and short reads (<20 nt) were removed. Alignment of the processed reads was made using bwa version 0.7.4-r385, using the mem option and the parameters -k 20 -B 3 -O 3 -T 20 for seed in the length of the average primer and for considering the Ion typical error of small indel.
Alignment Process
Sequences were aligned to the mouse refseq database, and reads aligning to multiple loci were excluded from further analysis. Samtools mpileup was employed for aligned sequencing reads, and in-house scripts were employed to transfer the results to the genomic locations from the refseq loci followed by counting the number of different nucleotides in each genomic location that had a q score ≥20. To obtain high-confidence editing sites, only loci with >2,000 reads were included for further analysis, resulting in a total of 100 sites within the BLA and 105 sites within the mPFC (Supplemental Table 2). Data presented for each editing loci represent the total number of reads, and the calculated percentage of reads that have a “G” at the specified genomic location was conducted accordingly with the formula (# of “G” reads/[# of “G” reads + # of “A” reads]).
Statistical Analysis
Two-way repeated-measures analysis of variance (ANOVA) with Sidak post hoc comparison was used to compare body weights throughout the CSDS procedure and SI time in the SI test between control and stressed mice. One-way ANOVAs with Sidak post hoc comparisons were used to compare SI ratios between control, susceptible, and resilient mice as well as between groups at each time point for body weights at sacrifice, adrenal weights, corticosterone levels, qPCR expression levels, Htr2c editing, and isoform abundance. Editing levels were roughly normally distributed, and no normalization was applied such that paired t tests were employed for each RNA editing site. Significance was accepted as p < 0.05. Data are presented as mean ± standard error of the mean (SEM) unless otherwise stated.
Results
Adult male mice were subjected to 21 days of CSDS with no significant differences in body weight observed throughout the stress procedure as revealed by a main effect of time [F(3,117) = 45.83, p < 0.001] but not of treatment or an interaction between factors (Figure 1A). CSDS significantly decreased SI with a novel CD1 mouse as evidenced by a main effect of trial [F(1,37) = 5.252] and a treatment by trial interaction [F(2,37) = 14.47] whereby susceptible mice spent significantly decreased time in the interaction zone in the CD1 compared to the habituation trial (p < 0.001, Figure 1B). For the SI ratio, there was a main effect of treatment [F(2,37) = 13.72, p < 0.001] and significantly decreased SI ratio in susceptible mice and an increased SI ratio in resilient mice compared to controls (p < 0.05, Figure 1C). There were no differences in body weight between groups 2 or 8 days following the final defeat (Figure 1D). However, CSDS induced adrenal hypertrophy as evidenced by a main effect of treatment at 2 days [F(2,19) = 5.42, p = 0.014] with significantly increased adrenal weights in both susceptible and resilient mice compared to controls at 2 days (p < 0.01) but not 8 days [F(2,16) = 2.524, p = 0.111] following the final defeat (Figure 1E). No differences in basal corticosterone were observed at either time point following CSDS (Figure 1F).
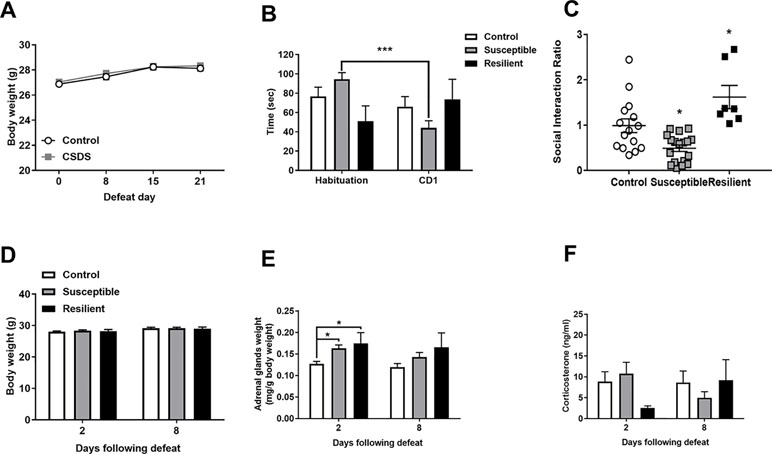
Figure 1 Chronic social defeat stress (CSDS) induces social avoidance and adrenal hypertrophy without changes in basal corticosterone levels. Body weight throughout the stress procedure did not differ between groups (A). Following CSDS, susceptible mice spent significantly less time investigating a novel CD1 mouse than an empty cage in the social interaction (SI) test (B) and significantly decreased the SI ratio in susceptible and increased SI ratio in resilient mice compared to controls (C). CSDS did not alter body weight at 2 or 8 days following the final defeat (D) but induced an increase in adrenal weight in both susceptible and resilient mice at 2 days but not 8 days (E) with no changes in basal corticosterone (F). ***p < 0.001, two-way repeated measures (RM) ANOVA with Sidak post hoc comparisons; *p < 0.05, susceptible and resilient groups compared to control, one-way ANOVA with Sidak post hoc comparisons.
As stress is known to alter A-to-I editing and ADAR levels within the rodent brain, we initially analyzed mRNA expression of transcripts encoding ADAR enzymes within the BLA and mPFC following CSDS. No significant difference in Adar mRNA expression levels was observed in the BLA 2 days following the final defeat, although a trend toward increased Adar transcript variant 3 mRNA encoding the shorter ADAR1 p110 protein and Adarb1 mRNA expression was evident in both susceptible and resilient mice, yet this did not reach statistical significance (Figure 2A). In contrast, CSDS induced a significant decrease in Adar transcript variant 2 mRNA (p = 0.018) encoding the longer ADAR1 p150 protein and Adarb1 mRNA expression (p = 0.002) specifically in the mPFC of susceptible mice 2 days following the final defeat (Figure 2C). No differences in expression levels of Adar mRNAs were observed 8 days following CSDS (Figure 2).
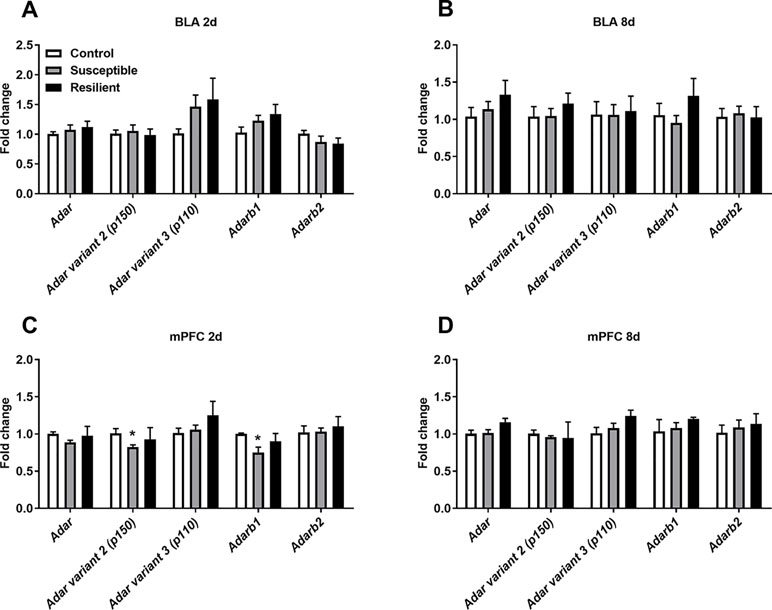
Figure 2 CSDS acutely alters transcripts encoding adenosine deaminases acting on RNA (ADARs) within the medial prefrontal cortex (mPFC) of susceptible mice. Following CSDS, no changes in Adar, Adarb1, or Adarb2 mRNA levels were observed within the basolateral amygdala (BLA) at 2 days (A) or 8 days (B). CSDS significantly decreased the expression levels of Adar variant 2 (p150) and Adarb1 mRNA in the mPFC only in mice susceptible to the CSDS procedure (C). No changes were observed within the mPFC 8 days following CSDS (D). Control 2 days, n = 7; susceptible 2 days, n = 10; resilient 2 days, n = 4; control 8 days, n = 5–7; susceptible 8 days, n = 7; resilient 8 days, n = 3. *p < 0.05, one-way ANOVA with Sidak post hoc comparisons.
To assess CSDS-induced A-to-I RNA editing within the mPFC and BLA, we employed an established mmPCR-seq approach for sensitive and accurate quantification of RNA editing (19). Employing high-confidence editing sites (see Methods), we quantified editing at 100 sites within the mouse BLA and 105 sites within the mouse mPFC. Sequencing analysis revealed that global levels of A-to-I editing within the BLA did not differ at either 2 days [Control mice (Con), 22.96% ± 0.23%; susceptible mice (SUS), 23.10% ± 0.46%; resilient mice (RES), 23.06% ± 0.33%] or 8 days (Con, 23.42% ± 0.29%; SUS, 23.09% ± 0.08%; RES, 23.39% ± 0.19%) following CSDS. Similarly, no differences were observed in the mPFC at 2 days (Con, 24.65% ± 0.24%; SUS, 24.66% ± 0.34%, RES, 25.03% ± 0.40%) or 8 days following CSDS (Con, 24.93% ± 0.25%; SUS, 24.65% ± 0.20%; RES, 24.62% ± 0.34%). Considering the lack of differences in global editing levels between groups, we pooled all samples in each region and found a significantly increased global editing level within the mPFC (24.74% ± 0.12%) compared to BLA (23.20% ± 0.14%, p < 0.001). This finding and global editing levels are in line with those previously reported for the rodent brain with mmPCR-seq (5, 27).
In contrast, differential A-to-I editing was observed at four editing sites in susceptible mice and one site in resilient mice within the BLA 2 days following CSDS with another seven differentially edited sites identified in susceptible mice and six sites in resilient mice 8 days following CSDS within this region (Table 1). Dynamic regulation of editing within the Zfp324 transcript was observed within the BLA of susceptible mice with decreased and increased editing observed at 2 and 8 days, respectively. No other persistent changes were observed in this region. Within the mPFC, four sites were differentially edited in susceptible mice and another four sites were differentially edited in resilient mice 2 days following CSDS with a further seven sites in susceptible mice and four sites in resilient mice differentially edited 8 days following CSDS (Table 2). Increased editing at the Commd2 locus encoding the Copper metabolism Murr1 domain-containing protein 2 (COMMD2) was observed at both 2 and 8 days following CSDS with no other persistent changes observed. Moreover, RNA editing of a nonsynonymous recoding site within the Nova1 transcript encoding the RNA binding protein (RBP) NOVA Alternative Splicing Regulator 1 (NOVA1) was also observed in both the BLA and mPFC 8 days following CSDS, although in opposite directions. Differential editing of several sites following CSDS did not alter mRNA expression levels of candidate transcripts in either brain region (Figure 3).
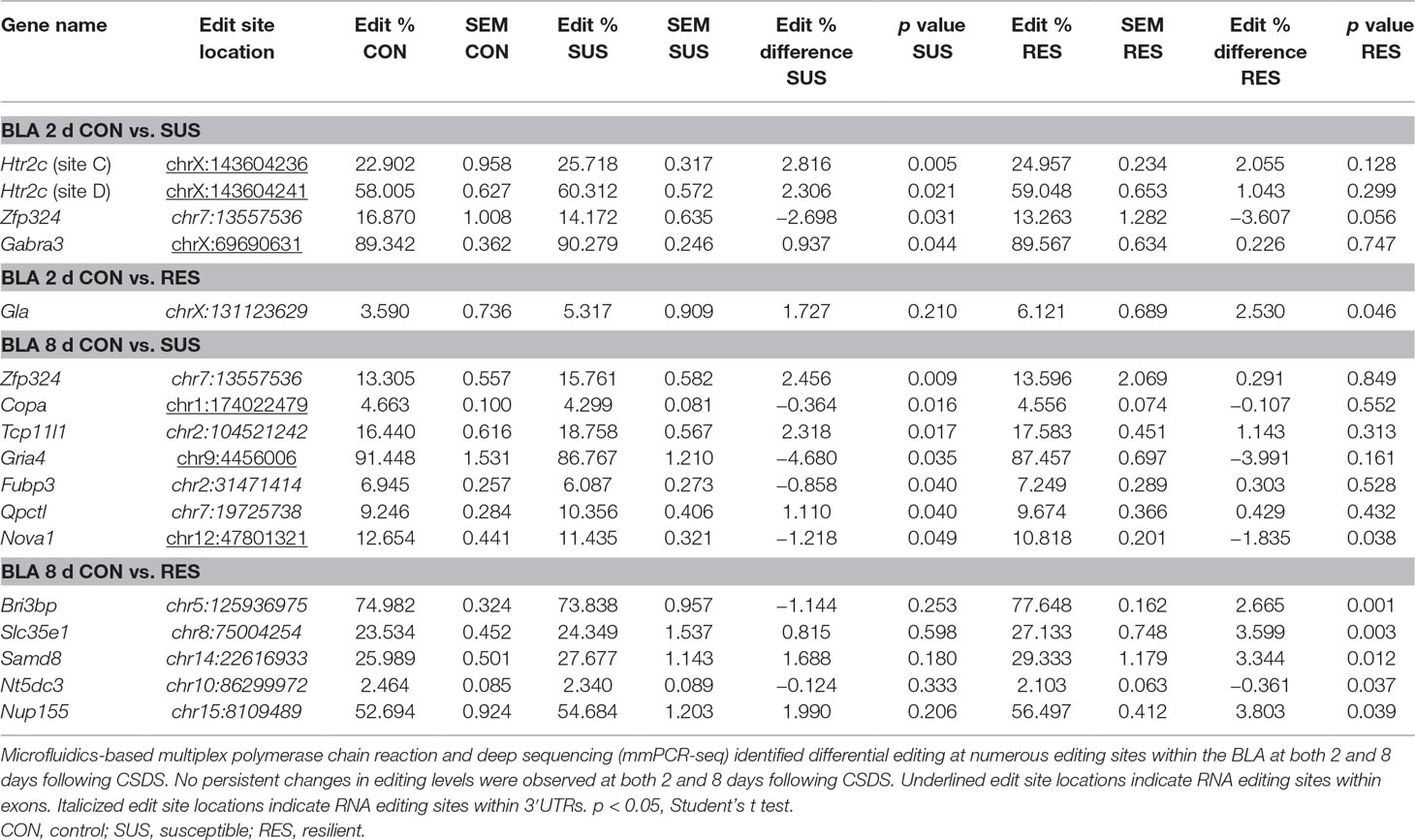
Table 1 Differentially editied loci within the basolateral amygdala (BLA) following chronic social defeat stress (CSDS).
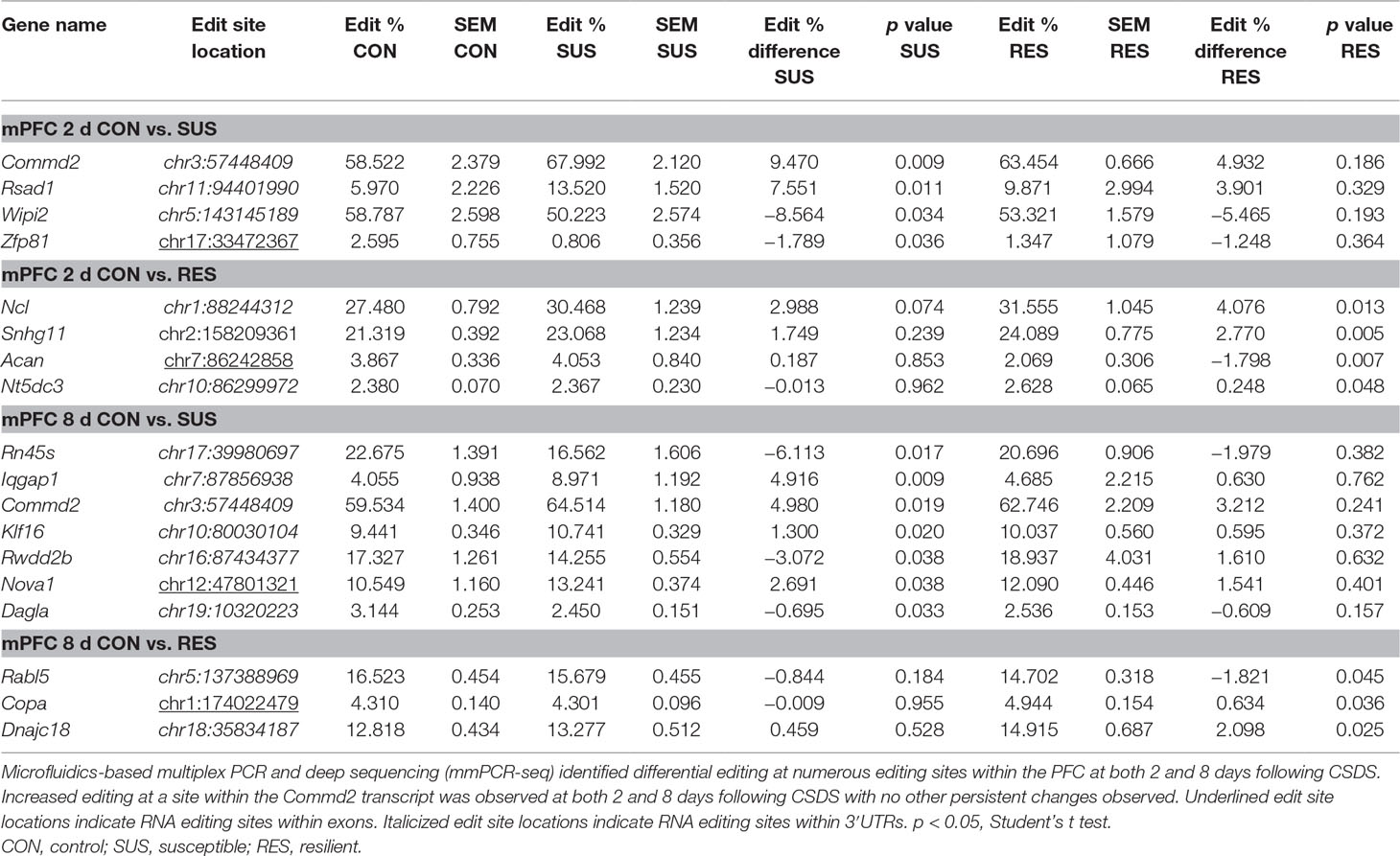
Table 2 Differentially editied loci within the medial prefrontal cortex (mPFC) following CSDS.
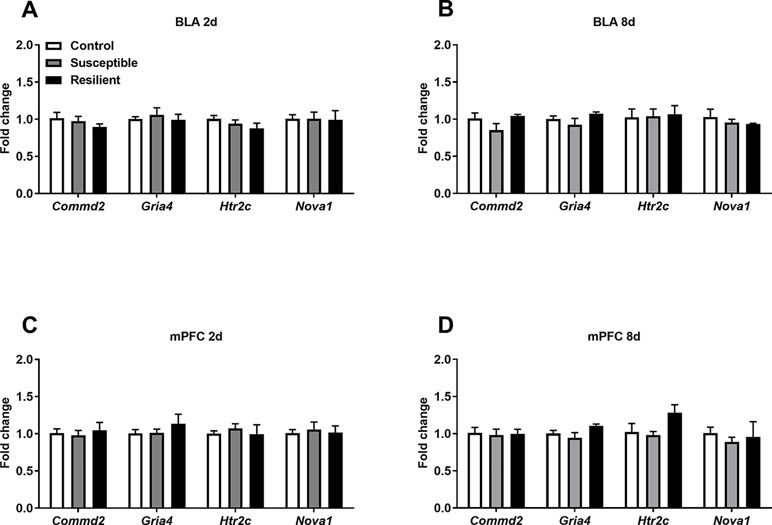
Figure 3 Expression levels of differentially edited transcripts are unaltered following CSDS. No significant changes in the expression levels of several differentially edited transcripts were observed with the BLA (A) or mPFC (B) at 2 days or within the BLA (C) or mPFC (D) 8 days following the final defeat. Control 2 days, n = 7; susceptible 2 days, n = 10; resilient 2 days, n = 4; control 8 days, n = 5–7; susceptible 8 days, n = 7; resilient 8 days, n = 3.
Well-established A-to-I editing of five sites within the Htr2c transcript, known as A, B, C, D, and E, results in the generation of 32 mRNA variants generating up to 24 different protein isoforms of the HTR2C receptor with varying biochemical properties (28). As expected, we detected high-confidence editing at sites A, B, C, and D but only minimal editing at site E within the mouse BLA and mPFC (Figure 4). CSDS induced a modest increase in editing at both the C and D sites within the BLA of susceptible mice but not resilient mice 2 days following the final defeat (Figure 4A) with no other changes observed at 8 days (Figure 4B) or within the mPFC at either time point (Figure 4C and D).
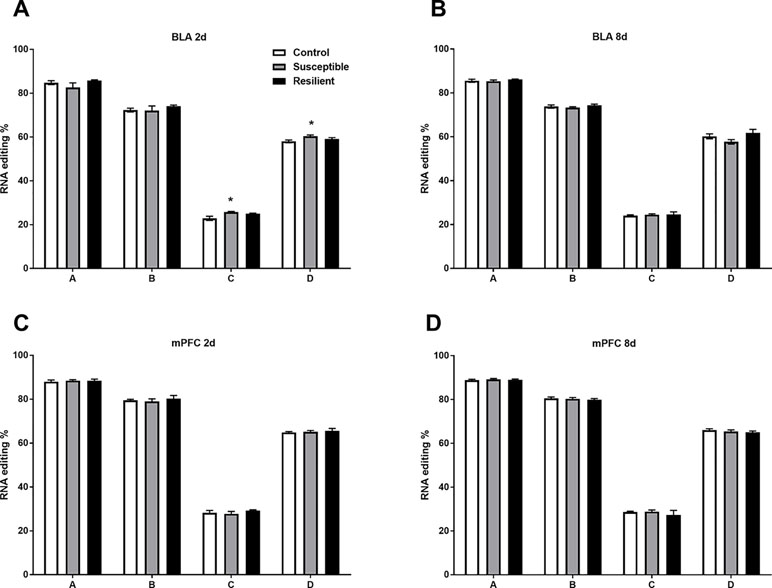
Figure 4 RNA editing of the Htr2c transcript is altered within the BLA of susceptible mice 2 days following CSDS. Sequencing analysis revealed modest increases of RNA editing at Htr2c sites C and D within the BLA of susceptible but not resilient mice 2 days following CSDS (A) with no other changes observed at 8 days (B) or at 2 days (C) or 8 days within the mPFC (D). Control 2 days, n = 6–8; susceptible 2 days, n = 10; resilient 2 days, n = 4; control 8 days, n = 7–8; susceptible 8 days, n = 7; resilient 8 days, n = 3. *p < 0.05, one-way ANOVA with Sidak post hoc comparisons.
Editing at these loci induces recoding events, ultimately generating different HTR2C protein isoforms. Thus, we next quantified the relative abundance of Htr2c mRNA variants within the BLA and mPFC following CSDS. As expected, there was a similar distribution of Htr2c mRNA variants within the BLA and mPFC, with the edited VNV isoform being most abundant in both regions (Figure 5), as previously reported in the rodent brain (27). Following CSDS, we observed a trend toward an effect of treatment for the VNI variant [F(2,18) = 3.068, p = 0.071] mainly due to a trend toward decreased VNI abundance in susceptible mice at this time point (p = 0.058, Figure 5A). No other changes were observed within the BLA at 8 days (Figure 5B) or within the mPFC (Figure 5C and D).
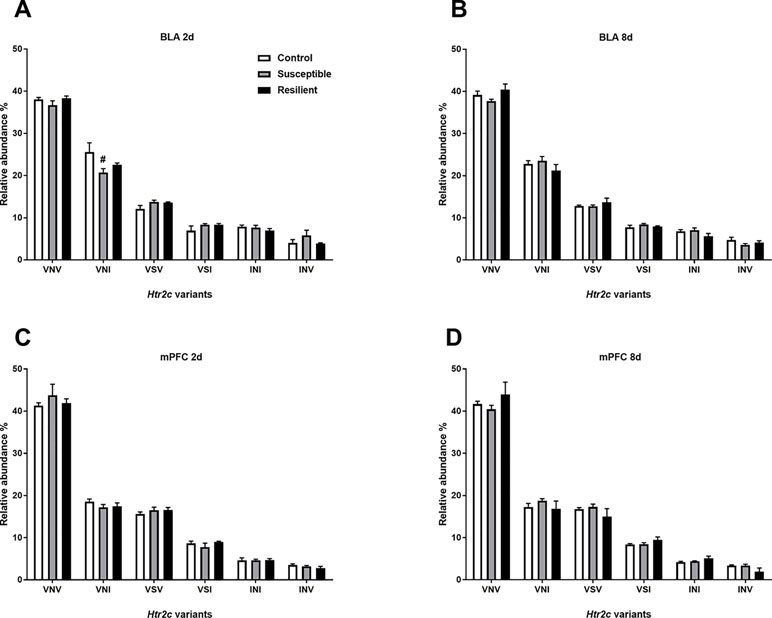
Figure 5 Relative abundance of Htr2c variants is minimally altered within the BLA 2 days following CSDS. CSDS induced a nominally significant decrease of the VNI Htr2c transcript variant in the BLA of susceptible mice 2 days following CSDS (A) with no other changes observed at 8 days (B) or at 2 days (C) or 8 days within the mPFC (D). Control 2 days, n = 6–8; susceptible 2 days, n = 10; resilient 2 days, n = 4; control 8 days, n = 7–8; susceptible 8 days, n = 7; resilient 8 days, n = 3. #p = 0.058, one-way ANOVA with Sidak post hoc comparisons.
Discussion
In this study, we demonstrated that CSDS in adult mice induces a moderate degree of differential editing in a subset of novel transcripts within the BLA and mPFC, including modest regulation of editing within the Htr2c transcript and thus isoform abundance previously demonstrated to be sensitive to stress-induced regulation. Our results further emphasize the sensitivity of RNA editing to stress and suggest that both acute and chronic changes in editing, although moderate, may contribute to behavioral deficits observed following CSDS in adult mice. Moreover, differential regulation in susceptible and resilient mice suggests that RNA editing may be a novel molecular mechanism involved in resiliency and susceptibility in this model requiring further investigation.
To our knowledge, this is the first study to investigate a large subset of RNA editing sites within the mouse brain in response to chronic stress and the first report of A-to-I editing following CSDS. Interestingly, minimal changes in transcripts encoding ADAR enzymes were identified following CSDS. Specific reduction of the interferon-inducible Adar variant 2 (p150) and Adarb1 mRNA levels was observed within the mPFC of susceptible mice only without changes in global RNA editing in these mice. RNA editing within the rodent brain is relatively stable upon induction of ADAR p150, such that decreased levels within the mPFC are unlikely to explain observed effects in this study (29). As ADARB1 is primarily responsible for editing at recoding sites (5), decreased expression in susceptible mice may mediate decreased editing at specific ADARB1 target sites. However, we observed both increased editing within Commd2 and Rsad1 and decreased editing in Wipi2 and Zfp81 within the mPFC of susceptible mice. Moreover, editing differences were also observed following CSDS in the absence of Adar mRNA expression changes, suggesting that differential RNA editing is unlikely to be mediated by the Adarb1 mRNA expression changes observed. This is in line with evidence suggesting complex regulation of RNA editing activity in a tissue-specific and cell-type-specific manner independent of ADAR family expression levels including interaction with RBPs, such as fragile X mental retardation protein (2, 5, 30, 31). Thus, our results suggest that differential RNA editing at the sites identified in this study is more likely mediated by site-specific regulation of editing activity opposed to CSDS-induced changes in the levels of ADAR family enzymes.
Although we identified differential editing at novel editing sites within the mouse brain, notable differential editing was observed within the Htr2c transcript specifically within the BLA of susceptible mice. Considering the well-established regulation of editing within this transcript in rodent models of acute and chronic stress as well as within the brain of MDD patients (13–16, 32), these results further implicate editing at this transcript in susceptibility to CSDS and support the model’s relevance to stress-related psychiatric disorders. However, it must be noted that stress-induced editing of the Htr2c transcript and variant abundance is context-dependent based on species, strain, stress modality, brain region, and developmental age (27, 28). RNA editing of the Htr2c transcript reduces both receptor/Gαq-protein coupling and constitutive activity of the 5HT2C receptor (12, 33, 34). Transgenic mice exclusively expressing the fully edited VGV isoform also display anxiogenic and aggressive behaviors, with altered 5-HTR2C receptor signaling within discrete brain regions in these mice (35). Thus, editing at this locus may mediate altered 5HTR2C signaling within the BLA of susceptible mice following CSDS, yet further investigation is needed to assess the functional consequences of CSDS-induced editing at the Htr2c locus in this context.
Editing within transcripts encoding Gamma-Aminobutyric Acid Type A (GABAA) and AMPA receptor subunits were also affected following CSDS. Differential editing of sites in the Gabra3 and Gria4 transcripts were identified within the BLA 2 and 8 days following CSDS, respectively. The Gabra3 transcript encodes the α3 GABAA receptor subunit with editing at this highly conserved site resulting in an isoleucine-to-methionine change in the third transmembrane domain. This site is developmentally regulated and mediates receptor trafficking and GABA sensitivity whereby increased editing is thought to decrease GABAA receptor signaling (36, 37). Editing at this site was also increased following chronic mild stress within the PFC of adult female rats, suggesting that this site may be sensitive to various stress modalities in different contexts (27). Moreover, modulation of GABAA signaling within the BLA impairs SI in rats (38). Alterations in GABAA signaling via RNA editing may contribute to the social avoidance phenotype observed in susceptible mice in this model, although the functional consequences of stress-induced Gabra3 editing need to be established following CSDS.
Glutamatergic signaling may also be affected by stress-induced RNA editing as increased editing within the Gria4 transcript encoding the AMPAR α4 subunit in the BLA of susceptible mice at 8 days is indicative of more persistent changes in the RNA editome. Editing at this Gria4 site confers differences in AMPA receptorchannel kinetics due to regulation of Gria4 splicing variants, which is sensitive to neuronal stimulation in rat primary cortical neurons (39). Thus, decreased editing in susceptible mice may mediate AMPAR signaling deficits in the BLA in part via RNA editing. Indeed, differential AMPAR signaling has been reported within the PFC (40) and hippocampus (41) following CSDS in mice. Further work is required to assess the role of AMPAR signaling, as well as GABAergic signaling, in stress susceptibility following CSDS.
Apart from changes in the aforementioned established editing sites, we aimed to identify novel editing sites sensitive to stress-induced regulation following CSDS. One such example is the Nova1 transcript encoding the RBP NOVA1. Editing at this Nova1 recoding site results in a serine-to-glycine exchange, which stabilizes the NOVA1 protein by decreasing proteasome-mediated degradation (42). Interestingly, we identified modest changes in Nova1 editing in both the BLA and mPFC 8 days following CSDS with increased and decreased editing observed, respectively. Within the BLA, Nova1 was similarly edited in both susceptible and resilient mice with differential editing only observed within the mPFC of susceptible mice, suggesting that Nova1 editing is regulated in a region-specific manner following chronic stress. Considering the effects of RNA editing upon NOVA1 protein stability and the lack of CSDS-induced changes in Nova1 mRNA levels, it would be of interest to assess NOVA1 protein levels within the BLA and mPFC following CSDS. Moreover, considering the established role of NOVA1 as an important RBP within the brain, which mediates both alternative splicing (43) and miRNA activity (44), NOVA1 is an interesting novel candidate requiring further investigation for its role in stress-induced regulation of the transcriptome.
Apart from such nonsynonymous recoding sites, the majority of differential editing sites identified in the current study were located within the 3′UTR of various transcripts such that the protein coding capacity of these mRNAs remains unaltered. However, RNA editing within 3′UTRs regulates mRNA availability and translation efficiency due to the editing of miRNA binding sites, which can induce differential miRNA-mediated regulation of edited transcripts (6). Persistent increases in editing at a site within the 3′UTR of Commd2 in the mPFC of susceptible mice did not, however, alter mRNA levels in this brain region, suggesting that miRNA activity at this site is likely unaffected by editing at the site examined in this context. Further studies are required to investigate the consequences of CSDS-induced editing in 3′UTRs in this study including those sites identified in resilient mice such as in the 3′UTR of Ncl encoding the eukaryotic nucleolar phosphoprotein Nucelolin, which interestingly interacts with the brain-specific small nucleolar RNA MBII-52, known to regulate Htr2c editing within the mammalian brain (45). Furthermore, many differentially edited sites were identified within transcripts encoding proteins with poorly understood functions, particularly in resilient mice (e.g., Bri3bp and Slc35e1), which should be the focus of further investigation.
Several caveats to this study must be noted, including the small sample size for resilient groups as well as the mainly modest changes in editing levels observed. Repetition of mmPCR-seq analysis in a larger CSDS cohort would likely enable identification of further stress-sensitive editing sites, particularly those associated with resiliency. Moreover, utilizing RNA-seq would enable transcriptome-wide analysis of stress-induced editing and mRNA levels. Despite this, the A-to-I editing changes observed in this study are similar with the editing level changes observed in the rat PFC and amygdala following chronic stress employing a similar mmPCR-seq technique (27). Such modest changes of RNA editing in bulk brain tissue are also likely explained by cellular heterogeneity as recent advances in single-cell transcriptomics have demonstrated that A-to-I editing is indeed cell type specific with changes even observed between different cells of given cellular population within the mammalian brain (46, 47). Thus, it would be of interest in the future to study stress-induced changes in RNA editing using single cell transcriptomics.
In conclusion, the current study has identified A-to-I editing as another molecular mechanism of likely relevance to stress resiliency and susceptibility to CSDS in adult mice, in line with the growing appreciation for stress-induced regulation of RNA metabolism within the brain (21, 48, 49). Further investigation of the consequences of these editing changes is required at both the mRNA and protein levels to decipher the functional consequences of RNA editing following chronic stress.
Ethics Statement
This study was carried out in accordance with the recommendations of the European Communities; Council Directive 2010/63/EU. The protocols were approved by the Animal Care and Use Committee of the Government of Upper Bavaria, Munich, Germany.
Author Contributions
AD, KK, EL, and AC conceptualized and designed the experiments and wrote the manuscript. AD and EP conducted all animal experiments analyzed by AD. AD and KK conducted and analyzed all mmPCR-seq experiments. AD and FS conducted qPCR experiments analyzed by AD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank L. Tietze, B. Schmid, and D. Harbich for their technical support and A. Varga and the animal care team for their assistance with animal care. AC is the head of the Max Planck Society–Weizmann Institute of Science Laboratory for Experimental Neuropsychiatry and Behavioral Neurogenetics. This work is supported by an FP7 Grant from the European Research Council (260463, AC); a research grant from the Israel Science Foundation (1565/15, AC); the ERANET Program, supported by the Chief Scientist Office of the Israeli Ministry of Health (3-11389, AC); the project funded by the Federal Ministry of Education and Research under the funding code 01KU1501A (AC); research support from Roberto and Renata Ruhman (AC); research support from Bruno and Simone Licht; I-CORE Program of the Planning and Budgeting Committee and The Israel Science Foundation (grant no. 1916/12 to AC); the Nella and Leon Benoziyo Center for Neurological Diseases (AC); the Henry Chanoch Krenter Institute for Biomedical Imaging and Genomics (AC); the Perlman Family Foundation, founded by Louis L. and Anita M. Perlman (AC); the Adelis Foundation (AC); the Marc Besen and the Pratt Foundation (AC); and the Irving I. Moskowitz Foundation (AC).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2019.00277/full#supplementary-material
References
1. Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem (2002) 71:817–46. doi: 10.1146/annurev.biochem.71.110601.135501
2. Eisenberg E, Levanon EY. A-to-I RNA editing—immune protector and transcriptome diversifier. Nat Rev Genet (2018) 19(8):473–90. doi: 10.1038/s41576-018-0006-1
3. Savva YA, Rieder LE, Reenan RA. The ADAR protein family. Genome Biol (2012) 13(12):252. doi: 10.1186/gb-2012-13-12-252
4. Patterson JB, Samuel CE. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol (1995) 15(10):5376–88. doi: 10.1128/MCB.15.10.5376
5. Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, et al. Dynamic landscape and regulation of RNA editing in mammals. Nature (2017) 550(7675):249–54. doi: 10.1038/nature24041
6. Pinto Y, Buchumenski I, Levanon EY, Eisenberg E. Human cancer tissues exhibit reduced A-to-I editing of miRNAs coupled with elevated editing of their targets. Nucleic Acids Res (2018) 46(1):71–82. doi: 10.1093/nar/gkx1176
7. Wang Y, Xu X, Yu S, Jeong KJ, Zhou Z, Han L, et al. Systematic characterization of A-to-I RNA editing hotspots in microRNAs across human cancers. Genome Res (2017) 27(7):1112–25. doi: 10.1101/gr.219741.116
8. Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science (2007) 315(5815):1137–40. doi: 10.1126/science.1138050
9. Hwang T, Park CK, Leung AK, Gao Y, Hyde TM, Kleinman JE, et al. Dynamic regulation of RNA editing in human brain development and disease. Nat Neurosci (2016) 19(8):1093–9. doi: 10.1038/nn.4337
10. Rosenthal JJ, Seeburg PH. A-to-I RNA editing: effects on proteins key to neural excitability. Neuron (2012) 74(3):432–9. doi: 10.1016/j.neuron.2012.04.010
11. Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, et al. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature (2000) 406(6791):78–81. doi: 10.1038/35017558
12. Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, et al. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature (1997) 387(6630):303–8. doi: 10.1038/387303a0
13. Bhansali P, Dunning J, Singer SE, David L, Schmauss C. Early life stress alters adult serotonin 2C receptor pre-mRNA editing and expression of the alpha subunit of the heterotrimeric G-protein G q. J Neurosci (2007) 27(6):1467–73. doi: 10.1523/JNEUROSCI.4632-06.2007
14. Bombail V, Qing W, Chapman KE, Holmes MC. Prevention of 5-hydroxytryptamine2C receptor RNA editing and alternate splicing in C57BL/6 mice activates the hypothalamic-pituitary-adrenal axis and alters mood. Eur J Neurosci (2014) 40(11):3663–73. doi: 10.1111/ejn.12727
15. Gurevich I, Tamir H, Arango V, Dwork AJ, Mann JJ, Schmauss C. Altered editing of serotonin 2C receptor pre-mRNA in the prefrontal cortex of depressed suicide victims. Neuron (2002) 34(3):349–56. doi: 10.1016/S0896-6273(02)00660-8
16. Weissmann D, van der Laan S, Underwood MD, Salvetat N, Cavarec L, Vincent L, et al. Region-specific alterations of A-to-I RNA editing of serotonin 2c receptor in the cortex of suicides with major depression. Transl Psychiatry (2016) 6(8):e878. doi: 10.1038/tp.2016.121
17. Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S. Glutamate receptors: RNA editing and death of motor neurons. Nature (2004) 427(6977):801. doi: 10.1038/427801a
18. Srivastava PK, Bagnati M, Delahaye-Duriez A, Ko JH, Rotival M, Langley SR, et al. Genome-wide analysis of differential RNA editing in epilepsy. Genome Res (2017) 27(3):440–50. doi: 10.1101/gr.210740.116
19. Khermesh K, D’Erchia AM, Barak M, Annese A, Wachtel C, Levanon EY, et al. Reduced levels of protein recoding by A-to-I RNA editing in Alzheimer’s disease. RNA (2016) 22(2):290–302. doi: 10.1261/rna.054627.115
20. Silberberg G, Lundin D, Navon R, Ohman M. Deregulation of the A-to-I RNA editing mechanism in psychiatric disorders. Hum Mol Genet (2012) 21(2):311–21. doi: 10.1093/hmg/ddr461
21. Bagot RC, Cates HM, Purushothaman I, Lorsch ZS, Walker DM, Wang J, et al. Circuit-wide transcriptional profiling reveals brain region-specific gene networks regulating depression susceptibility. Neuron (2016) 90(5):969–83. doi: 10.1016/j.neuron.2016.04.015
22. Golden SA, Covington HE II, Berton O, Russo SJ. A standardized protocol for repeated social defeat stress in mice. Nature protocols (2011) 6(8):1183–91. doi: 10.1038/nprot.2011.361
23. Zhang R, Li X, Ramaswami G, Smith KS, Turecki G, Montgomery SB, et al. Quantifying RNA allelic ratios by microfluidic multiplex PCR and sequencing. Nat Methods (2014) 11(1):51–4. doi: 10.1038/nmeth.2736
24. Hartmann J, Wagner KV, Gaali S, Kirschner A, Kozany C, Ruhter G, et al. Pharmacological inhibition of the psychiatric risk factor FKBP51 has anxiolytic properties. J Neurosci (2015) 35(24):9007–16. doi: 10.1523/JNEUROSCI.4024-14.2015
25. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods (2001) 25(4):402–8. doi: 10.1006/meth.2001.1262
26. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc (2008) 3(6):1101–8. doi: 10.1038/nprot.2008.73
27. Zaidan H, Ramaswami G, Golumbic YN, Sher N, Malik A, Barak M, et al. A-to-I RNA editing in the rat brain is age-dependent, region-specific and sensitive to environmental stress across generations. BMC Genomics (2018) 19(1):28. doi: 10.1186/s12864-017-4409-8
28. Hackler EA, Airey DC, Shannon CC, Sodhi MS, Sanders-Bush E. 5-HT(2C) receptor RNA editing in the amygdala of C57BL/6J, DBA/2J, and BALB/cJ mice. Neurosci Res (2006) 55(1):96–104. doi: 10.1016/j.neures.2006.02.005
29. Hood JL, Morabito MV, Martinez CR 3rd, Gilbert JA, Ferrick EA, Ayers GD, et al. Reovirus-mediated induction of ADAR1 (p150) minimally alters RNA editing patterns in discrete brain regions. Mol Cell Neurosci (2014) 61:97–109. doi: 10.1016/j.mcn.2014.06.001
30. Shamay-Ramot A, Khermesh K, Porath HT, Barak M, Pinto Y, Wachtel C, et al. Fmrp interacts with Adar and regulates RNA editing, synaptic density and locomotor activity in zebrafish. PLoS Genet (2015) 11(12):e1005702. doi: 10.1371/journal.pgen.1005702
31. Wahlstedt H, Daniel C, Enstero M, Ohman M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res (2009) 19(6):978–86. doi: 10.1101/gr.089409.108
32. Barbon A, Orlandi C, La Via L, Caracciolo L, Tardito D, Musazzi L, et al. Antidepressant treatments change 5-HT2C receptor mRNA expression in rat prefrontal/frontal cortex and hippocampus. Neuropsychobiology (2011) 63(3):160–8. doi: 10.1159/000321593
33. McGrew L, Price RD, Hackler E, Chang MS, Sanders-Bush E. RNA editing of the human serotonin 5-HT2C receptor disrupts transactivation of the small G-protein RhoA. Mol Pharmacol (2004) 65(1):252–6. doi: 10.1124/mol.65.1.252
34. Niswender CM, Copeland SC, Herrick-Davis K, Emeson RB, Sanders-Bush E. RNA editing of the human serotonin 5-hydroxytryptamine 2C receptor silences constitutive activity. J Biol Chem (1999) 274(14):9472–8. doi: 10.1074/jbc.274.14.9472
35. Martin CB, Ramond F, Farrington DT, Aguiar AS Jr., Chevarin C, Berthiau AS, et al. RNA splicing and editing modulation of 5-HT(2C) receptor function: relevance to anxiety and aggression in VGV mice. Mol Psychiatry (2013) 18(6):656–65. doi: 10.1038/mp.2012.171
36. Nimmich ML, Heidelberg LS, Fisher JL. RNA editing of the GABA(A) receptor alpha3 subunit alters the functional properties of recombinant receptors. Neurosci Res (2009) 63(4):288–93. doi: 10.1016/j.neures.2009.01.003
37. Daniel C, Wahlstedt H, Ohlson J, Bjork P, Ohman M. Adenosine-to-inosine RNA editing affects trafficking of the gamma-aminobutyric acid type A (GABA(A)) receptor. J Biol Chem (2011) 286(3):2031–40. doi: 10.1074/jbc.M110.130096
38. Paine TA, Swedlow N, Swetschinski L. Decreasing GABA function within the medial prefrontal cortex or basolateral amygdala decreases sociability. Behav Brain Res (2017) 317:542–52. doi: 10.1016/j.bbr.2016.10.012
39. Orlandi C, La Via L, Bonini D, Mora C, Russo I, Barbon A, et al. AMPA receptor regulation at the mRNA and protein level in rat primary cortical cultures. PLoS One (2011) 6(9):e25350. doi: 10.1371/journal.pone.0025350
40. Zhang RX, Han Y, Chen C, Xu LZ, Li JL, Chen N, et al. EphB2 in the medial prefrontal cortex regulates vulnerability to stress. Neuropsychopharmacology (2016) 41(10):2541–56. doi: 10.1038/npp.2016.58
41. Li MX, Zheng HL, Luo Y, He JG, Wang W, Han J, et al. Gene deficiency and pharmacological inhibition of caspase-1 confers resilience to chronic social defeat stress via regulating the stability of surface AMPARs. Mol Psychiatry (2018) 23(3):556–68. doi: 10.1038/mp.2017.76
42. Irimia M, Denuc A, Ferran JL, Pernaute B, Puelles L, Roy SW, et al. Evolutionarily conserved A-to-I editing increases protein stability of the alternative splicing factor Nova1. RNA Biol (2012) 9(1):12–21. doi: 10.4161/rna.9.1.18387
43. Jelen N, Ule J, Zivin M, Darnell RB. Evolution of Nova-dependent splicing regulation in the brain. PLoS Genet (2007) 3(10):1838–47. doi: 10.1371/journal.pgen.0030173
44. Storchel PH, Thummler J, Siegel G, Aksoy-Aksel A, Zampa F, Sumer S, et al. A large-scale functional screen identifies Nova1 and Ncoa3 as regulators of neuronal miRNA function. EMBO J (2015) 34(17):2237–54. doi: 10.15252/embj.201490643
45. Soeno Y, Taya Y, Stasyk T, Huber LA, Aoba T, Huttenhofer A. Identification of novel ribonucleo-protein complexes from the brain-specific snoRNA MBII-52. RNA (2010) 16(7):1293–300. doi: 10.1261/rna.2109710
46. Gal-Mark N, Shallev L, Sweetat S, Barak M, Billy Li J, Levanon EY, et al. Abnormalities in A-to-I RNA editing patterns in CNS injuries correlate with dynamic changes in cell type composition. Sci Rep (2017) 7:43421. doi: 10.1038/srep43421
47. Picardi E, Horner DS, Pesole G. Single-cell transcriptomics reveals specific RNA editing signatures in the human brain. RNA (2017) 23(6):860–5. doi: 10.1261/rna.058271.116
48. Dias C, Feng J, Sun H, Shao NY, Mazei-Robison MS, Damez-Werno D, et al. beta-catenin mediates stress resilience through Dicer1/microRNA regulation. Nature (2014) 516(7529):51–5. doi: 10.1038/nature13976
Keywords: A-to-I RNA editing, chronic social defeat stress, microfluidics-based multiplex polymerase chain reaction, adenosine deaminases acting on RNA, ADAR, chronic stress
Citation: Dick ALW, Khermesh K, Paul E, Stamp F, Levanon EY and Chen A (2019) Adenosine-to-Inosine RNA Editing Within Corticolimbic Brain Regions Is Regulated in Response to Chronic Social Defeat Stress in Mice. Front. Psychiatry 10:277. doi: 10.3389/fpsyt.2019.00277
Received: 17 August 2018; Accepted: 10 April 2019;
Published: 26 April 2019.
Edited by:
George P. Chrousos, National and Kapodistrian University of Athens, GreeceReviewed by:
Tomoshige Kino, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), United StatesDimitrios P. Vlachakis, Agricultural University of Athens, Greece
Copyright © 2019 Dick, Khermesh, Paul, Stamp, Levanon and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alon Chen YWxvbl9jaGVuQHBzeWNoLm1wZy5kZQ==