Abstract
Globally, psychiatric disorders, such as anxiety disorder, bipolar disorder, schizophrenia, depression, autism spectrum disorder, and attention-deficit/hyperactivity disorder (ADHD) are becoming more prevalent. Although the exact pathological alterations are not yet clear, recent studies have demonstrated that widespread changes of very complex metabolic pathways may partially underlie the pathophysiology of many psychiatric diseases. Thus, more attention should be directed to metabolic-based therapeutic interventions in the treatment of psychiatric disorders. Emerging evidence from numerous studies suggests that administration of exogenous ketone supplements, such as ketone salts or ketone esters, generates rapid and sustained nutritional ketosis and metabolic changes, which may evoke potential therapeutic effects in cases of central nervous system (CNS) disorders, including psychiatric diseases. Therefore, the aim of this review is to summarize the current information on ketone supplementation as a potential therapeutic tool for psychiatric disorders. Ketone supplementation elevates blood levels of the ketone bodies: D-β-hydroxybutyrate (βHB), acetoacetate (AcAc), and acetone. These compounds, either directly or indirectly, beneficially affect the mitochondria, glycolysis, neurotransmitter levels, activity of free fatty acid receptor 3 (FFAR3), hydroxycarboxylic acid receptor 2 (HCAR2), and histone deacetylase, as well as functioning of NOD-like receptor pyrin domain 3 (NLRP3) inflammasome and mitochondrial uncoupling protein (UCP) expression. The result of downstream cellular and molecular changes is a reduction in the pathophysiology associated with various psychiatric disorders. We conclude that supplement-induced nutritional ketosis leads to metabolic changes and improvements, for example, in mitochondrial function and inflammatory processes, and suggest that development of specific adjunctive ketogenic protocols for psychiatric diseases should be actively pursued.
Introduction
With an increasing global prevalence, psychiatric disorders can present as serious medical conditions composed of emotional, cognitive, social, behavioral, and functional impairments (1). Lifetime onset of major depressive disorders in the general population is up to 11–16% (2, 3), with bipolar disorder present in 1% (4, 5), schizophrenia in 1% (6, 7), and anxiety disorder in 5–31% (1). In relation to attention-deficit/hyperactivity disorder (ADHD), worldwide prevalence of this disease in children/adolescence and adults is about 5.3% and 2.5%, respectively (8, 9), while about 1 in 68 children were diagnosed with autism in the United States in 2012 (10). It has been demonstrated that not only genetic factors but also environmental factors (e.g., infections, early traumas, and drugs), age, sociodemographic factors (e.g., ethnicity and socioeconomic status), and a complex interplay between these factors have a role in the pathophysiology of different psychiatric diseases, such as anxiety disorder (1, 11), bipolar disorder (5), schizophrenia (6, 12), major depressive disorder (2, 13, 14), autism spectrum disorder (15), and ADHD (16). Close association between different psychiatric disorders, such as anxiety disorder and major depressive disorder, has been demonstrated (5, 17–21).
However, while symptoms, characteristics, and classification of different psychiatric disorders are adequately described (1, 5, 7, 15, 16, 22), the pathophysiology of psychiatric diseases is not yet fully understood. Nevertheless, recent studies have demonstrated that the disturbance in the monoaminergic (23–26) and other neurotransmitter systems (e.g., glutamatergic, purinergic, and GABAergic) (27–34), in addition to widespread changes of very complex and connected metabolic pathways, may partially explain the general condition. For example, it has been suggested that mitochondrial dysfunction could play a major role (35). Mitochondrial dysfunction may decrease energy/ATP production, impair calcium homeostasis, increase levels of reactive oxygen species (ROS), and alter apoptotic pathways, inflammatory processes, neurotransmission, synaptic plasticity, and neuronal activity and connectivity (35, 36). Moreover, changes in hypothalamic–pituitary–adrenal (HPA) axis activity were also demonstrated in patients with psychiatric diseases, in which alterations may influence mitochondrial functions: a chronic increase in glucocorticoid levels may decrease mitochondrial energy production (35, 37). Membrane lipid dysregulation may affect the levels of pro-inflammatory cytokines, as well as the function of mitochondria, ion channels, and neurotransmitter systems implicated in the pathophysiology of psychiatric diseases (38, 39). In addition, changes in membrane fatty acid composition may alter the function of different cell-surface receptors, ion pumps, and special enzymes, such as 5’-nucleotidase, adenylate cyclase, and Na+/K+-ATPase (38, 40). Increased activity of the inflammatory system and redox pathways may enhance oxidative and nitrosative stress, mitochondrial dysfunction, neurodegeneration and neuronal death, production of pro-inflammatory cytokines, and activity of the HPA axis, whereas it may decrease neurogenesis and serotonin levels (35, 37). In addition, functional brain imaging studies demonstrated abnormalities in regional cerebral glucose metabolism in the prefrontal cortex in patients with mood disorders, providing evidence of persistent hypometabolism, particularly in the frontal gyrus, in depressed patients (41). Recent transcriptomic, proteomic, and metabolomics studies have also highlighted an abnormal cerebral glucose and energy metabolism as one of the potential pathophysiological mechanisms of schizophrenia, raising the possibility that a metabolically based intervention might have therapeutic value in the management of the disease (42).
Consequently, different metabolic changes and their downstream effects may generate complex, interlinked molecular and cellular processes, which may lead to different psychiatric diseases. It can be concluded that alterations in multiple interactive metabolic pathways and their effects on different physiological processes may largely underlie the pathophysiology in patients with psychiatric diseases. Indeed, if defective metabolism is the cause of such pathologies, then utilization of therapies designed to address deficiencies of metabolism (known as metabolic therapies) would be a rational approach for the treatment of these diseases.
In a process known as ketogenesis, the ketone bodies [D-β-hydroxybutyrate (βHB), acetoacetate (AcAc), and acetone] are catabolized under normal physiological conditions by the liver from fatty acids as a source of fuel (43–45). Higher levels of ketones are produced during starvation, fasting, and neonatal development (46, 47). Moreover, although most of βHB, which is used as an energy source in the brain, is synthesized by the liver, ketone body synthesis and release by astrocytes have also been demonstrated (48, 49). Ketone bodies can transport to the bloodstream from the liver, cross the blood–brain barrier (BBB), enter brain cells through monocarboxylic transporters, convert to acetyl CoA in the mitochondria, and enter the Krebs cycle (43–44, 45, 50). Through this process, ketosis (increased ketone body levels in the blood) provides energy by metabolism of ketone bodies to acetyl-CoA and synthesis of ATP for cells in the central nervous system (CNS) (43, 51, 52). It has been demonstrated in animal—and/or human studies—that ketogenic diets and supplements may have metabolism-based therapeutic potential in the treatment of several diseases, such as Alzheimer’s disease (53–57), Parkinson’s disease (54, 58–60), glucose transporter type 1-deficiency syndrome (61–63), amyotrophic lateral sclerosis (60, 64), cancer (44, 58, 65, 66), epilepsy (54, 67, 68), schizophrenia (42, 69–74), anxiety (55, 75–77), autism spectrum disorder (78–81), and depression (69, 77, 82).
Ketogenic diets are high-fat, adequate protein and very low carbohydrate diets that may have an alleviating role on psychiatric diseases (69, 73), likely through bioenergetics, ketone metabolism, and signaling, as well as their effects on, for example, neuronal activity, neurotransmitter balance, and inflammatory processes (43, 52, 83–91). Strict patient compliance to the KD is the primary factor in achieving therapeutic ketosis, and this is often difficult or impossible in the psychiatric population (69). Therefore, the administration of exogenous ketone supplements including medium chain triglycerides (MCTs), ketone salt (KS), ketone ester (KE), and their combination with MCT oil (e.g., KSMCT) presents a strategy to circumvent dietary restriction to rapidly induce and sustain nutritional ketosis (65, 75, 84, 92). Ketone bodies not only enhance cell energy metabolism through anaplerotic effects but also suppress oxidative stress, decrease inflammatory processes, and regulate functions of ion channels and neurotransmitter systems (45, 93, 94)—all processes implicated in the pathophysiology of psychiatric diseases (1, 5, 6, 15, 16, 22). Therefore, the rationale exists for the use of exogenous ketone supplementation, which induces a nutritional ketotic state similar to that derived from the ketogenic diet and may mimic the effects of ketogenic diet on several CNS diseases through ketone body-evoked metabolic and signaling alterations (54, 55, 67, 75, 95–99) and epigenetic effects (100).
In contrast to diabetic ketosis, which can induce pathological levels of blood βHB (ranging >25 mM) and potentially lead to life-threatening acidosis, nutritional ketosis elevates blood βHB from the normal range (0.1–0.2 mM) to a safe and—in many cases—therapeutic range (1–7 mM: therapeutic ketosis) (44, 54, 101). While rigorous adherence to ketogenic diets is typically difficult to follow and requires clear medical guidance and strong motivation, consumption of exogenous ketogenic agents effectively induces ketosis with little difficulty (65, 75, 84, 92, 102). Moreover, prolonged consumption of ketogenic diets may generate side effects, such as weight loss, alteration of mentation, growth retardation, nephrolithiasis, nausea, constipation, gastritis, hyperlipidemia, hypoglycemia, hyperuricemia, and ulcerative colitis (44, 69, 103, 104). Consequently, developing a safer alternative method using ketone body precursors and exogenous ketone supplements, such as KSs or KEs, to circumvent dietary restriction is appealing.
Recent research has demonstrated that it is possible to rapidly increase and maintain blood levels of ketone bodies in a dose-dependent manner in both animals and humans (54, 84, 99) for the treatment of several CNS diseases (55, 64, 67, 75). Thus, it is possible that exogenous ketone supplementation-induced ketosis may be an effective therapeutic tool against psychiatric diseases. Indeed, exogenous ketone supplements have a modulatory influence on behavior and anxiolytic effect in animal studies (55, 75, 83). Moreover, in contrast to ketogenic diets, exogenous ketone supplements are relatively well-tolerated and can be formulated and titrated to minimize or avoid side effects (56, 65, 75, 84, 99, 105, 106).
There is limited evidence to support the beneficial effects of exogenous ketone supplements in psychiatric diseases at the moment [e.g., Refs. (55, 75, 76)], but the use of exogenous ketone supplements may be a viable alternative or adjuvant to pharmacotherapy in the treatment of these disorders. Consequently, in the following major section, we provide a short overview of the metabolism of exogenous ketone supplements, which results in rapid and safe mild therapeutic ketosis and, as a consequence, may be an alternative method to ketogenic diets for the treatment of psychiatric disorders. In the next major sections, therapeutic potential of exogenous ketone supplements in the treatment of each psychiatric disease is summarized. This is followed by a brief conclusions section with perspective and future outlook.
Metabolism of Exogenous Ketone Supplements: Generation of Therapeutic Ketosis
Under typical (high carbohydrate) diet conditions, glycogen-derived glucose is the main energy source of brain cells (43, 107). However, ketogenic diets, starvation, and fasting result in an increased reliance of the brain on fat-derived ketones for fuel (43, 44, 108). Free fatty acids are converted into acyl-CoA in the liver cells, and subsequently, acyl-CoA is metabolized to acetyl-CoA by mitochondrial β-oxidation (Figure 1A). Acetyl-CoA may generate energy (via Krebs cycle: tricarboxylic acid cycle/TCA cycle) or it gets converted into ketone bodies (43–44, 45, 50). As hepatocytes are not able to utilize the high levels of acetyl-CoA derived from ketogenic diet-, starvation-, and fasting-evoked increase in fatty acids, under these conditions, a large portion of acetyl-CoA can be converted to ketone bodies (44, 45, 107). Two acetyl-CoA molecules fuse into one acetoacetyl-CoA molecule by acetoacetyl-CoA-thiolase. Subsequently, hydroxymethylglutaryl-CoA-synthase (HMGS) condenses the third acetyl-CoA molecule with acetoacetyl-CoA to form hydroxymethylglutaryl-CoA (HMG-CoA) (this process, catalyzed by HMGS, is the rate-limiting step of ketogenesis) (43–44, 45, 50). AcAc is liberated from HMG-CoA by hydroxymethylglutaryl-CoA-lyase (HMGL). AcAc may reduce to βHB by a NADH molecule in a βHB dehydrogenase (β-OHBD) catalyzed reaction, or, in lesser amounts, a part of AcAc may metabolize to acetone by the spontaneous, non-enzymatic decarboxylation of AcAc (43–44, 45, 50). The major circulating water-soluble ketone body is βHB (44, 50). AcAc is a chemically unstable molecule, and acetone is a very volatile compound (eliminated mainly via respiration from the lungs) (44, 50). As the metabolic enzyme succinyl-CoA:3-ketoacid CoA transferase (SCOT) is not expressed in the liver, hepatocytes are not able to consume ketone bodies as an energy substrate (45, 50, 52); thus, AcAc and βHB can exit the liver, enter the bloodstream, and be distributed to various tissues, including the brain, after transport through monocarboxylate transporters (43–44, 45, 50). In the mitochondria of brain cells, ketone bodies are converted back to acetyl-CoA (Figure 1A) (43–44, 45, 50). As the first step of this metabolic pathway, βHB oxidizes to AcAc by NAD+ and β-OHBD. AcAc is then metabolized to acetoacetyl-CoA, which converts to two acetyl-CoA molecules (by SCOT and acetoacetyl-CoA-thiolase, respectively). Finally, acetyl-CoA molecules enter the Krebs cycle as an energy source for ATP synthesis (43–44, 45, 50).
Figure 1
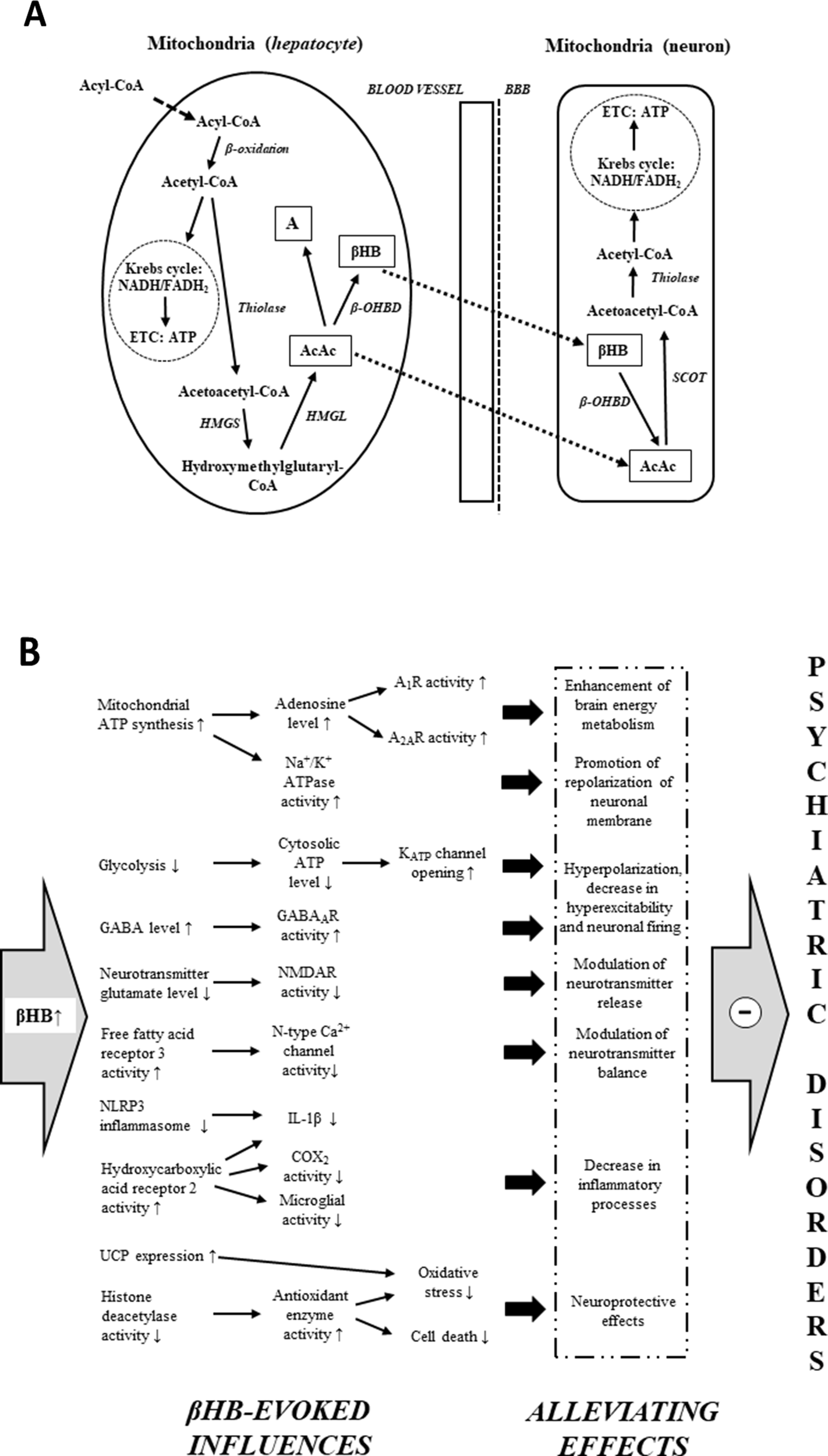
Mitochondrial ketone body metabolism: ketogenesis in liver cells (hepatocytes) and ketolysis in brain cells (neuron) (A). Main βHB-evoked metabolic effects and their consequences, which may evoke alleviating effects on different psychiatric diseases (B) (see text for more detailed putative mechanisms by which βHB may evoke alleviating effects on psychiatric diseases). Abbreviations: A, acetone; A1R and A2AR, different types of adenosine receptors; AcAc, acetoacetate; ATP, adenosine triphosphate; BBB, blood–brain barrier; βHB, D-beta-hydroxybutyrate (R-3-hydroxybutyrate); β-OHBD, βHB dehydrogenase; COX-2, cyclooxygenase-2; ETC, electron transport chain; GABA, gamma-aminobutyric acid; HMGL, hydroxymethylglutaryl-CoA-lyase; HMGS, hydroxymethylglutaryl-CoA-synthase; IL-1β, interleukin-1β; NADH/FADH2, nicotinamide adenine dinucleotide/flavin adenine dinucleotide; NLRP3, NOD-like receptor pyrin domain 3; NMDAR, N-methyl-D-aspartate receptor; SCOT, succinyl-CoA:3-ketoacid CoA transferase; thiolase, acetoacetyl-CoA-thiolase; UCP, uncoupling proteins.
While a ketogenic diet could potentially confer numerous benefits to patients suffering from psychiatric disorders, compliance to the diet would likely be low. Reasons include the lack of knowledge, support, palatability, and different adverse effects such as gastrointestinal side effects (69, 103, 104). Most importantly, ketogenic diets must continuously restrict carbohydrates (typically 20 g/day) to sustain ketogenesis through elevated long-chain fatty acid oxidation (109). Nevertheless, the production of ketone bodies from KSs or KEs (e.g., by liver alcohol dehydrogenase and/or hydrolysis in the small intestine) is not inhibited by carbohydrates; thus, ketone supplements may be usable while maintaining a normal diet (105) to generate therapeutic ketosis.
After consumption or gavage administration, KEs are fully hydrolyzed in the small intestine by esterases, which can be transported to the systemic bloodstream, and converted to 1,3-butanediol. Following this, 1,3-butanediol is metabolized to AcAc and βHB in the liver by alcohol and aldehyde dehydrogenase (106, 110, 111). Moreover, MCTs/MCT oils are hydrolyzed to medium chain fatty acids (e.g., decanoic and octanoic acid) by lipases in the gastrointestinal tract, which are metabolized to ketone bodies in the liver (112). Thus, similar to ketogenic diets, metabolism of exogenous ketone supplements may result in increased levels of blood ketone bodies, which may serve the energy needs of brain cells (Figure 1A). For example, KS supplementation significantly increased the mitochondrial activity of both β-OHBD and acetoacetyl-CoA-thiolase in the brain of rats (83), and oral administration of exogenous ketone supplements is able to evoke and maintain rapid and safe mild ketosis in both animals and human (54, 64, 65, 75, 84, 92, 99, 101, 106, 108).
Unfortunately, MCTs are often not well tolerated because of their gastrointestinal side effects (e.g., diarrhea, dyspepsia, and flatulence) and supplementation of MCTs generates relatively low levels of ketone bodies in the blood (113). Oral administration of KEs fully metabolizes to βHB and AcAc and, as a consequence, more effectively increases ketone body levels compared to MCTs (56). KEs, such as (R)-3-hydroxybutyl-(R)-3-hydroxybutyrate and R,S-1,3-butanediol AcAc diester, are well-tolerated, safe, and efficient ketogenic agents in both animals and humans (56, 99, 105, 106). Moreover, it was demonstrated that a proper dose of KS alone (99) or in combination with other exogenous ketone supplements, such as KE and MCT (KEKS and KSMCT, respectively), may be a safe and efficacious way to achieve ketosis (65, 75, 84, 99). Thus, exogenous ketone supplements may be an effective alternative to ketogenic diets for therapeutic ketosis.
Therapeutic Potential of Exogenous Ketone Supplements in the Treatment of Psychiatric Diseases
Although there has been remarkable progress in our knowledge on the biological effects and mechanisms of action of exogenous ketone supplements, their exact mechanisms on CNS diseases are largely unknown. It has been demonstrated that an increase in ketone body/βHB concentration may modulate neurotransmitter balance and release (43, 52, 85), decrease hyperexcitability, reduce firing rates of neurons (43, 84, 86), decrease neuroinflammation (43, 91), enhance brain energy metabolism (43, 50, 83, 84, 87), and provide neuroprotective effects (43, 45, 84, 88, 90), which together may protect different physiological processes under pathological conditions resulting in CNS diseases, such as psychiatric disorders (35–36, 37, 58, 69). Thus, it is possible that exogenous ketone supplement-evoked ketosis (65, 75, 84) and its significant metabolic effects, as well as their consequences, may have both preventive and therapeutic potential as a metabolic-based therapy in patients with psychiatric diseases (Figure 1B). In spite of the several metabolic alterations, the mechanism of action of exogenous ketone supplement-evoked ketosis on different psychiatric diseases was not investigated comprehensively. As a result, we have only limited results in relation to exact links between alleviating effects of ketone supplement-generated ketosis and pathological changes in psychiatric diseases. Nevertheless, both recent literature results on basic pathomechanisms of psychiatric diseases and mechanisms of therapeutic effects of exogenous ketone supplement-evoked ketosis strongly support the hypothesis that exogenous ketone supplement-evoked ketosis may modulate the background pathophysiological processes of psychiatric diseases. Indeed, an MCT diet caused anxiolytic effects (76) and βHB decreased anxiety-related and depressive behaviors in rats and mice (114, 115). It has also been demonstrated that sub-chronic (7 days) oral administration of exogenous ketone supplements, such as KE, KS, and KSMCT, evoked an anxiolytic effect in normal rats (Sprague–Dawley/SPD rats) and diseased rats (Wistar Albino Glaxo/Rijswijk rats: WAG/Rij rats; a rat model of human absence epilepsy) on elevated plus maze (EPM) test in correlation with increased levels of βHB (75, 95). Elevated ketone body levels were demonstrated in schizophrenic patients, suggesting that the energy supply of brain shifts from glucose towards ketone bodies in this disease (116). Based on correlation between βHB plasma levels and symptoms it was suggested that βHB may have a protective effect on executive functions in patients treated with schizophrenia (117). Other studies presented cases of patients with chronic schizoaffective disorders where the KD begin helping with mood and psychotic symptoms within 1 month or lead to remission of psychotic symptoms (73, 74). It has also been suggested that plasma level of βHB is associated with severity of depression in human and that βHB-evoked antidepressant-like effects may be in relation to its inhibitory effect on NOD-like receptor pyrin domain 3 (NLRP3)-induced neuro-inflammatory processes. The authors also suggested that modification of βHB levels by diet may be a novel therapeutic target for the treatment of mood disorders, such as depression (115, 118). In addition, ketosis (induction of βHB) may be the primary mediator of the therapeutic effect of the ketogenic diet and exogenous ketone supplements on different CNS diseases. From this viewpoint, the effect of exogenous ketone supplements mimics the ketogenic diet (43, 44, 51, 52, 54, 58, 72, 94, 96, 101, 119). Thus, ketogenic diet-evoked effects on psychiatric diseases may result (at least partly) from beneficial metabolic effects of βHB, for example, on mitochondrial functions, neuronal activity, neurotransmitter release, and inflammatory processes (43, 50, 52, 86, 91). Indeed, administration of a ketogenic diet not only increased the ketone body level but also was associated with improvements in anxiety disorder (75, 77), bipolar disorder (120), schizophrenia (42, 70, 73, 74, 121), depression (77, 122), autism spectrum disorder (78, 80, 123), and ADHD (124, 125) in animal models and/or humans, suggesting the beneficial effects of exogenous ketone supplement-induced ketosis on psychiatric diseases (Figure 1B).
However, thorough investigation of signaling pathways by which exogenous ketone supplement-evoked ketosis exerts beneficial effects on psychiatric diseases is needed. In the following subsection, we provide an overview of the main putative basic mechanisms, by which ketone supplement-evoked ketosis may alleviate different pathophysiological processes involved in psychiatric disorders.
Ketosis-Generated Effects on Mitochondrial Functions, Neurotransmitter Systems, Inflammatory Processes, and Their Consequences: Putative Alleviating Influences on Psychiatric Diseases
It has been demonstrated that ketone bodies serve as alternative fuel for brain cells when the glucose supply is insufficient: ketone bodies improve mitochondrial respiration and enhance mitochondrial ATP synthesis (Figure 1B) (47, 126). Increased mitochondrial ATP production may promote the repolarization of neuronal membrane after stimulation by means of Na+/K+ ATPase and may modulate the neurotransmitter levels (119). In addition, βHB may inhibit vesicular glutamate transporters (127). This effect, together with increased ATP production, decreases glutamate loading to vesicles and glutamate release and, as a consequence, suppresses neuronal excitability (68, 119, 127).
It was recently demonstrated that βHB inhibits the activity of N-type Ca2+ channels in sympathetic nerve terminals and may decrease the release of noradrenaline via activation of its G-protein-coupled receptor free fatty acid receptor 3 (FFAR3) (128). Increased levels of ketone bodies, such as βHB, may evoke other changes in metabolic pathways, such as inhibition of glycolysis (43). An inhibition of glycolysis may result in decreased levels of cytosolic ATP and, as a consequence, increased activity of ATP-sensitive potassium (KATP) channels generating hyperpolarization of neuronal membrane and decrease in neuronal activity (43, 129). As it was demonstrated, ketosis not only decreases glutamate release and extracellular glutamate levels and enhances the GABAergic effects by means of increased GABA levels and GABAA receptor activity (43, 68) but also increases adenosine levels (130) and may modulate metabolism of monoamines (Figure 1B). For example, increased levels of noradrenaline in mice brain (131) and decreased levels of metabolites of monoamine dopamine and serotonin (homovanillic acid/HVA and 5-hydroxyindole acetic acid/5-HIAA, respectively) in the human cerebrospinal fluid (132) were demonstrated under a ketotic state. Increased levels of extracellular adenosine lead to increased activity of adenosine receptors and may decrease hyperexcitability via A1Rs, increase hyperpolarization of neuronal membrane, and decrease neuronal activity (133, 134). In addition, adenosine decreases the energy demand of brain tissue (e.g., via A1R and A2AR) (135), modulates immune system functions (e.g., activation of A2AR decreases the inflammation-induced cytokine production from microglial cells) (136), and has a neuroprotective effect (e.g., evokes a decrease in oxidative stress and attenuates the harmful influence of ROS on brain cells via A1R) (137, 138).
β-Hydroxybutyrate may exert its effects on numerous targets, including oxidative stress mediators (e.g., by inhibition of histone deacetylases and increased activity of antioxidant enzymes) and metabolic rate (e.g., increased NAD+–NAD+/NADH ratio) directly and/or indirectly via its G-protein-coupled receptors, such as hydroxycarboxylic acid receptor 2 (HCAR2, also known as PUMA-G or GPR109 receptor) (45, 90, 139, 140). As an endogenous ligand, βHB activates the HCAR2 receptor expressed on, for example, microglial cells (141). HCAR2 mediates the inhibitory effects of βHB on neurodegeneration, microglial activation, and inflammatory processes [e.g., decreases the expression/level of interleukins, such as interleukin-1β (IL-1β), and lipopolysaccharide/LPS-induced increase in cyclooxygenase-2/COX-2 activity and interleukin levels] (141–143) (Figure 1B). NOD-like receptor pyrin domain 3 inflammasome is a multiprotein complex, which may evoke cleavage of pro-IL-1β to its active form (IL-1β) for secretion by caspase-1 (144, 145). It was demonstrated that βHB decreases inflammatory processes likely through inhibition of NLRP3: βHB decreased not only the expression of NLRP3 and caspase-1 but also the level/release of proinflammatory cytokines, such as IL-1β (91, 146).
In general, oxidative stress damages proteins, lipids, and nucleic acids. One putative downstream effect of this damage is the opening of the mitochondrial permeability transition (mPT) pore and, as a consequence, activation of the apoptotic cascade processes pursuant to release of cytochrome c to the cytoplasm (147). It was demonstrated that increased production of ROS may activate mPT pore (97, 147). Ketone bodies decreased oxidative stress and ROS formation by enhancing complex I (NADH dehydrogenase)-driven mitochondrial respiration (140). It has also been demonstrated that KE increased both ketone body levels and expression of mitochondrial uncoupling proteins (UCPs; e.g., UCP 4 and UCP 5 in rat brain), which can decrease the production of ROS (50, 148) (Figure 1B). In addition, it was suggested that βHB not only prevents neuronal loss but also preserves synaptic function: βHB mitigates effects, which may evoke cell death/apoptosis (e.g., glutamate excitotoxicity, enhanced ROS production, impaired mitochondrial energetic functions, pathogenic mutations on mitochondrial DNA, and activation of mPT pore) (44, 97, 119, 149), and βHB may restore impairment of hippocampal long-term potentiation (150).
The changes induced by ketosis may lead to enhanced brain energy metabolism, promotion of repolarization of neuronal membrane, neuronal hyperpolarization, decreased hyperexcitability and neuronal firing, modulation of neurotransmitter release/balance, neuroprotective effects, and decreased inflammatory processes (Figure 1B). Downstream effects may include increased GABA and ATP/adenosine levels, decreased levels of glutamate and IL-1β, and reductions in neuronal excitability and ROS formation. Based on these putative alleviating effects, which may have therapeutic potential in the treatment of different psychiatric diseases, this subsection is followed by a brief overview of the main pathological changes in different psychiatric diseases, which may be modulated or improved by ketosis-evoked beneficial effects and their consequences. Currently, we lack detailed information for understanding the exact mechanisms by which ketosis evokes beneficial effects on psychiatric disorders. However, we can be reasonably confident that the alleviating effects of exogenous ketone supplements on these disorders affect several interacting factors, including mitochondrial function, neurotransmitter levels, and inflammatory processes.
Anxiety Disorders
An increasing body of evidence suggests that dysregulation of the glutamatergic, serotonergic, purinergic, and GABAergic systems plays a role in the pathophysiology of anxiety disorders (33, 34, 151–153). For example, inhibition of NMDA and AMPA receptors by their antagonists (e.g., DL-2-amino-5-phosphonovaleric acid/APV and 6-cyano-7-nitroquinoxaline-2,3-dione/CNQX, respectively) fully or partially blocked the expression and/or acquisition of fear conditioning (30, 154). Activation of the serotonergic system (e.g., via increased levels of serotonin by selective serotonin reuptake inhibitors/SSRIs and activation of serotonin 5-HT1A receptors by buspirone or tandospirone) and increased activity of adenosinergic system (e.g., via activation of A1 type of adenosine receptors/A1R) have an anxiolytic effect (34, 155). Moreover, enhanced GABAergic neurotransmission evoked an anxiolytic effect, whereas decreased GABAergic transmission generated anxiogenic responses in animals (151, 153, 156). Altered functions are present in many regions, including the extended amygdala, ventromedial prefrontal cortex, hippocampus, hypothalamus, and the midbrain, and changed connections between these areas are implicated in the pathophysiology of anxiety disorders (157–159). Specific changes, such as underactivation (e.g., in ventromedial prefrontal cortex), overactivaton (e.g., in amygdala), and deficient functional connectivity (e.g., between hippocampus and amygdala), have also been demonstrated (157, 158, 160, 161). Changes in gray matter volume (e.g., in the right orbitofrontal cortex, amygdala, and hippocampus) (160, 162, 163), as well as dysfunction or hyperactivation of HPA axis and inflammatory system (e.g., increased level of proinflammatory cytokines) (14, 164), may have a role in pathophysiology of anxiety disorders. It was also demonstrated that mitochondrial dysfunctions and oxidative stress may be key factors in the emergence of anxiety disorders (165, 166).
Schizophrenia
It has been demonstrated that alterations in the neurotransmitter systems governed by GABA, glutamate, and the monoamines are involved in the development of schizophrenia (7, 23, 27, 32, 167–169). For example, in the prefrontal cortex, which partially mediates the negative symptoms of schizophrenia, low serotonin and dopamine levels were detected (7, 23). Cognitive symptoms may be linked with decreased level of GABA and serotonin (e.g., in the dorsolateral prefrontal cortex) (7, 170). Moreover, decrease in serotonin level was demonstrated in amygdala, which may lead to aggressive symptoms (7). It was concluded that, among others, hypofunction of the inhibitory GABAergic interneurons and changes in activity of implicated brain areas (e.g., because of decreased activity of inhibitory effects and imbalance between inhibitory/excitatory processes) have a role in the pathophysiology of schizophrenia (7, 167). Another recent study using an acute NMDA receptor hypofunction model of schizophrenia showed that feeding C57BL/6 mice a low carbohydrate/high-fat KD for 7 weeks prevented a variety of behavioral abnormalities induced by pharmacological inhibition of NMDA glutamate receptors (42). In the study, they found a lack of correlation between the measured prepulse inhibition of startle and body weight changes, providing evidence against the role of calorie restriction in its mechanism of action (42). Case studies on human patients with schizophrenia also supported the efficacy of using KD to improve symptoms (73, 74). Reduction in the volume of brain areas encompassing cortical gray and white matter (e.g., in amygdala and hippocampus/sensorimotor and dorsolateral prefrontal cortices) (171–173), gliosis (174), and increased neuronal apoptosis (7, 175) were also demonstrated in patients with schizophrenia. A great deal of evidence suggests that microglial activation, oxidative stress (e.g., increase in ROS activity), and mitochondrial dysfunction (e.g., changes in activity of complex I and cytochrome-c-oxidase/IV of electron transport chain) may also be involved in the pathophysiology of schizophrenia (167, 176–178). Increased activation of HPA axis by psychological stress, inflammatory processes, and increased level of cytokines (e.g., tumor necrosis factor alpha/TNF-α and IL-1β), as well as enhanced levels of glutamate and dopamine auto-oxidation, could lead to enhanced production of ROS and subsequently neurodegeneration and apoptosis (7, 167, 178–180).
Major Depressive Disorder
Structural brain alterations, such as decreased volume and cell number of brain areas (e.g., in hippocampus and several cortical areas) (3, 181–183) and abnormalities in activation or connectivity of brain structures and networks (e.g., chronic hyperactivity of limbic centers and brainstem) (13, 22, 184–186), may underlie the functional and behavioral changes observed in depressed patients. It has been demonstrated that changes in several components, including the glutamatergic system (e.g., increased glutamate level) (29), monoaminergic system (e.g., decrease in the level of serotonin, noradrenaline, and dopamine) (3, 13, 24, 187, 188), GABAergic system (e.g., reduced plasma and cerebrospinal fluid GABA levels) (189, 190), and purinergic system (e.g., overexpression of A2A type of adenosine receptors/A2AR) (28) have a role in the pathophysiology of major depressive disorder. Activation of microglia and astrocytes and inflammatory pathways (14, 164, 191, 192) may be associated with major depressive disorder. For example, increased activation and expression of NLRP3 inflammasome and interleukins (e.g., IL-1β) were revealed in both animal models and patients with depression (13, 193, 194). Hyperactivity of HPA system was also demonstrated (195). Neurodegeneration and neuronal death (e.g., through increased oxidative/nitrosative stress) and alterations in mitochondrial functions (e.g., decreased ATP production as well as enhanced apoptosis and oxidative stress) (35, 177, 196) also play a role in the emergence of major depressive disorder. It has been demonstrated that enhancement of inflammatory processes is associated with depression by modulation of different neurotransmitter systems: for example, inflammatory cytokines (e.g., IL-1β) reduce synaptic availability of monoamines and increase excitotoxicity (via extrasynaptic NMDA receptors) by increasing levels of extracellular glutamate (164, 197, 198). Moreover, cytokines may evoke decreased motivation and anhedonia via different pathways (e.g., by decreased release of dopamine in the basal ganglia) (164, 199).
Bipolar Disorder
It has been demonstrated that imbalance in monoaminergic neurotransmitter system (e.g., serotonergic, dopaminergic, and noradrenergic) (200–202), GABAergic system (e.g., decreased GABAergic transmission) (190), purinergic system (e.g., increased level of uric acid and reduced adenosinergic activity at A1Rs) (31), and glutamatergic system (e.g., increased glutamate levels and NMDA receptor activity) (29) are associated with bipolar disorder. These alterations may be associated with mitochondrial dysfunction (e.g., deficit in activity of complexes I and IV), apoptosis, increase in ROS, oxidative damage, hyperexcitability (5, 177, 203, 204), and, as a consequence, decrease in glial cell or neuron number and gray matter, as well as changes in connectivity between implicated brain areas (e.g., hippocampus, prefrontal cortex, and amygdala) (205–207). Changes in endocrine functions (e.g., dysregulation of HPA axis) and inflammatory processes (e.g., increased proinflammatory cytokine levels, such as IL-1β) were demonstrated in association with bipolar disorder (203, 208).
Autism Spectrum Disorder
It has been demonstrated that agenesis of corpus callosum, changes in brain volume, thinning of several brain cortical areas (e.g., in the frontal parietal lobe), and decreased functional connectivity between brain areas (e.g., within frontal cortex) contribute to pathophysiology of autism spectrum disorder (209–212). It was also demonstrated that dysfunction in glutamatergic system (e.g., exaggerated signaling) (213–215) and GABAergic system (e.g., decreased GABA receptor expression and GABA-evoked inhibitory effects) (215, 216) may have a role in the pathophysiology of autism spectrum disorder by alterations in the excitation/inhibition balance. In addition, decreased level of serotonin/adenosine in implicated brain areas (e.g., medial frontal cortex) have also been demonstrated/suggested in this disease (25, 217–220). Impaired immune response, inflammation, and oxidative stress may be causative factors of autism spectrum disorder (15, 221). In fact, recent studies suggest that autism spectrum disorder is associated with inflammation (e.g., activation of glial cells and increased levels of cytokines) (222–224), mitochondrial dysfunction, and oxidative stress (e.g., increased ROS activity) (79, 225–227).
Attention Deficit/Hyperactivity Disorder
Reduction of brain volume and gray matter (e.g., in putamen and caudate nucleus) and underactivation or hyperactivation of different brain networks (e.g., in the frontoparietal and ventral attention network and the somatomotor system) were demonstrated in patients with ADHD (228, 229). Numerous studies have shown that increased glutamatergic tone/glutamate level (230), dopamine hypofunction (e.g., decreased stimulation-evoked release of dopamine) (26), and changes in GABAergic (e.g., decrease in GABA level) (230, 231), noradrenergic, and serotonergic system (16, 232–235) in the implicated brain areas may be causative factors of ADHD. Furthermore, increased oxidative stress (e.g., enhanced production of ROS) was demonstrated in a rat model of ADHD (236).
Conclusion
The effects of nutritional ketosis on CNS diseases, whether through diet or supplementation, have not been fully investigated. Consequently, only limited results have demonstrated the existence of alleviating effects of exogenous ketone supplement administration on animal models of psychiatric diseases and patients with psychiatric disorders. Nevertheless, there are several common pathophysiological metabolic alterations, such as changes in neurotransmitter release, increased inflammatory processes, abnormal cerebral glucose metabolism, and decreased mitochondrial-associated brain energy metabolism, which may have a role in the emergence of psychiatric diseases. Consequently, ketogenic interventions that can modulate a broad array of metabolic and signaling changes underlying the pathophysiology of psychiatric diseases may alleviate the onset of symptoms.
Based on our review of the literature, we hypothesize that utilizing exogenous ketone supplements alone or with ketogenic diet, either as a primary or an adjunctive therapy for selected psychiatric disorders, may potentially be an effective treatment. Thus, adding ketone supplements as an additional agent to the therapeutic regimen may alleviate symptoms of psychiatric diseases via modulation of different metabolic routes implicated in psychiatric disorders. Therefore, detailed investigation of exogenous ketone supplement-evoked direct and/or indirect alterations in molecular pathways and signaling processes associated with psychiatric diseases is needed.
The use of exogenous ketone supplements in psychiatric diseases is only in its infancy. Nevertheless, our increasing understanding of how exogenous ketone supplement-evoked ketosis/βHB exerts its effects on CNS diseases, combining with new results on pathophysiology of psychiatric diseases and their complex interplay with each other, suggests that exogenous ketone supplements may be ideal and effective adjuvants to drugs used in the treatment of psychiatric diseases. Thus, because exogenous ketone supplements modulate endogenous processes, their administration is a safe method to promote disease-alleviating effects without considerable risk, as well as minimal or no side effects compared to pharmacological treatments. Consequently, exogenous ketone supplements may help to both manage the side effects and increase the efficacy of drugs used in psychiatric diseases, especially in cases of treatment resistance.
Future research should explore the effects of exogenous ketones on the metabolic processes that underlie the diseases leading to psychiatric disorders in order to restore abnormal cerebral glucose and energy metabolism. Moreover, new studies are needed to investigate the effects, therapeutic efficacy, and exact mechanism(s) of action of exogenous ketone supplements alone or in combination with a ketogenic diet not only on animal models of psychiatric diseases, but also on patients with different psychiatric disorders. Future studies are needed to reveal which factors (e.g., age, sex, lifestyle, drugs, other diseases, and so on) can modify the effects of exogenous ketone supplements on psychiatric diseases; to develop new, more effective, and safe ketone supplements, which can be used in special ketogenic foods for treatment of CNS disorders, including psychiatric diseases. There is urgent need to develop therapeutic strategies and broadly accepted protocols guiding the administration of different types and combinations of exogenous ketone supplements. As a result of new studies in the near future, a better understanding of the pathophysiology of different psychiatric diseases and the connections between the underlying metabolic/signaling pathways may promote the development of novel metabolism-based adjuvant therapies, such as the administration of exogenous ketone supplements against psychiatric diseases.
Abbreviations
A1R and A2AR, different types of adenosine receptors; AcAc, acetoacetate; ADHD, attention-deficit/hyperactivity disorder; BBB, blood–brain barrier; βHB, D-beta-hydroxybutyrate (R-3-hydroxybutyrate); β-OHBD, βHB dehydrogenase; CNS, central nervous system; COX-2, cyclooxygenase-2; FFAR3, free fatty acid receptor 3; HCAR2, hydroxycarboxylic acid receptor 2; HPA, hypothalamic-pituitary-adrenal; HMG-CoA, hydroxymethylglutaryl-CoA; HMGL, hydroxymethylglutaryl-CoA-lyase; HMGS, hydroxymethylglutaryl-CoA-synthase; KE, ketone ester; KS, ketone salt; IL-1β, interleukin-1β; MCT, medium chain triglyceride; NLRP3, NOD-like receptor pyrin domain 3; ROS, reactive oxygen species; SCOT: succinyl-CoA:3-ketoacid CoA transferase; SSRI, selective serotonin reuptake inhibitor; UCP, uncoupling proteins; WAG/Rij, Wistar Albino Glaxo/Rijswijk.
Statements
Author contributions
ZK contributed to the conception of the manuscript, comprehensive search of the electronic databases, and writing of the manuscript. DPD contributed to the writing of the manuscript. DD, MK, and CR were in charge of revising the manuscript. CA was in charge of writing and revising the manuscript.
Acknowledgments
This work was supported by OTKA K124558 Research Grant (to Zsolt Kovács) and ONR Grant N000141310062 and GLUT1D Foundation Grant #6143113500 (to Dominic D`Agostino). We thank Quest Nutrition LLC for supporting ongoing studies on this topic (to Csilla Ari).
Conflict of interest
International Patent #PCT/US2014/031237, University of South Florida, D.P. D’Agostino, S. Kesl, P. Arnold, “Compositions and Methods for Producing Elevated and Sustained Ketosis.” Non-provisional patents: #62289749, University of South Florida, C. Ari, D.P. D’Agostino, “Exogenous ketone supplements for reducing anxiety-related behavior”; Ari, C., Arnold P., D’Agostino, D.P. Technology Title: “Elevated blood ketone levels by ketogenic diet or exogenous ketone supplements induced increased latency of anesthetic induction” USF Ref. No. 16A018PR; Ari, C., Arnold P., D’Agostino, D.P. Technology Title: “Exogenous ketone supplementation improved motor function in Sprague–Dawley rats.” USF Ref. No: 16A019; Ari, C., Arnold P., D’Agostino, D.P. Technology Title: “Lowering of blood glucose in exercising and non-exercising rats following administration of exogenous ketones and ketone formulas.” USF Ref. No: 16A049; Ari, C., Arnold P., D’Agostino, D.P. Technology Title: “Ketone supplementation elevates blood ketone level and improves motor function in GLUT1 deficiency syndrome mice.” USF Ref. No: 16B116 (provisional patent); Ari, C., Arnold P., D’Agostino, D.P. Technology Title: “Neuroregeneration improved by ketone.” USF Ref. No: 16B128 (provisional patent); Ari, C., D’Agostino, D.P. Dean, J.B. Technology Title: “Delaying latency to seizure by combinations of ketone supplements.” USF Ref. No: 16B138PR. D.P. D’Agostino and C. Ari are co-owners of the company Ketone Technologies LLC, providing scientific consulting and public speaking engagements about ketogenic therapies. The company obtained an option agreement from the University of South Florida on the non-provisional patent no. 62/310,302 “Methods of increasing latency of anesthetic induction using ketone supplementation.” These interests have been reviewed and managed by the University in accordance with its Institutional and Individual Conflict of Interest policies. All authors declare that there are no additional conflicts of interest.
References
1
CraskeMGSteinMBEleyTCMiladMRHolmesARapeeRMet al. Anxiety disorders. Nat Rev Dis Primers (2017) 3:17024. doi: 10.1038/nrdp.2017.24
2
BrometEAndradeLHHwangISampsonNAAlonsoJde GirolamoGet al. Cross-national epidemiology of DSM-IV major depressive episode. BMC Med (2011) 9:90. doi: 10.1186/1741-7015-9-90
3
KupferDJFrankEPhillipsML. Major depressive disorder: new clinical, neurobiological, and treatment perspectives. Lancet (2012) 379(9820):1045–55. doi: 10.1016/S0140-6736(11)60602-8
4
MerikangasKRJinRHeJPKesslerRCLeeSSampsonNAet al. Prevalence and correlates of bipolar spectrum disorder in the world mental health survey initiative. Arch Gen Psychiatry (2011) 68(3):241–51. doi: 10.1001/archgenpsychiatry.2011.12
5
VietaEBerkMSchulzeTGCarvalhoAFSuppesTCalabreseJRet al. Bipolar disorders. Nat Rev Dis Primers (2018) 4:18008. doi: 10.1038/nrdp.2018.8
6
KahnRSSommerIEMurrayRMMeyer-LindenbergAWeinbergerDRCannonTDet al. Schizophrenia. Nat Rev Dis Primers (2015) 1:15067. doi: 10.1038/nrdp.2015.67
7
PatelSSharmaDKaliaKTiwariV. Crosstalk between endoplasmic reticulum stress and oxidative stress in schizophrenia: the dawn of new therapeutic approaches. Neurosci Biobehav Rev (2017b) 83:589–603. doi: 10.1016/j.neubiorev.2017.08.025
8
PolanczykGde LimaMSHortaBLBiedermanJRohdeLA. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry (2007) 164(6):942–8. doi: 10.1176/appi.ajp.164.6.942
9
SimonVCzoborPBálintSMészárosABitterI. Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br J Psychiatry (2009) (3):204–11. doi: 10.1192/bjp.bp.107.048827
10
ChristensenDLBaioJVan Naarden BraunKBilderDCharlesJConstantinoJNet al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2012. MMWR Surveill Summ (2016) 65(3):1–23. doi: 10.15585/mmwr.ss6503a1
11
CraskeMGSteinMB. Anxiety. Lancet (2016) 388(10063):3048–59. doi: 10.1016/S0140-6736(16)30381-6
12
LichtensteinPYipBHBjörkCPawitanYCannonTDSullivanPFet al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet (2009) 373(9659):234–9. doi: 10.1016/S0140-6736(09)60072-6
13
DeanJKeshavanM. The neurobiology of depression: an integrated view. Asian J Psychiatr (2017) 27:101–11. doi: 10.1016/j.ajp.2017.01.025
14
HodesGEKanaVMenardCMeradMRussoSJ. Neuroimmune mechanisms of depression. Nat Neurosci (2015) 18(10):1386–93. doi: 10.1038/nn.4113
15
BhatSAcharyaURAdeliHBairyGMAdeliA. Autism: cause factors, early diagnosis and therapies. Rev Neurosci (2014) 25(6):841–50. doi: 10.1515/revneuro-2014-0056
16
FaraoneSVAshersonPBanaschewskiTBiedermanJBuitelaarJKRamos-QuirogaJAet al. Attention-deficit/hyperactivity disorder. Nat Rev Dis Primers (2015) 1:15020. doi: 10.1038/nrdp.2015.20
17
BrownKASamuelSPatelDR. Pharmacologic management of attention deficit hyperactivity disorder in children and adolescents: a review for practitioners. Transl Pediatr (2018a) 7(1):36–47. doi: 10.21037/tp.2017.08.02
18
MoffittTEHarringtonHCaspiAKim-CohenJGoldbergDGregoryAMet al. Depression and generalized anxiety disorder: cumulative and sequential comorbidity in a birth cohort followed prospectively to age 32 years. Arch Gen Psychiatry (2007) 64(6):651–60. doi: 10.1001/archpsyc.64.6.651
19
PatelDRFeuchtCBrownKRamsayJ. Pharmacological treatment of anxiety disorders in children and adolescents: a review for practitioners. Transl Pediatr (2018) 7(1):23–35. doi: 10.21037/tp.2017.08.05
20
WhiteheadCMossSCardnoALewisG. Antidepressants for the treatment of depression in people with schizophrenia: a systematic review. Psychol Med (2003) 33(4):589–99. doi: 10.1017/S0033291703007645
21
WilczynskiSMMenousekKHunterMMudgalD. Individualized education programs for youth with autism spectrum disorders. Psychol Schools (2007) 44(7):653. doi: 10.1002/pits.20255
22
OtteCGoldSMPenninxBWParianteCMEtkinAFavaMet al. Major depressive disorder. Nat Rev Dis Primers (2016) 2:16065. doi: 10.1038/nrdp.2016.65
23
BrischRSaniotisAWolfRBielauHBernsteinHGSteinerJet al. The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: old fashioned, but still in vogue. Front Psychiatry (2014) 5:47. doi: 10.3389/fpsyt.2014.00047
24
DelgadoPL. Depression: the case for a monoamine deficiency. J Clin Psychiatry (2000) 61 (Suppl 6):7–11. doi: 10.4088/JCP.v61n0103
25
NakamuraKSekineYOuchiYTsujiiMYoshikawaEFutatsubashiMet al. Brain serotonin and dopamine transporter bindings in adults with high-functioning autism. Arch Gen Psychiatry (2010) 67(1):59–68. doi: 10.1001/archgenpsychiatry.2009.137
26
RussellVA. Dopamine hypofunction possibly results from a defect in glutamate-stimulated release of dopamine in the nucleus accumbens shell of a rat model for attention deficit hyperactivity disorder–the spontaneously hypertensive rat. Neurosci Biobehav Rev (2003) 27(7):671–82. doi: 10.1016/j.neubiorev.2003.08.010
27
ChiapponiCPirasFPirasFCaltagironeCSpallettaG. GABA system in schizophrenia and mood disorders: a mini review on third-generation imaging studies. Front Psychiatry (2016) 7:61. doi: 10.3389/fpsyt.2016.00061
28
CoelhoJEAlvesPCanasPMValadasJSShmidtTBatalhaVLet al. Overexpression of adenosine A2A receptors in rats: effects on depression, locomotion, and anxiety. Front Psychiatry (2014) 5:67. doi: 10.3389/fpsyt.2014.00067
29
HashimotoKSawaAIyoM. Increased levels of glutamate in brains from patients with mood disorders. Biol Psychiatry (2007) 62(11):1310–6. doi: 10.1016/j.biopsych.2007.03.017
30
KimMCampeauSFallsWADavisM. Infusion of the non-NMDA receptor antagonist CNQX into the amygdala blocks the expression of fear-potentiated startle. Behav Neural Biol (1993) 59(1):5–8. doi: 10.1016/0163-1047(93)91075-X
31
Machado-VieiraRLaraDRSouzaDOKapczinskiF. Purinergic dysfunction in mania: an integrative model. Med Hypotheses (2002) 58(4):297–304. doi: 10.1054/mehy.2001.1543
32
MoghaddamBJavittD. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology (2012) 37(1):4–15. doi: 10.1038/npp.2011.181
33
MöhlerH. The GABA system in anxiety and depression and its therapeutic potential. Neuropharmacology (2012) 62(1):42–53. doi: 10.1016/j.neuropharm.2011.08.040
34
VincenziFRavaniAPasquiniSMerighiSGessiSRomagnoliRet al. Positive allosteric modulation of A1 adenosine receptors as a novel and promising therapeutic strategy for anxiety. Neuropharmacology (2016) 111:283–92. doi: 10.1016/j.neuropharm.2016.09.015
35
AdzicMBrkicZBulajicSMiticMRadojcicMB. Antidepressant action on mitochondrial dysfunction in psychiatric disorders. Drug Dev Res (2016) 77(7):400–6. doi: 10.1002/ddr.21332
36
ZuccoliGSSaia-CeredaVMNascimentoJMMartins-de-SouzaD. The energy metabolism dysfunction in psychiatric disorders postmortem brains: focus on proteomic evidence. Front Neurosci (2017) 11:493. doi: 10.3389/fnins.2017.00493
37
MaesMYirmyiaRNorabergJBreneSHibbelnJPeriniGet al. The inflammatory & neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab Brain Dis (2009) 24(1):27–53. doi: 10.1007/s11011-008-9118-1
38
MurphyMG. Dietary fatty acids and membrane protein function. J Nutr Biochem (1990) 1(2):68–79. doi: 10.1016/0955-2863(90)90052-M
39
MüllerCPReichelMMühleCRheinCGulbinsEKornhuberJ. Brain membrane lipids in major depression and anxiety disorders. Biochim Biophys Acta (2015) (8):1052–65. doi: 10.1016/j.bbalip.2014.12.014
40
MockingRJTAssiesJRuhéHGScheneAH. Focus on fatty acids in the neurometabolic pathophysiology of psychiatric disorders. J Inherit Metab Dis (2018) (4):597–611. doi: 10.1007/s10545-018-0158-3
41
HosokawaTMomoseTKasaiK. Brain glucose metabolism difference between bipolar and unipolar mood disorders in depressed and euthymic states. Prog Neuropsychopharmacol Biol Psychiatry (2009) 33(2):243–50. doi: 10.1016/j.pnpbp.2008.11.014
42
KraeuterAKvan den BuuseMSarnyaiZ. Ketogenic diet prevents impaired prepulse inhibition of startle in an acute NMDA receptor hypofunction model of schizophrenia. Schizophr Res (2018) 206:244–500. doi: 10.1016/j.schres.2018.11.011
43
AchantaLBRaeCD. β-Hydroxybutyrate in the brain: one molecule, multiple mechanisms. Neurochem Res (2017) 42:35–49. doi: 10.1007/s11064-016-2099-2
44
BrancoAFFerreiraASimõesRFMagalhães-NovaisSZehowskiCCopeEet al. Ketogenic diets: from cancer to mitochondrial diseases and beyond. Eur J Clin Invest (2016) 46(3):285–98. doi: 10.1111/eci.12591
45
NewmanJCVerdinE. Ketone bodies as signaling metabolites. Trends Endocrinol Metab (2014) 25:42–52. doi: 10.1016/j.tem.2013.09.002
46
AugustinKKhabbushAWilliamsSEatonSOrfordMCrossJHet al. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol (2018) 17(1):84–93. doi: 10.1016/S1474-4422(17)30408-8
47
VanItallieTBNufertTH. Ketones: metabolism’s ugly duckling. Nutr Rev (2003) (10):327–41. doi: 10.1301/nr.2003.oct.327-341
48
GuzmánMBlázquezC. Ketone body synthesis in the brain: possible neuroprotective effects. Prostaglandins Leukot Essent Fatty Acids (2004) 70(3):287–92. doi: 10.1016/j.plefa.2003.05.001
49
Le FollCLevinBE. Fatty acid-induced astrocyte ketone production and the control of food intake. Am J Physiol Regul Integr Comp Physiol (2016) 310(11):R1186–92. doi: 10.1152/ajpregu.00113.2016
50
KoppelSJSwerdlowRH. Neuroketotherapeutics: a modern review of a century-old therapy. Neurochem Int (2018) 117:114–125. doi: 10.1016/j.neuint.2017.05.019
51
VeechRL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids (2004) 70:309–19. doi: 10.1016/j.plefa.2003.09.007
52
YudkoffMDaikhinYMeløTMNissimISonnewaldUNissimI. The ketogenic diet and brain metabolism of amino acids: relationship to the anticonvulsant effect. Annu Rev Nutr (2007) 27:415–30. doi: 10.1146/annurev.nutr.27.061406.093722
53
BroomGMShawICRucklidgeJJ. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition (2019) 60:118–21. doi: 10.1016/j.nut.2018.10.003
54
HashimSAVanItallieTB. Ketone body therapy: from the ketogenic diet to the oral administration of ketone ester. J Lipid Res (2014) 55:1818–26. doi: 10.1194/jlr.R046599
55
KashiwayaYBergmanCLeeJHWanRKingMTMughalMRet al. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol Aging (2013) 34(6):1530–9. doi: 10.1016/j.neurobiolaging.2012.11.023
56
NewportMTVanItallieTBKashiwayaYKingMTVeechRL. A new way to produce hyperketonemia: use of ketone ester in a case of Alzheimer’s disease. Alzheimers Dement (2015) 11:99–103. doi: 10.1016/j.jalz.2014.01.006
57
PenkeBParagiGGeraJBerkeczRKovácsZCrulTet al. The role of lipids and membranes in the pathogenesis of Alzheimer’s disease: a comprehensive view. Curr Alzheimer Res (2018) 15(13):1191–212. doi: 10.2174/1567205015666180911151716
58
StafstromCERhoJM. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front Pharmacol (2012) 3:59. doi: 10.3389/fphar.2012.00059
59
VanitallieTBNonasCDi RoccoABoyarKHyamsKHeymsfieldSB. Treatment of Parkinson disease with diet-induced hyperketonemia: a feasibility study. Neurology (2005) 64(4):728–30. doi: 10.1212/01.WNL.0000152046.11390.45
60
Veyrat-DurebexCReynierPProcaccioVHergesheimerRCorciaPAndresCRet al. How can a ketogenic diet improve motor function? Front Mol Neurosci (2018) 11:15. doi: 10.3389/fnmol.2018.00015
61
BekkerYACLambrechtsDAVerhoevenJSvan BoxtelJTroostCKamsteegEJet al. Failure of ketogenic diet therapy in GLUT1 deficiency syndrome. Eur J Paediatr Neurol (2019) (in press). doi: 10.1016/j.ejpn.2019.02.012
62
KlepperJLeiendeckerB. Glut1 deficiency syndrome and novel ketogenic diets. J Child Neurol (2013) 28(8):1045–8. doi: 10.1177/0883073813487600
63
VeggiottiPDe GiorgisV. Dietary treatments and new therapeutic perspective in GLUT1 deficiency syndrome. Curr Treat Options Neurol (2014) 16(5):291. doi: 10.1007/s11940-014-0291-8
64
AriCPillaRD’AgostinoD. Nutritional/metabolic therapies in animal models of amyotrophic lateral sclerosis, Alzheimer’s disease, and seizures. In: Bioactive Nutraceuticals and Dietary Supplements in Neurological and Brain Disease. Elsevier, Chapter 47 (2015). p. 449–59. doi: 10.1016/B978-0-12-411462-3.00047-3
65
PoffAMWardNSeyfriedTNArnoldPD’AgostinoDP. Non-toxic metabolic management of metastatic cancer in VM mice: novel combination of ketogenic diet, ketone supplementation, and hyperbaric oxygen therapy. PLoS One (2015) 10:e0127407. doi: 10.1371/journal.pone.0127407
66
SremanakovaJSowerbuttsAMBurdenS. A systematic review of the use of ketogenic diets in adult patients with cancer. J Hum Nutr Diet (2018) 31(6):793–802. doi: 10.1111/jhn.12587
67
KovácsZD’AgostinoDPDobolyiAAriC. Adenosine A1 receptor antagonism abolished the anti-seizure effects of exogenous ketone supplementation in Wistar Albino Glaxo Rijswijk rats. Front Mol Neurosci (2017) 10:235. doi: 10.3389/fnmol.2017.00235
68
McNallyMAHartmanAL. Ketone bodies in epilepsy. J Neurochem (2012) 121:28–35. doi: 10.1111/j.1471-4159.2012.07670.x
69
BostockECKirkbyKCTaylorBV. The current status of the ketogenic diet in psychiatry. Front Psychiatry (2017) 8:43. doi: 10.3389/fpsyt.2017.00043
70
KraftBDWestmanEC. Schizophrenia, gluten, and low-carbohydrate, ketogenic diets: a case report and review of the literature. Nutr Metab (Lond) (2009) 6:10. doi: 10.1186/1743-7075-6-10
71
PachecoAEasterlingWSPryerMW. A pilot study of the ketogenic diet in schizophrenia. Am J Psychiatry (1965) 121:1110–1. doi: 10.1176/ajp.121.11.1110
72
WłodarczykAWigluszMSCubałaWJ. Ketogenic diet for schizophrenia: nutritional approach to antipsychotic treatment. Med Hypotheses (2018) 118:74–7. doi: 10.1016/j.mehy.2018.06.022
73
PalmerCM. Ketogenic diet in the treatment of schizoafective disorder: two case studies. Schizophr Res (2017) 189:208–9. doi: 10.1016/j.schres.2017.01.053
74
PalmerCMGilbert-JaramilloJWestmanEC. The ketogenic diet and remission of psychotic symptoms in schizophrenia: two case studies. Schizophr Res (2019) (in press). doi: 10.1016/j.schres.2019.03.019
75
AriCKovácsZJuhaszGMurdunCGoldhagenCRKoutnikAPet al. Exogenous ketone supplements reduce anxiety-related behavior in Sprague-Dawley and Wistar Albino Glaxo/Rijswijk rats. Front Mol Neurosci (2016) 9:137. doi: 10.3389/fnmol.2016.00137
76
HollisFMitchellESCantoCWangDSandiC. Medium chain triglyceride diet reduces anxiety-like behaviors and enhances social competitiveness in rats. Neuropharmacology (2018) 138:245–56. doi: 10.1016/j.neuropharm.2018.06.017
77
SussmanDGermannJHenkelmanM. Gestational ketogenic diet programs brain structure and susceptibility to depression & anxiety in the adult mouse offspring. Brain Behav (2015) 5(2):e00300. doi: 10.1002/brb3.300
78
AhnYNarousMTobiasRRhoJMMychasiukR. The ketogenic diet modifies social and metabolic alterations identified in the prenatal valproic acid model of autism spectrum disorder. Dev Neurosci (2014) 36(5):371–80. doi: 10.1159/000362645
79
ChengNRhoJMMasinoSA. Metabolic dysfunction underlying autism spectrum disorder and potential treatment approaches. Front Mol Neurosci (2017) 10:34. doi: 10.3389/fnmol.2017.00034
80
EvangeliouAVlachonikolisIMihailidouHSpiliotiMSkarpalezouAMakaronasNet al. Application of a ketogenic diet in children with autistic behavior: pilot study. J Child Neurol (2003) 18(2):113–8. doi: 10.1177/08830738030180020501
81
RuskinDNMurphyMISladeSLMasinoSA. Ketogenic diet improves behaviors in a maternal immune activation model of autism spectrum disorder. PLoS One (2017b) 12(2):e0171643. doi: 10.1371/journal.pone.0171643
82
BrietzkeEMansurRBSubramaniapillaiMBalanzá-MartínezVVinbergMGonzález-PintoAet al. Ketogenic diet as a metabolic therapy for mood disorders: evidence and developments. Neurosci Biobehav Rev (2018) 94:11–6. doi: 10.1016/j.neubiorev.2018.07.020
83
BrownlowMLJungSHMooreRJBechmannNJankordR. Nutritional ketosis affects metabolism and behavior in Sprague-Dawley rats in both control and chronic stress environments. Front Mol Neurosci (2017) 10:129. doi: 10.3389/fnmol.2017.00129
84
D’AgostinoDPillaRHeldHLandonCPuchowiczMBrunengraberHet al. Therapeutic ketosis with ketone ester delays central nervous system oxygen toxicity seizures in rats. Am J Phys Reg Integr Comp Phys (2013) 304:829–36. doi: 10.1152/ajpregu.00506.2012
85
ErecińskaMNelsonDDaikhinYYudkoffM. Regulation of GABA level in rat brain synaptosomes: fluxes through enzymes of the GABA shunt and effects of glutamate, calcium, and ketone bodies. J Neurochem (1996) 67(6):2325–34. doi: 10.1046/j.1471-4159.1996.67062325.x
86
MaWBergJYellenG. Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J Neurosci (2007) 27:3618–25. doi: 10.1523/JNEUROSCI.0132-07.2007
87
MarosiKKimSWMoehlKScheibye-KnudsenMChengACutlerRet al. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J Neurochem (2016) 139(5):769–81. doi: 10.1111/jnc.13868
88
PuchalskaPCrawfordPA. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab (2017) 25(2):262–84. doi: 10.1016/j.cmet.2016.12.022
89
SamoilovaMWeisspapirMAbdelmalikPVelumianAACarlenPL. Chronic in vitro ketosis is neuroprotective but not anti-convulsant. J Neurochem (2010) (4):826–35. doi: 10.1111/j.1471-4159.2010.06645.x
90
ShimazuTHirscheyMDNewmanJHeWShirakawaKLe MoanNet al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science (2013) 339(6116):211–4. doi: 10.1126/science.1227166
91
YoumYHNguyenKYGrantRWGoldbergELBodogaiMKimDet al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med (2015) 21(3):263–9. doi: 10.1038/nm.3804
92
KeslSLPoffAMWardNPFiorelliTNAriCVan PuttenAJet al. Effects of exogenous ketone supplementation on blood ketone, glucose, triglyceride, and lipoprotein levels in Sprague-Dawley rats. Nutr Metab (Lond) (2016) 13:9. doi: 10.1186/s12986-016-0069-y
93
BoughKJRhoJM. Anticonvulsant mechanisms of the ketogenic diet. Epilepsia (2007) 48:43–58. doi: 10.1111/j.1528-1167.2007.00915.x
94
RogawskiMALöscherWRhoJM. Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harb Perspect Med (2016) 6:1–28. doi: 10.1101/cshperspect.a022780
95
KovácsZD’AgostinoDPAriC. Anxiolytic effect of exogenous ketone supplementation is abolished by adenosine A1 receptor inhibition in Wistar Albino Glaxo/Rijswijk rats. Front Behav Neurosci (2018) 12:29. doi: 10.3389/fnbeh.2018.00029
96
LutasAYellenG. The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci (2013) 36(1):32–40. doi: 10.1016/j.tins.2012.11.005
97
MaaloufMRhoJMMattsonMP. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res Rev (2009) 59(2):293–315. doi: 10.1016/j.brainresrev.2008.09.002
98
SimeoneTASimeoneKAStafstromCERhoJM. Do ketone bodies mediate the anti-seizure effects of the ketogenic diet? Neuropharmacology (2018) 133:233–41. doi: 10.1016/j.neuropharm.2018.01.011
99
StubbsBJCoxPJEvansRDSanterPMillerJJFaullOKet al. On the metabolism of exogenous ketones in humans. Front Physiol (2017) 8:848. doi: 10.3389/fphys.2017.00848
100
BenjaminJSPilarowskiGOCarossoGAZhangLHusoDLGoffLAet al. A ketogenic diet rescues hippocampal memory defects in a mouse model of Kabuki syndrome. Proc Natl Acad Sci U S A (2017) 114(1):125–30. doi: 10.1073/pnas.1611431114
101
VeechRLChanceBKashiwayaYLardyHACahillGFJr. Ketone bodies, potential therapeutic uses. IUBMB Life (2001) 51(4):241–7. doi: 10.1080/152165401753311780
102
KovácsZD’AgostinoDPDiamondDMAriC. Exogenous ketone supplementation decreased the lipopolysaccharide-induced increase in absence epileptic activity in Wistar Albino Glaxo Rijswijk rats. Front Mol Neurosci (2019) 12:45. doi: 10.3389/fnmol.2019.00045
103
HartmanALViningEP. Clinical aspects of the ketogenic diet. Epilepsia (2007) 48(1):31–42. doi: 10.1111/j.1528-1167.2007.00914.x
104
NordliDRJrKurodaMMCarrollJKoenigsbergerDYHirschLJBrunerHJet al. Experience with the ketogenic diet in infants. Pediatrics (2001) 108(1):129–33. doi: 10.1542/peds.108.1.129
105
BrunengraberH. Potential of ketone body esters for parenteral and oral nutrition. Nutrition (1997) 13(3):233–5. doi: 10.1016/S0899-9007(96)00409-1
106
ClarkeKTchabanenkoKPawloskyRCarterETodd KingMMusa-VelosoKet al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul Toxicol Pharmacol (2012a) 63(3):401–8. doi: 10.1016/j.yrtph.2012.04.008
107
CahillGF. Starvation in man. Clin Endocrinol Metab (1976) 5(2):397–415. doi: 10.1016/S0300-595X(76)80028-X
108
CahillGF. Fuel metabolism in starvation. Annu Rev Nutr (2006) 26:1–22. doi: 10.1146/annurev.nutr.26.061505.111258
109
McDonaldTJWHenry-BarronBJFeltonEAGutierrezEGBarnettJFisherRet al. Improving compliance in adults with epilepsy on a modified Atkins diet: a randomized trial. Seizure (2018) 60:132–8. doi: 10.1016/j.seizure.2018.06.019
110
ClarkeKTchabanenkoKPawloskyRCarterEKnightNSMurrayAJet al. Oral 28-day and developmental toxicity studies of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate. Regul Toxicol Pharmacol (2012b) 63(2):196–208. doi: 10.1016/j.yrtph.2012.04.001
111
TateRLMehlmanMATobinRB. Metabolic fate of 1,3-butanediol in the rat: conversion to -hydroxybutyrate. J Nutr (1971) 101(12):1719–26. doi: 10.1093/jn/101.12.1719
112
SchönfeldPWojtczakL. Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J Lipid Res (2016) 57(6):943–54. doi: 10.1194/jlr.R067629
113
HendersonST. Ketone bodies as a therapeutic for Alzheimer’s disease. Neurotherapeutics (2008) 5(3):470–80. doi: 10.1016/j.nurt.2008.05.004
114
ChenLMiaoZXuX. β-hydroxybutyrate alleviates depressive behaviors in mice possibly by increasing the histone3-lysine9-β-hydroxybutyrylation. Biochem Biophys Res Commun (2017) 490(2):117–22. doi: 10.1016/j.bbrc.2017.05.184
115
YamanashiTIwataMKamiyaNTsunetomiKKajitaniNWadaNet al. Beta-hydroxybutyrate, an endogenic NLRP3 inflammasome inhibitor, attenuates stress-induced behavioral and inflammatory responses. Sci Rep (2017) 7(1):7677. doi: 10.1038/s41598-017-08055-1
116
YangJChenTSunLZhaoZQiXZhouKet al. Potential metabolite markers of schizophrenia. Mol Psychiatry (2013) 18(1):67–78. doi: 10.1038/mp.2011.131
117
HuangYCLinPYLeeYHungCFHsuSTWuCCet al. Serum levels of β-hydroxybutyrate and pyruvate, metabolic changes and cognitive function in patients with schizophrenia during antipsychotic treatment: a preliminary study. Neuropsychiatr Dis Treat (2018) 14:799–808. doi: 10.2147/NDT.S157055
118
SetoyamaDKatoTAHashimotoRKunugiHHattoriKHayakawaKet al. Plasma metabolites predict severity of depression and suicidal ideation in psychiatric patients—a multicenter pilot analysis. PLoS One (2016) 11(12):e0165267. doi: 10.1371/journal.pone.0165267
119
SimeoneTASimeoneKARhoJM. Ketone bodies as anti-seizure agents. Neurochem Res (2017) 42:2011–8. doi: 10.1007/s11064-017-2253-5
120
PhelpsJRSiemersSVEl-MallakhRS. The ketogenic diet for type II bipolar disorder. Neurocase (2013) 19(5):423–6. doi: 10.1080/13554794.2012.690421
121
KraeuterAKLoxtonHLimaBCRuddDSarnyaiZ. Ketogenic diet reverses behavioral abnormalities in an acute NMDA receptor hypofunction model of schizophrenia. Schizophr Res (2015) 169(1–3):491–3. doi: 10.1016/j.schres.2015.10.041
122
MurphyPLikhodiiSNylenKBurnhamWM. The antidepressant properties of the ketogenic diet. Biol Psychiatry (2004) 56(12):981–3. doi: 10.1016/j.biopsych.2004.09.019
123
RuskinDNFortinJABisnauthSNMasinoSA. Ketogenic diets improve behaviors associated with autism spectrum disorder in a sex-specific manner in the EL mouse. Physiol Behav (2017a) 168:138–45. doi: 10.1016/j.physbeh.2016.10.023
124
MurphyPBurnhamWM. The ketogenic diet causes a reversible decrease in activity level in Long-Evans rats. Exp Neurol (2006) 201(1):84–9. doi: 10.1016/j.expneurol.2006.03.024
125
PackerRMLawTHDaviesEZanghiBPanYVolkHA. Effects of a ketogenic diet on ADHD-like behavior in dogs with idiopathic epilepsy. Epilepsy Behav (2016) 55:62–8. doi: 10.1016/j.yebeh.2015.11.014
126
SatoKKashiwayaYKeonCATsuchiyaNKingMTRaddaGKet al. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J (1995) 9(8):651–8. doi: 10.1096/fasebj.9.8.7768357
127
JugeNGrayJAOmoteHMiyajiTInoueTHaraCet al. Metabolic control of vesicular glutamate transport and release. Neuron (2010) 68:99–112. doi: 10.1016/j.neuron.2010.09.002
128
WonYJLuVBPuhlHL3rdIkedaSR. β-Hydroxybutyrate modulates N-type calcium channels in rat sympathetic neurons by acting as an agonist for the G-protein-coupled receptor FFA3. J Neurosci (2013) 33(49):19314–25. doi: 10.1523/JNEUROSCI.3102-13.2013
129
LundTMPlougKBIversenAJensenAAJansen-OlesenI. The metabolic impact of β-hydroxybutyrate on neurotransmission: reduced glycolysis mediates changes in calcium responses and KATP channel receptor sensitivity. J Neurochem (2015) 132(5):520–31. doi: 10.1111/jnc.12975
130
SharmaAKRaniEWaheedARajputSK. Pharmacoresistant epilepsy: a current update on non-conventional pharmacological and non-pharmacological interventions. J. Epilepsy Res (2015) 5:1–8. doi: 10.14581/jer.15001
131
OtaniKYamatodaniAWadaHMimakiTYabuuchiH. Effect of ketogenic diet on the convulsive threshold and brain amino acid and monoamine levels in young mice. No To Hattatsu (1984) 16(3):196–204. doi: 10.11251/ojjscn1969.16.196
132
DahlinMMånssonJEÅmarkP. CSF levels of dopamine and serotonin, but not norepinephrine, metabolites are influenced by the ketogenic diet in children with epilepsy. Epilepsy Res (2012) 99(1–2):132–8. doi: 10.1016/j.eplepsyres.2011.11.003
133
HaasHLGreeneRW. Adenosine enhances afterhyperpolarization and accommodation in hippocampal pyramidal cells. Pflugers Arch (1984) 402:244–7. doi: 10.1007/BF00585506
134
KovácsZKékesiKAJuhászGDobolyiA. The antiepileptic potential of nucleosides. Curr Med Chem (2014) 21:788–821. doi: 10.2174/1381612819666131119154505
135
PoulsenSAQuinnRJ. Adenosine receptors: new opportunities for future drugs. Bioorg Med Chem (1998) 6(6):619–41. doi: 10.1016/S0968-0896(98)00038-8
136
Van der PuttenCZuiderwijk-SickEAvan StraalenLde GeusEDBovenLAKondovaIet al. Differential expression of adenosine A3 receptors controls adenosine A2A receptor-mediated inhibition of TLR responses in microglia. J Immunol (2009) 182(12):7603–12. doi: 10.4049/jimmunol.0803383
137
AlmeidaCGde MendonçaACunhaRARibeiroJA. Adenosine promotes neuronal recovery from reactive oxygen species induced lesion in rat hippocampal slices. Neurosci Lett (2003) 339(2):127–30. doi: 10.1016/S0304-3940(02)01478-7
138
HuSDongHZhangHWangSHouLChenSet al. Noninvasive limb remote ischemic preconditioning contributes neuroprotective effects via activation of adenosine A1 receptor and redox status after transient focal cerebral ischemia in rats. Brain Res (2012) 1459:81–90. doi: 10.1016/j.brainres.2012.04.017
139
ElaminMRuskinDNMasinoSASacchettiP. Ketone-based metabolic therapy: is increased NAD+ a primary mechanism? Front Mol Neurosci (2017) 10:377. doi: 10.3389/fnmol.2017.00377
140
MaaloufMSullivanPGDavisLKimDYRhoJM. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience (2007) 145(1):256–64. doi: 10.1016/j.neuroscience.2006.11.065
141
FuSPWangJFXueWJLiuHMLiuBRZengYLet al. Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J Neuroinflammation (2015) 12:9. doi: 10.1186/s12974-014-0230-3
142
GraffECFangHWandersDJuddRL. Anti-inflammatory effects of the hydroxycarboxylic acid receptor 2. Metabolism (2016) 65(2):102–13. doi: 10.1016/j.metabol.2015.10.001
143
YangXChengB. Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J Mol Neurosci (2010) 42(2):145–53. doi: 10.1007/s12031-010-9336-y
144
LevyMThaissCAElinavE. Taming the inflammasome. Nat Med (2015) 21(3):213–5. doi: 10.1038/nm.3808
145
PatelMNCarrollRGGalván-PeñaSMillsELOldenRTriantafilouMet al. Inflammasome priming in sterile inflammatory disease. Trends Mol Med (2017a) 23(2):165–80. doi: 10.1016/j.molmed.2016.12.007
146
BaeHRKimDHParkMHLeeBKimMJLeeEKet al. β-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget (2016) 7(41):66444–54. doi: 10.18632/oncotarget.12119
147
EmeritJEdeasMBricaireF. Neurodegenerative diseases and oxidative stress. Biomed Pharmacother (2004) 58(1):39–46. doi: 10.1016/j.biopha.2003.11.004
148
KashiwayaYPawloskyRMarkisWKingMTBergmanCSrivastavaSet al. A ketone ester diet increases brain malonyl-CoA and Uncoupling proteins 4 and 5 while decreasing food intake in the normal Wistar Rat. J Biol Chem (2010) 285(34):25950–6. doi: 10.1074/jbc.M110.138198
149
XiaoXQZhaoYChenGQ. The effect of 3-hydroxybutyrate and its derivatives on the growth of glial cells. Biomaterials (2007) 28(25):3608–16. doi: 10.1016/j.biomaterials.2007.04.046
150
KimDYSimeoneKASimeoneTAPandyaJDWilkeJCAhnYet al. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann Neurol (2015) 78(1):77–87. doi: 10.1002/ana.24424
151
LiX. Using the conditioned fear stress (CFS) animal model to understand the neurobiological mechanisms and pharmacological treatment of anxiety. Shanghai Arch Psychiatry (2012) 24(5):241–9. doi: 10.3969/j.issn.1002-0829.2012.05.001
152
NagyJZámbóKDecsiL. Anti-anxiety action of diazepam after intra-amygdaloid application in the rat. Neuropharmacology (1979) 18(6):573–6. doi: 10.1016/0028-3908(79)90104-7
153
TangHHMcNallyGPRichardsonR. The effects of FG7142 on two types of forgetting in 18-day-old rats. Behav Neurosci (2007) 121(6):1421–5. doi: 10.1037/0735-7044.121.6.1421
154
LeeHKimJJ. Amygdalar NMDA receptors are critical for new fear learning in previously fear-conditioned rats. J Neurosci (1998) 18(20):8444–54. doi: 10.1523/JNEUROSCI.18-20-08444.1998
155
NishikawaHInoueTMasuiTIzumiTKoyamaT. Effects of cytochrome P450 (CYP) 3A4 inhibitors on the anxiolytic action of tandospirone in rat contextual conditioned fear. Prog Neuropsychopharmacol Biol Psychiatry (2007) 31(4):926–31. doi: 10.1016/j.pnpbp.2007.02.010
156
HelmstetterFJBellgowanPS. Effects of muscimol applied to the basolateral amygdala on acquisition and expression of contextual fear conditioning in rats. Behav Neurosci (1994) 108(5):1005–9. doi: 10.1037/0735-7044.108.5.1005
157
DiamondDMZoladzPR. Dysfunctional or hyperfunctional? The amygdala in posttraumatic stress disorder is the bull in the evolutionary China shop. J Neurosci Res (2016) 94(6):437–44. doi: 10.1002/jnr.23684
158
LeDouxJEPineDS. Using neuroscience to help understand fear and anxiety: a two-system framework. Am J Psychiatry (2016) 173(11):1083–93. doi: 10.1176/appi.ajp.2016.16030353
159
ZoladzPRDiamondDM. Current status on behavioral and biological markers of PTSD: a search for clarity in a conflicting literature. Neurosci Biobehav Rev (2013) 37(5):860–95. doi: 10.1016/j.neubiorev.2013.03.024
160
EtkinAPraterKESchatzbergAFMenonVGreiciusMD. Disrupted amygdalar subregion functional connectivity and evidence of a compensatory network in generalized anxiety disorder. Arch Gen Psychiatry (2009) 66(12):1361–72. doi: 10.1001/archgenpsychiatry.2009.104
161
TeicherMHSamsonJAAndersonCMOhashiK. The effects of childhood maltreatment on brain structure, function and connectivity. Nat Rev Neurosci (2016) 17(10):652–66. doi: 10.1038/nrn.2016.111
162
ThomaesKDorrepaalEDraijerNde RuiterMBvan BalkomAJSmitJHet al. Reduced anterior cingulate and orbitofrontal volumes in child abuse-related complex PTSD. J Clin Psychiatry (2010) 71(12):1636–44. doi: 10.4088/JCP.08m04754blu
163
WoonFLSoodSHedgesDW. Hippocampal volume deficits associated with exposure to psychological trauma and posttraumatic stress disorder in adults: a meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry (2010) 34(7):1181–8. doi: 10.1016/j.pnpbp.2010.06.016
164
MillerAHRaisonCL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol (2016) 16(1):22–34. doi: 10.1038/nri.2015.5
165
EinatHYuanPManjiHK. Increased anxiety-like behaviors and mitochondrial dysfunction in mice with targeted mutation of the Bcl-2 gene: further support for the involvement of mitochondrial function in anxiety disorders. Behav Brain Res (2005) (2):172–80. doi: 10.1016/j.bbr.2005.06.012
166
SchiavoneSTrabaceL. Pharmacological targeting of redox regulation systems as new therapeutic approach for psychiatric disorders: a literature overview. Pharmacol Res (2016) 107:195–204. doi: 10.1016/j.phrs.2016.03.019
167
KahnRSSommerIE. The neurobiology and treatment of first-episode schizophrenia. Mol Psychiatry (2015) 20(1):84–97. doi: 10.1038/mp.2014.66
168
McCullumsmithREClintonSMMeador-WoodruffJH. Schizophrenia as a disorder of neuroplasticity. Int Rev Neurobiol (2004) 59:19–45. doi: 10.1016/S0074-7742(04)59002-5
169
YoonJHMaddockRJRokemASilverMAMinzenbergMJRaglandJDet al. GABA concentration is reduced in visual cortex in schizophrenia and correlates with orientation-specific surround suppression. J Neurosci (2010) 30(10):3777–81. doi: 10.1523/JNEUROSCI.6158-09.2010
170
WassefABakerJKochanLD. GABA and schizophrenia: a review of basic science and clinical studies. J Clin Psychopharmacol (2003) 23(6):601–40. doi: 10.1097/01.jcp.0000095349.32154.a5
171
HaijmaSVVan HarenNCahnWKoolschijnPCHulshoff PolHEKahnRS. Brain volumes in schizophrenia: a meta-analysis in over 18 000 subjects. Schizophr Bull (2013) 39(5):1129–38. doi: 10.1093/schbul/sbs118
172
ShentonMEDickeyCCFruminMMcCarleyRW. A review of MRI findings in schizophrenia. Schizophr Res (2001) 49(1–2):1–52. doi: 10.1016/S0920-9964(01)00163-3
173
ThompsonPMVidalCGieddJNGochmanPBlumenthalJNicolsonRet al. Mapping adolescent brain change reveals dynamic wave of accelerated gray matter loss in very early-onset schizophrenia. Proc Natl Acad Sci U S A (2001) 98(20):11650–5. doi: 10.1073/pnas.201243998
174
SchniederTPDworkAJ. Searching for neuropathology: gliosis in schizophrenia. Biol Psychiatry (2011) 69(2):134–9. doi: 10.1016/j.biopsych.2010.08.027
175
CattsVSCattsSVMcGrathJJFéronFMcLeanDCoulsonEJet al. Apoptosis and schizophrenia: a pilot study based on dermal fibroblast cell lines. Schizophr Res (2006) 84(1):20–8. doi: 10.1016/j.schres.2006.03.016
176
Ben-ShacharDLaifenfeldD. Mitochondria, synaptic plasticity, and schizophrenia. Int Rev Neurobiol (2004) 59:273–96. doi: 10.1016/S0074-7742(04)59011-6
177
HolperLBen-ShacharDMannJJ. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology (2019) 44:837–49. doi: 10.1038/s41386-018-0090-0
178
KimYKNaKS. Neuroprotection in schizophrenia and its therapeutic implications. Psychiatry Investig (2017) 14(4):383–91. doi: 10.4306/pi.2017.14.4.383
179
MillerBJBuckleyPSeaboltWMellorAKirkpatrickB. Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry (2011) 70(7):663–71. doi: 10.1016/j.biopsych.2011.04.013
180
YeginAAyNAydinOYargiciNErenEYilmazN. Increased oxidant stress and inflammation in patients with chronic schizophrenia. Intl J Clin Med (2012) 3(5):368–76. doi: 10.4236/ijcm.2012.35070
181
RajkowskaGMiguel-HidalgoJJWeiJDilleyGPittmanSDMeltzerHYet al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry (1999) 45(9):1085–98. doi: 10.1016/S0006-3223(99)00041-4
182
SchmaalLVeltmanDJvan ErpTGSämannPGFrodlTJahanshadNet al. Subcortical brain alterations in major depressive disorder: findings from the ENIGMA Major Depressive Disorder working group. Mol Psychiatry (2016) 21(6):806–12. doi: 10.1038/mp.2015.69
183
SchmaalLHibarDPSämannPGHallGBBauneBTJahanshadNet al. Cortical abnormalities in adults and adolescents with major depression based on brain scans from 20 cohorts worldwide in the ENIGMA Major Depressive Disorder Working Group. Mol Psychiatry (2017) (6):900–9. doi: 10.1038/mp.2016.60
184
HamiltonJPEtkinAFurmanDJLemusMGJohnsonRFGotlibIH. Functional neuroimaging of major depressive disorder: a meta-analysis and new integration of base line activation and neural response data. Am J Psychiatry (2012) 169(7):693–703. doi: 10.1176/appi.ajp.2012.11071105
185
MaybergHS. Positron emission tomography imaging in depression: a neural systems perspective. Neuroimaging Clin N Am (2003) 13(4):805–15. doi: 10.1016/S1052-5149(03)00104-7
186
SatterthwaiteTDKableJWVandekarLKatchmarNBassettDSBaldassanoCFet al. Common and dissociable dysfunction of the reward system in bipolar and unipolar depression. Neuropsychopharmacology (2015) 40(9):2258–68. doi: 10.1038/npp.2015.75
187
FeerH. The catecholamine hypothesis of depressions: further arguments. Compr Psychiatry (1967) 8(1):1–6. doi: 10.1016/S0010-440X(67)80008-7
188
MüllerNSchwarzMJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry (2007) 12(11):988–1000. doi: 10.1038/sj.mp.4002006
189
KrystalJHSanacoraGBlumbergHAnandACharneyDSMarekGet al. Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol Psychiatry (2002) 7 (Suppl 1):S71–80. doi: 10.1038/sj.mp.4001021
190
PettyF. GABA and mood disorders: a brief review and hypothesis. J Affect Disord (1995) 34(4):275–81. doi: 10.1016/0165-0327(95)00025-I
191
SuWJZhangYChenYGongHLianYJPengWet al. NLRP3 gene knockout blocks NF-κB and MAPK signaling pathway in CUMS-induced depression mouse model. Behav Brain Res (2017) 322(Pt A):1–8. doi: 10.1016/j.bbr.2017.01.018
192
ZhuWCaoFSFengJChenHWWanJRLuQet al. NLRP3 inflammasome activation contributes to long-term behavioral alterations in mice injected with lipopolysaccharide. Neuroscience (2017) 343:77–84. doi: 10.1016/j.neuroscience.2016.11.037
193
Alcocer-GómezEde MiguelMCasas-BarqueroNNúñez-VascoJSánchez-AlcazarJAFernández-RodríguezAet al. NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder. Brain Behav Immun (2014) 36:111–7. doi: 10.1016/j.bbi.2013.10.017
194
KaufmannFNCostaAPGhisleniGDiazAPRodriguesALSPeluffoHet al. NLRP3 inflammasome-driven pathways in depression: clinical and preclinical findings. Brain Behav Immun (2017) 64:367–83. doi: 10.1016/j.bbi.2017.03.002
195
StetlerCMillerGE. Depression and hypothalamic–pituitary–adrenal activation: a quantitative summary of four decades of research. Psychosom Med (2011) (2):114–26. doi: 10.1097/PSY.0b013e31820ad12b
196
BrownGMMcIntyreRSRosenblatJHardelandR. Depressive disorders: processes leading to neurogeneration and potential novel treatments. Prog Neuropsychopharmacol Biol Psychiatry (2018b) 80(Pt C):189–204. doi: 10.1016/j.pnpbp.2017.04.023
197
HardinghamGEFukunagaYBadingH. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci (2002) (5):405–14. doi: 10.1038/nn835
198
ZhuCBLindlerKMOwensAWDawsLCBlakelyRDHewlettWA. Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology (2010) 35(13):2510–20. doi: 10.1038/npp.2010.116
199
CapuronLPagnoniGDrakeDFWoolwineBJSpiveyJRCroweRJet al. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry (2012) 69(10):1044–53. doi: 10.1001/archgenpsychiatry.2011.2094
200
GoodwinFKJamisonKR. Manic-depressive illness: bipolar disorders and recurrent depression. New York: Oxford University Press (2007), ISBN: 978-0-19-513579-4.
201
KuritaMNishinoSNumataYOkuboYSatoT. The noradrenaline metabolite MHPG is a candidate biomarker between the depressive, remission, and manic states in bipolar disorder I: two long-term naturalistic case reports. Neuropsychiatr Dis Treat (2015) 11:353–8. doi: 10.2147/NDT.S74550
202
YathamLNKennedySHParikhSVSchafferABondDJFreyBNet al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) 2018 guidelines for the management of patients with bipolar disorder. Bipolar Disord (2018) 20(2):97–170. doi: 10.1111/bdi.12609
203
BerkMKapczinskiFAndreazzaACDeanOMGiorlandoFMaesMet al. Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci Biobehav Rev (2011) 35(3):804–17. doi: 10.1016/j.neubiorev.2010.10.001
204
MertensJWangQWKimYYuDXPhamSYangBet al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature (2015) 527(7576):95–9. doi: 10.1038/nature15526
205
BlumbergHPKaufmanJMartinAWhitemanRZhangJHGoreJCet al. Amygdala and hippocampal volumes in adolescents and adults with bipolar disorder. Arch Gen Psychiatry (2003) 60(12):1201–8. doi: 10.1001/archpsyc.60.12.1201
206
CaoBPassosICMwangiBBauerIEZunta-SoaresGBKapczinskiFet al. Hippocampal volume and verbal memory performance in late-stage bipolar disorder. J Psychiatr Res (2016) 73:102–7. doi: 10.1016/j.jpsychires.2015.12.012
207
HibarDPWestlyeLTDoanNTJahanshadNCheungJWChingCRKet al. Cortical abnormalities in bipolar disorder: an MRI analysis of 6503 individuals from the ENIGMA Bipolar Disorder Working Group. Mol Psychiatry (2018) 23(4):932–42. doi: 10.1038/mp.2017.73
208
RaoJSHarryGJRapoportSIKimHW. Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry (2010) 15(4):384–92. doi: 10.1038/mp.2009.47
209
LangeNTraversBGBiglerEDPriggeMBFroehlichALNielsenJAet al. Longitudinal volumetric brain changes in autism spectrum disorder ages 6-35 years. Autism Res. (2015) 8(1):82–93. doi: 10.1002/aur.1427
210
PaulLKCorselloCKennedyDPAdolphsR. Agenesis of the corpus callosum and autism: a comprehensive comparison. Brain (2014) 137(Pt 6):1813–29. doi: 10.1093/brain/awu070
211
TyszkaJMKennedyDPPaulLKAdolphsR. Largely typical patterns of resting-state functional connectivity in high-functioning adults with autism. Cereb Cortex (2014) 24(7):1894–905. doi: 10.1093/cercor/bht040
212
ZielinskiBAPriggeMBNielsenJAFroehlichALAbildskovTJAndersonJSet al. Longitudinal changes in cortical thickness in autism and typical development. Brain (2014) 137(Pt 6):1799–812. doi: 10.1093/brain/awu083
213
BearMFHuberKMWarrenST. The mGluR theory of fragile X mental retardation. Trends Neurosci (2004) 27(7):370–7. doi: 10.1016/j.tins.2004.04.009
214
EricksonCAMullettJEMcDougleCJ. Open-label memantine in fragile X syndrome. J Autism Dev Disord (2009) 39(12):1629–35. doi: 10.1007/s10803-009-0807-3
215
GreenJGargS. Annual Research Review: the state of autism intervention science: progress, target psychological and biological mechanisms and future prospects. J Child Psychol Psychiatry (2018) 59(4):424–43. doi: 10.1111/jcpp.12892
216
LemonnierEVilleneuveNSonieSSerretSRosierARoueMet al. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl Psychiatry (2017) 7(3):e1056. doi: 10.1038/tp.2017.10
217
ChandanaSRBehenMEJuhászCMuzikORothermelRDMangnerTJet al. Significance of abnormalities in developmental trajectory and asymmetry of cortical serotonin synthesis in autism. Int J Dev Neurosci (2005) 23(2–3):171–82. doi: 10.1016/j.ijdevneu.2004.08.002
218
ChefferACastilloARGCorrêa-VellosoJGonçalvesMCBNaaldijkYNascimentoICet al. Purinergic system in psychiatric diseases. Mol Psychiatry (2018) 23(1):94–106. doi: 10.1038/mp.2017.188
219
MasinoSAKawamuraMJr.PlotkinLMSvedovaJDiMarioFJJrEigstiIM. The relationship between the neuromodulator adenosine and behavioral symptoms of autism. Neurosci Lett (2011) 500(1):1–5. doi: 10.1016/j.neulet.2011.06.007
220
MasinoSAKawamuraMCoteJLWilliamsRBRuskinDN. Adenosine and autism: a spectrum of opportunities. Neuropharmacology (2013) 68:116–21. doi: 10.1016/j.neuropharm.2012.08.013
221
RodriguezJIKernJK. Evidence of microglial activation in autism and its possible role in brain underconnectivity. Neuron Glia Biol (2011) 7(2–4):205–13. doi: 10.1017/S1740925X12000142
222
HughesHKMills KoERoseDAshwoodP. Immune dysfunction and autoimmunity as pathological mechanisms in autism spectrum disorders. Front Cell Neurosci (2018) 12:405. doi: 10.3389/fncel.2018.00405
223
VargasDLNascimbeneCKrishnanCZimmermanAWPardoCA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol (2005) 57(1):67–81. doi: 10.1002/ana.20315
224
ZimmermanAWJyonouchiHComiAMConnorsSLMilstienSVarsouAet al. Cerebrospinal fluid and serum markers of inflammation in autism. Pediatr Neurol (2005) 33(3):195–201. doi: 10.1016/j.pediatrneurol.2005.03.014
225
FilipekPAJuranekJSmithMMaysLZRamosERBocianMet al. Mitochondrial dysfunction in autistic patients with 15q inverted duplication. Ann Neurol (2003) 53(6):801–4. doi: 10.1002/ana.10596
226
FryeRERossignolDA. Treatments for biomedical abnormalities associated with autism spectrum disorder. Front Pediatr (2014) 2:66. doi: 10.3389/fped.2014.00066
227
RossignolDAFryeRE. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front Physiol (2014) 5:150. doi: 10.3389/fphys.2014.00150
228
CorteseSKellyCChabernaudCProalEDi MartinoAMilhamMPet al. Toward systems neuroscience of ADHD: a meta-analysis of 55 fMRI studies. Am J Psychiatry (2012) 169(10):1038–55. doi: 10.1176/appi.ajp.2012.11101521
229
GrevenCUBraltenJMennesMO’DwyerLvan HulzenKJRommelseNet al. Developmentally stable whole-brain volume reductions and developmentally sensitive caudate and putamen volume alterations in those with attention-deficit/hyperactivity disorder and their unaffected siblings. JAMA Psychiatry (2015) 72(5):490–9. doi: 10.1001/jamapsychiatry.2014.3162
230
CourvoisieHHooperSRFineCKwockLCastilloM. Neurometabolic functioning and neuropsychological correlates in children with ADHD-H: preliminary findings. J Neuropsychiatry Clin Neurosci (2004) 16(1):63–9. doi: 10.1176/appi.neuropsych.16.1.63
231
EddenRACrocettiDZhuHGilbertDLMostofskySH. Reduced GABA concentration in attention-deficit/hyperactivity disorder. Arch Gen Psychiatry (2012) 69(7):750–3. doi: 10.1001/archgenpsychiatry.2011.2280
232
BriarsLToddT. A review of pharmacological management of attention-deficit/hyperactivity disorder. J Pediatr Pharmacol Ther (2016) 21(3):192–206. doi: 10.5863/1551-6776-21.3.192
233
GizerIRFicksCWaldmanID. Candidate gene studies of ADHD: a meta-analytic review. Hum Genet (2009) 126(1):51–90. doi: 10.1007/s00439-009-0694-x
234
OadesRDSlusarekMVellingSBondyB. Serotonin platelet-transporter measures in childhood attention-deficit/hyperactivity disorder (ADHD): clinical versus experimental measures of impulsivity. World J Biol Psychiatry (2002) 3(2):96–100. doi: 10.3109/15622970209150607
235
SowinskiHKarpawichPP. Management of a hyperactive teen and cardiac safety. Pediatr Clin North Am (2014) 61(1):81–90. doi: 10.1016/j.pcl.2013.09.021
236
LeffaDTBellaverBde OliveiraCde MacedoICde FreitasJSGrevetEHet al. Increased oxidative parameters and decreased cytokine levels in an animal model of attention-deficit/hyperactivity disorder. Neurochem Res (2017) 42(11):3084–92. doi: 10.1007/s11064-017-2341-6
Summary
Keywords
psychiatric diseases, exogenous ketone supplements, ketosis, mitochondrial dysfunction, inflammation
Citation
Kovács Z, D’Agostino DP, Diamond D, Kindy MS, Rogers C and Ari C (2019) Therapeutic Potential of Exogenous Ketone Supplement Induced Ketosis in the Treatment of Psychiatric Disorders: Review of Current Literature. Front. Psychiatry 10:363. doi: 10.3389/fpsyt.2019.00363
Received
21 August 2018
Accepted
10 May 2019
Published
23 May 2019
Volume
10 - 2019
Edited by
Lourdes Martorell, Institut Pere Mata, Spain
Reviewed by
Hiromasa Funato, Toho University, Japan; Yuri Zilberter, INSERM U1106 Institut de Neurosciences des Systèmes, France; Zoltan Sarnyai, James Cook University, Australia
Updates
Copyright
© 2019 Kovács, D’Agostino, Diamond, Kindy, Rogers and Ari.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Csilla Ari, csari2000@yahoo.com
This article was submitted to Molecular Psychiatry, a section of the journal Frontiers in Psychiatry
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.