 Norbert Müller1,2*
Norbert Müller1,2*- 1Department of Psychiatry and Psychotherapy, Ludwig-Maximilians-Universität Munich, Munich, Germany
- 2Marion von Tessin Memory Center, Munich, Germany
Inflammatory processes associated with persistent (chronic) infection have long been discussed as etiological factors in psychiatric disorders. Studies have found that people with major depression have higher levels of pro-inflammatory cytokines, for example, IL-1, IL-6, and tumor necrosis factor-alpha, and C-reactive protein. In schizophrenia, many reports have described raised levels of cytokines, for example, IL-6; and meta-analyses have confirmed these findings. Microglia cells are important in inflammatory processes, and positron emission tomography studies have shown microglia activation in both depression and schizophrenia.As a consequence of the above findings, immunomodulation is widely discussed as a potential treatment approach in both major depression and schizophrenia. The COX-2 inhibitor celecoxib was found to have a significant positive effect on major depression, not only in single studies but also in meta-analyses. Celecoxib has also been studied in schizophrenia and has shown efficacy, in particular, in early disease stages. The mixed COX inhibitor aspirin (acetylsalicylic acid) seems to have both protective and therapeutic effects on schizophrenia.This paper discusses the hypothesized role of inflammation in major depression and schizophrenia, including markers of inflammation; pertinent studies on celecoxib and aspirin; and additional immunomodulatory therapeutic strategies.
Introduction
Studies in animal models have provided evidence that both early infection and immune activation can affect several neurodevelopmental processes, including serotonergic (1) and dopaminergic and glutamatergic neurotransmission (2, 3). Human studies on infections (4) and a cohort study on bacterial infections (5) also indicated that infection and activation of the immune system are associated with neurodevelopment. Furthermore, studies found that higher cytokine and C-reactive protein (CRP) levels in childhood increased the risk to develop depression (6) and schizophrenia (7). In meta-analyses, cytokine levels were altered in adults with major depression (MD), schizophrenia, and bipolar disorder (8, 9).
Major Depression
Inflammation and Depression Risk
A population-based, prospective cohort study in Denmark followed a total of 3.56 million people for 24 years and found that those who were hospitalized for an infection or visited a hospital for treatment of an autoimmune disease had a significantly higher risk for developing a depressive disorder (10). If people were hospitalized for infection, the risk increased by 62% [incidence rate ratio (IRR) 1.62]; and if they visited the hospital because of an autoimmune disease, it increased by 45% (IRR 1.45). People with a history of both risk factors had an even higher risk of developing mood disorders (IRR 2.35), indicating that the two factors interact. In this study, the risk for later mood disorders was lower after infections of the central nervous system (CNS; IRR 1.65) than after peripheral infections, for example, hepatitis (IRR 2.82) and sepsis (IRR 2.06). Interesting findings were that the shorter the time since the infection, the greater the risk of developing a mood disorder and that the highest risk was found in the 12 months after the infection (IRR 2.70). If the study had assessed all kinds of infections—not only those for which people visited a hospital—the risk of developing a mood disorder might have been even higher (10).
Another Danish population-based register study evaluated whether the use of anti-inflammatory agents, including aspirin (acetylsalicylic acid [ASA], an inhibitor of both COX-1 and COX-2), is associated with a lower rate of depression (11). The study found a dose-dependent risk; that is, continued use of low-dose aspirin reduced the risk for incident depression, whereas the use of non-steroidal anti-inflammatory drugs (NSAIDs) and high-dose aspirin increased the rate.
Anti-Inflammatory Treatment Approaches in Major Depression
Non-Steroidal Anti-Inflammatory Drugs
NSAIDs act by inhibiting COX-1 and COX-2. These cyclooxygenase enzymes are necessary for the synthesis of some of the prostaglandins involved in inflammation. COX-2 inhibitors have direct effects on the CNS serotonergic system and can also affect it indirectly via immune processes in the CNS. A study in rats showed higher serotonin levels in both the frontal and temporoparietal cortices after administration of rofecoxib, a COX-2 selective NSAID (12). Consequently, the authors hypothesized that COX-2 inhibitors may have antidepressant effects. In a study in bulbectomized rats (a model for depression), chronic administration of the COX-2 inhibitor celecoxib decreased cytokine levels and changed the animals’ behavior (13). Brunello et al. (14) studied ASA in rats with the chronic escape deficit model and found that ASA accelerated the antidepressant effect of fluoxetine. In people with MD, a randomized, double-blind pilot study compared reboxetine plus celecoxib with reboxetine plus placebo and found a significant therapeutic effect of celecoxib (15). One interesting finding of this study was that the kynurenine/tryptophan ratio, which reflects the activity of indoleamine 2,3-dioxygenase (IDO), a pro-inflammatory, cytokine-driven enzyme, predicted the antidepressant response to celecoxib; that is, celecoxib had better effects on patients with a high level of IDO activity (16). A double-blind, randomized controlled trial (RCT) in MD (n = 50 patients) compared fluoxetine plus celecoxib with fluoxetine plus placebo and found a significantly better outcome in the group receiving adjunctive celecoxib (17). Similar results were found in two studies of sertraline plus celecoxib or placebo in MD (n = 40 and n = 30) (18, 19), where Hamilton Depression Rating Scale scores decreased significantly more in the celecoxib group; in one of the studies (18), serum IL-6, a pleiotropic immune-activating cytokine that primarily promotes innate and B- and T-cellular immunity and plays an important role in inflammation, correlated with the decrease in the depression rating score.
The efficacy of adjunctive treatment with an NSAID in MD was evaluated in a meta-analysis of four celecoxib studies in a total of 150 patients (20). The analysis concluded that celecoxib may be a potential treatment in this disorder, although the authors stated the benefit and safety of celecoxib and other NSAIDs need to be confirmed in larger studies of longer duration (20).
The findings of another meta-analysis on inflammation-related therapeutic approaches in MD are also of great interest (21). This analysis evaluated data from 14 studies (10 on NSAIDs, n = 4,258; 4 on cytokine inhibitors, n = 2,004) and found that the anti-inflammatory treatments had positive effects compared with placebo [standardized mean difference (SMD), −0.34; 95% confidence interval (CI), −0.57 to −0.11; I2 = 90%], both in depression (SMD, −0.54; 95% CI, −1.08 to −0.01; I2 = 68%) and in depressive symptoms (SMD, −0.27; 95% CI, −0.53 to −0.01; I2 = 68%). The type of depression (clinical depression vs. depressive symptoms) or the agent (NSAID vs. cytokine inhibitor) did not explain the heterogeneity of the studies with respect to differences in treatment regimens and patient populations. Sub-analyses provided support for the positive effects of celecoxib (SMD, −0.29; 95% CI, −0.49 to −0.08; I2 = 73%) on both remission [odds ratio (OR), 7.89; 95% CI, 2.94 to 21.17; I2 = 0%] and response (OR, 6.59; 95% CI, 2.24 to 19.42; I2 = 0%). Six of the studies reported adverse effects but found no difference in the rate of gastrointestinal or cardiovascular side effects at 6 weeks or infections at 12 weeks between the active treatments and placebo. The meta-analysis suggested that treatment with an anti-inflammatory agent, particularly celecoxib, can ameliorate symptoms of depression but does not carry a higher risk of side effects (21). Other studies, however, reported higher rates of adverse cardiovascular events with COX-2 inhibitors (22).
ASA has also been studied in depression. In an 8-week RCT in MD, patients were assigned to 160 mg of aspirin add-on to sertraline (n = 50) or placebo add-on (n = 50); the groups were matched for age, gender, and severity of depression (23). After 4 and 8 weeks of treatment, depression scores were significantly lower than at baseline only in the sertraline plus aspirin group. These results indicate that aspirin has positive effects on depression, although the unusually low responder rate in the sertraline (plus placebo) group may indicate that treatment resistance affected the results. A complex, placebo-controlled study of patients with bipolar depression found a significantly higher response rate in the aspirin group; however, the comparison of the mean values found no significant difference between aspirin and placebo (24).
Another recent meta-analysis included additional studies on NSAIDs and differentiated between studies on patients with a diagnosis of MD and patients with “classical” inflammatory diseases, such as arthritis or psoriasis, who also had depressive symptoms (25). The analysis found highly significant effects on both MD and depressive symptoms and a highly significant overall effect on the combined analysis of both indications. In the analysis of different substance classes and mechanisms of action of the anti-inflammatory agents, classical NSAIDs were significantly superior to placebo. Other anti-inflammatory compounds, such as cytokine inhibitors, glucocorticoids (two RCTs), and minocycline, also showed a significant advantage than did placebo. The overall effect of all anti-inflammatory substances was p = 0.00001.
Table 1 gives an overview of studies on selective COX-2 inhibitors and ASA in MD. Despite the limitations of the studies described above, they provide important information on the effects of anti-inflammatory treatment and, in particular, COX-2 inhibition in MD. Additional studies are needed in larger samples. Furthermore, studies need to consider the high placebo response, particularly in add-on studies with an effective antidepressant, and the severity of depression.
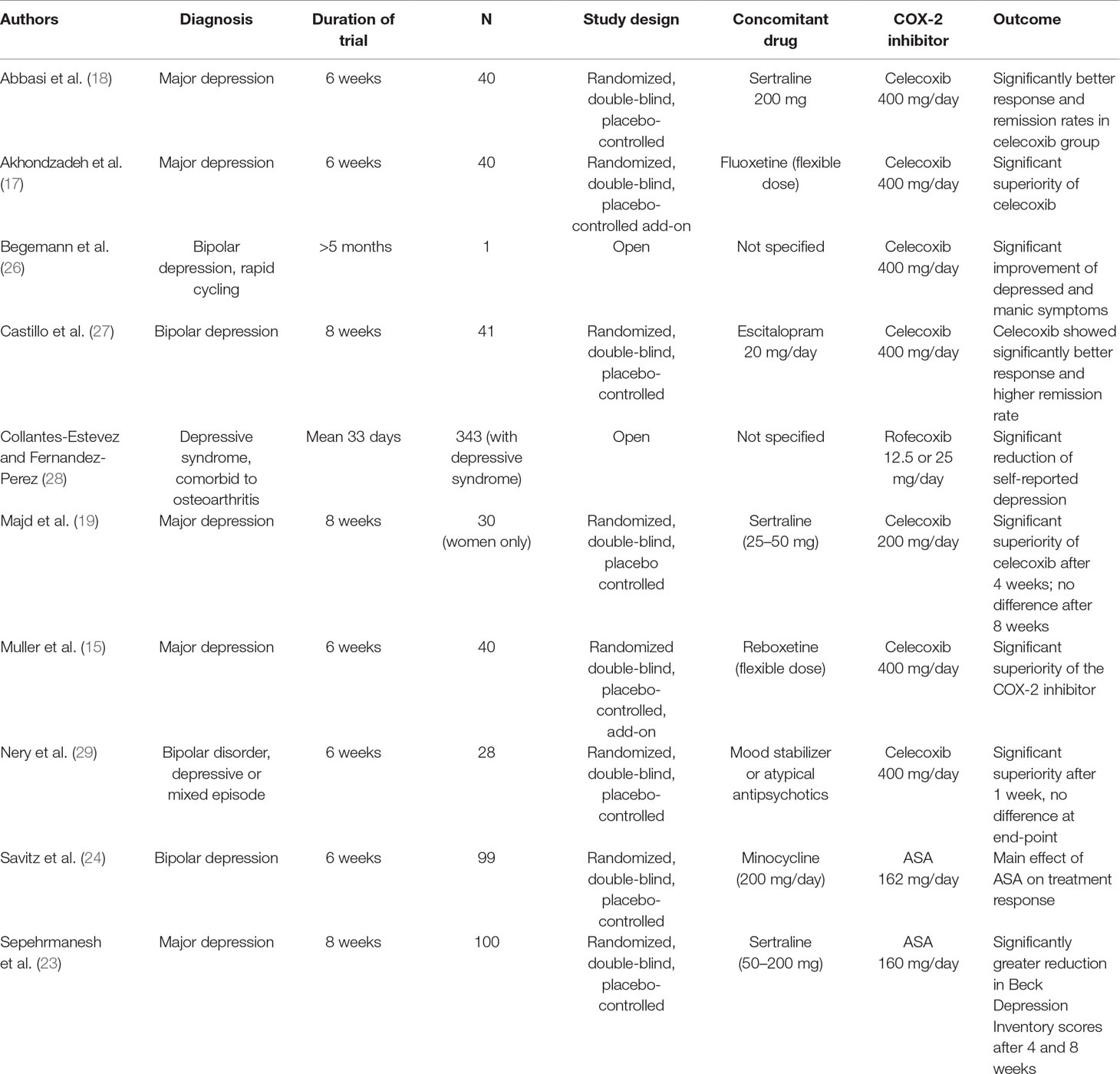
Table 1 Clinical studies of selective COX-2 inhibitors and the mixed COX-1/COX-2 inhibitor acetyl salicylic acid (ASA) in major depression.
Drugs Targeting Cytokines
Tumor necrosis factor-alpha (TNF-a) promotes the activation of the innate and adaptive immune response and is a key molecule in inflammation. The anti-TNF-a antibody infliximab prevents the cytokine TNF-a from interacting with receptors on the cell surface and has an anti-inflammatory effect. TNF-a was initially designed as a treatment for inflammatory joint disorders and psoriasis but was then found to significantly improve symptoms of depression in patients with psoriasis (30).
In a random-effect meta-analysis of seven placebo-controlled RCTs, anti-cytokine treatment had significantly greater effects on depressive symptoms than had placebo (anti-cytokine drug: n = 1,309; placebo: n = 1,061; SMD = 0.40, 95% CI, 0.22 to 0.59) (31). Five of the seven studies were on anti-TNF-a agents, that is, adalimumab, etanercept, and infliximab (SMD = 0.33; 95% CI, 0.06 to 0.60). Similar, small-to-medium effect sizes for anti-cytokine therapy were obtained in the analyses of the two RCTs on adjunctive anti-cytokine treatment (SMD = 0.19; 95% CI, 0.00 to 0.37) and the eight non-randomized and/or non-placebo studies (SMD = 0.51; 95% CI, 0.34 to 0.67). The three anti-TNF-a drugs and the anti-IL-6 antibody (tocilizumab) significantly improved symptoms of depression. A meta-regression showed that baseline symptom severity was a predictor of antidepressant effect (p = 0.018), but sex, age, study duration, and improvement in the primary physical illness were not. An important limitation of these studies was that besides having an inflammatory disease, such as atopic dermatitis, psoriasis, Crohn’s disease, or rheumatoid arthritis, the patients had concomitant symptoms of anxiety or depression or both. In addition, their overall symptoms of depression were mild to moderate, and they did not have a formal diagnosis of depression.
To my knowledge, only one 12-week, placebo-controlled study has evaluated an anti-TNF-a antibody in treatment-resistant MD (n = 60). The participants, who were either taking an antidepressant (n = 37) or partly medication free (n = 23), received three infusions of infliximab (n = 30) or placebo (n = 30). Infliximab was not superior to placebo, but the study found a significant interaction between time, treatment, and baseline levels of CRP (≤5 mg/L); that is, the response rate to infliximab (62%) was higher than that to placebo (33%) in patients who had higher CRP levels at baseline. In addition, participants who responded to infliximab had significantly higher baseline concentrations of TNF-a, sTNFR1, and sTNFR2 (p ≤ 0.01) and significantly larger decreases in CRP (p ≤ 0.01) than had those who did not respond (32). This study is encouraging, in particular because treatment-resistant patients with MD represent a negative selection for treatment outcome, and it indicates that CRP—the most widespread clinical marker for inflammation—may be a biomarker of anti-TNF-a antibody treatment outcome.
Drugs Targeting the IL-6 Complex
As mentioned above, studies have found that in patients with depression, IL-6 levels in the peripheral blood and cerebrospinal fluid (CSF) are higher. Consequently, the IL-6 complex has been proposed as a target for anti-cytokine treatment. The above-mentioned random-effect meta-analysis of seven RCTs (31) included two open studies of the anti-IL-6 antibody tocilizumab (33, 34), both of which showed improvements in patients with concomitant anxiety and depression. Despite these findings, we still need valid, reliable data on the effects of anti-IL-6 treatment in MD and the correct target for such treatment. Because higher levels of IL-6 are found in the CSF than in the blood and in patients with depression than in controls, IL-6 in the CSF was hypothesized to be the most promising therapeutic target. Further support for this hypothesis was given by the finding that IL-6 is overexpressed in the pre-clinical model of chronically stressed rats (35). However, IL-6 levels in the periphery are not completely independent of those in the CSF or brain but are closely connected to them. For this reason, researchers have proposed that targeting peripheral IL-6 may be a potential treatment approach in depression (35). This proposal is supported by the findings in an animal model of depression that significantly higher serum IL-6 levels were found in learned helplessness rats, which were susceptible to chronic inescapable electric stress, than in control and non-learned helplessness rats, which were resilient to the same stress (36). Supporting evidence was also provided by the finding that in depression, serum levels of IL-6 predict patients’ response to ketamine (37). As an alternative, more promising strategy to inhibit IL-6, Maes et al. (38) proposed that administering soluble glycoprotein 130 (sgp 130), a component of the IL-6 complex, would increase inhibition of IL-6 trans-signaling and consequently maintain IL-6 receptor (IL-6R) signaling. In addition to tocilizumab, sirukumab, another monoclonal antibody to IL-6, has also been proposed as a potential treatment for depression (39). It acts on the signaling pathway of IL-6 and can inhibit its pro-inflammatory and anti-inflammatory effects; studies have shown beneficial effects of sirukumab in the inflammatory diseases lupus erythematosus and rheumatoid arthritis, among others (39).
Other Immune-Related Substances
Microglia cells act as macrophages in the brain and are important in inflammatory processes in the CNS. Positron emission tomography (PET) studies showed activation of microglia in MD (40, 41). Minocycline, an antibiotic, can cross the blood–brain barrier into the CNS, where it inhibits the activation of microglia. Consequently, it was proposed as a potential treatment in MD. A recent meta-analysis identified 18 clinical studies (a case report, RCTs, and open and ongoing trials) on minocycline in depression; however, only three RCTs (in a total of 158 participants) were suitable for analysis (42). The analysis observed a large, statistically significant antidepressant effect of minocycline compared with placebo, although limitations of the study included the small number and sample size of the trials included, their heterogeneity, and potential publication bias.
Although statins are primarily used to lower cholesterol, they also have direct anti-inflammatory effects (43, 44). For example, a population-based study in users of selective serotonin reuptake inhibitors (SSRIs; N = 872,216), n = 113,108 of whom were also taking a statin, found that the risk for (a relapse to) depression was clearly lower in participants using both SSRIs and statins than in those using an SSRI alone (43).
Methodological Issues
Although a “gold standard” study in MD would be an RCT that directly compares the anti-inflammatory agent, for example, a COX-2 inhibitor or an anti-cytokine antibody, with placebo, it would not be ethical. Therefore, in almost all RCTs, the anti-inflammatory agent is administered as an adjunct to an antidepressant. As a result of this approach, the anti-inflammatory agent must have a large effect to show an advantage over the antidepressant alone or in combination with placebo. This problem is further exacerbated by the high placebo rate of up to 40% that is commonly seen in studies of antidepressants. Nevertheless, although diverse anti-inflammatory therapeutic approaches show a statistically significant beneficial effect, much further research is needed, and possible subgroups of responders and non-responders have to be identified.
It is well known that in more severe cases of depression, antidepressants show more efficacy than does placebo. This might also be true for anti-inflammatory agents. If possible, clinical studies should therefore include more severely depressed patients.
The pathology of depression is unlikely to be identical in every patient, and, consequently, an inflammatory process is probably not always present, although this hypothesis requires further research. Patients with treatment-resistant MD had higher levels of pro-inflammatory cytokines, which was taken as an indication that these patients’ depression might have an inflammatory origin (45). If a clinical study of anti-inflammatory compounds has a large proportion of patients with an inflammatory pathology, it is more likely to find a positive treatment effect. A goal for future research is to identify a valid, reliable marker for an inflammatory process in depression so that we can apply targeted anti-inflammatory treatment in those patients who would benefit from it.
Schizophrenia
Inflammation and the Rrisk for Schizophrenia
Numerous studies, including recent genetic data (46), show that an immune process and inflammation play a role in at least a subgroup of patients with schizophrenia. The results of these studies gave rise to the vulnerability–stress–inflammation hypothesis of schizophrenia (see Figure 1). One study in Denmark linked population-based registers nationwide and identified autoimmune disorders and severe infections that required admission to hospital as risk factors for schizophrenia and schizophrenia spectrum disorders; the risk was greatest in patients who had an autoimmune disorder and a severe infection (47). No evidence was found, however, that parental infections increased risk in the offspring (47, 48). Although the study was large, it did not have high sensitivity, and the authors consequently described the identified risk factors for schizophrenia as only the “tip of the iceberg” (48).
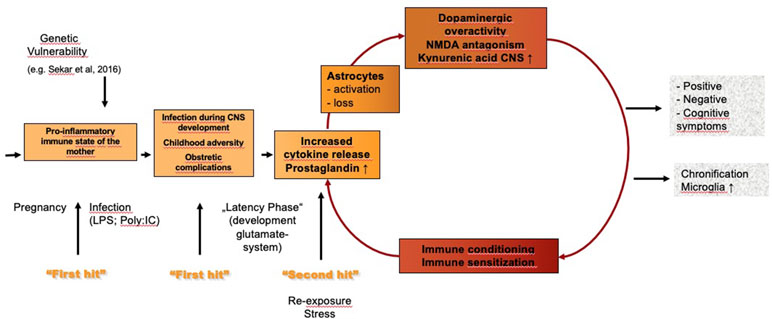
Figure 1 Vulnerability–stress–inflammation hypothesis of schizophrenia. LPS, lipopolysaccharides; poly:IC, polyinosinic–polycytidylic acid.
Another population-based study in Denmark identified all incident patients receiving antipsychotics for schizophrenia (n = 16,235) (49). Of these patients, n = 1480 (9.1%) were using concomitant NSAIDs, and n = 767 (4.7%) were using concomitant paracetamol. The risk of relapse was higher in the patients using NSAIDs [hazard rate ratio (HRR) = 1.21; 95% CI, 1.11 to 1.31], particularly ASA and diclofenac, but not in those using paracetamol (HRR = 0.97; 95% CI, 0.87 to 1.08). The authors analyzed subgroups of patients and found that among patients taking NSAIDs, the relapse risk was higher in those with a comorbid physical illness and lower in those who had been diagnosed with musculoskeletal disease (HRR = 0.82; 95% CI, 0.71 to 0.94) (49).
Anti-Inflammatory Treatment in Schizophrenia
Non-Steroidal Anti-Inflammatory Drugs
Perhaps the most convincing evidence for an involvement of inflammation in schizophrenia is provided by the finding that anti-inflammatory medication is useful in schizophrenia (see Table 2). Noteworthy in this context is the paper by Sommer et al. (50), which reviews the effects of various anti-inflammatory treatments, including celecoxib, in schizophrenia.
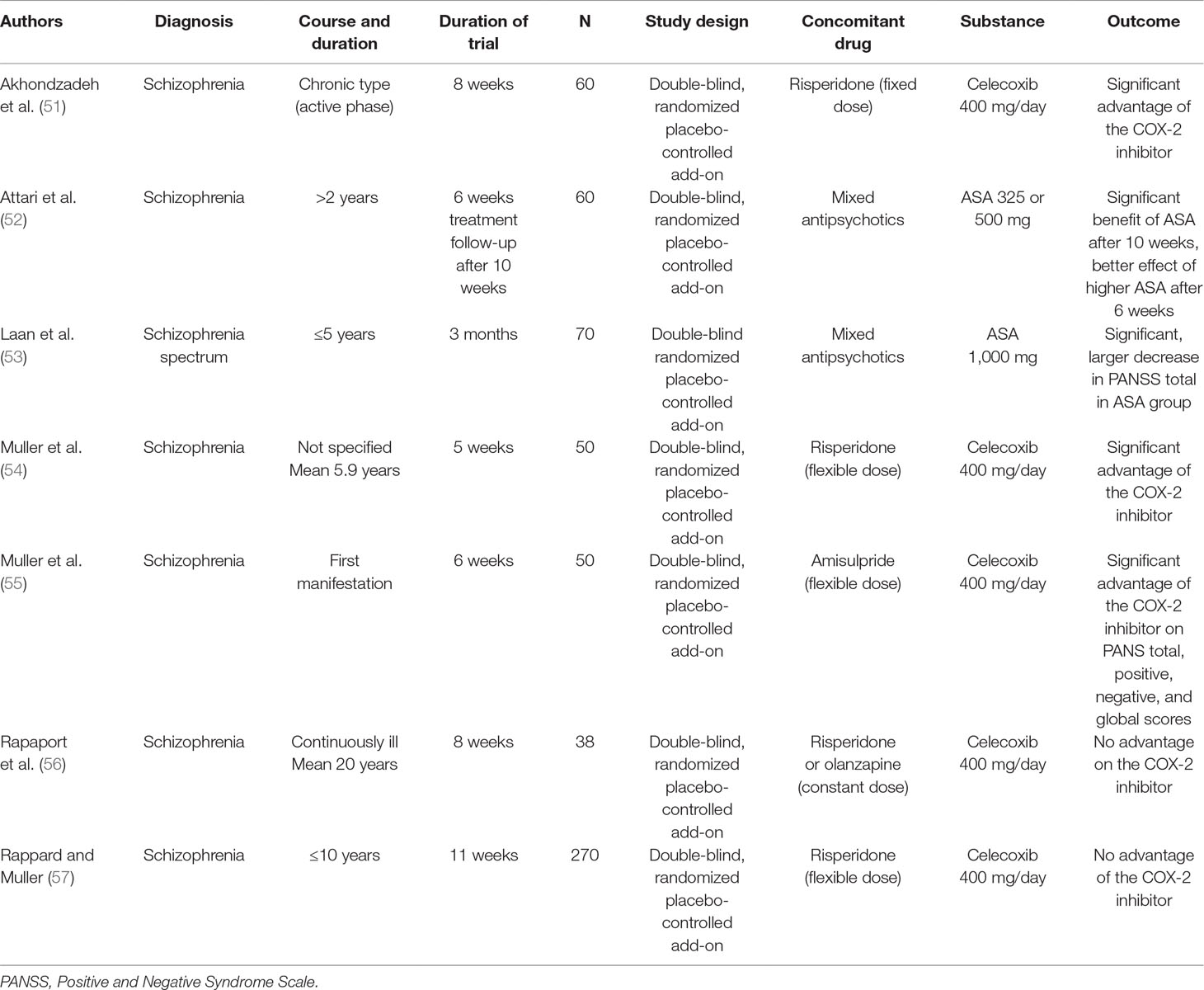
Table 2 Clinical studies of the selective COX-2 inhibitor celecoxib and the mixed COX-1/COX-2 inhibitor acetyl salicylic acid (ASA) in schizophrenia.
My group performed a 6-week double-blind RCT in acute schizophrenia to compare risperidone plus celecoxib with risperidone alone and risperidone plus placebo (54). Outcome was significantly better in the celecoxib add-on group (n = 25) than in the group receiving risperidone alone (n = 25) (54), particularly regarding cognition (58). Data from this study and another 6-week risperidone/celecoxib study were pooled (total N = 90), and the analysis found a benefit of celecoxib add-on if the illness duration was 24 months or less but no such benefit in more chronic cases. A large study that included a broad spectrum of schizophrenia patients found no advantage of celecoxib over placebo (57), whereby the disease duration of up to 10 years in many patients, some of whom had chronic schizophrenia, may have contributed to the negative result. Another study in chronic schizophrenia showed no advantage of add-on celecoxib in patients with chronic schizophrenia (56), providing further support for differential effects of COX-2 inhibitors in acute and chronic phases of the disease. To further test this hypothesis, my group performed a double-blind study of amisulpride with adjunct celecoxib or placebo in first-episode schizophrenia (55); the celecoxib group showed more improvement than did the placebo group on the positive, negative, total, and general psychopathology scores of the Positive and Negative Syndrome Scale (PANSS) (55, 59).
Similar benefits have been found for ASA in schizophrenia spectrum disorders. A double-blind RCT studied 70 inpatients and outpatients with schizophrenia spectrum disorders treated with antipsychotics and add-on aspirin 1 g/day or placebo (53). The total and positive PANSS scores decreased significantly more in patients taking aspirin add-on than in those taking placebo, and the more altered the immune function, the greater the effect on the total PANSS score. Another double-blind, placebo-controlled study evaluated 6-week adjunctive treatment with ASA in 60 inpatients with schizophrenia (52): In addition to their antipsychotic, one group (n = 20) received aspirin 325 mg/day; one (n = 20), aspirin 500 mg/day; and one (n = 20), placebo. At the end of the study, the PANSS positive and negative symptom scores and general psychopathology score were significantly lower in the aspirin groups than in the placebo group.
A meta-analysis of data from n = 264 patients from five double-blind, randomized, placebo-controlled studies on NSAIDs (celecoxib, four studies; ASA, one study) in schizophrenia found that the drugs had significant positive effects on total symptom severity and positive and negative symptom severity (60). A meta-analysis of data from n = 774 patients from eight studies (celecoxib, six; ASA, two) found that the drugs had significant effects only in patients with a first manifestation of schizophrenia and not in those with chronic schizophrenia and in inpatients but not outpatients (61).
Other Immune-Related Treatment
PET studies have shown that microglia cells are also activated in schizophrenia (62). As mentioned in the section Other Immune-Related Substances, the antibiotic minocycline can inhibit microglia activation, and studies in animal models of schizophrenia found that minocycline improves cognition (63). Double-blind, placebo-controlled, add-on studies of minocycline in schizophrenia patients found positive effects on negative and cognitive symptoms (64), and case reports described benefits for symptoms overall (65).
Other potential anti-inflammatory agents have shown some benefit in schizophrenia. For example, a meta-analysis of 26 double-blind RCTs found significant effects for N-acetylcysteine and estrogen (50). Positive effects were also found for interferon-gamma (IFN-γ), a cytokine responsible for the monocytic type 1 immune response (66). IFN-γ can have severe side effects, such as hallucinations, seizures, and immunosuppression, so patients must be monitored carefully.
The initial studies on monoclonal antibodies against pro-inflammatory cytokines indicate that they may also be a potential treatment approach in schizophrenia, but more studies are needed (67).
Two interesting case reports shed light on a theoretical concept that is interesting but will probably never become an alternative for schizophrenia treatment: In one case, schizophrenia seems to have transferred by bone marrow transplantation to a leukemia patient (68); and in another contrasting case, schizophrenia was remitted by bone marrow transplantation to a patient with schizophrenia (69).
Special Issues Regarding Anti-Inflammatory Treatment in Schizophrenia
On the basis of the above, we can conclude that anti-inflammatory agents may be beneficial in schizophrenia, but their usefulness depends on the stage of the disease; that is, their efficacy is lower in chronic than in acute schizophrenia; this difference may be associated with the neuroprogression of the disease. In schizophrenia, chronification is known to negatively impact outcome. Long-term treatment with anti-inflammatory agents has not yet been studied, although it may have more beneficial effects (51). This is the case in chronic inflammatory diseases, where short-term treatment with anti-inflammatory agents has only weak effects.
Further research is required on predictors of better response to anti-inflammatory treatment in schizophrenia. In all studies to date, response to antipsychotics was worse if levels of inflammation were higher (70, 71). The question whether the outcome of anti-inflammatory treatment is better in patients with high levels of inflammation, as was shown for anti-TNF-α treatment and celecoxib in MD (18, 32), remains open. As in MD, in schizophrenia, no markers have been identified that could help predict the outcome of treatment with anti-inflammatory agents.
Studies have shown probable non-immune-mediated effects for several NSAIDs, including COX-2 inhibitors (72), although their effects on schizophrenia are most likely due to their anti-inflammatory effects. Clearer evidence that inflammation is involved in the pathophysiology of schizophrenia is provided by studies on monoclonal antibodies, which do not affect neurotransmitters in any other way. However, further research is needed on this topic.
Conclusion
Studies and meta-analyses showing increased levels of pro-inflammatory cytokines and CRP, for example, indicate that the development of some psychiatric disorders, including MD and schizophrenia, may involve inflammation. Further support is provided by the positive effects of immunomodulatory agents in treating these disorders. Statistically significant therapeutic effects have been shown for the COX-2 inhibitor celecoxib in MD and early-stage schizophrenia, and protective and therapeutic effects have been shown for the mixed COX-1 and COX-2 inhibitor ASA (aspirin) in schizophrenia. Statistical significance, however, does not necessarily equate to clinical significance and high effect sizes. Therefore, further studies are warranted, including stand-alone studies with immunomodulators. A further limitation is that studies of these compounds in psychiatric indications lasted only several weeks, and these agents have not been evaluated in long-term studies. Celecoxib is known to be associated with an increase in cardiovascular side effects after treatment lasting about 18 months. ASA and, to a lesser extent, COX-2 inhibitors can have gastrointestinal side effects, including gastrointestinal bleeding. Nevertheless, celecoxib and, in particular, ASA are well described and, in general, are well tolerated, in a dose-dependent manner, in many different clinical indications. Another limitation is that no specific dose–response relationship studies have so far been performed in psychiatric indications, and doses have been chosen on the basis of those used in non-psychiatric indications. Different dose regimens may optimize the benefits of these substances in psychiatric indications. Many other interesting substances, such as anti-cytokines, anti-Il-6 complex substances, minocycline, and statins, that target various components of the immune system may be beneficial in MD or schizophrenia or both.
Author Contributions
NM is the sole author of this review and performed all tasks related to preparation of the manuscript.
Funding
The work was supported by the Foundation “Immunität und Seele.”
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author thanks Jacquie Klesing, Board-certified Editor in the Life Sciences (ELS), for editing assistance with the manuscript.
References
1. Goeden N, Velasquez J, Arnold KA, Chan Y, Lund BT, Anderson GM, et al. Maternal inflammation disrupts fetal neurodevelopment via increased placental output of serotonin to the fetal brain. J Neurosci (2016) 36(22):6041–9. doi: 10.1523/JNEUROSCI.2534-15.2016
2. Kabiersch A, Furukawa H, del Rey A, Besedovsky HO. Administration of interleukin-1 at birth affects dopaminergic neurons in adult mice. Ann N Y Acad Sci (1998) 840:123–7. doi: 10.1111/j.1749-6632.1998.tb09556.x
3. Meehan C, Harms L, Frost JD, Barreto R, Todd J, Schall U, et al. Effects of immune activation during early or late gestation on schizophrenia-related behaviour in adult rat offspring. Brain Behav Immun (2017) 63:8–20. doi: 10.1016/j.bbi.2016.07.144
4. Brown AS, Schaefer CA, Wyatt RJ, Goetz R, Begg MD, Gorman JM, et al. Maternal exposure to respiratory infections and adult schizophrenia spectrum disorders: a prospective birth cohort study. Schizophr Bull (2000) 26(2):287–95. doi: 10.1093/oxfordjournals.schbul.a033453
5. Sorensen HJ, Mortensen EL, Reinisch JM, Mednick SA. Association between prenatal exposure to bacterial infection and risk of schizophrenia. Schizophr Bull (2009) 35(3):631–7. doi: 10.1093/schbul/sbn121
6. Cattaneo A, Macchi F, Plazzotta G, Veronica B, Bocchio-Chiavetto L, Riva MA, et al. Inflammation and neuronal plasticity: a link between childhood trauma and depression pathogenesis. Front Cell Neurosci (2015) 9:40. doi: 10.3389/fncel.2015.00040
7. Metcalf SA, Jones PB, Nordstrom T, Timonen M, Maki P, Miettunen J, et al. Serum C-reactive protein in adolescence and risk of schizophrenia in adulthood: a prospective birth cohort study. Brain Behav Immun (2017) 59:253–9. doi: 10.1016/j.bbi.2016.09.008
8. Goldsmith DR, Rapaport MH. Miller BJ. A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol Psychiatry (2016) 21(12):1696–709. doi: 10.1038/mp.2016.3
9. Wang AK, Miller BJ. Meta-analysis of cerebrospinal fluid cytokine and tryptophan catabolite alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder, and depression. Schizophr Bull (2018) 44(1):75–83. doi: 10.1093/schbul/sbx035
10. Benros ME, Waltoft BL, Nordentoft M, Ostergaard SD, Eaton WW, Krogh J, et al. Autoimmune diseases and severe infections as risk factors for mood disorders: a nationwide study. JAMA Psychiatry (2013) 70(8):812–20. doi: 10.1001/jamapsychiatry.2013.1111
11. Kessing LV, Rytgaard HC, Gerds TA, Berk M, Ekstrom CT, Andersen PK. New drug candidates for depression—a nationwide population-based study. Acta Psychiatr Scand (2019) 139(1):68–77. doi: 10.1111/acps.12957
12. Sandrini M, Vitale G, Pini LA. Effect of rofecoxib on nociception and the serotonin system in the rat brain. Inflamm Res (2002) 51(3):154–9. doi: 10.1007/PL00000287
13. Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B. Kynurenine pathway in major depression: evidence of impaired neuroprotection. J Affect Disord (2007) 98(1–2):143–51. doi: 10.1016/j.jad.2006.07.013
14. Brunello N, Alboni S, Capone G, Benatti C, Blom JM, Tascedda F, et al. Acetylsalicylic acid accelerates the antidepressant effect of fluoxetine in the chronic escape deficit model of depression. Int Clin Psychopharmacol (2006) 21(4):219–25. doi: 10.1097/00004850-200607000-00004
15. Muller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Muller B, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry (2006) 11(7):680–4. doi: 10.1038/sj.mp.4001805
16. Krause D, Myint AM, Schuett C, Musil R, Dehning S, Cerovecki A, et al. High kynurenine (a tryptophan metabolite) predicts remission in patients with major depression to add-on treatment with celecoxib. Front Psychiatry (2017) 8:16. doi: 10.3389/fpsyt.2017.00016
17. Akhondzadeh S, Jafari S, Raisi F, Nasehi AA, Ghoreishi A, Salehi B, et al. Clinical trial of adjunctive celecoxib treatment in patients with major depression: a double blind and placebo controlled trial. Depress Anxiety (2009) 26(7):607–11. doi: 10.1002/da.20589
18. Abbasi SH, Hosseini F, Modabbernia A, Ashrafi M, Akhondzadeh S. Effect of celecoxib add-on treatment on symptoms and serum IL-6 concentrations in patients with major depressive disorder: randomized double-blind placebo-controlled study. J Affect Disord (2012) 141(2–3):308–14. doi: 10.1016/j.jad.2012.03.033
19. Majd M, Hashemian F, Hosseini SM, Vahdat Shariatpanahi M, Sharifi A. A randomized, double-blind, placebo-controlled trial of celecoxib augmentation of sertraline in treatment of drug-naive depressed women: a pilot study. Iran J Pharm Res (2015) 14(3):891–9.
20. Na KS, Lee KJ, Lee JS, Cho YS, Jung HY. Efficacy of adjunctive celecoxib treatment for patients with major depressive disorder: a meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry (2014) 48:79–85. doi: 10.1016/j.pnpbp.2013.09.006
21. Kohler O, Benros ME, Nordentoft M, Farkouh ME, Iyengar RL, Mors O, et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry (2014) 71(12):1381–91. doi: 10.1001/jamapsychiatry.2014.1611
22. Walker C, Biasucci LM. Cardiovascular safety of non-steroidal anti-inflammatory drugs revisited. Postgrad Med (2018) 130(1):55–71. doi: 10.1080/00325481.2018.1412799
23. Sepehrmanesh Z, Fahimi H, Akasheh G, Davoudi M, Gilasi H, Ghaderi A. The effects of combined sertraline and aspirin therapy on depression severity among patients with major depressive disorder: a randomized clinical trial. Electron Physician (2017) 9(11):5770–7. doi: 10.19082/5770
24. Savitz JB, Teague TK, Misaki M, Macaluso M, Wurfel BE, Meyer M, et al. Treatment of bipolar depression with minocycline and/or aspirin: an adaptive, 2 × 2 double-blind, randomized, placebo-controlled, phase IIA clinical trial. Transl Psychiatry (2018) 8(1):27. doi: 10.1038/s41398-017-0073-7
25. Kohler-Forsberg O, Lydholm C, Hjorthoj C, Nordentoft M, Mors O, Benros ME. Efficacy of anti-inflammatory treatment on major depressive disorder or depressive symptoms: meta-analysis of clinical trials. Acta Psychiatr Scand (2019) 39(5):404–19. doi: 10.1111/acps.13016
26. Begemann M, Sargin D, Rossner MJ, Bartels C, Theis F, Wichert SP, et al. Episode-specific differential gene expression of peripheral blood mononuclear cells in rapid cycling supports novel treatment approaches. Mol Med (2008) 14(9–10):546–52. doi: 10.2119/2008-00053.Begemann
27. Castillo MFR, Murata S, Schwarz MJ, Schutze G, Moll N, Weidinger E, et al. Celecoxib augmentation of escitalopram in treatment-resistant bipolar depression and effects of quinolinic acid. Neurol Psychiat Br (2019) 32:22–9. doi: 10.1016/j.npbr.2019.03.005
28. Collantes-Estevez E, Fernandez-Perez C. Improved control of osteoarthritis pain and self-reported health status in non-responders to celecoxib switched to rofecoxib: results of PAVIA, an open-label post-marketing survey in Spain. Curr Med Res Opin (2003) 19(5):402–10. doi: 10.1185/030079903125001938
29. Nery FG, Monkul ES, Hatch JP, Fonseca M, Zunta-Soares GB, Frey BN, et al. Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: a double-blind, randomized, placebo-controlled study. Hum Psychopharmacol (2008) 23(2):87–94. doi: 10.1002/hup.912
30. Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A, et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet (2006), 367(9504):29–35. doi: 10.1016/S0140-6736(05)67763-X
31. Kappelmann N, Lewis G, Dantzer R, Jones PB, Khandaker GM. Antidepressant activity of anti-cytokine treatment: a systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol Psychiatry (2018) 23(2):335–43. doi: 10.1038/mp.2016.167
32. Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry (2013) 70(1):31–41. doi: 10.1001/2013.jamapsychiatry.4
33. Gossec L, Steinberg G, Rouanet S, Combe B. Fatigue in rheumatoid arthritis: quantitative findings on the efficacy of tocilizumab and on factors associated with fatigue. Clin Exp Rheumatol (2015) 33(5):664–70.
34. Traki L, Rostom S, Tahiri L, Bahiri R, Harzy T, Abouqal R, et al. Responsiveness of the EuroQol EQ-5D and Hospital Anxiety and Depression Scale (HADS) in rheumatoid arthritis patients receiving tocilizumab. Clin Rheumatol (2014) 33(8):1055–60. doi: 10.1007/s10067-014-2609-z
35. Yang C, Hashimoto K. Peripheral IL-6 signaling: a promising therapeutic target for depression? Expert Opin Investig Drugs (2015) 24(7):989–90. doi: 10.1517/13543784.2015.1055669
36. Yang C, Shirayama Y, Zhang JC, Ren Q, Hashimoto K. Peripheral interleukin-6 promotes resilience versus susceptibility to inescapable electric stress. Acta Neuropsychiatr (2015) 27(5):312–6. doi: 10.1017/neu.2015.36
37. Yang JJ, Wang N, Yang C, Shi JY, Yu HY, Hashimoto K. Serum interleukin-6 is a predictive biomarker for ketamine’s antidepressant effect in treatment-resistant patients with major depression. Biol Psychiatry (2015) 77(3):e19–20. doi: 10.1016/j.biopsych.2014.06.021
38. Maes M, Anderson G, Kubera M, Berk M. Targeting classical IL-6 signalling or IL-6 trans-signalling in depression? Expert Opin Ther Targets (2014) 18(5):495–512. doi: 10.1517/14728222.2014.888417
39. Zhou AJ, Lee Y, Salvadore G, Hsu B, Fonseka TM, Kennedy SH, et al. Sirukumab: a potential treatment for mood disorders? Adv Ther (2017) 34(1):78–90. doi: 10.1007/s12325-016-0455-x
40. Holmes SE, Hinz R, Conen S, Gregory CJ, Matthews JC, Anton-Rodriguez JM, et al. Elevated translocator protein in anterior cingulate in major depression and a role for inflammation in suicidal thinking: a positron emission tomography study. Biol Psychiatry (2018) 83(1):61–9. doi: 10.1016/j.biopsych.2017.08.005
41. Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry (2015) 72(3):268–75. doi: 10.1001/jamapsychiatry.2014.2427
42. Rosenblat JD, McIntyre RS. Efficacy and tolerability of minocycline for depression: a systematic review and meta-analysis of clinical trials. J Affect Disord (2018) 227:219–25. doi: 10.1016/j.jad.2017.10.042
43. Kohler O, Gasse C, Petersen L, Ingstrup KG, Nierenberg AA, Mors O, et al. The effect of concomitant treatment with SSRIs and statins: a population-based study. Am J Psychiatry (2016) 173(8):807–15. doi: 10.1176/appi.ajp.2016.15040463
44. Weitz-Schmidt G. Statins as anti-inflammatory agents. Trends Pharmacol Sci (2002) 23(10):482–6. doi: 10.1016/S0165-6147(02)02077-1
45. Lanquillon S, Krieg JC, Bening-Abu-Shach U, Vedder H. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology (2000) 22(4):370–9. doi: 10.1016/S0893-133X(99)00134-7
46. Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature (2016) 530(7589):177–83. doi: 10.1038/nature16549
47. Benros ME, Nielsen PR, Nordentoft M, Eaton WW, Dalton SO, Mortensen PB. Autoimmune diseases and severe infections as risk factors for schizophrenia: a 30-year population-based register study. Am J Psychiatry (2011) 168(12):1303–10. doi: 10.1176/appi.ajp.2011.11030516
48. Benros ME, Mortensen PB, Eaton WW. Autoimmune diseases and infections as risk factors for schizophrenia. Ann N Y Acad Sci (2012) 1262:56–66. doi: 10.1111/j.1749-6632.2012.06638.x
49. Kohler O, Petersen L, Benros ME, Mors O, Gasse C. Concomitant NSAID use during antipsychotic treatment and risk of 2-year relapse—a population-based study of 16,253 incident patients with schizophrenia. Expert Opin Pharmacother (2016) 17(8):1055–62. doi: 10.1517/14656566.2016.1168400
50. Sommer IE, van Westrhenen R, Begemann MJ, de Witte LD, Leucht S, Kahn RS. Efficacy of anti-inflammatory agents to improve symptoms in patients with schizophrenia: an update. Schizophr Bull (2014) 40(1):181–91. doi: 10.1093/schbul/sbt139
51. Akhondzadeh S, Tabatabaee M, Amini H, Ahmadi Abhari SA, Abbasi SH, Behnam B. Celecoxib as adjunctive therapy in schizophrenia: a double-blind, randomized and placebo-controlled trial. Schizophr Res (2007) 90(1–3):179–85. doi: 10.1016/j.schres.2006.11.016
52. Attari A, Mojdeh A, Soltani FASK, Najarzadegan MR. Aspirin inclusion in antipsychotic treatment on severity of symptoms in schizophrenia: a randomized clinical trial. Iran J Psychiatry Behav Sci (2017) 11(1):e5848. doi: 10.5812/ijpbs.5848
53. Laan W, Grobbee DE, Selten JP, Heijnen CJ, Kahn RS, Burger H. Adjuvant aspirin therapy reduces symptoms of schizophrenia spectrum disorders: results from a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry (2010) 71(5):520–7. doi: 10.4088/JCP.09m05117yel
54. Muller N, Riedel M, Scheppach C, Brandstatter B, Sokullu S, Krampe K, et al. Beneficial antipsychotic effects of celecoxib add-on therapy compared to risperidone alone in schizophrenia. Am J Psychiatry (2002) 159(6):1029–34. doi: 10.1176/appi.ajp.159.6.1029
55. Muller N, Krause D, Dehning S, Musil R, Schennach-Wolff R, Obermeier M, et al. Celecoxib treatment in an early stage of schizophrenia: results of a randomized, double-blind, placebo-controlled trial of celecoxib augmentation of amisulpride treatment. Schizophr Res (2010) 121(1–3):118–24. doi: 10.1016/j.schres.2010.04.015
56. Rapaport MH, Delrahim KK, Bresee CJ, Maddux RE, Ahmadpour O, Dolnak D. Celecoxib augmentation of continuously ill patients with schizophrenia. Biol Psychiatry (2005) 57(12):1594–6. doi: 10.1016/j.biopsych.2005.02.024
57. Rappard F, Muller N. Celecoxib add-on therapy does not have beneficial antipsychotic effects over risperidone alone in schizophrenia. Neuropsychopharmacology (2004) 29(Suppl 1):S222.
58. Muller N, Riedel M, Schwarz MJ, Engel RR. Clinical effects of COX-2 inhibitors on cognition in schizophrenia. Eur Arch Psychiatry Clin Neurosci (2005) 255(2):149–51. doi: 10.1007/s00406-004-0548-4
59. Muller N. COX-2 inhibitors as antidepressants and antipsychotics: clinical evidence. Curr Opin Investig Drugs (2010) 11(1):31–42.
60. Sommer IE, de Witte L, Begemann M, Kahn RS. Nonsteroidal anti-inflammatory drugs in schizophrenia: ready for practice or a good start? A meta-analysis. J Clin Psychiatry (2012) 73(4):414–9. doi: 10.4088/JCP.10r06823
61. Nitta M, Kishimoto T, Muller N, Weiser M, Davidson M, Kane JM, et al. Adjunctive use of nonsteroidal anti-inflammatory drugs for schizophrenia: a meta-analytic investigation of randomized controlled trials. Schizophr Bull (2013) 39(6):1230–41. doi: 10.1093/schbul/sbt070
62. De Picker LJ, Morrens M, Chance SA, Boche D. Microglia and brain plasticity in acute psychosis and schizophrenia illness course: a meta-review. Front Psychiatry (2017) 8:238.
63. Mizoguchi H, Takuma K, Fukakusa A, Ito Y, Nakatani A, Ibi D, et al. Improvement by minocycline of methamphetamine-induced impairment of recognition memory in mice. Psychopharmacology (Berl) (2008) 196(2):233–41. doi: 10.1007/s00213-007-0955-0
64. Chaudhry IB, Hallak J, Husain N, Minhas F, Stirling J, Richardson P, et al. Minocycline benefits negative symptoms in early schizophrenia: a randomised double-blind placebo-controlled clinical trial in patients on standard treatment. J Psychopharmacol (2012) 26(9):1185–93. doi: 10.1177/0269881112444941
65. Ahuja N, Carroll BT. Possible anti-catatonic effects of minocycline in patients with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2007) 31(4):968–9. doi: 10.1016/j.pnpbp.2007.01.018
66. Gruber L, Bunse T, Weidinger E, Reichard H, Muller N. Adjunctive recombinant human interferon gamma-1b for treatment-resistant schizophrenia in 2 patients. J Clin Psychiatry (2014) 75(11):1266–7. doi: 10.4088/JCP.14l09005
67. Miller BJ, Buckley PF. The case for adjunctive monoclonal antibody immunotherapy in schizophrenia. Psychiatr Clin North Am (2016) 39(2):187–98. doi: 10.1016/j.psc.2016.01.003
68. Sommer IE, van Bekkum DW, Klein H, Yolken R, de Witte L, Talamo G. Severe chronic psychosis after allogeneic SCT from a schizophrenic sibling. Bone Marrow Transplant (2015) 50(1):153–4. doi: 10.1038/bmt.2014.221
69. Miyaoka T, Wake R, Hashioka S, Hayashida M, Oh-Nishi A, Azis IA, et al. Remission of psychosis in treatment-resistant schizophrenia following bone marrow transplantation: a case report. Front Psychiatry (2017) 8:174. doi: 10.3389/fpsyt.2017.00174
70. Mondelli V, Ciufolini S, Belvederi Murri M, Bonaccorso S, Di Forti M, Giordano A, et al. Cortisol and inflammatory biomarkers predict poor treatment response in first episode psychosis. Schizophr Bull (2015) 41(5):1162–70. doi: 10.1093/schbul/sbv028
71. Zhang XY, Zhou DF, Cao LY, Wu GY, Shen YC. Cortisol and cytokines in chronic and treatment-resistant patients with schizophrenia: association with psychopathology and response to antipsychotics. Neuropsychopharmacology (2005) 30(8):1532–8. doi: 10.1038/sj.npp.1300756
Keywords: Inflammation, major depression, psychoneuroimmunology, COX-2 inhibition, aspirin, psychiatry, schizophrenia
Citation: and Müller N (2019) COX-2 Inhibitors, Aspirin, and Other Potential Anti-Inflammatory Treatments for Psychiatric Disorders. Front. Psychiatry 10:375. doi: 10.3389/fpsyt.2019.00375
Received: 03 December 2018; Accepted: 13 May 2019;
Published: 31 May 2019.
Edited by:
Marion Leboyer, Université Paris - Est Créteil Val de Marne, FranceReviewed by:
Stefania Schiavone, University of Foggia, ItalyCarlos M. Opazo, The University of Melbourne, Australia
Copyright © 2019 Müller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Norbert Müller, Ti5NdWVsbGVyQG12dC16ZW50cnVtLmRl