 Kevin W. Hoffman1
Kevin W. Hoffman1 Jakleen J. Lee1
Jakleen J. Lee1 Cheryl M. Corcoran1,2
Cheryl M. Corcoran1,2 David Kimhy1,2
David Kimhy1,2 Thorsten M. Kranz3
Thorsten M. Kranz3 Dolores Malaspina1*
Dolores Malaspina1*- 1Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 2James J. Peters VA Medical Center, Mental Illness Research, Education and Clinical Centers (MIRECC), New York, NY, United States
- 3Department of Psychiatry, Psychosomatic Medicine and Psychotherapy, University Hospital, Goethe University, Frankfurt, Germany
Early life adversity and prenatal stress are consistently associated with an increased risk for schizophrenia, although the exact pathogenic mechanisms linking the exposures with the disease remain elusive. Our previous view of the HPA stress axis as an elegant but simple negative feedback loop, orchestrating adaptation to stressors among the hypothalamus, pituitary, and adrenal glands, needs to be updated. Research in the last two decades shows that important bidirectional signaling between the HPA axis and intestinal mucosa modulates brain function and neurochemistry, including effects on glucocorticoid hormones and brain-derived neurotrophic factor (BDNF). The intestinal microbiome in earliest life, which is seeded by the vaginal microbiome during delivery, programs the development of the HPA axis in a critical developmental window, determining stress sensitivity and HPA function as well as immune system development. The crosstalk between the HPA and the Microbiome Gut Brain Axis (MGBA) is particularly high in the hippocampus, the most consistently disrupted neural region in persons with schizophrenia. Animal models suggest that the MGBA remains influential on behavior and physiology across developmental stages, including the perinatal window, early childhood, adolescence, and young adulthood. Understanding the role of the microbiome on critical risk related stressors may enhance or transform of understanding of the origins of schizophrenia and offer new approaches to increase resilience against stress effects for preventing and treating schizophrenia.
Introduction
Schizophrenia presents an enormous burden to individuals, families, communities, and public health, but the mechanisms underlying its pathogenesis, presentation, and course remain largely enigmatic, with no interventions known to prevent or cure the disease. New perspectives are necessary to overcome this roadblock. The microbiome, which broadly refers to the collection of genomes of the commensal microbes inhabiting our bodies, influences our health in broad and complex ways. The emerging science of the microbiome is a promising new domain that could shed light on crucial disparate features of schizophrenia, including its association with prenatal and life course stressors, neurodevelopmental underpinnings, inflammatory neuropathology, particularly of the hippocampus and its metabolic comorbidity.
The Microbiome
The microbiome comprises a dynamic ecological community of commensal microorganisms that inhabit our body where it interfaces with the environment. These specific microbes, which are collectively referred to as the microbiota, consist of bacteria, viruses, fungi, and protozoa; approximately equal our own cells in number; and combined pose over 200 times the number of genes as the human genome (1, reviewed in 2). Recent advances in high-throughput genetic sequencing and computational abilities reveal the richness, complexity, and essential role of the microbiome in human health. Its composition varies by anatomic region, with the gut microbiome in the distal large intestine considered the most influential for health.
After being seeded at birth by maternal vaginal bacteria in the birth canal, the neonate gut microbiota develops in a phasic manner, largely due to feeding. The gut is initially colonized by microaerophilic Proteobacteria and facultative anaerobic Actinobacteria, which consume oxygen and create a suitable niche for subsequent obligate anaerobes like Bacteroides, Clostridium, and Bifidobacterium spp. (3, 4) Breast milk stimulates the growth of bifidabacteria, but weaning results in the emergence of Firmicutes and Bacteroidetes (5). These phyla proliferate with the introduction of solid foods and eventually come to dominate the gut microbiota (5). By 2.5 to 3 years of age, the infant gut microbiota structure stabilizes and resembles the adult gut microbiota, which is also dominated by Firmicutes and Bacteroidetes (3). The developmental dynamics of the infant gut microbiota are shaped by host genes, host immunity and environmental factors, such as diet, medications, and climate (6–8).
Over the last decade, it has emerged that the human microbiome highly influences the development of the central nervous system (CNS) and the immune system. The microbiome is shaped by stress exposures from early life and, in turn, influences stress responsivity (9). Given this new information, our models of the endocrine modulation of the stress response should be updated to account for the microbiome.
The bidirectional influence of the gut microbiome and CNS occurs through the “gut-brain axis” (GBA), components of which include the vagal nerve, gut hormone signaling, immune system, tryptophan metabolism, and microbial metabolites, such as short-chain fatty acids (reviewed in 10). Activity along the GBA intersects with the HPA axis (Figure 1) and may influence many psychiatric disorders, as evidenced by the association of gut dysbiosis with autism, depression, and anxiety disorders as well as functional gastrointestinal disorders (11–16). Given the purported inflammatory underpinnings for schizophrenia and its severe comorbidities with other microbiome-linked metabolic diseases, associations between schizophrenia and the microbiome are of great interest.
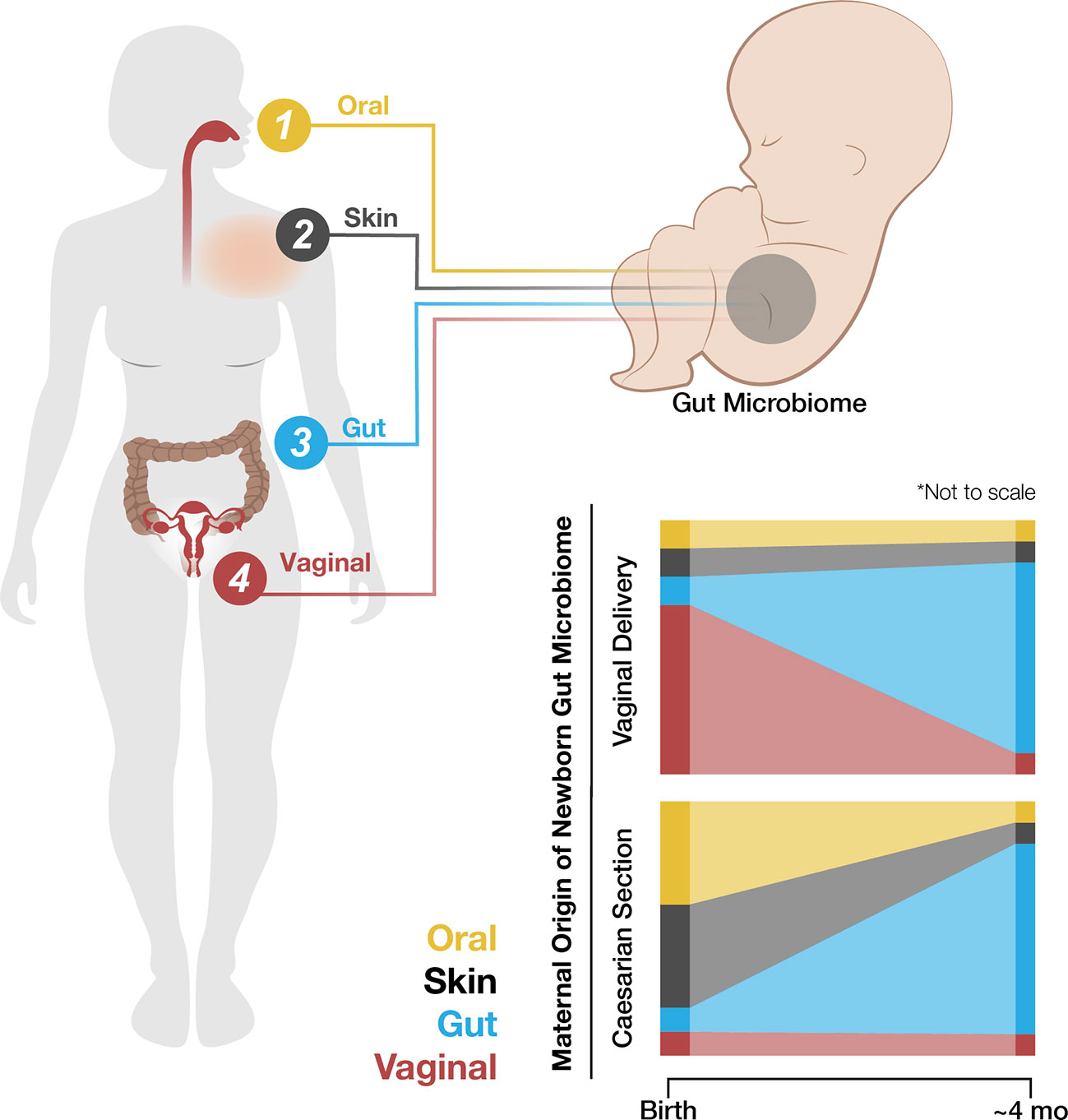
Figure 1 The hypothalamic-pituitary-adrenal (HPA) axis regulates the response to stress (red lines). Stress activates the hypothalamus to secrete cortisol-releasing hormone (CRH), which induces the anterior pituitary gland to release adenocorticotropin hormone (ACTH; solid red lines). ACTH stimulates the adrenal gland cortex to produce cortisol (solid red line), which negatively regulates CRH production to terminate the stress response cascade (dashed red line). Excess or chronic stress can disturb normal HPA axis function via altered neuroendocrine signaling and gut dysbiosis (blue). Under excess or chronic stress, the hypothalamus is hyperactivated, leading to upregulation of the anterior pituitary gland and adrenal gland activation (plus signs) as well as downregulation of CRH inhibition (minus sign). Consequently, abnormally high levels of cortisol result in increased hippocampal signaling, which may overactivate the hippocampus, cause inflammation, and alter the crosstalk equilibrium between cortisol and BDNF in the hippocampus. Excess or chronic stress causes gut dysbiosis, which alters gut hormone and microbe metabolite signaling from the gut to the brain through the vagus nerve, i.e., the GBA.
Stress Response and the Microbiome
Awareness of the overlap of stress signaling and the microbiome began in 2004 with the report that germ-free mice had an exaggerated hypothalamus-pituitary-adrenal (HPA) responses to stress in comparison to non-germ-free mice (17). The “microbiome-gut-brain axis” (MGBA) refers to bidirectional signaling between the gut flora and CNS. Acute and chronic stressors that activate the HPA axis also influence the microbiome and gut epithelium which participate in behavioral and systemic stress effects. The gut and brain communicate through the vagal (parasympathetic) nerve, which is a cholinergic anti-inflammatory pathway associated with slowed heart and respiratory rates and digestive function. Under stress, the sympathetic nervous system predominates and vagal function is reduced. The gut microbiome produces neurotransmitters that influence behavior, including acetylcholine, catecholamines, γ-aminobutyric acid, histamine, melatonin, and serotonin, all of which are also essential for regulating gastrointestinal peristalsis and sensation. Thus, the HPA axis and GBA are intersecting, co-dependent loops for managing stress and inflammation as part of their physiological function.
In this review, we illuminate aspects of the stress response and the microbiome as the GBA, with respect to schizophrenia. The impact of stress exposures on the brain will almost certainly entail signaling with the microbiome. Some factors that are associated with an increased risk for schizophrenia are considered across developmental stages, including the perinatal window, early childhood, adolescence, and young adulthood.
The HPA Axis, Hippocampus, Neurotrophins, and Schizophrenia
The neurobiology of the stress cascade and its potential for toxicity is well described. The HPA axis is the stress response system through which stress hormones and the CNS interact. Early dysregulation of the HPA axis is associated with adult stress-related disorders, including schizophrenia (18–20). Mechanistically, HPA axis dysregulation is considered to be linked to schizophrenia risk via glucocorticoid (GC) overproduction, especially during vulnerable phases of neurodevelopment. Cortisol-releasing hormone (CRH) is released from the paraventricular nucleus of the hypothalamus following physical or psychological stressors. CRH binds receptors on the anterior pituitary gland, driving release of adrenocorticotropic hormone (ACTH). This stimulates the adrenal cortex to release cortisol, the human GC hormone. Under physiological conditions increasing cortisol levels inhibit CRH release, terminating this stress cascade through a negative feedback loop. However, excess and chronic stress hyperactivate the HPA axis and cause abnormally high GC levels (21–24).
The effect of elevated GC levels on the hippocampus, the essential structure for memory and contextualizing new information, may be relevant. The hippocampus is the most commonly abnormal brain region in groups of schizophrenia cases, with progressive hippocampal volume loss a common observation (25). Increased activation, metabolism, and inflammation of the anterior hippocampus are associated with psychotic symptoms (26, 27) (reviewed in 28). The hippocampus possesses a high concentration of GC receptors that promote threat appraisal and help organize the stress response. Increased GC levels may drive overactivation and inflammation of the hippocampus and thereby promote schizophrenia (reviewed in 29–31).
GCs may also influence schizophrenia through interaction with neurotrophin pathways. Neurotrophins are growth factors responsible for neuron growth, differentiation, and formation of new synapses (32). Brain-derived neurotrophic factor (BDNF), the most abundant neurotrophin, is highly active in the hippocampus, cortex, and basal forebrain, where it binds its receptor, tyrosine kinase receptor B (TrkB), to play a key role in synaptic plasticity and long-term memory formation (33). Because GC receptors and TrkB are co-expressed in the hippocampus, important crosstalk between GCs and BDNF occurs here, as threat appraisal relies on both current stress and appropriate context from memory (34). As such, GC and BDNF equilibrium remains crucial for stress response regulation throughout life. Impairment of GC receptors and TrkB in the hippocampus favors vulnerability to stress-related disorders, including schizophrenia (reviewed in 35).
These pathways are influenced by the microbiome. Gut dysbiosis can indirectly influence cortisol release and sensitivity via chronic cytokine-mediated inflammation (36–38). This proinflammatory state may be driven by microbes crossing the intestinal barrier, releasing microbial byproducts such as lipopolysaccharide (LPS), or be moderated through bacterial metabolites, such as short-chain fatty acids (39–43) (reviewed in 44). The microbiome further influences the structure and function of the amygdala, which is critical for emotion learning and social behavior, especially responses linked to anxiety and/or fear (45, 46). Studies of germ-free mice show that the absence of the microbiome during early critical developmental windows leads to chronic cortisol elevation and altered hippocampal BDNF levels (17, 47). Depleting the microbiome of previously healthy mice through antibiotics disrupts the HPA axis (36, 48, 49). Taken together, these findings suggest that a healthy microbiome is an important component of HPA axis development and that early alterations of the microbiome can affect neuroendocrine pathways throughout life.
Resilience in Schizophrenia
Identifying factors to increase resilience against stress is an area of active research that may be addressed through MGBA research. Anxiety and depression-like symptoms in germ free animas as well as the transference of a depression phenotype from a human patient to a rats through fecal microbiota support the feasibility of this approach (50). Mice deficient in the CRH1 receptor and those with increased GR activity display more resilient behaviors (51–54) and these hormones can be modulating by the gut microbiome (11, 50). Likewise, the expression of serotonergic, glutamatergic, and GABA, which are dysregulated in association with poor resilience (55), are modulated through microbiome effects in animal models (reviewed in 56). A healthy microbiome may also contribute to resilience through emotion regulation that manifests as positive emotions and optimism, cognitive flexibility, and healthy interpersonal function, attributes that are associated with active coping styles (reviewed in 57). There may be treatment role for nutritional supplementation, as stress-related behaviors and HPA dysfunction in socially isolated male mice was remedied by dietary supplementation with DHA (58) and a rat study even demonstrated that stress sensitivity from early life trauma might be remediated through long-term supplementation with an eicosapentaenoic acid (EPA)/DHA mixture (59). The overlap of findings on the M-GBA with neuroendocrine and behavioral measures with those implicated for resilience indicate opportunities to modify the impact of stress exposures and augment resilience by targeting the microbiome.
Perinatal Development
Introduction
In 1934, Rosanoff and colleagues published “The Etiology of So-Called Schizophrenic Psychosis” in the American Journal of Psychology (60). This manuscript, which examined 142 pairs of twins either concordant or discordant for schizophrenia, was the first to associate birth complications with schizophrenia. In subsequent decades, schizophrenia risk during pregnancy, birth, and the neonatal period was broadly examined. Many risk factors were identified that occurred in important early developmental stages, including maternal infection, stress, and medical complications during pregnancy, fetal growth restrictions, and hypoxia during pregnancy and birth. Overall, early-life exposures have the greatest impact on the development and function of central neural circuits and the immune system (46).
Missing from this well-developed story is the impact of maternal exposures on her microbiome and the potential for vaginal dysbiosis (61, 62). The newborn’s gut microbiome is seeded by the maternal vaginal microbiome during passage through the vaginal canal (8, 63). Disruptions in maternal microbiome may cause the newborn to be seeded with a more inflammatory gut microbiome (64, 65). It is this newborn microbiome that appears to have a strong influence in driving the development of the immune system and directing neurodevelopment (17, 66–70). These important contributions to fetal development must now be included is considering the action of schizophrenia risk factors in the perinatal period.
Maternal Infection
Maternal infection during pregnancy is associated with the risk for schizophrenia and is a maternal stressor. A 1988 study reported an increased rate for persons who were in utero during the 1957 influenza epidemic (71). Subsequent studies replicated this finding and suggested the second trimester as the gestational risk period for schizophrenia from influenza infection, although other evidence points to the first trimester (72–74). Other maternal infections associated with the offspring’s risk for schizophrenia include rubella, varicella zoster virus, herpes simplex virus, and Toxoplasma gondii, known as TORCH agents, which can cross the placental barrier and directly infect the fetus, as can measles, polio, bacterial bronchopneumonias, and infections of the genitals and reproductive tract (75–77). Taken as a whole, infection with this group of pathogens during pregnancy is relatively common and may be an important factor for psychiatric disorder risk.
As to mechanism, there are several possibilities. One of these is direct invasion, which is consistent with the very high rate of schizophrenia following prenatal rubella, up to 20%, given rubella’s well-known propensity for neural invasion in the developing fetus (76). Supporting invasion, a mouse model of influenza infection showed persistence of influenza RNA in the brains of offspring of infected pregnant mice (78). Another possibility is indirect damage driven by maternal inflammation. During maternal infection, inflammatory cytokine levels are elevated (75) and these may disrupt fetal neurodevelopment and potentially drive schizophrenia risk. For instance, the proinflammatory cytokine IL-1β negatively regulates hippocampal neurogenesis, suggesting a possible mechanism through which chronic inflammation could affect schizophrenia susceptibility (79). Notably, maternal inflammation correlates with later childhood psychiatric symptoms (80). Other potential risk pathways include effects from maternal fever, maternal antibodies crossing the placenta and medications, such as analgesics and anti-inflammatories, taken by the mother during infection, all of which may impact fetal neurodevelopment (81–83).
However, maternal infections also alter her microbiome, potentially leading to increased production of inflammatory products released by her gut, as well as to disrupted seeding of the neonatal microbiome at birth (64, 65). Neonates born to mothers with ongoing HIV infection show decreased gut microbiome diversity including reduced levels of Prevotella, a bacterial genus linked to inflammatory regulation of stressor (84). It is possible that dysbiosis secondary to maternal infection sensitizes the neonate to further stress-related injury, including elevated schizophrenia risk. Given the data demonstrating the impact of maternal inflammation on offspring schizophrenia risk and the microbiome’s potential contributions to this inflammation, the microbiome may be a key player in schizophrenia pathogenesis.
Maternal Stress
Maternal stressors, such as depression, unwanted pregnancy, death of a partner, and exposure to war and disasters, are associated with schizophrenia in offspring (19, 85–88). For female fetuses, these external stressors are most strongly correlated with schizophrenia when they occur during the first trimester; however, male fetuses demonstrated increased schizophrenia risk through the second trimester, suggesting sex differences in critical periods (87, 88). Importantly, maternal stress during the first six months of postnatal life is associated with worse behavioral outcomes in children, suggesting that disrupted caregiving may also be a component to the schizophrenia risk posed by maternal stress (89). Additionally, prenatal nutritional deficiencies, including gross calorie deficits during famine and micronutrient deficiencies in homocysteine and vitamin D, are associated with both schizophrenia and the above-mentioned stressors (90–94), which certainly impact the microbiome composition. The short-term effects of maternal stress may act through adverse pregnancy outcomes, while the long-term effects on neurodevelopment may involve altered neonatal stress programming and gut dysbiosis (95). Maternal stress increases fetal and neonatal exposure to maternal cortisol, altering growth and behavior in humans and animal models (reviewed in 96). Stress also has well-documented effects on the microbiome, which may in turn alter inflammation and neurodevelopment in a developing neonate (62, 97–103). As an example, maternal perinatal stress increases offspring susceptibility to allergic diseases, which suggests interactivity between maternal GCs, perinatal immune development, and possible maternal dysbiosis (79). In a mouse model, prenatal maternal stress led to dysbiosis in both mother and offspring, increased IL-1β in utero, and a corresponding decrease in BDNF in offspring (104). Other experiments have shown antibiotics alter BDNF levels in dysbiotic mice, suggesting that interventions in the gut microbiome may be important in modifying risk (105).
Exploring how the maternal stress influences her microbiome for fetal effects relevant to schizophrenia risk may enhance our understanding of the disease and suggest new treatments or prophylactics through probiotic use (reviewed in 106). Mechanistically, the microbiome-driven effects of stress may manifest through alterations of the HPA axis during key developmental stages (107), impaired development of small intestine immune tissue and IgA production (108, 109), or alterations in gut-metabolites leading to aberrant development (110). Given that many of these downstream events are linked with schizophrenia risk, future work should aim to elicit the microbiome contributions of schizophrenia risk secondary to maternal stress.
Fetal Hypoxia
Many obstetric complications can lead to fetal hypoxia, which carries well-known risks to medial temporal regions. With regards to schizophrenia, fetal hypoxia may be the most significant risk factor among obstetric complications, in addition to maternal infections and fetal growth restriction (111, 112). Multiple studies report increased exposure to fetal hypoxia among persons with schizophrenia (113–115). One study show fetal hypoxia predicts the risk for early onset schizophrenia even after controlling for prenatal infection and fetal growth restriction (116). Further, fetal hypoxia is associated with reduced gray matter and ventricular enlargement in cases with schizophrenia and their non-ill siblings, although not in unrelated controls (117). Mechanistically, hypoxia may have an additive effect with genetic factors hastening the onset of schizophrenia in susceptible individuals (118). Certainly hypoxia may influence the composition and function of the gut microbiome (119, 120). As described above with infection and stress, these alterations increase future susceptibility to stress by influencing systemic inflammation, stress pathways, and BDNF production. Additionally, maternal microbes may invade the fetal brain following a hypoxic episode, as has been shown in sheep (121).
Fetal Growth Restriction
In 1966, a small but significant reduction in birth weight was observed in schizophrenic patients when compared to their siblings (122), prompting consideration that fetal growth restriction was a schizophrenia risk factor. Some, but not all studies associated lower birth weight, reduced head circumference, and congenital malformations with increased schizophrenia risk (123). There are heterogeneous causes of fetal growth restriction, only some of which may be associated with the risk for schizophrenia (124).
Maternal Complications
Other perinatal obstetric complications include maternal bleeding, maternal diabetes, preeclampsia, and caesarean section birth complications (125–127) (reviewed in 128). These perinatal traumas—along with the aforementioned factors of maternal infection, maternal stress, fetal hypoxia, and fetal growth restriction—altogether present a compelling argument for a close connection between the early window of neural development and schizophrenia risk. Recent advances indicate that the vaginal microbiome suggest that it may be a key player in this relationship. After all, these traumas occur during the perinatal period, when initial microbiota seeding of the newborn’s gut by the maternal vaginal microbiome occurs during fetal passage through the birth canal.
Neuroendocrine Pathways
Cortisol, the primary human “stress” hormone, is also of central relevance for the developing fetus, promoting the maturation of vital organs, including the lungs, gastrointestinal tract, liver, heart, and brain. As such, the fetal HPA axis is tightly regulated, and is responsive to minute changes in fetal plasma levels of cortisol, which easily crosses the placental barrier (reviewed in 129). Due to their high cortisol sensitivity, developing fetuses rely on the placental enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) to inactivate maternal cortisol by converting it to less active cortisone, beginning in the second trimester (130). Thus, in early gestation, before placental 11β-HSD2 is induced, maternal hypercortisolemia has potent effects on developmental gene expression. Even after the induction of 11β-HST2, some cortisone can be reactivated through 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), which converts cortisone back to cortisol (reviewed in 131). This effect can be heightened by factors like maternal protein malnutrition, which diminish 11β-HSD2 gene expression (132). The detrimental effects of elevated exposure to maternal cortisol go beyond fetal development to influence emotional and behavioral disturbances during infancy and childhood and in later life (85) possibly including the perinatal schizophrenia risk pathways described above (reviewed in 133). Beyond the direct association between maternal stress and schizophrenia, elevated maternal cortisol may enhance other risks. In one study, elevated maternal cortisol during the second trimester enhanced the risk for adolescent onset depression in the offspring of mothers who experienced infections during pregnancy (134).
The neurotrophin BDNF is also critical for neurodevelopment. Elevated levels of BDNF are reported in fetuses with severe growth restriction as well as those with macrosomia in the context of maternal diabetes (135). Mechanistically, it is proposed that BDNF is neuroprotective in the developing fetus through anti-inflammatory mechanisms (136). In vivo animal models demonstrate that BDNF can reduce hypoxic brain injury through modulation of inflammatory cytokines and promotion of microglial activation (137). Given BDNF’s protective role in the developing brain, it is possible that downregulation of BDNF could exacerbate schizophrenia risk in the perinatal window.
The Microbiome
Colonization of a newborn neonate gut is normally seeded by the vaginal microbiome during birth, as described, along with maternal vaginal, skin, and oral and fecal bacterial strains (8, 63, 138). These vaginal contributions are transient and by four months post-birth, the infant’s gut microbiome is more similar to the maternal gut microbiome (Figure 2) (139, reviewed in 140). Neonates born via caesarean section lack exposure to the maternal vaginal microbiome and demonstrate a higher prevalence of maternal oral and skin microbes. They are also more likely to develop immune-related disorders (8, 64, 65, 141, 142). Disruptions of the maternal vaginal microbiome via infection, stress, or other pathways may lead to neonatal dysbiosis (65, reviewed in 143). Pre-term birth, caesarean sections, steroid use, and antibiotic use are also associated with dysbiosis in the newborn infant (144, 145).
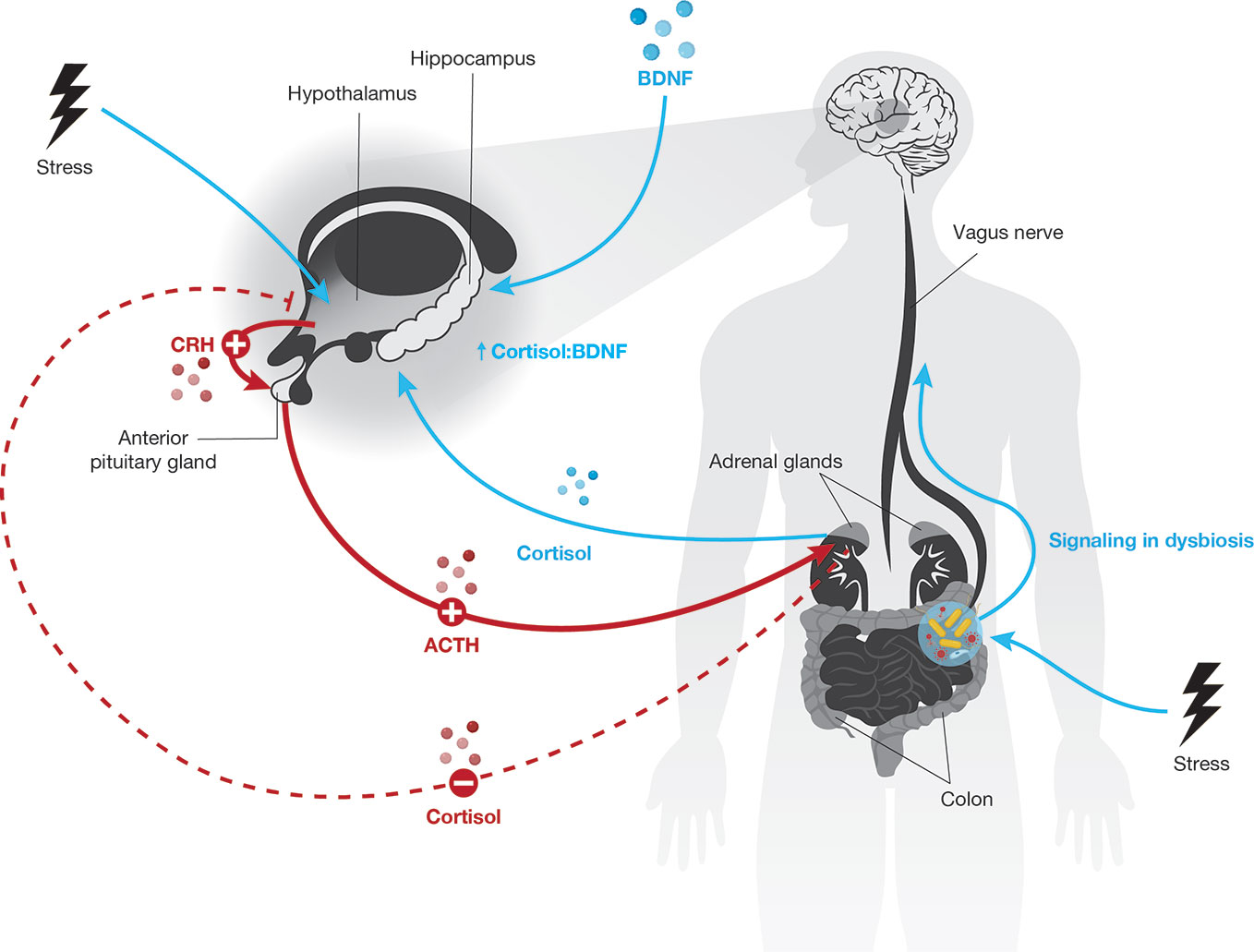
Figure 2 The newborn neonate gut initially contains bacterial strains from the mother’s oral, skin, gut, and vaginal microbiomes. The maternal source of initial colonization varies by the method of fetal delivery, i.e., vaginal birth or caesarean section. Although the newborn neonate gut microbiome stabilizes to resemble the mother’s gut microbiome by about four months of age, this early and transient variability may have long-term impacts on childhood development.
The initial development of the microbiome, including its seeding at birth and development through very early childhood, is important for the development of a healthy core microbiome that is resistant to later perturbation. Given that dysregulation of the microbiome can cause pathogenic inflammation, dysbiosis in the perinatal window may lead to long-term inflammatory dysregulation (146–148). Further studies are needed to determine how maternal flora may influence immune development and schizophrenia risk in their offspring.
Early Childhood
Introduction
Childhood onset of schizophrenia is rare, but a number of neurologic and psychiatric features are already present in childhood. Likewise, a number of traumatic exposures in childhood are associated with schizophrenia risk. The possibility that these presentations reflect the interactions of stress signaling and neurotrophic effects that may be influenced or modulate by the microbiome effects, which is currently being explored.
Signs Across Neurodevelopment
Schizophrenia is highly heterogeneous and no one developmental trajectory can describe the risk pathway for all cases. However, the literature does note certain clusters of behavioral features, including gross motor dysfunction and lower verbal intelligence (149–153). As children develop, personality traits, social behaviors, and mood symptoms may emerge that are more imminently related to the risk for psychosis (reviewed in 154).
During the first two years of life, infants undergo rapid neurodevelopment achieving important motor milestones, like walking, running, pointing, stacking blocks; language milestones, like simple sentences and phrases from a vocabulary of hundreds of words; and social milestones, like beginning self-sufficiency, responding to requests, recognizing self in photographs (reviewed in 155). As failure to achieve milestones raises concern for neurologic development, this developmental stage has been studied in the offspring of affected mothers, in whom a 10% recurrence risk is anticipated. “Pandysmaturation’ was identified as a risk predictor in these “high risk” offspring, which involves a delay in cranial development and visual-motor development and disorganized motor performance (156, 157). Other studies identified passive infants with short attention spans, absence of stranger anxiety, poor communication competence, or abnormal use of language, and lower reactivity as signs of increased schizophrenia risk (158–162).
As children grow into early childhood, they typically become more coordinated, speak fluently, begin to learn reading and writing, and form friendships and social circles. Here again motor difficulties including clumsiness, poor coordination, and poor balance are predictive of higher schizophrenia risk (163, 164). Academically, learning disabilities like dyslexia are associated with higher schizophrenia risk (162). Among children of individuals with schizophrenia, relative decreases in coherence and complexity of language are associated with later schizophrenia risk (165). Socially, isolation, impaired affection, disturbed behavior, hyperactivity, impulsivity, and mood dysregulation including depressive signs and emotional lability are concerning for increased schizophrenia risk (158, 161, 162, 164).
In later childhood before transitioning into adolescence, children continue to improve in athletic, academic, and social behavior. Motor impairment of coordination and balance may become more striking in children with high schizophrenia risk (166–168). Additionally, high risk children may display learning difficulties in attention, concentration, memory, and thought as well as behavioral and mood dysfunctions such as increased aggression, problematic interpersonal relations, social isolation, low self-esteem, offending behaviors, poor affective control, and depression (152, 153, 162, 169–176).
Taken together, childhood impairments in neurologic development, marked by motor, cognitive, and behavioral disturbances, appear be on the trajectory toward schizophrenia, although most children with these features will not become psychotic. Notably, many of these factors entail stress effects on neuroendocrine function and neural plasticity. New research tracking the microbiome over development is showing its role in neurodevelopment and behavioral responses.
Exposure to Trauma
Traumatic experiences, ranging from abuse to accidental injuries, serious infections, and hospitalizations, may increase risk for schizophrenia (reviewed in 177). Trauma that occurs in childhood and adolescence is associated with psychosis and other psychiatric outcomes. Neurobiological studies demonstrate a stress hyporesponsive period in humans during the 6th through 12th postnatal months. Adverse experiences of newborns during this period can have lasting effects on HPA axis modulation (178, 179) from a long term elevation of basal GC secretion. Early life stress (ELS) rodent experiments demonstrate that maternal separation effects on stress sensitivity are mediated through GC-dependent mechanisms (reviewed in 180).
BDNF genetic variants may also influence sensitivity to trauma. There are many variants to the human BDNF gene, however, relatively few common variants fall within coding regions (181). Among these the BDNFVal66Met variant is the most studied overall and has been specifically investigated with regards to schizophrenia risk (reviewed in 182). The BDNFVal66Met polymorphism disrupts episodic memory in humans as a hippocampus-dependent memory function. Extensive studies in both animal models and humans have explored the effects of this polymorphism on numerous psychiatric disorders (reviewed in 183). Regarding schizophrenia risk, the 66Met allele decreases BDNF release probability (184), producing lower efficiency in neurotrophic activity, which is required for neurogenesis and neuroplasticity (185). It is associated with impaired episodic memory and lesser hippocampal activation (186). 66Met carriers with schizophrenia spectrum or bipolar disorders exposed to childhood sexual abuse show reduced grey matter volumes, consistent with the reduced BDNF mRNA levels in 66Met carriers who were exposed to childhood sexual abuse (186).
The higher sensitivity to trauma among 66Met carriers may be explained through the physiopathology of stress-induced changes in neural systems. BDNF plays a key role in neuronal plasticity (32, 187). BDNF-signaling is impaired by ELS; early traumas can evoke significant memory impairments in adulthood in association with reduced BDNF levels (188). This reduction, explained by hypermethylation of the BDNF promoters, can interact with genetic susceptibility, as in the BDNF 66Met carriers (189).
ELS prepares an organism, over the modulation of the HPA axis, for similar adversities during life. This way, a mismatching environment results in an increased susceptibility to psychopathology (131) such as major depression, panic and other co-morbidities. Epigenetics seem to make limbic system structures—mainly the hippocampus and amygdala—more rigid and prone to react depressively and protectively through adulthood. Of clinical significance, a higher occurrence of co-morbidities is usually related to a higher severity of positive and negative symptoms, suicidality, and poorer outcomes (190, 191).
ELS exposure is a negative regulator of BDNF and glucocorticoid receptors (GR) expression in the hippocampus, in the long term, favoring the vulnerability to develop neuropsychiatric disorders, especially upon additional stress exposures (192, 193). An alternative consideration is whether reduced neural capacity leads to a compensatory brain activation that might produce or activate trauma memories. A study of spatial working memory monitored by fMRI found that subjects with schizophrenia had to recruit more cortical regions for the task (194). In this same study, false memory errors were also associated with greater bilateral prefrontal activation. It is plausible that neural strategies to compensate for deficits of perceptual organization, working memory and visuospatial function may lead to a higher recognition of new stimuli as (false) memories. False trauma memory is more frequent among adolescents with posttraumatic stress disorder (PTSD) who experienced childhood sexual abuse (195). Combined, psychosis and childhood sexual abuse may greatly amplify false memories.
It is possible that some of these traumatic experiences are related to PTSD or stress symptoms, as is likely in many cases of abuse. Alternatively, they may be related to direct brain injury, as is likely in many cases of meningitis and encephalitis (196). Most studies examining trauma in schizophrenia risk do not distinguish between events that occur in early childhood versus adolescence, instead identifying events that occur before a determined age (e.g., 16 or 18 years old). However, examination of the timing of trauma suggests that puberty is an important window for distinguishing between anxious and depressive outcomes (197). Future studies examining the timing of traumatic exposures against puberty onset can better elucidate schizophrenia risk in these two populations.
Abuse
History of sexual and physical abuse is strongly correlated with greater psychotic symptom severity among adolescents and young adults in clinical high-risk (CHR) cohorts for schizophrenia. Patients from one such cohort reporting sexual abuse as children or adolescents had increased likelihood of transitioning to psychosis (198). Overall, sexual abuse history is more prevalent in these high-risk individuals than the general population (198–204). Physical abuse is also commonly reported by CHR individuals and may be linked with cognitive defects (205–209). Early physical trauma may lead to hyperarousal of the stress response and chronically elevated cortisol levels (210).
Emotional abuse in childhood, including neglect and maltreatment, has negative effects on mental health (211). Perceived discrimination significantly predicts the transition to psychosis, and emotional trauma and bullying are associated with depression, anxiety, and low self-esteem in CHR individuals (205). These various emotional traumas may impair cognitive function by denying a positive, stimulating environment for the developing brain (212, 213).
Physical and emotional trauma in childhood appears to alter stress response. Adults who reported childhood trauma demonstrate blunted cortisol responses, likely an adaptive response to chronic cortisol elevation (211, 214). In schizophrenia, increased stress sensitivity is a potential causal factor (133, reviewed in 215). Mechanistically, chronically increased cortisol may make the hippocampus vulnerable to injury via cortisol-induced dendritic restructuring or altered cortisol receptor levels (216–221). Cytokines like IL-6 and TNF-α are elevated in children exposed to trauma and can alter cortisol responses (222, 223). Additionally, the BDNF pathway may be relatively inhibited from chronically elevated cortisol, further promoting hippocampal injury and schizophrenia risk (224).
The gut microbiome is influenced by early childhood trauma and likely influences schizophrenia risk in turn (225). Gastrointestinal distress is frequently associated with early adversity in children, and the gut microbiome appears to influence stress programming in animal models (226–229) (reviewed in 230). Recent studies describe altered microbial patterns in children subjected to adversity, with elevations in Lachnospiraceae spp. suggestive of a potential influence on stress sensitivity (231). Additionally, childhood adversity is associated with altered gut microbiota during pregnancy, and may influence observed alterations in inflammatory and GC response to stress, thus contributing to propagation of schizophrenia risk across generations (232). Mechanistically, microglia have an important role in neuroplasticity and neurogenesis and are also sensitive to peripheral inflammation. Gut dysbiosis may negatively influence neurodevelopment through altered microglia activation (228, 233). Future work examining gut microbiome, inflammation, and effects of probiotics in CHR patients may help further elucidate connections between the microbiome, early trauma, and schizophrenia.
Infections
Childhood infections are another important risk factor for schizophrenia onset, especially viral CNS infections (234–236) implicating the microbiome. Childhood infections increase schizophrenia risk in a dose-dependent manner and familial liability for infection also increases schizophrenia risk (237). Additionally, hospitalization for severe infection and even outpatient antibiotic treatment in children are related to increased risk for future psychiatric hospitalizations, suggesting a broad impact of childhood infections on mental health (238).
Mechanistically, direct CNS damage from infection or indirect inflammatory damage may drive the increased schizophrenia risk following childhood infections (238). Antibiotic use in response to infection may also drive risk. Several antibiotics including fluoroquinolones are associated with neurotoxicity and psychosis risk (239). In addition to neurotoxic effects, infections and antibiotics can elevate cortisol levels, potentially affecting the stress cascade (240).
The microbiome also likely influences infection risk in schizophrenia. Studies of germ-free mice show that the gut microbiome primes microglia, stimulating viral specific immunity and reducing viral-driven demyelination via a TLR4-mediated process (241). Dysbiosis driven by antibiotic use or other factors may therefore increase CNS damage from neuroinvasive viruses and thereby increase schizophrenia risk. Interestingly, one study showed antibiotic treatment during adolescence in mice reduced anxiety-like behavior (99). However, cognitive deficits were shown along with reduced hippocampal BDNF and hypothalamic oxytocin and vasopressin expression so the reduction in anxiety-like behavior is suggestive of negative symptoms.
Adolescence
Adolescence is the transition from childhood into adulthood that begins with puberty and ends with cessation of physical growth and neural development in the early 20s (242). Puberty broadly impacts mental health, neuroendocrinology, and the microbiome (reviewed in 243). Neurologically, adolescence encompasses improved abstract thinking, reasoning, and knowledge while also seeing a trend toward increased risk-taking behavior. Schizophrenia most frequently develops during adolescence and young adulthood, and the changes that occur during this developmental stage likely participate in shaping schizophrenia risk. As with early childhood, there are concerning signs and exposures during adolescence that are linked to schizophrenia.
Adolescent Signs
As with early childhood, broad impairments in neuromotor development, cognitive function, and behavior often mark individuals at risk for schizophrenia (reviewed in 154). As the adolescent matures, poor coordination, balance, and perceptual-motor and visual-motor functioning may become more apparent in a subgroup of cases (152, 168, 173). Cognitively, lower intelligence and especially a decrease in intellectual function mark schizophrenia risk (151, 153, 169, 244). There is impairment of individual domains including arithmetic and spelling, formal thought disorders, attention difficulties, increased distractibility, poor executive functioning, and general learning and memory difficulties (152, 153, 169, 173, 245). Behaviorally, aggression, withdrawal, and generally poor social competence and peer relations are also concerning, with psychiatric symptoms including affective flattening and anxiety often present (149, 151, 174, 175, 246, 247).
Risk Exposures
As discussed earlier, studies of exposures do not usually distinguish between pre-pubescent children and post-pubescent adolescents. The aforementioned exposures of sexual, physical, and emotional abuse as well as infection similarly convey schizophrenia risk among adolescents. However, trauma may have different long-term outcomes post-puberty, and its potential effect on schizophrenia risk merits further study. Additionally, the increased risk-taking behavior exhibited at this stage may be influenced by early trauma and influence further trauma exposures. New exposures, such as recreational drug use, may also contribute to schizophrenia risk.
Recreational Drug Use
Recreational drugs exploration is frequent in adolescence and many carry a significant risk for psychosis, particularly cannabis. By their first psychotic episode, approximately half of patients will have a history of cannabis use and one-third meet criteria for cannabis use disorder (248). Alcohol use is similarly high among individuals who have experienced their first psychotic episode, and there is elevated use of cocaine, amphetamine, barbiturate, and other drugs. Cause and effect associations of cannabis and psychosis are well described, although some schizophrenia-susceptible individuals may self-medicate to reduce the anxiety surrounding the presentation of schizophrenia symptoms, with this drug-seeking behavior may further exacerbate their risk for the disorder (249). Chronic exposure to tetrahydrocannabinol (THC), an active ingredient in cannabis, can disrupt neurodevelopmental maturation dependent on endocannabinoid pathways and may lead to overactivation of a pro-hallucinogenic pathway of 5-HT2A receptors, which may promote schizophrenia onset in susceptible individuals (250).
Substance abuse can dysregulate the HPA axis. Alcohol and nicotine use induce cortisol production, and long-term use can cause chronic cortisol elevation and dysregulation similarly to previously described trauma (251–254). Additionally, the gut microbiome is dysregulated by psychostimulants, alcohol, and opioids (255–259) (reviewed in 260). Microbiome influences on addiction are an active area of research. Microglial function is shaped by the microbiome and altered by drugs of abuse (233, 261). Likewise, BDNF dysregulation by dysbiosis is associated with altered behavioral response to cocaine and alcohol (256, 262, 263). While more work is needed to establish causal relationships, these findings suggest multiple ways in which the microbiome may influence addiction behaviors.
Young Adulthood
The transition from adolescence to adulthood occurs during the 20s (242). This transition is typically marked by completion of education and transition to complete independence, which can increase stress in a young adult’s life. Onset of schizophrenia typically occurs around this life transition, peaking at 18 to 25 years old in men and 25 to 35 years old in women, with 80% of cases initially presenting before 40 years of age (264–266). The age of schizophrenia onset may be related to immune activation and stress. Interestingly, inflammatory diseases including inflammatory bowel disease, multiple sclerosis, and some autoimmune diseases tend to initially present in young adulthood (264–266). Gut dysbiosis and cortisol dysregulation are observed in many autoimmune diseases and disruptions to these systems in early adulthood likely influence schizophrenia onset as well (reviewed in 267, 268). First-episode schizophrenia patients have well-documented inflammatory disturbances, such as cytokine elevations and microglial activation (reviewed in 269).
Metabolic disturbances, including glucose intolerance, insulin resistance, and hyperglycemia, also frequently present in this age group and are more common among antipsychotic and naïve first-episode schizophrenic patients compared to the general population (270, 271). These changes may promote schizophrenia onset through persistent inflammatory effects. Stress-related cortisol elevations and gut dysbiosis both contribute to metabolic disturbances, suggesting alternative pathways that influence schizophrenia risk (272, 273). The microbiota also regulate adult neuroplasticity and microglia activation (233, 274).
Aerobic Exercise: A Potentially Protective Factor
While a number of risk factors for schizophrenia are identified, recent evidence points to protective factors. Specifically, aerobic exercise (AE) is hypothesized to play an important protective role against stress induced effects. AE induces a cascade of molecular and cellular processes that support brain plasticity and growth of new vasculature and trigger the processes through which neurotrophins mediate neural plasticity (reviewed in 275–278). Among neurotrophins, BDNF is the most susceptible to regulation by physical exercise (279–281), with synthesis and release into the blood circulation increasing in a dose-response manner (282, 283). Consistent with these findings, Voss et al. (284, 285) found increased connectivity between the bilateral parahippocampus and the bilateral middle temporal gyrus was linked to BDNF increase in AE subjects. A recent meta-analysis (286) of 29 studies (N = 1111 healthy subjects) examined the effect of exercise on BDNF in three exercise paradigms: 1) a single session of exercise; 2) a session of exercise following a program of regular exercise; and 3) resting BDNF levels following a program of regular exercise. Results demonstrated a moderate effect size for increases in BDNF following a single session of exercise (Hedges’ g = .46, p < .001). Further, regular exercise intensified the effect of a session of exercise on BDNF levels (Hedges’ g = .59, p = .02). Finally, results indicated a small effect of regular exercise on resting BDNF levels (Hedges’ g = .27, p = .005). Examination of moderator effects across paradigms found that subjects’ age was not significantly related to changes in BDNF following exercise, but sex significantly moderated the effect of exercise on BDNF levels, such that studies with more women showed less BDNF change resulting from exercise.
Consistent with these reports, findings indicate individuals with schizophrenia tend to have highly sedentary lifestyle characterized by low aerobic fitness which was highly correlated with poor cognitive functioning and symptoms (287). These findings parallel reports among individuals at clinical high risk for psychosis indicating lower levels of fitness, less physical activity, as well as more barriers to exercise (288–292). Yet, a pilot AE RCT indicated engagement in AE led to 11.0% increase aerobic fitness (293) as well as BDNF vs. a 1.9% in the TAU subjects (294) (reviewed in 295). A hierarchical multiple regression analysis indicated that, after controlling for age, sex, changes in anti-psychotic and SSRIs, and changes in menstrual cycle phase, BDNF changes independently predicted changes in cognitive function (b = .38, t = 2.06, p = .05) (296). Notably, improvements in cognitive functioning were associated with intensity of AE activity (294).
Exercise alters the composition and functional capacity of the gut microbiome independent of diet (reviewed by 28). As the effects of AE on BDNF production are further studied in schizophrenia, examination of how the microbiome influences this pathway may be illuminating.
Potential Mechanisms
Although some stress exposure is essential for growth and development, stress that overwhelms adaptive capacities has adverse physiological consequences, as initially described in 1938 by Hans Selye (297). The initial stress axis model included direct and feedback interactions among the hypothalamus (release of corticotropin-releasing factor), pituitary (ACTH), and adrenal glands (cortisol), which was then expanded by Sapolsky’s “glucocorticoid cascade hypothesis” (298) to encompass catecholamines and other interacting mediators of adaptation in addition to GCs. This model must now be widened to include the central influence of the microbiota on the initial programming of the stress axis and ongoing bidirectional effects that influence stress responding. The communication pathways between the gut and brain includes the vagal nerve, through which some microbial species invoke anxiolytic effects of some species (299). Enteroendocrine cells secrete biologically active peptides, including galanin, which stimulates the central HPA axis leading to increased adrenal cortisol secretion, and ghrelin which has similar effects linked to nutritional and metabolic conditions (300, 301) (reviewed in 302, 303). Reciprocally, even short durations of stress impact the relative proportions of phyla in the microbiota mediated through neuroendocrine and autonomic nervous system activity (304). The neuro-immuno-endocrine pathways linking the gut and brain include afferent and efferent neural pathways, immune effects, bi-directional neuroendocrine signaling and by alterations in intestinal permeability, critically influenced by relative proportions of microbiota species, as shown in Figure 1.
Examined as a whole, broad pathways through which the gut may influence stress and schizophrenia risk include cytokine-driven global inflammatory modifications, stress hormone metabolism, microglial activation, neuroplastic regulation, direct infection, and other nervous system activity as described above. Given schizophrenia risks at key developmental stages also coincide with microbiome development and associated changes, examining these pathways across development may be especially poignant. During the perinatal period, as the brain and HPA axis develop, dysbiosis in mother and child is influenced by multiple factors including infection and stress and in turn may influence the brain and HPA axis. As the child continues to grow and develop, the microbiome continues to adapt and change. While stressors including psychic and physical trauma, recreational substance use, inflammatory diseases, metabolic disturbances, and AE have been previously understood in context of neuroendocrine pathways, these events also affect the microbiome which in turn likely feed back into stress and neurodevelopment pathways. When viewed as one interconnected system, the ways microbial, endocrine, and neurological pathways influence each other across development should improve our understanding of schizophrenia risk and perhaps offer novel treatment methods. While current knowledge rests largely on germ free, antibiotic treated or probiotic supplemented animal models, the field is finally advancing to human studies.
Conclusion
Our understanding of schizophrenia risk has evolved over the past century as technological improvements have made better research methods possible. Recent decades demonstrate the profound impact that neuroendocrine pathways have on schizophrenia risk across human development. The microbiome represents one of the newest frontiers in research that is broadly impacting healthcare. Recent work has already demonstrated many interactions between schizophrenia risk, neuroendocrinology, and the microbiome, but there are unexplored areas throughout development where further interactions likely occur. Thus, future work examining schizophrenia risk must continue to incorporate the crosstalk between the neuroendocrine pathways and the microbiome.
Author Contributions
KH, JL, CC, DK, TK, and DM all contributed to writing and editing manuscript.
Funding
This work is supported by NIMH R01 MH110623 and P50MH115843 (DK); NIMH R01 MH107558 and R01 MH115332 (CC); NIMH R01 MH110418 (DM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, et al. Structure, function and diversity of the healthy human microbiome. Nature (2012) 486:207–14. doi: 10.1038/nature11234
2. Kamada N, Seo SU, Chen GY, Núñez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol (2013) 13:321–35. doi: 10.1038/nri3430
3. Milani C, Duranti S, Bottacini F, Casey E, Turroni F, Mahony J, et al. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol Mol Biol Rev (2017) 81:e00036–17. doi: 10.1128/MMBR.00036-17
4. Rodríguez JM, Murphy K, Stanton C, Ross RP, Kober OI, Juge N, et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb Ecol Health Dis (2015) 26:26050. doi: 10.3402/mehd.v26.26050
5. Moore RE, Townsend SD. Temporal development of the infant gut microbiome. Open Biol (2019) 9:190128. doi: 10.1098/rsob.190128
6. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature (2012) 486:222–7. doi: 10.1038/nature11053
7. Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U.S.A. (2011) 108:4578–85. doi: 10.1073/pnas.1000081107
8. Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U.S.A. (2010) 107:11971–5. doi: 10.1073/pnas.1002601107
9. Stewart CJ, Ajami NJ, O’Brien JL, Hutchinson DS, Smith DP, Wong MC, et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature (2018) 562:583–8. doi: 10.1038/s41586-018-0617-x
10. Cryan JF, Dinan TG. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci (2012) 13:701–12. doi: 10.1038/nrn3346
11. Bravo JA, Forsythe P, Chew MV, Escaravage E, Savignac HM, Dinan TG, et al. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci U.S.A. (2011) 108:16050–5. doi: 10.1073/pnas.1102999108
12. Jiang H, Ling Z, Zhang Y, Mao H, Ma Z, Yin Y, et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav Immun (2015) 48:186–94. doi: 10.1016/j.bbi.2015.03.016
13. Naseribafrouei A, Hestad K, Avershina E, Sekelja M, Linløkken A, Wilson R, et al. Correlation between the human fecal microbiota and depression. Neurogastroenterol Motil (2014) 26:1155–62. doi: 10.1111/nmo.12378
14. Neufeld KM, Kang N, Bienenstock J, Foster JA. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol Motil (2011) 23:255–64. doi: 10.1111/j.1365-2982.2010.01620.x
15. Simreń M, Barbara G, Flint HJ, Spiegel BMR, Spiller RC, Vanner S, et al. Intestinal microbiota in functional bowel disorders: A Rome foundation report. Gut (2013) 62:159–76. doi: 10.1136/gutjnl-2012-302167
16. Song Y, Liu C, Finegold SM. Real-time PCR quantitation of clostridia in feces of autistic children. Appl Environ Microbiol (2004) 70:6459–65. doi: 10.1128/AEM.70.11.6459-6465.2004
17. Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu XN, et al. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol (2004) 558:263–75. doi: 10.1113/jphysiol.2004.063388
18. Daskalakis NP, Cohen H, Cai G, Buxbaum JD, Yehuda R. Expression profiling associates blood and brain glucocorticoid receptor signaling with trauma-related individual differences in both sexes. Proc Natl Acad Sci U.S.A. (2014) 111:13529–34. doi: 10.1073/pnas.1401660111
19. Yehuda R, Daskalakis NP, Lehrner A, Desarnaud F, Bader HN, Makotkine I, et al. Influences of maternal and paternal PTSD on epigenetic regulation of the glucocorticoid receptor gene in Holocaust survivor offspring. Am J Psychiatry (2014) 171:872–80. doi: 10.1176/appi.ajp.2014.13121571
20. Ruby E, Rothman K, Corcoran C, Goetz RR, Malaspina D. Influence of early trauma on features of schizophrenia. Early Interv Psychiatry (2017) 11:322–33. doi: 10.1111/eip.12239
21. Lawrence MS, Sapolsky RM. Glucocorticoids accelerate ATP loss following metabolic insults in cultured hippocampal neurons. Brain Res (1994) 646:303–6. doi: 10.1016/0006-8993(94)90094-9
22. Sapolsky RM. Why stress is bad for your brain. Science (80-) (1996) 273:749–50. doi: 10.1126/science.273.5276.749
23. Branchi I, Karpova NN, D’Andrea I, Castrén E, Alleva E. Epigenetic modifications induced by early enrichment are associated with changes in timing of induction of BDNF expression. Neurosci Lett (2011) 495:168–72. doi: 10.1016/j.neulet.2011.03.038
24. Bremner JD, Vythilingam M, Vermetten E, Southwick SM, McGlashan T, Nazeer A, et al. MRI and PET study of deficits in hippocampal structure and function in women with childhood sexual abuse and posttraumatic stress disorder. Am J Psychiatry (2003) 160:924–32. doi: 10.1176/appi.ajp.160.5.924
25. Osimo EF, Beck K, Reis Marques T, Howes OD. Synaptic loss in schizophrenia: a meta-analysis and systematic review of synaptic protein and mRNA measures. Mol Psychiatry (2019) 24:549–61. doi: 10.1038/s41380-018-0041-5
26. McHugo M, Talati P, Armstrong K, Vandekar SN, Blackford JU, Woodward ND, et al. Hyperactivity and Reduced Activation of Anterior Hippocampus in Early Psychosis. Am J Psychiatry (2019) 176:1030–8. doi: 10.1176/appi.ajp.2019.19020151
27. Velakoulis D, Wood SJ, Wong MTH, McGorry PD, Yung A, Phillips L, et al. Hippocampal and amygdala volumes according to psychosis stage and diagnosis: A magnetic resonance imaging study of chronic schizophrenia, first-episode psychosis, and ultra-high-risk individuals. Arch Gen Psychiatry (2006) 63:139–49. doi: 10.1001/archpsyc.63.2.139
28. Mailing LJ, Allen JM, Buford TW, Fields CJ, Woods JA. Exercise and the Gut Microbiome. Exerc Sport Sci Rev (2019) 47:75–85. doi: 10.1249/JES.0000000000000183
29. De Kloet ER, Joëls M, Holsboer F. Stress and the brain: From adaptation to disease. Nat Rev Neurosci (2005) 6:463–75. doi: 10.1038/nrn1683
30. de Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M. Brain Corticosteroid Receptor Balance in Health and Disease 1. Endocr Rev (1998) 19:269–301. doi: 10.1210/edrv.19.3.0331
31. McEWEN BS. Stress, Adaptation, and Disease: Allostasis and Allostatic Load. Ann N Y Acad Sci (1998) 840:33–44. doi: 10.1111/j.1749-6632.1998.tb09546.x
32. Ghosh A, Greenberg ME. Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron (1995) 15:89–103. doi: 10.1016/0896-6273(95)90067-5
33. Bekinschtein P, Cammarota M, Katche C, Slipczuk L, Rossato JI, Goldin A, et al. BDNF is essential to promote persistence of long-term memory storage. Proc Natl Acad Sci U.S.A. (2008) 105:2711–6. doi: 10.1073/pnas.0711863105
34. Jeanneteau FD, Lambert WM, Ismaili N, Bath KG, Lee FS, Garabedian MJ, et al. BDNF and glucocorticoids regulate corticotrophin-releasing hormone (CRH) homeostasis in the hypothalamus. Proc Natl Acad Sci U.S.A. (2012) 109:1305–10. doi: 10.1073/pnas.1114122109
35. Jeanneteau F, Chao MV. Are BDNF and glucocorticoid activities calibrated? Neuroscience (2013) 239:173–95. doi: 10.1016/j.neuroscience.2012.09.017
36. Fröhlich EE, Farzi A, Mayerhofer R, Reichmann F, Jačan A, Wagner B, et al. Cognitive impairment by antibiotic-induced gut dysbiosis: Analysis of gut microbiota-brain communication. Brain Behav Immun (2016) 56:140–55. doi: 10.1016/j.bbi.2016.02.020
37. Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, et al. Type i interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med (2016) 22:586–97. doi: 10.1038/nm.4106
38. Serrats J, Schiltz JC, García-Bueno B, van Rooijen N, Reyes TM, Sawchenko PE. Dual Roles for Perivascular Macrophages in Immune-to-Brain Signaling. Neuron (2010) 65:94–106. doi: 10.1016/j.neuron.2009.11.032
39. Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U.S.A. (2014) 111:2247–52. doi: 10.1073/pnas.1322269111
40. Farzi A, Reichmann F, Meinitzer A, Mayerhofer R, Jain P, Hassan AM, et al. Synergistic effects of NOD1 or NOD2 and TLR4 activation on mouse sickness behavior in relation to immune and brain activity markers. Brain Behav Immun (2015) 44:106–20. doi: 10.1016/j.bbi.2014.08.011
41. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504:446–50. doi: 10.1038/nature12721
42. Mayerhofer R, Fröhlich EE, Reichmann F, Farzi A, Kogelnik N, Fröhlich E, et al. Diverse action of lipoteichoic acid and lipopolysaccharide on neuroinflammation, blood-brain barrier disruption, and anxiety in mice. Brain Behav Immun (2017) 60:174–87. doi: 10.1016/j.bbi.2016.10.011
43. Usami M, Kishimoto K, Ohata A, Miyoshi M, Aoyama M, Fueda Y, et al. Butyrate and trichostatin A attenuate nuclear factor κB activation and tumor necrosis factor α secretion and increase prostaglandin E2 secretion in human peripheral blood mononuclear cells. Nutr Res (2008) 28:321–8. doi: 10.1016/j.nutres.2008.02.012
44. De Punder K, Pruimboom L. Stress induces endotoxemia and low-grade inflammation by increasing barrier permeability. Front Immunol (2015) 6:223. doi: 10.3389/fimmu.2015.00223
46. Stilling RM, Ryan FJ, Hoban AE, Shanahan F, Clarke G, Claesson MJ, et al. Microbes & neurodevelopment - Absence of microbiota during early life increases activity-related transcriptional pathways in the amygdala. Brain Behav Immun (2015) 50:209–20. doi: 10.1016/j.bbi.2015.07.009
47. Roceri M, Hendriks W, Racagni G, Ellenbroek BA, Riva MA. Early maternal deprivation reduces the expression of BDNF and NMDA receptor subunits in rat hippocampus. Mol Psychiatry (2002) 7:609–16. doi: 10.1038/sj.mp.4001036
48. Ait-Belgnaoui A, Durand H, Cartier C, Chaumaz G, Eutamene H, Ferrier L, et al. Prevention of gut leakiness by a probiotic treatment leads to attenuated HPA response to an acute psychological stress in rats. Psychoneuroendocrinology (2012) 37:1885–95. doi: 10.1016/j.psyneuen.2012.03.024
49. Scheer S, Medina TS, Murison A, Taves MD, Antignano F, Chenery A, et al. Early-life antibiotic treatment enhances the pathogenicity of CD4+ T cells during intestinal inflammation. J Leukoc Biol (2017) 101:893–900. doi: 10.1189/jlb.3MA0716-334RR
50. Liu S, Guo R, Liu F, Yuan Q, Yu Y, Ren F. Gut microbiota regulates depression-like behavior in rats through the neuroendocrine-immune-mitochondrial pathway. Neuropsychiatr Dis Treat (2020) 16:859–69. doi: 10.2147/NDT.S243551
51. Jochems J, Teegarden SL, Chen Y, Boulden J, Challis C, Ben-Dor GA, et al. Enhancement of stress resilience through histone deacetylase 6-mediated regulation of glucocorticoid receptor chaperone dynamics. Biol Psychiatry (2015) 77:345–55. doi: 10.1016/j.biopsych.2014.07.036
52. Hartmann J, Wagner KV, Liebl C, Scharf SH, Wang XD, Wolf M, et al. The involvement of FK506-binding protein 51 (FKBP5) in the behavioral and neuroendocrine effects of chronic social defeat stress. Neuropharmacology (2012) 62(1):332–9. doi: 10.1016/j.neuropharm.2011.07.041
53. Ridder S, Chourbaji S, Hellweg R, Urani A, Zacher C, Schmid W, et al. Mice with genetically altered glucocorticoid receptor expression show altered sensitivity for stress-induced depressive reactions. J Neurosci (2005) 25:6243–50. doi: 10.1523/JNEUROSCI.0736-05.2005
54. Smith GW, Aubry JM, Dellu F, Contarino A, Bilezikjian LM, Gold LH, et al. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron (1998) 20:1093–102. doi: 10.1016/S0896-6273(00)80491-2
55. Faye C, Mcgowan JC, Denny CA, David DJ. Neurobiological Mechanisms of Stress Resilience and Implications for the Aged Population. Curr Neuropharmacol (2018) 16:234–70. doi: 10.2174/1570159x15666170818095105
56. Saulnier DM, Ringel Y, Heyman MB, Foster JA, Bercik P, Shulman RJ, et al. The intestinal microbiome, probiotics and prebiotics in neurogastroenterology. Gut Microbes (2013) 4:17–27. doi: 10.4161/gmic.22973
57. Southwick S, Vythilingam M, Charney D. The Psychobiology of Depression and Resilience to Stress: Implications for Prevention and Treatment. Annu Rev Clin Psychol (2005) 1:255–91. doi: 10.1146/annurev.clinpsy.1.102803.143948
58. Davis DJ, Hecht PM, Jasarevic E, Beversdorf DQ, Will MJ, Fritsche K, et al. Sex-specific effects of docosahexaenoic acid (DHA) on the microbiome and behavior of socially-isolated mice. Brain Behav Immun (2017) 59:38–48. doi: 10.1016/j.bbi.2016.09.003
59. Pusceddu MM, El Aidy S, Crispie F, O’Sullivan O, Cotter P, Stanton C, et al. N-3 polyunsaturated fatty acids (PUFAs) reverse the impact of early-life stress on the gut microbiota. PloS One (2015) 10:e0139721. doi: 10.1371/journal.pone.0139721
60. Rosanoff AJ, Handy LM, Plesset IR, Brush S. The etiology of so-called schizophrenic psychoses. Am J Psychiatry (1934) 91:247–86. doi: 10.1176/ajp.91.2.247
61. Brosnahan AJ, Vulchanova L, Witta SR, Dai Y, Jones BJ, Brown DR. Norepinephrine potentiates proinflammatory responses of human vaginal epithelial cells. J Neuroimmunol (2013) 259:8–16. doi: 10.1016/j.jneuroim.2013.03.005
62. Jašarević E, Howerton CL, Howard CD, Bale TL. Alterations in the vaginal microbiome by maternal stress are associated with metabolic reprogramming of the offspring gut and brain. Endocrinology (2015) 156:3265–76. doi: 10.1210/en.2015-1177
63. Ferretti P, Pasolli E, Tett A, Asnicar F, Gorfer V, Fedi S, et al. Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host Microbe (2018) 24:133–145.e5. doi: 10.1016/j.chom.2018.06.005
64. Borre YE, Moloney RD, Clarke G, Dinan TG, Cryan JF. The impact of microbiota on brain and behavior: Mechanisms & therapeutic potential. Adv Exp Med Biol (2014) 817:373–403. doi: 10.1007/978-1-4939-0897-4_17
65. Hočevar K, Maver A, Vidmar Šimic M, Hodžić A, Haslberger A, Premru Seršen T, et al. Vaginal Microbiome Signature Is Associated With Spontaneous Preterm Delivery. Front Med (2019) 6:201. doi: 10.3389/fmed.2019.00201
66. Ly NP, Ruiz-Pérez B, Onderdonk AB, Tzianabos AO, Litonjua AA, Liang C, et al. Mode of delivery and cord blood cytokines: A birth cohort study. Clin Mol Allergy (2006) 4:13. doi: 10.1186/1476-7961-4-13
67. Sudo N, Sawamura S, Tanaka K, Aiba Y, Kubo C, Koga Y. The requirement of intestinal bacterial flora for the development of an IgE production system fully susceptible to oral tolerance induction. J Immunol (1997) 159:1739–45.
68. Bager P, Wohlfahrt J, Westergaard T. Caesarean delivery and risk of atopy and allergic disesase: Meta-analyses. Clin Exp Allergy (2008) 38:634–42. doi: 10.1111/j.1365-2222.2008.02939.x
69. Kummeling I, Stelma FF, Dagnelie PC, Snijdersa BEP, Penders J, Huber M, et al. Early life exposure to antibiotics and the subsequent development of eczema, wheeze, and allergic sensitization in the first 2 years of life: The KOALA Birth Cohort Study. Pediatrics (2007) 119:e225–31. doi: 10.1542/peds.2006-0896
70. Souza DG, Vieira AT, Soares AC, Pinho V, Nicoli JR, Vieira LQ, et al. The Essential Role of the Intestinal Microbiota in Facilitating Acute Inflammatory Responses. J Immunol (2004) 173:4137–46. doi: 10.4049/jimmunol.173.6.4137
71. Mednick SA, Machon RA, Huttunen MO, Bonett D. Adult Schizophrenia Following Prenatal Exposure to an Influenza Epidemic. Arch Gen Psychiatry (1988) 45:189–92. doi: 10.1001/archpsyc.1988.01800260109013
72. Byrne M, Agerbo E, Bennedsen B, Eaton WW, Mortensen PB. Obstetric conditions and risk of first admission with schizophrenia: a Danish national register based study. Schizophr Res (2007) 97:51–9. doi: 10.1016/j.schres.2007.07.018
73. Machón RA, Mednick SA, Huttunen MO. Adult major affective disorder after prenatal exposure to an influenza epidemic. Arch Gen Psychiatry (1997) 54:322–8. doi: 10.1001/archpsyc.1997.01830160040006
74. Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, et al. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry (2004) 61:774–80. doi: 10.1001/archpsyc.61.8.774
75. Boksa P. Maternal infection during pregnancy and schizophrenia. J Psychiatry Neurosci (2008) 33:183–5.
76. Brown AS, Cohen P, Harkavy-Friedman J, Babulas V, Malaspina D, Gorman JM, et al. Prenatal rubella, premorbid abnormalities, and adult schizophrenia. Biol Psychiatry (2001) 49:473–86. doi: 10.1016/S0006-3223(01)01068-X
77. Mortensen PB, Nørgaard-Pedersen B, Waltoft BL, Sørensen TL, Hougaard D, Torrey EF, et al. Toxoplasma gondii as a Risk Factor for Early-Onset Schizophrenia: Analysis of Filter Paper Blood Samples Obtained at Birth. Biol Psychiatry (2007) 61:688–93. doi: 10.1016/j.biopsych.2006.05.024
78. Aronsson F, Lannebo C, Paucar M, Brask J, Kristensson K, Karlsson H. Persistence of viral RNA in the brain of offspring to mice infected with influenza A/WSN/33 virus during pregnancy. J Neurovirol (2002) 8:353–7. doi: 10.1080/13550280290100480
79. Moustaki M, Tsabouri S, Priftis KN, Douros K. Prenatal Stress Enhances Susceptibility to Allergic Diseases of Offspring. Endocrine Metab Immune Disord Drug Targets (2017) 17:255–63. doi: 10.2174/1871530317666170912160646
80. Mac Giollabhui N, Breen EC, Murphy SK, Maxwell SD, Cohn BA, Krigbaum NY, et al. Maternal inflammation during pregnancy and offspring psychiatric symptoms in childhood: Timing and sex matter. J Psychiatr Res (2019) 111:96–103. doi: 10.1016/j.jpsychires.2019.01.009
81. Khan VR, Brown IR. The effect of hyperthermia on the induction of cell death in brain, testis, and thymus of the adult and developing rat. Cell Stress Chaperones (2002) 7:73–90. doi: 10.1379/1466-1268(2002)007<0073:TEOHOT>2.0.CO;2
82. Sørensen HJ, Mortensen EL, Reinisch JM, Mednick SA. Association between prenatal exposure to analgesics and risk of schizophrenia. Br J Psychiatry (2004) 185:366–71. doi: 10.1192/bjp.185.5.366
83. Wright P, Takei N, Rifkin L, Murray RM. Maternal influenza, obstetric complications, and schizophrenia. Am J Psychiatry (1995) 152:1714–20. doi: 10.1176/ajp.152.12.1714
84. Bender JM, Li F, Martelly S, Byrt E, Rouzier V, Leo M, et al. Maternal HIV infection influences the microbiome of HIV-uninfected infants. Sci Transl Med (2016) 8:349ra100. doi: 10.1126/scitranslmed.aaf5103
85. Davis EP, Sandman CA. The timing of prenatal exposure to maternal cortisol and psychosocial stress is associated with human infant cognitive development. Child Dev (2010) 81:131–48. doi: 10.1111/j.1467-8624.2009.01385.x
86. Huttunen MO, Niskanen P. Prenatal Loss of Father and Psychiatric Disorders. Arch Gen Psychiatry (1978) 35:429–31. doi: 10.1001/archpsyc.1978.01770280039004
87. Malaspina D, Corcoran C, Kleinhaus KR, Perrin MC, Fennig S, Nahon D, et al. Acute maternal stress in pregnancy and schizophrenia in offspring: A cohort prospective study. BMC Psychiatry (2008) 8:71. doi: 10.1186/1471-244X-8-71
88. Van Os J, Selten J-P. Prenatal exposure to maternal stress and subsequent schizophrenia. Br J Psychiatry (1998) 172:324–6. doi: 10.1192/bjp.172.4.324
89. Meijer A. Child psychiatric sequelae of maternal war stress. Acta Psychiatr Scand (1985) 72:505–11. doi: 10.1111/j.1600-0447.1985.tb02647.x
90. McGrath J. Hypothesis: Is low prenatal vitamin D a risk-modifying factor for schizophrenia? Schizophr Res (1999) 40:173–7. doi: 10.1016/S0920-9964(99)00052-3
91. Brown AS, Bottiglieri T, Schaefer CA, Quesenberry CP, Liu L, Bresnahan M, et al. Elevated prenatal homocysteine levels as a risk factor for schizophrenia. Arch Gen Psychiatry (2007) 64:31–9. doi: 10.1001/archpsyc.64.1.31
92. Xu MQ, Sun WS, Liu BX, Feng GY, Yu L, Yang L, et al. Prenatal malnutrition and adult Schizophrenia: Further evidence from the 1959-1961 chinese famine. Schizophr Bull (2009) 35:568–76. doi: 10.1093/schbul/sbn168
93. St Clair D, Xu M, Wang P, Yu Y, Fang Y, Zhang F, et al. Rates of adult schizophrenia following prenatal exposure to the Chinese famine of 1959-1961. J Am Med Assoc (2005) 294:557–62. doi: 10.1001/jama.294.5.557
94. Susser E, Neugebauer R, Hoek HW, Brown AS, Lin S, Labovitz D, et al. Schizophrenia after prenatal famine further evidence. Arch Gen Psychiatry (1996) 53:25–31. doi: 10.1001/archpsyc.1996.01830010027005
95. Rakers F, Rupprecht S, Dreiling M, Bergmeier C, Witte OW, Schwab M. Transfer of maternal psychosocial stress to the fetus. Neurosci Biobehav Rev (Forthcoming 2016). doi: 10.1016/j.neubiorev.2017.02.019
96. Lu A, Petrullo L, Carrera S, Feder J, Schneider-Crease I, Snyder-Mackler N. Developmental responses to early-life adversity: Evolutionary and mechanistic perspectives. Evol Anthropol (2019) 28:249–66. doi: 10.1002/evan.21791
97. Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain Behav Immun (2011) 25:397–407. doi: 10.1016/j.bbi.2010.10.023
98. Clarke G, Grenham S, Scully P, Fitzgerald P, Moloney RD, Shanahan F, et al. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol Psychiatry (2013) 18:666–73. doi: 10.1038/mp.2012.77
99. Desbonnet L, Clarke G, Traplin A, O’Sullivan O, Crispie F, Moloney RD, et al. Gut microbiota depletion from early adolescence in mice: Implications for brain and behaviour. Brain Behav Immun (2015) 48:165–73. doi: 10.1016/j.bbi.2015.04.004
100. Golubeva AV, Crampton S, Desbonnet L, Edge D, O’Sullivan O, Lomasney KW, et al. Prenatal stress-induced alterations in major physiological systems correlate with gut microbiota composition in adulthood. Psychoneuroendocrinology (2015) 60:58–74. doi: 10.1016/j.psyneuen.2015.06.002
101. Hyland NP, O’Mahony SM, O’Malley D, O’Mahony CM, Dinan TG, Cryan JF. Early-life stress selectively affects gastrointestinal but not behavioral responses in a genetic model of brain-gut axis dysfunction. Neurogastroenterol Motil (2015) 27:105–13. doi: 10.1111/nmo.12486
102. Jašarević E, Howard CD, Morrison K, Misic A, Weinkopff T, Scott P, et al. The maternal vaginal microbiome partially mediates the effects of prenatal stress on offspring gut and hypothalamus. Nat Neurosci (2018) 21:1061–71. doi: 10.1038/s41593-018-0182-5
103. Zaneveld JR, McMinds R, Thurber RV. Stress and stability: Applying the Anna Karenina principle to animal microbiomes. Nat Microbiol (2017) 2:1–8. doi: 10.1038/nmicrobiol.2017.121
104. Gur TL, Shay L, Palkar AV, Fisher S, Varaljay VA, Dowd S, et al. Prenatal stress affects placental cytokines and neurotrophins, commensal microbes, and anxiety-like behavior in adult female offspring. Brain Behav Immun (2017) 64:50–8. doi: 10.1016/j.bbi.2016.12.021
105. Soto M, Herzog C, Pacheco JA, Fujisaka S, Bullock K, Clish CB, et al. Gut microbiota modulate neurobehavior through changes in brain insulin sensitivity and metabolism. Mol Psychiatry (2018) 23:2287–301. doi: 10.1038/s41380-018-0086-5
106. Scott LV, Clarke G, Dinan TG. The brain-gut axis: A target for treating stress-related disorders. Inflammation Psychiatry (2013) 28:90–9. doi: 10.1159/000343971
107. Heijtz RD, Wang S, Anuar F, Qian Y, Björkholm B, Samuelsson A, et al. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci U.S.A. (2011) 108:3047–52. doi: 10.1073/pnas.1010529108
108. Macpherson AJ, Hunziker L, McCoy K, Lamarre A. IgA responses in the intestinal mucosa against pathogenic and non-pathogenic microorganisms. Microbes Infect (2001) 3:1021–35. doi: 10.1016/S1286-4579(01)01460-5
109. Moreau MC, Ducluzeau R, Guy-Grand D, Muller MC. Increase in the population of duodenal immunoglobulin A plasmocytes in axenic mice associated with different living or dead bacterial strains of intestinal origin. Infect Immun (1978) 21:532–9. doi: 10.1128/iai.21.2.532-539.1978
110. Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci U.S.A. (2008) 105:16767–72. doi: 10.1073/pnas.0808567105
111. McNeil TF. Obstetric factors and perinatal injuries. In: Tsuang MF, Simpson JC, editors. Handbook of Schizophrenia, Vol. 3: Nosology, Epidemiology and Genetics. New York: Elsevier (1988) p. 319–44.
112. Buka SL, Tsuang MT, Lipsitt LP. Pregnancy/Delivery Complications and Psychiatric Diagnosis: A Prospective Study. Arch Gen Psychiatry (1993) 50:151–6. doi: 10.1001/archpsyc.1993.01820140077009
113. Dalman C, Thomas HV, David AS, Gentz J, Lewis G, Allebeck P. Signs of asphyxia at birth and risk of schizophrenia: Population-based case-control study. Br J Psychiatry (2001) 179:403–8. doi: 10.1192/bjp.179.5.403
114. Zornberg GL, Buka SL, Tsuang MT. Hypoxic-ischemia-related fetal/neonatal complications and risk of schizophrenia and other nonaffective psychoses: A 19-year longitudinal study. Am J Psychiatry (2000) 157:196–202. doi: 10.1176/appi.ajp.157.2.196
115. Geddes JR, Verdoux H, Takei N, Lawrie SM, Bovet P, Eagles JM, et al. Schizophrenia and complications of pregnancy and labor: An individual patient data meta-analysis. Schizophr Bull (1999) 25:413–23. doi: 10.1093/oxfordjournals.schbul.a033389
116. Rosso IM, Cannon TD, Huttunen T, Huttunen MO, Lönnqvist J, Gasperoni TL. Obstetric risk factors for early-onset schizophrenia in a Finnish birth cohort. Am J Psychiatry (2000) 157:801–7. doi: 10.1176/appi.ajp.157.5.801
117. Cannon TD, Van Erp TGM, Rosso IM, Huttunen M, Lönnqvist J, Pirkola T, et al. Fetal hypoxia and structural brain abnormalities in schizophrenic patients, their siblings, and controls. Arch Gen Psychiatry (2002) 59:35–41. doi: 10.1001/archpsyc.59.1.35
118. Van Erp TGM, Saleh PA, Rosso IM, Huttunen M, Lönnqvist J, Pirkola T, et al. Contributions of genetic risk and fetal hypoxia to hippocampal volume in patients with schizophrenia or schizoaffective disorder, their unaffected siblings, and healthy unrelated volunteers. Am J Psychiatry (2002) 159:1514–20. doi: 10.1176/appi.ajp.159.9.1514
119. Moreno-Indias I, Torres M, Montserrat JM, Sanchez-Alcoholado L, Cardona F, Tinahones FJ, et al. Intermittent hypoxia alters gut microbiota diversity in a mouse model of sleep apnoea. Eur Respir J (2015) 45:1055–65. doi: 10.1183/09031936.00184314
120. Tripathi A, Melnik AV, Xue J, Poulsen O, Meehan MJ, Humphrey G, et al. Intermittent Hypoxia and Hypercapnia, a Hallmark of Obstructive Sleep Apnea, Alters the Gut Microbiome and Metabolome. mSystems (2018) 3:e00020–18. doi: 10.1128/msystems.00020-18
121. Zarate MA, Rodriguez MD, Chang EI, Russell JT, Arndt TJ, Richards EM, et al. Post-hypoxia Invasion of the fetal brain by multidrug resistant Staphylococcus. Sci Rep (2017) 7:6458–68. doi: 10.1038/s41598-017-06789-6
122. Lane EA, Albee GW. Comparative Birth Weights of Schizophrenics and Their Siblings. J Psychol Interdiscip Appl (1966) 64:227–31. doi: 10.1080/00223980.1966.10544847
123. Nilsson E, Stålberg G, Lichtenstein P, Cnattingius S, Olausson PO, Hultman CM. Fetal growth restriction and schizophrenia: A Swedish twin study. Twin Res Hum Genet (2005) 8:402–8. doi: 10.1375/1832427054936727
124. Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: Historical and meta-analytic review. Am J Psychiatry (2002) 159:1080–92. doi: 10.1176/appi.ajp.159.7.1080
125. Pasamanick B, Rogers ME, Lilienfeld AM. Pregnancy experience and the development of behavior disorders in children. Am J Psychiatry (1956) 112:613–8. doi: 10.1176/ajp.112.8.613
126. Schaefer CA, Brown AS, Wyatt RJ, Kline J, Begg MD, Bresnahan MA, et al. Maternal prepregnant body mass and risk of schizophrenia in adult offspring. Schizophr Bull (2000) 26:275–86. doi: 10.1093/oxfordjournals.schbul.a033452
127. Verdoux H, Geddes JR, Takei N, Lawrie SM, Bovet P, Eagles JM, et al. Obstetric complications and age at onset in schizophrenia: An international collaborative meta-analysis of individual patient data. Am J Psychiatry (1997) 154:1220–7. doi: 10.1176/ajp.154.9.1220
128. Clarke MC, Harley M, Cannon M. The role of obstetric events in schizophrenia. Schizophr Bull (2006) 32:3–8. doi: 10.1093/schbul/sbj028
129. Wood CE, Keller-Wood M. The critical importance of the fetal hypothalamus-pituitary-adrenal axis. F1000Research (2016) 5:115–22. doi: 10.12688/f1000research.7224.1
130. Shams M, Kilby MD, Somerset DA, Howie AJ, Gupta A, Wood PJ, et al. 11β-hydroxysteroid dehydrogenase type 2 in human pregnancy and reduced expression in intrauterine growth restriction. Hum Reprod (1998) 13(4):799–804. doi: 10.1093/humrep/13.4.799
131. Van Bodegom M, Homberg JR, Henckens MJAG. Modulation of the hypothalamic-pituitary-adrenal axis by early life stress exposure. Front Cell Neurosci (2017) 11:87–120. doi: 10.3389/fncel.2017.00087
132. Cottrell EC, Holmes MC, Livingstone DE, Kenyon CJ, Seckl JR. Reconciling the nutritional and glucocorticoid hypotheses of fetal programming. FASEB J (2012) 26:1866–74. doi: 10.1096/fj.12-203489
133. Corcoran C, Walker E, Huot R, Mittal V, Tessner K, Kestler L, et al. The Stress Cascade and Schizophrenia: Etiology and Onset. Schizophr Bull (2003) 29(4):671–92. doi: 10.1093/oxfordjournals.schbul.a007038
134. Murphy S, Breen E, Cohn B, Kringbaum N, Cirillo P, Perez C, et al. F157. Infection and Increased Cortisol During Pregnancy and Risk for Adolescent Depression. Biol Psychiatry (2018) 83:S299. doi: 10.1016/j.biopsych.2018.02.771
135. Antonakopoulos N, Iliodromiti Z, Mastorakos G, Iavazzo C, Valsamakis G, Salakos N, et al. Association between brain-derived neurotrophic factor (BDNF) levels in 2 nd trimester amniotic fluid and fetal development. Mediators Inflammation (2018) 2018:1–7. doi: 10.1155/2018/8476217
136. Saha RN, Liu X, Pahan K. Up-regulation of BDNF in astrocytes by TNF-α: A case for the neuroprotective role of cytokine. J Neuroimmune Pharmacol (2006) 1:212–22. doi: 10.1007/s11481-006-9020-8
137. Jiang Y, Wei N, Lu T, Zhu J, Xu G, Liu X. Intranasal brain-derived neurotrophic factor protects brain from ischemic insult via modulating local inflammation in rats. Neuroscience (2011) 172:398–405. doi: 10.1016/j.neuroscience.2010.10.054
138. Yassour M, Jason E, Hogstrom LJ, Arthur TD, Tripathi S, Siljander H, et al. Strain-Level Analysis of Mother-to-Child Bacterial Transmission during the First Few Months of Life. Cell Host Microbe (2018) 24:146–154.e4. doi: 10.1016/j.chom.2018.06.007
139. Korpela K, Costea P, Coelho LP, Kandels-Lewis S, Willemsen G, Boomsma DI, et al. Selective maternal seeding and environment shape the human gut microbiome. Genome Res (2018) 28:561–8. doi: 10.1101/gr.233940.117
140. Mohammadkhah AI, Simpson EB, Patterson SG, Ferguson JF. Development of the Gut Microbiome in Children, and Lifetime Implications for Obesity and Cardiometabolic Disease. Children (2018) 5:160. doi: 10.3390/children5120160
141. Darabi B, Rahmati S, HafeziAhmadi MR, Badfar G, Azami M. The association between caesarean section and childhood asthma: an updated systematic review and meta-analysis. Allergy Asthma Clin Immunol (2019) 15:62. doi: 10.1186/s13223-019-0367-9
142. Li H-T, Ye R, Achenbach T, Ren A, Pei L, Zheng X, et al. Caesarean delivery on maternal request and childhood psychopathology: a retrospective cohort study in China. BJOG Int J Obstet Gynaecol (2011) 118:42–8. doi: 10.1111/j.1471-0528.2010.02762.x
143. Dunn AB, Jordan S, Baker BJ, Carlson NS. The Maternal Infant Microbiome: Considerations for Labor and Birth. MCN Am J Matern Nurs (2017) 42:318–25. doi: 10.1097/NMC.0000000000000373
144. Wandro S, Osborne S, Enriquez C, Bixby C, Arrieta A, Whiteson K. The Microbiome and Metabolome of Preterm Infant Stool Are Personalized and Not Driven by Health Outcomes, Including Necrotizing Enterocolitis and Late-Onset Sepsis. mSphere (2018) 3:e00104–18. doi: 10.1128/mSphere.00104-18
145. Chernikova DA, Koestler DC, Hoen AG, Housman ML, Hibberd PL, Moore JH, et al. Fetal exposures and perinatal influences on the stool microbiota of premature infants. J Matern Neonatal Med (2016) 29:99–105. doi: 10.3109/14767058.2014.987748
146. De Agüero MG, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, et al. The maternal microbiota drives early postnatal innate immune development. Science (80-) (2016) 351:1296–302. doi: 10.1126/science.aad2571
147. Fulde M, Sommer F, Chassaing B, van Vorst K, Dupont A, Hensel M, et al. Neonatal selection by Toll-like receptor 5 influences long-term gut microbiota composition. Nature (2018) 560:489–93. doi: 10.1038/s41586-018-0395-5
148. Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science (80-) (2012) 336:489–93. doi: 10.1126/science.1219328
149. Dworkin RH, Cornblatt BA, Friedmann R, Kaplansky LM, Lewis JA, Rinaldi A, et al. Childhood precursors of affective vs. social deficits in adolescents at risk for schizophrenia. Schizophr Bull (1993) 19:563–77. doi: 10.1093/schbul/19.3.563
150. Iacono WG, Clementz BA. A strategy for elucidating genetic influences on complex psychopathological syndromes (with special reference to ocular motor functioning and schizophrenia). Prog Exp Pers Psychopathol Res (1993) 16:11–65.
151. Mednick SA, Schulsinger F. Some premorbid characteristics related to breakdown in children with schizophrenic mothers. J Psychiatr Res (1968) 6:267–91. doi: 10.1016/0022-3956(68)90022-8
152. Sohlberg SC, Yaniv S. Social adjustment and cognitive performance of high-risk children. Schizophr Bull (1985) 11:61–5. doi: 10.1093/schbul/11.1.61
153. Weintraub S. Risk factors in schizophrenia: The Stony Brook high-risk project. Schizophr Bull (1987) 13:439–50. doi: 10.1093/schbul/13.3.439
154. Niemi LT, Suvisaari JM, Tuulio-Henriksson A, Lönnqvist JK. Childhood developmental abnormalities in schizophrenia: Evidence from high-risk studies. Schizophr Res (2003) 60:239–58. doi: 10.1016/S0920-9964(02)00234-7
155. Scharf RJ, Scharf GJ, Stroustrup A. Developmental milestones. Pediatr Rev (2016) 37:25–38. doi: 10.1542/pir.2014-0103
156. McNeil TF, Fish B, Schubert EW. Prospective study of pandysmaturation and adult mental disorder in high-risk and normal-risk offspring. J Psychiatr Res (2011) 45:561–7. doi: 10.1016/j.jpsychires.2010.09.010
157. Fish B, Marcus J, Hans SL, Auerbach JG, Perdue S. Infants at Risk for Schizophrenia: Sequelae of a Genetic Neurointegrative Defect: A Review and Replication Analysis of Pandysmaturation in the Jerusalem Infant Development Study. Arch Gen Psychiatry (1992) 49:221–35. doi: 10.1001/archpsyc.1992.01820030053007
158. Sameroff AJ, Barocas R, Seifer R. The early development of children born to mentally ill women. In: Children at risk for schizophrenia: A longitudinal perspective. New York, NY, US: Cambridge University Press. (1984) p. 482–514.
159. Parnas J, Schulsinger F, Schulsinger H, Mednick SA, Teasdale TW. Behavioral Precursors of Schizophrenia Spectrum: A Prospective Study. Arch Gen Psychiatry (1982) 39:658–64. doi: 10.1001/archpsyc.1982.04290060020005
160. Näslund B, Persson-Blennow I, McNeil T, Kaij L, Malmquist-Larsson A. Offspring of women with nonorganic psychosis: fear of strangers during the first year of life. Acta Psychiatr Scand (1984) 69:435–44. doi: 10.1111/j.1600-0447.1984.tb02516.x
161. Goodman SH. Emory University Project on Children of Disturbed Parents. Schizophr Bull (1987) 13:411–23. doi: 10.1093/schbul/13.3.411
162. Fish B. Infant predictors of the longitudinal course of schizophrenic development. Schizophr Bull (1987) 13:395–409. doi: 10.1093/schbul/13.3.395
163. McNeil TF, Harty B, Blennow G, Cantor-Graae E. Neuromotor deviation in offspring of psychotic mothers: A selective developmental deficiency in two groups of children at heightened psychiatric risk? J Psychiatr Res (1993) 27:39–54. doi: 10.1016/0022-3956(93)90048-7
164. Rieder RO, Nichols PL. Offspring of Schizophrenics III: Hyperactivity and Neurological Soft Signs. Arch Gen Psychiatry (1979) 36:665–74. doi: 10.1001/archpsyc.1979.01780060055006
165. Gooding DC, Ott SL, Roberts SA, Erlenmeyer-Kimling L. Thought disorder in mid-childhood as a predictor of adulthood diagnostic outcome: Findings from the New York High-Risk Project. Psychol Med (2013) 43:1003–12. doi: 10.1017/S0033291712001791
166. Hans SL, Marcus J, Nuechterlein KH, Asarnow RF, Styr B, Auerbach JG. Neurobehavioral deficits at adolescence in children at risk for schizophrenia: The Jerusalem Infant Development Study. Arch Gen Psychiatry (1999) 56:741–8. doi: 10.1001/archpsyc.56.8.741
167. Marcus J, Hans SL, Auerbach JG, Auerbach AG. Children at Risk for Schizophrenia: The Jerusalem Infant Development Study: II. Neurobehavioral Deficits at School Age. Arch Gen Psychiatry (1993) 50:797–809. doi: 10.1001/archpsyc.1993.01820220053006
168. Marcus J, Hans SL, Lewow E, Wilkinson L, Burack CM. Neurological findings in high-risk children: childhood assessment and 5-year followup. Schizophr Bull (1985) 11:85–100. doi: 10.1093/schbul/11.1.85
169. Hodges A, Byrne M, Grant E, Johnstone E. People at risk of schizophrenia. Sample characteristics of the first 100 cases in the Edinburgh high-risk study. Br J Psychiatry (1999) 174:547–53. doi: 10.1192/bjp.174.6.547
170. Garmezy N, Devine V. Project Competence: The Minnesota studies of children vulnerable to psychopathology. In: Children at risk for schizophrenia: A longitudinal perspective. New York, NY, US: Cambridge University Press. (1984) p. 289–303.
171. Erlenmeyer-Kimling L, Rock D, Roberts SA, Janal M, Kestenbaum C, Cornblatt B, et al. Attention, memory, and motor skills as childhood predictors of schizophrenia-related psychoses: The New York high-risk project. Am J Psychiatry (2000) 157:1416–22. doi: 10.1176/appi.ajp.157.9.1416
172. Bolinskey PK, Gottesman II, Nichols DS, Shapiro BM, Roberts SA, Adamo UH, et al. A new MMPI-derived indicator of liability to develop schizophrenia: Evidence from the New York High-Risk Project. Assessment (2001) 8:127–43. doi: 10.1177/107319110100800202
173. Lifshitz M, Kugelmass S, Karov M. Perceptual-motor and memory performance of high-risk children. Schizophr Bull (1985) 11:74–84. doi: 10.1093/schbul/11.1.74
174. Nagler S, Glueck Z. The clinical interview. Schizophr Bull (1985) 11:38–47. doi: 10.1093/schbul/11.1.38
175. Weintraub S, Neale JM. The Stony Brook High-Risk Project. In: . Children at risk for schizophrenia: A longitudinal perspective. New York, NY, US: Cambridge University Press. (1984) p. 243–63.
176. Johnstone EC, Abukmeil SS, Byrne M, Clafferty R, Grant E, Hodges A, et al. Edinburgh high risk study–findings after four years: demographic, attainment and psychopathological issues. Schizophr Res (2000) 46:1–15. doi: 10.1016/s0920-9964(99)00225-x
177. Redman SL, Corcoran CM, Kimhy D, Malaspina D. Effects of early trauma on psychosis development in clinical high-risk individuals and stability of trauma assessment across studies: a review. Arch Psychol (Chicago Ill) (2017) 1:28–49. [Accessed January 22, 2020].
178. Ramsay DS, Lewis M. Developmental Change in Infant Cortisol and Behavioral Response to Inoculation. Child Dev (1994) 65:1491–502. doi: 10.1111/j.1467-8624.1994.tb00831.x
179. Gunnar MR, Brodersen L, Krueger K, Rigatuso J. Dampening of Adrenocortical Responses during Infancy: Normative Changes and Individual Differences. Child Dev (1996) 67:877–89. doi: 10.1111/j.1467-8624.1996.tb01770.x
180. Daskalakis NP, Kloet ER, De, Yehuda R, Malaspina D, Kranz TM. Early life stress effects on glucocorticoid—BDNF interplay in the hippocampus. Front Mol Neurosci (2015) 8:1–13. doi: 10.3389/fnmol.2015.00068
181. Notaras M, van den Buuse M. Neurobiology of BDNF in fear memory, sensitivity to stress, and stress-related disorders. Mol Psychiatry (2020) doi: 10.1038/s41380-019-0639-2
182. Notaras M, Hill R, Van den Buuse M. A role for the BDNF gene Val66Met polymorphism in schizophrenia? A comprehensive review. Neurosci Biobehav Rev (2015) 51:15–30. doi: 10.1016/j.neubiorev.2014.12.016
183. Notaras M, Hill R, Van Den Buuse M. The BDNF gene Val66Met polymorphism as a modifier of psychiatric disorder susceptibility: Progress and controversy. Mol Psychiatry (2015) 20:916–30. doi: 10.1038/mp.2015.27
184. Jing D, Lee FS, Ninan I. The BDNF Val66Met polymorphism enhances glutamatergic transmission but diminishes activity-dependent synaptic plasticity in the dorsolateral striatum. Neuropharmacology (2017) 112:84–93. doi: 10.1016/j.neuropharm.2016.06.030
185. Pattwell SS, Bath KG, Perez-Castro R, Lee FS, Chao MV, Ninan I. The BDNF Val66Met polymorphism impairs synaptic transmission and plasticity in the infralimbic medial prefrontal cortex. J Neurosci (2012) 32:2410–21. doi: 10.1523/JNEUROSCI.5205-11.2012
186. Aas M, Haukvik UK, Djurovic S, Bergmann Ø, Athanasiu L, Tesli MS, et al. BDNF val66met modulates the association between childhood trauma, cognitive and brain abnormalities in psychoses. Prog Neuropsychopharmacol Biol Psychiatry (2013) 46:181–8. doi: 10.1016/j.pnpbp.2013.07.008
187. Lee J, Duan W, Mattson MP. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J Neurochem (2002) 82:1367–75. doi: 10.1046/j.1471-4159.2002.01085.x
188. Grassi-Oliveira R, Ashy M, Stein LM. Psychobiology of childhood maltreatment: Effects of allostatic load? Rev Bras Psiquiatr (2008) 30:60–8. doi: 10.1590/S1516-44462008000100012
189. Elzinga BM, Molendijk ML, Oude Voshaar RC, Bus BAA, Prickaerts J, Spinhoven P, et al. The impact of childhood abuse and recent stress on serum brain-derived neurotrophic factor and the moderating role of BDNF Val 66Met. Psychopharmacol (Berl) (2011) 214:319–28. doi: 10.1007/s00213-010-1961-1
190. Bermanzohn PC, Porto L, Arlow PB, Pollack S, Stronger R, Siris SG. At Issue: Hierarchical Diagnosis in Chronic Schizophrenia: A Clinical Study of Co-occurring Syndromes. Schizophr Bull (2000) 26:517–25. doi: 10.1093/oxfordjournals.schbul.a033472
191. Mehl S, Landsberg MW, Schmidt AC, Cabanis M, Bechdolf A, Herrlich J, et al. Why do bad things happen to me? Attributional style, depressed mood, and persecutory delusions in patients with schizophrenia. Schizophr Bull (2014) 40:1338–46. doi: 10.1093/schbul/sbu040
192. Grassi-Oliveira R, Stein LM, Lopes RP, Teixeira AL, Bauer ME. Low Plasma Brain-Derived Neurotrophic Factor and Childhood Physical Neglect Are Associated with Verbal Memory Impairment in Major Depression-A Preliminary Report. Biol Psychiatry (2008) 64:281–5. doi: 10.1016/j.biopsych.2008.02.023
193. Liu D, Diorio J, Day JC, Francis DD, Meaney MJ. Maternal care, hippocampal synaptogenesis and cognitive development in rats. Nat Neurosci (2000) 3:799–806. doi: 10.1038/77702
194. Lee J, Folley BS, Gore J, Park S. Origins of spatial working memory deficits in schizophrenia: An event-related fMRI and near-infrared spectroscopy study. PloS One (2008) 3:e1760. doi: 10.1371/journal.pone.0001760
195. Goodman GS, Ogle CM, Block SD, Harris LS, Larson RP, Augusti EM, et al. False memory for trauma-related Deese-Roediger-McDermott lists in adolescents and adults with histories of child sexual abuse. Dev Psychopathol (2011) 23:423–38. doi: 10.1017/S0954579411000150
196. Strakowski SM, Keck PE, McElroy SL, Lonczak HS, West SA. Chronology of comorbid and principal syndromes in first-episode psychosis. Compr Psychiatry (1995) 36:106–12. doi: 10.1016/s0010-440x(95)90104-3
197. Marshall AD. Developmental timing of trauma exposure relative to puberty and the nature of psychopathology among adolescent girls. J Am Acad Child Adolesc Psychiatry (2016) 55:25–32.e1. doi: 10.1016/j.jaac.2015.10.004
198. Thompson A, Nelson B, McNab C, Simmons M, Leicester S, McGorry PD, et al. Psychotic symptoms with sexual content in the “ultra high risk” for psychosis population: Frequency and association with sexual trauma. Psychiatry Res (2010) 177:84–91. doi: 10.1016/j.psychres.2010.02.011
199. Kraan T, Velthorst E, Smit F, de Haan L, van der Gaag M. Trauma and recent life events in individuals at ultra high risk for psychosis: Review and meta-analysis. Schizophr Res (2015) 161:143–9. doi: 10.1016/j.schres.2014.11.026
200. Falukozi E, Addington J. Impact of trauma on attenuated psychotic symptoms. Psychosis (2012) 4:203–12. doi: 10.1080/17522439.2011.626867
201. Bechdolf A, Thompson A, Nelson B, Cotton S, Simmons MB, Amminger GP, et al. Experience of trauma and conversion to psychosis in an ultra-high-risk (prodromal) group. Acta Psychiatr Scand (2010) 121:377–84. doi: 10.1111/j.1600-0447.2010.01542.x
202. Russo DA, Stochl J, Painter M, Dobler V, Jackson E, Jones PB, et al. Trauma history characteristics associated with mental states at clinical high risk for psychosis. Psychiatry Res (2014) 220:237–44. doi: 10.1016/j.psychres.2014.08.028
203. Thompson JL, Kelly M, Kimhy D, Harkavy-Friedman JM, Khan S, Messinger JW, et al. Childhood trauma and prodromal symptoms among individuals at clinical high risk for psychosis. Schizophr Res (2009) 108:176–81. doi: 10.1016/j.schres.2008.12.005
204. Thompson A, Marwaha S, Nelson B, Wood SJ, McGorry PD, Yung AR, et al. Do affective or dissociative symptoms mediate the association between childhood sexual trauma and transition to psychosis in an ultra-high risk cohort? Psychiatry Res (2016) 236:182–5. doi: 10.1016/j.psychres.2016.01.017
205. Stowkowy J, Liu L, Cadenhead KS, Cannon TD, Cornblatt BA, McGlashan TH, et al. Early traumatic experiences, perceived discrimination and conversion to psychosis in those at clinical high risk for psychosis. Soc Psychiatry Psychiatr Epidemiol (2016) 51:497–503. doi: 10.1007/s00127-016-1182-y
206. Stowkowy J, Addington J. Predictors of a clinical high risk status among individuals with a family history of psychosis. Schizophr Res (2013) 147:281–6. doi: 10.1016/j.schres.2013.03.030
207. Şahin S, Yüksel Ç, Güler J, Karadayi G, Akturan E, Göde E, et al. The history of childhood trauma among individuals with ultra high risk for psychosis is as common as among patients with first-episode schizophrenia. Early Interv Psychiatry (2013) 7:414–20. doi: 10.1111/eip.12022
208. Yung AR, McGorry PD. The initial prodrome in psychosis: Descriptive and qualitative aspects. Aust N Z J Psychiatry (1996) 30:587–99. doi: 10.3109/00048679609062654
209. Üçok A, Kaya H, Uğurpala C, Çikrikçili U, Ergül C, Yokuşoğlu Ç, et al. History of childhood physical trauma is related to cognitive decline in individuals with ultra-high risk for psychosis. Schizophr Res (2015) 169:199–203. doi: 10.1016/j.schres.2015.08.038
210. Trotman HD, Holtzman CW, Walker EF, Addington JM, Bearden CE, Cadenhead KS, et al. Stress exposure and sensitivity in the clinical high-risk syndrome: Initial findings from the North American Prodrome Longitudinal Study (NAPLS). Schizophr Res (2014) 160:104–9. doi: 10.1016/j.schres.2014.09.017
211. Carpenter LL, Tyrka AR, Ross NS, Khoury L, Anderson GM, Price LH. Effect of Childhood Emotional Abuse and Age on Cortisol Responsivity in Adulthood. Biol Psychiatry (2009) 66:69–75. doi: 10.1016/j.biopsych.2009.02.030
212. van Dam DS, Korver-Nieberg N, Velthorst E, Meijer CJ, de Haan L. Childhood maltreatment, adult attachment and psychotic symptomatology: a study in patients, siblings and controls. Soc Psychiatry Psychiatr Epidemiol (2014) 49:1759–67. doi: 10.1007/s00127-014-0894-0
213. Heins M, Simons C, Lataster T, Pfeifer S, Versmissen D, Lardinois M, et al. Childhood trauma and psychosis: A case-control and case-sibling comparison across different levels of genetic liability, psychopathology, and type of trauma. Am J Psychiatry (2011) 168:1286–94. doi: 10.1176/appi.ajp.2011.10101531
214. Suzuki A, Poon L, Papadopoulos AS, Kumari V, Cleare AJ. Long term effects of childhood trauma on cortisol stress reactivity in adulthood and relationship to the occurrence of depression. Psychoneuroendocrinology (2014) 50:289–99. doi: 10.1016/j.psyneuen.2014.09.007
215. Ruby E, Polito S, McMahon K, Gorovitz M, Corcoran C, Malaspina D. Pathways Associating Childhood Trauma to the Neurobiology of Schizophrenia. Front Psychol Behav Sci (2014) 3:1–17.
216. Kitraki E, Kremmyda O, Youlatos D, Alexis MN, Kittas C. Spatial performance and corticosteroid receptor status in the 21-day restraint stress paradigm. Ann New York Acad Sci (2004) 1018(1):323–7. doi: 10.1196/annals.1296.039
217. Magariños AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: Involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience (1995) 69:89–98. doi: 10.1016/0306-4522(95)00259-L
218. Sunanda, Rao MS, Raju TR. Effect of chronic restraint stress on dendritic spines and excrescences of hippocampal CA3 pyramidal neurons-a quantitative study. Brain Res (1995) 694:312–7. doi: 10.1016/0006-8993(95)00822-8
219. Tata DA, Marciano VA, Anderson BJ. Synapse loss from chronically elevated glucocorticoids: Relationship to neuropil volume and cell number in hippocampal area CA3. J Comp Neurol (2006) 498:363–74. doi: 10.1002/cne.21071
220. Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci (2002) 22:6810–8. doi: 10.1523/jneurosci.22-15-06810.2002
221. Wright RL, Lightner EN, Harman JS, Meijer OC, Conrad CD. Attenuating corticosterone levels on the day of memory assessment prevents chronic stress-induced impairments in spatial memory. Eur J Neurosci (2006) 24:595–605. doi: 10.1111/j.1460-9568.2006.04948.x
222. Baumeister D, Akhtar R, Ciufolini S, Pariante CM, Mondelli V. Childhood trauma and adulthood inflammation: A meta-analysis of peripheral C-reactive protein, interleukin-6 and tumour necrosis factor-α. Mol Psychiatry (2016) 21:642–9. doi: 10.1038/mp.2015.67
223. Slopen N, Kubzansky LD, McLaughlin KA, Koenen KC. Childhood adversity and inflammatory processes in youth: A prospective study. Psychoneuroendocrinology (2013) 38:188–200. doi: 10.1016/j.psyneuen.2012.05.013
224. Benedetti F, Ambrée O, Locatelli C, Lorenzi C, Poletti S, Colombo C, et al. The effect of childhood trauma on serum BDNF in bipolar depression is modulated by the serotonin promoter genotype. Neurosci Lett (2017) 656:177–81. doi: 10.1016/j.neulet.2017.07.043
225. Hemmings SMJ, Malan-Müller S, Van Den Heuvel LL, Demmitt BA, Stanislawski MA, Smith DG, et al. The Microbiome in Posttraumatic Stress Disorder and Trauma-Exposed Controls: An Exploratory Study. Psychosom Med (2017) 79:936–46. doi: 10.1097/PSY.0000000000000512
226. Bradford K, Shih W, Videlock EJ, Presson AP, Naliboff BD, Mayer EA, et al. Association Between Early Adverse Life Events and Irritable Bowel Syndrome. Clin Gastroenterol Hepatol (2012) 10:385–90. doi: 10.1016/j.cgh.2011.12.018
227. García-Ródenas CL, Bergonzelli GE, Nutten S, Schumann A, Cherbut C, Turini M, et al. Nutritional approach to restore impaired intestinal barrier function and growth after neonatal stress in rats. J Pediatr Gastroenterol Nutr (2006) 43:16–24. doi: 10.1097/01.mpg.0000226376.95623.9f
228. O’Mahony SM, Marchesi JR, Scully P, Codling C, Ceolho AM, Quigley EMM, et al. Early Life Stress Alters Behavior, Immunity, and Microbiota in Rats: Implications for Irritable Bowel Syndrome and Psychiatric Illnesses. Biol Psychiatry (2009) 65:263–7. doi: 10.1016/j.biopsych.2008.06.026
229. Park SH, Videlock EJ, Shih W, Presson AP, Mayer EA, Chang L. Adverse childhood experiences are associated with irritable bowel syndrome and gastrointestinal symptom severity. Neurogastroenterol Motil (2016) 28:1252–60. doi: 10.1111/nmo.12826
230. Chitkara DK, Van Tilburg MAL, Blois-Martin N, Whitehead WE. Early life risk factors that contribute to irritable bowel syndrome in adults: A systematic review. Am J Gastroenterol (2008) 103:765–74. doi: 10.1111/j.1572-0241.2007.01722.x
231. Callaghan BL, Fields A, Gee DG, Gabard-Durnam L, Caldera C, Humphreys KL, et al. Mind and gut: Associations between mood and gastrointestinal distress in children exposed to adversity. Dev Psychopathol (2019) 32:309–28. doi: 10.1017/S0954579419000087
232. Hantsoo L, Jašarević E, Criniti S, McGeehan B, Tanes C, Sammel MD, et al. Childhood adversity impact on gut microbiota and inflammatory response to stress during pregnancy. Brain Behav Immun (2019) 75:240–50. doi: 10.1016/j.bbi.2018.11.005
233. Erny D, De Angelis ALH, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci (2015) 18:965–77. doi: 10.1038/nn.4030
234. Rantakallio P, Jones P, Moring J, Von Wendt L. Association between central nervous system infections during childhood and adult onset schizophrenia and other psychoses: A 28-year follow-up. Int J Epidemiol (1997) 26:837–43. doi: 10.1093/ije/26.4.837
235. Leask SJ, Done DJ, Crow TJ. Adult psychosis, common childhood infections and neurological soft signs in a national birth cohort. Br J Psychiatry (2002) 181:387–92. doi: 10.1192/bjp.181.5.387
236. Dalman C, Allebeck P, Gunnell D, Harrison G, Kristensson K, Lewis G, et al. Infections in the CNS during childhood and the risk of subsequent psychotic illness: A cohort study of more than one million Swedish subjects. Am J Psychiatry (2008) 165:59–65. doi: 10.1176/appi.ajp.2007.07050740
237. Debost JC, Larsen JT, Munk-Olsen T, Mortensen PB, Agerbo E, Petersen LV. Childhood infections and schizophrenia: The impact of parental SES and mental illness, and childhood adversities. Brain Behav Immun (2019) 81:341–7. doi: 10.1016/j.bbi.2019.06.031
238. Köhler-Forsberg O, Petersen L, Gasse C, Mortensen PB, Dalsgaard S, Yolken RH, et al. A Nationwide Study in Denmark of the Association between Treated Infections and the Subsequent Risk of Treated Mental Disorders in Children and Adolescents. JAMA Psychiatry (2019) 76:271–9. doi: 10.1001/jamapsychiatry.2018.3428
239. Bhattacharyya S, Darby RR, Raibagkar P, Castro LNG, Berkowitz AL. Antibiotic-associated encephalopathy. Neurology (2016) 86:963–71. doi: 10.1212/WNL.0000000000002455
240. Torpy DJ, Ho JT. Value of free cortisol measurement in systemic infection. Hormone Metab Res (2007) 39(6):439–44. doi: 10.1055/s-2007-980200
241. Brown DG, Soto R, Yandamuri S, Stone C, Dickey L, Gomes-Neto JC, et al. The microbiota protects from viral- induced neurologic damage through microglia-intrinsic TLR signaling. Elife (2019) 8:e47117. doi: 10.7554/eLife.47117
242. Jaworska N, MacQueen G. Adolescence as a unique developmental period. J Psychiatry Neurosci (2015) 40:291–3. doi: 10.1503/jpn.150268
243. McVey Neufeld KA, Luczynski P, Dinan TG, Cryan JF. Reframing the teenage wasteland: Adolescent microbiota-gut-brain axis. Can J Psychiatry (2016) 61:214–21. doi: 10.1177/0706743716635536
244. Worland J, Weeks DG, Weiner SM, Schechtman J. Longitudinal, Prospective Evaluations of Intelligence in Children at Risk. Schizophr Bull (1982) 8:135–41. doi: 10.1093/schbul/8.1.135
245. Erlenmeyer-Kimling L, Cornblatt BA. A summary of attentional findings in the New York High-Risk Project. J Psychiatr Res (1992) 26:405–26. doi: 10.1016/0022-3956(92)90043-n
246. Ayalon M, Merom H. The teacher interview. Schizophr Bull (1985) 11:117–20. doi: 10.1093/schbul/11.1.117
247. Kugelmass S, Faber N, Frenkel E, Ingraham LJ, Mirsky AF, Nathan M, et al. Reanalysis of SCOR and anxiety measures in the Israeli high-risk study. Schizophr Bull (1995) 21:205–17. doi: 10.1093/schbul/21.2.205
248. Wisdom JP, Manuel JI, Drake RE. Substance Use Disorder Among People With First-Episode Psychosis: A Systematic Review of Course and Treatment. Psychiatr Serv (2011) 62:1007. doi: 10.1176/appi.ps.62.9.1007
249. Asher CJ, Gask L. Reasons for illicit drug use in people with schizophrenia: Qualitative study. BMC Psychiatry (2010) 10:94. doi: 10.1186/1471-244X-10-94
250. Ibarra-Lecue I, Mollinedo-Gajate I, Meana JJ, Callado LF, Diez-Alarcia R, Urigüen L. Chronic cannabis promotes pro-hallucinogenic signaling of 5-HT2A receptors through Akt/mTOR pathway. Neuropsychopharmacology (2018) 43:2028–35. doi: 10.1038/s41386-018-0076-y
251. Al’Absi M. Hypothalamic-pituitary-adrenocortical responses to psychological stress and risk for smoking relapse. Int J Psychophysiol (2006) 59:218–27. doi: 10.1016/j.ijpsycho.2005.10.010
252. Mendelson JH, Ogata M, Mello NK. Adrenal function and alcoholism. I. Serum cortisol. Psychosom Med (1971) 33:145–57. doi: 10.1097/00006842-197103000-00006
253. Junghanns K, Junghanns K, Tietz U, Dibbelt L, Kuether M, Jurth R, et al. Attenuated salivary cortisol secretion under cue exposure is associated with early relapse. Alcohol Alcohol (2005) 40(1):80–5. Available at: http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.583.4191 [Accessed May 29, 2020]. doi: 10.1093/alcalc/agh107
254. Junghanns K, Backhaus J, Tietz U, Lange W, Bernzen J, Wetterling T, et al. Impaired serum cortisol stress response is a predictor of early relapse. Alcohol Alcohol (2003) 38:189—193. doi: 10.1093/alcalc/agg052
255. de Timary P, Leclercq S, Stärkel P, Delzenne N. A dysbiotic subpopulation of alcohol-dependent subjects. Gut Microbes (2015) 6:388–91. doi: 10.1080/19490976.2015.1107696
256. Kiraly DD, Walker DM, Calipari ES, Labonte B, Issler O, Pena CJ, et al. Alterations of the host microbiome affect behavioral responses to cocaine. Sci Rep (2016) 6:1–12. doi: 10.1038/srep35455
257. Lee K, Vuong HE, Nusbaum DJ, Hsiao EY, Evans CJ, Taylor AMW. The gut microbiota mediates reward and sensory responses associated with regimen-selective morphine dependence. Neuropsychopharmacology (2018) 43:2606–14. doi: 10.1038/s41386-018-0211-9
258. Ning T, Gong X, Xie L, Ma B. Gut microbiota analysis in rats with methamphetamine-induced conditioned place preference. Front Microbiol (2017) 8:1620–9. doi: 10.3389/fmicb.2017.01620
259. Peterson VL, Jury NJ, Cabrera-Rubio R, Draper LA, Crispie F, Cotter PD, et al. Drunk bugs: Chronic vapour alcohol exposure induces marked changes in the gut microbiome in mice. Behav Brain Res (2017) 323:172–6. doi: 10.1016/j.bbr.2017.01.049
260. Wang F, Roy S. Gut Homeostasis, Microbial Dysbiosis, and Opioids. Toxicol Pathol (2017) 45:150–6. doi: 10.1177/0192623316679898
261. Meckel KR, Kiraly DD. A potential role for the gut microbiome in substance use disorders. Psychopharmacol (Berl) (2019) 236:1513–30. doi: 10.1007/s00213-019-05232-0
262. Xu Z, Wang C, Dong X, Hu T, Wang L, Zhao W, et al. Chronic alcohol exposure induced gut microbiota dysbiosis and its correlations with neuropsychic behaviors and brain BDNF/Gabra1 changes in mice. BioFactors (2019) 45:187–99. doi: 10.1002/biof.1469
263. Xiao Hw, Ge C, Feng Gx, Li Y, Luo D, Dong J, et al. Gut microbiota modulates alcohol withdrawal-induced anxiety in mice. Toxicol Lett (2018) 287:23–30. doi: 10.1016/j.toxlet.2018.01.021
264. Galderisi S, Bucci P, Üçok A, Peuskens J. No gender differences in social outcome in patients suffering from schizophrenia. Eur Psychiatry (2012) 27:406–8. doi: 10.1016/j.eurpsy.2011.01.011
265. Gureje O. Gender and schizophrenia: age at onset and sociodemographic attributes. Acta Psychiatr Scand (1991) 83:402–5. doi: 10.1111/j.1600-0447.1991.tb05564.x
266. Goldstein JM, Tsuang MT, Faraone SV. Gender and schizophrenia: Implications for understanding the heterogeneity of the illness. Psychiatry Res (1989) 28:243–53. doi: 10.1016/0165-1781(89)90205-9
267. De Luca F, Shoenfeld Y. The microbiome in autoimmune diseases. Clin Exp Immunol (2019) 195:74–85. doi: 10.1111/cei.13158
268. Stojanovich L, Marisavljevich D. Stress as a trigger of autoimmune disease. Autoimmun Rev (2008) 7:209–13. doi: 10.1016/j.autrev.2007.11.007
269. Müller N, Weidinger E, Leitner B, Schwarz MJ. The role of inflammation in schizophrenia. Front Neurosci (2015) 9:372–81. doi: 10.3389/fnins.2015.00372
270. Ryan MCM, Collins P, Thakore JH. Impaired fasting glucose tolerance in first-episode, drug-naive patients with schizophrenia. Am J Psychiatry (2003) 160:284–9. doi: 10.1176/appi.ajp.160.2.284
271. Saddichha S, Manjunatha N, Ameen S, Akhtar S. Metabolic syndrome in first episode schizophrenia - A randomized double-blind controlled, short-term prospective study. Schizophr Res (2008) 101:266–72. doi: 10.1016/j.schres.2008.01.004
272. He Y, Wu W, Wu S, Zheng HM, Li P, Sheng HF, et al. Linking gut microbiota, metabolic syndrome and economic status based on a population-level analysis. Microbiome (2018) 6:172. doi: 10.1186/s40168-018-0557-6
273. Brunner EJ, Hemingway H, Walker BR, Page M, Clarke P, Juneja M, et al. Adrenocortical, autonomic, and inflammatory causes of the metabolic syndrome: Nested case-control study. Circulation (2002) 106:2659–65. doi: 10.1161/01.CIR.0000038364.26310.BD
274. Ogbonnaya ES, Clarke G, Shanahan F, Dinan TG, Cryan JF, O’Leary OF. Adult Hippocampal Neurogenesis Is Regulated by the Microbiome. Biol Psychiatry (2015) 78:e7–9. doi: 10.1016/j.biopsych.2014.12.023
275. van Praag H. Exercise and the brain: something to chew on. Trends Neurosci (2009) 32:283–90. doi: 10.1016/j.tins.2008.12.007
276. Van Praag H. Neurogenesis and exercise: Past and future directions. NeuroMolecular Med (2008) 10:128–40. doi: 10.1007/s12017-008-8028-z
277. Hennigan A, O’Callaghan RM, Kelly ÁM. Neurotrophins and their receptors: Roles in plasticity, neurodegeneration and neuroprotection. Biochem Soc Trans (2007) 35(2):424–7. doi: 10.1042/BST0350424
278. Johnston MV. Plasticity in the developing brain: implications for rehabilitation. Dev Disabil Res Rev (2009) 15:94–101. doi: 10.1002/ddrr.64
279. Farmer J, Zhao X, Van Praag H, Wodtke K, Gage FH, Christie BR. Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male sprague-dawley rats in vivo. Neuroscience (2004) 124:71–9. doi: 10.1016/j.neuroscience.2003.09.029
280. Tong L, Shen H, Perreau VM, Balazs R, Cotman CW. Effects of exercise on gene-expression profile in the rat hippocampus. Neurobiol Dis (2001) 8:1046–56. doi: 10.1006/nbdi.2001.0427
281. Neeper SA, Góauctemez-Pinilla F, Choi J, Cotman C. Exercise and brain neurotrophins. Nature (1995) 373:109. doi: 10.1038/373109a0
282. Seifert T, Brassard P, Wissenberg M, Rasmussen P, Nordby P, Stallknecht B, et al. Endurance training enhances BDNF release from the human brain. Am J Physiol Regul Integr Comp Physiol (2010) 298:R372–7. doi: 10.1152/ajpregu.00525.2009
283. Rasmussen P, Brassard P, Adser H, Pedersen MV, Leick L, Hart E, et al. Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp Physiol (2009) 94:1062–9. doi: 10.1113/expphysiol.2009.048512
284. Voss MW, Vivar C, Kramer AF, van Praag H. Bridging animal and human models of exercise-induced brain plasticity. Trends Cognit Sci (2013) 17:525–44. doi: 10.1016/j.tics.2013.08.001
285. Voss MW, Erickson KI, Prakash RS, Chaddock L, Kim JS, Alves H, et al. Neurobiological markers of exercise-related brain plasticity in older adults. Brain Behav Immun (2013) 28:90–9. doi: 10.1016/j.bbi.2012.10.021
286. Szuhany KL, Bugatti M, Otto MW. A meta-analytic review of the effects of exercise on brain-derived neurotrophic factor. J Psychiatr Res (2015) 60:56–64. doi: 10.1016/j.jpsychires.2014.10.003
287. Kimhy D, Vakhrusheva J, Khan S, Chang RW, Hansen MC, Ballon JS, et al. Emotional granularity and social functioning in individuals with schizophrenia: An experience sampling study. J Psychiatr Res (2014) 53:141–8. doi: 10.1016/j.jpsychires.2014.01.020
288. Deighton S, Addington J. Exercise practices of young people at their first episode of psychosis. Schizophr Res (2014) 152:311–2. doi: 10.1016/j.schres.2013.10.045
289. Hodgekins J, French P, Birchwood M, Mugford M, Christopher R, Marshall M, et al. Comparing time use in individuals at different stages of psychosis and a non-clinical comparison group. Schizophr Res (2015) 161:188–93. doi: 10.1016/j.schres.2014.12.011
290. Koivukangas J, Tammelin T, Kaakinen M, Mäki P, Moilanen I, Taanila A, et al. Physical activity and fitness in adolescents at risk for psychosis within the Northern Finland 1986 Birth Cohort. Schizophr Res (2010) 116:152–8. doi: 10.1016/j.schres.2009.10.022
291. Newberry RE, Dean DJ, Sayyah MD, Mittal VA. What prevents youth at clinical high risk for psychosis from engaging in physical activity? An examination of the barriers to physical activity. Schizophr Res (2018) 201:400–5. doi: 10.1016/j.schres.2018.06.011
292. Mittal VA, Vargas T, Juston Osborne K, Dean D, Gupta T, Ristanovic I, et al. Exercise Treatments for Psychosis: a Review. Curr Treat Options Psychiatry (2017) 4:152–66. doi: 10.1007/s40501-017-0112-2
293. Armstrong HF, Bartels MN, Paslavski O, Cain D, Shoval HA, Ballon JS, et al. The impact of aerobic exercise training on cardiopulmonary functioning in individuals with schizophrenia. Schizophr Res (2016) 173:116–7. doi: 10.1016/j.schres.2016.03.009
294. Kimhy D, Gill KE, Brucato G, Vakhrusheva J, Arndt L, Gross JJ, et al. The impact of emotion awareness and regulation on social functioning in individuals at clinical high risk for psychosis. Psychol Med (2016) 46:2907–18. doi: 10.1017/S0033291716000490
295. Vakhrusheva J, Marino B, Stroup TS, Kimhy D. Aerobic Exercise in People with Schizophrenia: Neural and Neurocognitive Benefits. Curr Behav Neurosci Rep (2016) 3:165–75. doi: 10.1007/s40473-016-0077-2
296. Kimhy D, Vakhrusheva J, Bartels MN, Armstrong HF, Ballon JS, Khan S, et al. The Impact of Aerobic Exercise on Brain-Derived Neurotrophic Factor and Neurocognition in Individuals With Schizophrenia: A Single-Blind, Randomized Clinical Trial. Schizophr Bull (2015) 41:859–68. doi: 10.1093/schbul/sbv022
298. Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: The glucocorticoid cascade hypothesis. Endocr Rev (1986) 7:284–301. doi: 10.1210/edrv-7-3-284
299. Bercik P, Park AJ, Sinclair D, Khoshdel A, Lu J, Huang X, et al. The anxiolytic effect of Bifidobacterium longum NCC3001 involves vagal pathways for gut-brain communication. Neurogastroenterol Motil (2011) 23:1132–9. doi: 10.1111/j.1365-2982.2011.01796.x
300. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature (1999) 402:656–60. doi: 10.1038/45230
301. Muccioli G, Papotti M, Locatelli V, Ghigo E, Deghenghi R. Binding of 125I-labeled ghrelin to membranes from human hypothalamus and pituitary gland. J Endocrinol Invest (2001) 24:7–9. doi: 10.1007/BF03343831
302. Giordano R, Pellegrino M, Picu A, Bonelli L, Balbo M, Berardelli R, et al. Neuroregulation of the hypothalamus-pituitary-adrenal (HPA) axis in humans: Effects of GABA-, mineralocorticoid-, and GH-secretagogue-receptor modulation. Sci World J (2006) 6:1–11. doi: 10.1100/tsw.2006.09
303. Tortorella C, Neri G, Nussdorfer GG. Galanin in the regulation of the hypothalamic-pituitary-adrenal axis (review). Int J Mol Med (2007) 19:639–47. doi: 10.3892/ijmm.19.4.639
Keywords: schizophrenia, microbiome, brain-derived neurotrophic factor, development, stress, cortisol
Citation: Hoffman KW, Lee JJ, Corcoran CM, Kimhy D, Kranz TM and Malaspina D (2020) Considering the Microbiome in Stress-Related and Neurodevelopmental Trajectories to Schizophrenia. Front. Psychiatry 11:629. doi: 10.3389/fpsyt.2020.00629
Received: 20 March 2020; Accepted: 16 June 2020;
Published: 03 July 2020.
Edited by:
Mary V. Seeman, University of Toronto, CanadaReviewed by:
Rachel Anne Hill, Monash University, AustraliaEldin Jasarevic, University of Pennsylvania, United States
Copyright © 2020 Hoffman, Lee, Corcoran, Kimhy, Kranz and Malaspina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dolores Malaspina, ZG9sb3Jlcy5tYWxhc3BpbmFAbXNzbS5lZHU=