 Shengtao Yang
Shengtao Yang Hyojung Seo1,2
Hyojung Seo1,2 Min Wang
Min Wang Amy F. T. Arnsten
Amy F. T. Arnsten- 1Department of Neuroscience, Yale University School of Medicine, New Haven, CT, United States
- 2Department of Psychiatry, Yale University School of Medicine, New Haven, CT, United States
The dorsolateral prefrontal cortex (dlPFC) generates the mental representations that are the foundation of abstract thought, and provides top-down regulation of emotion through projections to the medial PFC and cingulate cortices. Physiological recordings from dlPFC Delay cells have shown that the generation of mental representations during working memory relies on NMDAR neurotransmission, with surprisingly little contribution from AMPAR. Systemic administration of low “antidepressant” doses of the NMDAR antagonist, ketamine, erodes these representations and reduces dlPFC Delay cell firing. In contrast to the dlPFC, V1 neuronal firing to visual stimuli depends on AMPAR, with much less contribution from NMDAR. Similarly, neurons in the dlPFC that respond to sensory events (cue cells, response feedback cells) rely on AMPAR, and systemic ketamine increases their firing. Insults to NMDAR transmission, and the impaired ability for dlPFC to generate mental representations, may contribute to cognitive deficits in schizophrenia, e.g., from genetic insults that weaken NMDAR transmission, or from blockade of NMDAR by kynurenic acid. Elevated levels of kynurenic acid in dlPFC may also contribute to cognitive deficits in other disorders with pronounced neuroinflammation (e.g., Alzheimer's disease), or peripheral infections where kynurenine can enter brain (e.g., delirium from sepsis, “brain fog” in COVID19). Much less is known about NMDAR actions in the primate cingulate cortices. However, NMDAR neurotransmission appears to process the affective and visceral responses to pain and other aversive experiences mediated by the cingulate cortices, which may contribute to sustained alterations in mood state. We hypothesize that the very rapid, antidepressant effects of intranasal ketamine may involve the disruption of NMDAR-generated aversive mood states by the anterior and subgenual cingulate cortices, providing a “foot in the door” to allow the subsequent return of top-down regulation by higher PFC areas. Thus, the detrimental vs. therapeutic effects of NMDAR blockade may be circuit dependent.
Introduction
The recent discovery that the NMDA receptor (NMDAR) antagonist, ketamine, can produce rapid, antidepressant actions has stirred interest in the possible mechanisms underlying these therapeutic effects, and why blockade of NMDAR can produce such a swift change in mood. The current review discusses how NMDAR-calcium mechanisms are needed for sustained neural representations, e.g., such as the persistent representation of visual space in working memory by circuits in the dorsolateral prefrontal cortex (dlPFC), and suggests that parallel mechanisms in the cingulate circuits mediating mood and emotion may be overactivated in depression, and aided by NMDAR blockade (1, 2).
NMDAR are heterotetramers composed of GluN1 and GluN2 (A-D) or GluN3 (A-B) subunits- usually with two GluN1 and two GluN2 subunits (3). The GluN2B subunit, also known as the NR2B subunit, has been of particular interest, as it closes more slowly than the common, GluN2A subunit, and fluxes high levels of calcium into the neuron (4). Although previous research in rodent classic circuits had found that NMDA-GluN2B were mostly at extra-synaptic locations (5), or played a role only in immature neurons (6), more recent research has shown that GluN2B play a critical, synaptic role in the primate cortical circuits mediating higher cognition, providing the synaptic events that generate sustained representations of visual space in working memory in the dlPFC (7, 8). The high levels of calcium influx into spines may be especially important for maintaining a depolarized post-synaptic membrane, permitting continued neural firing needed to sustain representations over long time periods (9). Recent research has also shown that expression of NMDAR with GluN2B subunits encoded by the GRIN2B gene expands across primate cortical evolution (10), and across the cortical hierarchy in humans, with especially high levels in association and limbic cortices such as the anterior cingulate cortex (11). The following paper explores the hypothesis that the critical role of GluN2B in generating sustained representations in dlPFC may extend to the generation of aversive mood state by the anterior and subgenual cingulate cortices, and that NMDAR blockade by ketamine may be helpful by relieving this self-perpetuating, aversive network activity.
The paper will briefly review PFC circuits in primates and their regulation of the cingulate cortices, and then discuss the critical role of NMDAR for generating mental representations in dlPFC, the expansion in NMDAR-GluN2B transmission across the cortical hierarchy and across cortical evolution, and the role of NMDAR-GluN2B in the cingulate cortices mediating affective pain responses and depression. It will briefly discuss how stress exposure impairs higher PFC regulation, and will close with an exploration of the idea that ketamine's rapid antidepressant actions may involve blocking mental representations of aversive mood state in the cingulate cortices.
Primate Prefrontal Cortical Circuits
The PFC greatly expands and differentiates over brain evolution, allowing representations of information in the absence of sensory stimulation. The primate PFC is topographically organized across multiple dimensions, e.g., with “simpler” representative functions found more caudally and more complex (e.g., metacognition) more rostrally in the frontal pole (12, 13). There are also topographic differences across the dorsolateral to ventromedial dimensions (14), where the dlPFC represents the outer world (e.g., with inputs from parietal areas that process visual space, Figure 1A), while the ventral and medial PFC regions represent the inner world, including taste and olfaction combining to represent flavor in orbital (ventral) PFC, and projections from the medial thalamus to the medial anterior cingulate cortex (ACC, BA24) mediating the emotional aspects of pain (Figure 1A). Neurons in the dorsomedial PFC also can represent persistent signatures of loss during a competitive game (15), and anterior cingulate neurons respond to errors (16), suggesting these regions are also activated by aversive psychological events. This information is relayed to the subgenual cingulate (BA25) that has extensive visceromotor connections to induce the physical aspects of the emotional response to pain [Figure 1A; (14)]. For example, BA25 projects to the amygdala, and the hypothalamus and brainstem to effect the autonomic nervous system and facial expression, and to the periaqueductal gray and medial subthalamic nucleus to alter behavioral response (14, 17–19), e.g., “freezing” behavior in response to a threat.
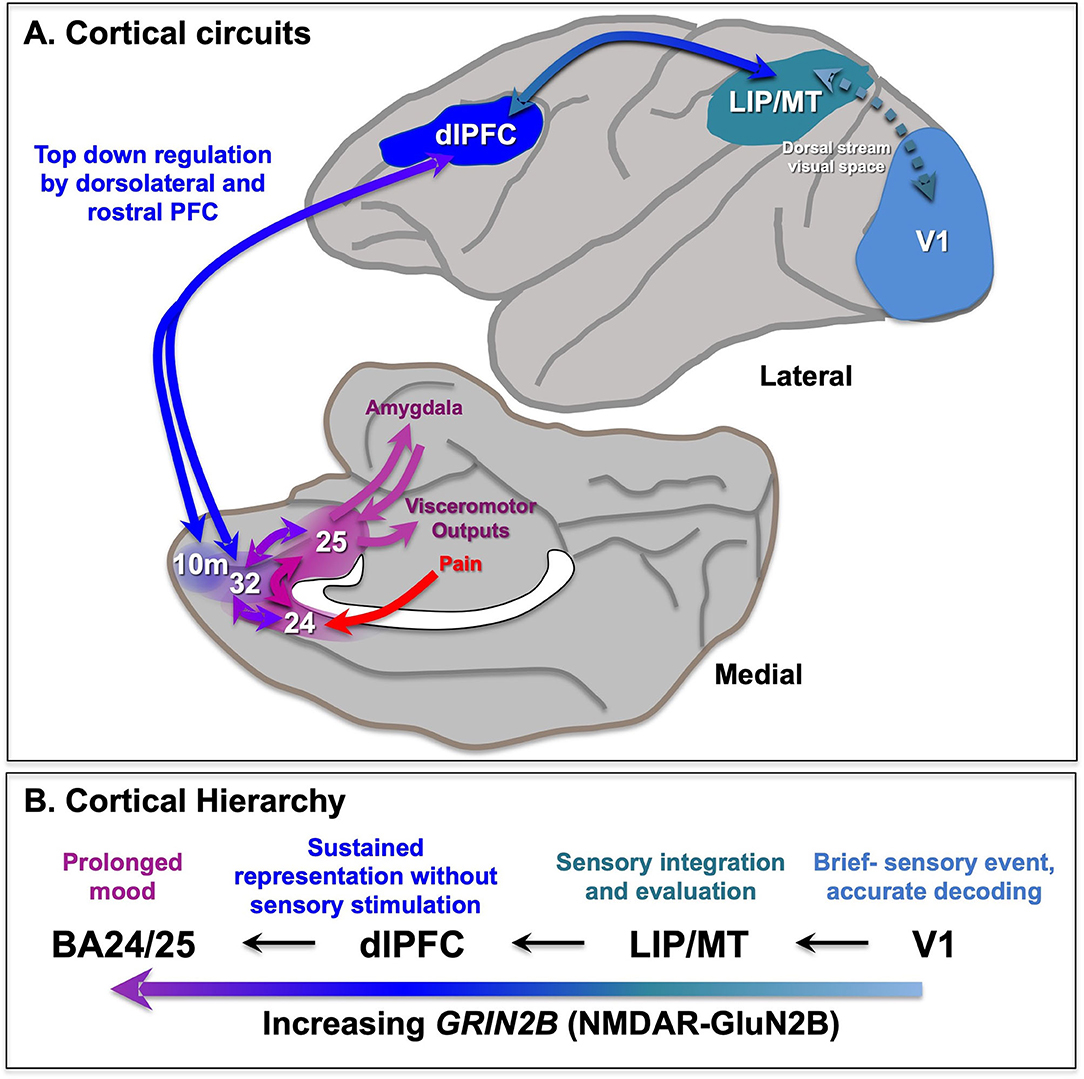
Figure 1. Primate cortical circuits. (A) Schematic diagram of circuits in the rhesus monkey cortex, where the lateral surface represents the outer world, and the medial and orbital surface represents inner state. The dorsal stream is shown on the lateral surface, where dlPFC represents visual space in working memory, and generates the goals for top-down regulation of emotion. The medial surface shows the pathways mediating the emotional response to pain, arising from medial thalamic projections to the insular cortex (not shown) and the anterior cingulate cortex BA24, which both project to BA25 (subgenual cingulate). BA25 is a major center for visceromotor outputs, e.g., to the amygdala, brainstem, and hypothalamus to alter heart rate. These cingulate cortices are often overactive in depression, and a target of DBS treatments. The dlPFC provides top-down regulation of emotion through indirect projections to BA25 via areas BA10m and BA32, and direct projections to BA24 (not shown). (B) The increasing timescales across the primate cortical hierarchy, and their relationship to GRIN2B expression. Based on (11) and (9). LIP, lateral intraparietal cortex; MT, middle temporal visual cortex.
The more newly evolved, rostral and lateral areas of PFC provide top-down regulation of the more primitive medial and caudal areas. For example, the dlPFC can regulates emotion via direct projections to BA24 (20, 21), and indirect projections to BA25 via BA10m or BA32 to BA25 (22, 23) (Figure 1A). The pathways from dlPFC to BA32 and then to BA25 are now known in great detail at the ultrastructural level (23–25), showing how dlPFC and BA32 are positioned to either inhibit or activate emotional responses by BA25.
An important note about species differences: rodents do not have rostral PFC areas (e.g., frontal pole) or a dlPFC, and even the medial and orbital PFC areas they do have are much less developed and differentiated than those in primates (26). Indeed, the dorsal to ventral topography of medial PFC subregions appears to be reversed from rodent to monkey, with the most ventral BA25 activating the stress response in monkeys, but inhibiting it in rodents (27). This may be due to the medial PFC being less differentiated in rodents, with a dorsal-ventral gradient in many medial PFC connections (28). Thus, the actual circuit connections, e.g., with excitatory vs. inhibitory neurons in amygdala, need to be identified for proper interpretation.
The Critical Role of NMDAR-GluN2B in the Generation of Mental Representations by the dlPFC
The primate dlPFC has the remarkable ability to generate and sustain mental representations without sensory stimulation, the foundation of abstract thought (29). dlPFC “Delay cells” are able to maintain persistent firing across the delay period in a working memory task, sustaining representations over many seconds e.g., remembering a position in visual space (30). “Delay cells” appear to reside in pyramidal cell microcircuits in deep layer III of the dlPFC that have extensive recurrent excitatory connections [Figure 2A; (29, 31)], as well as lateral inhibition from parvalbumin-containing interneurons to refine spatial tuning (29, 32). The persistent firing of Delay cells across the delay period depends on NMDAR stimulation (7), a finding predicted by computational models (33). Thus, iontophoresis (local electrical application) of low doses of NMDAR antagonists, including antagonists that selectively block those with GluN2A or GluN2B subunits, markedly reduces Delay cell firing (7). An example is shown in Figure 2B, where under control conditions a Delay cell can sustain the representation of the cue that had been flashed at 270° over many seconds in working memory. However, the Delay cell is no longer able to represent spatial information in working memory following the local iontophoretic blockade of NMDAR GluN2B with the antagonist, TCN237.
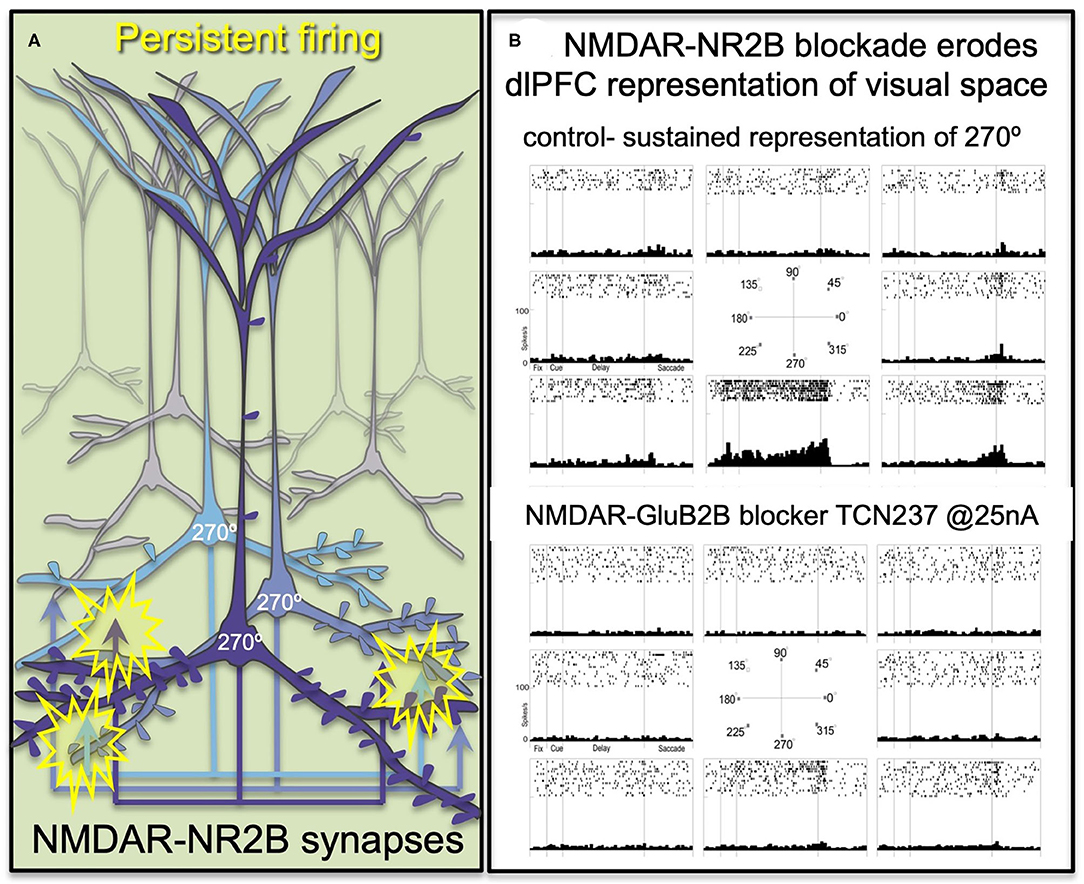
Figure 2. The persistent firing of dlPFC Delay cells depends on NMDAR with GluN2B subunits. (A) Schematic illustration of the recurrent excitatory microcircuits in deep layer III of dlPFC that generate persistent firing. (B) A dlPFC Delay cell that represents the spatial position of 270° during a spatial working memory task, maintaining firing across the delay period for only that preferred location. Iontophoresis of the selective NMDAR- GluN2B antagonist, TCN237, completely blocks the ability of the neuron to generate representations of visual space.
Immunoelectron microscopy (immunoEM) showed that NMDAR-GluN2B are expressed exclusively within the post-synaptic density (PSD) in layer III dlPFC spines, and are not extra-synaptic, consistent with their direct mediation of neurotransmission (7). The ability of GluN2B subunits to flux large amounts of calcium may be a key aspect of why they support persistent firing in computational models (33) and in Delay cells (7).
In contrast to NMDAR, blockade of AMPAR has remarkably subtle effects on Delay cell firing (7) (Figures 3A,B). This finding was initially confusing, as it is generally thought that AMPAR are essential to depolarize the PSD membrane and relieve the magnesium (Mg2+) block within the NMDAR pore, permitting NMDAR actions (Figure 3C). However, in dlPFC, this key permissive role appears to be played by acetylcholine acting at Nic-α7R and muscarinic M1R within the glutamate synapse (34, 35) which may depolarize the PSD to support persistent firing (Figures 3A,B). M1R may depolarize the PSD via closing of KCNQ channels localized in the PSD, and/or by enhancing levels of internal calcium release. These physiological data are consistent with behavioral data showing that Ach depletion from dlPFC is as deleterious as removing the cortex itself (36). As acetylcholine is released during wakefulness but not deep sleep, these mechanisms also help to coordinate cognitive state with arousal state, permitting conscious experience during wakefulness, but may render us unconscious during deep sleep when there is no acetylcholine release. Thus, as summarized in Figures 3A,B, Delay cell firing in dlPFC depends on NMDAR stimulation, including those with GluN2B subunits, with permissive actions by acetylcholine and more limited contributions from AMPAR.
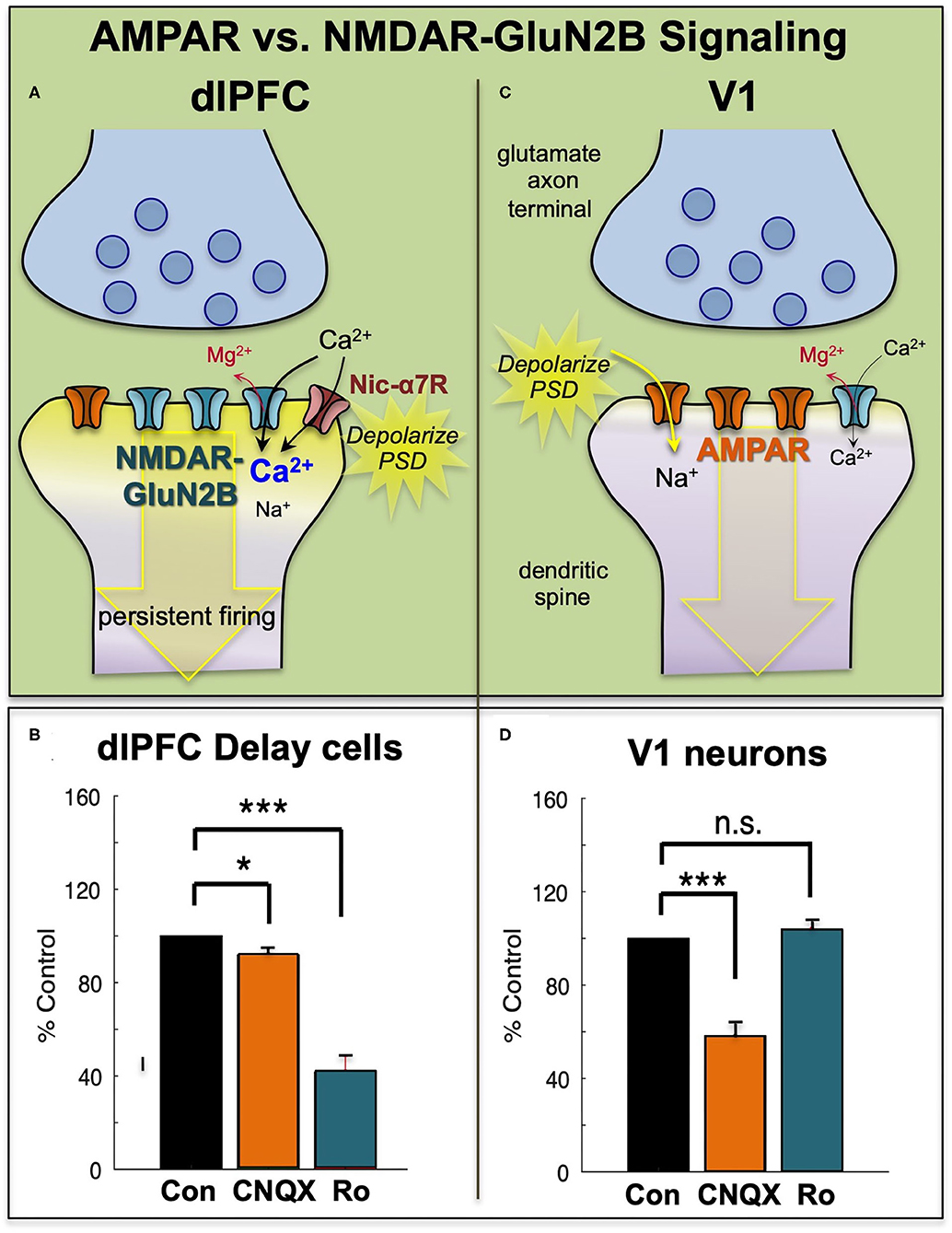
Figure 3. The primate dlPFC and primary visual cortex (V1) have very different neurotransmission. (A) The dlPFC depends on NMDAR neurotransmission, including those with slowly closing GluN2B subunits, that are exclusively within the PSD. The permissive excitatory effects to relieve the magnesium (Mg2+) block of the NMDAR ion channel are provided by acetylcholine (including Nic-a7R), with a surprisingly small influence from AMPAR. (B) Iontophoresis of the AMPAR antagonist, CNQX, has only subtle effects on dlPFC Delay cell firing, while blockade of NMDAR- GluN2B with Ro25-6981 (Ro) markedly reduces Delay cell firing. (C) Neurons in primate V1 show a more classic profile, relying heavily on AMPAR neurotransmission, with less influence by NMDAR. (D) Iontophoresis of low doses of the AMPAR antagonist, CNQX, markedly reduces V1 neuronal firing, while blockade of NMDAR- GluN2B with Ro has little effect. Adapted from (9) and (8). *p < 0.05, ***p < 0.001.
AMPAR neurotransmission does play an important role in some dlPFC neurons that respond to sensory events, i.e., dlPFC Cue cells, and dlPFC response feedback cells that are thought to convey the corollary discharge back to dlPFC that the intended motor response has occurred (7). As these events require accurate timing, it is logical that they would have more of a reliance on rapid AMPAR neurotransmission.
Systemic ketamine treatment has differential effects on dlPFC neuronal firing depending upon their reliance on AMPAR vs. NMDAR neurotransmission. Consistent with their reliance on NMDAR neurotransmission, dlPFC Delay cells show decreased firing following systemic administration of the NMDAR antagonist, ketamine, at low doses used to treat intractable depression (7). This is only seen during cognitive performance and is not evident at rest. In contrast, systemic ketamine administration increases the spontaneous firing of response feedback neurons that rely on AMPAR (7), which resembles the increased firing seen with deep layer neurons in rat mPFC following NMDAR blockade, the basis for the “glutamate surge” (37). Some of this heterogeneity may arise from the balance of NMDAR on pyramidal cells vs. GABAergic interneurons, where pyramidal cell circuits with extensive recurrent NMDAR excitation may show loss of firing, while those circuits with extensive NMDAR on interneurons (e.g., in the primary sensory cortices) may have an overall increase in glutamate signaling. These data caution that ketamine's actions are heterogeneous, and that methods that average the response of large populations of neurons under resting conditions (e.g., resting fMRI, multi-electrode recording) may miss critical ketamine actions such as the loss of representations during working memory. The fact that ketamine's effects are circuit-specific creates a complicated picture, confounding our ability to identify the specific actions relevant to its antidepressant effects, distinguished from its actions that lead to cognitive disorder.
The importance of NMDAR transmission to the generation of mental representations needed for working memory and abstract thought may have relevance to a number of conditions where NMDAR are blocked or genetically weakened. The data from monkeys help to explain the profound cognitive alterations that can occur in the encephalitis arising from anti-NMDAR antibodies (38). The loss of mental representations with NMDAR blockade also helps to explain the profound cognitive impairments in schizophrenia where there can be genetic mutations that weaken NMDAR signaling (39), and/or blockade of NMDAR by kynurenic acid, especially under conditions of inflammation (40). Blockade of NMDAR by kynurenic acid may also contribute to cognitive deficits in Alzheimer's disease (41), given the importance of inflammatory signaling in early stages of disease. It is also possible that systemic infection may impair higher brain functions through the uptake of kyrurenine across the blood brain barrier (42). For example, the pervasive cognitive deficits in delirium might arise from high levels of kyrurenine crossing into the brain during systemic infection (43), and that the residual “brain fog” from infections such as COVID19 (44–46), which also leads to systemic kynurenine production, may also involve sustained blockade of NMDAR in higher cortical circuits by kynurenic acid. As there are pharmacological tools to reduce kynurenine production that may relieve NMDAR blockade, these are important areas for future research.
NMDAR-GluN2B Expression Increases Across the Primate Cortical Hierarchy and Across Primate Evolution
There are multiple differences in function and physiology across the cortical hierarchy from primary sensory cortices, to association cortices to limbic cortices (Figure 1B). For example, there are increasing time scales in neuronal firing across the cortical hierarchy in rhesus monkeys (47) and in gray/white matter ratios in humans that correspond to transcriptional expression patterns (11). In particular, there is increasing expressing of the NMDAR GluN2B gene, GRIN2B, across the cortical hierarchy in humans, with low levels in primary visual cortex, high levels in dlPFC, and higher levels still in anterior cingulate cortices (11). As GRIN2B expression in dlPFC also increases across primate evolution, it suggests that this receptor plays an increasing role in primate mental experience.
Physiological studies in rodents (48) and monkeys are consistent with this hypothesis, as NMDAR-GluN2B has a much larger role in neurotransmission in the PFC than in the primary visual cortex, area V1. In rat medial PFC, the recurrent excitatory connections in layer V depend on NMDAR-GluN2B neurotransmission, while neurons in V1 showed much less reliance on these receptors (48). Similar results were seen in rhesus monkey dlPFC vs. V1. Neurons in V1 respond to the presentation of visual stimuli of a preferred orientation in their receptive field. These neurons have a great reliance on AMPAR transmission, where even low doses of AMPAR blockers such as CNQX markedly reduce stimulus-related firing (8) (Figures 3C,D). In contrast, high doses of NMDAR blockers are needed to reduce V1 neuronal firing [(8), Figures 3C,D]. A reliance on AMPAR stimulation is consistent with the function of V1 neurons, as the rapid kinetics of these receptors, in addition to their membrane properties (49), would allow accurate timing to encode the onset and offset of a sensory event. Thus, NMDAR transmission is not uniform across the primate cortex, and may be a feature of neurons requiring sustained neuronal firing for cognitive and possibly affective functions.
The very high levels of GRIN2B expression in the human anterior cingulate cortex (11) suggests that these receptor subtypes may be particularly important for the functioning of the cingulate cortices, e.g., in error detection, affective pain processing, and visceral affective responding. These limbic cortices and their corresponding connections are part of the neural networks that create “mood,” a sustained brain state. Given the role of NMDAR-GluN2B in mediating sustained firing in dlPFC, it is possible that these receptors have a parallel role in anterior and subgenual cingulate cortex. Although there are currently no direct iontophoretic recordings from primate anterior or subgenual cingulate cortex examining the role of GluN2B in cingulate physiology, this will be an important arena for future research. The following section outlines the importance of these receptors to cingulate processing of pain and visceral responding.
The Role of NMDAR-GluN2B in the Cingulate Cortices Mediating Affective Pain Responses and Depression
The anterior cingulate (BA24) and subgenual cingulate (BA25) cortices mediate the emotional responses to pain [(14), reviewed in (2)], and are overactive in depression (50, 51). For example, the ACC is overactive in chronic pain and is a common ablation site for neurosurgical alleviation of intractable pain (52). In particular, BA25 in particular overactive in depression and a focus of deep brain stimulation (DBS) to relieve intractable depression (51). As described below, there is accumulating evidence that the emotional responses of the anterior and subgenual cingulate cortices rely on NMDAR-GluN2B neurotransmission, and that these aversive responses are reduced by ketamine administration in the treatment of chronic pain and depression.
Increasing evidence indicates that the response to pain in the rodent ACC (BA24) is mediated by NMDAR, including those with GluR2B subunits (53). GluR2B upregulate in response to chronic pain (54, 55), and long-term potentiation in the anterior cingulate cortex in response to painful stimuli is mediated by NMDAR-calcium-cAMP signaling, including NMDAR with GluR2B subunits, consistent with the sensitized response to chronic pain [reviewed in (56, 57)]. Systemic administration of ketamine, or of its active enatiomer, esketamine, reduces the response to pain as well as accompanying depressive symptoms in both rodents (58) and humans (59–62).
The subgenual cingulate (BA25) has extensive subcortical projections to mediate the emotional and visceral response to pain or other affective experiences (14), including to the lateral habenula (63), a nucleus activated by aversive events (64). Recent studies in marmosets have illuminated its functional role and relationship to ketamine treatment. These studies showed that pharmacological inactivation of BA25 decreased the autonomic and behavioral correlates of negative emotion expectation, while inactivation of BA32 increased them via generalization (27), consistent with BA32 providing top-down regulation of BA25. Conversely, activation of BA25 in marmosets induced an anhedonic state and reduced willingness to work for reward that was reversed by systemic administration of ketamine (65). 18F-FDG PET imaging of the marmosets showed that activation of BA25 was accompanied by activation of BA24 and insular cortex, while systemic ketamine treatment reduced the activation of these cortical areas (65). Over-activation of BA25 in marmosets also reduced vagal tone and heart rate variability, reduced the extinction of an aversive response and potentiated cortisol release during threat (66). Activation of BA25 in this study was associated with increased activity in the amygdala, the hypothalamus, and the temporal association area TH (66), but decreased the activity of the frontopolar cortex area 9, the dlPFC area 46, the central orbitofrontal cortex area13, and the lateral caudate (66). However, in this study, systemic ketamine did not reverse the effects of threat, suggesting that primitive responses to threat (e.g., in amygdala) may still control network activity. These data suggest that ketamine treatment may be most effective under conditions of safety. Research is still needed to determine how local infusion of ketamine into BA24 and/or BA25 alters emotional responding.
Uncontrollable Stress Impairs Higher PFC Functions
The findings from the Roberts lab that activation of BA25 in marmoset reduces the activity of the rostral PFC and the dlPFC are consistent with a long line of research showing that these more newly evolved PFC areas are weakened by exposure to uncontrollable stress. As described above, under control conditions the dlPFC and rostral PFC can regulate emotion via projections to BA25 (Figures 1A, 4A), which in turn can control the activity of the brain's emotional circuits, including the amygdala, hypothalamus and brainstem (23, 25). A recent imaging study observed these rapid dynamics in human brain, where uncontrollable stress exposure initially reduced the activity of BA32, which then normalized in correspondence with reducing the stress response, and BA32 increased its functional connectivity with the dlPFC (67).
The more primitive cingulate and amygdala circuits may remove the top-down regulation by higher PFC circuits through activation of catecholamine neurons in the brainstem, which can weaken PFC connectivity. The PFC and cingulate cortices receive catecholamine innervation (68) and can also regulate the activity of the monoamine nuclei in the brainstem (18, 63, 69). The dlPFC requires moderate levels of catecholamines to function, but high levels of catecholamines released during even mild uncontrollable stress rapidly take the dlPFC “offline” [reviewed in (9, 70)]. Studies in rodents have shown that psychological stressors or threatening stimuli activate projections from the amygdala, e.g., to the locus coeruleus, increasing catecholamine release in the medial PFC (71–76). High levels of catecholamines in dlPFC drive feedforward calcium-cAMP signaling, opening nearby potassium (K+) channels on spines to rapidly weaken synaptic efficacy. This reduces the recurrent excitation underlying the persistent neuronal firing needed for mental representations [reviewed in (77, 78)]. High levels of glucocorticoids, released due to hypothalamic-pituitary-adrenal (HPA) actions, can also impair PFC working memory function (79), and may do so in part by blocking the extraneuronal catecholamine transporters on glia, which normally serve to reduce catecholamine levels in the extracellular space (80). In contrast to the dlPFC, high levels of catecholamines and glucocorticoids enhance the affective functioning of the amygdala (81–83), thus flipping the brain from a reflective to reflexive state. The rapid loss of dlPFC executive and working memory functions from a hypercatecholaminergic state has now been documented in humans (84–86) in addition to the original studies in rodents and monkeys (9, 77, 78). Thus, BA25 and amygdala can rapidly remove their regulation from higher order PFC circuits through activation of excessive catecholamine release in these higher PFC regions (Figure 4B). The cingulate cortices may also inhibit dlPFC by activating inhibitory GABAergic interneurons in the dlPFC (87).
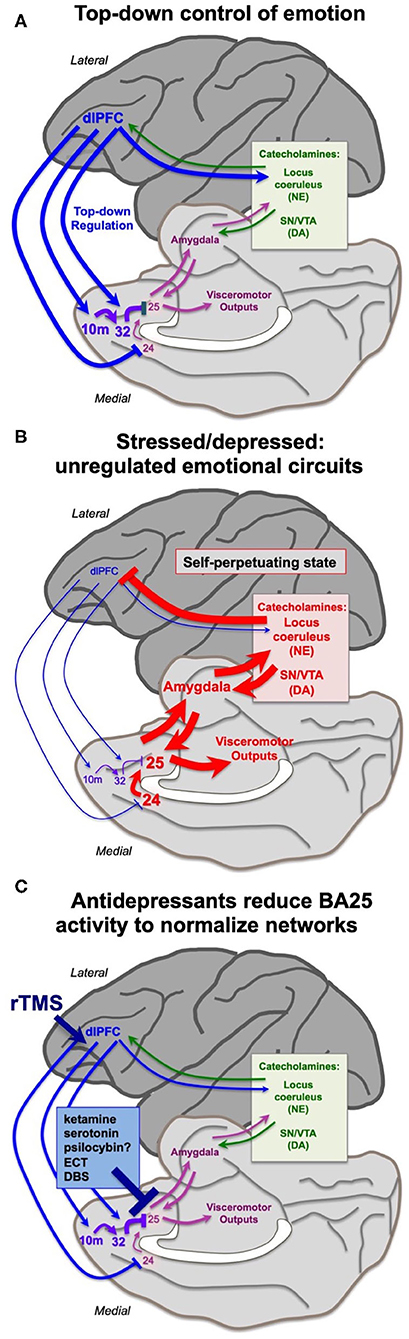
Figure 4. Hypothesis regarding the state of cortical circuits under conditions of health vs. depression, and their normalization by antidepressant treatments. (A) Under healthy conditions, the dlPFC and rostral medial PFC areas provide top-down regulation of the cingulate cortices via medial PFC connections, reducing BA25 activation of the stress response. The dlPFC also projects directly to the monoamine nuclei in the brainstem to regulate catecholamine release. (B) Under conditions of stress or depression, elevated activity in the cingulate cortices can activate the amygdala, and very high levels of catecholamine release in cortex takes higher PFC areas such as dlPFC “offline.” Thus, there is a self-perpetuating, unregulated state, where primitive circuits prevail. (C) Many antidepressant treatments reduce the activity of BA25. This may give the cortex a “foot in the door” to restore top-down regulation, especially when treatments promote dendritic spine restoration in higher PFC circuits. Other treatments may directly enhance the top-down regulation by the left dlPFC, e.g., rTMS and insight therapies.
This state of weakened higher PFC circuits and stronger BA24/BA25/amygdala control of brain responding is codified by chronic stress, which induces spine loss and dendritic retraction in PFC neurons which correlate with impaired working memory and attention regulation (88–90). Much of this research has been done in rats, where it is important to identify the projections of the neurons under study. Shansky's (91) elegant studies have shown that chronic stress exposure causes atrophy of cortico-cortical projecting mPFC neurons, but expands the dendrites of PFC neurons that activate the amygdala (i.e. those that are similar to primate BA25). Weaker connectivity and reduced gray matter in higher PFC circuits following chronic stress exposure has also been documented in humans (92, 93). Thus, chronic stress can create a self-perpetuating state where high levels of BA25/amygdala activity maintain a high catecholamine state, which simultaneously strengthens the amygdala but weakens higher PFC areas, removing inhibitory regulation of emotional response (Figure 4B). It is not known how catecholamines alter the activity of BA24 or BA25 in primates; this would be an important area for future research. Studies in rats have shown that the spine loss and dendritic retraction caused by chronic stress exposure can reverse with substantial time spent in a non-stressed state, at least in young animals, indicating a plastic dendritic response (94).
Hypothesis: The Rapid Antidepressant Actions of Ketamine May Arise from Blockade of Mental Representations Generating Aversive Mood State in Cingulate Cortices
The loss of rostral PFC and dlPFC activity in concert with increased cingulate and amygdala activation would shift mental state from an outward, cognitively-engaged frame of mind to one focused inwardly on aversive experience. This is common in depression, where there is often loss of perspective, reduced empathy for others, anhedonia, and an urgent need for relief of mental anguish (95). Symptoms such as loss of motivation and psychomotor paralysis might also arise from BA25 activation of the peri-aqueductal gray and subthalamic nucleus that are positioned to reduce motor, cognitive and affective actions. Thus, the overactive subgenual cingulate must be inhibited to give more rostral PFC and dlPFC areas a “foot in the door” to regain regulation of the brain, including the regrowth of spines in higher PFC areas (96, 97), to restore top-down higher network connections.
We have hypothesized that ketamine interrupts the self-perpetuating cycle of primitive circuit activity that is sustained by BA25 overactivity, allowing higher PFC circuits the opportunity to restore more normal functioning [Figures 4B,C; (2)]. As noted by Mayberg (51), all effective antidepressant treatments, whether pharmacological (selective serotonin reuptake inhibitors (SSRIs), possibly psilocybin?), electrical (ECT, DBS) or cognitive (talk therapy, CBT), reduce BA25 hyperactivity in depressed patients (Figure 4C). rTMS (repetitive transcranial magnetic stimulation) to strengthen the functioning of the left dlPFC may also help to restore regulation of the cingulate cortices (Figure 4C), as the efficacy of this treatment correlates with reduced activity of the anterior cingulate cortex (98), and weaker connectivity of the subgenual cingulate cortex (99). The antidepressant effects of SSRIs may be related to the very high levels of serotonin transporters in BA25 (100), although research is still needed to determine the receptor mechanisms by which serotonin can inhibit BA25 neuronal firing. We have proposed that ketamine's therapeutic effects may arise from ultra-rapid inhibition of BA25 neurons (2). As described above, systemic ketamine administration can overcome the deleterious effects of BA25 over-activation in marmosets (65), and can also normalize BA25 hyperactivity in depressed subjects (101), which may involve blockade of NMDAR transmission in the cingulate circuits representing a sustained, aversive state. Ketamine also reduces burst firing in the habenula, which may also contribute to its ultrarapid therapeutic effects (64).
Intranasal ketamine or esketamine administration may produce ultra-rapid antidepressant effects by delivering the drug directly to the anterior and subgenual cingulate cortices, which reside directly caudal to the nasal epithelium (2). Ultra-rapid effects have been documented following this route of administration, with significant improvement at 40 min (102), maximal improvement at 24 h, with therapeutic effects waning, but still evident at 48 h post-administration (102). We have proposed that the initial improvement at 40 min would arise from NMDAR blockade of excessive neuronal firing in the anterior and subgenual cingulate cortices, allowing a restoration of regulation by higher PFC areas, where spine growth would provide more sustained antidepressant actions (2).
Support for this hypothesis comes from a remarkable recent rodent study, where dendritic spine changes in medial PFC could be monitored in vivo (103). Prolonged exposure to chronic unpredictable stress increased “depressive-like behaviors” in the mice, and caused a retraction of dendritic spines in the mPFC, while systemic administration of ketamine normalized behavior and restored spine density (103). However, this study found that ketamine improved behavior prior to spine re-emergence (103), suggesting that the initial beneficial effects may arise from alterations in neuronal firing, while the longer-term, sustained antidepressant response requires regrowth of spines in PFC circuits that provide top-down regulation. Finally, our data from the dlPFC in monkeys would suggest that ketamine levels would need to dissipate before full dlPFC function could be restored, given the reliance of layer III dlPFC circuits on NMDAR-GluN2B neurotransmission. This hypothesis would be consistent with the maximal therapeutic effects observed 24 h after ketamine administration.
In closing, we are learning that NMDAR transmission is especially important for persistent neuronal firing. It is possible that the sustained neuronal activity underlying mood state, and particularly an aversive mental state, similarly relies on NMDAR transmission, and thus is relieved by NMDAR blockade from ketamine.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was1 funded by R01 MH108643-01 to AA and R01 MH093354-05 to MW.
Conflict of Interest
AA and Yale University receive royalties from Shire/Takeda from the USA sales of Intuniv. They do not receive royalties from generic or nonUSA sales.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ACC, anterior cingulate cortex; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, an ionotropic glutamate receptor; BA24, Brodmann's area 24, part of the anterior cingulate cortex; BA25, Brodmann's area 25, also known as the subgenual cingulate cortex; BA32, Brodmann's area 32, part of the ventromedial cortex; BA46, Brodmann's area 46, part of the dorsolateral prefrontal cortex; dlPFC, dorsolateral prefrontal cortex; GABA, Gamma-AminoButyric Acid, an inhibitory neurotransmitter; GluN2B, a subunit of the NMDAR, which closes slowly and fluxes high levels of calcium; HPA axis, Hypothalamus Pituitary Adrenal gland axis for control of cortisol release from the adrenal cortex (corticosterone in rodents); immunoEM, Immunoelectron microscopy; PFC, prefrontal cortex; LIP, lateral intraparietal cortex specialized for analyzing visual space; M1R, cholinergic muscarinic M1 receptor; mPFC, medial prefrontal cortex; MT, middle temporal visual cortical area specialized for analyzing visual motion; Nic, α7R, cholinergic nicotinic α7 receptor; NMDAR, N-methyl-D-aspartate receptor, an ionotropic glutamate receptor; PFC, prefrontal cortex; PSD, postsynaptic density; V1, primary visual cortex.
References
1. Arnsten AF, Murray JD, Seo H, Lee D. Ketamine's antidepressant actions: potential mechanisms in the primate medial prefrontal circuits that represent aversive experience. Biol Psychiatry. (2016) 79:713–5. doi: 10.1016/j.biopsych.2016.02.014
2. Opler LA, Opler MG, Arnsten AFT. Ameliorating treatment-refractory depression with intranasal ketamine: potential NMDA receptor actions in the pain circuitry representing mental anguish. CNS Spectrums. (2016) 21:12–22. doi: 10.1017/S1092852914000686
3. Qiu S, Li XY, Zhuo M. Post-translational modification of NMDA receptor GluN2B subunit and its roles in chronic pain and memory. Semin Cell Dev Biol. (2011) 22:521–9. doi: 10.1016/j.semcdb.2011.06.003
4. Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol. (2005) 563:345–58. doi: 10.1113/jphysiol.2004.080028
5. Goebel-Goody SM, Davies KD, Alvestad Linger RM, Freund RK, Browning MD. Phospho-regulation of synaptic and extrasynaptic N-methyl-d-aspartate receptors in adult hippocampal slices. Neuroscience. (2009) 158:1446–59. doi: 10.1016/j.neuroscience.2008.11.006
6. Liu XB, Murray KD, Jones EG. Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J Neurosci. (2004) 24:8885–95. doi: 10.1523/JNEUROSCI.2476-04.2004
7. Wang M, Yang Y, Wang CJ, Gamo NJ, Jin LE, Mazer JA, et al. NMDA receptors subserve working memory persistent neuronal firing In dorsolateral prefrontal cortex. Neuron. (2013) 77:736–49. doi: 10.1016/j.neuron.2012.12.032
8. Yang ST, Wang M, Paspalas CP, Crimins JL, Altman MT, Mazer JA, et al. Core differences in synaptic signaling between primary visual and dorsolateral prefrontal cortex. Cereb Cortex. (2018) 28:1458–71. doi: 10.1093/cercor/bhx357
9. Arnsten AFT, Datta D, Wang M. The genie in the bottle-magnified calcium signaling in dorsolateral prefrontal cortex. Mol Psychiatry. (2020). doi: 10.1038/s41380-020-00973-3. [Epub ahead of print].
10. Muntané G, Horvath JE, Hof PR, Ely JJ, Hopkins WD, Raghanti MA, et al. Analysis of synaptic gene expression in the neocortex of primates reveals evolutionary changes in glutamatergic neurotransmission. Cereb Cortex. (2015) 25:1596–607. doi: 10.1093/cercor/bht354
11. Burt JB, Demirtas M, Eckner WJ, Navejar NM, Ji JL, Martin WJ, et al. Hierarchy of transcriptomic specialization across human cortex captured by structural neuroimaging topography. Nature Neurosci. (2018) 21:1251–9. doi: 10.1038/s41593-018-0195-0
12. Badre D, D'esposito M. Functional magnetic resonance imaging evidence for a hierarchical organization of the prefrontal cortex. J Cogn Neurosci. (2007) 19:2082–99. doi: 10.1162/jocn.2007.19.12.2082
13. Tsujimoto S, Genovesio A, Wise SP. Frontal pole cortex: encoding ends at the end of the endbrain. Trends Cogn Sci. (2011) 15:169–76. doi: 10.1016/j.tics.2011.02.001
14. Price DD. Psychological and neural mechanisms of the affective dimension of pain. Science. (2000) 288:1769–72. doi: 10.1126/science.288.5472.1769
15. Seo H, Lee D. Behavioral and neural changes after gains and losses of conditioned reinforcers. J Neurosci. (2009) 29:3627–41. doi: 10.1523/JNEUROSCI.4726-08.2009
16. Ito S, Stuphorn V, Brown JW, Schall JD. Performance monitoring by the anterior cingulate cortex during saccade countermanding. Science. (2003) 302:120–2. doi: 10.1126/science.1087847
17. An X, Bandler R, Ongür D, Price JL. Prefrontal cortical projections to longitudinal columns in the midbrain periaqueductal gray in macaque monkeys. J Comp Neurol. (1998) 401:455–79. doi: 10.1002/(SICI)1096-9861(19981130)401:4<455::AID-CNE3>3.0.CO;2-6
18. Freedman LJ, Insel TR, Smith Y. Subcortical projections of area 25 (subgenual cortex) of the macaque monkey. J Comp Neurol. (2000) 421:172–88. doi: 10.1002/(SICI)1096-9861(20000529)421:2<172::AID-CNE4>3.0.CO;2-8
19. Haynes WI, Haber SN. The organization of prefrontal-subthalamic inputs in primates provides an anatomical substrate for both functional specificity and integration: implications for Basal Ganglia models and deep brain stimulation. J Neurosci. (2013) 4804–14. doi: 10.1523/JNEUROSCI.4674-12.2013
20. Pandya DN, Dye P, Butters N. Efferent cortico-cortical projections of the prefrontal cortex in the rhesus monkey. Brain Res. (1971) 31:35–46. doi: 10.1016/0006-8993(71)90632-9
21. Pandya DN, Van Hoesen GW, Mesulam MM. Efferent connections of the cingulate gyrus in the rhesus monkey. Exp Brain Res. (1981) 42:319–30. doi: 10.1007/BF00237497
22. Barbas H, Pandya DN. Architecture and intrinsic connections of the prefrontal cortex in the rhesus monkey. J Comp Neurol. (1989) 286:353–75. doi: 10.1002/cne.902860306
23. Barbas H, Joyce MK, Garcia-Cabezas MA, John Y. Serial prefrontal pathways are positioned to balance cognition emotion in primates. J Neurosci. (2020). 40:8306–28. doi: 10.1523/JNEUROSCI.0860-20.2020
24. Barbas H, Wang J, Joyce MKP, García-Cabezas MÁ. Pathway mechanism for excitatory and inhibitory control in working memory. J Neurophysiol. (2018) 120:2659–78. doi: 10.1152/jn.00936.2017
25. Joyce MKP, Barbas H. Cortical connections position primate area 25 as a keystone for interoception, emotion, and memory. J Neurosci. (2018) 38:1677–98. doi: 10.1523/JNEUROSCI.2363-17.2017
26. Wise SP. Forward frontal fields: phylogeny and fundamental function. Trends Neurosci. (2008) 31:599–608. doi: 10.1016/j.tins.2008.08.008
27. Wallis CU, Cardinal RN, Alexander L, Roberts AC, Clarke HF. Opposing roles of primate areas 25 and 32 and their putative rodent homologs in the regulation of negative emotion. Proc Natl Acad Sci USA. (2017) 114:E4075–84. doi: 10.1073/pnas.1620115114
28. Heidbreder CA, Groenewegen HJ. The medial prefrontal cortex in the rat: evidence for a dorso-ventral distinction based upon functional and anatomical characteristics. Neurosci Biobehav Rev. (2003) 7:555–79. doi: 10.1016/j.neubiorev.2003.09.003
29. Goldman-Rakic PS. Cellular basis of working memory. Neuron. (1995) 14:477–85. doi: 10.1016/0896-6273(95)90304-6
30. Funahashi S, Bruce CJ, Goldman-Rakic PS. Mnemonic coding of visual space in the monkey's dorsolateral prefrontal cortex. J Neurophysiol. (1989) 61:331–49. doi: 10.1152/jn.1989.61.2.331
31. González-Burgos G, Barrionuevo G, Lewis DA. Horizontal synaptic connections in monkey prefrontal cortex: an in vitro electrophysiological study. Cereb Cortex. (2000) 10:82–92. doi: 10.1093/cercor/10.1.82
32. González-Burgos G, Krimer LS, Povysheva NV, Barrionuevo G, Lewis DA. Functional properties of fast spiking interneurons and their synaptic connections with pyramidal cells in primate dorsolateral prefrontal cortex. J Neurophysiol. (2005) 93:942–53. doi: 10.1152/jn.00787.2004
33. Wang XJ. Synaptic basis of cortical persistent activity: the importance of NMDA receptors to working memory. J Neurosci. (1999) 19:9587–603. doi: 10.1523/JNEUROSCI.19-21-09587.1999
34. Yang Y, Paspalas CD, Jin LE, Picciotto MR, Arnsten AFT, Wang M. Nicotinic α7 receptors enhance NMDA cognitive circuits in dorsolateral prefrontal cortex. Proc Nat Acad Sci USA. (2013) 110:12078–83. doi: 10.1073/pnas.1307849110
35. Galvin VC, Yang S-T, Paspalas CD, Yang Y, Jin LE, Datta D, et al. Muscarinic M1 receptors modulate working memory performance and activity via KCNQ potassium channels in primate prefrontal cortex. Neuron. (2020) 106:649–61. doi: 10.1016/j.neuron.2020.02.030
36. Croxson PL, Kyriazis DA, Baxter MG. Cholinergic modulation of a specific memory function of prefrontal cortex. Nat Neurosci. (2011) 14:1510–2. doi: 10.1038/nn.2971
37. Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. (2007) 27:11496–500. doi: 10.1523/JNEUROSCI.2213-07.2007
38. Lynch DR, Rattelle A, Dong YN, Roslin K, Gleichman AJ, Panzer JA. Anti-NMDA receptor encephalitis: clinical features and basic mechanisms. Adv Pharmacol. (2018) 82:235–60. doi: 10.1016/bs.apha.2017.08.005
39. Weickert CS, Fung SJ, Catts VS, Schofield PR, Allen KM, Moore LT, et al. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol Psychiatry. (2012). 18:1185–92. doi: 10.1038/mp.2012.137
40. Kindler J, Lim CK, Weickert CS, Boerrigter D, Galletly C, Liu D, et al. Dysregulation of kynurenine metabolism is related to proinflammatory cytokines, attention, and prefrontal cortex volume in schizophrenia. Mol Psychiatry. (2019) 25:2860–72. doi: 10.1038/s41380-019-0401-9
41. Jacobs KR, Lim CK, Blennow K, Zetterberg H, Chatterjee P, Martins RN, et al. Correlation between plasma and CSF concentrations of kynurenine pathway metabolites in Alzheimer's disease and relationship to amyloid-β and tau. Neurobiol Aging. (2019) 80:11–20. doi: 10.1016/j.neurobiolaging.2019.03.015
42. Fukui S, Schwarcz R, Rapoport SI, Takada Y, Smith QR. Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem. (1991) 56:2007–17. doi: 10.1111/j.1471-4159.1991.tb03460.x
43. Voils SA, Shoulders BR, Singh S, Solberg LM, Garrett TJ, Frye RF. Intensive care unit delirium in surgical patients is associated with upregulation in tryptophan metabolism. Pharmacotherapy. (2020) 40:500–6. doi: 10.1002/phar.2392
44. Cai Y, Kim DJ, Takahashi T, David I, Broadhurst DI, Ma S, Rattray NJW, et al. Kynurenic acid underlies sex-specific immune responses to COVID-19. medRxiv [Preprint]. (2020). doi: 10.1101/2020.09.06.20189159
45. Singh R, Salunke DB. Diverse chemical space of indoleamine-2,3-dioxygenase 1 (Ido1) inhibitors. Eur J Med Chem. (2020) 211:113071. doi: 10.1016/j.ejmech.2020.113071
46. Thomas T, Stefanoni D, Reisz JA, Nemkov T, Bertolone L, Francis RO, et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight. (2020) 5:e140327. doi: 10.1172/jci.insight.140327
47. Murray JD, Bernacchia A, Freedman DJ, Romo R, Wallis JD, Cai X, et al. A hierarchy of intrinsic timescales across primate cortex. Nat Neurosci. (2014) 17:1661–3. doi: 10.1038/nn.3862
48. Wang H, Stradtman GGR, Wang XJ, Gao WJ. A specialized NMDA receptor function in layer 5 recurrent microcircuitry of the adult rat prefrontal cortex. Proc Natl Acad Sci USA. (2008) 105:16791–6. doi: 10.1073/pnas.0804318105
49. Gilman JP, Medalla M, Luebke JI. Area-specific features of pyramidal neurons-a comparative study in mouse and rhesus monkey. Cereb Cortex. (2017) 27:2078–94. doi: 10.1093/cercor/bhw062
50. Drevets WC, Price JL, Simpson JRJ, Todd RD, Reich T, Vannier M, et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. (1997) 386:824–7. doi: 10.1038/386824a0
51. Mayberg HS, Lozano AM, Voon V, Mcneely HE, Seminowicz D, Hamani C, et al. Deep brain stimulation for treatment-resistant depression. Neuron. (2005) 45:651–60. doi: 10.1016/j.neuron.2005.02.014
52. Dougherty DD, Weiss AP, Cosgrove GR, Alpert NM, Cassem EH, Nierenberg AA, et al. Cerebral metabolic correlates as potential predictors of response to anterior cingulotomy for treatment of major depression. J Neurosurg. (2003) 99:1010–7. doi: 10.3171/jns.2003.99.6.1010
53. Yang LK, Lu L, Feng B, Wang XS, Yue J, Li XB, et al. FMRP acts as a key messenger for visceral pain modulation. Mol Pain. (2020) 16:1744806920972241. doi: 10.1177/1744806920972241
54. Fan J, Wu X, Cao Z, Chen S, Owyang C, Li Y. Up-regulation of anterior cingulate cortex NR2B receptors contributes to visceral pain responses in rats. Gastroenterology. (2009) 136:1732–40. doi: 10.1053/j.gastro.2009.01.069
55. Zhou L, Huang J, Gao J, Zhang G, Jiang J. NMDA and AMPA receptors in the anterior cingulate cortex mediates visceral pain in visceral hypersensitivity rats. Cell Immunol. (2014) 287:86–90. doi: 10.1016/j.cellimm.2013.12.001
56. Li Y. Synaptic plasticity and synchrony in the anterior cingulate cortex circuitry: a neural network approach to causality of chronic visceral pain and associated cognitive deficits. Adv Neurobiol. (2018) 21:219–45. doi: 10.1007/978-3-319-94593-4_8
57. Li XH, Miao HH, Zhuo M. NMDA Receptor Dependent Long-term Potentiation in Chronic Pain. Neurochem Res. (2019) 44:531–8. doi: 10.1007/s11064-018-2614-8
58. Humo M, Ayazgök B, Becker LJ, Waltisperger E, Rantamäki T, Yalcin I. Ketamine induces rapid and sustained antidepressant-like effects in chronic pain induced depression: Role of MAPK signaling pathway. Prog Neuropsychopharmacol Biol Psychiatry. (2020) 100:109898. doi: 10.1016/j.pnpbp.2020.109898
59. Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. (2013) 73:1133–41. doi: 10.1016/j.biopsych.2013.03.026
60. Persson J. Ketamine in pain management. CNS Neurosci Ther. (2013) 19:396–402. doi: 10.1111/cns.12111
61. Yeaman F, Meek R, Egerton-Warburton D, Rosengarten P, Graudins A. Sub-dissociative-dose intranasal ketamine for moderate to severe pain in adult emergency department patients. Emerg Med Australas. (2014) 26:237–42. doi: 10.1111/1742-6723.12173
62. Popova V, Daly EJ, Trivedi M, Cooper K, Lane R, Lim P, et al. Efficacy and safety of flexibly dosed esketamine nasal spray combined with a newly initiated oral antidepressant in treatment-resistant depression: a randomized double-blind active-controlled study. Am J Psychiatry. (2019) 176:428–38. doi: 10.1176/appi.ajp.2019.19020172
63. Chiba T, Kayahara K, Nakano K. Efferent projections of infralimbic and prelimbic areas of the medial prefrontal cortex in the Japanese monkey, Macaca fuscata. Brain Research. (2001) 888:83–101. doi: 10.1016/S0006-8993(00)03013-4
64. Cui Y, Hu S, Hu H. Lateral habenular burst firing as a target of the rapid antidepressant effects of ketamine. Trends Neurosci. (2019) 42:179–91. doi: 10.1016/j.tins.2018.12.002
65. Alexander L, Gaskin PLR, Sawiak SJ, Fryer TD, Hong YT, Cockcroft GJ, et al. Fractionating blunted reward processing characteristic of anhedonia by over-activating primate subgenual anterior cingulate cortex. Neuron. (2019) 101:307–20. doi: 10.1016/j.neuron.2018.11.021
66. Alexander L, Wood CM, Gaskin PLR, Sawiak SJ, Fryer TD, Hong YT, et al. Over-activation of primate subgenual cingulate cortex enhances the cardiovascular, behavioral and neural responses to threat. Nat Commun. (2020) 11:5386. doi: 10.1038/s41467-020-19167-0
67. Sinha R, Lacadie CM, Constable RT, Seo D. Dynamic neural activity during stress signals resilient coping. Proc Natl Acad Sci USA. (2016) 113:8837–42. doi: 10.1073/pnas.1600965113
68. Porrino LJ, Goldman-Rakic PS. Brainstem innervation of prefrontal and anterior cingulate cortex in the rhesus monkey revealed by retrograde transport of HRP. J Comp Neurol. (1982) 205:63–76. doi: 10.1002/cne.902050107
69. Arnsten AFT, Goldman-Rakic PS. Selective prefrontal cortical projections to the region of the locus coeruleus and raphe nuclei in the rhesus monkey. Brain Res. (1984) 306:9–18. doi: 10.1016/0006-8993(84)90351-2
70. Arnsten AFT. Stress signaling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. (2009) 10:410–22. doi: 10.1038/nrn2648
71. Deutch AY, Roth RH. The determinants of stress-induced activation of the prefrontal cortical dopamine system. Prog in Brain Res. (1990) 85:367–403. doi: 10.1016/S0079-6123(08)62691-6
72. Finlay JM, Zigmond MJ, Abercrombie ED. Increased dopamine and norepinephrine release in medial prefrontal cortex induced by acute and chronic stress: effects of diazepam. Neuroscience. (1995) 64:619–28. doi: 10.1016/0306-4522(94)00331-X
73. Goldstein LE, Rasmusson AM, Bunney SB, Roth RH. Role of the amygdala in the coordination of behavioral, neuroendocrine and prefrontal cortical monoamine responses to psychological stress in the rat. J Neurosci. (1996) 16:4787–98. doi: 10.1523/JNEUROSCI.16-15-04787.1996
74. Murphy BL, Arnsten AFT, Goldman-Rakic PS, Roth RH. Increased dopamine turnover in the prefrontal cortex impairs spatial working memory performance in rats and monkeys. Proc Nat Acad Sci USA. (1996) 93:1325–9. doi: 10.1073/pnas.93.3.1325
75. Valentino RJ, Curtis AL, Page ME, Pavcovich LA, Florin-Lechner SM. Activation of the locus coeruleus brain noradrenergic system during stress: circuitry, consequences, and regulation. Adv Pharmacol. (1998) 42:781–4. doi: 10.1016/S1054-3589(08)60863-7
76. Van Bockstaele EJ, Colago EE, Valentino RJ. Amygdaloid corticotropin-releasing factor targets locus coeruleus dendrites: substrate for the co-ordination of emotional and cognitive limbs of the stress response. J Neuroendocrinol. (1998) 10:743–57. doi: 10.1046/j.1365-2826.1998.00254.x
77. Arnsten AFT. The biology of feeling frazzled. Science. (1998) 280:1711–2. doi: 10.1126/science.280.5370.1711
78. Arnsten AF. Stress weakens prefrontal networks: molecular insults to higher cognition. Nat Neurosci. (2015) 18:1376–85. doi: 10.1038/nn.4087
79. Barsegyan A, Mackenzie SM, Kurose BD, Mcgaugh JL, Roozendaal B. Glucocorticoids in the prefrontal cortex enhance memory consolidation and impair working memory by a common neural mechanism. Proc Natl Acad Sci USA. (2010) 107:16655–60. doi: 10.1073/pnas.1011975107
80. Grundemann D, Schechinger B, Rappold GA, Schomig E. Molecular identification of the cortisone-sensitive extraneuronal catecholamine transporter. Nature Neuroscience. (1998) 1:349–51. doi: 10.1038/1557
81. Cahill L, Mcgaugh JL. Modulation of memory storage. Curr Opin Neurobiol. (1996) 6:237–42. doi: 10.1016/S0959-4388(96)80078-X
82. Ferry B, Roozendaal B, Mcgaugh JL. Involvement of alpha-1-adrenoceptors in the basolateral amygdala in modulation of memory storage. Eur J Pharmacol. (1999) 372:9–16. doi: 10.1016/S0014-2999(99)00169-7
83. Rodrigues SM, Ledoux JE, Sapolsky RM. The influence of stress hormones on fear circuitry. Annu Rev Neurosci. (2009) 32:289–313. doi: 10.1146/annurev.neuro.051508.135620
84. Qin S, Hermans EJ, Van Marle HJF, Lou J, Fernandez G. Acute psychological stress reduces working memory-related activity in the dorsolateral prefrontal cortex. Biological Psychiatry. (2009) 66:25–32. doi: 10.1016/j.biopsych.2009.03.006
85. Qin S, Cousijn H, Rijpkema M, Luo J, Franke B, Hermans EJ, et al. The effect of moderate acute psychological stress on working memory-related neural activity is modulated by a genetic variation in catecholaminergic function in humans. Front Integr Neurosci. (2012) 6:16. doi: 10.3389/fnint.2012.00016
86. Zareyan S, Zhang H, Wang J, Song W, Hampson E, Abbott D, et al. First demonstration of double dissociation between COMT-Met158 and COMT-Val158 cognitive performance when stressed and when calmer. Cereb Cortex. (2020) 31:1411–26. doi: 10.1093/cercor/bhaa276
87. Medalla M, Barbas H. Anterior cingulate synapses in prefrontal areas 10 and 46 suggest differential influence in cognitive control. J Neurosci. (2010) 30:16068–81. doi: 10.1523/JNEUROSCI.1773-10.2010
88. Liston C, Miller MM, Goldwater DS, Radley JJ, Rocher AB, Hof PR, et al. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci. (2006) 26:7870–4. doi: 10.1523/JNEUROSCI.1184-06.2006
89. Radley JJ, Rocher AB, Miller M, Janssen WG, Liston C, Hof PR, et al. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex. (2006) 16:313–20. doi: 10.1093/cercor/bhi104
90. Hains AB, Vu MA, Maciejewski PK, Van Dyck CH, Gottron M, Arnsten AF. Inhibition of protein kinase C signaling protects prefrontal cortex dendritic spines and cognition from the effects of chronic stress. Proc Natl Acad Sci USA. (2009) 106:17957–62. doi: 10.1073/pnas.0908563106
91. Shansky RM, Hamo C, Hof PR, Mcewen BS, Morrison JH. Stress-induced dendritic remodeling in the prefrontal cortex is circuit specific. Cereb Cortex. (2009) 106:17957–62. doi: 10.1093/cercor/bhp003
92. Liston C, Mcewen BS, Casey BJ. Psychosocial stress reversibly disrupts prefrontal processing and attentional control. Proc Nat Acad Sci USA. (2009) 106:912–7. doi: 10.1073/pnas.0807041106
93. Ansell EB, Rando K, Tuit K, Guarnaccia J, Sinha R. Cumulative adversity and smaller gray matter volume in medial prefrontal, anterior cingulate, and insula regions. Biol Psychiatry. (2012) 72:57–64. doi: 10.1016/j.biopsych.2011.11.022
94. Bloss EB, Janssen WG, Ohm DT, Yuk FJ, Wadsworth S, Saardi KM, et al. Evidence for reduced experience-dependent dendritic spine plasticity in the aging prefrontal cortex. J Neurosci. (2011) 31:7831–9. doi: 10.1523/JNEUROSCI.0839-11.2011
95. Association AP. Diagnostic and Statistical Manual of Mental Disorders (DSM-5). Washington, D.C: American Psychiatric Association (2013). doi: 10.1176/appi.books.9780890425596
96. Li N, Lee BT, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. (2010) 329:959–64. doi: 10.1126/science.1190287
97. Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. (2012) 338:68–72. doi: 10.1126/science.1222939
98. Mottaghy FM, Keller CE, Gangitano M, Ly J, Thall M, Parker JA, et al. Correlation of cerebral blood flow and treatment effects of repetitive transcranial magnetic stimulation in depressed patients. Psychiatry Res. (2002) 115:1–14. doi: 10.1016/S0925-4927(02)00032-X
99. Fox MD, Buckner RL, White MP, Greicius MD, Pascual-Leone A. Efficacy of transcranial magnetic stimulation targets for depression is related to intrinsic functional connectivity with the subgenual cingulate. Biol Psychiatry. (2012) 72:595–603. doi: 10.1016/j.biopsych.2012.04.028
100. Baldinger P, Kranz GS, Haeusler D, Savli M, Spies M, Philippe C, et al. Regional differences in SERT occupancy after acute and prolonged SSRI intake investigated by brain PET. Neuroimage. (2014) 88:252–62. doi: 10.1016/j.neuroimage.2013.10.002
101. Morris LS, Costi S, Tan A, Stern ER, Charney DS, Murrough JW. Ketamine normalizes subgenual cingulate cortex hyper-activity in depression. Neuropsychopharmacology. (2020) 45:975–81. doi: 10.1038/s41386-019-0591-5
102. Lapidus KA, Levitch CF, Perez AM, Brallier JW, Parides MK, Soleimani L, et al. A randomized controlled trial of intranasal ketamine in Major Depressive Disorder. Biol Psychiatry. (2014) 76:970–6. doi: 10.1016/j.biopsych.2014.03.026
Keywords: NMDAR (NMDA receptor), prefrontal cortex, cingulate cortex, working memory, depression
Citation: Yang S, Seo H, Wang M and Arnsten AFT (2021) NMDAR Neurotransmission Needed for Persistent Neuronal Firing: Potential Roles in Mental Disorders. Front. Psychiatry 12:654322. doi: 10.3389/fpsyt.2021.654322
Received: 15 January 2021; Accepted: 25 February 2021;
Published: 09 April 2021.
Edited by:
Nevena V. Radonjic, Upstate Medical University, United StatesReviewed by:
Mira Jakovcevski, Max Planck Institute of Psychiatry (MPI), GermanyWen-Jun Gao, Drexel University, United States
Copyright © 2021 Yang, Seo, Wang and Arnsten. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amy F. T. Arnsten, YW15LmFybnN0ZW5AeWFsZS5lZHU=