 Wenting Zhuang
Wenting Zhuang Tong Ye
Tong Ye Wei Wang
Wei Wang Weihong Song
Weihong Song Tao Tan
Tao Tan- 1Oujiang Laboratory (Zhejiang Lab for Regenerative Medicine, Vision and Brain Health), Key Laboratory of Alzheimer's Disease of Zhejiang Province, Institute of Aging, Wenzhou Medical University, Wenzhou, China
- 2Department of Neuroscience, Baylor College of Medicine, Houston, TX, United States
CTNNB1 is the gene that encodes β-catenin which acts as a key player in the Wnt signaling pathway and regulates cellular homeostasis. Most CTNNB1-related studies have been mainly focused on its role in cancer. Recently, CTNNB1 has also been found involved in neurodevelopmental disorders (NDDs), such as intellectual disability, autism, and schizophrenia. Mutations of CTNNB1 lead to the dysfunction of the Wnt signaling pathway that regulates gene transcription and further disturbs synaptic plasticity, neuronal apoptosis, and neurogenesis. In this review, we discuss a wide range of aspects of CTNNB1 and its physiological and pathological functions in the brain. We also provide an overview of the most recent research regarding CTNNB1 expression and its function in NDDs. We propose that CTNNB1 would be one of the top high-risk genes for NDDs. It could also be a potential therapeutic target for the treatment of NDDs.
1. Introduction
In humans, CTNNB1 (transcript number: NM_001904) is located at chromosome 3q 22.1 (chr3:41240942–41281939). Its typical transcript contains 14 protein-coding exons, which encode β-catenin consisting of 781 amino acids (1). In mice, Ctnnb1 is located at chromosome 9. β-catenin is a key component of the complex that mediates intercellular adhesion and is a critical downstream molecule of the canonical Wnt signaling pathway (2). CTNNB1 was initially described as a potential tumor suppressor gene as somatic Ctnnb1 deletion or mutation, which is accompanied by loss of gene expression, is linked to tumor development in a variety of cancers (3). Further studies also suggest its role in the CNS, especially in cortical development (4), neural stem cell proliferation and neurogenesis (5), neuronal apoptosis (6), cell adhesion (7), and synaptic connection (8).
Neurodevelopmental disorders (NDDs) are a class of highly heritable and heterogeneous disorders caused by defects during early brain development, including autism spectrum disorder (ASD), intellectual disability (ID), schizophrenia (SCZ), and epilepsy (9). The prevalence of ID is about 1% in the UK as reported in 2017 (10). And 1 in 44 children were diagnosed with ASD in the United States in 2021 (11). Although it occurs during childhood, the clinical manifestations of NDDs will last for life, which causes a serious socioeconomic burden. Risk factors of NDDs include genetic, epigenetic, and environmental factors, among which the genetic factors play an important role in the pathogenesis of NDDs (12). In recent years, abundant evidence has been accumulated to draw the causal relationship between CTNNB1 and NDDs (13).
In this review, we discuss a wide range of aspects of CTNNB1 and its physiological and pathological functions in the brain. We also provide an overview of the most recent research regarding CTNNB1 expression and its function in neurodevelopmental disorders.
2. CTNNB1 and Wnt/β-catenin signaling pathway
CTNNB1 encodes β-catenin which is a key player in the canonical Wnt/β-catenin signaling pathway. This pathway is highly conserved and plays a broad role in almost all tissues (14). The Wnt/β-catenin signaling pathway is activated (on-state, left panel of Figure 1) when Wnt binds to the receptor complex composed of 7-transmembrane helix Frizzled and low-density lipoprotein receptor-related proteins (LRP5/6) (15). It phosphorylates LRP6 and recruits Dishevelled (DVL), a proteins serve as a pivotal mediator of Wnt/β-catenin signaling (16), that restrains the destruction complex (GSK-3β, CKIα, Axin, and APC) to formulate. Following this, the destruction complex attaches itself to the cell surface and inhibits the phosphorylation of β-catenin, thereby preventing its ubiquitination. And the stabilized cytoplasmic β-catenin then translocate into the nucleus, where it works with the transcription factors, particularly TCF and LEF, to control the transcription of target genes (17).
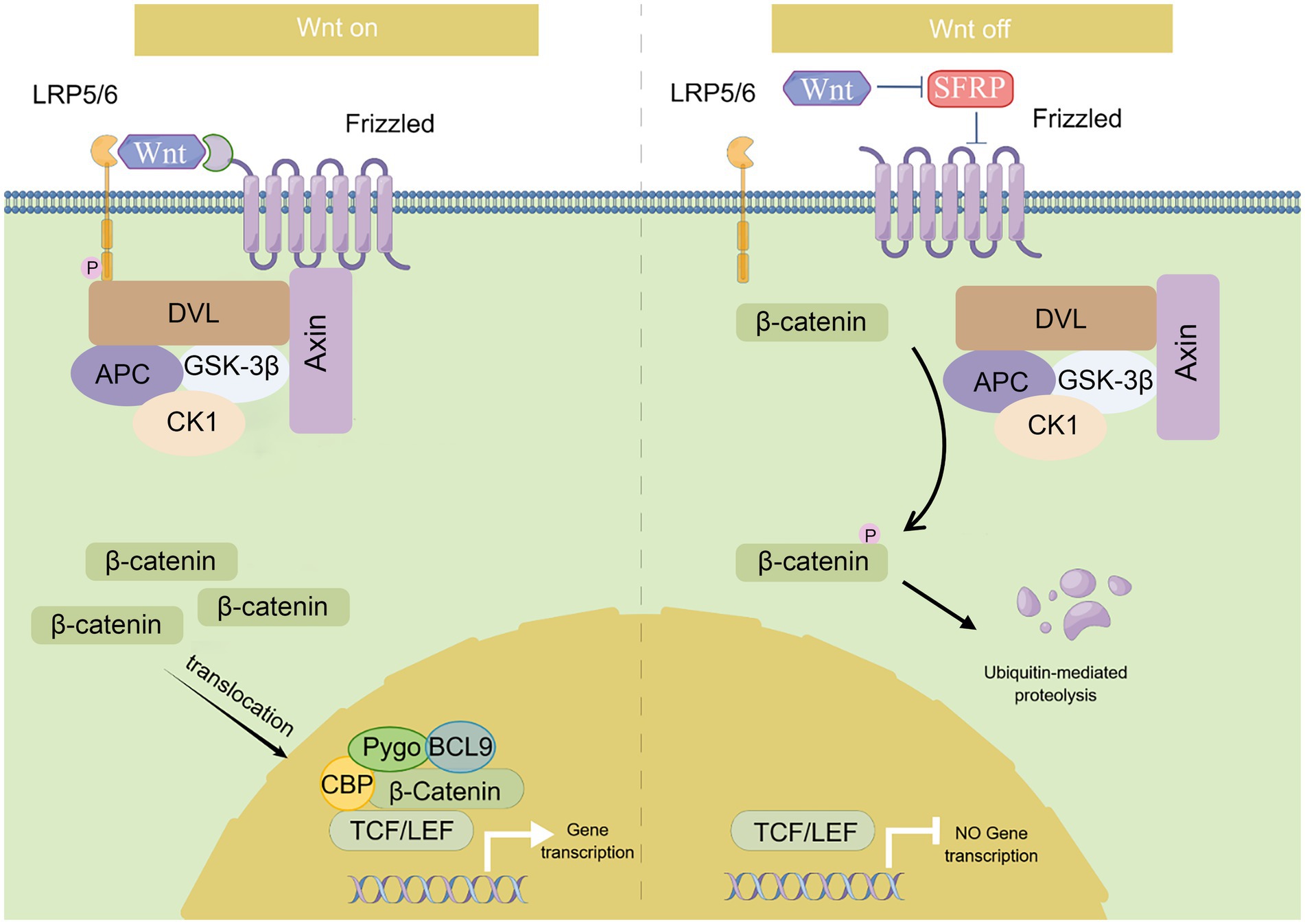
Figure 1. Summary of Wnt/β-catenin signaling pathway. This diagram depicts the canonical Wnt/β-catenin signaling pathway. Wnt/β-catenin signaling is activated (on-state) in the presence of Wnt ligand, which binds to Frizzled receptor and LRP5/6, and recruits Dishevelled (DVL) which restrains the destruction complex (GSK-3β, CK1, Axin, and APC) to formulate. β-catenin is accumulated in the cytoplasm and translocates into the nucleus where it facilitates gene transcription as a co-activator of LEF1/TCFs. While in the absence of the Wnt ligand (off-state), this signaling pathway is inactivated. β-catenin is phosphorylated by the destruction complex and then ubiquitinated by the SCF (Skp1/cullin/F-box) complex for proteasome degradation.
This pathway is inactivated (off-state, right panel for Figure 1) when Wnt is absent. The cytoplasmic destruction complex transports β-catenin to the proteasome for degradation (18). In the absence of β-catenin, the activity of the TCF/LEF transcription factor is suppressed in the nucleus. This pathway is primarily involved in regulating cell differentiation, proliferation, and inflammation (19–21). Early developmental studies in mice have also shown that Ctnnb1 regulates the Wnt signaling pathway at the transcriptional level in preimplantation embryonic development and gastrulation (22).
In addition to its role in the Wnt/β-catenin signaling pathway, β-catenin is also a crucial component in the formation and maintenance of adherens junctions (AJs). AJs are specific types of cell–cell junctions that provide tissue stability by binding to the cytoskeleton and forming extracellular bonds with neighboring cells (23, 24). They are essential for morphogenesis and remodeling of tissues and organs. AJs also play a significant role in the cortical development (25) and maintenance of the neuroepithelium’s structural integrity (26), particularly in the early telencephalon, where cell–cell adhesion is mediated by β-catenin. Moreover, β-catenin is also known to involve in cadherin-mediated adhesion. As a key component of cadherin adhesion complexes, β-catenin functions as a scaffolding protein that stabilizes the cadherin adhesion complex and anchors it to the actin cytoskeleton, thus promoting the formation of strong cell–cell contacts (26). The adhesion complex is present in both pre- and postsynaptic regions and has a significant impact on the modulation of synapse formation and synaptic plasticity (27).
3. CTNNB1 transcription in the central nervous system
CTNNB1 is widely expressed in multiple tissues and organs and is highly expressed in the brain, uterus, lung, bladder, and kidney et al. (28). In the adult primate brain, CTNNB1 is highly expressed in the gray matter of the dorsolateral prefrontal cortex (DLPFC) and hippocampus (29, 30). To evaluate the comparative temporospatial transcription of CTNNB1 in the human central nervous system (CNS), we searched the Human Brain Transcriptome (HBT) dataset (https://hbatlas.org/, search gene Ctnnb1), which is based on Affymetrix GeneChip arrays (31). As showing in Figure 2A, CTNNB1 transcription is comparable and uniform in multiple brain regions, including the neocortex (NCX), hippocampus (HIP), mediodorsal nucleus of the thalamus (MD), and cerebellar cortex (CBC). Relatively higher CTNNB1 transcription level was found in all brain regions during the early embryonic development stage. During fetal development, the expression of CTNNB1 decreased constantly. But in NCX and CBC, it increased gradually from late embryonic to infancy and remained stable from childhood to adulthood. We also noticed higher CTNNB1 transcription in CBC and MD, and lower expression in the amygdala (AMY), and striatum (STR), compared to other brain regions, which persisted from late embryonic throughout adulthood.
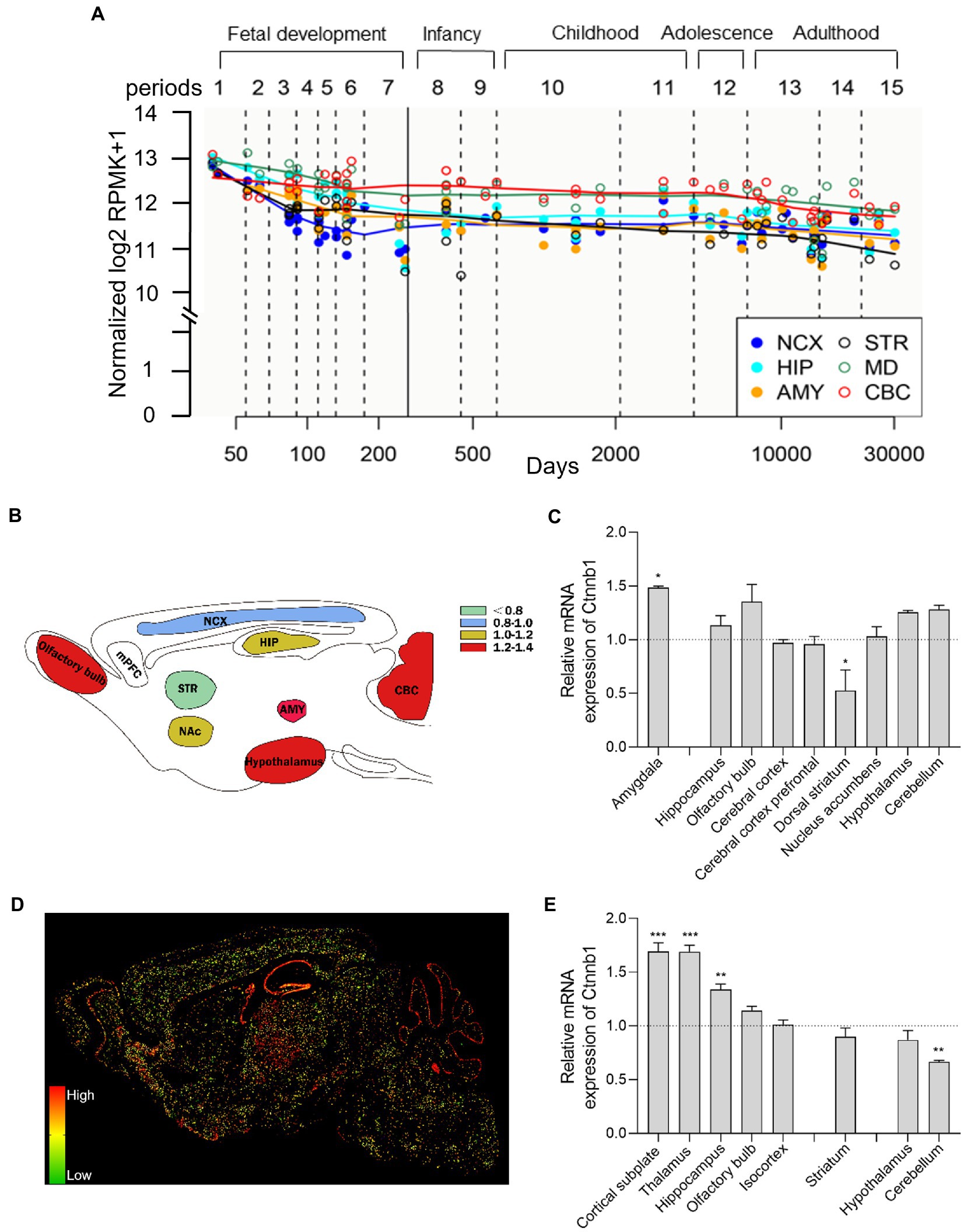
Figure 2. Brain-wide CTNNB1 expression and distribution. (A) Trajectory plot showing the expression of CTNNB1 during fetal development (period 1–7), early stages (period 8–9), youth (period 10–11), puberty (period 12), and adulthood (period 13–15) from different brain regions in human: NCX (the neocortex), HIP (hippocampus), AMY (amygdala), STR (striatum), MD (mediodorsal nucleus of the thalamus), and CBC (cerebellar cortex). Reprinted and modified from Human Brain Transcriptome dataset (https://hbatlas.org/, search gene Ctnnb1). (B,C) Schematic illustration and bar graph of Ctnnb1 expression in different brain regions of mice by using high-throughput gene expression profiling. Data obtained from BioGPS (http://biogps.org/#goto=welcome, search gene Ctnnb1). (D,E) The sagittal section and bar graph of the in-situ hybridization of Ctnnb1 mRNA signals in mice, obtained from Allen Institute ISH data (https://mouse.brain-map.org/, experiment 196, 197, 79904258). One–Way ANOVA with Holm-Sidak’s multiple comparisons test compared to whole-brain average expression, ***p < 0.001, **p < 0.01, *p < 0.05.
To explore the brain-wide Ctnnb1 transcription in mice, we searched the high-throughput gene expression profiling of mouse brains from bioGPS (http://biogps.org/#goto=welcome, search gene Ctnnb1). As shown in Figures 2B,C, Ctnnb1 is relatively highly expressed in the amygdala (fear condition and aggression (32)), cerebellum (responsible for balance and movement (33)), hypothalamus, and hippocampus (spatial memory and learning (34)), but below average expression level was found in the dorsal striatum (amygdala: p < 0.05, dorsal striatum: p < 0.05, One–Way ANOVA with Holm-Sidak’s multiple comparisons test compared to whole-brain average expression).
To further explore the spatial distribution of Ctnnb1 transcription, we searched the in-situ hybridization (ISH) data from the Allen Brain Atlas (https://mouse.brain-map.org, experiment 196, 197, 79904258). Consistent with the high-throughput gene data from the bulk sample, the ISH data also showed high Ctnnb1 mRNA levels in the cortical subplate, hippocampus, and olfactory bulb. Moreover, higher Ctnnb1 transcription was also confirmed in the thalamus [the relay station of the brain (35) (Figures 2D,E, cortical subplate and thalamus: p < 0.001, hippocampus: p < 0.01, One–Way ANOVA with Holm-Sidak’s multiple comparisons test compared to whole-brain average expression)]. However, CTNNB1 transcription level was found average in the striatum and hypothalamus, but lower in the cerebellum (p < 0.01, One–Way ANOVA with Holm-Sidak’s multiple comparisons test compared to whole-brain average expression). Interestingly, the ISH data also provide information on layer-specific distribution, which showed significantly higher expression in the pyramidal and granule cell layer of CA1 and DG of the hippocampus and Purkinje layer of the cerebellum (Figure 2D).
Overall, both human and mouse data demonstrate relatively higher gene transcription of CTNNB1 or Ctnnb1 in the hippocampus, cerebellum, and thalamus in a layer-specific manner.
To investigate the significant role of CTNNB1 in the etiology and progression of NDDs, we conducted a comprehensive literature search in PubMed from 1/1/2002 to 11/31/2022, utilizing the following search terms: [(Ctnnb1) OR (CTNNB1) OR (β-catenin)] AND {[(NDD) OR (neurodevelopmental disorder)] OR [(ASD) OR (autism spectrum disorder)] OR [(ID) OR (intellectual disability)] OR [(SCZ) OR (schizophrenia)]}. Only English language publications were considered for inclusion in our review.
4. CTNNB1-related NDDs in human studies
Since the first discovery of loss-of-function mutations in ID patients, CTNNB1 mutation/loss of function had been found in different kinds of NDDs (36). A summary of recent publications related to the clinical phenotypes of patients with CTNNB1 mutation is presented in Table 1. Multiple case reports have described the complicated abnormalities caused by different mutations in CTNNB1, including de novo nonsense mutation (47), haploinsufficiency (42), small de novo deletions (51), with behavioral anomalies, anxiety, impaired motor performance, cognitive impairment as major phenotypes.
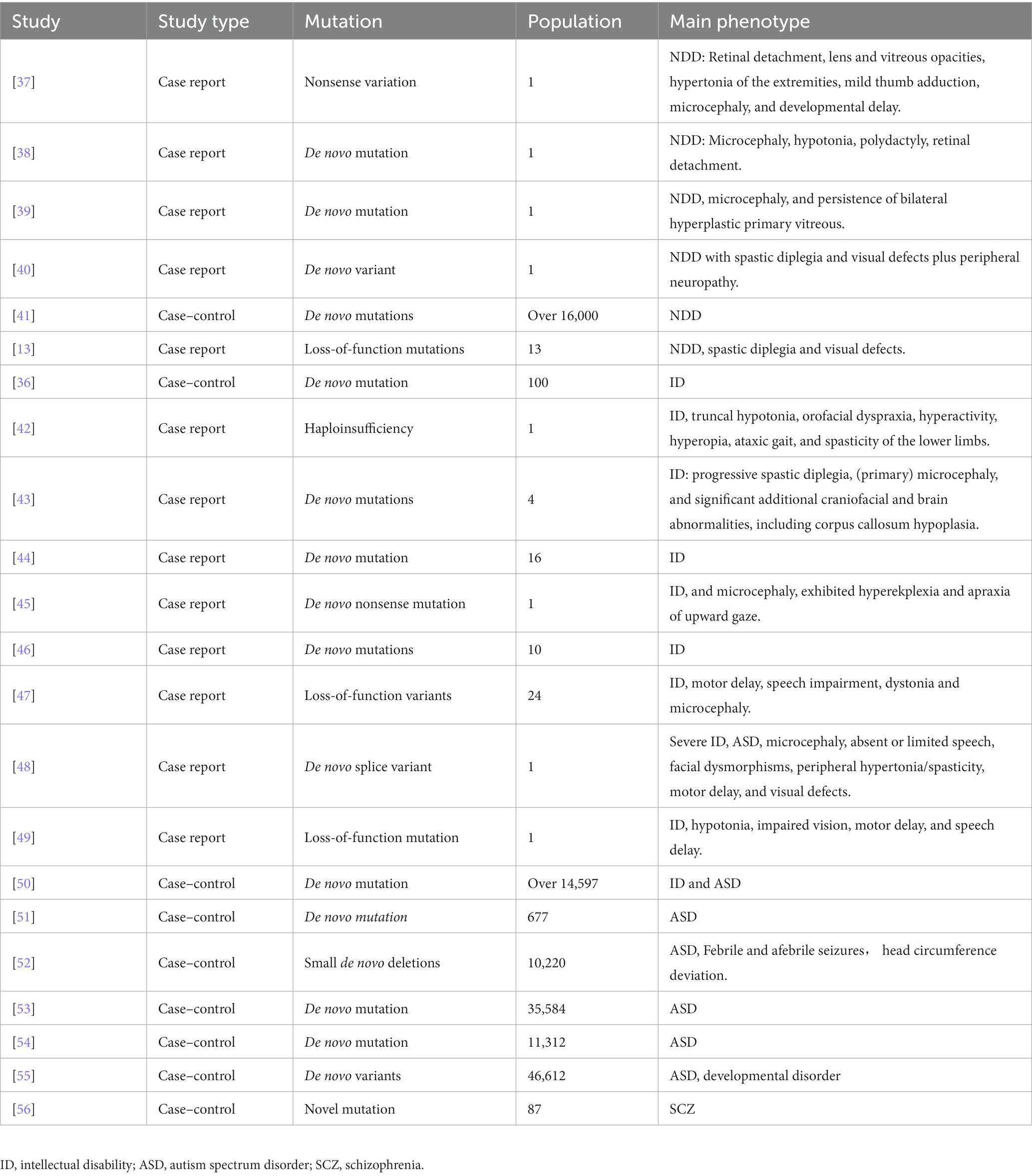
Table 1. Summary of recent CTNNB1-related NDDs in human studies.
4.1. Non-specific NDDs
Patients with the CTNNB1 mutation have been found to exhibit NDDs-like characteristics. Four case reports have described the NDDs features of patients with CTNNB1 mutations: a patient (a 15-month-old boy) with a novel heterozygous nonsense variation (c.1627C > T, p.Gln558X) in exon 11 of the CTNNB1 displayed retinal detachment, lens and vitreous opacities, hypertonia of the extremities, mild thumb adduction, microcephaly, and developmental delay (37); another patient (a 15-month-old girl) with a de novo mutation (c.1603C > T, p.R535X) showed spastic diplegia, visual defects, and severe language problems (38); two additional patients with de novo CTNNB1 mutations (39) and/or variants (40) have been reported in 2022, presenting with microcephaly, persistent bilateral hyperplastic primary vitreous, and visual defects; 13 Korean patients, who were found to have CTNNB1 pathogenic loss-of-function variants (specifically c.1867C > T, p.Gln623Ter and c.1420C > T, p.Arg474Ter) manifested developmental delay and ID (13). CTNNB1 has further been identified as one of the high-risk NDDs genes in a large population (16,000) of NDDs cases, compared to nonpsychiatric controls, through large-scale targeted sequencing (41). The evidence suggests a potential association between mutations or nonsense variations in the CTNNB1 gene and NDDs.
Furthermore, multiple studies also clearly claimed the ID, ASD, or SCZ-like phenotypes caused by various CTNNB1 mutations.
4.2. ID
CTNNB1 was first identified as a potential candidate gene for ID in 2012, through diagnostic exome sequencing of 100 ID patients (36). Subsequent case reports have described ID patients with various types of CTNNB1 mutations: including a 3-year-old girl with a de novo 333 kb microdeletion who presented with developmental delay and postnatal microcephaly (42); 4 ID patients with CTNNB1 haploinsufficiency had abnormal craniofacial development (43); 16 ID patients with de novo heterozygous CTNNB1 mutations (44); an 11-year-old boy with a de novo mutation of CTNNB1 who presented with ataxia, truncal hypotonia, and speech impairment (45); an Iranian 8-year-old girl, with a loss-of-function mutation (c.1014G > A, p.Trp338Ter) in exon 7 of the CTNNB1 gene, exhibited a range of clinical manifestations including ID, hypotonia, impaired vision, motor delay, and speech delay (49). A study of 10 ID patients suggests that CTNNB1 mutations may be more prevalent in females, with 7 out of the 10 patients being female (46).
4.3. ID with ASD
Meanwhile, both ID and ASD-like features have been reported in patients with CTNNB1 mutations. For example, a case study reported a 32-year-old female patient with a de novo splice variant in CTNNB1 who presented with microcephaly, absent or limited speech, facial dysmorphisms, peripheral hypertonia/spasticity, motor delay, and visual defects (48). Additionally, 13 patients carrying the de novo truncating variant in CTNNB1 displayed impaired motor performance, ophthalmologic problems, and hand stereotypy (13). Furthermore, CTNNB1 has been identified as one of 208 candidate risk genes from a cohort comprising over 11,730 individuals with ID/ASD and 2,867 controls (50).
4.4. ASD
Nonsense and missense mutations of CTNNB1 have also been identified in individuals with ASD. In 2012, O’Roak et al. conducted exome sequencing on 677 individuals from 209 families and identified 126 risk genes with severe or disruptive de novo mutations, 39% of which were highly connected to the β-catenin/chromatin remodeling protein network, including CTNNB1 (51). Similarly, Sanders et al. reported the presence of de novo mutations in CTNNB1 in ASD patients (n = 10,220 from 2,591 families) in the Simons Simplex Collection data (52). The largest exome sequencing study of ASD to date (11,986 individuals with ASD, 23,598 control individuals) identified CTNNB1 as one of the 102 ASD-associated genes at a false discovery rate (FDR) of 0.1 or less (FDRCTNNB1 = 0.001 to 0.0001) (53). It has been further categorized into gene expression regulation groups and more frequently mutated in NDDs-ascertained cohorts. In the latest release of whole-genome sequencing (WGS) data from the Autism Speaks MSSNG resource (https://research.mss.ng, 5,100 individuals with ASD and 6,212 non-ASD parents and siblings), CTNNB1 (FDR 0.001 to 0.0001) was further identified as one of the 134 ASD-associated genes (54). Based on another single-cell nuclei transcriptomic data (55), 615 NDDs candidate genes (FDR < 0.05) had been identified and CTNNB1 was recognized as high-risk ASD genes in 46,612 trios by de novo enrichment analysis. CTNNB1 is identified as a category 1 gene (strong candidate ASD gene) according to the Simons Foundation Autism Research Initiative (SFARI) database.1
4.5. SCZ
Limited studies have reported CTNNB1 mutations in schizophrenia. In 2015, through sequencing genes related to the Wnt signaling pathway, Levchenko et al. found a novel CTNNB1 mutation (1943A > G, p.N648S) in 87 schizophrenia patients (56).
To summarize, all these case reports and sequencing studies from different patients suggest that lost-of-function mutations in CTNNB1 would cause a wide range of behavioral abnormalities related to NDDs, which could be further mainly characterized into ID, ASD, or SCZ.
5. Ctnnb1 related animal studies
To gain a deeper understanding the roles of Ctnnb1 in NDDs, researchers have generated multiple mouse models with either insufficient or over-expressed levels of Ctnnb1, which has been summarized in Table 2, Figures 3, 4.
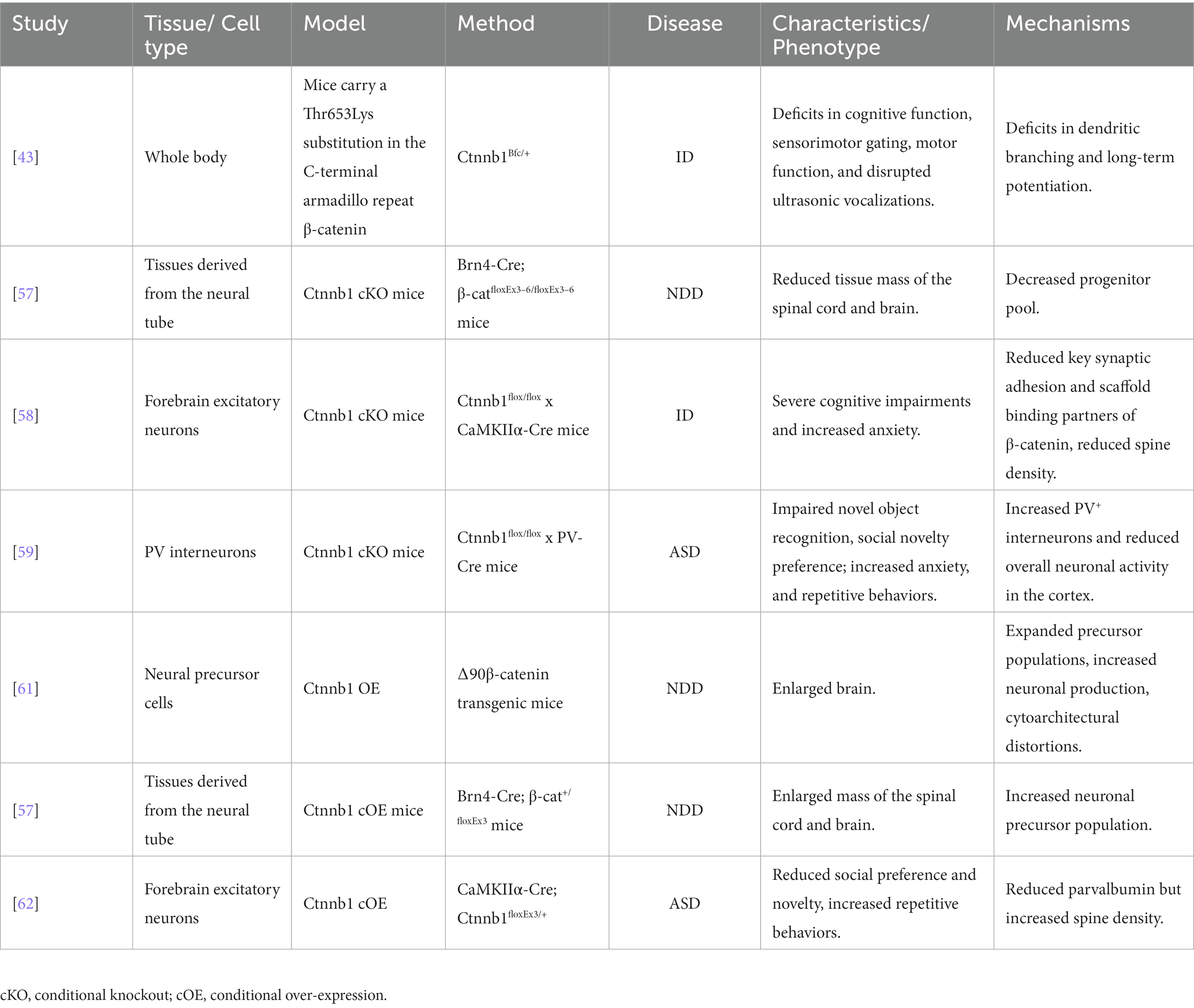
Table 2. Summary of Ctnnb1 deficiency and over-expression animal models.
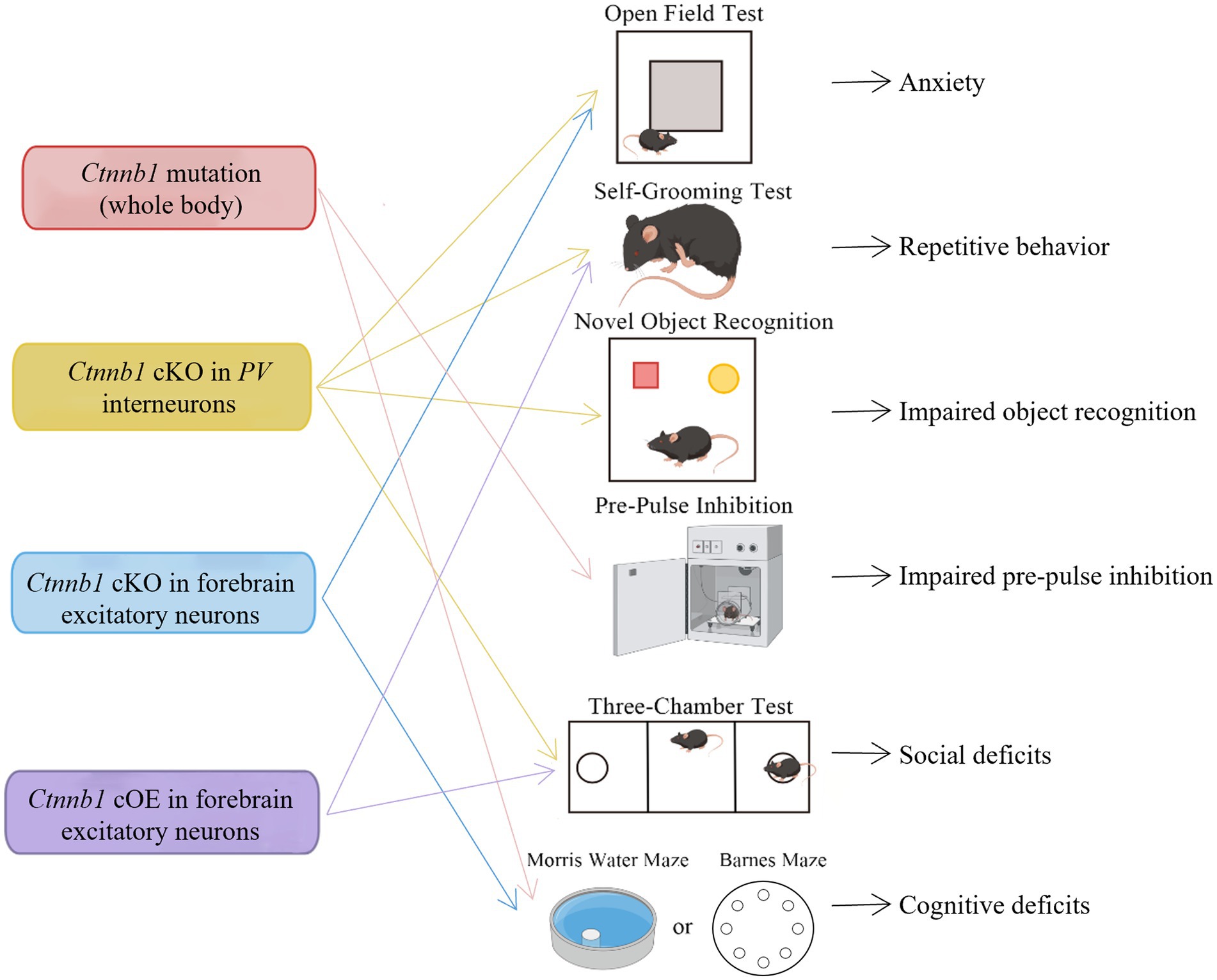
Figure 3. Behavioral deficits induced by Ctnnb1 loss of function or mutations in mice. Ctnnb1 conditional knockout (cKO) in parvalbumin (PV) interneurons induces anxiety, repetitive behavior, impaired object recognition, and social deficits. Ctnnb1 cKO in forebrain excitatory neurons causes repetitive behavior and cognitive impairment. Mice with whole-body Ctnnb1 mutations have impaired pre-pulse inhibition, repetitive behavior, as well as impaired social and cognition. Ctnnb1 cOE in excitatory neurons displayed repetitive behavior and social deficits.
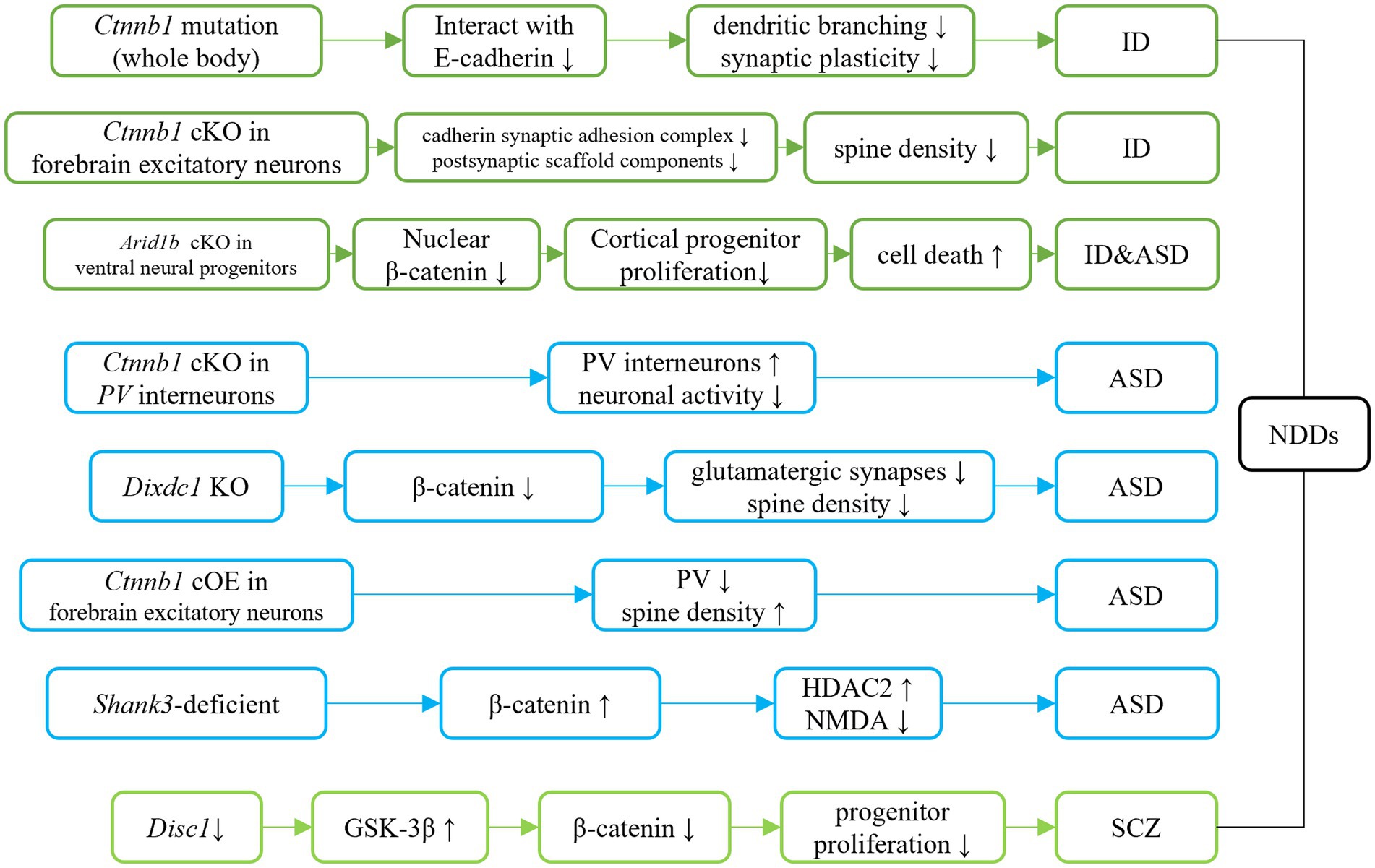
Figure 4. Ctnnb1 related signaling pathways in neurodevelopmental disorders. Dark green represents the direct/indirect link of Ctnnb1 to ID. Direct evidence of down-regulation of β-catenin suggests the β-catenin pathway involves ID. Indirect evidence indicates the Tcf4 mutation causes ID-like phenotypes in mice via down-regulation of β-catenin. Light blue represents the direct/indirect link of Ctnnb1 to ASD. Ctnnb1 deletion in PV interneuron results in social deficits in mice. Dixdc1 mutation and Shank3 mutation causes autism-like behaviors in mice via the suppressed Wnt/β-catenin signaling pathway. Disc1 deficiency impairs progenitor proliferation through the downregulation of β-catenin and introduces schizophrenia-like behaviors.
5.1. Ctnnb1 deficiency animal models
5.1.1. Whole body mutation
The Ctnnb1Bfc/+ mouse mutant (43), designated as “batface” (Bfc), exhibit abnormal craniofacial features due to a Thr653Lys substitution in the C-terminal armadillo repeat of β-catenin. This mutant also displayed ID-like behavior, including deficits in spatial learning and memory, fear conditioning, temporal cognition, and sensorimotor gating. Motor function and ultrasonic vocalizations were also disrupted in Bfc mutants (Table 2; Figure 3). The reduced affinity for membrane-associated cadherins, deficits in dendritic branching, and synaptic plasticity may contribute to the observed behavioral abnormalities (Figure 4).
5.1.2. Whole body conditional knock out
Ctnnb1 conditional knock out (cKO) mice (Brn4-Cre; β-catfloxEx3–6/floxEx3–6) (57) with a targeted deletion in tissues derived from the neural tube were generated using the Cre-Loxp system. Reduced tissue mass in the spinal cord and brain was observed in this model, potentially due to a decrease in the progenitor pool for proliferation and an increase in cell death (Table 2).
5.1.3. cKO in forebrain excitatory neurons
Mice with a Ctnnb1 cKO in forebrain excitatory neurons were generated by crossing Ctnnb1flox/flox with CaMKIIα-Cre mice (58). Severe cognitive impairments, including deficits in spatial learning and memory, as well as increased anxiety were found in this model (Table 2; Figure 3). In addition to these behavioral phenotypes, reduced levels of key synaptic adhesion and scaffold binding partners of β-catenin were observed, along with reduced spine density (Figure 4).
5.1.4. cKO in PV interneurons
The loss-of-function of Ctnnb1 in PV interneurons was generated through the crossing of Ctnnb1flox/flox with PV-Cre mice (59). These mice exhibited ASD-like behaviors, including impaired novel object recognition, reduced social novelty preference, increased repetitive behaviors, and anxiety (Table 2; Figure 3). Significantly reduced overall neuronal activity was also observed in the cortex of these cKO mice, accompanied by an increase in PV+ interneurons. These findings suggest that Ctnnb1 may play an important role in maintaining the excitatory/inhibitory balance (Figure 4).
5.1.5. Other models with indirect down-regulation of β-catenin
Instead of a mutation or knockout of Ctnnb1, several transgenic animal lines also exhibit indirect downregulation of β-catenin.
The GSK-3β/β-catenin signaling pathway was disrupted in a mouse model of schizophrenia with a loss of function of DISC1 (60). This disruption was characterized by the over-expression of GSK-3β and suppression of β-catenin. The mice exhibited behavioral deficits, including hyperlocomotion and depression-like behaviors (as demonstrated by increased immobility in the forced swimming test). These deficits were likely due to the impairment of progenitor proliferation caused by DISC1 knockdown (Figure 4). However, stable expression of β-catenin was able to override the defects in progenitor proliferation and rescue behavioral abnormalities in these mice.
Suppressed β-catenin has also been found in mice lacking DIX domain containing-1 (DIXDC1) (63), a protein involved in the intracellular Wnt/β-catenin signaling pathway, which exhibit abnormal anxiety, depression, and social deficits. Pyramidal neurons in the brains of these animals had reduced dendritic spines and glutamatergic synapses (Figure 4). Treatment with lithium or a GSK-3β inhibitor showed to correct the behavioral and neurodevelopmental phenotypes in these animals.
Decreased nucleus localization of β-catenin was found in mice with Arid1b cKO in ventral neural progenitors (Dlx-Cre; Arid1bflox/flox) (64), that displayed pronounced ID and ASD-like behaviors. Decreased proliferation of cortical and ventral neural progenitors, as well as altered cell cycle regulation and increased cell death, were also observed in this animal model (Figure 4).
5.2. Ctnnb1 over-expression animal models
Additionally, animal models with over-expression of Ctnnb1 have also been developed.
5.2.1. Whole body conditional over-expression
The Brn4-Cre; β-cat+/floxEx3 mice (57) were generated with conditional over-expression (cOE) of Ctnnb1. These mice with gain-of-function in β-catenin exhibited an enlarged mass of the spinal cord and brain, which may cause by an increased neuronal precursor population (Table 2).
5.2.2. cOE in forebrain excitatory neurons
Mice with cOE of β-catenin in forebrain excitatory neurons (CamKIIα-Cre; Ctnnb1floxEx3/+) (62) demonstrated reduced social and social novelty preference, as well as increased repetitive behaviors, such as repetitive circling (Table 2; Figure 3). Next-generation sequencing of RNA revealed dysregulation of several genes, including those involved in neuron projection development, neuron differentiation, canonical Wnt target genes, as well as several genes that have been implicated in human ASD. The underlying mechanisms are likely due to reduced parvalbumin at both the mRNA and protein levels, as well as increased dendritic spine density (Figure 4).
5.2.3. Other models with indirect up-regulation of β-catenin
In a widely utilized animal model of ASD, Shank3-deficient mice, both nucleus and cytosolic β-catenin were found to be upregulated (65). These mice also exhibited high expression of HDAC2 and suppressed NMDA function (Figure 4). Downregulation of β-catenin via β-catenin shRNA resulted in improved social preference in these mice.
The above findings suggest that disruptions in β-catenin expression, whether suppressed or overexpressed, can lead to abnormalities in neuronal proliferation and differentiation, which in turn contribute to the pathogenesis of NDDs such as ID, ASD, and SCZ. These findings highlight the importance of β-catenin in normal neurodevelopment and underscore the potential therapeutic value of targeting this protein in the treatment of NDDs.
6. Potential treatment in NDDs by targeting Wnt/β-catenin signaling pathway
As the canonical Wnt/β-catenin pathway is one of the major pathways involve in NDDs (66), targeting this signaling pathway would provide potential treatment for NDDs. For the following part, we continue summarizing different kinds of drugs that target the pathogenesis of NDDs due to dysregulation of the Wnt/β-catenin pathway. Several drugs have been identified potential treatment in NDDs by regulating β-catenin directly or indirectly.
6.1. Lithium
Lithium is a widely used drug that targets the Wnt/β-catenin signaling pathway to treat psychiatric disorders. As a classic mood stabilizer, lithium inhibits GSK-3β, a key enzyme in the Wnt signaling pathway, and up-regulates β-catenin (67). Although originally used to treat mania due to its tranquilizing effects (68), several studies have explored the potential therapeutic use of lithium in NDDs. For example, lithium treatment in ASD patients with SHANK3 mutations has been shown to improve behavioral symptoms and allow patients to recover their pre-catatonia level of functioning, with limited side effects (69).
In a Fragile X mouse model, lithium down-regulated GSK-3β activity and reduced the audiogenic seizures (70). Treatment with lithium rescued the behavioral performance in mice with Tbr1 haploinsufficiency, another high-risk gene for ASD, which lasts over 6 months. Chronic lithium treatment has also been shown to enhance spatial learning and memory (71), as well as to have anti-aggression (68) and anti-depression effects (72). However, it is important to note that prolonged maintenance of high serum lithium concentrations can lead to kidney disease (73).
6.2. SB216763
SB216763 is a highly potent and selective GSK-3β inhibitor with an IC50 of 0.2 μM and 96% inhibition at 10 μM. It has been successfully utilized to boost β-catenin by inhibiting GSK-3β in the treatment of various animal models with NDDs. For instance, the administration of 2 mg/kg SB216763 can rescue SCZ-like behavioral deficits caused by Disc1 KO (60). The same dose also proved to be effective in treating hippocampus-dependent learning deficits in Fmr1 KO mice (74). Furthermore, 10 mg/kg SB216763 administration has been shown to normalize the ASD-like behavioral outcomes in Dixdc1 KO (63). SB-216763 has been shown to effectively reduce audiogenic seizures and normalize open field behavior in Fragile X mice when administered at a dosage of 4–10 mg/kg (70).
6.3. Sulindac
As an FDA-approved anti-inflammatory drug, sulindac is another commonly used drug that serves as a β-catenin inhibitor (75). Sulindac has been shown to significantly inhibit β-catenin transcription in cancer cell studies (76). It has also been evaluated in valproic acid (VPA) induced ASD models and has been found to reduce VPA-induced stereotypic-like activity, anxiety (77), and other autism-like phenotypes in rats (78, 79). In cell culture studies, it has been demonstrated that Sulindac increases cell viability and reducing oxidative stress in VPA-treated primary neurons via suppressing β-catenin mRNA level (77). These findings suggest that small-molecule targeting of the Wnt/β-catenin pathway, such as sulindac, may be a promising approach for improving behavioral phenotypes in individuals with ASD.
6.4. PPARγ agonist
Another drug targeting to Wnt/β-catenin signaling pathway in NDDs would be a peroxisome proliferator-activated receptor gamma (PPARγ) agonist. PPARγ is a ligand-activated transcriptional factor, which interacts with other coactivator proteins to induce specific gene expression (80). In the Wnt/β-catenin pathway, PPARγ interacts with TCF/LEF domain and the β-catenin-binding domain (81). As an agonist of PPAR, pioglitazone showed anti-inflammation effects on ASD patients, which alleviated ASD-like behavior, such as irritability, lethargy, stereotypy, and hyperactivity (82). In the lipopolysaccharide-induced ASD rat model, pioglitazone treatment exhibited enhancement in social interaction and vocalization (83). Meanwhile, in a maternal immune SCZ rat model, pioglitazone, also mitigated the male offspring’s schizophrenia-like behaviors when given before parturition (84).
According to these clinical and preclinical studies, targeting the Wnt/β-catenin signaling pathway with lithium, SB216763 and sulindac, or targeting the transcriptional factor with PPARγ agonist may have therapeutic effects for NDDs.
7. Conclusion and future perspectives
Over the past decade, our understanding of the role of CTNNB1 and the Wnt/β-catenin signaling pathway in brain development has greatly increased following the identification of CTNNB1 as a risk gene for NDDs. Within the somatic system, CTNNB1 was initially identified as a potential tumor suppressor gene. Mutations in this gene can cause disruptions in the Wnt signaling pathway, leading to the formation of tumors in a variety of cancers (85, 86). Furthermore, mutations in CTNNB1 can result in decreased cell adhesion and epithelial to mesenchymal transition, allowing tumor cells to migrate and metastasize (87). However, within the central nervous system, CTNNB1 mutations can lead to defects in synaptic localization and stabilization, which can cause an imbalance between excitatory and inhibitory signaling, ultimately resulting in NDDs (88, 89). These findings suggest that CTNNB1 may have distinct roles within the somatic system and central nervous system, highlighting the need for further research in this area.
Studies have demonstrated the crucial role of CTNNB1 in central nervous system development. Individuals with CTNNB1 mutations exhibit NDDs-associated phenotypes. Moreover, various animal models with Ctnnb1 deletion/de novo mutations have been generated to replicate the core clinical features observed in NDDs patients. Several reagents and targets have been identified as potential treatments for diseases related to CTNNB1 mutations.
Based on high-throughput data from the human brain, it is known that CTNNB1 is highly expressed during embryonic stages and early childhood, suggesting that CTNNB1 plays an important role in brain development. Current transgenic animal models have covered all of these stages. It would be of scientific interest to examine the biological characteristics of high CTNNB1 expression during adolescence, as this would provide valuable insights into the role of CTNNB1 in postnatal psychiatric disorders.
As summarized previously, Ctnnb1 has been identified as highly expressed in the cerebellum, hippocampus, thalamus, and cerebral cortex. However, the specific effects of Ctnnb1 in these brain regions remain unknown, as previous studies have focused on the whole body or whole brain mutation of Ctnnb1. To further understand the function of Ctnnb1 in these high-expression regions, studies using local AAV infusion into the target region are necessary. In addition, ISH data suggest that Ctnnb1 expression exhibits a specific spatial distribution, including extremely high expression in the pyramidal and granule cell layers of the CA1 and DG of the hippocampus, and the Purkinje layer of the cerebellum. ISH data also indicate layer differences within the PFC. Advanced spatial transcriptome technologies such as MERFISH (90) and stereo-seq (91) could provide valuable information on the function of Ctnnb1 in different layers. Furthermore, the use of multi-patch techniques would be beneficial in exploring the microcircuit changes.
Interestingly, variable levels of Ctnnb1 expression were observed in the cerebellum using different techniques. High-throughput sequencing of mRNA extracted from brain tissue was conducted to determine the relative mRNA expression of Ctnnb1, as presented in Figures 2B,C. This method is prone to affected by cell numbers. And since the cerebellum possesses a considerably higher number of cells, especially in the granule layer, the average expression level is relatedly high. However, in situ RNA staining was performed in Figures 2D,E to determine the spatial distribution of Ctnnb1 in brain slices. The fluorescence intensity is indicative of Ctnnb1 expression levels and may be influenced by the total volume of the specific brain region. Despite the high expression of Ctnnb1 in the granule layer of the cerebellum, the average expression level was relatively low due to the lower Ctnnb1 expression in the molecular layer of the cerebellum.
With the development of various transgenic animals, scientists have studied the effects of knocking out the Ctnnb1 in specific cell types using the Cre-Loxp system. A previous study demonstrated that cKO of Ctnnb1 in forebrain excitatory neurons leads to severe cognitive impairments (58), indicating a significant role of Ctnnb1 in pyramidal neurons. However, there is a lack of research on its function in other types of excitatory neurons, such as VGLUT2, which is a primary cell type in the thalamus (92). Additionally, cKO of Ctnnb1 in PV interneurons has been found to result in ASD-like symptoms (59), further suggesting a crucial role for Ctnnb1 in interneurons. However, the function of Ctnnb1 in other types of interneurons, such as VIP and SST interneurons, remains to be further explored. Furthermore, the function of Ctnnb1 in non-neuronal cells, including astrocytes, glia, and microglia within the brain, has not been reported and warrants further investigation.
To date, most researches have been focused on characterizing the behavioral phenotypes of different Ctnnb1 KO animal models. However, the underlying mechanisms, particularly neuroanatomical and functional changes, have not been fully investigated. Electrophysiological studies, such as Patch-seq, could provide further evidence in this regard. Additionally, long-range circuit studies have been lacking. The use of optogenetics and chemogenetics could allow for exploration in cognitive or ASD-related circuits, such as within the hippocampal CA3-CA1, cortical-striatal, cortical-thalamic, and thalamic-striatal.
In the aspects of treatment, several drugs have been proposed with therapeutic effects for NDDs. These drugs either directly modulate β-catenin function (LiCl and sulforaphane) or indirectly modulate its expression (PPARγ). Future research utilizing proteomics, RNA-seq, and ATAC-seq may help to identify additional downstream molecular targets of β-catenin and epigenetic targets for Ctnnb1 transcription.
Overall, in-depth studies are required to investigate the impact of Ctnnb1 mutations in a specific manner by brain region, cell type, and layer. Neuroanatomical and functional neural activity and transmission studies should be conducted to understand the effects on micro-circuits and long-distance circuits. New high-throughput techniques such as proteomics, RNA-seq, and ATAC-seq could be utilized to identify potential therapeutic targets for Ctnnb1-induced NDDs.
Author contributions
WZ conducted the literature search and drafted the manuscript. WW and TY wrote part of the manuscript. WS and TT designed the outline of the review, supervised the entire process, and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by funding to WS from Oujiang Laboratory (OJQDJQ2022002), to TT from Oujiang Laboratory (OJQD2022002).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
1. Valenta, T, Hausmann, G, and Basler, K. The many faces and functions of β-catenin. EMBO J. (2012) 31:2714–36. doi: 10.1038/emboj.2012.150
2. Gao, C, Wang, Y, Broaddus, R, Sun, L, Xue, F, and Zhang, W. CTNNB1Exon 3 mutations of drive tumorigenesis: a review. Oncotarget. (2018) 9:5492–508. doi: 10.18632/oncotarget.23695
3. Saegusa, M, Hashimura, M, Yoshida, T, and Okayasu, I. Beta-catenin mutations and aberrant nuclear expression during endometrial tumorigenesis. Br J Cancer. (2001) 84:209–17. doi: 10.1054/bjoc.2000.1581
4. Zhang, L, Jing, H, Li, H, Chen, W, Luo, B, Zhang, H, et al. Neddylation is critical to cortical development by regulating Wnt/beta-catenin signaling. Proc Natl Acad Sci U S A. (2020) 117:26448–59. doi: 10.1073/pnas.2005395117
5. Bond, AM, Ming, GL, and Song, H. Adult mammalian neural stem cells and neurogenesis: five decades later. Cell Stem Cell. (2015) 17:385–95. doi: 10.1016/j.stem.2015.09.003
6. Rong, Y, Liu, W, Zhou, Z, Gong, F, Bai, J, Fan, J, et al. Harpagide inhibits neuronal apoptosis and promotes axonal regeneration after spinal cord injury in rats by activating the Wnt/β-catenin signaling pathway. Brain Res Bull. (2019) 148:91–9. doi: 10.1016/j.brainresbull.2019.03.014
7. el Khouri, E, Ghoumid, J, Haye, D, Giuliano, F, Drevillon, L, Briand-Suleau, A, et al. Wnt/β-catenin pathway and cell adhesion deregulation in CSDE1-related intellectual disability and autism spectrum disorders. Mol Psychiatry. (2021) 26:3572–85. doi: 10.1038/s41380-021-01072-7
8. Song, S, Zhang, B, Sun, H, Li, X, Xiang, Y, Liu, Z, et al. A Wnt-Frz/Ror-Dsh pathway regulates neurite outgrowth in Caenorhabditis elegans. PLoS Genet. (2010) 6:e1001056. doi: 10.1371/journal.pgen.1001056
9. Morris-Rosendahl, DJ, and Crocq, MA. Neurodevelopmental disorders-the history and future of a diagnostic concept. Dialogues Clin Neurosci. (2020) 22:65–72. doi: 10.31887/DCNS.2020.22.1/macrocq
10. Hughes-McCormack, LA, Rydzewska, E, Henderson, A, MacIntyre, C, Rintoul, J, and Cooper, SA. Prevalence of mental health conditions and relationship with general health in a whole-country population of people with intellectual disabilities compared with the general population. BJPsych Open. (2017) 3:243–8. doi: 10.1192/bjpo.bp.117.005462
11. Maenner, M., Shaw, K., Bakian, A., Bilder, D., Durkin, M., Esler, A., Furnier, S., Hallas, L., Hall-Lande, J., Hudson, A., et al. (2021). Prevalence and characteristics of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2018. Morbidity and mortality weekly report surveillance summaries. Washington, DC: 2002. 70, 1-16.
12. Reichard, J, and Zimmer-Bensch, G. The epigenome in neurodevelopmental disorders. Front Neurosci. (2021) 15:776809. doi: 10.3389/fnins.2021.776809
13. Lee, S, Jang, SS, Park, S, Yoon, JG, Kim, SY, Lim, BC, et al. The extended clinical and genetic spectrum of CTNNB1-related neurodevelopmental disorder. Front Pediatr. (2022) 10:960450. doi: 10.3389/fped.2022.960450
14. Kwan, V, Unda, B, and Singh, K. Wnt signaling networks in autism spectrum disorder and intellectual disability. J Neurodev Disord. (2016) 8:45. doi: 10.1186/s11689-016-9176-3
15. Nusse, R, and Clevers, H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cells. (2017) 169:985–99. doi: 10.1016/j.cell.2017.05.016
16. Shi, DL. Decoding Dishevelled-mediated Wnt signaling in vertebrate early development. Front Cell Dev Biol. (2020) 8:588370. doi: 10.3389/fcell.2020.588370
17. Caracci, MO, Avila, ME, Espinoza-Cavieres, FA, Lopez, HR, Ugarte, GD, and De Ferrari, GV. Wnt/beta-catenin-dependent transcription in autism spectrum disorders. Front Mol Neurosci. (2021) 14:764756. doi: 10.3389/fnmol.2021.764756
18. Stamos, JL, and Weis, WI. The beta-catenin destruction complex. Cold Spring Harb Perspect Biol. (2013) 5:a007898. doi: 10.1101/cshperspect.a007898
19. Perugorria, MJ, Olaizola, P, Labiano, I, Esparza-Baquer, A, Marzioni, M, Marin, JJG, et al. Wnt-beta-catenin signalling in liver development, health and disease. Nat Rev Gastroenterol Hepatol. (2019) 16:121–36. doi: 10.1038/s41575-018-0075-9
20. Vallée, A, and Lecarpentier, Y. Crosstalk between peroxisome proliferator-activated receptor gamma and the canonical WNT/β-catenin pathway in chronic inflammation and oxidative stress during carcinogenesis. Front Immunol. (2018) 9:745. doi: 10.3389/fimmu.2018.00745
21. Wray, J, and Hartmann, C. WNTing embryonic stem cells. Trends Cell Biol. (2012) 22:159–68. doi: 10.1016/j.tcb.2011.11.004
22. Clevers, H. Wnt/beta-catenin signaling in development and disease. Cells. (2006) 127:469–80. doi: 10.1016/j.cell.2006.10.018
23. Oda, H, and Takeichi, M. Evolution: structural and functional diversity of cadherin at the adherens junction. J Cell Biol. (2011) 193:1137–46. doi: 10.1083/jcb.201008173
24. van der Wal, T, and van Amerongen, R. Walking the tight wire between cell adhesion and WNT signalling: a balancing act for β-catenin. Open Biol. (2020) 10:200267. doi: 10.1098/rsob.200267
25. Veeraval, L, O’Leary, CJ, and Cooper, HM. Adherens junctions: guardians of cortical development. Front Cell Dev Biol. (2020) 8:6. doi: 10.3389/fcell.2020.00006
26. Junghans, D, Hack, I, Frotscher, M, Taylor, V, and Kemler, R. Beta-catenin-mediated cell-adhesion is vital for embryonic forebrain development. Dev Dynam. (2005) 233:528–39. doi: 10.1002/dvdy.20365
27. Brigidi, GS, and Bamji, SX. Cadherin-catenin adhesion complexes at the synapse. Curr Opin Neurobiol. (2011) 21:208–14. doi: 10.1016/j.conb.2010.12.004
28. Yu, X, and Malenka, RC. Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci. (2003) 6:1169–77. doi: 10.1038/nn1132
29. Joksimovic, M, and Awatramani, R. Wnt/beta-catenin signaling in midbrain dopaminergic neuron specification and neurogenesis. J Mol Cell Biol. (2014) 6:27–33. doi: 10.1093/jmcb/mjt043
30. Smith, A, Bourdeau, I, Wang, J, and Bondy, CA. Expression of catenin family members CTNNA1, CTNNA2, CTNNB1 and JUP in the primate prefrontal cortex and hippocampus. Brain Res Mol Brain Res. (2005) 135:225–31. doi: 10.1016/j.molbrainres.2004.12.025
31. Kang, HJ, Kawasawa, YI, Cheng, F, Zhu, Y, Xu, X, Li, M, et al. Spatio-temporal transcriptome of the human brain. Nature. (2011) 478:483–9. doi: 10.1038/nature10523
32. Adhikari, A, Lerner, T, Finkelstein, J, Pak, S, Jennings, J, Davidson, T, et al. Basomedial amygdala mediates top-down control of anxiety and fear. Nature. (2015) 527:179–85. doi: 10.1038/nature15698
33. Peterburs, J, and Desmond, J. The role of the human cerebellum in performance monitoring. Curr Opin Neurobiol. (2016) 40:38–44. doi: 10.1016/j.conb.2016.06.011
34. Hainmueller, T, and Bartos, M. Parallel emergence of stable and dynamic memory engrams in the hippocampus. Nature. (2018) 558:292–6. doi: 10.1038/s41586-018-0191-2
35. Moustafa, A, McMullan, R, Rostron, B, Hewedi, D, and Haladjian, H. The thalamus as a relay station and gatekeeper: relevance to brain disorders. Rev Neurosci. (2017) 28:203–18. doi: 10.1515/revneuro-2016-0067
36. de Ligt, J, Willemsen, MH, van Bon, BW, Kleefstra, T, Yntema, HG, Kroes, T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. (2012) 367:1921–9. doi: 10.1056/NEJMoa1206524
37. Li, N, Xu, Y, Li, G, Yu, T, Yao, RE, Wang, X, et al. Exome sequencing identifies a de novo mutation of CTNNB1 gene in a patient mainly presented with retinal detachment, lens and vitreous opacities, microcephaly, and developmental delay: case report and literature review. Medicine (Baltimore). (2017) 96:e6914. doi: 10.1097/MD.0000000000006914
38. Ke, Z, and Chen, Y. Case report: a de novo CTNNB1 nonsense mutation associated with neurodevelopmental disorder, retinal detachment, polydactyly. Front Pediatr. (2020) 8:575673. doi: 10.3389/fped.2020.575673
39. Zuluaga Gómez, L, Caballero Mojica, S, Vélez Rengifo, G, Bravo Acosta, J, and Montoya Villada, J. Mutación del gen CTNNB1 asociada a alteración del neurodesarrollo, microcefalia y persistencia del vítreo primario hiperplásico bilateral: reporte de un caso y revisión de la literatura. Arch Soc Esp Oftalmol. (2022) 97:44–7. doi: 10.1016/j.oftal.2020.11.028
40. Spagnoli, C, Salerno, GG, Rizzi, S, Frattini, D, Koskenvuo, J, and Fusco, C. Novel CTNNB1 variant leading to neurodevelopmental disorder with spastic diplegia and visual defects plus peripheral neuropathy: a case report. Am J Med Genet A. (2022) 188:3118–20. doi: 10.1002/ajmg.a.62902
41. Wang, T, Hoekzema, K, Vecchio, D, Wu, H, Sulovari, A, Coe, BP, et al. Large-scale targeted sequencing identifies risk genes for neurodevelopmental disorders. Nat Commun. (2020) 11:4932. doi: 10.1038/s41467-020-18723-y
42. Dubruc, E, Putoux, A, Labalme, A, Rougeot, C, Sanlaville, D, and Edery, P. A new intellectual disability syndrome caused by CTNNB1 haploinsufficiency. Am J Med Genet A. (2014) 164A:1571–5. doi: 10.1002/ajmg.a.36484
43. Tucci, V, Kleefstra, T, Hardy, A, Heise, I, Maggi, S, Willemsen, MH, et al. Dominant beta-catenin mutations cause intellectual disability with recognizable syndromic features. J Clin Invest. (2014) 124:1468–82. doi: 10.1172/JCI70372
44. Kuechler, A, Willemsen, MH, Albrecht, B, Bacino, CA, Bartholomew, DW, van Bokhoven, H, et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: expanding the mutational and clinical spectrum. Hum Genet. (2015) 134:97–109. doi: 10.1007/s00439-014-1498-1
45. Winczewska-Wiktor, A, Badura-Stronka, M, Monies-Nowicka, A, Nowicki, MM, Steinborn, B, Latos-Bielenska, A, et al. A de novo CTNNB1 nonsense mutation associated with syndromic atypical hyperekplexia, microcephaly and intellectual disability: a case report. BMC Neurol. (2016) 16:35. doi: 10.1186/s12883-016-0554-y
46. Kharbanda, M, Pilz, DT, Tomkins, S, Chandler, K, Saggar, A, Fryer, A, et al. Clinical features associated with CTNNB1 de novo loss of function mutations in ten individuals. Eur J Med Genet. (2017) 60:130–5. doi: 10.1016/j.ejmg.2016.11.008
47. Yan, D, Sun, Y, Xu, N, Yu, Y, and Zhan, Y, Mainland Chinese League of NRD. Genetic and clinical characteristics of 24 mainland Chinese patients with CTNNB1 loss-of-function variants. Mol Genet Genomic Med. (2022) 10:e2067.
48. Verhoeven, WMA, Egger, JIM, Jongbloed, RE, van Putten, MM, de Bruin-van Zandwijk, M, Zwemer, AS, et al. A de novo CTNNB1 novel splice variant in an adult female with severe intellectual disability. Int Med Case Rep J. (2020) Volume 13:487–92. doi: 10.2147/IMCRJ.S270487
49. Dashti, S, Salehpour, S, Ghasemi, M, Sadeghi, H, Rostami, M, Hashemi-Gorji, F, et al. Identification of a novel de novo mutation in the CTNNB1 gene in an Iranian patient with intellectual disability. Neurol Sci. (2022) 43:2859–63. doi: 10.1007/s10072-022-05904-4
50. Stessman, HA, Xiong, B, Coe, BP, Wang, T, Hoekzema, K, Fenckova, M, et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet. (2017) 49:515–26. doi: 10.1038/ng.3792
51. O'Roak, B, Vives, L, Girirajan, S, Karakoc, E, Krumm, N, Coe, B, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. (2012) 485:246–50. doi: 10.1038/nature10989
52. Sanders, SJ, He, X, Willsey, AJ, Ercan-Sencicek, AG, Samocha, KE, Cicek, AE, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. (2015) 87:1215–33. doi: 10.1016/j.neuron.2015.09.016
53. Satterstrom, F, Kosmicki, J, Wang, J, Breen, M, de Rubeis, S, An, J, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cells. (2020) 180:568–584.e23. doi: 10.1016/j.cell.2019.12.036
54. Trost, B, Thiruvahindrapuram, B, Chan, AJS, Engchuan, W, Higginbotham, EJ, Howe, JL, et al. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cells. (2022) 185:e4418:4409–4427.e18. doi: 10.1016/j.cell.2022.10.009
55. Wang, T, Kim, CN, Bakken, TE, Gillentine, MA, Henning, B, Mao, Y, et al. Integrated gene analyses of de novo variants from 46,612 trios with autism and developmental disorders. Proc Natl Acad Sci U S A. (2022) 119:e2203491119. doi: 10.1073/pnas.2203491119
56. Levchenko, A, Davtian, S, Freylichman, O, Zagrivnaya, M, Kostareva, A, and Malashichev, Y. Beta-catenin in schizophrenia: Possibly deleterious novel mutation. Psychiatry Res. (2015) 228:843–8. doi: 10.1016/j.psychres.2015.05.014
57. Zechner, D, Fujita, Y, Hülsken, J, Müller, T, Walther, I, Taketo, MM, et al. β-Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev Biol. (2003) 258:406–18. doi: 10.1016/S0012-1606(03)00123-4
58. Wickham, R, Alexander, J, Eden, L, Valencia-Yang, M, Llamas, J, Aubrey, J, et al. Learning impairments and molecular changes in the brain caused by β-catenin loss. Hum Mol Genet. (2019) 28:2965–75. doi: 10.1093/hmg/ddz115
59. Dong, F, Jiang, J, McSweeney, C, Zou, D, Liu, L, and Mao, Y. Deletion of CTNNB1 in inhibitory circuitry contributes to autism-associated behavioral defects. Hum Mol Genet. (2016) 25:2738–51. doi: 10.1093/hmg/ddw131
60. Mao, Y, Ge, X, Frank, CL, Madison, JM, Koehler, AN, Doud, MK, et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cells. (2009) 136:1017–31. doi: 10.1016/j.cell.2008.12.044
61. Chenn, A, and Walsh, CA. Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in beta-catenin overexpressing transgenic mice. Cereb Cortex. (2003) 13:599–606. doi: 10.1093/cercor/13.6.599
62. Alexander, J, Pirone, A, and Jacob, M. Excessive β-catenin in excitatory neurons results in reduced social and increased repetitive behaviors and altered expression of multiple genes linked to human autism. Front Synap Neurosci. (2020) 12:14. doi: 10.3389/fnsyn.2020.00014
63. Martin, PM, Stanley, RE, Ross, AP, Freitas, AE, Moyer, CE, Brumback, AC, et al. DIXDC1 contributes to psychiatric susceptibility by regulating dendritic spine and glutamatergic synapse density via GSK3 and Wnt/β-catenin signaling. Mol Psychiatry. (2018) 23:467–75. doi: 10.1038/mp.2016.184
64. Moffat, JJ, Jung, EM, Ka, M, Jeon, BT, Lee, H, and Kim, WY. Differential roles of ARID1B in excitatory and inhibitory neural progenitors in the developing cortex. Sci Rep. (2021) 11:3856. doi: 10.1038/s41598-021-82974-y
65. Qin, L, Ma, K, Wang, ZJ, Hu, Z, Matas, E, Wei, J, et al. Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat Neurosci. (2018) 21:564–75. doi: 10.1038/s41593-018-0110-8
66. Bae, SM, and Hong, JY. The Wnt signaling pathway and related therapeutic drugs in autism Spectrum disorder. Clin Psychopharmacol Neurosci. (2018) 16:129–35. doi: 10.9758/cpn.2018.16.2.129
67. Stambolic, V, Ruel, L, and Woodgett, J. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. (1996) 6:1664–9. doi: 10.1016/S0960-9822(02)70790-2
68. Kleindienst, N, and Greil, W. Lithium in the long-term treatment of bipolar disorders. Eur Arch Psychiatry Clin Neurosci. (2003) 253:120–5. doi: 10.1007/s00406-003-0429-2
69. Serret, S, Thummler, S, Dor, E, Vesperini, S, Santos, A, and Askenazy, F. Lithium as a rescue therapy for regression and catatonia features in two SHANK3 patients with autism spectrum disorder: case reports. BMC Psychiatry. (2015) 15:107. doi: 10.1186/s12888-015-0490-1
70. Min, WW, Yuskaitis, CJ, Yan, Q, Sikorski, C, Chen, S, Jope, RS, et al. Elevated glycogen synthase kinase-3 activity in fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology. (2009) 56:463–72. doi: 10.1016/j.neuropharm.2008.09.017
71. Nocjar, C, Hammonds, MD, and Shim, SS. Chronic lithium treatment magnifies learning in rats. Neuroscience. (2007) 150:774–88. doi: 10.1016/j.neuroscience.2007.09.063
72. Bersudsky, Y, Shaldubina, A, and Belmaker, R. Lithium's effect in forced-swim test is blood level dependent but not dependent on weight loss. Behav Pharmacol. (2007) 18:77–80. doi: 10.1097/FBP.0b013e32801416ed
73. Zaidan, M, Stucker, F, Stengel, B, Vasiliu, V, Hummel, A, Landais, P, et al. Increased risk of solid renal tumors in lithium-treated patients. Kidney Int. (2014) 86:184–90. doi: 10.1038/ki.2014.2
74. Guo, W, Murthy, AC, Zhang, L, Johnson, EB, Schaller, EG, Allan, AM, et al. Inhibition of GSK3beta improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum Mol Genet. (2012) 21:681–91. doi: 10.1093/hmg/ddr501
75. Han, A, Song, Z, Tong, C, Hu, D, Bi, X, Augenlicht, L, et al. Sulindac suppresses beta-catenin expression in human cancer cells. Eur J Pharmacol. (2008) 583:26–31. doi: 10.1016/j.ejphar.2007.12.034
76. Li, N, Xi, Y, Tinsley, H, Gurpinar, E, Gary, B, Zhu, B, et al. Sulindac selectively inhibits colon tumor cell growth by activating the cGMP/PKG pathway to suppress Wnt/β-catenin signaling. Mol Cancer Ther. (2013) 12:1848–59. doi: 10.1158/1535-7163.MCT-13-0048
77. Zhang, Y, Yang, C, Yuan, G, Wang, Z, Cui, W, and Li, R. Sulindac attenuates valproic acid-induced oxidative stress levels in primary cultured cortical neurons and ameliorates repetitive/stereotypic-like movement disorders in Wistar rats prenatally exposed to valproic acid. Int J Mol Med. (2015) 35:263–70. doi: 10.3892/ijmm.2014.1996
78. Qin, L, Dai, X, and Yin, Y. Valproic acid exposure sequentially activates Wnt and mTOR pathways in rats. Mol Cell Neurosci. (2016) 75:27–35. doi: 10.1016/j.mcn.2016.06.004
79. Zhang, Y, Sun, Y, Wang, F, Wang, Z, Peng, Y, and Li, R. Downregulating the canonical Wnt/beta-catenin signaling pathway attenuates the susceptibility to autism-like phenotypes by decreasing oxidative stress. Neurochem Res. (2012) 37:1409–19. doi: 10.1007/s11064-012-0724-2
80. Ahmadian, M, Suh, J, Hah, N, Liddle, C, Atkins, A, Downes, M, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. (2013) 19:557–66. doi: 10.1038/nm.3159
81. Lu, D, and Carson, D. Repression of beta-catenin signaling by PPAR gamma ligands. Eur J Pharmacol. (2010) 636:198–202. doi: 10.1016/j.ejphar.2010.03.010
82. Boris, M, Kaiser, C, Goldblatt, A, Elice, M, Edelson, S, Adams, J, et al. Effect of pioglitazone treatment on behavioral symptoms in autistic children. J Neuroinflammation. (2007) 4:3. doi: 10.1186/1742-2094-4-3
83. Kirsten, TB, Casarin, RC, Bernardi, MM, and Felicio, LF. Pioglitazone abolishes autistic-like behaviors via the IL-6 pathway. PLoS One. (2018) 13:e0197060. doi: 10.1371/journal.pone.0197060
84. De Felice, M, Melis, M, Aroni, S, Muntoni, AL, Fanni, S, Frau, R, et al. The PPARalpha agonist fenofibrate attenuates disruption of dopamine function in a maternal immune activation rat model of schizophrenia. CNS Neurosci Ther. (2019) 25:549–61. doi: 10.1111/cns.13087
85. Morin, P, Sparks, A, Korinek, V, Barker, N, Clevers, H, Vogelstein, B, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science (New York, NY). (1997) 275:1787–90. doi: 10.1126/science.275.5307.1787
86. Wang, J, Peng, K, Wu, V, Tseng, F, and Wu, K. CTNNB1 mutation in aldosterone producing adenoma. Endocrinol Metab. (2017) 32:332–8. doi: 10.3803/EnM.2017.32.3.332
87. Ledinek, Ž, Sobočan, M, and Knez, J. The role of CTNNB1 in endometrial cancer. Dis Markers. (2022) 2022:1–9. doi: 10.1155/2022/1442441
88. Arredondo, S, Valenzuela-Bezanilla, D, Mardones, M, and Varela-Nallar, L. Role of Wnt signaling in adult hippocampal neurogenesis in health and disease. Front Cell Dev Biol. (2020) 8:860. doi: 10.3389/fcell.2020.00860
89. Bamji, S, Shimazu, K, Kimes, N, Huelsken, J, Birchmeier, W, Lu, B, et al. Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron. (2003) 40:719–31. doi: 10.1016/S0896-6273(03)00718-9
90. Zhang, M, Eichhorn, S, Zingg, B, Yao, Z, Cotter, K, Zeng, H, et al. Spatially resolved cell atlas of the mouse primary motor cortex by MERFISH. Nature. (2021) 598:137–43. doi: 10.1038/s41586-021-03705-x
91. Pour, M, and Yanai, I. New adventures in spatial transcriptomics. Dev Cell. (2022) 57:1209–10. doi: 10.1016/j.devcel.2022.04.021
Keywords: CTNNB1, β-catenin, neurodevelopmental disorders, intellectual disability, autism spectrum disorder, schizophrenia
Citation: Zhuang W, Ye T, Wang W, Song W and Tan T (2023) CTNNB1 in neurodevelopmental disorders. Front. Psychiatry. 14:1143328. doi: 10.3389/fpsyt.2023.1143328
Edited by:
Michele H. Jacob, Tufts University, United StatesReviewed by:
Maria Marchese, Stella Maris Foundation (IRCCS), ItalyXiaokuang Ma, The University of Arizona College of Medicine – Phoenix, United States
Chuanliang Han, Shenzhen Institutes of Advanced Technology (CAS), China
Copyright © 2023 Zhuang, Ye, Wang, Song and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weihong Song, d2VpaG9uZ0B3bXUuZWR1LmNu; Tao Tan, dGFudGFvQG9qbGFiLmFjLmNu