 I. A. Roomaney
I. A. Roomaney S. Kabbashi
S. Kabbashi K. Beshtawi
K. Beshtawi S. Moosa
S. Moosa M. Y. Chothia
M. Y. Chothia M. Chetty
M. Chetty- 1Department of Craniofacial Biology, Pathology and Radiology, Faculty of Dentistry, University of Western Cape, Cape Town, South Africa
- 2Department of Dental Sciences, Faculty of Graduate Study, Arab American University, Jenin, Palestine
- 3Division of Molecular Biology and Human Genetics, Stellenbosch University Faculty of Medicine and Health Sciences, Cape Town, South Africa
- 4Medical Genetics, Tygerberg Hospital, Cape Town, South Africa
- 5Division of Nephrology, Department of Medicine, Faculty of Medicine and Health Sciences, Stellenbosch University and Tygerberg Hospital, Cape Town, South Africa
Enamel Renal Syndrome (ERS) (OMIM # 204690) is a rare genetic condition characterised by hypoplastic amelogenesis imperfecta, failed tooth eruption, intra-pulpal calcifications, gingival enlargement and occasionally nephrocalcinosis. In this case series, we report on four unrelated patients with a confirmed molecular diagnosis of ERS (FAM20A pathogenic variants) from Sub-Saharan Africa. The pathognomonic oral profile of ERS was mostly fulfilled in these patients, with the notable addition of an odontoma in one patient. The cases presented a spectrum of phenotypic severity both dentally and systemically. One patient presented with nephrocalcinosis and abnormal kidney function, one had reduced kidney size with normal kidney function, and two had no renal abnormalities. Patients presenting with the oral profile of ERS should receive a prompt referral to a nephrologist and a geneticist. They should receive long-term management from a multidisciplinary medical and dental team.
Introduction
Enamel Renal Syndrome (ERS) (OMIM # 204690) is a rare genetic condition characterised by hypoplastic amelogenesis imperfecta (AI), failed tooth eruption, intra-pulpal calcifications, gingival enlargement and, in some patients, nephrocalcinosis (1). In 1972, MacGibbon described two cases with generalised enamel hypoplasia and renal dysfunction (2). Several studies have since described conditions with a similar phenotypic oral presentation, with and without renal involvement. These include AI with nephrocalcinosis, AI syndrome, AI with inter-radicular dentine dysplasia, AI with gingival fibromatosis (AIGFS), MacGibbon syndrome, and Lubinsky-MacGibbon syndrome (3, 4). In 2011, a pathogenic variant in the FAM20A gene (FAMily with sequence similarity 20A) was implicated (5). This discovery was later confirmed in 2012 by a large international consortium, which observed 25 cases with the oral phenotype and nephrocalcinosis (6). The absence of strict characterisation of ERS has led to patients' renal status being overlooked and has resulted in an underestimation of the actual disease prevalence (4).
Patients with ERS are likely to seek dental care first due to the retention of primary teeth and failure of permanent tooth eruption (7). A review by de la Dure-Molla and colleagues (2014) suggested a distinctive pathognomonic oral profile for ERS patients (4); however, atypical features are being more frequently reported. These include sensorineural hearing loss, hypertrichosis (8), hypodontia (9), and periodontal disease (1, 10–12). A recent study also identified two unrelated patients with pathogenic FAM20A variants, one presenting with early eruption of permanent teeth and another with the normal eruption of all 32 teeth (8). Similarly, the prevalence of nephrocalcinosis in patients with ERS is unknown. Nephrocalcinosis has been reported in several studies (1, 6, 9–14); however, others have failed to identify renal involvement (8, 15–18). The renal phenotype, typically silent during childhood, is characterised by reduced calcium, phosphate, and citrate excretion with subsequent nephrocalcinosis (4).
There is a paucity of genetically confirmed cases of ERS from Sub-Saharan Africa. Case reports by Van Heerden et al. (1990), Peters et al. (1992), and Feller et al. (2006) are some of the earliest publications describing patients with oral profiles resembling that of ERS in South Africa, albeit under different nomenclatures [“Rough hypoplastic AI with Follicular hyperplasia”, “Enamel dysplasia with odontogenic fibroma-like hamartomas”, and “AI associated with multiple impactions and odontogenic fibromas (WHO) type”] (19–21). This suggests that the prevalence of ERS in the region may be higher than previously considered. Here we present four genetically confirmed cases of ERS from Sub-Saharan Africa and highlight the importance of the diagnosis in patient management.
Case description
This case series is a component of a project that was approved by the University of Western Cape (UWC) Biomedical Research Ethics Council (study number: BM21/7/16). The patients have provided written, informed consent for the publication of their data and images. The features of the patients found in this report are summarised in Table 1 and compared with the pathognomonic profile of ERS described by de la Dure-Molla et al. (13).

Table 1. Features of ERS in this case series compared to the pathognomonic oral profile to de la Dure-Molla et al. (4).
Case 1
A 26-year-old Shona-speaking female from Harare, Zimbabwe, presented to the UWC Dental Faculty with the main complaint of retained primary teeth. She was the only child of non-consanguineous parents. Two half-brothers from the father were unaffected. There was no family history of any genetic conditions. The patient's medical history revealed iron deficiency anaemia. She had been to several dentists previously who could not assist with her dental complaints.
The extra-oral and intra-oral images are presented in Figures 1A–C. The patient had normal growth and no further dysmorphic features. The intra-oral examination revealed thick, fibrous gingiva and bulbous maxillary and mandibular alveolar ridges. Pigmentation was present on the attached gingiva and multiple round-shaped, macular, pigmentations were also present on the hard palate mucosa (Figure 1C). Due to the relative microdontia and wear, the permanent incisors, and premolars present intra-orally were almost indistinguishable from primary teeth. During occlusion, the posterior alveolar ridges were closely aligned, causing the patient to utilise them for mastication. The primary teeth showed marked attrition resulting in a smooth brown/amber surface. Only the incisal third of the maxillary anterior teeth were visible due to the hyperplastic gingiva and alveolar bone. The permanent teeth were yellow with a smooth, thin enamel surface.

Figure 1. Clinical images of four patients with enamel renal syndrome. Case 1: (A): Profile showing no dysmorphic facial features. (B,C) Intra-oral images showing erupted primary and permanent teeth, fibrotic gingiva, and pigmentation on the attached and free-gingiva and the palatal mucosa. Case 2: (D) Lateral profile showing a mild Class III jaw relationship and no other dysmorphology. (E,F) Intra-oral images showing few erupted permanent teeth with variable enamel colourations, thick bone, and gingiva with no intermaxillary space posteriorly. Pigmentation is present on the attached gingiva. Case 3: (G): Profile view showing no dysmorphic facial features. (H,I): Intra-oral images showing mixed dentition, erupting premolar with no visible enamel and tooth wear. Case 4: (J) Profile picture showing vertical maxillary excess, thick arched brows, and protrusive ears. (K,L): Intraoral images showing severely thick, fibrotic gingiva and bulbous bone, severe tooth wear, and an absence of erupted maxillary incisors.
Radiologic examination (Figures 2A–D) showed multiple retained primary teeth (55, 53, 63, 64, 65, 75, 73 and 83), root remnants (26 and 85), infra-occlusion of the 15, and several impacted teeth (18, 17, 16, 13, 23, 24, 25, 27, 28, 38, 37, 36, 35, 33, 45, 47 and 48). Generalised, diffuse, intra-pulpal calcifications were noted, mainly in the coronal part of the primary and permanent teeth (Figure 2C). Teeth showed a delayed and aberrant eruption pattern. The molars had hypoplastic or absent enamel with flat cusps, and the roots were short and dilacerated. Enlarged dental follicles of teeth 18, 17, 24, 25, 27, 28, 38, 37, 33, 47, and 48 were noted. Pneumatisation of frontal sinuses (Figure 2A) and severe thinning of the upper and lower anterior alveolar ridges (Figure 2B) were present.
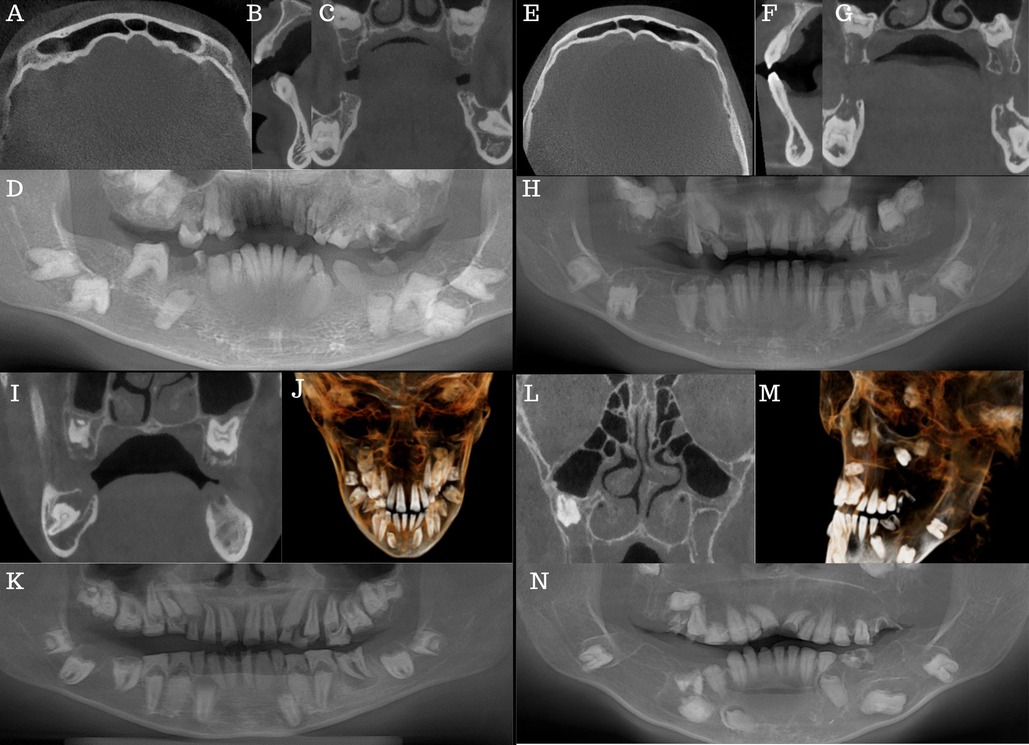
Figure 2. Case 1 (A–D): (A) axial view showing frontal sinus pneumatisation. (B) Sagittal view showing severe thinning of the lower anterior alveolar ridge. (C) Coronal view showing several impactions, pulp stones, and enlarged dental follicles stars. (D) CBCT reformatted panorama. Case 2 (E–H): (E) Axial view showing frontal sinus pneumatisation. (F) Sagittal view shows severe lower and upper anterior alveolar ridge thinning. (G): Coronal view showing several impactions, pulp stones and enlarged dental follicles stars. (H) CBCT reformatted panorama. Case 3 (I–K): (I) Coronal view showing several impactions, pulp stones, and enlarged dental follicles. (J) centre 3D model of the patient. (K) CBCT reformatted panorama. Case 4 (L–N): (L) Coronal view showing hypoplastic maxillary antra. (M) Left sides of the 3D model of the patient. (N) CBCT reformatted panorama.
After the initial examination, the patient was referred to the geneticist and the nephrologist for consultation. The medical genetics evaluation yielded no additional abnormalities or dysmorphology, specifically in relation to structures that originate from the endoderm. A pathogenic variant of FAM20A was confirmed via exome sequencing. Laboratory results revealed normal kidney function and nephrocalcinosis was absent on ultrasound examination.
Case 2
A 20-year-old Tsonga-speaking, female patient from Mpumalanga, South Africa, presented to the UWC Dental Faculty with the main complaint of difficulty eating due to a lack of teeth. She was the only child of non-consanguineous parents and had two unaffected half-siblings from the father. There was no significant family or medical history.
Extra-oral examination (Figures 1D–F) revealed a skeletal Class III jaw relationship with a protrusive mandible (Figure 1D). Her only erupted teeth were the 14, 11, 21, 22, 24,25 and the 42–32. The enamel showed inconsistent thickness, with some teeth appearing more yellow and others having a translucent, grey appearance (Figure 1E). The erupted 14 showed a complete absence of enamel (Figure 1F). Physiological pigmentation was visible on the attached gingiva and the alveolar ridge mucosa. The gingiva was thick and fibrous and showed no evidence of inflammatory changes. The ridges were thick and bulbous, particularly in the edentulous saddle region of the second quadrant (Figure 1E).
Radiologic examination (Figures 2E–H) showed missing teeth (16, 12, 26 and 46), a retained primary 53, and several tooth impactions (18, 17, 15, 13, 23, 27, 28, 35, 34, 38, 37, 44, 45, 47 and 48). Teeth 24,25, and 43 were not in occlusion. Intra-pulpal calcifications, delayed tooth eruption, hypoplastic enamel, and misshapen teeth with short, dilacerated roots were present (Figures 2G,H). Enlarged dental follicles of teeth 18, 17, 15, 13, 23, 27, 28, 38, 37, 35, 44, 45, 47 and 48 were noted. The frontal sinuses were pneumatised (Figure 2E), and severe thinning of the lower and upper anterior alveolar ridge was also noted (Figure 2F). The cortical outline of the right and left condyles was thin.
The medical genetic assessment found no additional dysmorphic features. A pathogenic variant of FAM20A was identified via exome sequencing. Although kidney sizes were small on ultrasound examination (left kidney: 8.5 cm and right kidney: 8.1 cm), no nephrocalcinosis was noted, and laboratory blood results revealed normal kidney function. A 24-hour urine collection revealed hypocitraturia of 0.2 mmol/day (reference: low urinary citrate <1.67 mmol/d) with normal urinary calcium-to-creatinine ratio of 0.35 mmol/mmol (reference range: 0.03–0.69 mmol/mmol) and urinary phosphate-to-creatinine ratio of 2.96 mmol/mmol (reference range: 0.13–6.47 mmol/mmol).
Case 3
A 14-year-old female of mixed ancestry from Cape Town, South Africa, presented with the main complaint of pain from a carious 36. She was the youngest of three daughters from non-consanguineous parents and the only affected family member. There was no significant medical or family history.
On extra-oral examination (Figures 1G–I), no abnormalities were detected. She had a borderline Class III skeletal profile and clinically insignificant facial asymmetry (Figure 1G). Intra-orally, the patient was in the mixed dentition phase with several retained primary teeth. The primary teeth were worn and very smooth, with the enamel clearly visible (Figure 1H). The erupted permanent teeth showed a smooth enamel surface and wear of the incisal edges. The erupted 36 was broken down at the sub-gingival level and decayed. No enamel was visible on the erupting 24 (Figure 1I). Fibrous gingiva was present; however, in this instance, the gingiva did not appear as thick, and the alveolar ridge was not as bulbous as in the other cases. No physiological pigmentation was visible.
Radiologic examination (Figures 2I–K) revealed mild mucosal thickening in the maxillary antra and an enlarged inferior, left turbinate (Figure 2I). Thinning of the cortical outline of the right and left condyles were noted. A well-defined, corticated low density was present periapically to the 36 (Figure 2K). Intra-pulpal calcifications, delayed tooth eruption, hypoplastic enamel, and misshapen teeth with short, dilacerated roots were present (Figure 2K). The tooth follicles surrounding the unerupted teeth exhibited enlargement, albeit not as significantly as observed in the other cases.
The medical genetic assessment identified no additional dysmorphic features and a pathogenic variant of FAM20A was identified via exome sequencing. Laboratory blood results revealed normal kidney function, and no nephrocalcinosis was noted on ultrasound examination.
Case 4
A 16-year-old male of mixed ancestry from Cape Town, South Africa, presented to the dental faculty with pain from a grossly carious 46. He was the only child of non-consanguineous parents and had no significant family history. His medical history revealed gastrointestinal and ophthalmological problems, which were being investigated and managed at the time. He reported progressive blurring of his vision with associated itching and dryness of the eyes. He suffered from recurring episodes of diarrhoea, abdominal cramps, and constipation. He also suffered from allergic rhinitis. He showed signs of mouth-breathing.
He presented with a vertical maxillary excess (Figures 1J–L) and an excessively gummy smile. He had a Class III malocclusion with a protrusive mandible and prominent chin. His ears were prominent, and he had thick, arched eyebrows (Figure 1J). Both his alveolar ridges were bulbous, with his maxillary ridge being significantly more affected, particularly on the palatal aspect where the ridges “crowded” the palate. This created the appearance of a very narrow, deep palate (Figure 1L). The gingiva was pale, thick, and smooth. Mild physiological pigmentation was visible on the attached gingiva of the mandibular ridge. The maxillary central incisors were unerupted. The teeth visible in the mouth were severely worn, microdontic, and spaced. The mandibular incisors were barrel-shaped, and the 36, the only permanent molar eruption, was also grossly carious. The erupted teeth had smooth enamel surfaces.
Hypoplastic maxillary antra and pneumatised frontal sinuses were present radiologically (Figure 2L). The inferior left and right turbinates were enlarged. Thinning of the cortical outline of the right and left condyles was noted. The 36 was extracted due to caries. Generalised foreshortening of teeth roots (particularly teeth 14, 15, 22, 25 and 34) was noted. The roots of 11 and 21 were severely dilacerated. The follicles around the mandibular molar teeth were enlarged and irregularly shaped.
The medical genetic assessment found no other dysmorphology, and a pathogenic variant of FAM20A was identified via exome sequencing. Laboratory blood results revealed mild kidney dysfunction with a serum creatinine concentration of 90 µmol/l [estimated glomerular filtration rate (modified Schwartz formula) of 67 ml/min/1.73 m2]. A 24-hour urine collection demonstrated hypocitraturia of 0.8 mmol/day, normal urinary calcium-to-creatinine ratio of 0.08 mmol/mmol, and normal urinary phosphate-to-creatinine ratio of 1.41 mmol/mmol. Nephrocalcinosis was present on ultrasound examination.
Discussion
ERS is a rare genetic condition which often results in a pathognomonic oral phenotype and sometimes has severe systemic effects. This case series presents four unrelated patients with ERS from Sub-Saharan Africa. These cases were identified by dental personnel with a high clinical index of suspicion for the diagnosis. They were referred to the medical geneticist and nephrologist for further assessment and testing. This highlights the importance of recognising the oral profile, as the dentist is often the patient's first contact with the healthcare system.
Although the pathognomonic oral profile by de la Dure Molla et al. (4) helps identify those potentially having ERS; it is important to note that not all features are required for diagnosis. The cases presented a spectrum of phenotypic severity. The anterior teeth and primary teeth clinically appeared to have enamel of variable thickness; however, the unerupted molars and premolars had very little to complete absence of enamel. In some instances, it appeared that the crowns of the teeth did not completely develop. Whether this is due to crown resorption, as mentioned in prior studies (4), is debatable and would require long-term monitoring. Molars also tended to be impacted more frequently than incisors, with only one of our cases (Case 4) having impacted incisors. In Cases 1 and 4, molars were more severely embedded and unfavourably angulated compared to Cases 2 and 3.
The cases described had several features not previously or infrequently reported to be associated with ERS. Cases 3 and 4 had thinning of the cortical outline of the condyles and enlargement of the turbinates. Cases 1, 2, and 4 had pneumatisation of the frontal sinuses. Case 1 also had an odontoma, a novel finding associated with ERS. Additionally, gingival pigmentation was noted in three patients. It is unclear whether the pigmentation is a physiological characteristic related to ethnicity or a feature specific to ERS. Although the regular-bordered, round-shaped, macular pigmentation on Case 1's palatal mucosa is not typical of physiological pigmentation. Further genotype-phenotype association studies and standardisation of reporting are required to elucidate whether the phenotypic diversity can be attributed to the FAM20A variation or other factors. A genotype-phenotype correlation study will be published in the future.
Although the oral profile of ERS is quite striking, the clinician needs to exclude conditions with overlapping characteristics. Non-syndromic amelogenesis imperfecta, for instance, can manifest with enamel defects, ranging from chalky-white to rough, yellow, and brown appearances due to quantitative or qualitative enamel defects. Chronic renal disease may also result In enamel hypoplasia and drug-induced gingival enlargement. (22) However, neither of these conditions are associated with the enlarged dental follicles and displaced unerupted teeth with which ERS is associated. In comparison, gingival enlargement linked to chronic kidney disease usually appears more inflamed, with enlarged interdental papillae (22), whereas ERS-associated gingiva generally lacks the signs of inflammation. ERS can also be confused with specific syndromes, particularly Jalili, Raine, and tricho-dento-osseous syndromes (4). Jalili syndrome presents with rod-cone dystrophy and amelogenesis imperfecta (23). There have been no reports of ERS associated with rod-cone dystrophy. Raine syndrome, caused by a pathogenic variant of FAM20C (a paralogue of FAM20A and FAM20B), shares a similar oral phenotype to ERS, including ectopic mineralizations of the pulp and gingiva. However, extraoral features such as choanal atresia, midface hypoplasia, neurologic and orthopaedic problems, and hypophosphatemic rickets distinguish Raine syndrome from ERS clinically (24–26). Tricho-deno-osseous syndrome is associated with AI and taurodontism, but nail defects, bone sclerosis, and hair described as curly or kinky are defining characteristics (27). Ultimately, genetic testing to identify the FAM20A pathogenic variant is the gold-standard method of diagnosing ERS.
Renal findings in ERS patients have been variable. A systematic review reported that 53% of patients who had laboratory evaluation had metabolic abnormalities (28). These included hypocalciuria (11.6%), raised serum creatinine concentrations (7.2%), hypocitraturia and hypophosphaturia (5.8%). The mechanisms underlying nephrocalcinosis are yet to be determined; however, studies in murine models suggest that the FAM20A gene mutation may cause altered phosphorylation of proteins in renal tubular cells predisposing to nephrocalcinosis (29). Two of our four patients had renal involvement. Case 2, a 20-year-old female, presented with kidneys which were smaller in size and hypocitraturia. Case 4, a 16-year-old male, displayed nephrocalcinosis with associated kidney dysfunction. There is uncertainty about whether nephrocalcinosis develops with increasing age since some reports have identified nephrocalcinosis in very young patients (10, 14, 30–33), including patients as young as six years old (14, 30). In 2012, Jaureguiberry et al. (6) investigated 25 patients from 16 families with the ERS oral profile and nephrocalcinosis and speculated that all individuals with biallelic FAM20A mutations would eventually show nephrocalcinosis; however, several authors failed to identify any renal involvement in patients presenting with the typical oral profile of ERS (8, 15, 17, 18, 34). There has also been no observed correlation between sex and the severity of the phenotype, with both males and females showing equivalent case numbers (28), but there has been no comparison of severity. Only larger cohorts and long-term follow-ups will clarify the natural history of the systemic manifestations of ERS.
In patients with ERS, tooth structures are compromised to the extent that crown preparation and bonding are unlikely to succeed. Hall et al. (35) attempted surgical exposure and restorations using composite material, and ultimately pulp necrosis ensued, necessitating endodontic treatment. Surgical exposure and orthodontic alignment of teeth have a highly questionable prognosis due to compromised periodontal ligaments and severely ectopically positioned teeth. Nitayavardhana et al. (8) advised that embedded teeth be removed due to the progressive embedding of teeth with age and the development of dental infection. Mauprivez et al. (36) were the only authors to detail the dental rehabilitation of a patient with ERS. Their treatment plan consisted of surgical removal of all non-viable teeth and alveolar bone recontouring to create space for a removable complete maxillary denture and an implant-supported mandibular denture. All our cases have undergone surgical removal of embedded teeth interfering with prosthesis placement, recontouring of the alveolar ridge and will have implant-supported dentures constructed. The dental management will be reported later. Due to limited resources, the dental treatment has been protracted; however, the patients have remained motivated to continue with treatment. Individualised dental management and continuation of care over the life course is required to determine which dental treatment modalities are most successful and feasible.
Currently, no guidelines are available for managing patients with ERS. Our patients consulted private general dental practices on several occasions before attending our clinic. They were unable to receive adequate care due to the complexity of their required management and financial constraints, resulting in frustration and reduced quality of life. When asked about their perspective of their management journey, one patient responded: “It's been a long journey. At first, l went for several consultations before anyone knew how to manage me. It took about three years for the hospital to finally start with treatment. I was excited for the treatment because l waited my entire life to get it done, and I was told it would take two years for me to get the results because l was going to get implants done. Unfortunately, due to doctors being changed, it will take more years, and it's not easy seeing specialists; it takes months or even a year before you get an appointment with them. My first surgery was done last year around October, and it was successful; l recovered very quickly. Also, due to my many scheduled appointments, and the distance I live from the hospital, it has cost me a lot of money. Every visit, I spend ZAR350,00–ZAR400,00 (USD15,00–USD20,00) on transport which takes a toll on me since I don’t earn that much. So, l hope in the future, the specialists and the hospital will come to a final decision which will not consume much time and more visits to the hospital.” This statement highlights the challenges encountered by patients being treated in low-resourced settings. With improved knowledge of this condition, more streamlined management approaches can be achieved.
The sequence of care received by our patients is presented in Table 2. Patients suspected of having ERS require a full physical examination and should be referred to a geneticist where possible. Although the presence of nephrocalcinosis appears to be variable, all patients presenting with ERS require renal examination and long-term monitoring of renal state to allow for earlier detection and decrease sequelae associated with deteriorating renal function. Increased medical-dental collaboration will lead to better characterisation of this condition and allow for more effective and efficient management of those affected.
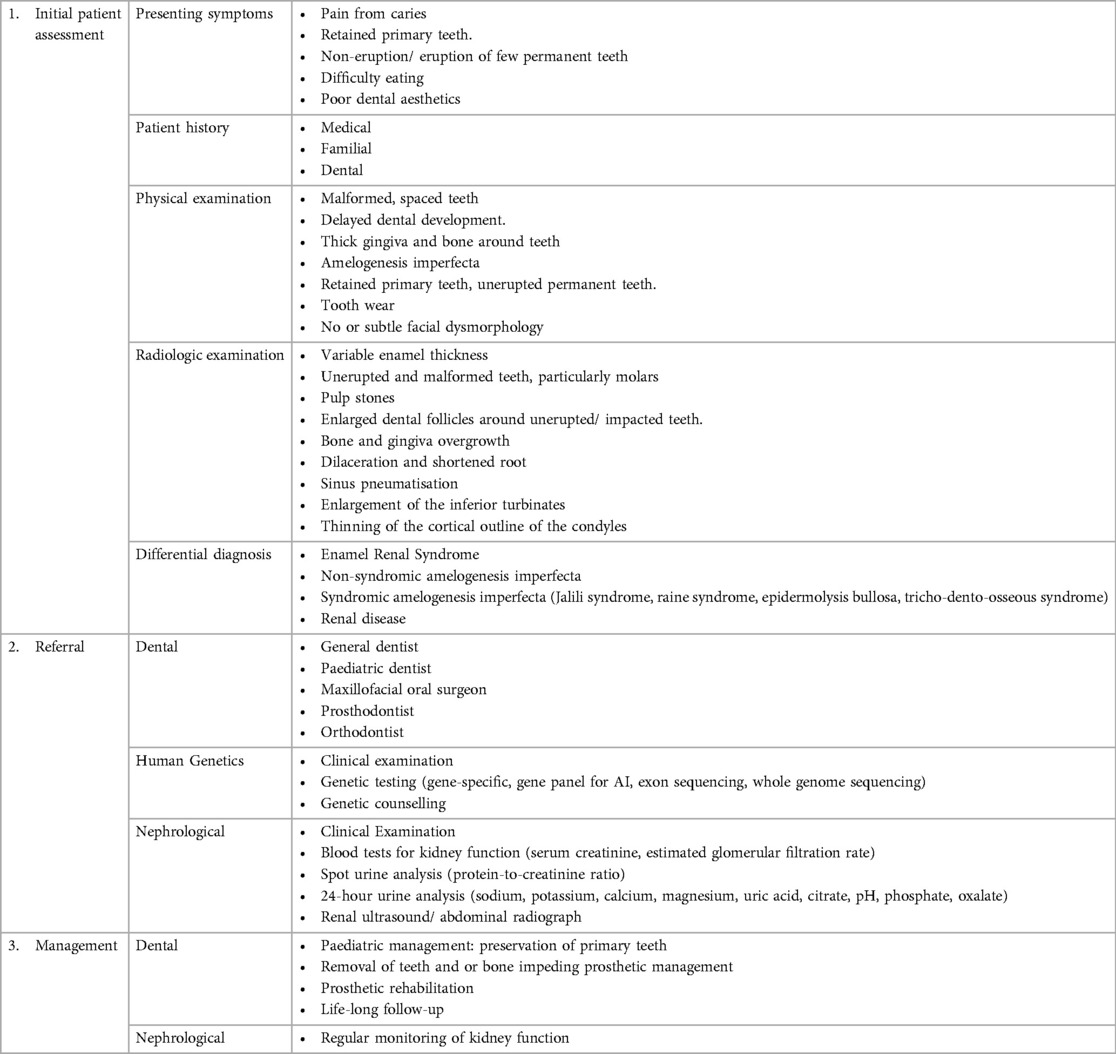
Table 2. Patient management according to CARE guidelines.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by University of Western Cape (UWC) Biomedical Research Ethics Council. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
IR and MC were responsible for this article's conceptualisation, drafting, and editing. SK, KB, MYC, and SM were responsible for analysing clinical data and drafting and editing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The research reported in this article was supported by the South African Medical Research Council (SAMRC) through its Division of Research Capacity Development under the Research Capacity Development Initiative from funding received from the South African National Treasury. The content and findings reported / illustrated are the sole deduction, view and responsibility of the researcher and do not reflect the official position and sentiments of the SAMRC. IR is supported by the SAMRC Clinician Researcher Development Programme. The work reported herein was also made possible through additional funding (SM) by the South African Medical Research Council through its Division of Research Capacity Development under the Early Investigators Programme from funding received from the South African National Treasury. The content hereof is the sole responsibility of the authors and does not necessarily represent the official views of the SAMRC. We gratefully acknowledge the Centre for High Performance Computing (CHPC, http://www.chpc.ac.za) and ILIFU (https://www.ilifu.ac.za) for computational resources.
Acknowledgments
The authors acknowledge the patients and their families for allowing the publication of this case report. They also acknowledge the clinicians involved in the patients' management, the bioinformaticists, and the photographer.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dourado MR, Dos Santos CRR, Dumitriu S, Iancu D, Albanyan S, Kleta R, et al. Enamel renal syndrome: a novel homozygous FAM20A founder mutation in 5 new Brazilian families. Eur J Med Genet. (2019) 62(11):103561. doi: 10.1016/j.ejmg.2018.10.013
2. MacGibbon D. Generalized enamel hypoplasia and renal dysfunction. Aust Dent J. (1972) 17(1):61–3. doi: 10.1111/j.1834-7819.1972.tb02747.x
3. Debnath K, Couthino A, Chatterjee A, Shenoy S. Enamel renal gingival syndrome: a rare case report. J Indian Soc Periodontol. (2019) 23(1):69–72. doi: 10.4103/jisp.jisp_532_18
4. de la Dure-Molla M, Quentric M, Yamaguti PM, Acevedo AC, Mighell AJ, Vikkula M, et al. Pathognomonic oral profile of enamel renal syndrome (ERS) caused by recessive FAM20A mutations. Orphanet J Rare Dis. (2014) 9(1):84–84. doi: 10.1186/1750-1172-9-84
5. O’Sullivan J, Bitu CC, Daly SB, Urquhart JE, Barron MJ, Bhaskar SS, et al. Whole-exome sequencing identifies FAM20A mutations as a cause of amelogenesis imperfecta and gingival hyperplasia syndrome. Am J Hum Genet. (2011) 88(5):616–20. doi: 10.1016/j.ajhg.2011.04.005
6. Jaureguiberry G, De la Dure-Molla M, Parry D, Quentric M, Himmerkus N, Koike T, et al. Nephrocalcinosis (enamel renal syndrome) caused by autosomal recessive FAM20A mutations. Nephron Physiol. (2012) 122(1–2):1–6. doi: 10.1159/000349989
7. Wang SK, Aref P, Hu Y, Milkovich RN, Simmer JP, El-Khateeb M, et al. FAM20A mutations can cause enamel-renal syndrome (ERS). PLoS Genet. (2013) 9(2):e1003302. doi: 10.1371/journal.pgen.1003302
8. Nitayavardhana I, Theerapanon T, Srichomthong C, Piwluang S, Wichadakul D, Porntaveetus T, et al. Four novel mutations of FAM20A in amelogenesis imperfecta type IG and review of literature for its genotype and phenotype spectra. Mol Genet Genomics. (2020) 295(4):923–31. doi: 10.1007/s00438-020-01668-8
9. Pêgo SPB, Coletta RD, Dumitriu S, Iancu D, Albanyan S, Kleta R, et al. Enamel-renal syndrome in 2 patients with a mutation in FAM20 A and atypical hypertrichosis and hearing loss phenotypes. Oral Surg Oral Med Oral Pathol Oral Radiol. (2017) 123(2):229. doi: 10.1016/j.oooo.2016.09.226
10. Ashkenazi M, Rafe Z, Sarnat H, Levin L. Nephrocalcinosis associated with continuous enamel hypoplasia and severe alveolar bone loss: a case report and literature review. Pediatr. Dent. (2014) 36:250–3. Available from: https://search.ebscohost.com/login.aspx?direct=true&db=a9h&AN=96386454&site=ehost-live&scope=site24960394
11. Patel A, Jagtap C, Bhat C, Shah R. Bilateral nephrocalcinosis and amelogenesis imperfecta: a case report. Contemp Clin Dent. (2015) 6(2):262–5. doi: 10.4103/0976-237X.156063
12. Simancas Escorcia V, Diarra A, Naveau A, Dessombz A, Felizardo R, Cannaya V, et al. Lack of FAM20A, ectopic gingival mineralization and chondro/osteogenic modifications in enamel renal syndrome. Front Cell Dev Biol. (2020) 8:605084. doi: 10.3389/fcell.2020.605084
13. Torres LHS, de-Azevedo-Vaz SL, Barroso DRC, Silva DN, Velloso TRG, de Barros LAP, et al. Enamel-renal-syndrome: case report. Spec Care Dentist. (2018) 38(3):172–5. doi: 10.1111/scd.12288
14. Koruyucu M, Seymen F, Gencay G, Gencay K, Tuna EB, Shin TJ, et al. Nephrocalcinosis in amelogenesis imperfecta caused by the FAM20A mutation. Nephron. (2018) 139(2):189–96. doi: 10.1159/000486607
15. Poulter JA, Smith CEL, Murrillo G, Silva S, Feather S, Howell M, et al. A distinctive oral phenotype points to FAM20A mutations not identified by sanger sequencing. Mol Genet Genomic Med. (2015) 3(6):543–9. doi: 10.1002/mgg3.164
16. Berès F, Lignon G, Rouzière S, Mauprivez C, Simon S, Berdal A, et al. Physicochemical analysis of human pulpal mineralization secondary to FAM20A mutations. Connect Tissue Res. (2018) 59:46–51. doi: 10.1080/03008207.2018.1435644
17. Cabral RM, Kurban M, Rothman L, Wajid M, Shimomura Y, Petukhova L, et al. Autosomal recessive gingival hyperplasia and dental anomalies caused by a 29-base pair duplication in the FAM20A gene. J Hum Genet. (2013) 58(8):566–7. doi: 10.1038/jhg.2013.44
18. Laouina S, Bloch Zupan A, El alloussi M. Enamel–renal syndrome with congenital heart defects and asthma: a rare association in a Moroccan child. Clin Dysmorphol. (2017) 26(2):114–6. doi: 10.1097/MCD.0000000000000146
19. Peters E, Cohen M, Altini M. Rough hypoplastic amelogenesis imperfecta with follicular hyperplasia. Oral Surgery, Oral Medicine, Oral Pathology. (1992) 74(1):87–92. doi: 10.1016/0030-4220(92)90220-K
20. Van Heerden WF, Raubenheimer EJ, Dreyer AF, Benn AM. Amelogenesis imperfecta: multiple impactions associated with odontogenic fibromas (WHO) type. J Dent Assoc S Afr. (1990) 45(11):467–71.2098937
21. Feller L, Jadwat Y, Bouckaert M, Buskin A, Raubenheimer EJ. Enamel dysplasia with odontogenic fibroma–like hamartomas: review of the literature and report of a case. Oral Surg Oral Med Oral Path Oral Radiol Endodontol. (2006) 101(5):620–4. doi: 10.1016/j.tripleo.2005.06.015
22. Gupta M, Gupta M. Oral conditions in renal disorders and treatment considerations—a review for pediatric dentist. Saudi Dent J. (2015) 27(3):113–9. doi: 10.1016/j.sdentj.2014.11.014
23. Luder HU, Gerth-Kahlert C, Ostertag-Benzinger S, Schorderet DF. Dental phenotype in jalili syndrome due to a c.1312 dupC homozygous mutation in the CNNM4 gene. PLoS One. (2013) 8(10):e78529. doi: 10.1371/journal.pone.0078529
24. Acevedo AC, Poulter JA, Alves PG, de Lima CL, Castro LC, Yamaguti PM, et al. Variability of systemic and oro-dental phenotype in two families with non-lethal raine syndrome with FAM20C mutations. BMC Med Genet. (2015) 16:8. doi: 10.1186/s12881-015-0154-5
25. Mameli C, Zichichi G, Mahmood N, Elalaoui SC, Mirza A, Dharmaraj P, et al. Natural history of non-lethal raine syndrome during childhood. Orphanet J Rare Dis. (2020) 15(1):1–9. doi: 10.1186/s13023-020-01373-0
26. Hung CY, Rodriguez M, Roberts A, Bauer M, Mihalek I, Bodamer O. A novel FAM20C mutation causes a rare form of neonatal lethal raine syndrome. Am J Med Genet Part A. (2019) 179(9):1866–71. doi: 10.1002/ajmg.a.61291
27. Al-Batayneh O. Tricho-dento-osseous syndrome: diagnosis and dental management. Int J Dent. (2012) 2012:514692. doi: 10.1155/2012/514692
28. Farias MLM, Ornela GO, de Andrade RS, Martelli DRB, Dias VO, Júnior HM. Enamel renal syndrome: a systematic review. Indian J Nephrol. (2021) 31(1):1–8. doi: 10.4103/ijn.IJN_27_19
29. Wang SK, Reid BM, Dugan SL, Roggenbuck JA, Read L, Aref P, et al. FAM20A Mutations associated with enamel renal syndrome. J Dent Res. (2014) 93(1):42–8. doi: 10.1177/0022034513512653
30. Choi S, Sohn YB, Ji S, Song S, Shin J, Kim S. Enamel renal syndrome: a case report of amelogenesis Imperfecta associated with nephrocalcinosis. Je Korean Acad Pedtatric Dent. (2020) 47(3):344–51. doi: 10.5933/JKAPD.2020.47.3.344
31. Hassib NF, Shoeib MA, ElSadek HA, Wali ME, Mostafa MI, Abdel-Hamid MS. Two new families with enamel renal syndrome: a novel FAM20A gene mutation and review of literature. Eur J Med Genet. (2020) 63(11):104045. doi: 10.1016/j.ejmg.2020.104045
32. Kala Vani SV, Varsha M, Sankar YU, Kala Vani SV, Varsha M, Sankar YU. Enamel renal syndrome: a rare case report. J Indian Soc Pedod Prev Dent. (2012) 30(2):169–72. doi: 10.4103/0970-4388.100006
33. Hasan RK, Ilyas AA, Mustafa OS. Enamel renal syndrome in a Bahraini child: a rare case report. J Bahrain Med Soc. (2020) 32(4):54–7. doi: 10.26715/jbms.32_2020_4_10
34. Cho SH, Seymen F, Lee KE, Lee SK, Kweon YS, Kim KJ, et al. Novel FAM20A mutations in hypoplastic amelogenesis imperfecta. Hum Mutat. (2012) 33(1):91–4. doi: 10.1002/humu.21621
35. Hall RK, Phakey P, Palamara J, McCredie DA. Amelogenesis imperfecta and nephrocalcinosis syndrome: case studies of clinical features and ultrastructure of tooth enamel in two siblings. Oral Surg Oral Med Oral Path Oral Radiol Endodontol. (1995) 79(5):583–92. doi: 10.1016/S1079-2104(05)80100-3
Keywords: enamel renal syndrome, fam20A, rare disease Africa, craniofacial manifestations, nephrocalcinosis
Citation: Roomaney IA, Kabbashi S, Beshtawi K, Moosa S, Chothia MY and Chetty M (2023) Case report: Enamel renal syndrome: a case series from sub-Saharan Africa. Front. Oral. Health 4:1228760. doi: 10.3389/froh.2023.1228760
Received: 25 May 2023; Accepted: 4 August 2023;
Published: 22 August 2023.
Edited by:
Ricardo D. Coletta, Campinas State University, BrazilReviewed by:
Erica Negrini Lia, University of Brasilia, BrazilSera Derelioğlu, Atatürk University, Türkiye
© 2023 Roomaney, Kabbashi, Beshtawi, Moosa, Chothia and Chetty. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: I. A. Roomaney aXJvb21hbmV5QHV3Yy5hYy56YQ==