 Jax D. Dallas
Jax D. Dallas Brent R. Westbrook
Brent R. Westbrook Ryan C. Fortenberry
Ryan C. Fortenberry- Department of Chemistry and Biochemistry, University of Mississippi, Oxford, MS, United States
The ethynol (HCCOH) molecule has recently been shown to be present in simulated astrochemical ices possibly linking it to molecular building blocks for interstellar complex organic molecules like amino acids. The proposed reaction mechanism suggests the simultaneous formation of both ketene and ethynol from mixed carbon monoxide/water ice in simulated interstellar conditions. Rigorous anharmonic spectral data within both the IR and microwave regions are needed for possible detection of ethynol in the interstellar medium. This study provides the first such data for this molecule from high-level quantum chemical computations where experiment is currently lacking. Ethynol has a
1 Introduction
Structural isomers of complex organic molecules (COMs) are telling of the chemical and physical traits of interstellar environments and are useful testers of molecular cloud chemical models (Turner et al., 2020). Therefore, the ability to accurately recognize the abundance of COM building blocks in interstellar environments is critical for the prediction of interstellar chemical pathways. Two promising reactants to form larger COMs are ethynol (Zasimov et al., 2019), also known as hydroxyacetylene, and ketene, both members of the C2H2O isomer family. Ketene has a carbon=carbon double bond as well as a carbon=oxygen double bond. Ethynol has a carbon–carbon triple bond and an accompanying hydroxyl group. Previous ab initio computations have shown ethynol to lie 150.9 kJ/mol above the more stable ketene (Tanaka and Yoshimine, 1980). A third C2H2O isomer, oxirene, is suspected to be a possible intermediate structure facilitating some isomerization between the more stable structures of ethynol and ketene (Turner et al., 2020). Visual representation of these three molecules can be seen in Figure 1. Of these three molecules, only ketene and its ketenyl radical have been observed in the interstellar medium (ISM) with the former first detected toward Sgr B2 (Turner, 1977). However, the downhill pathway for the formation of ethynol from the same starting materials as ketene implies that it should have non-negligible abundance. Furthermore, detection of ethynol giving the ketene/ethynol pair would imply that mixed CO/H2O ices could be the provenance of both molecules including the known ketene molecule.
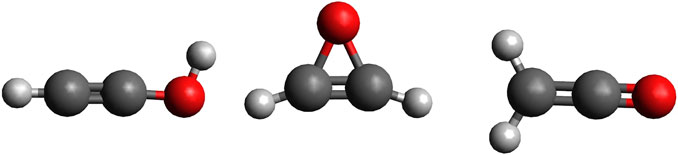
FIGURE 1. From left to right: ethynol, oxirene, and ketene.
In addition to ice experiments, part of the previous work by Turner et al. (2020) explored quantum chemical reaction schemes at the coupled cluster singles, doubles, and perturbative triples [CCSD(T)] level beginning with dicarbon monoxide in the presence of free hydrogen and found barrier free pathways producing both ketene and ethynol. The ketenyl radical is an intermediate along this pathway. The addition of hydrogen to the carbon of the ketenyl radical is strongly preferred and leads to the formation of ketene. The addition of hydrogen to the oxygen of the radical is feasible, however extremely disfavored, and leads to the formation of ethynol. This barrier free pathway was then tested experimentally and resulted in the first identification of ethynol in interstellar ice analogs (Turner et al., 2020). Preceding work has also experimentally shown the reversible conversion of ketene into ethynol via photolysis in an argon matrix (Hochstrasser and Wirz, 1990). However, energy must be put into the system for this conversion to take place. Regardless, considering these reaction pathways and that ketene and the ketenyl radical have been previously observed in the ISM, ethynol is likely present in detectable quantities as even tautomers that are less stable than HCCOH compared to their global minima are known to persist in the ISM if their barriers to rearrangement are high enough (Dommen et al., 1987). Before it can be detected, however, highly-accurate anharmonic vibrational frequencies and/or rotational spectroscopic data for ethynol must be produced in order to provide the necessary reference data for comparison to any subsequent experiments or observations. This is the primary objective of the current work.
An early theoretical study on the rotational and vibrational characteristics of ethynol was carried out by DeFrees and McLean (1982). Using Hartree-Fock and a 3–21G basis set, they obtained harmonic vibrational frequencies that they suggested could be scaled by a previously-derived empirical factor (Pople et al., 1981) to obtain values comparable to experiment. They further computed CISD+Q/DZ+P equilibrium rotational constants and accompanying scaling factors derived from the experimental rotational constants of HNCO and HN3 (DeFrees et al., 1982). Later work on ethynol offered rotational constants at the third-order Møller-Plesset perturbation theory (MP3) level with a 6–31G** basis set (Brown et al., 1985). The MP3 values were then scaled using factors obtained from experimental ketene data, yielding constants that were in good agreement with those computed by DeFrees and McLean (Brown et al., 1985).
Later theoretical work by Dommen et al. (1987) produced vibrational frequencies based on an MP2/6–31G** harmonic force field, with the force constants scaled based on empirical corrections from similar computations on methanol and acetylene (Dommen et al., 1987). Dommen et al. (1987) also questioned the proposed multiplicative scaling scheme of DeFrees and McLean, instead suggesting that the equilibrium rotational constants should be divided by the scaling factors (Dommen et al., 1987). The scaled vibrational frequencies and re-scaled rotational constants presented by Dommen and coworkers are the most accurate theoretical data currently available for ethynol, revealing the need for new, high-level computational data. In the meantime, some progress has been made on the experimental characterization of ethynol, with five of its vibrational frequencies observed in an argon matrix in 1989 (Hochstrasser and Wirz, 1989). More recently, its ground-state C rotational constant was detected at 9553 MHz in 2019 (Martin-Drumel et al., 2019). The authors therein also report scaled CCSD(T)/cc-pVQZ principle rotational constants for comparison (Martin-Drumel et al., 2019). In spite of these previous computational results, more rigorous theoretical treatment is necessary to complement further experimental and observational work in the gas phase.
The current approach to obtaining high-level, theoretical vibrational frequencies is via a fourth-order Taylor series expansion to the internuclear potential with composite CCSD(T) energies making considerations for complete basis set extrapolation, core electron correlation, and scalar relativity (Huang and Lee, 2008; Huang and Lee, 2009; Huang et al., 2011). Such a scheme is referred to as “CcCR”. The rigor of this method makes it very attractive, with it producing vibrational frequencies within 1.0 cm−1 of gas-phase experimental values in many cases (Huang and Lee, 2008; Huang and Lee, 2009; Huang et al., 2011; Fortenberry et al., 2012b; Huang et al., 2013; Fortenberry et al., 2014; Zhao et al., 2014; Fortenberry et al., 2015; Kitchens and Fortenberry, 2016; Bizzocchi et al., 2017; Fortenberry, 2017). However, its high computational cost makes this method less viable as molecules become larger and more complex. For this reason, a less complete but commensurately less computationally demanding method utilizing explicit correlation with a triple-ζ basis set is used as a point of comparison (Huang et al., 2010; Agbaglo et al., 2019; Agbaglo and Fortenberry, 2019a; Agbaglo and Fortenberry, 2019b), as it typically provides vibrational frequencies to within 7.0 cm−1 of gas-phase experimental values. The rotational constants, on the other hand, still require the more expensive composite approach in order to provide high-accuracy values (Agbaglo et al., 2019; Agbaglo and Fortenberry, 2019a; Agbaglo and Fortenberry, 2019b).
Cheaper methods do exist for determining relatively accurate rotational constants, such as the procedures outlined by Lee and McCarthy (2020). However, even in the best case outlined therein of the MP2/cc-pVQZ B rotational constants, the average deviation from experiment is 0.65%, while the performance of CcCR relative to experiment for the B and C rotational constants was recently shown to be 0.12% (Gardner et al., 2020), albeit for a much smaller selection of molecules. Additionally, the rotational constants from the CcCR methodology come at virtually no additional cost with the fundamental vibrational frequencies, making even marginal accuracy gains in these constants worth the increased computational demand when anharmonic frequencies are desired.
Both the aforementioned CcCR and explicitly correlated procedures are utilized within the current work to produce accurate vibrational frequencies for ethynol. By using the high-level anharmonic vibrational frequencies proposed in the current work, the search for interstellar ethynol may continue with more clarity in the regions in which ketene has been previously detected such as Sgr B2 (Turner, 1977); Orion KL (Johansson et al., 1984); and TMC-1 (Matthews and Sears, 1986).
2 Computational Details
Optimized geometries, dipole moments, and harmonic frequencies are computed using the MOLPRO 2015.1 software package (Werner et al., 2015) with canonical CCSD(T) and CCSD(T) within the F12 explicitly correlated construction [CCSD(T)-F12b] (Raghavachari et al., 1989; Crawford and Schaefer, 2000; Adler et al., 2007; Shavitt and Bartlett, 2009). As mentioned above, anharmonic frequencies are produced by a fourth-order Taylor series expansion of the internuclear portion of the Watson A-Reduced Hamiltonian referred to as a quartic force field (QFF). Two QFFs are utilized. The CCSD(T)-F12/cc-pVTZ-F12 version (Adler et al., 2007; Peterson et al., 2008; Yousaf and Peterson, 2008; Knizia et al., 2009) will be referred to as the F12-TZ QFF. The second QFF is the composite approach mentioned previously. This methodology is defined as the CcCR QFF since it includes complete basis set extrapolation (“C”) using a three-point formula from aug-cc-pVTZ, aug-cc-pVQZ, and aug-cc-pV5Z basis sets (Martin and Lee, 1996); core electron correlation (“cC”) using the Martin-Taylor (MT) core correlating basis set (Martin and Taylor, 1994); and scalar relativity (“R”) using Douglas-Kroll basis sets (Douglas and Kroll, 1974; de Jong et al., 2001). Recent results show that the F12-TZ QFF produces comparable anharmonic data to the CcCR QFF but with significantly lower computational costs (Agbaglo and Fortenberry, 2019a).
For both QFFs, an optimized reference geometry is required. For the F12-TZ QFF, this optimization is carried out at the CCSD(T)-F12/cc-pVTZ-F12 level. For the CcCR QFF, structural optimizations are determined at the canonical CCSD(T) level using the aug-cc-pV5Z basis set (Dunning, 1989; Kendall et al., 1992; Peterson and Dunning, 1995) as well as the MT core correlating basis set (Martin and Taylor, 1994). The difference in the bond lengths and angles between the CCSD(T)/MT computations with and without the core correction is then added to the respective variables within the optimized CCSD(T)/aug-cc-pV5Z geometry in order to correct for core correlation.
From the previously discussed reference geometries, displacements of 0.005 Å for bond lengths, 0.005 radians for bond angles, and 0.005 unitless displacements of LINX/LINY coordinates (Fortenberry et al., 2012b), which are necessary for the near-prolate structure of ethynol, are used to generate a QFF of 3,161 points using the following coordinate system:
From this coordinate system, single point energies are computed for the composite method using the definition described above. The F12-TZ method only relies on CCSD(T)-F12/cc-pVTZ-F12. Once the single point energies are computed for both of the QFFs, they are fit using a least squares procedure with a sum of squared residuals less than
Combinations of
3 Results and Discussion
3.1 Structure and Rotational Spectroscopic Constants
The optimized structures at both the F12-TZ and CcCR levels are given in Table 1. The optimized bond lengths computed at both levels of theory agree to within 0.02 Å when compared to one another and previous computations at the MP2/6–31G** level of theory (Dommen et al., 1987). The quartic and sextic A-reduced Hamiltonian spectroscopic constants of ethynol are given in Table 2 alongside dipole moments. Ethynol’s vibrationally-excited rotational constants are provided in Table 1 in the same order as the fundamental vibrational frequencies. Due to ethynol’s considerable dipole moment of 1.58 D, as shown in Table 2, its rotational transitions should be easily observable via rovibrational spectroscopy.
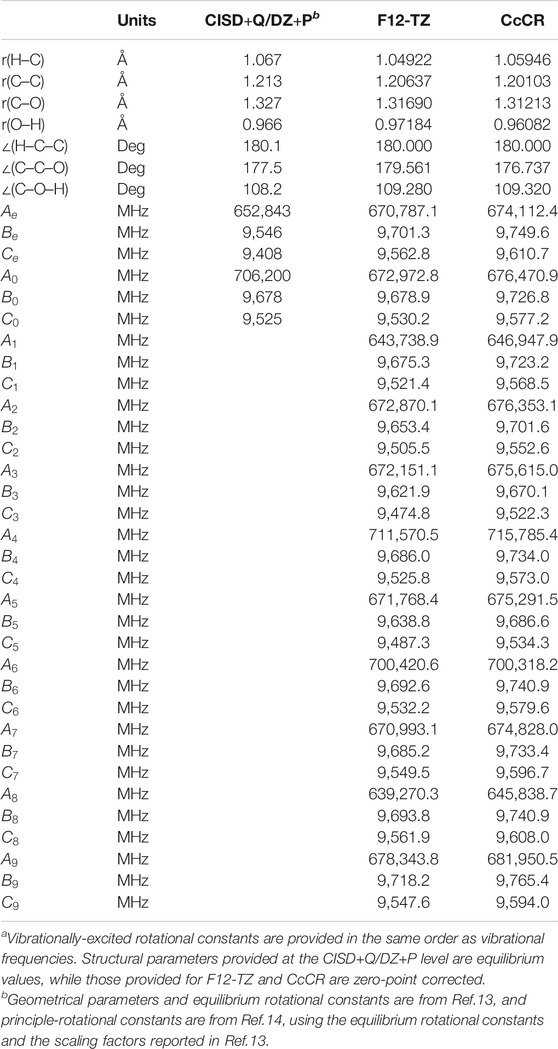
TABLE 1. Structural data and rotational constants for HCCOH.a
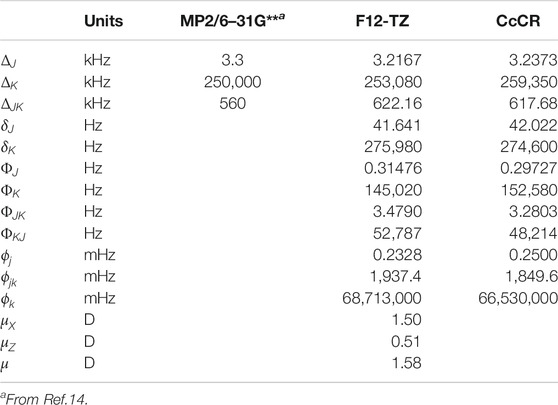
TABLE 2. Quartic and sextic spectroscopic constants.
A useful point of comparison for ethynol’s equilibrium rotational constants would be with those of acetonitrile. Acetonitrile is a prolate molecule that compares nicely to ethynol’s near-prolate structure and has a nearly equivalent molecular weight to that of ethynol. As seen in Table 1, ethynol has CcCR
3.2 Vibrational Frequencies
Vibrational frequencies for ethynol are given at the F12-TZ and CcCR levels of theory alongside experimental values in Table 3. F12-TZ and CcCR anharmonic frequencies compare well to each other, with a mean absolute error of less than 8 cm−1 between the two methods. Although gas-phase experimental data are currently unavailable for ethynol, argon matrix experimental data (Hochstrasser and Wirz, 1989) correlates in expected fashion with theoretical values presented in this work. Namely, all but
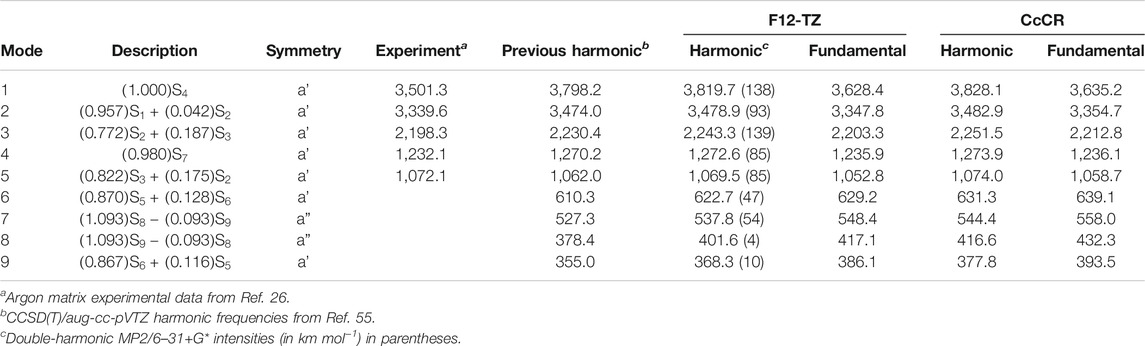
TABLE 3. Vibrational frequencies (in cm−1) of ethynol.
Of ethynol’s vibrational frequencies,
With only a slightly lower intensity than
Of the three brightest vibrational frequencies present in ethynol,
Positive anharmonicities are present for
The high intensity stretching modes
4 Conclusion
Considering that ethynol has already been shown to form in interstellar ice analogs and that the related ketene molecule (Turner et al., 2020) is known to be present in the ISM, the high-level vibrational and rotational data provided here may lead to the detection of ethynol in various astrophysical regions, especially those where ketene and its dehydrogenated radical are known to exist. Detection of ethynol through rotational spectroscopy should be possible due to its 1.58 D dipole moment. Further, detection is also feasible via mid-IR spectroscopy due to the bright nature of vibrational frequencies
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
JD and BW performed the computations. JD wrote the draft. JD, BW, and RF edited the manuscript. RF provided conceptualization, oversight, methodology, and provision of funds.
Funding
Additional funding provided by the Barry Goldwater Foundation. Funds from NASA and the NSF supported the student workers and provided much of the computational power utilized in this work. Open access publication fees are provided by the University of Mississippi.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
RF acknowledges support from NSF Grant OIA-1757220, NASA Grant NNX17AH15G, and the University of Mississippi for the provision of startup funds. JD recognizes support from the Goldwater Foundation. Additionally, the authors would like to thank Ralf I. Kaiser of the University of Hawaii for useful discussions related to the development of this manuscript and for initiating the research collaboration that led to the work reported herein.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fspas.2020.626407/full#supplementary-material.
References
Adler, T. B., Knizia, G., and Werner, H. J. (2007). A simple and efficient CCSD(T)-F12 approximation. J. Chem. Phys. 127, 221106. doi:10.1063/1.2817618
Agbaglo, D., and Fortenberry, R. C. (2019a). The performance of explicitly correlated methods for the computation of anharmonic vibrational frequencies. Int. J. Quant. Chem. 119, e25899. doi:10.1002/qua.25899
Agbaglo, D., and Fortenberry, R. C. (2019b). The performance of explicitly correlated wavefunctions [CCSD(T)-F12b] in the computation of anharmonic vibrational frequencies. Chem. Phys. Lett. 734, 136720. doi:10.1016/j.cplett.2019.136720
Agbaglo, D., Lee, T. J., Thackston, R., and Fortenberry, R. C. (2019). A small molecule with PAH vibrational properties and a detectable rotational spectrum: ?-(C)C3H2, cyclopropenylidenyl carbene. Astrophys. J. 871, 236. doi:10.3847/1538-4357/aaf85a
Allen, W. D.coworkers (2005). Is a general program written by W. D. Allen and coworkers, which performs vibrational analysis and higher-order non-linear transformations. Athens, Georgia; Center for Computational Quantum Chemistry.
Anderson, J. S., and Ogden, J. S. (1969). Matrix isolation studies of group-iv oxides. i. infrared spectra and structures of sio, sio, and sio. J. Chem. Phys. 51, 4189. doi:10.1063/1.1671778
Ball, J. A., Gottlieb, C. A., Lilley, A. E., and Radford, H. E. (1970). Detection of methyl alcohol in Sagittarius. Astrophys. J. Lett. 162, L203. doi:10.1086/180654
Bizzocchi, L., Lattanzi, V., Laas, J., Spezzano, S., Giuliano, B. M., Prudenzano, D., et al. (2017). Accurate sub-millimetre rest frequencies for HOCO and DOCO ions. Astron. Astrophys. 602, A34. doi:10.1051/0004-6361/201730638
Brown, R. D., Rice, E. H. N., and Rodler, M. (1985). Ab Initio studies of the structures and force fields of ketenimine and related molecules. Chem. Phys. 99, 347–356. doi:10.1016/0301-0104(85)80175-0
Crawford, T. D., and Schaefer, H. F. (2000). “An introduction to coupled cluster theory for computational chemists,” in Reviews in computational chemistry. Editors K. B. Lipkowitz, and D. B. Boyd (New York: Wiley), Vol. 14, 33–136.
de Jong, W. A., Harrison, R. J., and Dixon, D. A. (2001). Parallel Douglas-Kroll energy and gradients in nwchem: estimating scalar relativistic effects using douglas-kroll contracted basis sets. J. Chem. Phys. 114, 48–53. doi:10.1063/1.1329891
DeFrees, D. J., Loew, G. H., and McLean, A. D. (1982). The rotational spectra of hoco, hocn, hn, and hnco from quantum mechanical calculations. Astrophys. J. 254, 405–411. doi:10.1086/159745
DeFrees, D. J., and McLean, A. D. (1982). Rotational and vibrational spectra of ethynol from quantum-mechanical calculations. J. Phys. Chem. 86, 2835–2837. doi:10.1021/j100212a006
Dommen, J., Rodler, M., and Ha, T.-K. (1987). Vibrational spectrum of ethynol: ab initio calculation and matrix isolation studies. Chem. Phys. 117, 65–72. doi:10.1016/0301-0104(87)80097-6
Douglas, M., and Kroll, N. (1974). Quantum electrodynamical corrections to the fine structure of helium. Ann. Phys. 82, 89–155. doi:10.1016/0003-4916(74)90333-9
Dunning, T. H. (1989). Gaussian basis sets for use in correlated molecular calculations. i. the atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023. doi:10.1063/1.456153
Fortenberry, R. C., Huang, X., Francisco, J. S., Crawford, T. D., and Lee, T. J. (2012a). Fundamental vibrational frequencies and spectroscopic constants of HOCS+, HSCO+, and isotopologues via quartic force fields. J. Phys. Chem. 116, 9582–9590. doi:10.1021/jp3073206
Fortenberry, R. C., Huang, X., Francisco, J. S., Crawford, T. D., and Lee, T. J. (2012b). Quartic force field predictions of the fundamental vibrational frequencies and spectroscopic constants of the cations HOCO+ and DOCO+. J. Chem. Phys. 136, 234309. doi:10.1063/1.4729309
Fortenberry, R. C., Huang, X., Schwenke, D. W., and Lee, T. J. (2014). Limited rotational and rovibrational line lists computed with highly accurate quartic force fields and ab initio dipole surfaces. Spectrochim. Acta Mol. Biomol. Spectrosc. 119, 76–83. doi:10.1016/j.saa.2013.03.092
Fortenberry, R. C., Huang, X., Crawford, T. D., and Lee, T. J. (2013). High-accuracy quartic force field calculations for the spectroscopic constants and vibrational frequencies of ?-CH: a possible link to lines observed in the horsehead nebula pdr. Astrophys. J. 772, 39. doi:10.1088/0004-637x/772/1/39
Fortenberry, R. C., Lee, T. J., and Müller, H. S. P. (2015). Excited vibrational level rotational constants for SiC: a sensitive molecular diagnostic for astrophysical conditions. Molec. Astrophys. 1, 13–19. doi:10.1016/j.molap.2015.07.001
Fortenberry, R. C. (2017). Rovibrational characterization of the proton-bound, noble gas complexes: ArHNe, ArHAr, and NeHNe. ACS Earth Space Chem. 1, 60–69. doi:10.1021/acsearthspacechem.7b00003
Gardner, M. B., Westbrook, B. R., Fortenberry, R., and Lee, T. J. (2020). Highly-accurate quartic force fields for the prediction of anharmonic rotational constants and fundamental vibrational frequencies. Spectrochim. Acta A Mol. Biomol. Spectrosc. [Epub ahead of print]. doi:10.1016/j.saa.2020.119184
Gaw, J. F., Willets, A., Green, W. H., and Handy, N. C. (1991). “SPECTRO: a program for the derivation of spectrscopic constants from provided quartic force fields and cubic dipole fields,” in Advances in molecular vibrations and collision dynamics. Editors J. M. Bowman, and M. A. Ratner (Greenwich, Connecticut: JAI Press, Inc.), 170–185.
Herzberg, G. (1966). Electronic spectra and electronic structure of polyatomic molecules. New York: Van Nostrand).
Hochstrasser, R., and Wirz, J. (1989). Ethynol: photochemical generation in an argon matrix, ir spectrum, and photoisomerization to ketene. Angew. Chem. Int. Ed. 28, 181–183. doi:10.1002/anie.198901811
Hochstrasser, R., and Wirz, J. (1990). Reversible photoisomerization of ketene to ethynol. Angew. Chem. Int. Ed. 29, 411–413. doi:10.1002/anie.199004111
Huang, X., and Lee, T. J. (2008). A procedure for computing accurate ab initio quartic force fields: application to HO2+ and H2O. J. Chem. Phys. 129, 044312. doi:10.1063/1.2957488
Huang, X., Taylor, P. R., and Lee, T. J. (2011). Highly accurate quartic force fields, vibrational frequencies, and spectroscopic constants for cyclic and linear C3H3(+). J. Phys. Chem. 115, 5005–5016. doi:10.1021/jp2019704
Huang, X., Fortenberry, R. C., and Lee, T. J. (2013). Spectroscopic constants and vibrational frequencies for l-CH and isotopologues from highly-accurate quartic force fields: the detection of l-CH in the Horsehead Nebula PDR questioned. Astrophys. J. Lett. 768, 25. doi:10.1088/2041-8205/768/2/l25
Huang, X., and Lee, T. J. (2009). Accurate quartic force fields for NH and CCH and rovibrational spectroscopic constants for their isotopologs. J. Chem. Phys. 131, 104301. doi:10.1063/1.3212560
Huang, X., Valeev, E. F., and Lee, T. J. (2010). Comparison of one-particle basis set extrapolation to explicitly correlated methods for the calculation of accurate quartic force fields, vibrational frequencies, and spectroscopic constants: application to HO, NH, NO, and CH. J. Chem. Phys. 133, 244108. doi:10.1063/1.3506341
Johansson, L. E., Andersson, C., Ellder, J., Friberg, P., Hjalmarson, A., Hoglund, B., et al. (1984). Spectral scan of orion A and IRC+10216 from 72 to 91 GHz. Astron. Astrophys. 130, 227–256
Kitchens, M. J. R., and Fortenberry, R. C. (2016). The rovibrational nature of closed-shell third-row triatomics: HOX and HXO, X = Si, P, S, and Cl. Chem. Phys. 472, 119–127. doi:10.1016/j.chemphys.2016.03.018
Knizia, G., Adler, T. B., and Werner, H. J. (2009). Simplified CCSD(T)-F12 methods: theory and benchmarks. J. Chem. Phys. 130, 054104. doi:10.1063/1.3054300
Lee, K. L. K., and McCarthy, M. (2020). Bayesian analysis of theoretical rotational constants from low-cost electronic structure methods. J. Phys. Chem. 124, 898–910. doi:10.1021/acs.jpca.9b09982
Martin, J. M. L., and Lee, T. J. (1996). The atomization energy and proton affinity of NH. an calibration study. Chem. Phys. Lett. 258, 136–143. doi:10.1016/0009-2614(96)00658-6
Martin, J. M. L., and Taylor, P. R. (1994). Basis set convergence for geometry and harmonic frequencies. are h functions enough?. Chem. Phys. Lett. 225, 473–479. doi:10.1016/0009-2614(94)87114-0
Martin-Drumel, M.-A., Lee, K. L. K., Pirali, O., Guillemin, J.-C., and McCarthy, M. C. (2019). “Investigating isomers of astrophysical molecules by rotational spectroscopy: the case of [CHO] compounds,” in International symposium on molecular spectroscopy. [Dataset].
Matthews, H. E., and Sears, T. J. (1986). Interstellar molecular line searches at 1.5 centimeters. Astrophys. J. 300, 766. doi:10.1086/163852
Mills, I. M. (1972). “Vibration-rotation structure in asymmetric- and symmetric-top molecules,” in Molecular spectroscopy–modern research. Editors K. N. Rao, and C. W. Mathews (New York: Academic Press), 115–140.
Peeters, E., Allamandola, L. J., Hudgins, D. M., Hony, S., and Tielens, A. G. G. M. (2004). “The unidentified infrared features after ISO,” in Astrophysics of dust, ASP conference series. Editors A. N. Witt, G. C. Clayton, and B. T. Draine (San Francisco, CA: Astronomical Society of the Pacific), 309.
Peterson, K. A., Adler, T. B., and Werner, H. J. (2008). Systematically convergent basis sets for explicitly correlated wavefunctions: the atoms H, He, B-Ne, and Al-Ar. J. Chem. Phys. 128, 084102. doi:10.1063/1.2831537
Peterson, K. A., and Dunning, T. H. (1995). Benchmark calculations with correlated molecular wave functions. vii. binding energy and structure of the hf dimer. J. Chem. Phys. 102, 2032–2041. doi:10.1063/1.468725
Pople, J. A., Schlegel, H. B., Krishnan, R., DeFrees, D. J., Binkley, J. S., Frisch, M. J., et al. (1981). Molecular orbital studies of vibrational frequencies. Int. J. Quant. Chem. 15, 269–278
Raghavachari, K., Trucks, G. W., Pople, J. A., and Replogle, E. (1989). Highly correlated systems: structure, binding energy and harmonic vibrational frequencies of ozone. Chem. Phys. Lett. 158, 207–212. doi:10.1016/0009-2614(89)87322-1
Rydbeck, R. A., Dunning, T. H., and Harrison, R. J. (1992). Electron affinities of the first-row atoms revisited. systematic basis sets and wave functions. J. Chem. Phys. 96, 6796–6806. doi:10.1063/1.462569
Shavitt, I., and Bartlett, R. J. (2009). Many-body methods in chemistry and physics: MBPT and coupled-cluster theory. Cambridge: Cambridge University Press.
Shimanouchi, T. (1972). Tables of molecular vibrational frequencies. 39th Edn. Washington, DC: National Standards Reference Data System, Vol. 1.
Solomon, P. M., Jefferts, K. B., Penzias, A. A., and Wilson, R. W. (1971). Detection of millimeter emission lines from interstellar methyl cyanide. Astrophys. J. Lett. 168, L107. doi:10.1086/180794
Stephens, J., Yan, W.-B., Richnow, M. L., Solka, H., and Curl, R. (1988). Infrared kinetic spectroscopy of CH and CD. J. Mol. Struct. 190, 41–60. doi:10.1016/0022-2860(88)80269-2
Tanaka, K., and Yoshimine, M. (1980). An ab initio study on ketene, hydroxyacetylene, formylmethylene, oxirene, and their rearrangement paths. J. Am. Chem. Soc. 102, 7655–7662. doi:10.1021/ja00546a006
Tucker, K. D., Kutner, M. L., and Thaddeus, P. (1974). The ethynyl radical C2H–A new interstellar molecule. Astrophys. J. 193, L115–L119. doi:10.1086/181646
Turner, A. M., Koutsogiannis, A. S., Kleimeier, N. F., Bergantini, A., Zhu, C., Fortenberry, R. C., et al. (2020). An experimental and theoretical investigation into the formation of ketene (HCCO) and ethynol (HCCOH) in interstellar analog ices. Astrophys. J. 896, 88. doi:3847/1538-4357/ab8dbc
Turner, B. E. (1977). Microwave detection of interstellar ketene. Astrophys. J. Lett. 213, L75–L79. doi:10.1086/182413
Watson, J. K. G. (1977). “Aspects of quartic and sextic centrifugal effects on rotational energy levels,” in Vibrational spectra and structure. Editor J. R. During (Amsterdam: Elsevier), 1–89.
Werner, H.-J., Knowles, P. J., Knizia, G., Manby, F. R., Schütz, M., Celani, P., et al. (2015). Molpro, version 2015.1, a package of programs. Available at: http://www.molpro.net (Accessed February 22, 2015)
Yousaf, K. E., and Peterson, K. A. (2008). Optimized auxiliary basis sets for explicitly correlated methods. J. Chem. Phys. 129, 184108. doi:10.1063/1.3009271
Zasimov, P. V., Ryazantsev, S. V., Tyurin, D. A., and Feldman, V. I. (2019). Radiation-induced chemistry in the CH–HO system at cryogenic temperatures: a matrix isolation study. Mon. Not. Roy. Astron. Soc. 491, 5140–5150. doi:10.1093/mnras/stz3228
Keywords: astrochemistry, quantum chemistry, computational spectroscopy, coupled cluster theory, COM formation
Citation: Dallas JD, Westbrook BR and Fortenberry RC (2021) Anharmonic Vibrational Frequencies and Spectroscopic Constants for the Detection of Ethynol in Space. Front. Astron. Space Sci. 7:626407. doi: 10.3389/fspas.2020.626407
Received: 05 November 2020; Accepted: 07 December 2020;
Published: 15 January 2021.
Edited by:
Majdi Hochlaf, Université Paris Est Marne La Vallée, FranceReviewed by:
Brett McGuire, Massachusetts Institute of Technology, United StatesAlbert Rimola, Autonomous University of Barcelona, Spain
Copyright © 2021 Dallas, Westbrook and Fortenberry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryan C. Fortenberry, cjQxMEBvbGVtaXNzLmVkdQ==