 Silvia Alessandrini
Silvia Alessandrini Mattia Melosso
Mattia Melosso- 1Scuola Normale Superiore, Pisa, Italy
- 2Dipartimento di Chimica “Giacomo Ciamician”, Università di Bologna, Bologna, Italy
The escalating identification of new complex molecules in the interstellar medium claims for potential formation routes of such species. In this regard, the present work considers the reaction between oxirane and the CN radical as a feasible formation mechanism of species having the C3H3NO molecular formula. Indeed, the compounds of this family are elusive in the interstellar medium and suggestions on which species could be formed at low temperature and low pressure conditions might aid their discovery. The c-C2H4O + CN reaction has been investigated from the thermodynamic and kinetic points of view. The thermodynamic has been studied by means of a double-hybrid density functional and revealed the presence of several mechanisms submerged with respect to the reactants energy, with the potential formation of oxazole and cyanoacetaldehyde. However, the kinetic results suggest that the main reaction pathway is the H-extraction, leading to 2-oxiranyl radical and HCN. The formation of cyanoacetaldehyde + H and of H2CCN + H2CO is also possible with smaller rate constants, while the production of oxazole is negligible due to the presence of a high energy barrier.
1 Introduction
The evermore increasing discovery of new and somewhat complex interstellar species, such as propargylimine (Bizzocchi et al., 2020), ethanolamine (Rivilla et al., 2021), glycolonitrile (Zeng et al., 2019), but also cyclopentadiene and indene (Cernicharo et al., 2021), benzonitrile (McGuire et al., 2018) together with larger species such as 1 and 2-cyanocylopentadiene (Lee et al., 2021; McCarthy et al., 2021) or 1 and 2-cyanonaphthalene (McGuire et al., 2021), as well as chiral molecules like propylene oxide (McGuire et al., 2016), claims for a boost up on hypothetical formation routes of these species in the interstellar medium (ISM) and, more generally, for an intensification of the study of potential formation mechanisms for molecules of astrochemical interest.
From the experimental point of view, crossed molecular beams (Casavecchia, 2000) and CRESU studies (Sims and Smith, 1995; Potapov et al., 2017) can investigate gas-phase reactions that are likely to occur in conditions similar to those of the ISM, i.e., low temperatures and low number density of molecules. However, the interpretation of the results of such reactions often requires the assistance of computational simulations (Gupta et al., 2019; Recio et al., 2021). Computational modelling via suitable electronic structure methods offers a clear view of the pathways involved in bi-molecular reactions, straightforwardly cutting out all the paths above a threshold energy, which is usually that available from the reactants. Once all the accessible pathways have been explored, kinetics simulations can be used to retrieve rate coefficients of the reactions and a suitable Arrhenius equation for their approximation, thereby obtaining the branching ratio of each reactive channel (Puzzarini and Barone, 2020; Puzzarini and Barone, 2021a). In addition, considering the similarity with other established formation routes for species already detected in the ISM (Vazart et al., 2015; Vazart et al., 2016; Lupi et al., 2020), theoretical studies can also shed light on possible formation routes of species that have not been observed in the ISM yet (Alessandrini et al., 2021).
For example, despite the simple chemical formula and the fact that far more complex species have been identified in space (see, e.g., isopropyl cyanide (Belloche et al., 2014) or ethyl methyl ether (Tercero et al., 2015)), no members belonging to the C3H3NO family have ever been detected in the ISM. Indeed, both the search of propiolamide [HCCC(O)NH2] towards Sagittarius B2 (Alonso et al., 2021) as well as the tentative observation of cyanooxirane [c-C2H3OCN] in dark clouds and hot-core sources (Dickens et al., 1996; Wirström et al., 2007) resulted unsuccessful. As it is clear that the family of C3H3NO isomers remains elusive, it is even less clear which isomer is expected to be the most abundant one and whether any of these species can be efficiently formed in the ISM or not. On the one hand, answering the first question would require – at least, and assuming that the Minimum Energy Principle holds (Lattelais et al., 2009) – the computation of the relative isomeric energies, thus pointing out the thermodynamically favoured species. On the other hand, it has to be verified if feasible formation pathways exist for the production of C3H3NO isomers and if their kinetics would be able to account for reasonable molecular abundances. With the respect to the kinetic aspect, another important prerequisite is the fact that the reactants must be both abundant and widespread in the ISM. All these considerations led us to investigate theoretically the gas-phase reaction between oxirane (c-C2H4O, also known as ethylene oxide) and the cyano radical (CN, 2Σ+ ground electronic state), which possibly produce various C3H3NO isomers plus an H atom that can carry away any excess of energy. While the focus of this paper is on the gas-phase reaction of the aforementioned reactants, it should also be mentioned that this reaction could occur in the solid-phase. In this case, the reaction would have access to different paths thanks to the mediation of the surface that acts as a catalyst. This process cannot be neglected but is out of the scope of the present paper, since it would involve also the modelling of the interaction between the two reactants and the surface material.
Oxirane has been detected in several astronomic sources, including molecular clouds in the Central Molecular Zone (Dickens et al., 1997; Requena-Torres et al., 2008), other massive star-forming regions (Nummelin et al., 1998), and the low-mass protostar IRAS 16293 (Lykke et al., 2017). The CN radical is the second species that has ever been detected in space (McKellar, 1940) — even before its spectroscopic laboratory characterisation (Jefferts et al., 1970; McGuire, 2018) — and is considered quite ubiquitous in the ISM (Savage et al., 2002; McGuire, 2018). Therefore, the reaction c-C2H4O + CN appears to be a promising reactive pathway, considering that the CN radical has been proved to be a good reactant for barrierless reactions being involved in – for example – the formation of formyl cyanide (Tonolo et al., 2020), cyanomethanimine (Vazart et al., 2015), benzonitrile (Woon, 2006; Cooke et al., 2020), and cyanocyclopentadiene (McCarthy et al., 2021).
To tackle this study, the reactive potential energy surface (PES) has been investigated by means of an accurate double-hybrid density functional and the rate coefficients for the most promising path will be reported. The computational details of the dynamic and kinetic study are discussed in the next section (Section 2). Then, the results obtained will be reported in Section 3 with a focus on the reaction mechanisms involved. This will be followed by the discussion of such results (Section 4), underlying the useful information that could be of support for future experimental studies of the reaction.
2 Computational Details
To asses if a reaction can occur in the ISM, the first point to be addressed is the thermodynamic of the process. Indeed, the reaction should proceed through pathways that require, at most, the energy of the reactants, considering that the energy available to molecules is usually a few K. With all the thermodynamically-active channels pinpointed, the second aspect to consider is the evaluation of the rate coefficients of the reaction, thus highlighting the most favoured product of the reaction from the kinetic point of view.
2.1 Reactive Potential Energy Surface
The investigation of the PES for the reaction c-C2H4O + CN has been carried out by means of the double-hybrid density functional rev-DSD-PBEP86 with a partially augmented triple-ζ basis set, i.e., the jun-cc-pVTZ (Dunning, 1989; Papajak and Truhlar, 2011). Since density functional theory has renowned problems in describing dispersion energies, which strongly characterise transitions states (TSs) and pre-reactive complexes, the dispersion correction D3 by Grimme et al. (2010) with dumping function (BJ, Grimme et al., 2011) has been systematically employed through all the computations. This methodology is shortly denoted in the following as jun-rDSD.
The exploration of the PES was carried out with the following strategy: firstly, the focus was on the initial part of the reaction, where the CN radical is approaching the oxirane molecule. The initial wells of the reaction, i.e., when the CN radical is bonded to oxirane, were the first points obtained on the PES. Then, the TSs leading to the formation of these initial minima starting from the reagents were searched for. Once the latter had been obtained, the connection to the corresponding wells was ensured via intrinsic reaction coordinate (IRC) calculations (Fukui, 1981), thus highlighting also the presence of some pre-reactive complexes. The barrierless connection of these van der Waals complexes with the reactants was also ensured by means of a scans of the most appropriate internal coordinate.
With the initial part of the reaction laid down, the focus moved to the routes occurring after the initial wells. From these, several TSs have been hypothesised and if they were actually located on the PES, the pathway analysis continued adopting the approach “TS → IRC → new minimum”. The Hessian of each stationary point was evaluated at the jun-rDSD level of theory in order to assure its nature, either TS or minimum. This approach was assisted by a continuous evaluation of the relative energy of the new stationary point with respect to reactants, thereby removing paths that led to intermediates having a positive relative energy. This approach has been applied iteratively to the newly discovered minima, thus unveiling only the thermodynamically accessible products. From the evaluation of the Hessian also the vibrational zero-point energy (ZPE) corrections were obtained for each stationary point.
All the computations have been carried out with the Gaussian16 suits of programs, revision C.01 (Frisch et al., 2016).
2.2 Kinetic Modelling
Having outlined the dynamic part of the reaction, the following step was the estimation of global rate constants. This translates into resolving the Master Equation for a multi-well reaction with more than one bimolecular product. For this aim, the MESS (Master Equation System Solver) program package (Georgievskii et al., 2013; Georgievskii and Klippenstein, 2016) was employed to model 1) the barrierless entrance channels of interest with phase-space theory (PST, Pechukas and Light 1965; Chesnavich 1986) and 2) all the other channel involving TSs with transition state theory (TST) and accounting for tunnelling via the Eckart model (Eckart, 1930). In the present case, only one barrierless approach was of interest for the reaction simulated in the ISM conditions, i.e., the one with the C-end of the CN radical approaching one carbon atom of the oxirane ring (see Figure 1 and Section 3 for more details). Therefore, only the potential of this entrance channel was modelled to obtain the C6/R6 power law underlying the PST (Fernández-Ramos et al., 2006). The R coordinate has been chosen as distance between the two carbon atoms (N– C ⋯ C) and the potential energy was obtained by means of a rigid scan of such coordinate from 3 to 10 Å. Finally, the C6 parameter was obtained by fitting the potential to the function
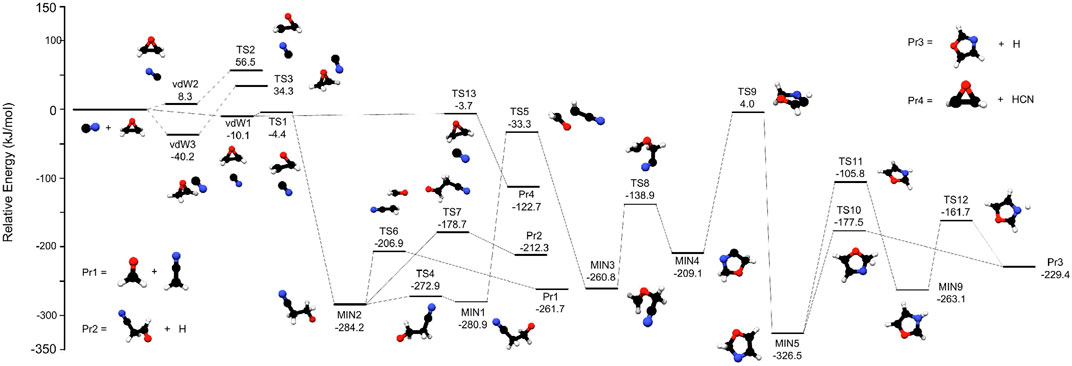
FIGURE 1. The most relevant stationary points located on the PES for the reaction between c-C2H4O and CN. Energies (in kJ mol−1) are given with the respect to the reactants.
After the approach of the two fragments, the reaction can be considered unimolecular and TST can been employed. The kinetic constant of a single step is expressed by:
where the main parameter is the partition function at the temperature T, i.e., Q(T), of the unimolecular species considered. The latter can be separated in rotational and vibrational partition functions and is directly computed by the MESS program considering – as input – the molecular structure, the corresponding harmonic frequencies, the electronic state, and the zero-point corrected energy with respect to the reactants. More details on the kinetic modelling are reported by Puzzarini and Barone (2021). In the present case, all the aforementioned quantities are derived from the preliminary thermodynamic study detailed in Section 2.1. The other quantities reported in Eq. 1 are: the Plank constant (h), the sum of states of the transition state (N‡(E)) involved, the Boltzmann constant (kB), and the energy (E) of the unimolecular species.
The global rate constants have been computed in the 60–200 K temperature range and for a pressure of 1 × 10–12 atm, that is in the low-pressure limit of the reaction, considering a energy step over temperature of 0.2 cm−1 and adopting the low-eigenvalue method available in MESS.
3 Results
The relevant reaction pathways for the reaction c-C2H4O + CN, obtained with the procedure explained in Section 2, are reported in Figure 1 while the energies of all the stationary points are summarised in Table 1.
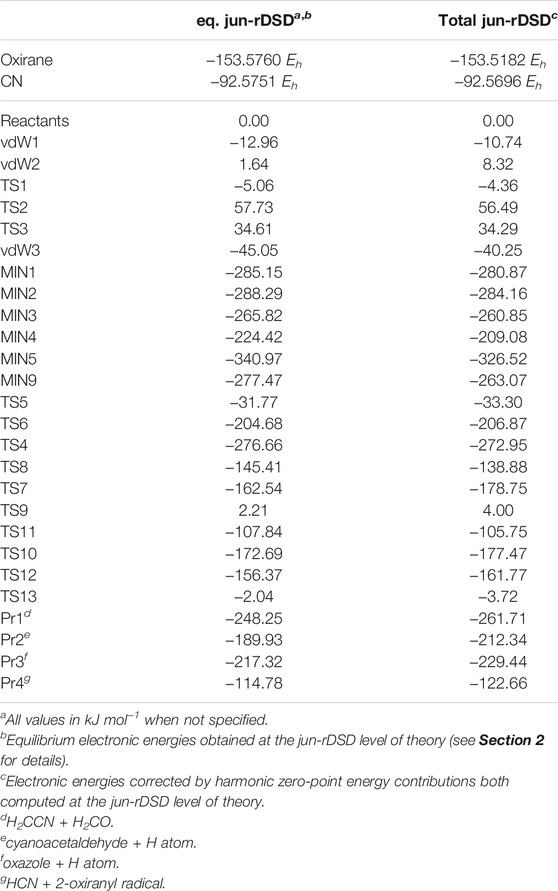
TABLE 1. Equilibrium and ZPE-corrected relative energies for the most relevant stationary points located on the PES and reported in Figure 1. For completeness the absolute energies (in Hartree units) of the two reactants are also reported.
The first possible approach of the CN radical on the ring is represented by vdW3 in Figure 1, where the C-end of the radical is approaching the oxygen atom. This pre-complex is located at −40.2 kJ mol−1 with respect to the reactants and it is the most stable pre-reactive complex on the PES. However, the following transition state (TS3) is 34.3 kJ mol−1 above the reactants, making this channel energetically-forbidden in astrochemical conditions; therefore, this path was not further investigated. Moreover, it should be noted that N-end approach of the CN radical on the oxygen atom could not be located on the PES.
The CN radical can also approach the carbon backbone of the oxirane ring either with its C-end or the N-end. The former attack leads to the formation of vdW1 which is 10.1 kJ mol−1 below the reactants, while the latter approach leads to vdW2, which is about 8 kJ mol−1 higher then the reactants. From vdW2 the reaction can proceed only via TS2, which is located 56.5 kJ mol−1 above the reactants; thus, also this channel was not further investigated. Although the paths proceeding via TS2 and TS3 are not of interest for the gas-phase reaction studied here, their importance in process occurring in the solid-phase (grains, ices, etc) cannot be ruled out and should be considered as well. Consequently, the reaction must proceed through vdW1: two energy-accessible TSs connected to this pre-reactive complex have been identified: TS1 (−4.4 kJ mol−1) and TS13 (−3.7 kJ mol−1). The latter regulates the H abstraction of the CN radical from the oxirane ring, forming 2-oxiranyl radical (C2H3O) + HCN, that are 122.7 kJ mol−1 more stable than the reactants (see Pr4 in Figure 1).
The other possibility for the reaction is to proceed via TS1, which brings to the formation of a C– C bond between the CN radical and oxirane, with a concomitant cleavage of the corresponding O-C bond. This is a nucleophilic approch that results in the opening of the ring with MIN2 as following stable well. This occurs with a small reaction barrier, i.e., 6 kJ mol−1 with MIN2 being also a deep well, located 284.2 kJ mol−1 below the reactants. MIN2 is a crucial point for the reaction, opening the route to the production of various bimolecular products. Indeed, it can decompose via TS6 to H2CCN + H2CO (Pr1 in Figure 1), with a barrier of 77.3 kJ mol−1 that corresponds to the cleavage of a C–C bond. Another accessible path leads to Pr2, i.e., cyanoacetaldehyde (NC–CH2–CHO) plus a H atom. Here, the H atom is detaching from the terminal C=O group, via TS7. The barrier for this step is 105 kJ mol−1 and the final products are 212.3 kJ mol−1 below the reactants. The latter possibility starting from MIN2 involves the formation of its conformer (MIN1) thanks to the rotation of the central CC bond. The results show that only MIN1 has the oxygen atom in the correct position to proceed the reaction via TS5, forming CH2OCH2CN that is MIN3. While the latter closely resembles cyanooxirane, the production of this species through the loss of an H atom requires too much energy and therefore it is not reported in Figure 1. However, MIN3 has the possibility of forming a 5-membered ring, with the terminal N atom being able to perform a nucleophilic attack on the CH2 end. This requires a barrier of 121.9 kJ mol−1 and results in MIN4, which is 51.7 kJ mol−1 higher in energy than MIN3. The energetically lower path from MIN4 involves an H-migration described by TS9, which is located 4 kJ mol−1 above the reactants and leads to MIN5, the most stable point of the PES reported in Figure 1. The possibility of forming a 4-membered ring from MIN3 has also been investigated but the TSs involved are all above the reactants energy by several kJ mol−1; therefore, this possibility was not further considered. However, also TS9 is above the reactants energy but considering the accuracy of our computations, it might as well be below the reference energy. Thus, also the products that could be formed after this point have been investigated. In particular, Figure 1 shows that MIN5 can evolve only to the products Pr3, i.e., oxazole + H atom, via a 1-step or a 2-step path. The former involves a direct cleavage of the C–H bond with the following removal of the H atom and occurs with a barrier of ∼150 kJ mol−1 (TS10). In contrast, the 2-step path begins with the cleavage of the same C–H bond, but the H atom migrates on the N atom (TS11) to form MIN9. Then, MIN9 forms Pr3 through the cleavage of the N–H bond described by TS12.
From the thermodynamic point of view, four products can be obtained on the PES for the reaction c-C2H4O + CN at low temperature and in vacuum conditions. However, those that appear to be the most favoured are Pr1, Pr2, and Pr4. To understand which one is the kinetically-favoured, the rate coefficients of the reaction have been computed in the 60–200 K temperature range and in the low pressure limit of the reaction. The temperature dependency of the rate coefficients is shown in Figure 2 and it can be seen that the rate constants for all the possible products decrease when increasing the temperature.
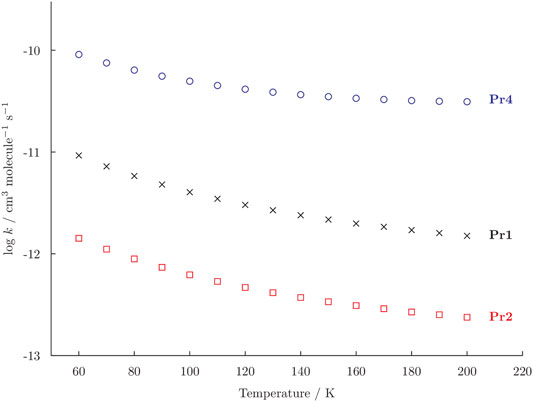
FIGURE 2. Rate constants (k) for the formation of Pr1 (black crosses), Pr2 (red squares), and Pr4 (blue dots) computed in the temperature range 60–200 K. The vertical axis is reported as logarithmic scale.
The formation of Pr4 has a rate constants that is generally one order of magnitude higher than that leading to Pr1. For example, at 60 K the rate coefficients for the reaction c-C2H4O + CN → Pr1 is 9.13 × 10–12 cm3 molecule−1 s−1 and the one to Pr4 is 9.08 × 10–11 cm3 molecule−1 s−1. Focusing only on Pr1 and Pr2, the rate constants of these two channels also differ for one order of magnitude, with the production of the former being favoured. However, in Figure 2 one can also observe how the production of Pr2 increase slightly more rapidly than the one of Pr1 at T below 100 K. Indeed, a 200 K the rate coefficient for the production of Pr1 is 1.5 × 10–12 cm3 molecule−1 s−1 and the one for Pr2 is 2.4 × 10–13 cm3 molecule−1 s−1 and at 60 K these values become 9.3 × 10–12 cm3 molecule−1 s−1 and 1.4 × 10–12 cm3 molecule−1 s−1. The production of Pr3 is always negligible, the rate constant being in the order of 1 × 10–24 cm3 molecule−1 s−1 for this reason it has not been reported in Figure 2. However, this result was expected considering that TS9 is found to be 4 kJ mol−1 above the reactants. Globally, the branching ratios at 60 K are 89.5% for Pr4, 9.1% for Pr1, and 1.4% for Pr2. The branching ratio to Pr4 increases when increasing the temperature, becoming as high as 94.5% at 200 K. At the latter temperature, the branching ratio to Pr1 is 4.8 and 0.8% for Pr3. Furthermore, it should also be noted that all the reverse reactions are negligible.
4 Discussion and Conclusion
Summarising, the investigation of the PES at the jun-rDSD level of theory indicates that several products could be formed starting from c-C2H4O + CN but the most probable reaction path is the extraction of one H atom from the oxirane ring, thus forming HCN and the 2-oxiranyl radical. If this occurs, a first point that would be interesting to investigate is the presence of such radical in the ISM. However, the rotational spectrum of this species has never been reported in the literature, thus preventing its astronomical detection. 2-oxiranyl radical is an highly unstable species, meaning that its study requires an in situ production from an appropriate starting species, as – for example – oxirane or a mixture of acetylene and oxygen. Not only, the measurements of the rotational spectrum of an open-shell species produced in situ can be challenging due to 1) the presence of several interfering species and 2) the hyperfine and fine splittings of the energy levels that turn out in a global loss of intensity and generate complicated spectral patterns. Therefore, experimental studies of 2-oxiranyl radical cannot avoid a theoretical computation of accurate spectroscopic parameters, needed to guide the identification and interpretation of the experimental spectrum. It is also worth of mention that 2-oxiranyl radical can likely undergo to recombination reactions with other species, among which the H2 molecule as it is by far the most abundant species in the ISM. This reaction would re-form oxirane, which could react again with the CN radical. In this view, the formation of Pr4 does not hamper severely the production of Pr1 and Pr2. Therefore, even considering that Pr4 is the most favoured product from the kinetic point of view, a clear picture of its abundance can only be obtained with a full chemical modelling accounting for related reactions and species.
Considering the title reaction as potential formation route of the C3H3NO isomers, the only species belonging to this family that can be produced are cyanoacetaldehyde and, quite unlikely from the kinetic point of view, oxazole. Although the rotational spectrum of cyanoacetaldehyde has been studied in the laboratory (Møllendal et al., 2012), this species has never been searched for in the ISM and its abundance in any astronomical source is unknown. As far as oxazole is concerned, its formation – which proceed through a ring expansion mechanism – involves a TS that at the current level of theory is above the energy of the reactants. Since our methodology has at least an error of ±5 kJ mol−1, the kinetic computations have also been simulated with the TS9 being 1 kJ mol−1 below the reactants. This turns out in a small increase of the rate constant, i.e., from 1 × 10–24 cm3 molecule−1 s−1 to 9 × 10–24 cm3 molecule−1 s−1, which however is still negligible with respect to the other available paths.
Moreover, differently from what has been reported in literature for many cyano-containing molecules (Vazart et al., 2015; Cooke et al., 2020; Tonolo et al., 2020), the title reaction does not form the cyano-derivative of the reaction partner, i.e., cyanooxirane, since the nucleophilic attack of the radical always leads to the opening of the ring. This result demonstrates that cyanooxirane cannot be formed efficiently from its parent oxirane and would explain why the astronomical searches of this species have resulted unsuccessful so far (Dickens et al., 1996; Wirström et al., 2007).
In conclusion, the present study has shown that the reaction c-C2H4O + CN most likely ends with the H-extraction for the oxirane ring, thus forming HCN and 2-oxiranyl radical. Being this the main reaction pathway, it is also likely that the C2H3O radical could be detected in the ISM if accurate measurements of its rotational spectrum will be reported. The reactive system could also undertake other pathways that would lead to H2CCN radical and H2CO, or cyanoacetaldehyde + H atom, with kinetic constants that are in the order of 10–12 cm3 molecule−1 s−1 in astronomical conditions, i.e., one order of magnitude below the rate constants for the production of the 2-oxiranyl radical. The production of oxazole is negligible according to the presence of a high energy barrier, involving the migration of an H atom.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
SA and MM contributed to conception and design of the study; SA carried out the computations required for the exploration of the PES while MM ran the kinetic simulations. Both authors prepared the figures and contributed to article revision, read, and approved the submitted version.
Funding
This study was supported by University of Bologna (RFO funds). The SMART@SNS Laboratory (http://smart.sns.it) is acknowledged for providing high-performance computing facilities.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Professor C. Puzzarini and Dr. L. Bizzocchi for the fruitful discussions and J. Lupi for his support with the MESS program.
References
Alessandrini, S., Tonolo, F., and Puzzarini, C. (2021). In Search of Phosphorus in Astronomical Environments: The Reaction between the CP Radical (X2Σ+) and Methanimine. J. Chem. Phys. 154, 054306. doi:10.1063/5.0038072
Alonso, E. R., Kolesniková, L., Belloche, A., Mata, S., Garrod, R. T., Jabri, A., et al. (2021). Rotational Spectroscopic Study and Astronomical Search for Propiolamide in Sgr B2(N). A&A 647, A55. doi:10.1051/0004-6361/202040211
Belloche, A., Garrod, R. T., Müller, H. S. P., and Menten, K. M. (2014). Detection of a Branched Alkyl Molecule in the Interstellar Medium: Iso-Propyl Cyanide. Science 345, 1584–1587. doi:10.1126/science.1256678
Bizzocchi, L., Prudenzano, D., Rivilla, V. M., Pietropolli-Charmet, A., Giuliano, B. M., Caselli, P., et al. (2020). Propargylimine in the Laboratory and in Space: Millimetre-Wave Spectroscopy and its First Detection in the ISM. A&A 640, A98. doi:10.1051/0004-6361/202038083
Casavecchia, P. (2000). Chemical Reaction Dynamics with Molecular Beams. Rep. Prog. Phys. 63, 355–414. doi:10.1088/0034-4885/63/3/203
Cernicharo, J., Agúndez, M., Cabezas, C., Tercero, B., Marcelino, N., Pardo, J. R., et al. (2021). Pure Hydrocarbon Cycles in TMC-1: Discovery of Ethynyl Cyclopropenylidene, Cyclopentadiene, and Indene. A&A 649, L15. doi:10.1051/0004-6361/202141156
Chesnavich, W. J. (1986). Multiple Transition States in Unimolecular Reactions. J. Chem. Phys. 84, 2615–2619. doi:10.1063/1.450331
Cooke, I. R., Gupta, D., Messinger, J. P., and Sims, I. R. (2020). Benzonitrile as a Proxy for Benzene in the Cold ISM: Low-Temperature Rate Coefficients for CN + C6H6. ApJ 891, L41. doi:10.3847/2041-8213/ab7a9c
Dickens, J. E., Irvine, W. M., Ohishi, M., Arrhenius, G., Pitsch, S., Bauder, A., et al. (1996). A Search for Interstellar Oxiranecarbonitrile (C3H3NO). Origins Life Evol. Biosph. 26, 97–110. doi:10.1007/bf01809850
Dickens, J. E., Irvine, W. M., Ohishi, M., Ikeda, M., Ishikawa, S., Nummelin, A., et al. (1997). Detection of Interstellar Ethylene Oxide (c‐C2H4O). ApJ 489, 753–757. doi:10.1086/304821
Dunning, T. H. (1989). Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms boron through Neon and Hydrogen. J. Chem. Phys. 90, 1007–1023. doi:10.1063/1.456153
Eckart, C. (1930). The Penetration of a Potential Barrier by Electrons. Phys. Rev. 35, 1303–1309. doi:10.1103/physrev.35.1303
Fernández-Ramos, A., Miller, J. A., Klippenstein, S. J., and Truhlar, D. G. (2006). Modeling the Kinetics of Bimolecular Reactions. Chem. Rev. 106, 4518–4584. doi:10.1021/cr050205w
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian16 Revision C.01. Wallingford CT: Gaussian Inc.
Fukui, K. (1981). The Path of Chemical Reactions - the IRC Approach. Acc. Chem. Res. 14, 363–368. doi:10.1021/ar00072a001
Georgievskii, Y., Miller, J. A., Burke, M. P., and Klippenstein, S. J. (2013). Reformulation and Solution of the Master Equation for Multiple-Well Chemical Reactions. J. Phys. Chem. A. 117, 12146–12154. doi:10.1021/jp4060704
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 132, 154104. doi:10.1063/1.3382344
Grimme, S., Ehrlich, S., and Goerigk, L. (2011). Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 32, 1456–1465. doi:10.1002/jcc.21759
Gupta, D., Cheikh Sid Ely, S., Cooke, I. R., Guillaume, T., Abdelkader Khedaoui, O., Hearne, T. S., et al. (2019). Low Temperature Kinetics of the Reaction between Methanol and the CN Radical. J. Phys. Chem. A. 123, 9995–10003. doi:10.1021/acs.jpca.9b08472
Jefferts, K. B., Penzias, A. A., and Wilson, R. W. (1970). Observation of the CN Radical in the Orion Nebula and W51. ApJ 161, L87. doi:10.1086/180576
Kelvin Lee, K. L., Changala, P. B., Loomis, R. A., Burkhardt, A. M., Xue, C., Cordiner, M. A., et al. (2021). Interstellar Detection of 2-cyanocyclopentadiene, C5H5CN, a Second Five-Membered Ring toward TMC-1. ApJL 910, L2. doi:10.3847/2041-8213/abe764
Lattelais, M., Pauzat, F., Ellinger, Y., and Ceccarelli, C. (2009). Interstellar Complex Organic Molecules and the Minimum Energy Principle. ApJ 696, L133–L136. doi:10.1088/0004-637x/696/2/l133
Lupi, J., Puzzarini, C., and Barone, V. (2020). Methanimine as a Key Precursor of Imines in the Interstellar Medium: The Case of Propargylimine. ApJ 903, L35. doi:10.3847/2041-8213/abc25c
Lykke, J. M., Coutens, A., Jørgensen, J. K., van der Wiel, M. H. D., Garrod, R. T., Müller, H. S. P., et al. (2017). The ALMA-PILS Survey: First Detections of Ethylene Oxide, Acetone and Propanal toward the Low-Mass Protostar IRAS 16293-2422. A&A 597, A53. doi:10.1051/0004-6361/201629180
McCarthy, M. C., Lee, K. L. K., Loomis, R. A., Burkhardt, A. M., Shingledecker, C. N., Charnley, S. B., et al. (2021). Interstellar Detection of the Highly Polar Five-Membered Ring Cyanocyclopentadiene. Nat. Astron. 5, 176–180. doi:10.1038/s41550-020-01213-y
McGuire, B. A. (2018). 2018 Census of Interstellar, Circumstellar, Extragalactic, Protoplanetary Disk, and Exoplanetary Molecules. ApJS 239, 17. doi:10.3847/1538-4365/aae5d2
McGuire, B. A., Burkhardt, A. M., Kalenskii, S., Shingledecker, C. N., Remijan, A. J., Herbst, E., et al. (2018). Detection of the Aromatic Molecule Benzonitrile (c-C6H5CN) in the Interstellar Medium. Science 359, 202–205. doi:10.1126/science.aao4890
McGuire, B. A., Carroll, P. B., Loomis, R. A., Finneran, I. A., Jewell, P. R., Remijan, A. J., et al. (2016). Discovery of the Interstellar Chiral Molecule Propylene Oxide (CH3CHCH2O). Science 352, 1449–1452. doi:10.1126/science.aae0328
McGuire, B. A., Loomis, R. A., Burkhardt, A. M., Lee, K. L. K., Shingledecker, C. N., Charnley, S. B., et al. (2021). Detection of Two Interstellar Polycyclic Aromatic Hydrocarbons via Spectral Matched Filtering. Science 371, 1265–1269. doi:10.1126/science.abb7535
McKellar, A. (1940). Evidence for the Molecular Origin of Some Hitherto Unidentified Interstellar Lines. Pasp 52, 187–192. doi:10.1086/125159
Møllendal, H., Margulés, L., Motiyenko, R. A., Larsen, N. W., and Guillemin, J.-C. (2012). Rotational Spectrum and Conformational Composition of Cyanoacetaldehyde, a Compound of Potential Prebiotic and Astrochemical Interest. J. Phys. Chem. A. 116, 4047–4056.
Nummelin, A., Dickens, J. E., Bergman, P., Hjalmarson, A., Irvine, W. M., Ikeda, M., et al. (1998). Abundances of Ethylene Oxide and Acetaldehyde in Hot Molecular Cloud Cores. Astron. Astrophys. 337, 275–286.
Papajak, E., and Truhlar, D. G. (2011). Convergent Partially Augmented Basis Sets for Post-Hartree−Fock Calculations of Molecular Properties and Reaction Barrier Heights. J. Chem. Theor. Comput. 7, 10–18. doi:10.1021/ct1005533
Pechukas, P., and Light, J. C. (1965). On Detailed Balancing and Statistical Theories of Chemical Kinetics. J. Chem. Phys. 42, 3281–3291. doi:10.1063/1.1696411
Potapov, A., Canosa, A., Jiménez, E., and Rowe, B. (2017). Uniform Supersonic Chemical Reactors: 30 Years of Astrochemical History and Future Challenges. Angew. Chem. Int. Ed. 56, 8618–8640. doi:10.1002/anie.201611240
Puzzarini, C., and Barone, V. (2020). A Never-Ending story in the Sky: The Secrets of Chemical Evolution. Phys. Life Rev. Burlington House, London: Royal Society of Chemistry 32, 59–94. doi:10.1016/j.plrev.2019.07.001
Puzzarini, C., and Barone, V. (2021).CHAPTER 10. Interstellar Complex Organic Molecules: A Step toward Biomolecule Building Blocks in the Skies. In Prebiotic Photochemistry: From Urey–Miller-like Experiments to Recent Findings. The Royal Society of Chemistry, 195–218. doi:10.1039/9781839164354-00195
Recio, P., Marchione, D., Caracciolo, A., Murray, V. J., Mancini, L., Rosi, M., et al. (2021). A Crossed Molecular Beam Investigation of the N(2D) + Pyridine Reaction and Implications for Prebiotic Chemistry. Chem. Phys. Lett. 779, 138852. doi:10.1016/j.cplett.2021.138852
Requena-Torres, M. A., Martin-Pintado, J., Martin, S., and Morris, M. R. (2008). The Galactic Center: The Largest Oxygen-Bearing Organic Molecule Repository. Astrophys. J. 672, 352–360.
Rivilla, V. M., Jiménez-Serra, I., Martín-Pintado, J., Briones, C., Rodríguez-Almeida, L. F., Rico-Villas, F., et al. (2021). Discovery in Space of Ethanolamine, the Simplest Phospholipid Head Group. Proc. Natl. Acad. Sci. U S A. 118. doi:10.1073/pnas.2101314118
Savage, C., Apponi, A. J., Ziurys, L. M., and Wyckoff, S. (2002). Galactic 12C/13C Ratios from Millimeter‐Wave Observations of Interstellar CN. ApJ 578, 211–223. doi:10.1086/342468
Sims, I. R., and Smith, I. W. M. (1995). Gas-Phase Reactions and Energy Transfer at Very Low Temperatures. Annu. Rev. Phys. Chem. 46, 109–138. doi:10.1146/annurev.pc.46.100195.000545
Tercero, B., Cernicharo, J., López, A., Brouillet, N., Kolesniková, L., Motiyenko, R. A., et al. (2015). Searching for Trans Ethyl Methyl Ether in Orion KL. A&A 582, L1. doi:10.1051/0004-6361/201526255
Tonolo, F., Lupi, J., Puzzarini, C., and Barone, V. (2020). The Quest for a Plausible Formation Route of Formyl Cyanide in the Interstellar Medium: a State-Of-The-Art Quantum-Chemical and Kinetic Approach. ApJ 900, 85. doi:10.3847/1538-4357/aba628
Vazart, F., Calderini, D., Puzzarini, C., Skouteris, D., and Barone, V. (2016). State-of-the-art Thermochemical and Kinetic Computations for Astrochemical Complex Organic Molecules: Formamide Formation in Cold Interstellar Clouds as a Case Study. J. Chem. Theor. Comput. 12, 5385–5397. doi:10.1021/acs.jctc.6b00379
Vazart, F., Latouche, C., Skouteris, D., Balucani, N., and Barone, V. (2015). Cyanomethanimine Isomers in Cold Interstellar Clouds: Insights from Electronic Structure and Kinetic Calculations. ApJ 810, 111. doi:10.1088/0004-637x/810/2/111
Wirström, E. S., Bergman, P., Hjalmarson, A., and Nummelin, A. (2007). A Search for Pre-biotic Molecules in Hot Cores. A&A 473, 177–180. doi:10.1051/0004-6361:20077535
Woon, D. E. (2006). Modeling Chemical Growth Processes in Titan's Atmosphere: 1. Theoretical Rates for Reactions between Benzene and the Ethynyl (C2H) and Cyano (CN) Radicals at Low Temperature and Pressure. Chem. Phys. 331, 67–76. doi:10.1016/j.chemphys.2006.09.028
Keywords: oxirane, cyanide radical, reactivity, astrochemistry, interstellar complex organic molecules, potential energy surface, kinetics
Citation: Alessandrini S and Melosso M (2021) Fate of the Gas-Phase Reaction Between Oxirane and the CN Radical in Interstellar Conditions. Front. Astron. Space Sci. 8:754977. doi: 10.3389/fspas.2021.754977
Received: 07 August 2021; Accepted: 07 September 2021;
Published: 20 September 2021.
Edited by:
Ryan C. Fortenberry, University of Mississippi, United StatesReviewed by:
Ankan Das, Indian Centre for Space Physics, IndiaDonghui Quan, Eastern Kentucky University, United States
Copyright © 2021 Alessandrini and Melosso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Alessandrini, c2lsdmlhLmFsZXNzYW5kcmluaUBzbnMuaXQ=