 Juliane Heitkämper1Sarah Suchaneck1Juan García de la Concepción2
Juliane Heitkämper1Sarah Suchaneck1Juan García de la Concepción2 Johannes Kästner1
Johannes Kästner1 Germán Molpeceres1,3*
Germán Molpeceres1,3*- 1Institute for Theoretical Chemistry, University of Stuttgart, Stuttgart, Germany
- 2Centro de Astrobiología (CSIC-INTA), Madrid, Spain
- 3Department of Astronomy, Graduate School of Science, The University of Tokyo, Tokyo, Japan
The recent detection of cyclic species in cold interstellar environments is an exciting discovery with yet many unknowns to be solved. Among them, the presence of aromatic heterocycles in space would act as an indirect evidence of the presence of precursors of nucleotides. The seeming absence of these species in the observations poses a fascinating conundrum that can be tackled with computational insights. Whilst many arguments can be given to explain the absence of heterocycles in space, one of the possible scenarios involves fast chemical conversion and formation of new species to be detected. We have tested this hypothesis for the reaction of pyridine with the CN radical to find possible scenarios in which the detectability of pyridine, as an archetypical heterocycle, could be enhanced or diminished via chemical conversions. Using a combination of ab-initio characterization of the reactive potential energy surface and kinetic and chemical simulations, we have established that pyridine does react very fast with CN radicals, estimating that the studied reactions is between 2.5–4.5 times faster in pyridine than in benzene, with a total loss rate constant of 1.33 × 10–9 cm3s−1 at 30 K, with an almost null temperature dependence in the (30–150) K range. Addition reactions forming 1,2,3-cyanopyridine are favored over abstraction reactions or the formation of isocyanides. Besides, for 1 and 2-cyanopyridine there is an increase in the total dipole moment with respect to pyridine, which can help in their detection. However, the reaction is not site specific, and equal amounts of 1,2,3-cyanopyridine are formed during the reaction, diluting the abundance of all the individual pyridine derivatives.
1 Introduction
Recent detections of cyclic and aromatic molecules in cold environments are revolutionizing astrochemistry. Between the 2018 census (McGuire, 2018) and the 2021 census (McGuire, 2022) of extraterrestrial molecules a total of 37 molecules have been detected. This corresponds to a rate of detections of 12.33 molecules/year (see Figure 1 of McGuire (2022)), surpassing older rates by a factor between two and three. It is thus fair to highlight that we are amidst an astrochemical revolution in term of detections, with experiments and simulations needing to keep up the pace. Amongst the molecules that have been detected recently, cyclic (aromatic or not) molecules in cold environments constitute a significant breakthrough. To date, several cyclic molecules containing five atoms or more in their structure have been detected, most of them in the Taurus Molecular Cloud (TMC-1) namely benzonitrile (c–C6H5–CN) (McGuire et al., 2018), cyano-cyclopentadiene (c–C5H4–CN) (McCarthy et al., 2020), cyano-napthalene, (McGuire et al., 2021), benzyne (Cernicharo et al., 2021c), cyclopentadiene (Cernicharo et al., 2021b), indene (Cernicharo et al., 2021b), or fulvenallene (Cernicharo, J. et al., 2022). From the last wave of detections numerous questions arise, for example, whether the molecules are formed from a bottom-up approach from simpler precursors or are produced from the fragmentation of, for example, graphene (Merino et al., 2014). Another fundamental question to be resolved is the apparent absence of heterocyclic species (molecules whose skeleton backbone contains other elements apart from carbon), for example pyridine, in relation with the bottom-up formation of cyclic structures and the possible conversion routes of these molecules once formed (Barnum et al., 2022).
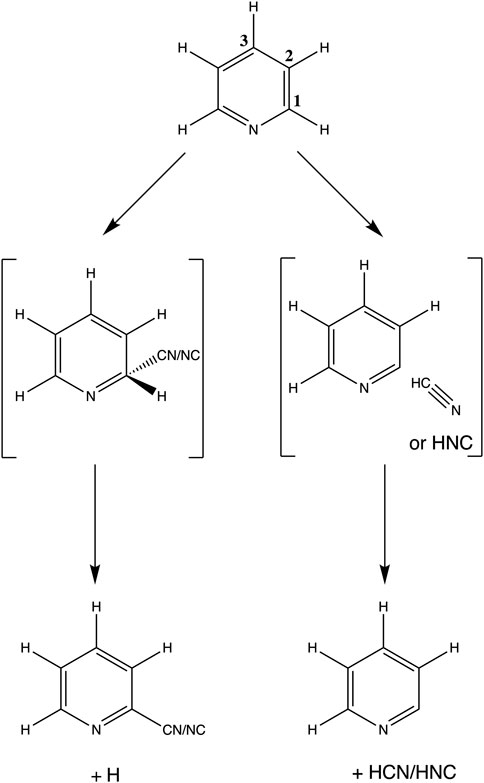
FIGURE 1. Scheme of the reactions considered in this work, as well as the different equivalent carbon positions for the reactions to occur. This example depicts reactions in C1.
In fact, nitrogen bearing polycyclic aromatic hydrocarbons (PANHs) are considered to be relatively abundant in the ISM, estimating that they account for 1%–2% of the total cosmic nitrogen (Hudgins et al., 2005). PANHs have been a subject of intense research because their presence could be linked with the presence of simpler precursors (pyridine, pyrimidine, triazine) that incidentally are constituents of prebiotic molecules like nucleobases. Besides, PANHs studied in the laboratory and with ab-initio simulations (Mattioda et al., 2003; Hudgins et al., 2005; McNaughton et al., 2007; Alvaro Galué et al., 2010; Canelo et al., 2018) show rich and distinguishable IR emission spectra, specially around the 6.2–6.3 μm region. These bands, in combination with others attributed to pure polycyclic aromatic hydrocarbon (PAH) ones, i.e., 3.3, 7.7, 8.6, 11.3, and 12.7 μm contribute to the so-called “Unidentified Infrared Emission” owing to the high blending of bands and the impossibility of disentangle the complex spectra. Conversely, such a limitation is not in place for radio astronomical observations, and specially considering that PANHs do possess a significant dipole moment which make them susceptible of detection (Mattioda et al., 2003; Malloci et al., 2007), and yet neither N-bearing heterocycles nor PANHs have been conclusively detected to date (Barnum et al., 2022).
There must be several reasons behind the lack of detection of heterocycles. Taking as an example one of the simplest N-bearing and more sought heterocycles, pyridine, we can find several possible explanations for its absence. First, current routes for the formation of pyridine, for example from cyanides (e.g., vynil cyanide condensation) do present emerged barriers (Parker et al., 2015) or alternative reaction products [e.g., formation of 1-cyano-1,3-butadiene in the reaction between CN + butadiene (Cooke and Sims, 2019; Barnum et al., 2022)]. This is in contrast with the majoritarian formation of benzene in the C2H + butadiene reaction (Jones et al., 2011), pointing to an essential different chemistry operating in the formation of heterocycles. Second, it was found that pyridine should not withstand the conditions of the diffuse interstellar medium and should be photolysed rather fast, in 10–100 years (Peeters et al., 2005). Under this assumption, all the pyridine that could be formed from, for example, fragmentation of large PANHs (Yang et al., 2020) should be destroyed by the time it reaches the dense molecular cloud and any new pyridine molecules should be formed bottom-up.
In addition to the competitive routes of reaction affecting the formation of heterocycles, another factor that heavily impacts the abundances of interstellar molecules are the routes of destruction of said molecules. Under this picture, a parent molecule reacting with some other species would not be detectable anymore, and rather the products of reaction would be. In fragmentation events, for example in reactions of molecules with
In this work we have carried out a systematic study of the interaction of CN radicals with the pyridine molecule in order to reveal more about the possible chemistry that this heterocycle experiences in the ISM, but also to determine the possibility of detecting it through cyanated proxies. Although pyridine itself has a net dipole moment, the addition of a CN group can, in some cases, increase its magnitude helping its detectability. However, and working against the previous argument, the reaction of pyridine with CN can dilute the already low abundance of pyridine. To shed some light in the topic we have studied the reactivity of the C5H5N + CN theoretically, characterizing the potential energy surface (PES) of the system and complementing the static study with kinetic simulations. The paper is distributed as follows, in Section 2 we summarize the methods used throughout the work. Section 3 is dedicated to a description of all the potential channels of reactions whereas in Section 4 we discern which ones are really contributing to the reaction. Section 5 is dedicated to discuss the results obtained during the work in light of the possible proxy effects emanating from the reactions. Finally we give some conclusion and outlook in Section 6.
2 Methods
2.1 Potential energy surface exploration
In this study we sampled both CN additions and H abstractions (e.g., formation of HCN/HNC) under the assumption that each reaction proceeds via a simplified kinetic model involving a single bimolecular reactant state (Pyridine + CN), a single reaction intermediate and a single product channel (Balucani et al., 1999; Trevitt et al., 2010), see Figure 1 for a schematic example of the reaction channels considered in this work in the single-well model. The intermediates may be connected with the reactant and product channel through a transition state or not (e.g., barrierless).
The potential energy surfaces for the different reaction channels were explored at the density functional theory level (DFT) using the M06-2X-D3 (Zhao and Truhlar, 2008; Grimme et al., 2010) functional, which is specially suited for kinetic studies, accompanied of a def2-TZVP basis set (Weigend and Ahlrichs, 2005). The electronic energies of our molecular geometries were then refined using a high-level correlation method, namely (U)CCSD(T)/cc-pVTZ (Dunning, 1989; Knowles et al., 1993). Hence, the level of theory that we used throughout the work can be abbreviated as CCSD(T)/cc-pVTZ//M06-2X-D3/def2-TZVP.
The exploration of the PES began with relaxed scans (e.g., sample a reaction coordinate optimizing at each step all the remaining degrees of freedom) to determine whether the entrance channel, either addition of abstraction, was barrierless. We performed relaxed scans in all the symmetry distinguishable positions of pyridine, both for additions and abstractions. In the cases where the entrance channel was barrierless we fitted the corresponding long-range decay to a function of the type, V(r) = −C/rn where C is a fitting constant employed in the subsequent rate constant calculations using phase space theory (See Section 2.2), r is the reaction coordinate sampled during the scan, and n = 6 in all cases. The potential energy scans (e.g., the values of r) were sampled in the ∼10–4 Å range. In cases where entrance barriers are present, we optimized the associated transitions states. Due to the great reactivity exhibited by the CN radical, we needed to do our PES scans constraining not only the reaction coordinate of interest, but also the angles between the atoms adjacent in the ring. Otherwise, every potential energy scan shows formation of non-wanted adducts. In some cases, this procedure led to small (submerged) spurious barriers due to the applied restraints. We confirmed that these barriers were an artifact by lifting the constraints and optimizing the structure to ensure the barrierless nature of the reactions. Finally, we note that restraining these degrees of freedom must have a minimal effect at long ranges, not affecting significantly our values of C.
The search for barrierless and activated steps was carried out for all subsequent steps of the reaction mechanism, determining all the stationary points of the PES (e.g., intermediate → TS → Exit channel, see Figure 1). We examined cyanide additions (-CN), isocyanide additions (-NC), hydrogen cyanide formation (HCN), and hydrogen isocyanide formation (HNC). Vibrational frequencies were calculated at each stationary point. We confirmed that each reactant, intermediate and product lacked any imaginary frequencies, characteristic of true minima of the PES, while transition states were checked to have exactly one imaginary vibrational frequency, corresponding to the reaction transition mode. All real frequencies below 100 cm−1 were raised to that number in the subsequent kinetic simulations for all species. Zero-point energies were corrected applying a correction factor of 0.971 to the harmonic value (Alecu et al., 2010). All the DFT calculations were performed with the ORCA code (Neese et al., 2020) (v. 5.0.1). Coupled cluster calculations employed Molpro (v. 2019.1.2) (Werner et al., 2019, 2012).
2.2 Kinetic simulations
As we introduced before, in our kinetic analysis we merged processes proceeding without an activation barrier for whose associated rate constants were calculated in the context of capture theory or phase-space theory (Pechukas and Light, 1965; Truhlar, 1969; Chesnavich, 1986) and activated processes (with submerged or emerged barriers) that are modelled in the context of the Rice-Ramsperger-Kassel-Marcus (RRKM) theory (Rice and Ramsperger, 1927; Kassel, 1928; Marcus, 1952). All the kinetic calculations used the MESS code (Georgievskii et al., 2013). Symmetry numbers (σ) and tunneling factors (κEckart) were introduced, where relevant. Tunneling factors are obtained from an asymmetric Eckart barrier. Using the same MESS framework, we carried out chemical simulations using a master equation approach implemented in such code. From these simulations we attained temperature (and pressure) dependent global rate constants for the reactions. Note that we did not explore pressure dependence in our rate constants, because we are interested in the limit of the very low pressures of the ISM. All our kinetic simulations use a pressure of 10–7 atm, following a previous work using this methodology (García de la Concepción et al., 2021). We did consider temperature dependence in our calculations, and evaluated the rate constants in the (30–360) K range.
3 Potential energy profiles
3.1 Addition of CN to pyridine
In analogy with the reaction of CN addition to benzene (Balucani et al., 1999; Woon, 2006; Trevitt et al., 2010) we started our exploration of reaction channels with the C5H5N + CN → C5H4NCN + H reactions, proceeding through the formation of a highly excited association complex that is subsequently relaxed by the release of atomic hydrogen from the same carbon where the CN is attached to. However, and comparing with the study on benzene, pyridine is a C2v molecule in contrast with the highly symmetric D6h benzene. This effectively means that not all carbon and nitrogen positions are equivalent for the approach of the CN radical, creating four different addition channels. All the additions proceed without a barrier. However, the addition of CN to the N atom (creating the -NCN moiety) does not have a clear exit. Thus, we only considered back dissociation to C5H5N + CN as possible exit channel for this addition.
3.1.1 Addition to C atoms
The PES for the selected reaction coordinates and the structures for the stationary points in this reaction channel are presented in Figure 2. Note that all internal energies U are referred with respect to the separated reactants asymptote.
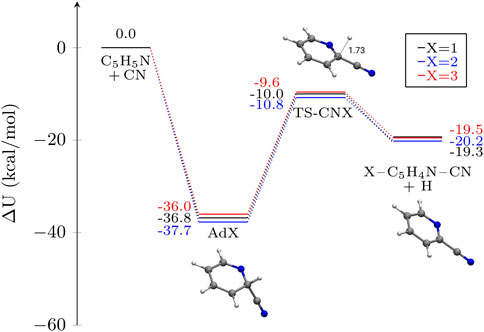
FIGURE 2. Potential energy profiles for the addition of CN to pyridine in different carbon atomic positions. The depicted magnitude is ΔU (electronic energy + ZPVE) in kcal mol−1. Molecular structures of the stationary points on the different PES other than pyridine are shown for X = 1 as an example. Distances are given in Å.
In all the sampled cases we observe a rather deep well, of around −37 kcal mol−1 that is accessed via a barrierless association to form the adducts ADX (with X = 1,2,3, e.g., ortho, meta and para, see Figure 1). These radicals are very stable and the relative energies between them are very similar, with differences of merely 1.7 kcal mol−1 in favor of AD2. These small differences do not affect the reaction kinetics in any way. We observe similar tiny differences for the products, with 2-C5H4NCN being the thermodynamically most favored product. As we show below, these slight differences in the PES do not translate into faster or slower reactions, with reaction rate constants for these systems being very similar for all the available carbon positions, especially at low temperatures. It is also worth noting that the activation energies for the barriers leading to the exit channel are almost isoenergetic, too, with reaction energies (exothermic) between 26–27 kcal mol−1 with respect to the PES well. A higher deviation between these barriers would enhance the importance of thermal effects in the reactions, enlarging the gap for the reaction rate constants and having a consequential effect in the branching ratios of reactions, with importance for the reactions at high temperatures, as we show in Section 5.
The reactivity of pyridine is very similar to the one in benzene presented in (Woon, 2006). The intermediate well depth for benzene is found to be −39.2 kcal mol−1 which is within our method uncertainty for pyridine. On a similar note, the submerged transition state for H detachment is −14.1 kcal mol−1, that is slightly below our predicted values for AdX, of around −10 kcal mol−1. Finally, the exothermicity of the reaction, of around 19.3.2–20.2 kcal mol−1 is in very good agreement with the 21.5 kcal mol−1 that is found for benzene.
3.1.2 Addition to the N atom of pyridine: Backdissociation
Apart from addition of the CN radical to the C atoms of pyridine, it is also possible that CN attaches barrierlessly to the N atom of pyridine, forming the NC–C5H5N adduct. This process, that proceeds without a barrier, evolves to back-dissociation to reactants, as we indicate in Section 4 and discuss in Section 5. This process effectively reduces the total loss rate constant of the process, and slows down the reaction. The relative energy of the stable adduct (AdN), not depicted in Figure 2 (but the geometry is provided in the Supplementary Materials) is ΔU = −38.6 kcal mol−1 with respect to the reactants, which makes it more stable than the other wells, discussed above.
3.2 Addition of NC to pyridine
The second type of additions that we considered in this work concern the formation of an isocyano complex with pyridine. Isocyanides are normally less stable than cyanides, evinced, for example, by the difference between hydrogen cyanide and hydrogen isocyanide of 14.7 kcal mol−1 (no ZPVE corrected) at the CCSD(T)/cc-pVTZ//M06-2X-D3/def2-TZVP level of theory. A similar behavior is found for the reactivity with pyridine, with wells depths in between −14–16 kcal mol−1 depending on the atomic position. These differ a lot from the wells we found for addition of CN to the pyridine backbone, of ∼ −37 kcal mol−1 (Section 3.1.1). The PES diagrams for isocyanide association can be found in Figure 3.
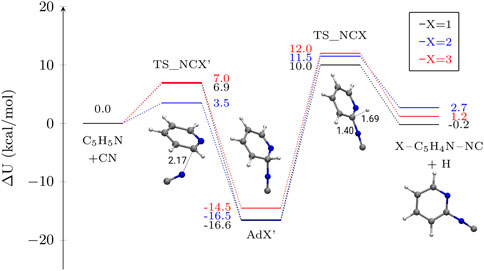
FIGURE 3. Potential energy profiles for the addition of NC (isocyanide) to pyridine in different atomic positions. The depicted magnitude is ΔU (electronic energy + ZPVE) in kcal mol−1. Molecular structures of the stationary points on the different PES other than pyridine are shown for X = 1 as an example. Distances are given in Å.
The most fundamental difference between the addition reactions of CN and NC can be found in the emerged nature of the entrance barrier in the case of the NC radical. These barriers, that vary between 3.5 and 7.0 kcal mol−1 essentially deem the reaction to be a negligible channel in comparison with CN additions. As we present in Section 4, the emergence of these barriers yield reaction rate constants in the order of 10–46–10−66 cm3s−1 at 30 K, that are many orders of magnitudes lower than the rate constants for the addition of CN (in the order of 10–10 cm3s−1). Similarly, the energy profile for NC addition shows that the H elimination to yield c–C5H4N–NC presents an emerged barrier of similar magnitude than the one for the c–C5H4N–CN formation, and of increased magnitude than the entrance barrier (see Figure 3). However, the kinetics of the system are dominated by the addition barrier. Involving a CN moiety, tunneling is therefore negligible and thermal effects drive all the dynamics. Thus, we conclude that the reaction is not competitive with CN addition, as evinced also by the associated rate constants.
When comparing with what was found for benzene, we confirm the presence of an emerged barrier for the H elimination step, that in the case of benzene is of 5.9 kcal mol−1, that is lower than our own results, of around 11 kcal mol−1. However, for benzene, it appears that the association step has a submerged barrier while we found that for pyridine that is both emerged and rate limiting. This discrepancy has no impact in the dynamics of both systems, with NC addition being a really minor route. However, it is interesting to distinguish these differences in the chemistry of both benzene and pyridine, because they can serve to rationalize and generate reaction trends with other complexes.
Unlike CN addition to the N position of pyridine, the isocyanide counterpart proceeds through a transition state with an activation energy at the CCSD(T)/cc-pVTZ//M06-2X-D3/def2-TZVP of 1.3 kcal mol−1. Because this reaction will be slow in comparison with additions and leads to back-dissociation to reactants, we have not deepened in its study.
3.3 Abstraction of H from pyridine: Hydrogen isocyanide formation
Summed to addition reactions, another possible reaction type affecting these molecules is the abstraction of a H atom from the skeletal backbone, releasing HCN (or HNC). This particular reaction is known to not proceed in benzene (Woon, 2006). For pyridine, we found a similar behavior, although the activation energies for the entrance barriers are very low, and dependant on the theoretical method. For example, for abstraction of H at the C1 atom, we found a transition state structure, with an energy of −1.3 kcal mol−1 with respect to the reactants at the M06-2X-D3 level of theory (e.g., without refining the electronic energies). When electronic energies are refined with a high level method, e.g., CCSD(T)/cc-pVTZ, such a barrier emerges, with an activation energy of ∼ 0.6 kcal mol−1. Contrarily, H abstraction reactions on C2 and C3 proceed with a non-submerged barrier both at the DFT level and also refining the electronic energies. The PES diagrams for the HCN abstraction can be found in Figure 4, note that we included a transient reaction intermediate in order to being able to apply the RRKM + Master equation kinetic protocol. The HCN abstraction reactions are less exothermic than CN additions but more exothermic than NC additions, with values of the reaction energy varying between ∼ 13–19 kcal mol−1. The emerged barriers for the reactions of H abstraction in C2 and C3 are lower than in the case of NC additions, of merely 0.6 kcal mol−1 for the abstraction of H on C1 and 1.5–1.3 kcal mol−1 for the abstraction in C2-C3. Even such small barriers completely dominate the chemistry at low temperatures and deem the reaction non competitive with CN addition. These very small barriers or lack of thereof dominating the kinetics of the formation of HCN pose a problem in the methodological side, especially for HCN abstraction at C1. With such a small activation energy, variations in the theoretical method under consideration may affect the activated or barrierless nature of the process, consequently affecting the predicted rate constants for the reaction. We did tests with kinetic calculations assuming HCN abstraction on C1 being barrierless, finding a very minor contribution in the total rate constants at low temperatures. At high temperatures, the effect of the presence or the lack of a barrier influences the kinetics more. However, in this study we are mostly interested in low-temperature and low-pressure rate constants, characteristic of the cold ISM.
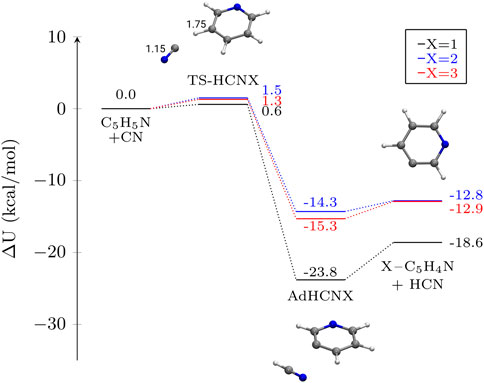
FIGURE 4. Potential energy profiles for the formation of HCN and X-pyridil (X–C5H4N) from pyridine and CN via a H abstraction in different atomic positions. The depicted magnitude is ΔU (electronic energy + ZPVE) in kcal mol−1. Molecular structures of the stationary points on the different PES other than pyridine are shown for X = 2 as an example. Distances are given in Å.
The most exothermic reaction for this set corresponds to the formation of 1-pyridyl. The rotational and vibrational spectra of pyridyl radicals was recently reported using vibrational perturbation theory (VPT2) (Meyer et al., 2022) using highly correlated coupled cluster methods (CCSD(T)/ANO0). In their study, the authors reported that the 1-pyridyl radical is more stable by 4.14–5.57 kcal mol−1 with respect to 2-3-pyridyl, in very good agreement with our values of ∼ 5.7 kcal mol−1. We interpret this energy difference as the key factor behind the apparent different reactivity with CN radicals in different positions of the pyridine structure. The abstraction on C1 generates an intramolecular cyanide (R-N-C-R’), similar to o-benzyne (c–C6H4), a molecule with positive detection in the ISM (Cernicharo et al., 2021a), which, in fact, can stabilize the 1-pyridyl radical.
3.4 Abstraction of H from pyridine: Hydrogen isocyanide formation
The last set of reactions under consideration pertains the formation of HNC, that is the abstraction of H from the pyridine molecule to form isocyanic acid. As in the case of the contrast between CN and NC addition, we found that the abstraction of HNC presents significant emerged barriers that deem the reaction to be unfeasible under astrophysical conditions. These barriers are in the order of 8.0–11.3 kcal mol−1 (see Figure 5). Thus, these activation energies are among the highest of this work. However, the magnitude of these values does not necessarily reflect into slower rate constants for abstraction, because quantum tunneling plays a greater role (the reaction involves a H atom) than in the case of NC additions. It is therefore important to note also the influence of the imaginary frequencies of the transition modes, that vary between 692i cm−1 for position C1 and 1334i and 1399i cm−1 for positions C2 and C3 respectively. That is why rate constants at lower temperatures are higher even considering significantly larger activation barriers between C1 and C2/C3. The reaction in C1 is slightly exothermic (−4.2 kcal mol−1) whereas the abstraction reactions in C2 and C3 are endothermic by around ∼ 1.5 kcal mol−1. As in the case of HCN abstractions, the formation of 1-pyridyl is favored, because of the formation of the intracycle CN moeity. Overall, the reaction is clearly non competitive with all the other reactions considered in this work.
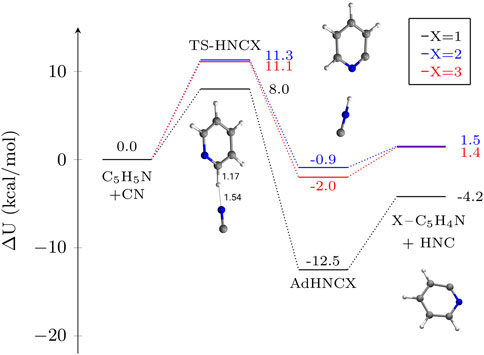
FIGURE 5. Potential energy profiles for the formation of HNC and X-pyridil (X–C5H4N) from pyridine and CN via a H abstraction in different atomic positions. The depicted magnitude is ΔU (electronic energy + ZPVE) in kcal mol−1. Molecular structures of the stationary points on the different PES other than pyridine are shown for X = 1 as an example. Distances are given in Å.
Finally, all the energetic quantities are collated (with respect to the separated reactants asymptote) in Table 1.
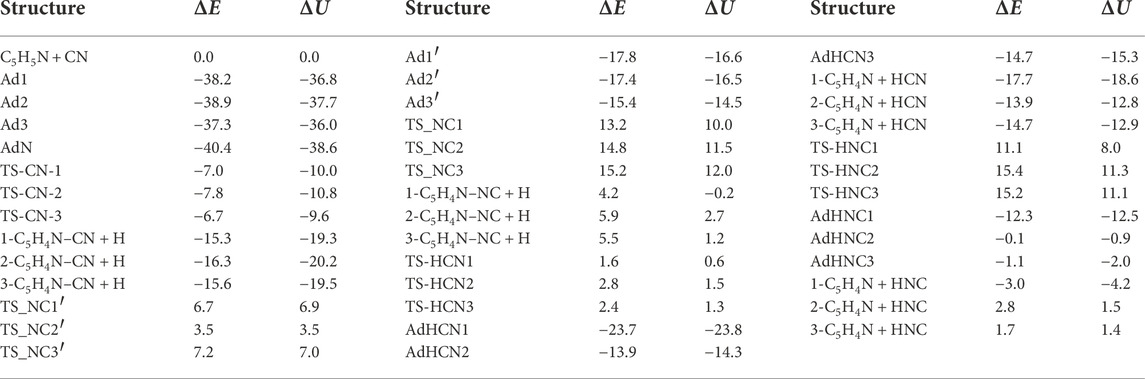
TABLE 1. Electronic energy difference with respect to the separated C5H5N + CN reactants without including ZPVE (ΔE, in kcal mol−1) and including ZPVE (ΔU, in kcal mol−1) for all species considered in this work. Figures 2–5 show the structures.
4 Kinetic modelling
From the above PES explorations we selected the most promising reaction channels to derive global rate constants and subsequently branching ratios of reactions (see Section 5). As we presented in Section 2 for processes without a barrier we fitted an expression of the type V(r) = −C/r6, obtaining the values of C required for the application of phase space theory. The values of these coefficients are collated in Table 2. These, in combination with the information of the different PES presented in the last section, allow us to attain global rate constants of reaction through our master equation calculations using MESS. The numerical values for the rate constants leading to new products are gathered in Table 3, whereas the rate constants for the back dissociation are gathered in Table 4. All temperature dependant reaction rate constants are represented in Figure 6.

TABLE 2. Reaction step and values of the capture coefficient C (in Eh/Bohr6) for the processes without a barrier studied in this work.
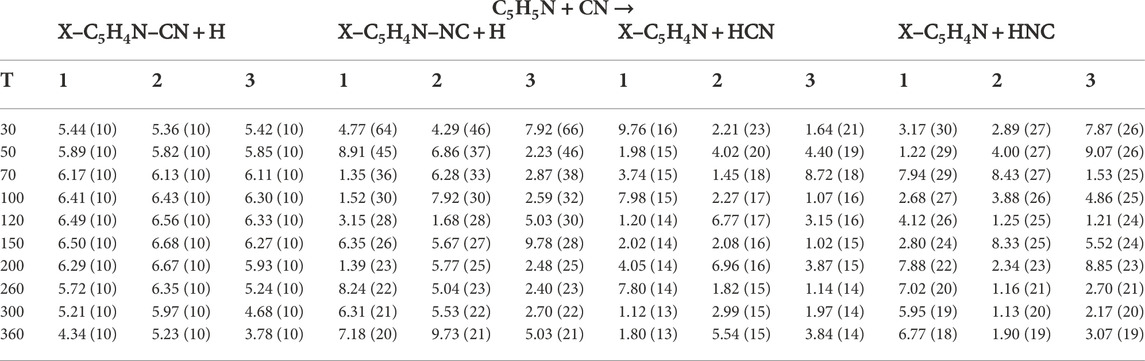
TABLE 3. Numerical values of the rate constants obtained in the present work for the different carbon and hydrogen positions in the (30–360) K range. The notation A (B) represents A × 10−B cm3 s−1.
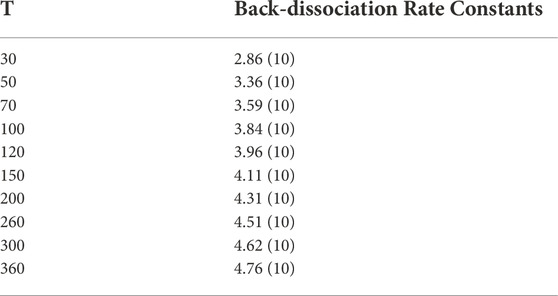
TABLE 4. Numerical values of the rate constants obtained in the present work for the back dissociation to pyridine (addition to the N position) in the (30–360) K range. The notation A (B) represents A × 10−B cm3 s−1.
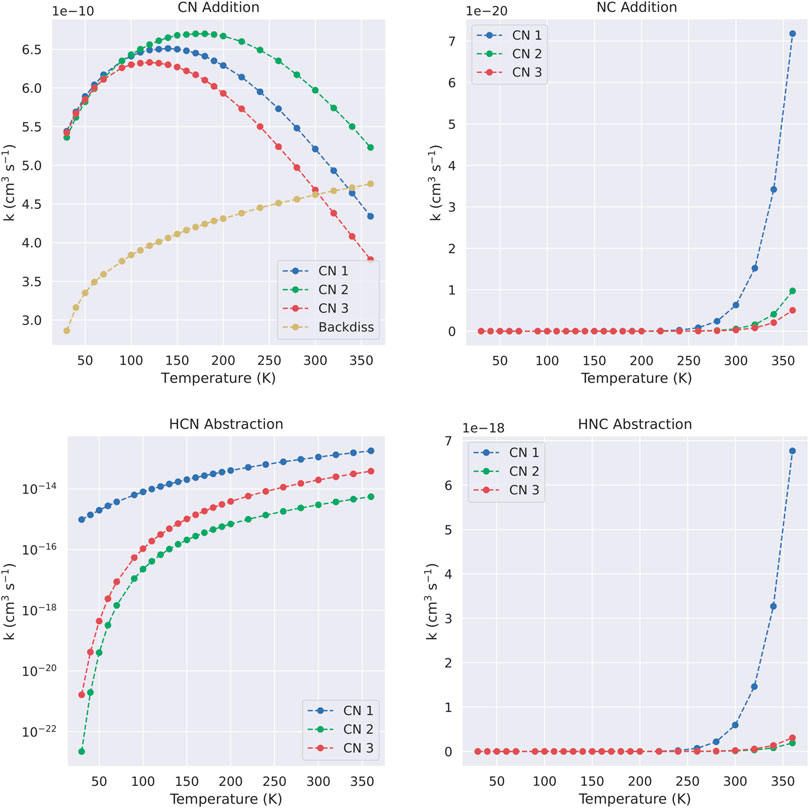
FIGURE 6. Global rate constants for the different reactions considered in this work. Note that for C5H5N + CN → C5H4N + HCN we employ a semilogarithmic scale.
Both the values and the graph confirm the picture obtained by the PES exploration, that is that the reaction proceeds via CN addition. All the latter are fast reactions with rate constants in the range of 10–10 cm3s−1 and with a very mild dependence on the temperature. Similarly, back-dissociation to pyridine via attachment of CN to the aromatic N atom of pyridine presents rate constants also in the order of 10–10 cm3s−1, and slightly smaller than addition to carbon positions at low temperatures. However, at higher temperatures, and thanks to the effect of thermal energy, back-dissociation rate constants monotonically increase, in contrast with CN addition reactions. For the latter, the rate constants present a competition of the capture event with tunneling in the H abstraction step, whereas for back-dissociation tunneling does not play a role at all. Rationalizing the trends of the rate constants for the rest of the considered reactions is complicated due to the interplay of thermal, energetic and tunneling contributions to the rate constant. For example, in the case of NC additions, a higher entrance barrier does not necessarily correlate with an overall lower rate constant in the whole range of temperatures because of the difference between the entrance and exit barriers for the different atomic position. Adopting a general explanation for each reaction would not provide a significant insight about the evolution of the reaction. Hence, we have not deepened in the discussion of the underlying values.
Finally, we have fitted our rate constants to a modified Arrhenius-Kooij expression of the type
to ease the inclusion of our global rate constants into astrochemical models. The values of α, β, and γ are gathered in Table 5. The fit for the C5H5N + CN → X − C5H4N + HNC reaction diverges at low temperature (not well fitted by a modified Arrhenius expression).
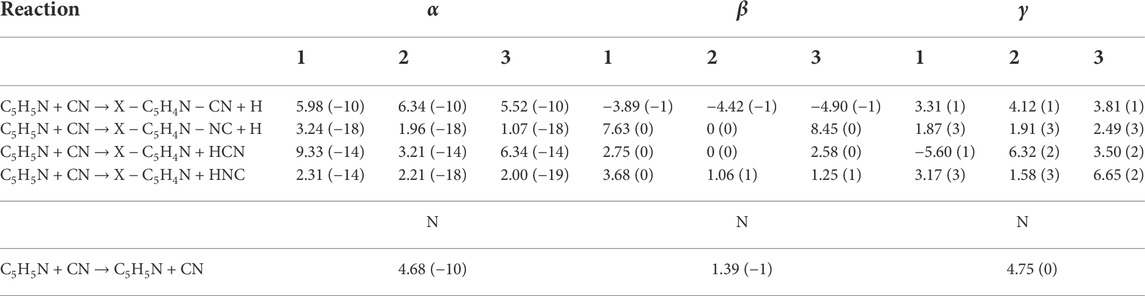
TABLE 5. Best fitting parameters α (in cm3 s−1), β (dimensionless), γ (in K) for the reactions in the carbon or hydrogen atoms considered in this work and for the back-dissociation reaction. The notation A(B) represents A × 10B units. Note that the fits for C5H5N + CN → X − C5H4N + HNC diverge at low temperature and are not well fitted by an Arrhenius-Kooij expression.
5 Discussion
5.1 Branching ratios of reaction
In order to determine the dilution of the parent molecule pyridine into more complex cyano derivatives, a key magnitude deriving from the reaction are the reaction branching ratios (BR). They represent the fraction of a given product of reaction weighted over all possible product channels. The BR for the products are obtained as the ratio between a particular rate constant (as obtained from Section 4) over the rate constants of all the possible exit channels, e.g:
We calculated the BR only considering the reaction channels that proceed without an entrance barrier, because the other ones contribution are negligible. Branching ratios are gathered in Figure 7 (left panel). For the purpose of discussion we gather here the values for a temperature of 30 K, being 0.29/0.28/0.28/0.15 for CN addition in 1, 2, 3 and back-dissociation to pyridine. Our results reveal that CN addition, is the most likely outcome (85%), in agreement with the values for benzene (Woon, 2006). The small emerged barriers for the formation of HCN via the abstraction of H on C1 may indicate that this reactions can be also barrierless, as we mentioned in Section 3.3. The contributions of this channel should remain minor though. Independent of the exact ratios, one of the main conclusions of the present article is that, similarly to what happened to benzene, the destruction of pyridine is indeed possible and fast via reactions with CN. The high reactivity of the cyano radical is a well known trait, responsible of the formation of many molecules in the gas phase of the ISM (Balucani et al., 1999; Woon, 2006; Vazart et al., 2015b; Sleiman et al., 2018; Puzzarini et al., 2020; Valença Ferreira de Aragão et al., 2020; Marchione et al.,2022).
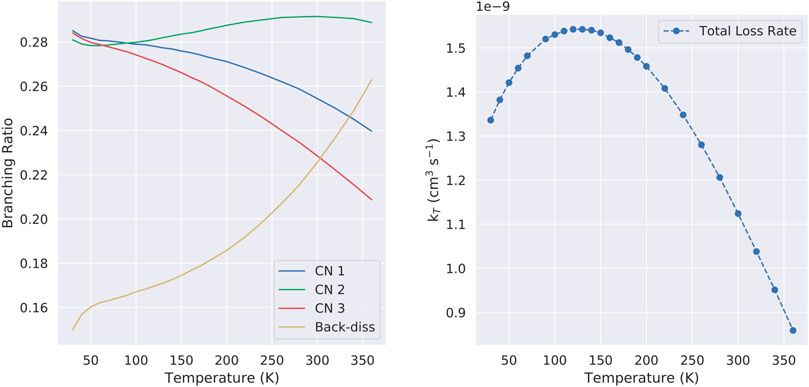
FIGURE 7. (Left) Branching ratios for the main reaction channels considered in this work (Right) Total loss rate constant for the title reaction.
In contrast with benzene, the formation of the different cyanated derivatives is diluted in three reaction channels, a consequence of the lower symmetry of the pyridine molecule with respect to benzene. Combining all relevant reactions, the total loss rate kT for the C5H5N + CN reaction is:
with ki,Add the individual CN addition reactions and kBack the back-dissociation rate constant. The total loss rate is 1.33 × 10−9 cm3s−1 at 30 K, a factor ∼ 4.3 higher than the loss rate of 3.15 × 10−10 cm3s−1 for benzene at that same temperature for theoretical calculations (Woon, 2006) and a factor between 2.5 and 3.4 (5.30 × 10−10–3.49 × 10−10 cm3s−1) with experiments (Trevitt et al., 2010; Cooke et al., 2020) at equivalent temperatures. We believe that the reason behind the increased reactivity of the pyridine in contrast with benzene is due to the different attractive forces implied in the CN capture event. While in the reaction CN + C6H6 the interaction potential is dominated by induced dipole interactions, in the case of CN + C5H5N the dominant interactions are dipole-dipole that are stronger and have a consequential effect in the capture (association of the CN radical) event. This higher loss rate may be partially behind the absence of pyridine in the ISM as discussed by (Barnum et al., 2022). At higher temperatures, and thanks to the gained importance of the back-dissociation channel, the loss rate constant for the CN + C5H5N reaction is decreased to a lower bound value of 8.59 × 10−10 cm3s−1 (See Figure 7, right panel).
5.2 Viable proxies of pyridine
While pyridine and benzene hold similarities in their reactivity with CN radicals, the situation concerning the detectability of pyridine and its cyano derivatives is different than with benzene. The dipole moment

TABLE 6. Magnitude of the dipole moments (
The combination of the increased loss rate of pyridine molecules with respect to benzene and the net dipole moment of pyridine are factors that go in different directions concerning the prevalence and detection of pyridine in the ISM. As we mentioned before, pyridine has a net dipole moment, and the absence in its detection can also be attributed to competitive formation routes out of the scope of the present work (Barnum et al., 2022). For example, the formation of carbon nitrogen bonds outside of the ring (as in the case of cyanocyclopentadiene (McCarthy et al., 2020)) might be favored over insertion of the CN moiety into the cyclic structure.
Although we cannot rule out the hypothesis of competitive routes of formation, the results of this study show that the destruction of pyridine from CN radicals proceeds through very fast reactions. The predicted smaller abundance of pyridine with respect to benzene (Barnum et al., 2022), summed to the dilution caused by the reactions with reactive radicals can be a cause of the lack of detection of this molecule in the ISM. The reduced symmetry of pyridine with respect to benzene also apply to other heterocyclic species target of current astronomical surveys, and the dilution caused by the different sub-products of reaction (benzene only has benzonitrile as product of reaction) has a direct impact on the potential observable abundances. A careful inspection of the reactivity of radicals with heterocycles, and possibly in a per-radical and per-heterocycle basis, is needed to suggest good proxies of heterocyclic species.
For the specific case of pyridine, the subject of this study, we suggest 1-cyano-pyridine (and in a lesser extent 2-cyano-pyridine) as good candidates to posses a significant “proxy effect” (Cooke et al., 2020) due to the increased dipole moment and the significant contribution to the branching ratio of reaction (see Table 6; Figure 7). We must emphasize however, that the dipole moment of pyridine should allow for a detection of a pure species in contrast with benzene.
6 Conclusion
In this work we presented a theoretical study of the reactivity of pyridine with the CN radical, one of the most abundant and reactive radicals in the ISM. Our results show that the main products of reaction are formed without an entrance barrier, leading to the formation of the addition products after a posterior H detachment. We did not find a preferential addition channel at low temperature in any of the carbons of the pyridine structure. The other only reaction that proceeds without an activation barrier is the back-dissociation to pyridine, that modulates the total reaction rate constant over our considered (30–360) K temperature range. At low temperatures, this channel is minoritary and accounts for a 15% of the total reaction, but this increases at higher temperatures. All the other reactions considered in this work (NC addition, HCN abstraction, and HNC abstraction) proceed via an entrance activation barrier. It is important to reiterate that, due to the small barrier for the C5H5N + CN → 1 − C5H4N + HCN there may be the possibility that alternative theoretical methods find this channel barrierless. If that would be the case, the total loss rate constant would increase, further strenghtening our conclusions regarding the detectability of pyridine. We attribute the very small value of the C5H5N + CN → 1 − C5H4N + HCN reaction to a stabilization effect arising from the formation of a benzyne like structure with an intramolecular nitrile. All the other reactions considered in this work are not competitive owing to the emergence of kinetic barriers.
Our kinetic study after the PES construction revealed a total loss rate in the order 1.33 × 10−9 cm3s−1 which was found to be between 2.5–4.5 times higher than the same rates obtained for the reaction of CN with benzene. We suggest that the reason for this behavior are the different interactions at play in benzene (an apolar molecule) and pyridine (a polar molecule). This is proof of a faster destruction of pure pyridine in the cold ISM by chemical agents. Moreover, the destruction pathways arising from this reaction lead to several branches that make a posterior detection of the considered molecules difficult. In favor of detection of molecular proxies, the reaction with CN leads to an increased dipole moment for 1-cyano-pyridine and 2-cyano-pyridine that should make the rotational transitions “brighter” and help the detectability of the molecule. Overall, the interplay of different factors affecting both the dilution of the parent species and the enhanced dipole of the products of reaction make it difficult to estimate the appearance of a proxy effect for pyridine. This may not be the case for other cyclic species.
The importance of the different formation routes of heterocycles in the apparent absence of this molecule in the ISM cannot be overruled (Cooke and Sims, 2019) and further molecular data is required to build a complete picture that could help astronomers to identify viable candidates for detection. We will continue this endeavor in the near future.
Data availability statement
The structures supporting this article can be found at: DOI: 10.5281/zenodo.7316129. Further inquiries can be directed to the corresponding author.
Author contributions
GM planed the calculations. GM and JH analyzed the results and wrote the initial manuscript. SS, JH, and GM performed the DFT calculations. JH, GM, and JG run the kinetic calculations. JK and GM supervised the project. All authors revised the first version of the manuscript.
Funding
GM thanks the Alexander von Humboldt Foundation and the Japanese Society for the Promotion of Science (grant: P22013) foundation for their patronage. JH acknowledges the financial support received in the form of a Ph.D. scholarship from the Studienstiftung des Deutschen Volkes (German National Academic Foundation). The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through Grant No. INST 40/575-1 FUGG (JUSTUS 2 cluster). JGdlC acknowledges support from Grant No. PID 2019-105552RB-C41 by the Spanish Ministry of Science and Innovation/State Agency of Research MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fspas.2022.1020635/full#supplementary-material
References
Alecu, I. M., Zheng, J., Zhao, Y., and Truhlar, D. G. (2010). Computational thermochemistry: Scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries. J. Chem. Theory Comput. 6, 2872–2887. doi:10.1021/ct100326h
Alvaro Galué, H., Pirali, O., and Oomens, J. (2010). Gas-phase infrared spectra of cationized nitrogen-substituted polycyclic aromatic hydrocarbons. Astron. Astrophys. 517, A15. doi:10.1051/0004-6361/201014050
Balucani, N., Asvany, O., Chang, A. H., Lin, S. H., Lee, Y. T., Kaiser, R. I., et al. (1999). Crossed beam reaction of cyano radicals with hydrocarbon molecules. I. Chemical dynamics of cyanobenzene (C6H5CN; X 1A1) and perdeutero cyanobenzene (C6D5CN; X 1A1) formation from reaction of CN(X 2Σ+) with benzene C6H6(X 1A1g), and d6-benzene C6D6(X 1A1g). J. Chem. Phys. 111, 7457–7471. doi:10.1063/1.480070
Barnum, T. J., Siebert, M. A., Lee, K. L. K., Loomis, R. A., Changala, P. B., Charnley, S. B., et al. (2022). A search for heterocycles in GOTHAM observations of TMC-1. J. Phys. Chem. A 126, 2716–2728. doi:10.1021/acs.jpca.2c01435
Canelo, C. M., Friaça, A. C. S., Sales, D. A., Pastoriza, M. G., and Ruschel-Dutra, D. (2018). Variations in the 6.2 μm emission profile in starburst-dominated galaxies: A signature of polycyclic aromatic nitrogen heterocycles (PANHs)? Mon. Not. R. Astron. Soc. 475, 3746–3763. doi:10.1093/mnras/stx3351
Cernicharo, J., Agúndez, M., Cabezas, C., Tercero, B., Marcelino, N., Fuentetaja, R., et al. (2021a). Discovery of HCCCO and C5O in TMC-1 with the QUIJOTE line survey. Astron. Astrophys. 656, L21. doi:10.1051/0004-6361/202142634
Cernicharo, J., Agúndez, M., Cabezas, C., Tercero, B., Marcelino, N., Pardo, J. R., et al. (2021b). Pure hydrocarbon cycles in TMC-1: Discovery of ethynyl cyclopropenylidene, cyclopentadiene, and indene. Astron. Astrophys. 649, L15. doi:10.1051/0004-6361/202141156
Cernicharo, J., Agúndez, M., Kaiser, R. I., Cabezas, C., Tercero, B., Marcelino, N., et al. (2021c). Discovery of benzyne, o-C6H4, in TMC-1 with the QUIJOTE line survey. A&A 652
Cernicharo, J., Fuentetaja, R., Agúndez, M., Kaiser, R. I., Cabezas, C., Marcelino, N., et al. (2022). Discovery of fulvenallene in tmc-1 with the quijote line survey. Astron. Astrophys. 663, L9. doi:10.1051/0004-6361/202244399
Chesnavich, W. J. (1986). Multiple transition states in unimolecular reactions. J. Chem. Phys. 84, 2615–2619. doi:10.1063/1.450331
Cooke, I. R., Gupta, D., Messinger, J. P., and Sims, I. R. (2020). Benzonitrile as a proxy for benzene in the cold ISM: Low-temperature rate coefficients for CN + C 6 H 6. Astrophys. J. 891, L41. doi:10.3847/2041-8213/ab7a9c
Cooke, I. R., and Sims, I. R. (2019). Experimental studies of gas-phase reactivity in relation to complex organic molecules in star-forming regions. ACS Earth Space Chem. 3, 1109–1134. doi:10.1021/acsearthspacechem.9b00064
Dunning, T. H. (1989). Gaussian basis sets for use in correlated molecular calculations. i. the atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023. doi:10.1063/1.456153
García de la Concepción, J., Puzzarini, C., Barone, V., Jiménez-Serra, I., and Roncero, O. (2021). formation of phosphorus monoxide (PO) in the interstellar medium: Insights from quantum-chemical and kinetic calculations. Astrophys. J. 922, 169. doi:10.3847/1538-4357/ac1e94
Georgievskii, Y., Miller, J. A., Burke, M. P., and Klippenstein, S. J. (2013). Reformulation and solution of the master equation for multiple-well chemical reactions. J. Phys. Chem. A 117, 12146–12154. doi:10.1021/jp4060704
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104. doi:10.1063/1.3382344
Hudgins, D. M., Bauschlicher, C. W., and Allamandola, L. J. (2005). Variations in the peak position of the 6.2 μm interstellar emission feature: A tracer of N in the interstellar polycyclic aromatic hydrocarbon population. Astrophys. J. 632, 316–332. doi:10.1086/432495
Jiménez-Serra, I., Vasyunin, A. I., Spezzano, S., Caselli, P., Cosentino, G., and Viti, S. (2021). The complex organic molecular content in the L1498 starless core. Astrophys. J. 917, 44. doi:10.3847/1538-4357/ac024c
Jones, B. M., Zhang, F., Kaiser, R. I., Jamal, A., Mebel, A. M., Cordiner, M. A., et al. (2011). Formation of benzene in the interstellar medium. Proc. Natl. Acad. Sci. U. S. A. 108, 452–457. doi:10.1073/pnas.1012468108
Kassel, L. S. (1928). Studies in homogeneous gas reactions I. J. Phys. Chem. 32, 225–242. doi:10.1021/j150284a007
Knowles, P. J., Hampel, C., and Werner, H. (1993). Coupled cluster theory for high spin, open shell reference wave functions. J. Chem. Phys. 99, 5219–5227. doi:10.1063/1.465990
Malloci, G., Joblin, C., and Mulas, G. (2007). On-line database of the spectral properties of polycyclic aromatic hydrocarbons. Chem. Phys. 332, 353–359. doi:10.1016/j.chemphys.2007.01.001
Marchione, D., Mancini, L., Liang, P., Vanuzzo, G., Pirani, F., Skouteris, D., et al. (2022). Unsaturated dinitriles formation routes in extraterrestrial environments: A combined experimental and theoretical investigation of the reaction between cyano radicals and cyanoethene (C2H3CN). J. Phys. Chem. A 126, 3569–3582. doi:10.1021/acs.jpca.2c01802
Marcus, R. A. (1952). Unimolecular dissociations and free radical recombination reactions. J. Chem. Phys. 20, 359–364. doi:10.1063/1.1700424
Mattioda, A. L., Hudgins, D. M., Bauschlicher, C. W., Rosi, M., and Allamandola, L. J. (2003). Infrared spectroscopy of matrix-isolated polycyclic aromatic compounds and their ions. 6. Polycyclic aromatic nitrogen heterocycles. J. Phys. Chem. A 107, 1486–1498. doi:10.1021/jp021938c
McCarthy, M. C., Lee, K. L. K., Loomis, R. A., Burkhardt, A. M., Shingledecker, C. N., Charnley, S. B., et al. (2020). Interstellar detection of the highly polar five-membered ring cyanocyclopentadiene. Nat. Astron. 5, 176–180. doi:10.1038/s41550-020-01213-y
McGuire, B. A. (2018). 2018 census of interstellar, circumstellar, extragalactic, protoplanetary disk, and exoplanetary molecules. Astrophys. J. Suppl. Ser. 239, 17. doi:10.3847/1538-4365/aae5d2
McGuire, B. A. (2022). 2021 census of interstellar, circumstellar, extragalactic, protoplanetary disk, and exoplanetary molecules. Astrophys. J. Suppl. Ser. 259, 30. doi:10.3847/1538-4365/ac2a48
McGuire, B. A., Burkhardt, A. M., Kalenskii, S., Shingledecker, C. N., Remijan, A. J., Herbst, E., et al. (2018). Detection of the aromatic molecule benzonitrile (c -C 6 H 5 CN) in the interstellar medium. Science 359, 202–205. doi:10.1126/science.aao4890
McGuire, B. A., Loomis, R. A., Burkhardt, A. M., Lee, K. L. K., Shingledecker, C. N., Charnley, S. B., et al. (2021). Detection of two interstellar polycyclic aromatic hydrocarbons via spectral matched filtering. Science 371, 1265–1269. doi:10.1126/science.abb7535
McNaughton, D., Godfrey, P. D., Brown, R. D., and Thorwirth, S. (2007). Millimetre wave spectroscopy of PANHs: Phenanthridine. Phys. Chem. Chem. Phys. 9, 591–595. doi:10.1039/B615485A
Merino, P., Švec, M., Martinez, J. I., Jelinek, P., Lacovig, P., Dalmiglio, M., et al. (2014). Graphene etching on SiC grains as a path to interstellar polycyclic aromatic hydrocarbons formation. Nat. Commun. 5, 3054. doi:10.1038/ncomms4054
Meyer, K. S., Westerfield, J. H., Johansen, S. L., Keane, J., Wannenmacher, A. C., and Crabtree, K. N. (2022). Rotational and vibrational spectra of the pyridyl radicals: A coupled-cluster study. J. Phys. Chem. A 126, 3185–3197. doi:10.1021/acs.jpca.2c01761
Miksch, A. M., Riffelt, A., Oliveira, R., Kästner, J., and Molpeceres, G. (2021). Hydrogenation of small aromatic heterocycles at low temperatures. Mon. Not. R. Astron. Soc. 505, 3157–3164. doi:10.1093/mnras/stab1514
Neese, F., Wennmohs, F., Becker, U., and Riplinger, C. (2020). The ORCA quantum chemistry program package. J. Chem. Phys. 152, 224108. doi:10.1063/5.0004608
Parker, D. S., Kaiser, R. I., Kostko, O., Troy, T. P., Ahmed, M., Sun, B. J., et al. (2015). On the formation of pyridine in the interstellar medium. Phys. Chem. Chem. Phys. 17, 32000–32008. doi:10.1039/c5cp02960k
Pechukas, P., and Light, J. C. (1965). On detailed balancing and statistical theories of chemical kinetics. J. Chem. Phys. 42, 3281–3291. doi:10.1063/1.1696411
Peeters, Z., Botta, O., Charnley, S. B., Kisiel, Z., Kuan, Y. J., and Ehrenfreund, P. (2005). Formation and photostability of N-heterocycles in space I. The effect of nitrogen on the photostability of small aromatic molecules. Astron. Astrophys. 433, 583–590. doi:10.1051/0004-6361:20042443
Puzzarini, C., Salta, Z., Tasinato, N., Lupi, J., Cavallotti, C., and Barone, V. (2020). A twist on the reaction of the CN radical with methylamine in the interstellar medium: New hints from a state-of-the-art quantum-chemical study. Mon. Not. R. Astron. Soc. 496, 4298–4310. doi:10.1093/mnras/staa1652
Rice, O. K., and Ramsperger, H. C. (1927). Theories of unimolecular gas reactions at low pressures. J. Am. Chem. Soc. 49, 1617–1629. doi:10.1021/ja01406a001
Sleiman, C., El Dib, G., Rosi, M., Skouteris, D., Balucani, N., and Canosa, A. (2018). Low temperature kinetics and theoretical studies of the reaction CN + CH3NH2: A potential source of cyanamide and methyl cyanamide in the interstellar medium. Phys. Chem. Chem. Phys. 20, 5478–5489. doi:10.1039/c7cp05746f
Trevitt, A. J., Goulay, F., Taatjes, C. A., Osborn, D. L., and Leone, S. R. (2010). Reactions of the CN radical with benzene and toluene: Product detection and low-temperature kinetics. J. Phys. Chem. A 114, 1749–1755. doi:10.1021/jp909633a
Truhlar, D. G. (1969). Statistical phase-space theory of the reaction C+ +D2 including threshold behavior. J. Chem. Phys. 51, 4617–4623. doi:10.1063/1.1671834
Valença Ferreira de Aragão, E., Faginas-Lago, N., Rosi, M., Mancini, L., Balucani, N., and Skouteris, D. (2020). A computational study of the reaction cyanoacetylene and cyano radical leading to 2-butynedinitrile and hydrogen radical. Cham 12251, 707–716. doi:10.1007/978-3-030-58808-3_51
Vazart, F., Latouche, C., Skouteris, D., Balucani, N., and Barone, V. (2015b). CYANOMETHANIMINE ISOMERS in COLD INTERSTELLAR CLOUDS: INSIGHTS from ELECTRONIC STRUCTURE and KINETIC CALCULATIONS. Astrophys. J. 810, 111. doi:10.1088/0004-637X/810/2/111
Weigend, F., and Ahlrichs, R. (2005). Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. doi:10.1039/b508541a
Werner, H.-J., Knowles, P. J., Knizia, G., Manby, F. R., Schütz, M., Celani, P., et al. (2019). MOLPRO, version 2019.2, a package of ab initio programs. Available at: https://www.molpro.net.
Werner, H. J., Knowles, P. J., Knizia, G., Manby, F. R., and Schütz, M. (2012). Molpro: A general-purpose quantum chemistry program package. WIREs. Comput. Mol. Sci. 2, 242–253. doi:10.1002/wcms.82
Woon, D. E. (2006). Modeling chemical growth processes in titan’s atmosphere: 1. Theoretical rates for reactions between benzene and the ethynyl (C2H) and cyano (CN) radicals at low temperature and pressure. Chem. Phys. 331, 67–76. doi:10.1016/j.chemphys.2006.09.028
Yang, Y., Hu, X., Zhang, D., Zhang, W., Liu, G., and Zhen, J. (2020). Laboratory formation and photochemistry of covalently bonded polycyclic aromatic nitrogen heterocycle (PANH) clusters in the gas phase. Mon. Not. R. Astron. Soc. 498, 1–11. doi:10.1093/mnras/staa2212
Zhao, Y., and Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241. doi:10.1007/s00214-007-0310-x
Keywords: proxy species, interstellar medium, astrochemistry, computational methods, heterocycles
Citation: Heitkämper J, Suchaneck S, García de la Concepción J, Kästner J and Molpeceres G (2022) The reactivity of pyridine in cold interstellar environments: The reaction of pyridine with the CN radical. Front. Astron. Space Sci. 9:1020635. doi: 10.3389/fspas.2022.1020635
Received: 16 August 2022; Accepted: 27 October 2022;
Published: 01 December 2022.
Edited by:
Ilsa Cooke, University of British Columbia, CanadaReviewed by:
Vincenzo Barone, Normal School of Pisa, ItalyDivita Gupta, I. Physikalisches Institut, Universität zu Köln, Germany
Copyright © 2022 Heitkämper, Suchaneck, García de la Concepción, Kästner and Molpeceres. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Germán Molpeceres, bW9scGVjZXJlc0Bhc3Ryb24ucy51LXRva3lvLmFjLmpw