 Sinalo Mani
Sinalo Mani Olayinka A. Aiyegoro
Olayinka A. Aiyegoro Matthew A. Adeleke
Matthew A. Adeleke- 1Gastrointestinal Microbiology and Biotechnology Unit, Agricultural Research Council - Animal Production, Irene, South Africa
- 2Discipline of Genetics, School of Life Sciences, College of Agriculture, Engineering and Science, University of KwaZulu-Natal, Durban, South Africa
- 3Research Unit for Environmental Sciences and Management, North-West University, Potchefstroom, South Africa
Supplementation of direct-fed microbials into ruminants' nutrition has shown great potential in manipulating rumen fermentation and enhancing productive animal performance. However, little is known about rumen microbial composition and diversity of Damara and Meatmaster sheep, breeds indigenous to South Africa. The study aimed at exploring and comparing the rumen microbiomes of two breeds with different feeding treatments as follows: no antibiotic, no probiotics (T1), only potential probiotic (T2), only potential probiotic (T3), the combination of potential probiotics (T4), antibiotic (T5); using a metagenomic approach. The results showed that based on the Shannon index, the microbial diversity of Damara was higher (p < 0.05) than Meatmaster, while treatment T4 was higher than treatment T1 (p < 0.05). The principal coordinate analysis showed no significant difference among treatments, while there were significant dissimilarities between sheep breeds and sample-day (p < 0.05). Canonical correspondence analysis (CCA) displayed the dispersion of microbial communities among treatments, where negative control (T1) was distinct from other treatments. Bacteroidetes and Firmicutes were the most abundant microbial phyla across treatments for both breeds. Negative control and the combination of potential probiotics showed lower proportions of Proteobacteria compared to other treatments. At the genus level, Prevotella and Clostridium were abundant across all treatments, while Pseudomonas was abundant only in T2, T3, and T5. In all treatments, Fibrobacter was detected after the feeding trials, while it was not detected in most treatments before trials. The results revealed that the rumen microbiome's structure and abundance were slightly altered by administering lactic acid as a putative probiotic.
Introduction
The rumen is an important digestive and metabolic site in ruminants, where rumen microbiota play a vital role in providing nutrients to the host in form of volatile acids and microbial proteins (1). The symbiotic microbial community is essential for the host health in several ways, such as in nutrient digestion, balancing the immune response, and mediating the host functioning. Furthermore, symbiosis enables the development of the latter's gastrointestinal tract (2). Consequently, alterations in ruminal microbial structure and function have shown an insightful impact on ruminants' health and productivity. Although rumen microbiota is relatively stable, it is greatly responsive to changes in the diet, host genetics, physiology, and environmental factors (3, 4).
Feed additives such as direct-fed microbials have shown great potential to manipulate rumen fermentation and enhance productive animal performance (5). Antibiotics as growth promoters in animals introduce residues and spread antibiotic resistance while enhancing growth performance (6). Antibiotic administration disrupts the ruminant's normal microbial balance, which might cause growth defects in animals (7). Probiotics are explored as safer alternatives to antibiotics to improve balance in the gastrointestinal microbiota and enhance ruminants' health and productivity (8). Previous studies have shown that the use of probiotics (direct-fed microbials), such as lactic acid bacteria, can improve nutrient digestibility and growth performance (9, 10); also decrease pathogen colonization in the gut (11).
Earlier studies reported that probiotics enhanced feed efficiency, increased weight gain, improved milk and meat production, reduced methane emission, and animal health (5, 12–15). However, limited studies have explored the rumen microbial community composition and diversity. A study by Zhang et al. (16) showed that Lactobacillus rhamnosus GG administration to neonatal calves increased their voluntary feed intake and growth performance. The ruminal pH of calves fed the probiotic was lower, which might be caused by increased volatile fatty acids (VFA) concentration in rumen fluids. The L. rhamnosus GG administration also varied the rumen microbial community composition and regulated the balance of rumen and intestinal microbes. The relative abundance and order of dominant bacteria families were inconsistent between the control and probiotic calves. Prevotellaceae (55.67%) had the highest relative abundance in the treated group of calves instead of Succinivibrionaceae, and it significantly increased while Succinivibrionaceae decreased drastically. The relative abundance of Lactobacillaceae increased in the rumen fluid after the GG administration in calves.
In recent years, high-throughput sequencing of the 16S rRNA gene has provided novel insights into the abundance and diversity of microbial communities. It has also expanded the possibilities for studying rumen microbial community and population dynamics, indicating their significant role in ruminal fermentation (17, 18). Knowledge of these microbiomes can further broaden our understanding of rumen microbial environments and ruminant productivity. Hence the current study aimed at exploring the rumen microbial diversity and abundance by describing and comparing the rumen microbiota of sheep breeds subjected to different feed supplements with lactic acid bacteria as direct-fed microbials.
Materials and Methods
Animals, Treatments, and Sampling
All the procedures involving animals were approved by the Agricultural Research Council - Animal Production Institute Ethics committee (APIEC17/21). The trials were done at the Agricultural Research Council (ARC), GI Microbiology and Biotechnology unit and the Small Stocks Unit in Irene, Gauteng province.
The lactic acid bacteria (putative probiotics) used in this study were isolated and characterized from fresh fecal samples of six Zulu sheep breed. These sheep have been described to possess great adaptation to harsh environmental conditions and resistance against parasites and diseases. Molecular sequencing and probiotic and technological properties such as antimicrobial activity, acid, and bile tolerance were used to identify the two potential probiotics. The potential probiotic bacteria were prepared in De Man, Rogosa, and Sharpe (MRS) broth (Oxoid, England) anaerobically and preserved in 25% glycerol in the ultra-low freezer. The two potential probiotic bacteria were then revived by inoculation in MRS broth. For suspension, MRS broth was inoculated with 1% (v/v) culture and incubated anaerobically at 37°C overnight prior to administering.
Sixty-four sheep, 32 Damara breed, and 32 Meatmaster breed were used in a 30-day trial. The animals were approximately 7 months old, with 16 males and 16 females per breed. Ten (10) animals were excluded from the experiment after the adaptation period before the trial commenced due to signs of sickness and four mortalities. The average initial weights of the animals were: Meatmaster males (24.6 ± 3.4 kg), Meatmaster females (21.5 ± 3.1 kg), Damara males (36.6 ± 8.3 kg), and Damara females (28.9 ± 6.9 kg). The animals were housed per treatment with males and females separated in open barn trial pens with ± 4 m2 shelters.
The sheep were randomly allocated to five treatment groups, 6–8 animals per treatment, considering the gender. The treatments were as follows: (a) Diet with no antibiotics, no probiotics (negative control) (T1); (b) Diet with no antibiotics, only L. rhamnosus PT9 (T2); (c) Diet with no antibiotics, only L. rhamnosus PT10 (T3); (d) Diet with the combination of L. rhamnosus PT9 and L. rhamnosus PT10 (T4); (e) Diet with antibiotics, no probiotics (positive control) (T5). The animals were fed on commercial pellet feed fortified with or without probiotics or antibiotics, and in case of the positive control, in-feed antibiotic rumensin was added. Hay and freshwater were supplied ad libitum. The experimental feed composition is presented in Table 1, feed composition is analyzed as follows: Dry matter (19), Moisture (19), Protein (19), NDF (20), ADF and NDL (21), Ash (19), Starch (22), Crude Fiber (19), Calcium and Phosphorus (23). The probiotic treatment groups were dosed once a week for the trial period using the dosing gun with 10 mL of the 24-h old LAB culture suspensions of approximately 2 × 109 CFU/mL. Animal weight was recorded, and rumen samples were collected before the trial commenced and at the end of the trial.
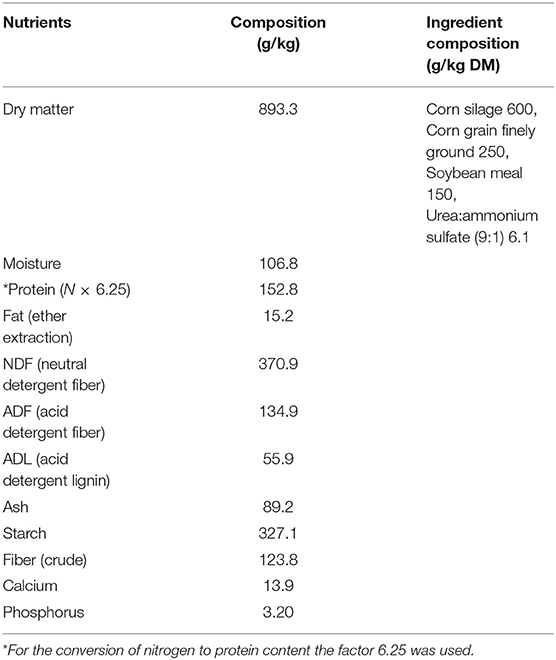
Table 1. Ingredient and chemical composition of experimental diet fed to sheep.
Rumen fluid samples were collected using a stomach tube, according to Shen et al. (24) procedures. About 40 mL of the collected rumen fluid contents were transferred to 50 mL centrifuge tubes after collection. The rumen pH was measured using a portable pH meter (Orion Star A121 Portable pH meter, Thermo Scientific, Singapore) immediately after sampling (before and after the trial). The pH meter was calibrated using fresh pH buffers that bracket expected sample pH using automatic buffer recognition according to manufacturer's guidelines. Before each sample reading, the pH meter was rinsed with distilled water and blot dried, rinsed again after each sample reading. Rumen samples were kept on ice until they were transferred to the lab, and they were stored at −80°C until further analysis.
DNA Extraction and 16S rRNA Amplification
Extraction of DNA from rumen samples (100 μl each) was done using QIAamp Fast DNA stool Mini Kit (Qiagen, Germany) according to the manufacturer's guidelines for pathogen detection, and DNA concentration was evaluated with Nanodrop 2000 (Thermo Electron Corporation, USA). The DNA samples were pooled according to their respective treatments, considering the sex of the animals (males and females DNA pooled separately), leading to four samples per treatment (two females and two males), and 20 samples per breed. The pooled DNA quality was assessed by electrophoresis on 1.5% agarose gels and visualized with UVP BioSpectrum 310 Imaging System (FisherScientific, UK). The pooled DNA samples were used as templates for amplifying a partial 16S rRNA sequence using the following primers, which include Illumina overhang adapter sequences:
Forward = 5'TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG3', Reverse = 5'GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC3' targeting the highly variable V3–V4 region of the prokaryotic 16S rRNA gene. PCR was done in 25 μl reaction mixture comprising 5 μl template DNA, 1 μl of each primer, 5.5 μl nuclease-free water, and 12.5 μl of 2 × KAPA HiFi HotStart Ready Mix (KAPA Biosystems), with the following conditions: initial denaturation at 95°C for 3 min, 30 cycles of amplification at 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s and a final extension at 72°C for 5 min. PCR amplicons were visualized using agarose gel electrophoresis to verify the expected band size of 550 bp. PCR products were purified to remove any primer dimers with NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel, Germany).
Library Preparation and Sequencing
Sequencing libraries were generated, and index codes were added using Illumina MiSeq Nextera XT DNA Library Preparation Kit (Illumina, USA), according to manufacturer's guidelines. The library quality was evaluated using Qubit 2.0 Fluorometer (ThermoScientific, USA) and Agilent 2100 Bioanalyzer system. Sequencing was performed using Illumina MiSeq platform, and 300 bp paired-end reads were generated.
Data Analysis
The raw data sequences generated by MiSeq Illumina sequencer were trimmed using Trimmomatics version 0.36, where the low-quality sequence regions and Illumina universal adapter sequences were removed. Demultiplexed sequence files were imported to QIIME2 software (version 2018.8) (25) for analysis. The imported reads were denoised and trimmed using DADA2. Sequences with ≥97% similarity were assigned to the same operational taxonomic units (OTU), and the OTU feature table was generated; chimeric sequences were detected and removed from the representative OTU sequences. For taxonomic analysis, OTU representative sequences were aligned to the Greengenes database. All other statistical analyses were carried out using RStudio (version 3.5.3) with phyloseq package (version 1.24.2). OTUs at a relative abundance ≥ 0.05% of the total reads in at least one sample were retained and analyzed further in RStudio.
Alpha diversity metrics including species richness, Simpson and Shannon index were computed for all samples. Beta diversity was computed using distance matrices generated from unweighted UniFrac analysis, principal coordinates analysis (PCoA), and non-metric multidimensional scaling (NMDS). ANOVA was used to compare the univariate analysis of alpha diversity measures. The probability of error α was fixed at 5%.
A co-occurrence network was constructed to determine potential associations between microbial communities. A pairwise correlation was calculated between microbial abundances present in at least 25% of the samples and with abundance higher than 0.05%, using Spearman's co-efficient, using Pairwise_correlations.R and Network_Analysis.R scripts (26). Multiple testing correction was done to reduce the chances of obtaining false-positive results; the P-values were adjusted using the Benjamini–Hochberg standard false discovery rate correction (FDR-BH) method (27). Spearman's coefficient correlations (ρ) ≥ 0.7 (or ≤ -0.7), and p-value < 0.01 were considered significant. The interactive platform Gephi (28) was used to visualize the network structure, using undirected network and Fruchterman–Reingold layout. Modularity was calculated by the Louvain method (29), using the clustering algorithm implemented in Gephi; modules were then filtered to remove nodes with a degree < 2. Betweenness centrality was used to measure the centrality of each node in the network.
Sequences for the two potential probiotics have been deposited on NCBI; L. rhamnosus PT9 and L. rhamnosus PT10 accession numbers MT492059 and MT492060, respectively. Raw reads used in the present study were deposited to the National Center of Biotechnology Information (NCBI) Sequence Read Archive (SRA) database under BioProject PRJNA578022 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA578022).
Results
Our results revealed that treatment, breed, and their interactions affected body weight (p < 0.001) and pH (p < 0.05) of the sheep; only breed was significant (p < 0.005) on weight gain. In Table 2, the final bodyweight of the control group (T1) was lower (p < 0.05) than other treatment groups. On average, bodyweight in other treatment groups was higher, with about 4.85 kg relative to the control group. Ruminal pH was higher (p < 0.05) in T5 than the probiotic-supplemented groups, but statistically the same with the control group. Except for T2, animals in T3, T4, and T5 groups gained more weight, with the highest weight gained being 1.7 kg in T4 within the 30-day trial. Damara breed was 12 kg heavier, 1.3 kg higher, and 0.34 higher than Meatmaster for body weight, weight gain, and pH, respectively, as shown in Table 3.
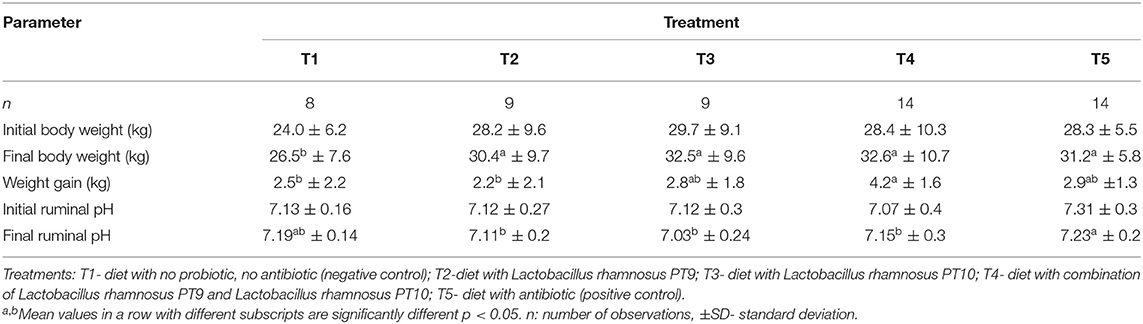
Table 2. Effect of treatment on body weight, weight gain and ruminal pH of sheep.
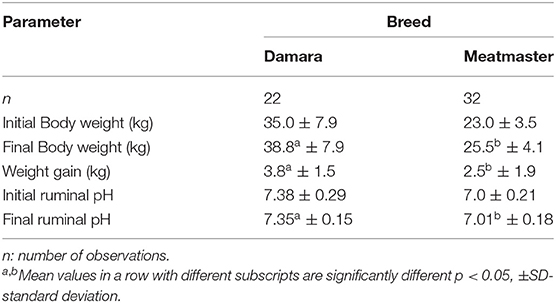
Table 3. Effect of breed on body weight, weight gain and ruminal pH of sheep.
After quality control, a total of 1,996,134 reads were obtained in 40 samples, with an average of 49,903 reads per sample. A total of 11 202 OTUs were obtained, out of which only 9,244 OTUs were classified at phylum level and were used for further analysis. The rest of 1,958 OTUs could be classified only up to kingdom level and they were discarded after normalization. The distinct variation by the two Damara samples was observed in the rarefaction curve (Figure 1), indicating a difference in of Damara and Meatmaster sheep breeds' microbial composition.
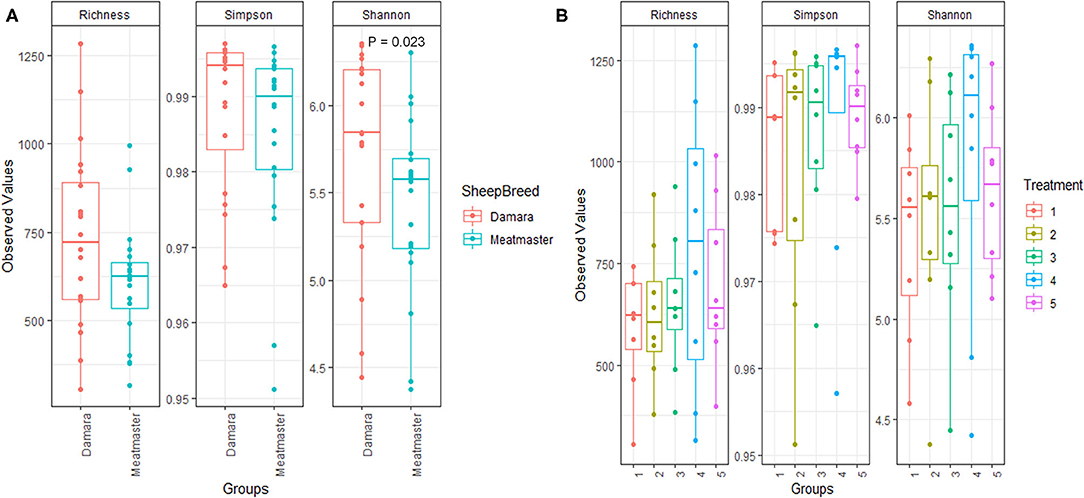
Figure 1. Comparison of alpha diversity metrics of communities by microbial richness, species diversity (Simpson and Shannon indexes). Treatments groups are as follows: 1. Diet with no antibiotics, no probiotics (negative control); 2. Diet with no antibiotics, only Lactobacillus rhamnosus PT9; 3. Diet with no antibiotics, only Lactobacillus rhamnosus PT10; 4. Diet with the combination of Lactobacillus rhamnosus PT9 and Lactobacillus rhamnosus PT10; 5. Diet with antibiotics, no probiotics (positive control). (A) Comparison of within community, grouped by sheep breed. (B) Comparison of within community, grouped by treatment. Shannon index was significant only between treatment 4 and treatment one, P = 0.042.
The alpha diversity (Richness, Simpson and Shannon indexes) is presented in Figure 1, showing indices across breed and treatment, respectively. Damara sheep had higher (p < 0.05) Shannon index than Meatmaster sheep (Figure 1A); while other indices showed no significant difference between the breeds, the microbial richness in Damara sheep was higher than in Meatmaster sheep. The combination of potential probiotics increased the microbial richness and diversity as treatment 4 had higher Shannon index than treatment 1 (p < 0.05, Figure 2B).
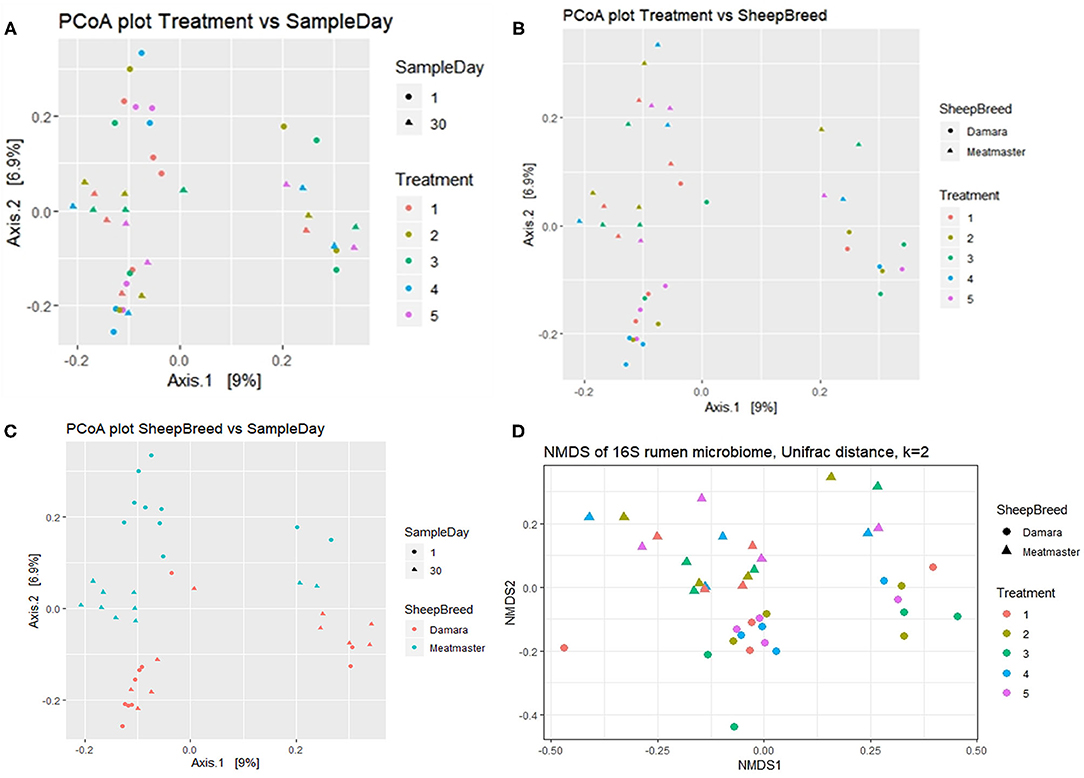
Figure 2. Beta diversity of rumen microbiome of Damara and Meatmaster sheep breeds. Treatment groups are as follows: 1. Diet with no antibiotics, no probiotics (negative control); 2. Diet with no antibiotics, only Lactobacillus rhamnosus PT9; 3. Diet with no antibiotics, only Lactobacillus rhamnosus PT10; 4. Diet with the combination of Lactobacillus rhamnosus PT9 and Lactobacillus rhamnosus PT10; 5. Diet with antibiotics, no probiotics (positive control). (A) Principal coordinate analysis (PCoA) defining the relationship between samples based on treatment and sample day. (B) PCoA defining the relationship between samples based on treatment and breed. (C) PCoA defining the relationship between samples based on sample day and breed. (D) Non-metric multidimensional scaling (NMDS) analysis based on the Bray-Curtis similarity index.
Beta diversity was calculated using the UniFrac dissimilarity and presented by PCoA (Figures 2A–C) and NMDS (Figure 2D). There were no dissimilarities (p > 0.05) detected in the rumen microbial community between the treatment groups and no clustering pattern was demonstrated between the treatments. Instead, dissimilarities were detectable between the sheep breeds (p < 0.05) and sample day (p < 0.05). The samples were clustered according to sample day or breed; few Meatmaster samples exhibited slightly higher diversity than other animals. The CCA (Figure 3) was used to identify the association between the treatment groups. Though the dissimilarities between the treatment groups were not significant, CCA showed that the microbial community from T1 was distinct from the microbial communities of other groups. The microbial communities for T2 and T4 were overlapping.
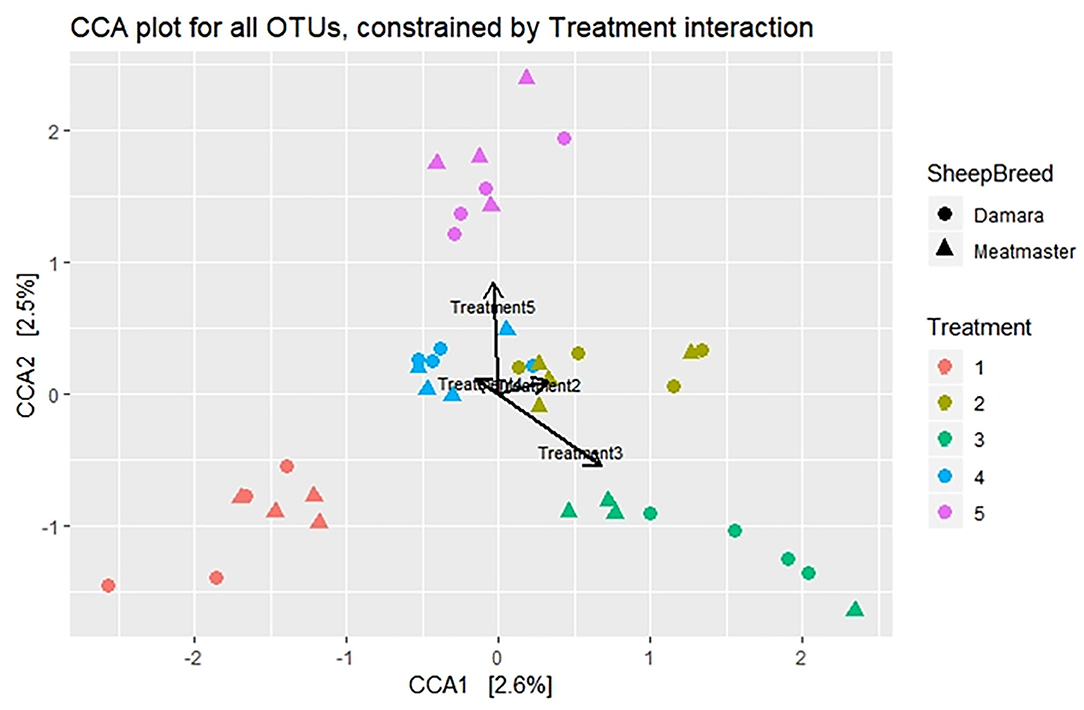
Figure 3. Canonical correspondence analysis (CCA) visualizing the correlation of the microbiome structures between treatments. Treatment groups are as follows: 1. Diet with no antibiotics, no probiotics (negative control); 2. Diet with no antibiotics, only Lactobacillus rhamnosus PT9; 3. Diet with no antibiotics, only Lactobacillus rhamnosus PT10; 4. Diet with the combination of Lactobacillus rhamnosus PT9 and Lactobacillus rhamnosus PT10; 5. Diet with antibiotics, no probiotics (positive control). Arrows show the relationship between treatments.
The abundance of microbiota and its taxonomic distribution was estimated in all treatments. Fifteen (15) bacterial phyla and 1 archaeal phylum were detected across all samples at a relative abundance of ≥0.05%. At phylum level; after the trials, Bacteroidetes were the highest abundant phenotype across all 5 treatment groups (1–5) accounted for 51%; 46.3%; 41.9%; 46.2% and 43.95% of relative abundance, respectively, followed by Firmicutes that accounted for 30.5%; 29.9%; 28.6%; 31.7% and 25.5% of relative abundance; and Proteobacteria that accounted for 4.77%; 13.83%; 19.82%; 4.92% and 17.41% of relative abundance. Treatments 1 and 4 had lower numbers of Proteobacteria compared to other treatments. Euryarchaeota abundance accounted for 2.31%; 1.7%; 1.78%; 3.02% and 1.82% respectively (Figure 4A). The supplementation of the combination of probiotics increased the percentage of Lentisphaerae (2.24%) in the rumen. At genus level, a total of 102 genera were identified in the rumen (Figure 4B). Prevotella and Clostridium were abundant across all treatment groups, while Pseudomonas was abundant only in treatments 2, 3, and 5. The relative Fibrobacter was detected in all treatment groups (3.98%, 3.47%, 5.74%, 5.97% and 6.33%, respectively) after the feeding trials, whereas it was detected in few treatment groups before the trials. Microbial relative abundance was diverse across treatments.
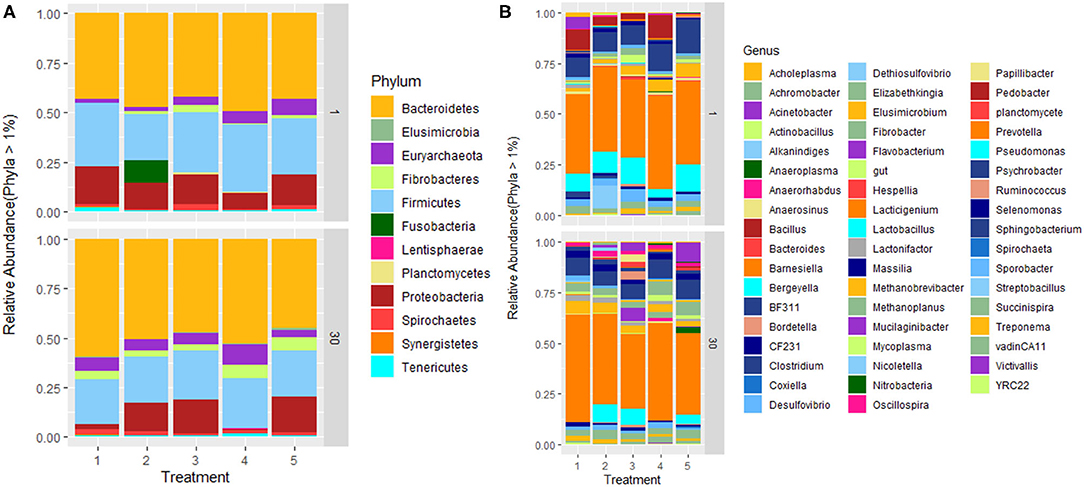
Figure 4. Relative abundance of rumen microbial communities at (A) phylum and (B) genus level. Treatment groups are as follows: 1. Diet with no antibiotics, no probiotics (negative control); 2. Diet with no antibiotics, only Lactobacillus rhamnosus PT9; 3. Diet with no antibiotics, only Lactobacillus rhamnosus PT10; 4. Diet with the combination of Lactobacillus rhamnosus PT9 and Lactobacillus rhamnosus PT10; 5. Diet with antibiotics, no probiotics (positive control).
Microbial abundance was further evaluated using heatmap analysis for top 30 microbial community at class (Figure 5) and genus (Figure 6) levels for Damara and Meatmaster sheep breeds as well as for sample period (Figure 7). At class level, a total of 32 classes were identified. Bacteroidia and Clostridia were most abundant in both breeds. Gammaproteobacteria accounted for 7.3% in Damara and 8.9% in Meatmaster sheep. TM7-3 remained unchanged in Damara sheep, and it increased in Meatmaster from 1.4 to 2.35% after the trials. Fibrobacteria and Verruco-5 were steady in Damara sheep, while they increased in Meatmaster (0.6–2.17% and 0.78–1.49%, respectively). Bacilli decreased in both breeds after the 30-day trial; 2.04–0.76% in Damara and 5.33–0.2% in Meatmaster.
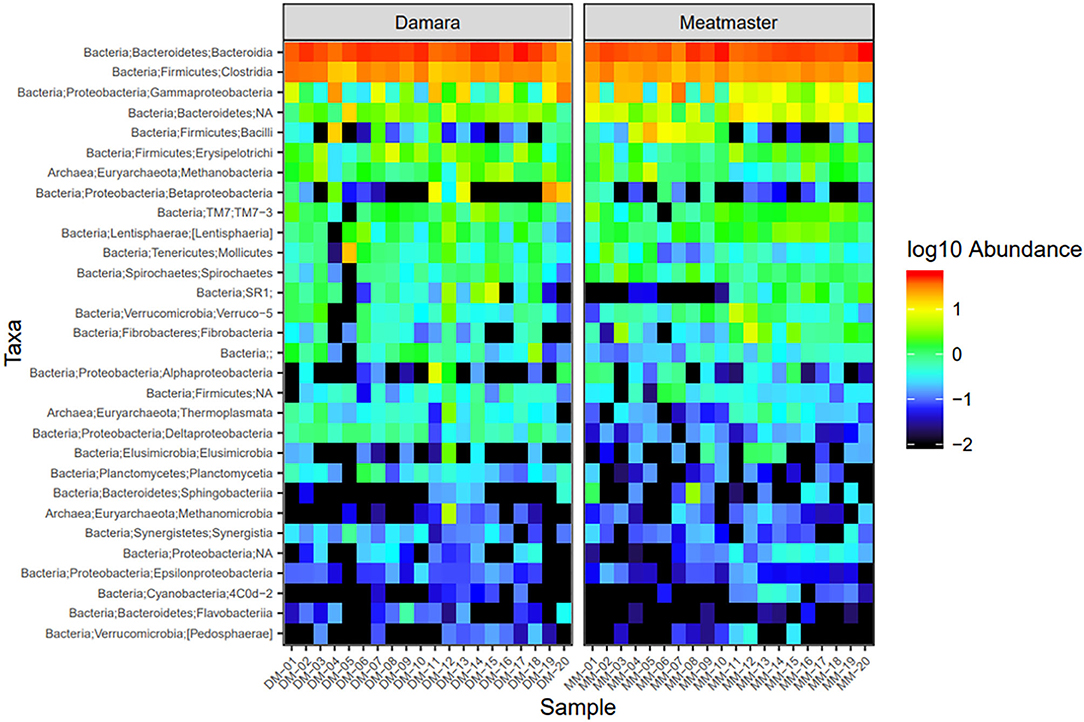
Figure 5. Heatmap of the top 30 most abundant taxa by class between the two sheep breeds. DM1-DM10 and MM1-MM10 are samples before the treatments were administered, DM11-DM20 and MM11-MM20 are samples after the trial. Relative values for each genus depicted by color intensity according to the legend provided on the scale of −2 to 1.
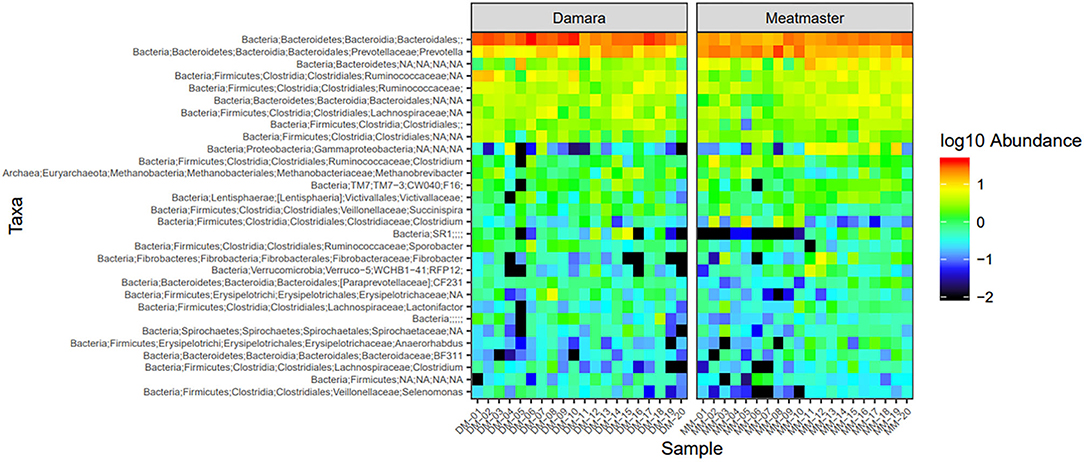
Figure 6. Heatmap of the top 30 most abundant taxa by genus between the two sheep breeds. DM1-DM10 and MM1-MM10 are samples before the treatments were administered, DM11-DM20 and MM11-MM20 are samples after the trial. Relative values for each genus depicted by color intensity according to the legend provided on the scale of −2 to 1.
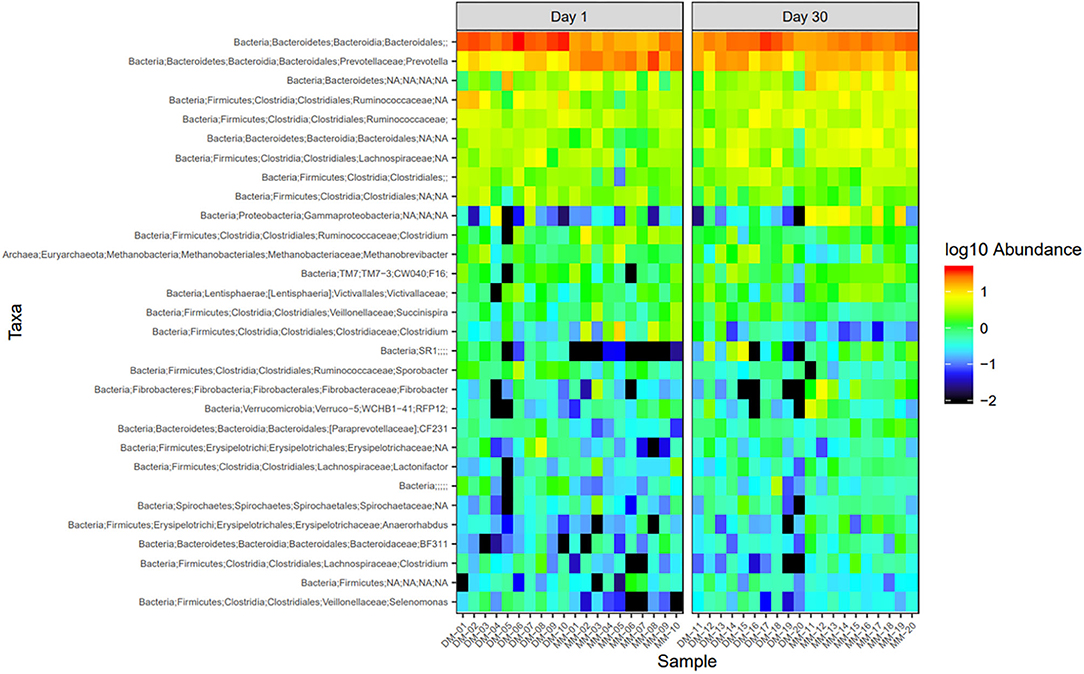
Figure 7. Heatmap of the top 30 most abundant taxa by genus between sampling days. DM1-DM10 and MM1-MM10 are samples before the treatments were administered, DM11-DM20 and MM11-MM20 are samples after the trial. Relative values for each genus depicted by color intensity according to the legend provided on the scale of −2 to 1.
At genus level, Bacteroidales and Prevotella were the most abundant taxa across the two breeds. Pseudomonas accounted for 15.1% in Meatmaster, while Damara had 5.8%. Proteobacteria phylum was more abundant in Meatmaster as compared to Damara sheep. This phylum consists of most pathogenic bacteria and Damara sheep are known to be resistant to parasites, which could be the reason for low quantities of Proteobacteria in Damara sheep.
A co-occurrence network was observed for microbial communities using Spearman correlation ( ≤ -0.7 or ≥0.7 and p-value of 0.01). The network had 158 nodes with a significant number of positive correlations compared to negative correlations. Each node represented a taxon with the node size comparative to its relative abundance in the sample (Figure 8A). The network displayed a modular structure with modularity of 0.532. A module depicts a microbial taxa subnetwork that potentially interacts or shares the same ecological niche without direct interaction. Three major modules comprising correlated microbial taxa centered by unclassified SR1, Clostridiales and unclassified Bacteroidetes were observed (Figure 8B). These microbial taxa were identified based on their betweenness centrality within the network. Module III being the only module showing negative correlations with about 78 pairs. Firmicutes were the most source taxa with members that showed a strong negative correlation with other taxa, mostly with Bacteroidetes.
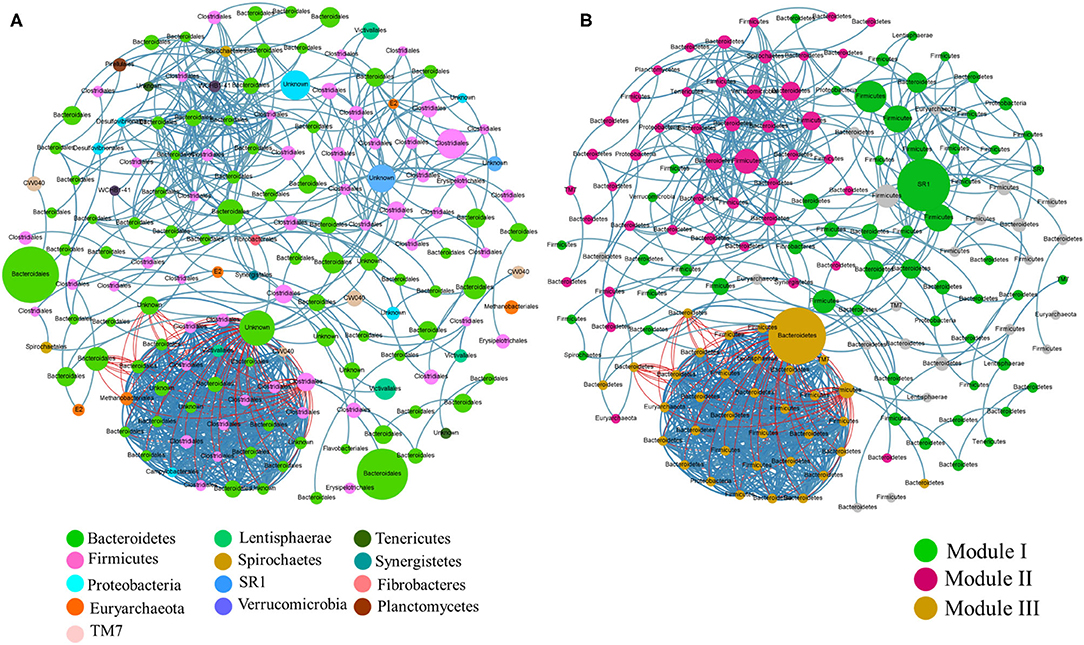
Figure 8. A Network analysis of rumen microbial communities. A connection stands for Spearman correlation with a value above 0.7 between microbial abundances. Edge color represents positive (blue) and negative (red) correlations and the edge thickness is equivalent to the correlation values. (A) Node color corresponds to the phylum taxonomic classification and the size of the node is proportional to the abundance of the order. (B) Node color corresponds to the modularity class and the node size is proportional to betweenness centrality.
Discussion
In recent years, the relationship between rumen microbiota and their host has been shown to play a significant role in the host's function and health (30). Previous studies have shown that certain microorganisms as direct-fed microbials can have beneficial effects on the host, thus improving the health and production performance of dairy cows, sheep and goats (31–34).
The supplementation of probiotics influenced the sheep's weight improvement as the negative control showed lower body weight than other treatment groups; T3 and T4 recorded the heaviest bodyweight, which implies that the probiotics may impact the digestion of nutrients. Treatment with the combination of probiotics (T4) showed greater weight gain than the control and T2. The observed comparative increase of body weight gain on microbial treated groups in this study cannot be explained by excess dry matter intake, as all experimental animals were given the same type and amount of feed. Instead, the body weight gain could be associated with nutrient digestion's effectiveness stimulated by probiotics with microbiota interactions. A study by Roodposhti AND Dabiri (35) reported that average weight gain was significantly greater for the synbiotic, prebiotic and probiotic treatments than the control. They observed that synbiotic-fed calves had greater weight gain than other treatments and no significant difference between prebiotic and probiotic treatments. Damara breed had heavier body weight gain than Meatmaster breed. The effect of breed on the sheep's body weight gain was also reported by Wilkes et al. (36), where they recorded a difference in weight gain of the breeds with the Damara gaining on average of 142 g/day and Merino at 80 g/day. However, no significant interaction was found between diet and breed, yet Damara sheep gained faster than Merino sheep in both diets.
Ruminal pH is the key guide signifying the internal balance in the rumen environment; hence it is vital to maintain a moderately stable pH to ensure efficiency in ruminal fermentation. In the present study, treatment and breed had a significant effect (p < 0.05) on ruminal pH. Damara breed recorded a higher ruminal pH value than Meatmaster breed; this might be because Damara can retain more fibrous constituents in the rumen for a longer period, permitting the cellulolytic microorganisms to degrade the feed's fibrous constituents thoroughly. Cellulolytic bacteria's ability to digest fibers is maintained by optimal ruminal conditions, where pH between 6 and 9 is best (37). The high ruminal pH recorded in the antibiotic-treated group (T5) in this study might be associated with a decrease of some lactic acid bacteria due to antibiotic supplementation, which may have increased the pH. Probiotics stabilized ruminal pH, which may have enriched microbial ecology and may have also led to increased nutrient absorption and lactate, resulting in improved weight gain. A study by Nocek et al. (38) reported that bacterial probiotics consisting of Lactobacillus plantarum and Enterococcus faecium prompted alterations in the ruminal pH of high grain-fed cows. Yet, the mechanism of the effects of the bacterial probiotics on rumen fermentation was unclear.
In the present study, we evaluated and compared rumen microbial abundance and diversity of five (5) treatment groups of Damara and Meatmaster sheep breeds. The results revealed that the structure and abundance of rumen microbial communities were altered by administering potential probiotics and antibiotics.
Alpha diversity results within treatments revealed that the combination of potential probiotics (T4) had higher microbial diversity than other treatments. This might be because other treatments were also treated with feed additives, even though they were not as effective as the synergetic effect of T4. Wang et al. (39) reported that the increased nutrient levels in the diet affected microbial richness, while no significant differences were detected in the diversity across the treatments. Jia et al. (40) showed no significant difference in microbial richness and diversity among the five treatment groups. Breed variation affected the alpha diversity also, as Damara had higher microbial richness and diversity than Meatmaster. These findings show that the host genotype does have a significant role in regulating the rumen microbial community structure. The results are in accordance with the reports from other ruminants that investigated rumen microbial diversity. Huang et al. (41) reported that rumen bacterial diversity in Tibetan sheep was greater than Gansu sheep based on the Shannon-Weiner and Simpson indexes. Another study also showed that water deer had the highest bacterial diversity than reindeer and goat (42).
Beta diversity showed no significant dissimilarities among the treatments, which could mean that there was no distinct diversity in the microbial communities of the treatment groups, whereas sample time and breed variation displayed significant diversity. These results agree with a study by Noel et al. (43), where they observed that only breed had a significant effect on methanogen community structure, while diet had no significant effect. A study by Xu et al. (44) observed a structural difference between before and after the probiotic intervention. Furthermore, the association among treatment groups was explored using CCA, which displayed that microbial community from the negative control (T1) was further distant from other treatments, while there was a close interaction among the probiotic treatment groups. This suggests that the probiotic used in treatment 2 was more effective in interacting with the rumen microbiota than the one from treatment 3, as the distance of treatment 2 with the combination group is lesser than treatment 3. Though the treatment groups were not diverse statistically, their interaction shows differences in the microbial communities and probiotic treated groups were more similar compared to the antibiotic-treated group and the negative control. Similar results were observed in a study by Xu et al. (44), where the microbial community of the control group formed a distinct cluster, suggesting that microbiota from the control differed from those of the probiotic treated group.
Microbial abundance slightly varied across treatments, with Bacteroidetes and Firmicutes being the most abundant phyla in all treatments. The results were similar to that of Peng et al. (45), where they observed Bacteroidetes (57%) being the most abundant phylum in the rumen, followed by Firmicutes (35%) and Proteobacteria (3.9%). Another study observed that Bacteroidetes were predominant in the rumen followed by Firmicutes and the findings suggested that a high concentrate diet provided a conducive environment for Bacteroidetes survival, though there was no significant difference between their treatments (33). The richness of phylum Lentisphaerae, Tenericutes, and Fibrobacteres was varied among the treatments, indicating that the treated groups affected the distribution of bacteria in the rumen.
Typically, Proteobacteria proportion is < 4% of the entire ruminal population, and an increase in the Proteobacteria population may cause subacute ruminal acidosis (46, 47). Proteobacteria was the most prevalent in T2, T3, and T5, while it was less abundant in T1 and T4. The low Proteobacteria abundance in the combination of the potential probiotic treated group indicates that the synergistic probiotics might be contributing to the balance of rumen microbial community at the phylum level. This is in accordance with results in a study by Pinloche et al. (48), as they observed a significant decrease in Proteobacteria occurrence in the highest level of yeast fed to the lactating cows. There is no clear indication of the cause of the decrease in Proteobacteria in the negative control. A closer look at both breeds in the negative control indicated that Damara accounted for about 14.5% of Proteobacteria before the trial, and at the end of 30 days, it only accounted for 2.8%, while Meatmaster had a slight decrease from 9.42–6.7%. This phylum consists of most pathogenic bacteria and the decrease might be because Damara sheep are known to be very adaptive to harsh conditions and resistant to parasites (49). On the contrary, in T2, T3, and T5, where Proteobacteria was most abundant, Damara sheep was the dominating breed as it resulted in the increase of this phylum after 30 days in these treatment groups, as compared to Meatmaster, which showed a decrease in all treatments. Even though the combination of potential probiotics treatment did not lead to any significant change of the core rumen microbial composition, the combination of potential probiotics (T4) was found to have stabilized the rumen microbiota and reduced the risk of pathogen colonization as compared to the singular probiotic groups and antibiotic group.
Genera Prevotella, Clostridium, CF231, BF311, Fibrobacter, Methanobrevibacter, Succinispira, and Treponema were found in all treatments and these genera belong to Firmicutes, Bacteroidetes, Fibrobacteres, Proteobacteria, Spirochaetes, and Euryarchaeota. Prevotella assist by utilizing feed proteins in the rumen and they are known to work in combination with cellulolytic species Fibrobacters in hemicellulose utilization (50, 51). Previous studies reported that Treponema is a genus of the primary bacterial community in the rumen and it is said that the genus break down plant polysaccharides from ingested feed (52). Methanobrevibacter produces CH4 by utilizing formate, CO2, H2 as substrates (53).
Antibiotic treated group (T5) and L. rhamnosus PT10 group (T3) exhibited aerobic bacterial genus Acinetobacter. Previous studies mostly found this genus in the small intestine mucosa, as it shows the presence of oxygen in the intestinal epithelial cell (54) and it represents a possible exclusion mechanism of strictly anaerobic and oxygen-sensitive microbes (55). The results suggest that the combination of potential probiotics was most effective in balancing the rumen microbial flora and improving the composition of the microbiota.
As revealed by the co-occurrence network, different rumen modules present a complex synergistic and unwieldy relationship. Three microbial taxa were observed to interact with other communities in their respective modules, suggesting that they may be keystone species of the rumen microbiota. Among the three keystone species observed, two (unclassified Bacteroidetes and Clostridiales) belong to the two most abundant phyla in rumen environment and had been widely studied through literature (2, 56, 57). However, the third keystone is from candidate phylum SR1; currently, there are no cultivated representatives available of rather extremely difficult to attain; only 16S rRNA gene information available (58). Nevertheless, this species occurrence is interesting as they appeared to present a significant shift in the network. Co-occurrence networks represent associations between microbial communities and help us understand the microbial ecology (59). The associations between microbial taxa may follow a sequence of universal dynamics as recently anticipated in the human microbiota (58).
The results from this study revealed that the administration of feed additives slightly altered rumen microbial community structure and abundance in the form of lactic acid as putative probiotics as well as increase in body weight gain. Although treatment groups were not diverse, their interactions showed that there were variations in the microbial communities. The synergism of probiotics demonstrated an improved microbial composition, which shows that probiotic administration can help maintain the balance of gut microbiota and enhance ruminal bacterial composition and structure (48). Breed variation affected rumen microbial composition and structure. This variation can be further investigated and explored to improve fitness traits in sheep production and breeding.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA578022.
Ethics Statement
The animal study was reviewed and approved by Agricultural Research Council - Animal Production Institute Ethics committee (APIEC17/21). The trials were done at the Agricultural Research Council (ARC) GI Microbiology and Biotechnology unit and the Small Stocks Unit in Irene, Gauteng province.
Author Contributions
SM conducted the study for her PhD research and drafted the manuscript. OA and MA designed, supervised, and proofread the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors will like to acknowledge the Agricultural Research Council – Animal Production, Irene, South Africa; National Research Foundation (NRF) of South Africa and University of KwaZulu Natal, Discipline of Genetics for funds and provision of research facilities.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2020.570074/full#supplementary-material
Supplementary Figure 1. Rarefaction curves of operational taxonomic units (OTUs at 97% sequence similarity), based on the 16S rRNA gene sequencing.
References
1. Liu J, Li H, Zhu W, Mao S. Dynamic changes in rumen fermentation and bacterial community following rumen fluid transplantation in a sheep model of rumen acidosis: implications for rumen health in ruminants. FASEB J. (2019) 33:8453–67. doi: 10.1096/fj.201802456R
2. Wang J, Fan H, Han Y, Zhao J, Zhou Z. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian Aust J Anim Sci. (2017) 30:100–10. doi: 10.5713/ajas.16.0166
3. Singh KM, Bagath M, Chikara SK, Joshi CG, Kothari RK. Metagenomic approaches in understanding the rumen function and establishing the rumen microbial diversity. In: Sejian V, Gaughan J, Baumgard L, Prasad C, editors. Climate Change Impact on Livestock: Adaptation and Mitigation. New Delhi: Springer (2015) 221–37. doi: 10.1007/978-81-322-2265-1_14
4. Huang Q, Holman DB, Alexander T, Hu T, Jin L, Xu Z, et al. Fecal microbiota of lambs fed purple prairie clover (Dalea purpurea Vent.) and alfalfa (Medicago sativa). Arch Microbiol. (2018) 200:137–45. doi: 10.1007/s00203-017-1427-5
5. Elghandour MMY, Salem AZM, Castañeda JSM, Camacho LM, Kholif AE, Chagoyán JCV. Direct-fed microbes: a tool for improving the utilization of low quality roughages in ruminants. J Integr Agric. (2015) 14:526–33. doi: 10.1016/S2095-3119(14)60834-0
6. Simoneit C, Burow E, Tenhagen BA, Käsbohrer A. Oral administration of antimicrobials increase antimicrobial resistance in E. coli from chicken - A systematic review. Prev Vet Med. (2015) 118:1–7. doi: 10.1016/j.prevetmed.2014.11.010
7. Lupindu AM, Dalsgaard A, Msoffe PLM, Ngowi HA, Mtambo MM, Olsen JE. Transmission of antibiotic-resistant Escherichia coli between cattle, humans and the environment in peri-urban livestock keeping communities in Morogoro, Tanzania. Prev Vet Med. (2015) 118:477–82. doi: 10.1016/j.prevetmed.2014.12.005
8. Ford AC, Quigley EMM, Lacy BE, Lembo AJ, Saito YA, Schiller LR, et al. Efficacy of prebiotics, probiotics, and synbiotics in irritable bowel syndrome and chronic idiopathic constipation: systematic review and meta-analysis. Am J Gastroenterol. (2014) 109:1547–62. doi: 10.1038/ajg.2014.202
9. Salem AZM, Gado HM, Colombatto D, Elghandour MMY. Effects of exogenous enzymes on nutrient digestibility, ruminal fermentation and growth performance in beef steers. Livest Sci. (2013) 154:69–73. doi: 10.1016/j.livsci.2013.02.014
10. Adjei-fremah S, Ekwemalor K, Worku M, Ibrahim S. Probiotics and Ruminant Health. Intech. (2018) 133–50. doi: 10.5772/intechopen.72846
11. Uyeno Y, Shigemori S, Shimosato T. Effect of probiotics/prebiotics on cattle health and productivity. Microbes Environ. (2015) 30:126–32. doi: 10.1264/jsme2.ME14176
12. Sun P, Wang JQ, Deng LF. Effects of Bacillus subtilis natto on milk production, rumen fermentation and ruminal microbiome of dairy cows. Animal. (2013) 7:216–22. doi: 10.1017/S1751731112001188
13. Fon FN, Nsahlai I V. Effect of direct-fed microbial consortia on ruminal fermentation of maize stover in sheep. Small Rumin Res. (2013) 111:71–5. doi: 10.1016/j.smallrumres.2012.09.016
14. Souza VL, Lopes NM, Zacaroni OF, Silveira VA, Pereira RAN, Freitas JA. Lactation performance and diet digestibility of dairy cows in response to the supplementation of Bacillus subtilis spores. Livest Sci. (2017) 200:35–9. doi: 10.1016/j.livsci.2017.03.023
15. Deng KD, Xiao Y, Ma T, Tu Y, Diao QY, Chen YH, et al. Ruminal fermentation, nutrient metabolism, and methane emissions of sheep in response to dietary supplementation with Bacillus licheniformis. Anim Feed Sci Technol. (2018) 241:38–44. doi: 10.1016/j.anifeedsci.2018.04.014
16. Zhang L, Jiang X, Liu X, Zhao X, Liu S, Li Y, et al. Growth, health, rumen fermentation, and bacterial community of Holstein calves fed Lactobacillus rhamnosus GG during the preweaning stage1. J Anim Sci. (2019) 97:2598–608. doi: 10.1093/jas/skz126
17. Sinclair L, Osman OA, Bertilsson S, Eiler A. Microbial community composition and diversity via 16S rRNA gene amplicons: evaluating the illumina platform. PLoS ONE. (2015) 10:e0116955. doi: 10.1371/journal.pone.0116955
18. Xue D, Chen H, Zhao X, Xu S, Hu L, Xu T, et al. Rumen prokaryotic communities of ruminants under different feeding paradigms on the Qinghai-Tibetan Plateau. Syst Appl Microbiol. (2017) 40:227–36. doi: 10.1016/j.syapm.2017.03.006
19. Association of Official Analytical Chemists. In: Official Methods of Analysis. Washington, DC (1990).
20. Van Soest PJ, Wine RH. Use of detergents in the analysis of fibrous feeds. IV. Determination of plant cell-wall constituents. J Assoc Off Anal Chem. (1967) 50:50–5. doi: 10.1093/jaoac/50.1.50
21. Goering HK, Van Soest PJ. Forage Fibre Analyses. Agricultural Handbook No 379. Washington, DC: A.R.S., U.S. Department of Agriculture (1970).
22. Association of Official Analytical Chemists (AOAC). Official Methods of Analysis. 14th ed. Arlington, VA: Association of Official Analytical Chemists, Inc. (1984).
23. Methods of Analysis for Fertilizers Ameliorants and Farm Feeds. 11 Compiled by F Bessinger & TF Bosch. Method no 3031 for Preparation, 3054 for EDTA Titration. Information Bulletin. Series: Routine Analysis Pretoria, South Africa: Soil and Irrigation Research Institute, Department of Agriculture, Pretoria, South Africa (1985).
24. Shen JS, Chai Z, Song LJ, Liu JX, Wu YM. Insertion depth of oral stomach tubes may affect the fermentation parameters of ruminal fluid collected in dairy cows1. J Dairy Sci. (2012) 95:5978–84. doi: 10.3168/jds.2012-5499
25. Bolyen E, Dillon M, Bokulich N, Abnet C, Al-Ghalith G, Alexander H, et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. Nature biotech. (2019) 37:852–7. doi: 10.1038/s41587-019-0209-9
26. Ju F, Xia Y, Guo F, Wang Z, Zhang T. Taxonomic relatedness shapes bacterial assembly in activated sludge of globally distributed wastewater treatment plants. Environ Microbiol. (2014) 16:2421–32. doi: 10.1111/1462-2920.12355
27. Benjamini Y, Krieger AM, Yekutieli D. Adaptive linear procedures that control the false discovery rate. Biometrika. (2006) 93:491–507. doi: 10.1093/biomet/93.3.491
28. Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks. Int AAAI Conf Weblogs Soc Media. (2009) 8:361–2. Available online at: https://www.aaai.org/ocs/index.php/ICWSM/09/paper/view/154
29. Blondel VD, Guillaume JL, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. J Stat Mech Theory Exp. (2008) 10:P10008. doi: 10.1088/1742-5468/2008/10/P10008
30. Jiao J, Huang J, Zhou C, Tan Z. Taxonomic identification of ruminal epithelial bacterial diversity during rumen development in goats. Appl Environ Microbiol. (2015) 81:3502–9. doi: 10.1128/AEM.00203-15
31. Qadis AQ, Goya S, Ikuta K, Yatsu M, Kimura A, Nakanishi S, et al. Effects of a bacteria-based probiotic on ruminal pH, volatile fatty acids and bacterial flora of holstein calves. J Vet Med Sci. (2014) 76:877–85. doi: 10.1292/jvms.14-0028
32. Long M, LI P, Zhang Y, Chen X, Gao Z, Liu G. Evaluation of the protective effect of the acid-tolerant engineered bacterial strain M. elsdenii H6F32 as a probiotic fed to sheep duringthe lactic acidosis challenge. Indian J Anim Res. (2016) 50:330–4. doi: 10.18805/ijar.9632
33. Thomas M, Webb M, Ghimire S, Blair A, Olson K, Fenske GJ, et al. Metagenomic characterization of the effect of feed additives on the gut microbiome and antibiotic resistome of feedlot cattle. Sci Rep. (2017) 7:1–13. doi: 10.1038/s41598-017-12481-6
34. Payandeh S, Kafilzadeh F, Juarez M, de la Fuente MA, Ghadimi D, Martinez Marin AL. Probiotic supplementation effects on milk fatty acid profile in ewes. J Dairy Res. (2017) 84:128–31. doi: 10.1017/S0022029917000115
35. Roodposhti PM, Dabiri N. Effects of probiotic and prebiotic on average daily gain, fecal shedding of Escherichia coli, and immune system status in newborn female calves. Asian Aust J Anim Sci. (2012) 25:1255–61. doi: 10.5713/ajas.2011.11312
36. Wilkes MJ, Hynd PI, Pitchford WS. Damara sheep have higher digestible energy intake than Merino sheep when fed low-quality or high-quality feed. Anim Prod Sci. (2012) 52:30–4. doi: 10.1071/AN11033
37. Castillo-Gonzalez A, Burrola-Barraza M, Dominguez-Viveros J, Chavez-Martinez A. Rumen microorganisms and fermentation. Arch Med Vet. (2014) 46:349–61. doi: 10.4067/S0301-732X2014000300003
38. Nocek JE, Kautz WP, Leedle JAZ, Allman JG. Ruminal supplementation of direct-fed microbials on diurnal pH variation and in situ digestion in dairy cattle. J Dairy Sci. (2002) 85:429–33. doi: 10.3168/jds.S0022-0302(02)74091-5
39. Wang Y, Cao P, Wang L, Zhao Z, Chen Y, Yang Y. Bacterial community diversity associated with different levels of dietary nutrition in the rumen of sheep. Appl Microbiol Biotechnol. (2017) 101:3717–28. doi: 10.1007/s00253-017-8144-5
40. Jia P, Cui K, Ma T, Wan F, Wang W, Yang D, et al. Influence of dietary supplementation with Bacillus licheniformis and Saccharomyces cerevisiae as alternatives to monensin on growth performance , antioxidant , immunity , ruminal fermentation and microbial diversity of fattening lambs. Sci Rep. (2018) 8:1–10. doi: 10.1038/s41598-018-35081-4
41. Huang XD, Martinez-Fernandez G, Padmanabha J, Long R, Denman SE, McSweeney CS. Methanogen diversity in indigenous and introduced ruminant species on the Tibetan Plateau. Archaea. (2016) 2016:5916067. doi: 10.1155/2016/5916067
42. Li Z, Wright AG, Yang Y, Si H, Li G. Unique bacteria community composition and co-occurrence in the milk of different ruminants. Nat Publ Gr. (2017) 7:1–9. doi: 10.1038/srep40950
43. Noel SJ, Olijhoek DW, Mclean F, Løvendahl P, Lund P, Højberg O. Rumen and fecal microbial community structure of holstein and jersey dairy cows as affected by breed, diet, and residual feed intake. Animals. (2019) 9:498. doi: 10.3390/ani9080498
44. Xu H, Huang W, Hou Q, Kwok LY, Sun Z, Ma H, et al. The effects of probiotics administration on the milk production, milk components and fecal bacteria microbiota of dairy cows. Sci Bull. (2017) 62:767–74. doi: 10.1016/j.scib.2017.04.019
45. Peng S, Yin J, Liu X, Jia B, Chang Z, Lu H, et al. First insights into the microbial diversity in the omasum and reticulum of bovine using Illumina sequencing. J Appl Genet. (2015) 56:393–401. doi: 10.1007/s13353-014-0258-1
46. Khafipour E, Li S, Plaizier JC, Krause DO. Rumen microbiome composition determined using two nutritional models of subacute ruminal acidosis. Appl Environ Microbiol. (2009) 75:7115–24. doi: 10.1128/AEM.00739-09
47. Pitta DW, Pinchak WE, Dowd SE, Osterstock J, Gontcharova V, Youn E, et al. Rumen bacterial diversity dynamics associated with changing from bermudagrass hay to grazed winter wheat diets. Microb Ecol. (2010) 59:511–22. doi: 10.1007/s00248-009-9609-6
48. Pinloche E, McEwan N, Marden JP, Bayourthe C, Auclair E, Newbold CJ. The effects of a probiotic yeast on the bacterial diversity and population structure in the rumen of cattle. PLoS ONE. (2013) 8:e67824 doi: 10.1371/journal.pone.0067824
49. Almeida AM. The Damara in the context of Southern Africa fat-tailed sheep breeds. Trop Anim Health Prod. (2011) 43:1427–41. doi: 10.1007/s11250-011-9868-3
50. Xu J, Gordon JI. Honor thy symbionts. Proc Natl Acad Sci USA. (2003) 100:10452–9. doi: 10.1073/pnas.1734063100
51. Zeng Y, Zeng D, Ni X, Zhu H, Jian P, Zhou Y, et al. Microbial community compositions in the gastrointestinal tract of Chinese Mongolian sheep using Illumina MiSeq sequencing revealed high microbial diversity. AMB Express. (2017) 7:1–10. doi: 10.1186/s13568-017-0378-1
52. Bekele AZ, Koike S, Kobayashi Y. Phylogenetic diversity and dietary association of rumen Treponema revealed using group-specific 16S rRNA gene-based analysis. FEMS Microbiol Lett. (2011) 316:51–60. doi: 10.1111/j.1574-6968.2010.02191.x
53. Danielsson R, Schnürer A, Arthurson V, Bertilsson J. Methanogenic population and CH4 production in swedish dairy cows fed different levels of forage. Appl Environ Microbiol. (2012) 78:6172–9. doi: 10.1128/AEM.00675-12
54. Mao S, Zhang M, Liu J, Zhu W. Characterising the bacterial microbiota across the gastrointestinal tracts of dairy cattle: membership and potential function. Sci Rep. (2015) 5:1–14. doi: 10.1038/srep16116
55. Pédron T, Mulet C, Dauga C, Frangeul L, Chervaux C, Grompone G, et al. A crypt-specific core microbiota resides in the mouse colon. MBio. (2012) 3:1–7. doi: 10.1128/mBio.00116-12
56. Præsteng KE, Pope PB, Cann IKO, Mackie RI, Mathiesen SD, Folkow LP, et al. Probiotic dosing of Ruminococcus flavefaciens affects rumen microbiome structure and function in reindeer. Microb Ecol. (2013) 66:840–9. doi: 10.1007/s00248-013-0279-z
57. Jewell KA, Mccormick CA, Odt CL, Weimer PJ, Suen G. Ruminal bacterial community composition in dairy cows is dynamic over the course of two lactations and correlates with feed efficiency. Appl Environ Microbiol. (2015) 81:4697–710. doi: 10.1128/AEM.00720-15
58. Ricci S, Sandfort R, Pinior B, Mann E, Wetzels SU, Stalder G. Impact of supplemental winter feeding on ruminal microbiota of roe deer Capreolus capreolus. Wildlife Biol. (2019) 1:1–11. doi: 10.2981/wlb.00572
Keywords: 16S rRNA gene sequencing, lactic acid bacteria, sheep breed, microbial community, rumen
Citation: Mani S, Aiyegoro OA and Adeleke MA (2021) Characterization of Rumen Microbiota of Two Sheep Breeds Supplemented With Direct-Fed Lactic Acid Bacteria. Front. Vet. Sci. 7:570074. doi: 10.3389/fvets.2020.570074
Received: 06 June 2020; Accepted: 15 December 2020;
Published: 15 January 2021.
Edited by:
Amlan Kumar Patra, West Bengal University of Animal and Fishery Sciences, IndiaReviewed by:
Yuxin Yang, Northwest A and F University, ChinaAnju Kala, Indian Veterinary Research Institute (IVRI), India
Jinzhen Jiao, Chinese Academy of Sciences, China
Copyright © 2021 Mani, Aiyegoro and Adeleke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olayinka A. Aiyegoro, QWl5ZWdvcm9vQGFyYy5hZ3JpYy56YQ==