 John I. Alawneh1,2*†
John I. Alawneh1,2*† Ben Vezina3†
Ben Vezina3† Hena R. Ramay4Hulayyil Al-Harbi1,2Ameh S. James2
Hena R. Ramay4Hulayyil Al-Harbi1,2Ameh S. James2 Martin Soust5
Martin Soust5 Robert J. Moore6
Robert J. Moore6 Timothy W. J. Olchowy1,7
Timothy W. J. Olchowy1,7- 1Good Clinical Practice Research Group, School of Veterinary Science, The University of Queensland, Gatton, QLD, Australia
- 2School of Veterinary Science, The University of Queensland, Gatton, QLD, Australia
- 3Centre for Cell Factories and Biopolymers, Griffith Institute for Drug Discovery, Griffith University, Nathan, QLD, Australia
- 4International Microbiome Centre, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
- 5Terragen Biotech Pty Ltd., Coolum Beach, QLD, Australia
- 6School of Science, RMIT University, Melbourne, VIC, Australia
- 7Faculty of Veterinary Medicine, University of Calgary, Calgary, AB, Canada
Escherichia coli is frequently associated with mastitis in cattle. “Pathogenic” and “commensal” isolates appear to be genetically similar. With a few exceptions, no notable genotypic differences have been found between commensal and mastitis-associated E. coli. In this study, 24 E. coli strains were isolated from dairy cows with clinical mastitis in three geographic regions of Australia (North Queensland, South Queensland, and Victoria), sequenced, then genomically surveyed. There was no observed relationship between sequence type (ST) and region (p = 0.51). The most common Multi Locus Sequence Type was ST10 (38%), then ST4429 (13%). Pangenomic analysis revealed a soft-core genome of 3,463 genes, including genes associated with antibiotic resistance, chemotaxis, motility, adhesion, biofilm formation, and pili. A total of 36 different plasmids were identified and generally found to have local distributions (p = 0.02). Only 2 plasmids contained antibiotic resistance genes, a p1303_5-like plasmid encoding multidrug-resistance (trimethoprim, quaternary ammonium, beta-lactam, streptomycin, sulfonamide, and kanamycin) from two North Queensland isolates on the same farm, while three Victorian isolates from the same farm contained a pCFSAN004177P_01-like plasmid encoding tetracycline-resistance. This pattern is consistent with a local spread of antibiotic resistance through plasmids of bovine mastitis cases. Notably, co-occurrence of plasmids containing virulence factors/antibiotic resistance with putative mobilization was rare, though the multidrug resistant p1303_5-like plasmid was predicted to be conjugative and is of some concern. This survey has provided greater understanding of antibiotic resistance within E. coli-associated bovine mastitis which will allow greater prediction and improved decision making in disease management.
Introduction
Bovine mastitis is the major production limiting disease in the dairy industry worldwide (1–3). In addition to the negative impact on animal welfare and farm economics, the extensive use of antimicrobials to treat and manage mastitis is a major concern to public health (4). A gram-negative opportunistic environmental bacterium, Escherichia coli is the coliform bacteria most frequently associated with mastitis in cattle (5–7). Many high quality genomic sequences of mastitis-associated E. coli are publicly available (8). Genomic analyses of “pathogenic” and “commensal” strains has shown that they appear to be genetically similar. With a few exceptions, no definitive differences in virulence factors, traits, or genotypes were found specific to mastitis-associated E. coli (9–11). However, several identified loci (22 genes) are either enriched or implicated in mastitis-associated E. coli. These include the putative proteins ymdE and ycdU, 10 genes from the phenylacetic acid degradation operon (feaR, feaB, paaFGHIJKXY), seven genes of the ferric citrate uptake system (fecIRABCDE) (10) and eprI (11). Addition of the fec locus to a non-pathogenic dairy farm E. coli caused it to produce intramammary inflammation in dairy cows (12). Similarly, knocking out the fec locus in a mastitis-causing E. coli strain resulted in the loss of its ability to cause mastitis. Milking hygiene and wild bird transmission have been implicated as a reservoir of mastitis-associated E. coli (7). However, the relatively low genetic diversity between disease-associated and commensal strains indicates the issue is more complex. It is likely that mastitis-associated E. coli are opportunistic pathogens originating from the bovine gastrointestinal tract because the virulence-associated genes implicated in mastitis are also found in commensal E. coli strains (11).
Antimicrobial agents are important in the treatment of bovine clinical mastitis (13). The widespread use of antimicrobials to manage mastitis in herds (14, 15) creates selection for, and progressive spread of resistance through sharing of conjugative plasmids or pathogenicity islands (16) as is evident from the reports of antibiotic resistant E. coli isolates found in dairy cows with mastitis (17–19).
Whole genome sequencing of E. coli isolates from mastitis cases provides valuable information regarding mobile genetic elements, antimicrobial resistance (AMR), or virulence traits and can contribute to epidemiological investigations (20–22). Incorporation of this information into targeted surveillance programs that aim to improve antimicrobial stewardship can reduce the risk of AMR.
The versatility of E. coli genomes allows for the acquisition of different combinations of virulence factors or divergent clades that are associated with disease of various severity (10, 11, 23–27). Evidence of geographical source variation of E. coli isolates, particularly for E. coli O157, exist (28–30). However, there is paucity in the literature investigating the possible geographical disposition of the genetically diverse E. coli isolates associated with mastitis. Therefore, the main objectives of this study were to examine and genomically characterize and compare E. coli isolates cultured from dairy cows with clinical mastitis in three distinctly different geographic and climatic regions of Australia to identify antibiotic resistance, virulence factors, and mobile genetic elements which could then inform the agricultural antimicrobial stewardship programs. A secondary objective was to examine the diversity of the isolates, in particular their specific antimicrobial resistance profiles, from these different regions to identify geographic or climatic associations that may exist as a potential basis for development of more effective and sustainable dairy mastitis treatments.
Materials and Methods
Sample Collection
This cross-sectional study was conducted between March and June 2019 using milk samples (n = 430) collected from 29 dairy herds located in Queensland (North Queensland [NQLD], n = 9 herds; Southeast Queensland [SQLD], n = 9 herds), and Victoria ([VICT], n = 11 herds) (Figure 1). Herd selection was based on willingness of dairy producers to participate, ease of access to the farm location, and the cooperation of the producer's associated veterinary practice. The study was conducted in accordance with the University of Queensland Animal Ethics and National Guidelines (animal ethics approvals: SVS/ANRFA/540/18 and SVS/043/18/TERRAGEN).
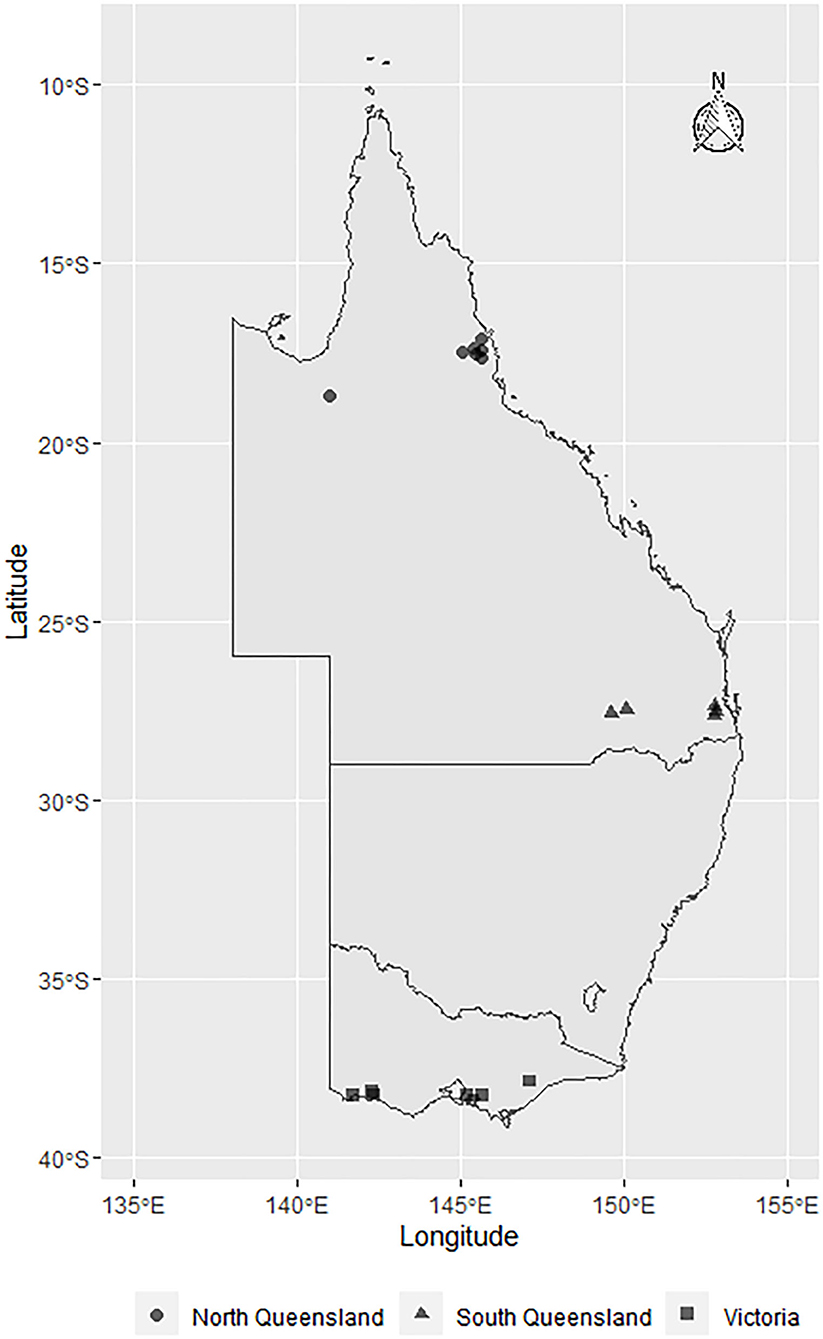
Figure 1. Map showing the geographical distribution of E. coli isolates sourced from farms across Australia: North Queensland farms (circles), South Queensland farms (triangles), and Victorian farms (squares). The symbols represent the townships centroids were the farms are located.
Milk samples were collected from eligible dairy cows with clinical mastitis. An enrolment eligible case of clinical mastitis case was defined as an apparently healthy lactating dairy cow of any age, breed, and stage of lactation that was experiencing a new clinical mastitis at the time of enrolment in the study and had not received systemic or intramammary antimicrobials, anti-inflammatory medications, or topical treatments in the 2 weeks prior to developing mastitis. Chronic mastitis cases (apparently healthy cow with lumps palpable in the udder, and mild changes to milk) and subclinical mastitis cases (apparently healthy cow with no observable changes in the udder, and significant elevated SCC) were not eligible for enrolment. A new clinical mastitis event was defined as either the first occurrence of a mastitis event in the current lactation or a mastitis event occurring at least after 21 days following a previous mastitis event that has clinically resolved or achieved a clinical cure (31).
Milk samples were collected aseptically from individual quarters after teats were cleaned. Briefly, each sampled teat was washed dried using a single-use paper towel and the teat end scrubbed with 70% ethanol until clean. Two to three foremilk streams were discarded before ~30 mL of milk was collected into a sterile tube which was immediately capped and placed in −20°C freezer. Collected samples were delivered frozen to the veterinary microbiology laboratory of the University of Queensland for bacterial culture.
Escherichia coli Isolation
Standard microbiological methods (gram stain, viable total plate count using Sheep Blood Agar (SBA, P2133 Sheep Blood Columbia Agar Plates, Thermofisher) and total viable gram negative count using MacConkey agar (MCA, PP2130 MacConkey No 3 Agar Plates, Thermofisher) were used to quantify the microorganisms present in all of the milk samples. Assessment of the haemolysis pattern (presence or absence of a clear haemolysis zone; E. coli 46B, 115C, and 143B were haemolytic) and biochemical tests were used to further characterize and identify the cultured bacteria. Individual colonies were sub-cultured on SBA and MCA plates and incubated aerobically at 37°C for 18–24 h. Pure isolates were then incubated in 2 mL of Brain Heart Infusion (BHI) broth, subsequently mixed with 20% glycerol and stored at −80°C. Matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS; Bruker™ Daltonik, Bremen, Germany) was then used to confirm the identification.
DNA Extraction
E. coli isolates were cultivated in BHI broth (37°C, orbital shaker at 300 rpm), 18–24 h. Genomic DNA was extracted using DNeasy PowerFood Microbial Kit (QIAGEN) with minor modifications. Eight mL of culture liquid was centrifuged (15 min at 4°C, 20,000 × g) to pellet the bacteria. The pellet was resuspended in 450 μL lysis buffer and incubated for 10 min at 65°C. Thereafter, the whole component was transferred to the Powerbead tubes and secured horizontally to a vortex adapter (Vortex-2 Genie®) and vortexed at a maximum speed for 10 min. After washing steps to remove protein and other inhibitors, purified DNA was eluted and the cconcentration and purity of the isolated genomic DNA evaluated using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). A sample of DNA was considered acceptable if the A260/280 ratio was ~1.8.
Whole Genome Sequencing
The New England Biolabs NEBNext Ultra II FS DNA Library Prep Kit for Illumina for multiplexing was used according to the manufacturer's instructions (Illumina) to construct whole genome sequencing libraries. Libraries were sequenced (Illumina NextSeq 500 instrument, 300 cycle mid-output kit; 2 × 150 bp paired end) to obtain an average coverage depth of >100 and a read retention of >98%. The draft genome sequences of the 24 E. coli isolates have been deposited with GenBank under Bio project PRJNA644956.
Whole Genome Assembly and Annotation
Primer sequences were removed and reads were quality trimmed using cutAdapt (32). The Nullarbor pipeline (33) was used to process the samples. SKESA assembler (version 2.3.0) (34) was used for de-novo assembly. Assemblies were annotated using prokka (version 1.14.5) (35) for loop with the following command.
for file in *.fna; do tag=$file%.fna; prokka –prefix “$tag” –locustag “$tag” –genus Escherichia –strain “$tag” –outdir “$tag”_prokka –force –addgenes “$file”; done.
Strain Typing
Assemblies were taxonomically assigned, Multi Locus Sequence Type (MLST) (36, 37) and scored for genome distance using Type (Strain) Genome Server (TYGS) (https://tygs.dsmz.de) (38) (date accessed: 19/5/2020). DNA-DNA hybridization values were measured based on formula 2. Pairwise comparison of genome sequences among the set of genomes were conducted by calculating precise distances using the Genome BLAST Distance Phylogeny approach (GBDP) under the algorithm “coverage” and distance formula d5 (39). These distances were used to determine the genome similarities for each of the genome pairs. 100 distance replicates were calculated each. Digital DDH values and confidence intervals were calculated using the recommended settings of the GGDC 2.1 (39).
Pangenome Analyses
Roary version 3.13.0 (40) was used for pangenome analysis, with the following arguments.
roary -e –mafft -i 90 -v -z -s -o
This list was then annotated and characterized via screening sequences against NCBI and Gene Ontology (41) using the PANNZER2 server (42). Signal sequences were obtained from the core genome using SignalP 5.0 (Linux x86_64) (43, 44), with the following arguments.
signalp -fasta input.fasta -format short -mature -org gram- -verbose.
Phylogenetic Analysis
A core genome alignment generated by roary was performed, including an outgroup Escherichia fergusonii ATCC 35469 (accession GCA_000026225.1) and ingroup E. coli K-12 MG1655 (accession GCF_000005845.2). RAxML (raxmlHPC-PTHREADS-SSE3 version 8.2.10) (45) was used with the following parameters, for maximum likelihood and rapid bootstrap analysis with 1,000 replicates.
raxmlHPC-PTHREADS-SSE3 -f a -x 123 -p 123 -N 1,000 -m GTRGAMMA -O -n.
The bipartitions output file was visualized in FigTree version 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) and bootstraps visualized. Inkscape version 0.92 (https://www.inkscape.org) was used to prepare the figure.
Plasmid Identification and Analysis
Mob-suite version 3.0.0 (46) was used to identify plasmids using the mob recon function, then visualized in ClustVis (https://biit.cs.ut.ee/clustvis/) (47). No scaling was performed on the data and clustering was performed on rows and columns using the “correlation” distance measure. Putative mobilization of plasmids was determined using the mob_typer function.
Identification of Putative Virulence Factors
ABRicate version 1.0.1 (https://github.com/tseemann/abricate) (48) was used, along with the virulence factor database (VFDB) (49) (date accessed 1/5/20) and a custom gene list identified from the literature (Supplementary Data Sheet 3), to identify the presence of putative virulence factors in E. coli. Both chromosomal and plasmids were analyzed.
Antibiotic Resistance Determination
Antimicrobial resistance prediction was performed separately on plasmid and chromosomal contigs using AMRFinderPlus version 3.8 (50). Database 2020-05-04.1 was used.
Bacteriophage and Prophages Identification
The PHASTER (https://phaster.ca/) (51) web server was used to identify bacteriophage and prophage regions using the API tool. “Questionable” phages (score of 70–90) were grouped with “incomplete.”
Transposon/Insertion Sequence Identification
ISESCAN version 1.7.2 (https://github.com/xiezhq/ISEScan) (52) was used to identify transposons and insertion sequences.
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas Identification
CRISPRCasFinder (https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index) (53) was used to identify CRISPR-Cas regions within the genomes.
Statistical Analysis
Fisher's exact test was performed to identify significance in the proportion of strain types, virulence genes, and plasmids from isolates recovered from NQLD, SQLD and VICT. A two sided p-value obtained by Monte-Carlo simulation (n= 2,000) of at least 0.05 was considered to be significant. Statistical analysis was conducted using stats package implement in R (54).
Results and Discussion
E. coli Mastitis Isolates, Sequence Types, and Phylogenetic Relationships
In this multi-regional Australian study, E. coli bacteria were isolated from dairy cows with clinical mastitis (Figure 1). There were 10 isolates from 8 farms in VICT, 9 isolates from 7 farms in SQLD, and 5 isolated from one farm in NQLD utilized in this study.
A total of 24 isolates were sequenced and confirmed as E. coli (Table 1). A variety of MLST sequence types (ST) were found. The most common strain type was ST10 (38%), followed by ST4429 (13%). The frequency of ST10 within these isolates was consistent with previously published findings (11). However, not all E. coli ST10 are mastitis-associated E. coli. Some ST10 can also be isolated as commensals from the gastrointestinal tract (11). The remaining STs had only one occurrence within this dataset which reflects the degree of genetic diversity of mastitis-associated E. coli (11, 55, 56).
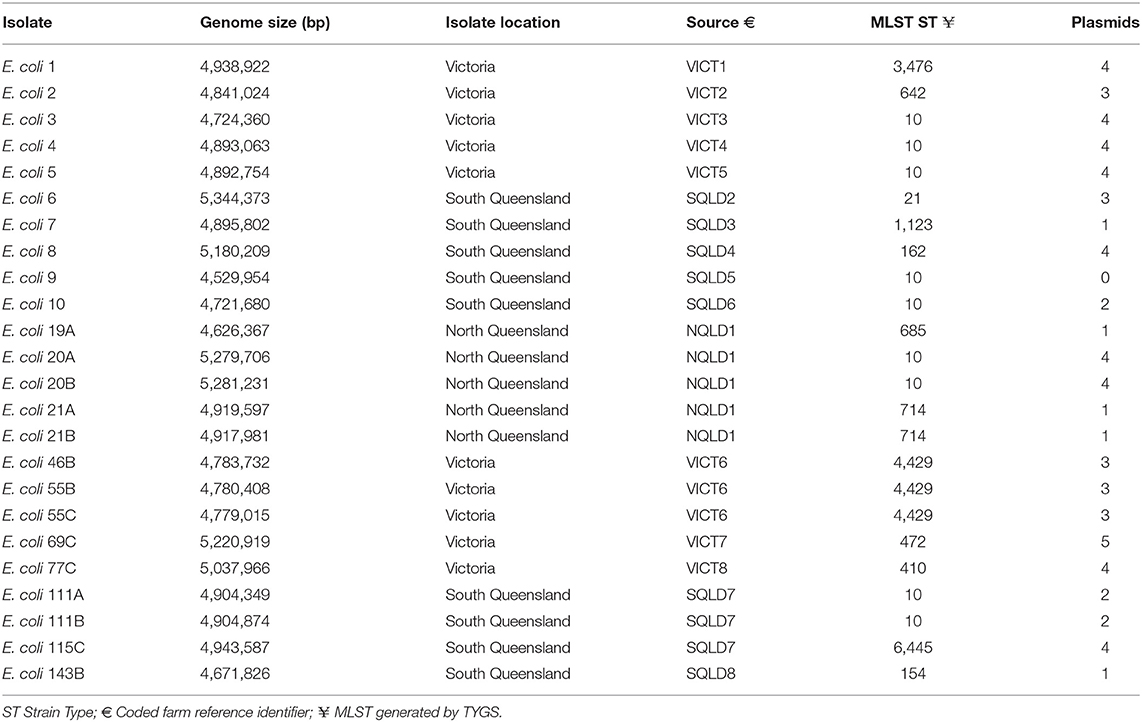
Table 1. Escherichia coli isolates used in this study.
A phylogenetic tree based on a core genome alignment was generated (Figure 2). The high bootstrap values reflect the robustness of the E. coli MLST scheme and the well-clustered strain types. There was no relationship between ST and location (p = 0.51) with ST10 being found in all study regions. E. coli isolates from within the same farm tended to be clonal as they produced identical alignments. This is evident at VICT6, NQLD1, and SQLD7. It is notable that VICT4 and VICT5 were also identical, despite being isolated from different farms. There was no notable clustering between NQLD, SQLD, and VICT (Figure 2). This finding is further supported by the genome-to-genome comparisons (Table 2). Genome-to-genome comparisons were made for all isolates according to their genetic properties or pairs from one region. The calculation of the intergenomic distance and DNA-DNA hybridization (Table 2) showed that most genomes belong to the same genomic subspecies (results not shown). Lower DNA-DNA hybridization values can be accounted for due to presence of a diverse accessory genome, (Table 3). Isolates NQLD 19A, NQLD 20A, VICT 1, VICT 2, VICT 4, VICT 10, VICT 46B, VICT 55C, VICT 69C, VICT 77C, SQLD 5, SQLD 6, SQLD 7, SQLD 8, SQLD 21B, and SQLD 1438 were similar but slightly spaced from each other.
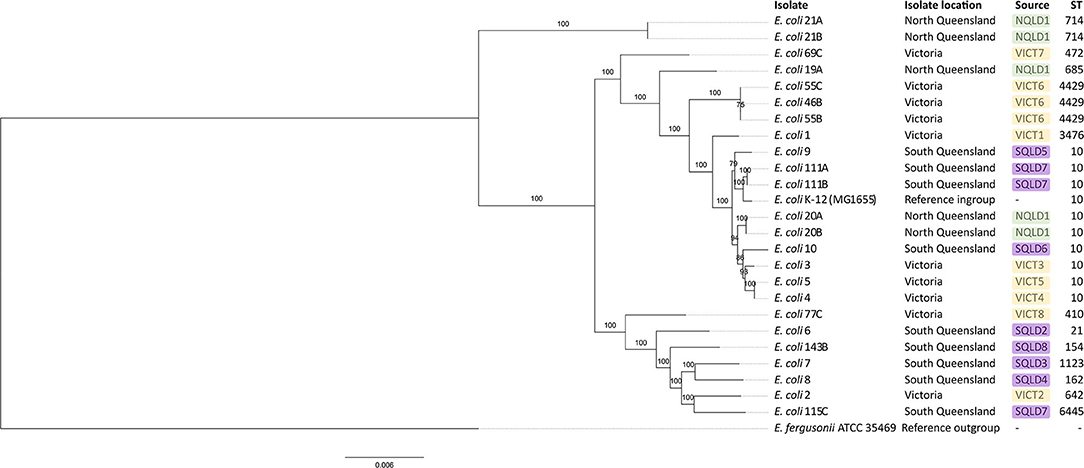
Figure 2. Maximum likelihood phylogenetic tree based on core genome of E. coli isolates identified in this study. Escherichia fergusonii ATCC 35469 was used as an outgroup and E. coli K-12 (MG1655) was used as an ingroup. Bootstrap values are shown on branches. Green labels show North Queensland isolates. Purple labels show South Queensland isolates. Yellow labels show Victorian isolates. ST stands for sequence type. Core genome alignment was performed in roary and maximum likelihood tree was built in RAxML with 1,000 replicates. Tree visualized in FigTree.
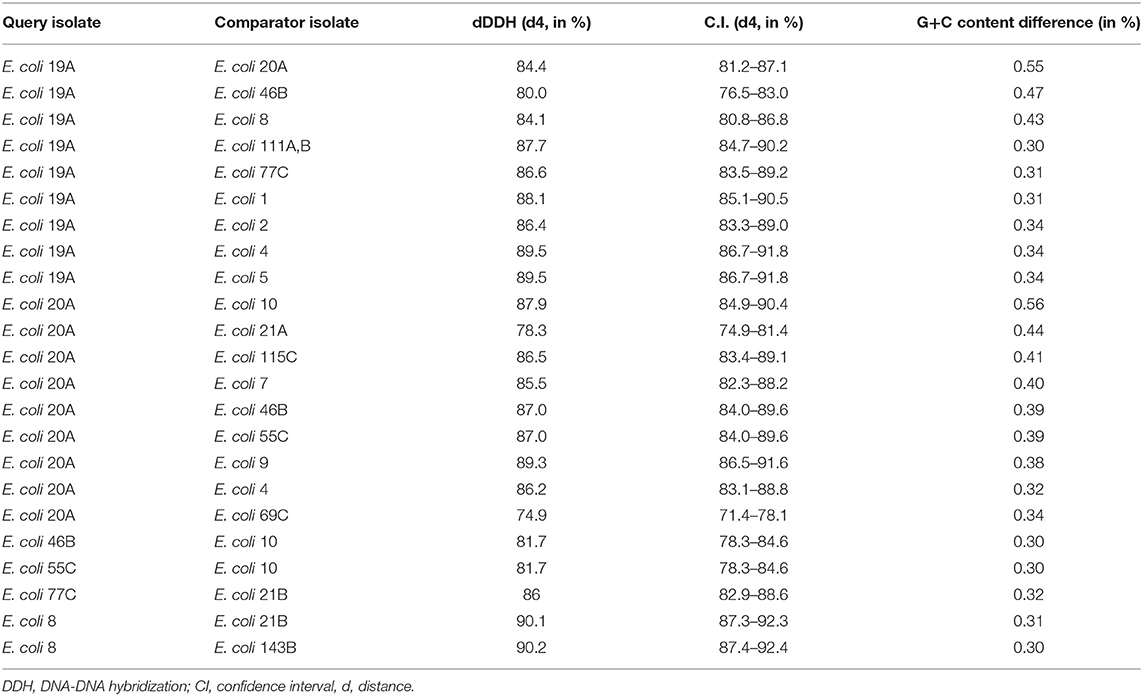
Table 2. Genome-to-genome comparison; pairwise analysis of Escherichia coli genomes.
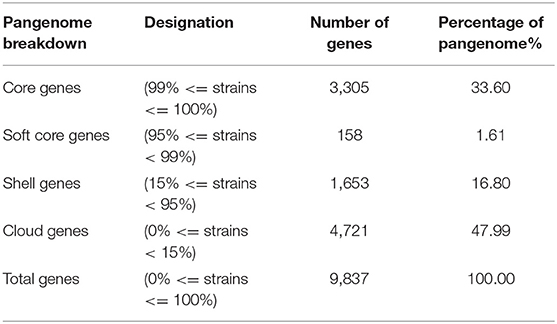
Table 3. Pangenome Analysis showing the total number of genes and percent of total pangenome.
Pangenome Analysis, Gene Content, and Chemotaxis
To uncover putative virulence factors in genes shared by all isolates, pangenomic analysis was performed. A pangenome of 9,837 genes was found, with a core genome of 3,305 genes (27.69%) (Table 3). This was consistent with previous studies of mastitis-associated E. coli which found 3,492 core genes from 66 isolates (10), 3,842 core orthologous groups from eight isolates (11), 1,976 genes from 20 isolates (55), and six fecal isolates (with a 70% rather than 90% cut-off) (11).
The core and soft core genome (3,463 genes) were further analyzed to identify any notable characteristics. Of the soft core genome, 3,166 out of 3,463 genes (91.4%) could be assigned a Gene Ontology (GO) identity and descriptors. This reflected the high degree to which E. coli has been studied and characterized. The most abundant localization of proteins appeared to be an integral component of the membrane (790), followed by cytoplasm (544) (Figure 3, Supplementary Data Sheet 6).
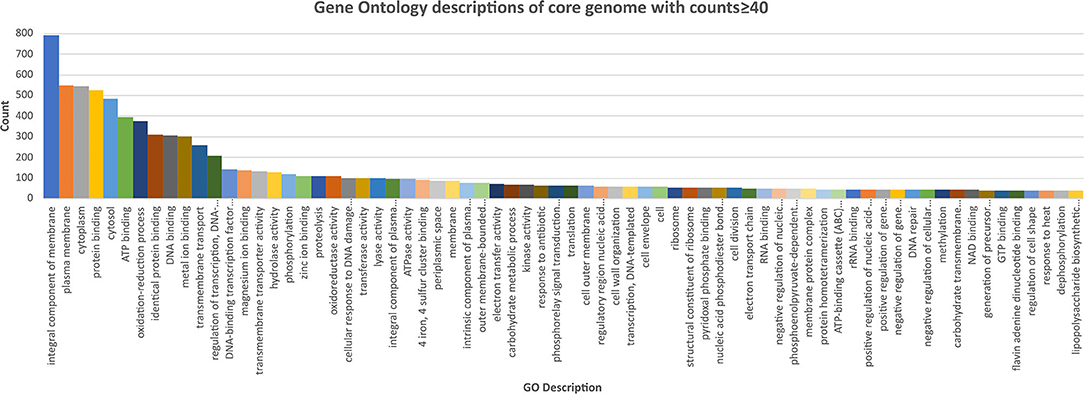
Figure 3. Chart of the most abundant Gene Onology (GO; core genome with counts ≥40) descriptions of the core genome of the E. coli isolates, ranked highest to lowest. Further GO annotations can be found in Supplementary Data Sheet 6.
Several genes were worthy of note. These were genes associated with response to antibiotics (74), chemotaxis (13), bacterial-type flagellum-dependent cell motility (28), cellular adhesion (17), biofilm formation (11) and pilus (14). No microcins, colicins or other bacteriocins were identified in the pangenome. Out of the soft core genome, 452 were predicted to be secreted (Supplementary Data Sheet 6), 37 were associated with proteolysis, 15 with cell adhesion, 14 with pilus, and 8 with heme binding. To further investigate the behavior of mastitis-associated E. coli, the core genome was screened for chemotaxis-associated genes. The screening included genes encoding maltose/maltodextrin binding (malE), flagellar genes (fliGJLMNO), signal transducer for aerotaxis sensory (aer), D-galactose/D-glucose-binding (mglB), heme binding dipeptide ABC transporter (dppA), NAD-dependent protein deacylase/protein desuccinylation (cobB), D-ribose substrate-binding (rbsB), methyl-accepting chemotaxis protein II (tsr), methyl-accepting chemotaxis protein (trg), and acetyl-coenzyme A synthetase (acs). A negative chemotaxis nickel-binding gene (nikA) was also identified. These chemotaxis-associated-genes may play a role in the pathogenesis of mastitis-associated E. coli and partially explain the presence of E. coli within the mammary glands. D-galactose and glucose are the components of lactose, the main sugar present in bovine milk, suggesting a beneficial role for the mglB gene. Similarly, the presence of the aer aerotaxis gene is consistent with E. coli's preference of oxygen (presumably at higher levels when compared to the relatively anaerobic bovine gastrointestinal tract) which may also make milk and the mammary gland a favorable bacterial environment. The presence of dppA, (encodes a heme-binding protein) in combination with the core hemolysin gene (ytjB) may favor a tissue environment (over the gastrointestinal tract) because of the proximity to hemoglobin-containing red blood cells.
Plasmids, Mobility, Virulence, and Antimicrobial Resistance
A total of 36 different plasmids were identified based on mash designations from mob-suite (Figure 4). The plasmid distribution pattern was associated with geographical location (p = 0.02). Of the 36 plasmids, 50% (18/36) were unique to VICT isolates, 28% (10/36) unique to SQLD, and 6% (2/36) unique to NQLD. A number of plasmids were shared between study regions: 6% between VICT and SQLD (2/36), 6% between VICT, SQLD and NQLD (2/36), and 6% between SQLD and NQLD (2/36). Supplementary Data Sheet 1 contains further information along with accession numbers of the most similar plasmids. Bacterial strains contained 0–5 plasmids, with E. coli 9 containing no plasmids, and E. coli 69C containing 5. The origins of the plasmids varied: 26 were E. coli plasmids, 8 were Salmonella enterica plasmids, 1 was a Shigella flexneri, and 1 was a Shigella boydii plasmid.
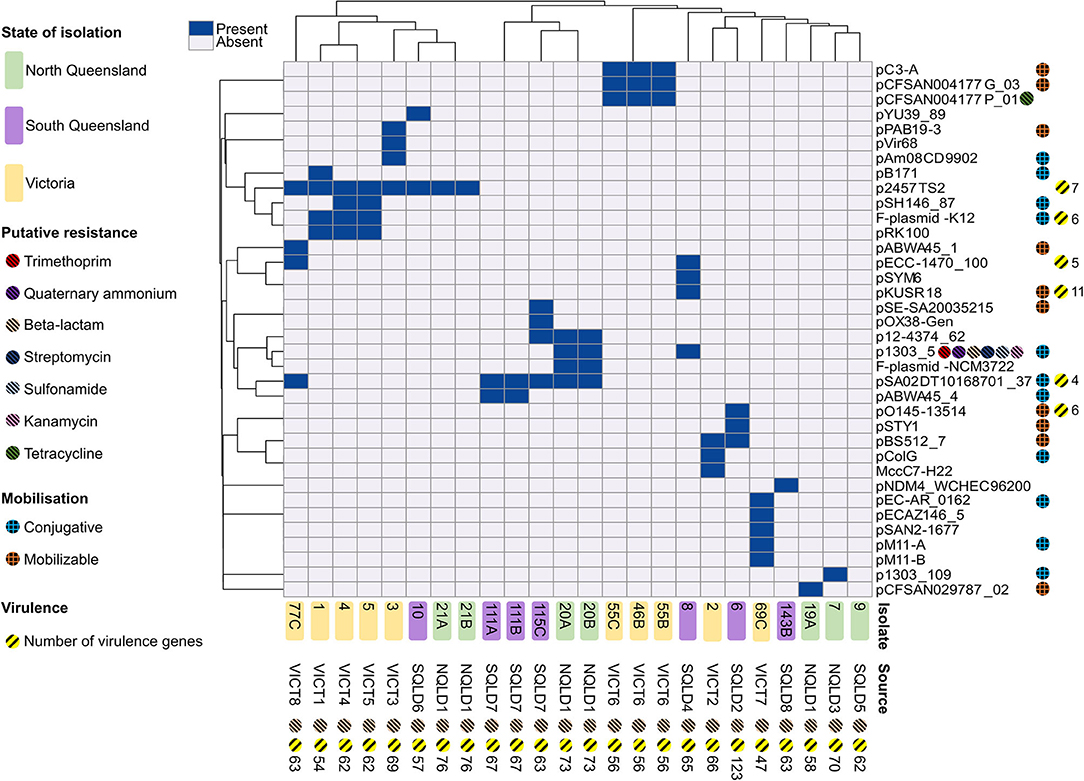
Figure 4. Heatmap of plasmid presence/absence within each E. coli isolate based on the correlation between isolates (x-axis) and plasmids (y-axis). Dark blue rectangles indicate presence of a corresponding -like plasmid. Isolate 9 contained no plasmids and was excluded from the heatmap. Victorian isolates are colored in yellow, North Queensland in green, South Queensland in purple. Plasmid elements are shown on the right y-axis. Chromosomal elements are shown along the x-axis. Putative resistance genes are shown for plasmids and chromosomal DNA for each isolate, represented by striped circles; red shows trimethoprim, purple show quaternary ammonium, peach shows beta-lactam, dark blue shows streptomycin, light blue shows sunfonamide, pink shows kanamycin, green shows tetracycline. Putative plasmid mobilization represented by hashed circles; blue shows putatively conjugative, orange shows putatively mobilizable. Yellow-striped circles and the corresponding number represent presence and number of putative virulence factors. Heatmap produced using ClustVis.
The most commonly identified plasmid (p2457TS2) was found in eight isolates across Victoria and Queensland. In general, there appeared to be a local geographical relationship between the specific isolation farm of a strain and the plasmids it carried (p < 0.001). Isolates from VICT6 all shared the same three plasmids. Two of four isolates from the NQLD1 farm shared four plasmids. Victorian isolates from VICT6, VICT 7, and VICT 8 shared three plasmids, and four out of VICT1–VICT5 isolates contained the p2457TS2 plasmid. VICT4 and VICT5 both shared the same four plasmids, despite being from different farms.
Plasmids were examined for putative virulence factors by screening against the VFDB and a custom database (Supplementary Data Sheet 3) created from previous genomic analyses (10–12). Putative virulence factors were present in 16.67% of plasmids (Table 4). A notable feature found in E. coli isolates E. coli 4, E. coli 5, E. coli 6, and E. coli 77C was the presence of RTX toxin cluster which included the hemolysin genes hlyABCD. HlyA has been shown to enhance the pathogenicity of extra-intestinal E. coli (ExPEC). A similar RTX toxin, TosA, has been implicated in uropathogenic E. coli (UPEC) (9, 55, 57). It is therefore possible that the RTX cluster present in these isolates is associated with or directly produces a cytotoxic effect in the mammary gland leading to tissue injury, damage and subsequently to the clinically observed signs of peracute mastitis observed in dairy cows infected by this strain. E. coli 4 contained two plasmids (p2457TS2-like and F-plasmid-K12-like) which contained virulence genes fecIRABDE; yjhV, and faeCDEFHI.
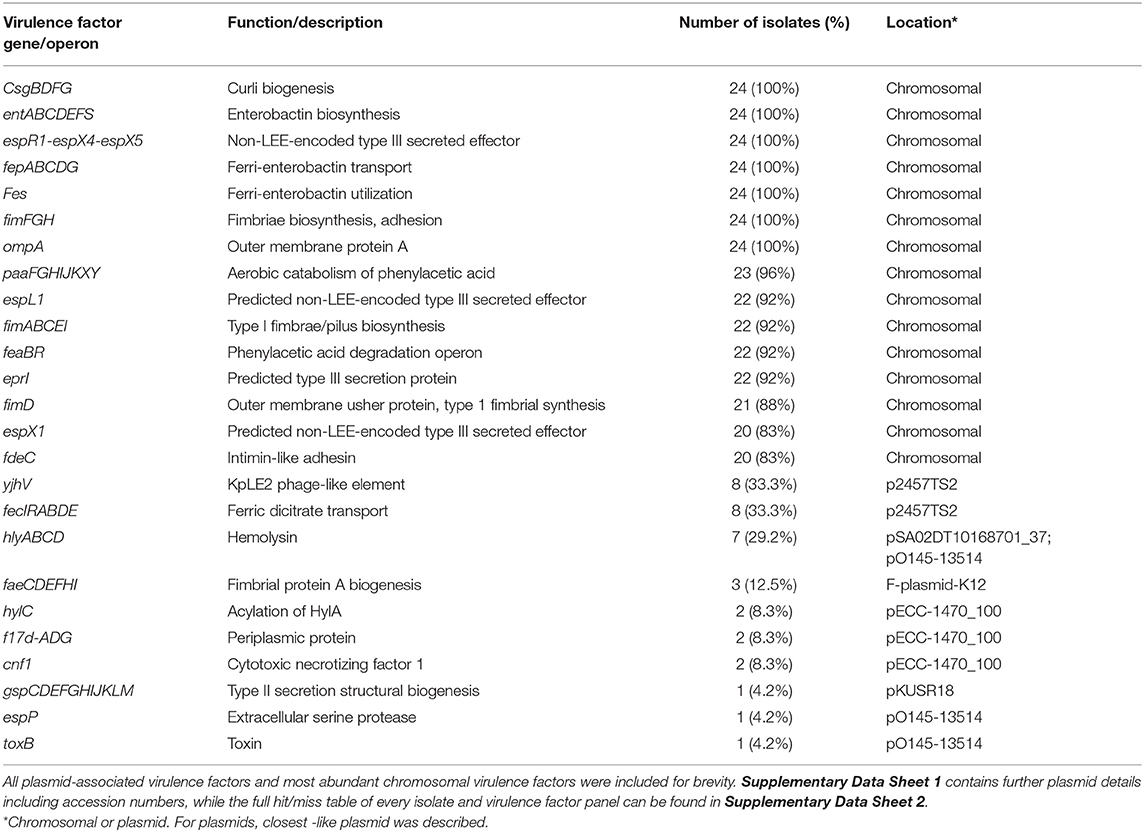
Table 4. Summary selection of virulence factors from bovine mastitis-associated E. coli isolated in this study.
Only a few antibiotic resistance genes were found in these isolates, with the exception that all isolates contained the beta-lactam resistance gene blaEC within their chromosome (Figure 4, Supplementary Data Sheet 2). Antibiotic resistance genes were located primarily in plasmids which correlated with the accessory genome from the pangenomic results. Two plasmids were identified with antimicrobial resistance genes, the p1303_5-like and the pCFSAN004177P_01-like plasmids. The p1303_5-like plasmid was found in two isolates from NQLD1 in North Queensland. It is a multi-drug resistant plasmid, containing trimethoprim, quaternary ammonium, beta-lactam, streptomycin, sulfonamide and kanamycin resistance genes. The pCFSAN004177P_01-like plasmid was found in 3 isolates from VICT6 in Victoria and contained a tetracycline resistance gene. With consideration to this limited dataset, the pattern is consistent with a local spread of antibiotic resistance through plasmids of bovine mastitis cases.
This analysis was limited by the small sample size. Larger sampling may uncover geographical relationships, if any exist. Furthermore, the proposed larger study should be combined with long-read genomic sequencing to allow analysis of complete plasmids. Plasmid lineages could be directly analyzed with less ambiguity.
Conjugative plasmids contain all the genes required for self-transfer. Whereas, mobilizable plasmids only contain a portion of these components, such as relaxosomal components oriT comprised of a relaxase gene and nicking proteins (58). In the presence of conjugative plasmids, mobilizable plasmids can piggyback off conjugative machinery to spread throughout a population.
To examine this possible mechanism of spread, plasmids were screened for presence of putative conjugative and mobilizable elements to predict potential mobility. Eleven (11) plasmids were conjugative, 10 were mobilizable, and the remaining 15 were non-mobilizable (Figure 4). Mobilizable plasmids were geographically clustered (p < 0.03). Four plasmids from six isolates were unique to VICT, three plasmids from three isolates were unique to SQLD while two plasmids from two isolates were unique to NQLD. Of all the E. coli isolates, 7 (29.17%) contain both putative conjugative and mobilizable plasmids which could allow the spread of mobilizable plasmids throughout the microbial community (Supplementary Data Sheet 1). If conjugative plasmids are able to spread throughout the communities and are acquired by strains which also contain mobilizable plasmids, this may assist in the communal dissemination of these mobilizable elements. Ongoing strain isolation and genomic surveillance should be undertaken to monitor this relationship and the potential risk.
Plasmid mobility did not co-occur with putative antibiotic resistance genes or virulence plasmids, except in 5/36 plasmids. However, these plasmids were found in a total of 10/24 isolates, indicating 41.7% contained mobilizable plasmids with associated virulence factors. The pKUSR18-like plasmid was putatively mobilizable and contained 11 virulence factors, found in a single isolate (E. coli 8) (Figure 4).
The p1303_5-like plasmid is of particular note. It is a multidrug resistant plasmid carrying six different antibiotic resistance genes and conjugative elements which creates the potential for spread throughout a population. This plasmid was found in 2 of 5 NQLD1 isolates and in the SQLD4 isolate suggesting that spread was occurring. Those 3 isolates contained multiple antibiotic resistance and more virulence genes compared to isolates which carried a single antibiotic resistance gene, a finding compatible with the previous studies (59–61). Moreover, tetracyclines and sulphonamides are preferred antibiotics for treatment of mastitis caused by gram-negative pathogens (62, 63). Internationally, it has been shown in Turkish cow herds where mastitis is managed with the fluoroquinolones (danofloxacin and enrofloxacin), E. coli was shown to have notable resistance to these antibiotics (61). In China, mastitis-associated E. coli have been shown to have notable resistance to sulfamonomethoxine and sulfamethoxazole, both used in management of intestinal infections (19). In contrast, within USA, antibiotic resistance has been shown to not correlate with antibiotic usage between 1985 and 1987 compared to 2009 (15).
Therefore, this observation may be associated with the widespread use of these antibiotics as part of a generic approach to the treatment of mastitis in cows, or farms not practicing herd culling (62–64), which may explain the presence of this multidrug resistant plasmid. However, 3 out of 5 NQLD isolates did not contain the plasmid. There may be an evolutionary disadvantage and energy burden associated with maintaining and expressing genes from this large 270,000 bp plasmid. In addition, it is also possible not every member of the microbial community must carry this plasmid for its effect to be mutually beneficial to the p1303_5-negative strains (65, 66). The pSA02DT10168701_37-like plasmid was both putatively conjugative and contained 4 virulence factors. It was found in 25% of all isolates, 3/3 from the SQLD7 farm, 2/4 from the NQLD1 farm and 1/1 from VICT8. The pO145-13514-like plasmid was putatively mobilizable and contained six virulence factors.
Antibiotic resistance-containing non-mobilizable plasmids such as the tetracycline resistance pCFSAN004177P_01-like plasmid was found in three isolates from the VICT6 farm, confirming clonality between these 3 isolates. Management of mastitis at VICT6 included a number of first-line antimicrobial treatments (62–64) providing selection pressure likely to be the responsible for the spread of the tetracycline resistance pCFSAN004177P_01-like plasmid at this farm (17–19). E. coli are frequently associated with mobile genetic elements containing AMR genes that have the potential to carry resistance to antimicrobials that are of importance in human medicine (67). As a member of the Enterobacteriaceae family, E. coli are omnipresent bacteria capable of rapidly mounting resistance to almost all first-line antibiotics (68). Our results are consistent with published studies on Enterobacteriaceae isolated from cattle (17, 19) and likely stem from widespread use or misuse of antimicrobials (26, 27, 69), selection pressure or horizontal gene transfer from intrinsically resistance commensals (19, 70).
The most common and also putatively non-mobilizable p2457TS2-like plasmid (8/24 isolates) contained the fec locus which consisting of 7 putative virulence factors. Knockout studies in a bovine mastitis disease model have demonstrated the fec locus to be an essential virulence factor (12). It is noteworthy that when the fec locus was not identified in a plasmid, it was detected in the chromosome of 10 isolates, a population representing 75% of all isolates. It is possible this locus is not required by every isolate, and mosaic presence within a community may be enough to cause disease.
Chromosomal Virulence Factors
Previous comparative genomic analysis concluded highly similar virulence factor profiles are present in both mastitis-associated E. coli isolates and the commensal E. coli present in the enteric microflora. The finding indicates these genes provide a selective advantage in the gastrointestinal tract and defines mastitis-associated E. coli as an opportunistic pathogen (11). To examine the isolates identified in this study, chromosomes were screened for putative virulence factors using the VFDB and a custom database (Supplementary Data Sheet 3).
Twenty-four genes across 8 chromosomal loci were identified as core virulence factors (Table 4). This included the csgBDFG, entABCDEFS, espR1-espX4-espX5, fepABCDG, fes, fimFGH, and ompA genes. fimFGH genes are associated with epithelial cell adherence. However, knocking out fimH demonstrated it is important but not essential in the adherence of E. coli (71). The chromosomal paaFGHIJKXY locus was present in 23/24 strains. It was absent from E. coli 9, which was also the only strain to not contain a plasmid. Virulence factors fimABCDEI and espL1- espX1 were found in ≥92% of strains. Previously implicated mastitis-associated virulence factors, feaBR (10) and eprI (11) were found in 92% of isolates.
Isolate E. coli 6 contains the most virulence factors, at 129, including 6 found on the putatively mobilizable pO145-13514-like plasmid, while E. coli 69C contains the least at with 47 (Supplementary Data Sheets 1, 2 contain the full virulence factor hit-miss tables). On average, isolates contained 70.25 ± [standard deviation; σ = 15.42] putative virulence factors.
Prophages and Mobilizable Elements
The E. coli isolates contained a total of 26 distinct intact prophages (Figure 5, Supplementary Data Sheet 4). The E. coli 9 isolate was unusual in that it appeared to be missing several characteristics shared by all other strains. It lacked plasmids, the chromosomal paaFGHIJKXY locus, and intact chromosomal prophages. Enterobacteria phage P88-like prophage was the most commonly found present across a number of sites in both Victoria and Queensland, located in nine strains and twice within E. coli 2. There appears to be a limited local geographical correlation between intact prophages within strains at the farm level, while there is no discernible correlation across the 3 study regions. Three out of 3 VICT6 Victorian strains contained 2 prophages unique to them and 2/5 NQLD1 isolates shared 4 prophages. The Salmonella phage SJ46-like prophage (NC_031129) was found on the putatively mobilizable pCFSAN004177G_03-like plasmid in all three VICT6 isolates, while the remaining intact prophages were found on the chromosome (Supplementary Data Sheet 4).
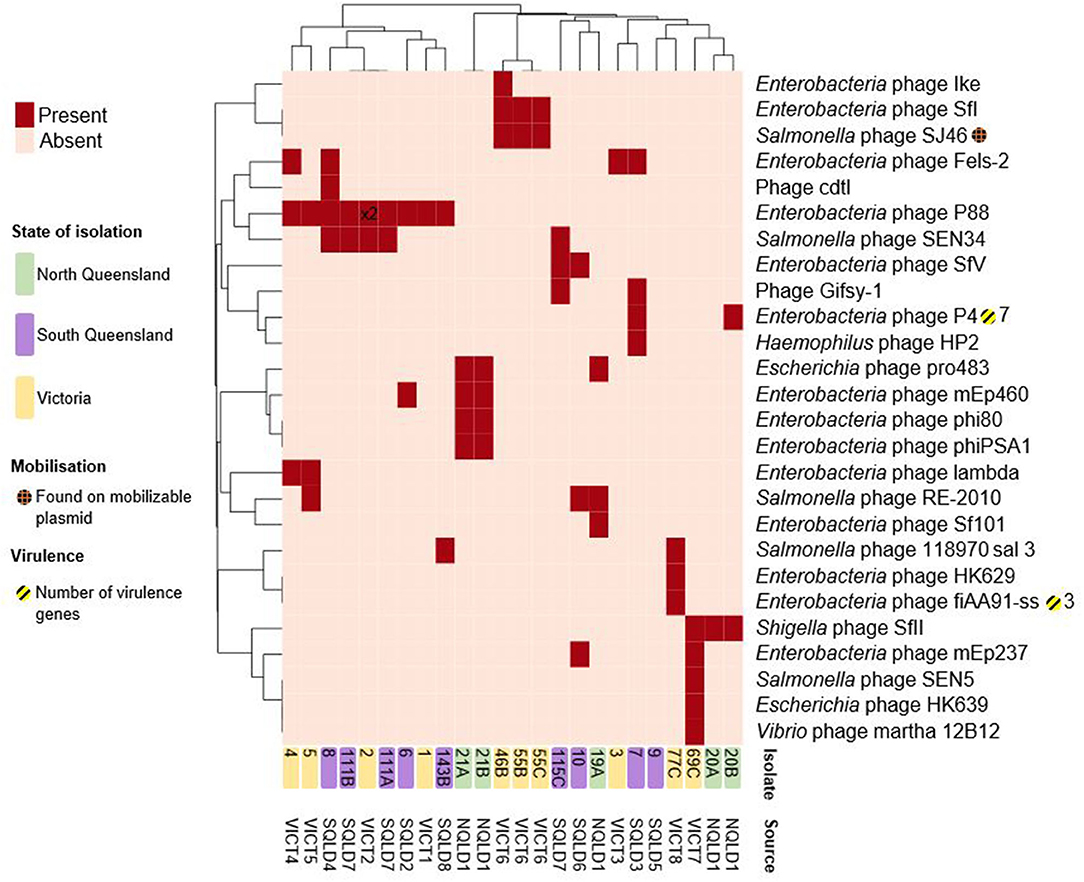
Figure 5. Heatmap of presence/absence of putatively intact prophages within the E. coli isolates. Clusters were based on correlation between prophages (x-axis) and isolates (y-axis). Dark red rectangles show hits to -like phages. Further information including accession numbers can be found in Supplementary Data Sheet 4. Clustering shown on rows and columns using the “correlation” distance measure. Victorian isolates are colored in yellow, North Queensland in green, South Queensland in purple. Presence on a putatively mobilizable plasmid represented by orange-hashed circles. Yellow-striped circles and the corresponding number represent presence and number of putative virulence factors. Number shown on heatmap (x2) indicate multiple intact prophages within the same genome. Heatmap produced using ClustVis.
These intact prophage regions were screened for virulence factors. Some prophages in E. coli have been shown to carry virulence factors (72). Amongst the 24 characterized isolates only two intact prophages had associated virulence factors. The Enterobacteria phage P4-like prophage (NC_001609), found in two Queensland isolates, carried the fecIRABDE and yjhV virulence-associated genes (Figure 5, Supplementary Data Sheet 4). The Enterobacteria phage fiAA91-ss-like prophage (NC_022750), found in one Victorian isolate, contained the cdtABC locus.
The genomes were interrogated to identify clustered regularly interspaced short palindromic repeats (CRISPR). CRISPR-Cas is a complex system that play a major role in host adaptive immune response and virulence (73, 74). They are found in most bacterial taxa, including E. coli, and also in most archaea. At least one sub-type of Cas1 gene (Cas-type I-E) was identified in 63% (15/24) isolates (Supplementary Data Sheet 7). All CRISPR-Cas systems were type I-E subtype (75), with 8 isolates across VICT, NQLD and SQLD containing intact Cas1, Cas2, Cas3, Cas5, Cas6, Cas7, Cse1, and Cse2. Isolate E. coli 1 only contained Cas2 and Cas3; signature genes that are associated with Cas1 and may not be functional due to lacking Cas1. The remaining nine isolates contained no CRISPR-Cas system genes. Among the isolates, we did not identify any Cas gene from six isolates from Victoria, one from South Queensland and 2 from North Queensland. The identified Cas from our study all belong to type 1-E sub-type. With exception to VICT6 isolate, all the remaining isolates without Cas1 gene had relatively higher virulence factors and moblisable elements (Supplementary Data Sheet 2, Figure 4). Although we did not directly determine the virulence or pathogenicity of isolates, our results are in agreement with the literature supporting the hypothesis of a negative correlation between the presence of CRISPR-Cas system and the number of predicted E.coli pathogenicity or virulence genes (76, 77). The absence or deletion of Cas1 in the 9 isolates we observed, indicates potential vulnerability to invaders or foreign nucleic acids, and possibly impaired chromosomal segregation (78).
The genomes were screened for the presence of transposons and insertion sequences (Figure 6, Supplementary Data Sheet 5). Isolates had 18.6 ± SD = 4.9 transposons within their genomes, with E. coli 6 having the most at 31, and E. coli 115C the fewest at 11. The most common family of transposons were the IS3 and ISNCY families, represented by ≥78 unique sequence across all isolates. There were few virulence factors associated with these transposable elements. astA and east1 were associated with the individual IS256-family ISs in E. coli 6, yjhV was associated with a single IS3-family IS in E. coli isolates 1 and 7.
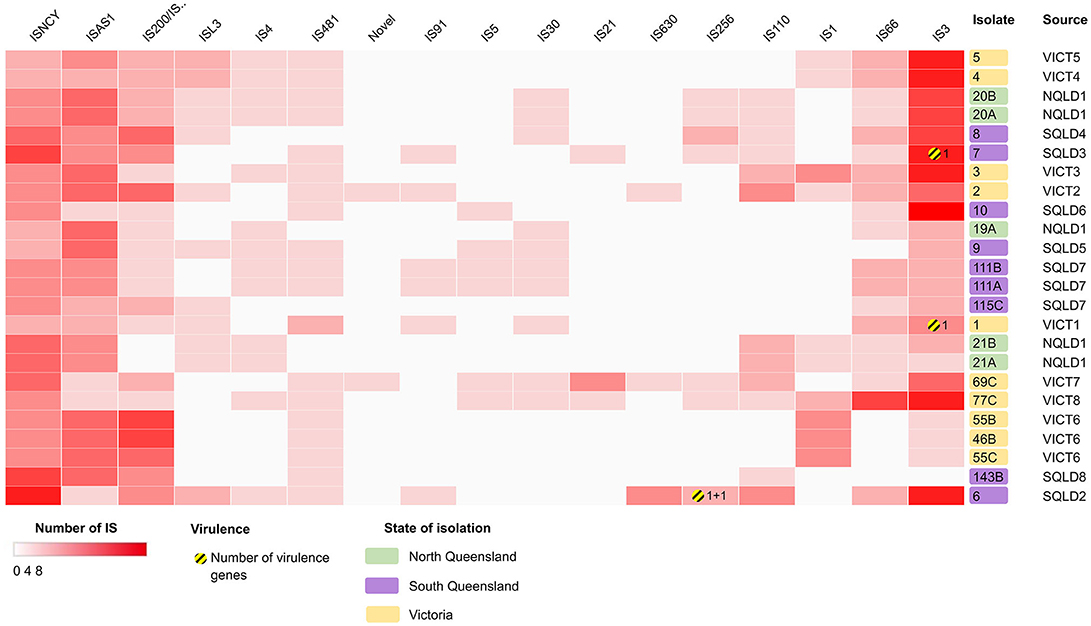
Figure 6. Heatmap showing the presence/absence of putatively intact insertion sequences (IS) within the E. coli isolates, clustered based on correlation between IS (x-axis) and isolates (y-axis). Darker red rectangles show more unique IS within the corresponding isolate. Further information can be found in Supplementary Data Sheet 5. Clustering shown on rows and columns using Euclidean correlation distance measure. Victorian isolates are colored in yellow, North Queensland in green, South Queensland in purple. Yellow-striped circles and the corresponding number represent presence and number of putative virulence factors. Heatmap produced using Clustergrammer.
Conclusions
The results in this study are consistent with prior genomic analysis of bovine mastitis-associated E. coli isolates in that there were only a few conserved core virulence factors, all of which are also present in commensal E. coli located within the bovine gastrointestinal tract. The comparison of mastitis strains collected from different geographical regions in Australia allowed the identification of common genes that could provide a partial explanation for the pathogenic effects in the mammary gland. Comparison of strains between regions demonstrated a high degree of similarity between isolates at the whole genome level. Analyses of the isolate genomes suggested there is no clear pathogenicity signature associated with the general metabolic, physiological, immunogenic, or resistance features of E. coli and the observed pathology in the E. coli infected mammary gland is not necessarily dependent on novel and unknown virulence factors specifically targeted at the mammary gland tissue. Culturing various strains of E. coli from clinical cases of bovine mastitis without reproducing disease in animal models has inherent risk. Koch's postulates are unfulfilled. Plasmids may be lost during the culture process directly affecting the resulting gene pool and subsequent conclusions from any analysis. Given there is likely no evidence for a bovine mastitis E. coli pathotype, alternative analyses such as in vivo RNA sequencing and metagenomics should be utilized to identify any relationships between gene content and bovine mastitis. Generation of such information would be invaluable for the development of diagnostic tools and provide opportunity for a greater understanding, prediction and improved decision making in the management of E. coli-associated bovine mastitis.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repositories and accession numbers can be found below:
https://www.ncbi.nlm.nih.gov/nuccore/JACCHD000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCHE000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGV000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGT000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGS000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGQ000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGR000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGN000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGO000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGP000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGM000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGL000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCHI000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCHF000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCHG000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCHC000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCHH000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCHA000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCHB000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGZ000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGY000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGW000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGX000000000
https://www.ncbi.nlm.nih.gov/nuccore/JACCGU000000000.
Ethics Statement
The animal study was reviewed and approved by The University of Queensland Animal Ethics and National Guidelines.
Author Contributions
JA: study design, sample collection, sample processing, and data analyses. BV: data analysis and interpretation. HA-H: sample collection and DNA extraction. TO: study design. HR: genome assembly and preliminary data analysis. AJ, MS, and RM: assisted with data interpretation. All authors contributed to drafting of the manuscript.
Funding
This work was funded by Science with Impact Scheme, the University of Queensland, Sub-tropical Dairy Board, Dairy Australia, and Terragen Biotech Ltd, Australia.
Conflict of Interest
MS was employed by the company Terragen Biotech Pty Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the help and cooperation of dairy farmers, veterinarians, Dr. Justine Gibson and dairy workers who helped and contributed to the current study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2020.582297/full#supplementary-material
References
1. Grohn YT, Wilson DJ, Gonzalez RN, Hertl JA, Schulte H, et al. Effect of pathogen-specific clinical mastitis on milk yield in dairy cows. J Dairy Sci. (2004) 87:3358–74. doi: 10.3168/jds.S0022-0302(04)73472-4
2. Halasa T, Huijps K, Osteras O, Hogeveen H. Economic effects of bovine mastitis and mastitis management: a review. Vet Q. (2007) 29:18–31. doi: 10.1080/01652176.2007.9695224
3. Hogeveen H, Huijps K, Lam TJGM. Economic aspects of mastitis: new developments. N Z Vet J. (2011) 59:16–23. doi: 10.1080/00480169.2011.547165
4. Erskine RJ, Walker RD, Bolin CA, Bartlett PC, White DG. Trends in antibacterial susceptibility of mastitis pathogens during a seven-year period. J Dairy Sci. (2002) 85:1111–8. doi: 10.3168/jds.S0022-0302(02)74172-6
5. Bradley AJ, Leach KA, Breen JE, Green LE, Green MJ. Survey of the incidence and aetiology of mastitis on dairy farms in England and Wales. Vet Rec. (2007) 160:253–8. doi: 10.1136/vr.160.8.253
6. Botrel MA, Haenni M, Morignat E, Sulpice P, Madec JY, Calavas D. Distribution and antimicrobial resistance of clinical and subclinical mastitis pathogens in dairy cows in Rhone-Alpes, France. Foodborne Pathog Dis. (2010) 7:479–87. doi: 10.1089/fpd.2009.0425
7. Fahim KM, Ismael E, Khalefa HS, Farag HS, Hamza DA. Isolation and characterization of E. coli strains causing intramammary infections from dairy animals and wild birds. Int J Vet Sci Med. (2019) 7:61–70. doi: 10.1080/23144599.2019.1691378
8. Leimbach A, Poehlein A, Witten A, Scheutz F, Schukken Y, Daniel R, et al. Complete genome sequences of Escherichia coli STRAINS 1303 and ECC-1470 isolated from bovine mastitis. Genome Announc. (2015) 3:e00182-15. doi: 10.1128/genomeA.00182-15
9. Blum SE, Heller ED, Sela S, Elad D, Edery N, Leitner G. Genomic and phenomic study of mammary pathogenic Escherichia coli. PLoS ONE. (2015) 10:e0136387. doi: 10.1371/journal.pone.0136387
10. Goldstone RJ, Harris S, Smith DGE. Genomic content typifying a prevalent clade of bovine mastitis-associated Escherichia coli. Sci Rep. (2016) 6:30115. doi: 10.1038/srep30115
11. Leimbach A, Poehlein A, Vollmers J, Görlich D, Daniel R, Dobrindt U. No evidence for a bovine mastitis Escherichia coli pathotype. BMC Genomics. (2017) 18:359. doi: 10.1186/s12864-017-3739-x
12. Blum SE, Goldstone RJ, Connolly JPR, Répérant-Ferter M, Germon P, Inglis NF, et al. Postgenomics characterization of an essential genetic determinant of mammary pathogenic Escherichia coli. MBio. (2018) 9:e00423-18. doi: 10.1128/mBio.00423-18
13. Bengtsson B, Unnerstad HE, Ekman T, Artursson K, Nilsson-Ost M, Waller KP. Antimicrobial susceptibility of udder pathogens from cases of acute clinical mastitis in dairy cows. Vet Microbiol. (2009) 136:142–9. doi: 10.1016/j.vetmic.2008.10.024
14. Jost BH, Field AC, Trinh HT, Songer JG, Billington SJ. Tylosin resistance in Arcanobacterium pyogenes is encoded by an Erm X determinant. Antimicrob Agents Chemother. (2003) 47:3519–24. doi: 10.1128/AAC.47.11.3519-3524.2003
15. Metzger SA, Hogan JS. Short communication: antimicrobial susceptibility and frequency of resistance genes in Escherichia coli isolated from bovine mastitis. J Dairy Sci. (2013) 96:3044–9. doi: 10.3168/jds.2012-6402
16. Boerlin P, White DG. Antimicrobial resistance and its epidemiology. In: Giguère S, Prescott JF, Dowling PM, editors. Antimicrobial Therapy in Veterinary Medicine. 5th ed. Hoboken, NJ: John Wiley and Sons, Inc (2013). p. 21–40. doi: 10.1002/9781118675014.ch3
17. Liu YX, Liu G, Liu WJ, Liu Y, Ali T, Chen W, et al. Phylogenetic group, virulence factors and antimicrobial resistance of Escherichia coli associated with bovine mastitis. Res Microbiol. (2014) 165:273–7. doi: 10.1016/j.resmic.2014.03.007
18. Fairbrother JH, Dufour S, Fairbrother JM, Francoz D, Nadeau E, Messier S. Characterization of persistent and transient Escherichia coli isolates recovered from clinical mastitis episodes in dairy cows. Vet Microbiol. (2015) 176:126–33. doi: 10.1016/j.vetmic.2014.12.025
19. Zhang D, Zhang Z, Huang C, Gao X, Wang Z, Liu Y, et al. The phylogenetic group, antimicrobial susceptibility, and virulence genes of Escherichia coli from clinical bovine mastitis. J Dairy Sci. (2018) 101:572–80. doi: 10.3168/jds.2017-13159
20. Quainoo S, Coolen JPM, van Hijum S, Huynen MA, Melchers WJG, van Schaik W, et al. Correction for Quainoo et al., “Whole-Genome Sequencing of Bacterial Pathogens: the Future of Nosocomial Outbreak Analysis”. Clin Microbiol Rev. (2018) 31:1015–63. doi: 10.1128/CMR.00082-17
21. Ludden C, Raven KE, Jamrozy D, Gouliouris T, Blane B, Coll F, et al. One health genomic surveillance of Escherichia coli demonstrates distinct lineages and mobile genetic elements in isolates from humans versus livestock. MBio. (2019) 10:e02693–18. doi: 10.1128/mBio.02693-18
22. Adator EH, Walker M, Narvaez-Bravo C, Zaheer R, Goji N, Cook SR, et al. Whole genome sequencing differentiates presumptive extended spectrum beta-lactamase producing Escherichia coli along segments of the one health continuum. Microorganisms. (2020) 8:448. doi: 10.3390/microorganisms8030448
23. Mora A, Blanco JE, Blanco M, Alonso MP, Dhabi G, Echeita A, et al. Antimicrobial resistance of Shiga toxin (verotoxin)-producing Escherichia coli O157:H7 and non-O157 strains isolated from humans, cattle, sheep and food in Spain. Res Microbiol. (2005) 156:793–806. doi: 10.1016/j.resmic.2005.03.006
24. Wick LM, Qi W, Lacher DW, Whittam TS. Evolution of genomic content in the stepwise emergence of Escherichia coli O157:H7. J Bacteriol. (2005) 187:1783–91. doi: 10.1128/JB.187.5.1783-1791.2005
25. Tenaillon O, Skurnik D, Picard B, Denamur E. The population genetics of commensal Escherichia coli. Nat Rev Microbiol. (2010) 8:207–17. doi: 10.1038/nrmicro2298
26. Suojala L, Pohjanvirta T, Simojoki H, Myllyniemi AL, Pitkala A, Pelkonen S, et al. Phylogeny, virulence factors and antimicrobial susceptibility of Escherichia coli isolated in clinical bovine mastitiss. Vet Microbiol. (2011) 147:383–8. doi: 10.1016/j.vetmic.2010.07.011
27. Lan T, Liu H, Meng L, Xing M, Dong L, Gu M, et al. Antimicrobial susceptibility, phylotypes, and virulence genes of Escherichia coli from clinical bovine mastitis in five provinces of China. Food Agr Immunol. (2020) 31:406–23. doi: 10.1080/09540105.2020.1736009
28. Kim J, Nietfeldt J, Ju J, Wise J, Fegan N, Desmarchelier P, et al. Ancestral divergence, genome diversification, and phylogeographic variation in subpopulations of sorbitol-negative, beta-glucuronidase-negative enterohemorrhagic Escherichia coli O157. J Bacteriol. (2001) 183:6885–97. doi: 10.1128/JB.183.23.6885-6897.2001
29. Whitworth J, Zhang Y, Bono J, Pleydell E, French N, Besser T. Diverse genetic markers concordantly identify bovine origin Escherichia coli O157 genotypes underrepresented in human disease. Appl Environ Microbiol. (2010) 76:361–5. doi: 10.1128/AEM.01761-09
30. Mellor GE, Besser TE, Davis MA, Beavis B, Jung W, Smith HV, et al. Multilocus genotype analysis of Escherichia coli O157 isolates from Australia and the United States provides evidence of geographic divergence. Appl Environ Microbiol. (2013) 79:5050–8. doi: 10.1128/AEM.01525-13
31. Santos J, Cerri R, Ballou M, Higginbotham G, Kirk J. Effect of timing of first clinical mastitis occurrence on lactational and reproductive performance of Holstein dairy cows. Anim Reprod Sci. (2004) 80:31–45. doi: 10.1016/S0378-4320(03)00133-7
32. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMB. (2011) 2011:1713. doi: 10.14806/ej.17.1.200
33. Seemann T, Goncalves SA, Bulach D, Schultz M, Kwong J, Howden B. Nullarbor [Online]. Github (2020). Available online at: https://github.com/tseemann/nullarbor (accessed August 08, 2020).
34. Souvorov A, Agarwala R, Lipman DJ. SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol. (2018) 19:153. doi: 10.1186/s13059-018-1540-z
35. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. (2014) 30:2068–9. doi: 10.1093/bioinformatics/btu153
36. Jolley K, Bray J, Maiden M. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications [version 1; peer review: 2 approved]. Wellcome Open Res. (2018) 3:124. doi: 10.12688/wellcomeopenres.14826.1
37. Zhou Z, Alikhan N-F, Mohamed K, Fan Y, Group AS, Achtman M. The EnteroBase user's guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny and Escherichia core genomic diversity. Genome Res. (2020) 30:138–52. doi: 10.1101/gr.251678.119
38. Meier-Kolthoff JP, Göker M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat Commun. (2019) 10:2182. doi: 10.1038/s41467-019-10210-3
39. Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. (2013) 14:60. doi: 10.1186/1471-2105-14-60
40. Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. (2015) 31:3691–3. doi: 10.1093/bioinformatics/btv421
41. Annon. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. (2004) 32:D258–61. doi: 10.1093/nar/gkh036
42. Törönen P, Medlar A, Holm L. PANNZER2: a rapid functional annotation web server. Nucleic Acids Res. (2018) 46:W84–8. doi: 10.1093/nar/gky350
43. Käll L, Krogh A, Sonnhammer EL. Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res. (2007) 35:W429–32. doi: 10.1093/nar/gkm256
44. Almagro Armenteros JJ, Tsirigos KD, Sonderby CK, Petersen TN, Winther O, Brunak S, et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. (2019) 37:420–3. doi: 10.1038/s41587-019-0036-z
45. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. (2014) 30:1312–3. doi: 10.1093/bioinformatics/btu033
46. Robertson J, Nash JHE. MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb Genom. (2018) 4:e000206. doi: 10.1099/mgen.0.000206
47. Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. (2015) 43:W566–70. doi: 10.1093/nar/gkv468
48. Seemann T. ABRicate [Online]. Github. Available online at: https://github.com/tseemann/abricate (accessed May 01, 2020).
49. Chen L, Zheng D, Liu B, Yang J, Jin Q. VFDB 2016: hierarchical and refined dataset for big data analysis−10 years on. Nucleic Acids Res. (2015) 44:D694–7. doi: 10.1093/nar/gkv1239
50. Feldgarden M, Brover V, Haft DH, Prasad AB, Slotta DJ, Tolstoy I, et al. Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob Agents Chemother. (2019) 63:e00483-19. doi: 10.1128/AAC.00483-19
51. Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. (2016) 44:W16–21. doi: 10.1093/nar/gkw387
52. Xie Z, Tang H. ISEScan: automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics. (2017) 33:3340–7. doi: 10.1093/bioinformatics/btx433
53. Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. (2018) 46:W246–51. doi: 10.1093/nar/gky425
54. R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing (2020).
55. Kempf F, Slugocki C, Blum SE, Leitner G, Germon P. Genomic comparative study of bovine mastitis Escherichia coli. PLoS ONE. (2016) 11:e0147954. doi: 10.1371/journal.pone.0147954
56. Nüesch-Inderbinen M, Käppeli N, Morach M, Eicher C, Corti S, Stephan R. Molecular types, virulence profiles and antimicrobial resistance of Escherichia coli causing bovine mastitis. Vet Rec. (2019) 6:e000369. doi: 10.1136/vetreco-2019-000369
57. Vigil PD, Alteri CJ, Mobley HL. Identification of in vivo-induced antigens including an RTX family exoprotein required for uropathogenic Escherichia coli virulence. Infect Immun. (2011) 79:2335–44. doi: 10.1128/IAI.00110-11
58. Smillie C, Garcillán-Barcia MP, Francia MV, Rocha EPC, de la Cruz F. Mobility of plasmids. Microbiol Mol Biol R. (2010) 74:434–52. doi: 10.1128/MMBR.00020-10
59. Nemeth J, Anne-Muckle C, Reggie YC. Serum resistance and the traT gene in bovine mastitis-causing Escherichia coli. Vet Microbiol. (1991) 28:343–51. doi: 10.1016/0378-1135(91)90069-R
60. Kobori D, Rigobelo EC, Macedo C, Marin JM, Avila FA. Virulence properties of shiga toxin-Producing Escherichia coli isolated from cases of bovine mastitis in Brazil. Revue Élev Méd vét Pays trop. (2004) 57:15–20. doi: 10.19182/remvt.9899
61. Cengiz S, Dinc G, Sogut MU. Detection of several virulence properties, antibiotic resistance and phylogenetic relationship in E. coli isolates originated from cow mastitis. Acta Vet. (2014) 64:413–25. doi: 10.2478/acve-2014-0039
62. MLA. Antimicrobials and the Cattle Industry. (2014). Available online at: https://www.mla.com.au/globalassets/mla-corporate/research-and-development/program-areas/food-safety/pdfs/antimicrobials-and-the-cattle-industry-fact-sheet.pdf (accessed June 01, 2020).
63. AVPG. Australian Veterinary Prescribing Guidelines [Online] (2020). Available online at: https://vetantibiotics.fvas.unimelb.edu.au/bovine-medicine-guidelines/mastitis/ (accessed June 1, 2020).
64. Steeneveld W, van Werven T, Barkema HW, Hogeveen H. Cow-specific treatment of clinical mastitis: an economic approach. J Dairy Sci. (2011) 94:174–88. doi: 10.3168/jds.2010-3367
65. Smalla K, Jechalke S, Top EM. Plasmid detection, characterization, and ecology. Microbiol Spectr. (2015) 3:PLAS-0038-2014. doi: 10.1128/microbiolspec.PLAS-0038-2014
66. Li L, Dechesne A, Madsen JS, Nesme J, Sørensen SJ, Smets BF. Plasmids persist in a microbial community by providing fitness benefit to multiple phylotypes. ISME J. (2020) 14:1170–81. doi: 10.1038/s41396-020-0596-4
67. Ramos S, Igrejas G, Capelo-Martinez JL, Poeta P. Antibiotic resistance and mechanisms implicated in fecal enterococci recovered from pigs, cattle and sheep in a Portuguese slaughterhouse. Ann Microbiol. (2012) 62:1485–94. doi: 10.1007/s13213-011-0402-7
68. Wichmann F, Udikovic-Kolic N, Andrew S, Handelsman J. Diverse antibiotic resistance genes in dairy cow manure. MBio. (2014) 5:e01017. doi: 10.1128/mBio.01017-13
69. Oliver SP, Murinda SE, Jayarao BM. Impact of antibiotic use in adult dairy cows on antimicrobial resistance of veterinary and human pathogens: a comprehensive review. Foodborne Pathog Dis. (2011) 8:337–55. doi: 10.1089/fpd.2010.0730
70. Munita JM, Arias CA. Mechanisms of antibiotic resistance. Microbiol Spectr. (2016) 4:10.1128/microbiolspec.VMBF-0016-2015. doi: 10.1128/microbiolspec.VMBF-0016-2015
71. Dogan B, Klaessig S, Rishniw M, Almeida RA, Oliver SP, Simpson K, et al. Adherent and invasive Escherichia coli are associated with persistent bovine mastitis. Vet Microbiol. (2006) 116:270–82. doi: 10.1016/j.vetmic.2006.04.023
72. Barondess JJ, Beckwith J. bor gene of phage lambda, involved in serum resistance, encodes a widely conserved outer membrane lipoprotein. J Bacteriol. (1995) 177:1247. doi: 10.1128/JB.177.5.1247-1253.1995
73. Jansen R, Embden JD, Gaastra W, Schouls LM. Identification of genes that are associated with DNArepeats in prokaryotes. Mol Microbiol. (2002) 43:1565–75. doi: 10.1046/j.1365-2958.2002.02839.x
74. Louwen R, Staals RH, Endtz HP, van Baarlen P, van der Oost J. The role of CRISPR-Cas systems in virulence of pathogenic bacteria. Microbiol Mol Biol Rev. (2014) 78:74–88. doi: 10.1128/MMBR.00039-13
75. Makarova KS, Koonin EV. Annotation and classification of CRISPR-Cas systems. Methods Mol Biol. (2015) 1311:47–75. doi: 10.1007/978-1-4939-2687-9_4
76. Dang TN, Zhang L, Zollner S, Srinivasan U, Abbas K, Marrs CF, et al. Uropathogenic Escherichia coli are less likely than paired fecal E. coli to have CRISPR loci. Infect Genet Evol. (2013) 19:212–8. doi: 10.1016/j.meegid.2013.07.017
77. Garcia-Gutierrez E, Almendros C, Mojica FJ, Guzman NM, Garcia-Martinez J. CRISPR content correlates with the pathogenic potential of Escherichia coli. PLoS ONE. (2015) 10:e0131935. doi: 10.1371/journal.pone.0131935
Keywords: cattle, genomics, virulence, plasmid, comparative genomics, antibiotic resistance, whole genome sequencing
Citation: Alawneh JI, Vezina B, Ramay HR, Al-Harbi H, James AS, Soust M, Moore RJ and Olchowy TWJ (2020) Survey and Sequence Characterization of Bovine Mastitis-Associated Escherichia coli in Dairy Herds. Front. Vet. Sci. 7:582297. doi: 10.3389/fvets.2020.582297
Received: 23 July 2020; Accepted: 05 November 2020;
Published: 07 December 2020.
Edited by:
Zhenhai Chen, Yangzhou University, ChinaReviewed by:
David Smith, Heriot-Watt University, United KingdomFabiana Horn, Federal University of Rio Grande do Sul, Brazil
Copyright © 2020 Alawneh, Vezina, Ramay, Al-Harbi, James, Soust, Moore and Olchowy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John I. Alawneh, ai5hbGF3bmVoQHVxLmVkdS5hdQ==
†These authors have contributed equally to this work