 Yukiko Nakamura1,2*
Yukiko Nakamura1,2* Kyoko Hayashida2,3Victoire Delesalle4
Kyoko Hayashida2,3Victoire Delesalle4 Yongjin Qiu2Ryosuke Omori2
Yongjin Qiu2Ryosuke Omori2 Martin Simuunza5
Martin Simuunza5 Chihiro Sugimoto2Boniface Namangala6
Chihiro Sugimoto2Boniface Namangala6 Junya Yamagishi2,3*
Junya Yamagishi2,3*- 1Graduate School of Infectious Diseases, Hokkaido University, Sapporo, Japan
- 2Research Center for Zoonosis Control, Hokkaido University, Sapporo, Japan
- 3International Collaboration Unit, Research Center for Zoonosis Control, Hokkaido University, Sapporo, Japan
- 4Melindika, Non-governmental Organization of International Solidarity, Itezhi-Tezhi, Zambia
- 5Department of Disease Control, School of Veterinary Medicine, The University of Zambia, Lusaka, Zambia
- 6Department of Para-Clinical Studies, School of Veterinary Medicine, The University of Zambia, Lusaka, Zambia
We clarified the genetic diversity of Trypanosoma spp. within the Kafue ecosystem, using PCR targeting the internal transcribed spacer 1 and the cathepsin L-like cysteine protease (CatL) sequences. The overall prevalence of Trypanosoma spp. in cattle and tsetse flies was 12.65 and 26.85%, respectively. Cattle positive for Trypanosoma vivax had a significantly lower packed cell volume, suggesting that T. vivax is the dominant Trypanosoma spp. causing anemia in this area. Among the 12 operational taxonomic units (OTUs) of T. vivax CatL sequences detected, one was from a known T. vivax lineage, two OTUs were from known T. vivax-like lineages, and nine OTUs were considered novel T. vivax-like lineages. These findings support previous reports that indicated the extensive diversity of T. vivax-like lineages. The findings also indicate that combining CatL PCR with next generation sequencing is useful in assessing Trypanosoma spp. diversity, especially for T. vivax and T. vivax-like lineages. In addition, the 5.42% prevalence of Trypanosoma brucei rhodesiense found in cattle raises concern in the community and requires careful monitoring of human African trypanosomiasis.
Introduction
Diseases caused by the infection of Salivaria Trypanosoma spp. are of medical and veterinary importance in sub-Saharan Africa. Human African trypanosomiasis (HAT) is caused by Trypanosoma brucei rhodesiense and T. b. gambiense in East and West Africa, respectively. African animal trypanosomosis (AAT) is caused by various Trypanosoma spp. from the subgenera Duttonella (Trypanosoma vivax), Nannomonas (Trypanosoma congolense, Trypanosoma simiae), and Trypanozoon (T. b. brucei). Salivaria Trypanosoma spp. are transmitted solely by tsetse flies (Glossina spp.), except for T. vivax, in which mechanical transmission by biting flies has been described (1).
African animal trypanosomosis, especially bovine trypanosomosis, is of national importance in Zambia. The prevalence of infection is generally high as a result of the continuous infestation of tsetse flies, abundant wildlife reservoirs, and cattle rearing in tsetse infested areas. As a result, bovine trypanosomosis remains a significant constraint to rural development in large areas of western, southern, and eastern Zambia (2). Zambia currently reports fewer than 100 new HAT cases annually, mainly from areas within the Luangwa River valley in eastern Zambia (3). Several HAT cases have been reported from re-emerging HAT foci outside the major HAT foci, such as the case in Kafue National Park in 2016, which had been more than 50 years since the last documented case (4). This suggests that the human infective T. b. rhodesiense is being maintained within this ecosystem, which puts the local community at the risk of HAT.
Bovine trypanosomosis in Zambia is mainly caused by T. congolense and T. vivax, with some cases caused by Trypanozoon (T. b. brucei). Previous studies have detected a high prevalence of these Trypanosoma spp. in the eastern and southern province of Zambia (33.5% prevalence, 29.3% prevalence among anemic cattle, respectively), where the majority of infections were attributed to T. congolense (5, 6). The clinical signs of bovine trypanosomosis include anemia, relapsing fever, enlarged lymph nodes, reduced milk yield, reduced productivity, decreased fertility, and, in severe cases, abortion, emaciation, and eventual death (7). Since these symptoms are non-specific, diagnosis of bovine trypanosomosis requires the detection of the parasite, its antigen, or antibodies against the parasite. The most widely used technique is parasite detection in light microscopy observation of wet blood films or Giemsa-stained thick and thin fixed blood films. Increased sensitivity can be obtained when hematocrit centrifugation technique or the buffy coat technique is used to concentrate the parasite load (8). However, these techniques cannot differentiate the Trypanosoma spp. intraspecifically, and are not sensitive enough to detect low parasite levels (9). Antibody detection methods, such as indirect ELISA, have been widely used in epidemiological studies. However, these methods do not prove on-going infection and cannot differentiate infection between Trypanosoma spp. (9). Molecular methods, such as PCR and loop-mediated isothermal amplification, have been used to establish species-specific and cross-species detection, and can detect intraspecific diversity. An example is PCR targeting the internal transcribed spacer (ITS) region of ribosomal genes (10, 11). This method has been commonly used due to its high sensitivity attributed to the high copy number and the feasibility of the inter-species length variation enabling visual discrimination by gel electrophoresis. Sequencing the amplicon can further elevate the sensitivity and allow intraspecific identification of T. congolense and, to some extent, T. vivax (12). However, to elucidate the highly complex disease epidemiology and genetic diversity of T. congolense and T. vivax, molecular methods with the resolution to distinguish beyond the T. congolense subgroups or T. vivax lineages are needed.
Trypanosoma congolense comprises of three subgroups (savannah, forest, and kilifi) that vary in virulence, pathogenicity, and geographical distribution (13). The three subgroups coexist in Zambia (14). In experimental infections of susceptible zebu cattle (Bos indicus), kilifi was non-pathogenic, forest was poorly pathogenic, and savannah was the most virulent subgroup (13). The savannah subgroup is reportedly the most genetically divergent and widespread across sub-Saharan Africa. Examinations of savannah phenotypes revealed markedly differing virulence and drug resistance, even in the same location (15, 16). The findings suggested that T. congolense undergoes genetic recombination in nature. This view is supported by previous population genetic studies based on microsatellites and population genomic research using whole-genome analyses (17, 18). Especially high diversity of T. congolense savannah was observed in Zambia, which was due to genetic exchange between phylogenetically distinct T. congolense savannah parasites (19).
The pathogenicity of T. vivax is known to differ between East and West Africa. West African T. vivax isolates are believed to be more pathogenic than East African isolates (20). However, severe hemorrhagic outbreaks with high mortality levels have also been reported in East Africa (7), and the correlation between the different isolates and disease severity in cattle remains unclear. While genetic homogeneity of isolates from West Africa and South America has been reported, greater genetic diversity among isolates from East Africa has been observed (21–30). In addition, T. vivax isolates that are genetic and morphologically distinct from the reference isolate T. vivax Y486 (isolated from Nigerian cattle) have been reported in East Africa (21–24). Therefore, parasites within the subgenus Duttonella separated by relative genetic distance from T. vivax Y486 have been widely termed as a “T. vivax-like” lineage (21, 23, 25–27). The taxonomic position of these trypanosomes is still unclear. It is possible that T. vivax-like trypanosomes correspond to other species within Duttonella, such as T. uniforme and T. vivax ellipsiprymni. Notably, the phylogenetic inference of T. vivax and T. vivax-like isolates from Mozambique suggested that a T. vivax-like lineage, TvL-Gorongosa, should be elevated to the species status (27).
Several genetic markers have been utilized to assess the genetic diversity of T. congolense and T. vivax at the intraspecific level. Representative markers include the glyceraldehyde 3-phosphate dehydrogenase gene (gGAPDH) sequence (31), fluorescent fragment length barcoding (FFLB) using the 18S and 28S ribosomal RNA regions (32), ITS rRNA sequences (ITS1, 5.8S, and ITS2) (24), and the cathepsin L-like cysteine protease (CatL) sequences (33), which all have their advantages and disadvantages. While gGAPDH sequences are valuable markers for identification of species and intraspecific diversity, the relationships within a subgroup of T. congolense or among the two T. vivax lineages (T. vivax and T. vivax-like) have not been well-resolved (27). Fluorescent fragment length barcoding displayed the highest sensitivity in detecting Duttonella trypanosomes from tsetse fly samples but could not distinguish within the two T. vivax lineages (32). ITS rRNA PCR and sequencing have high resolution and have been successful in illustrating the diversity within the T. vivax lineage and the T. vivax-like lineage. However, the sensitivity of this approach is inferior to FFLB and requires cloning and sequencing of several clones from each sample (27). CatL PCR displayed comparable sensitivity to FFLB (27), and has been used for genotyping a variety of Trypanosoma spp. (25, 33–36).
Kinetoplastid parasites express two C1 peptidases related to mammalian cathepsin B- and L (CatL)-like peptidases, which are either essential to survival or are important virulence factors contributing to disease pathogenesis (37). The pronounced genetic sequence diversity within the CatL sequences suggests that this region evolved early in evolution or has faster mutation rates to adapt to the parasites' divergent cellular functions (38). The high diversity in the CatL region between different Trypanosoma spp. has been proposed as a suitable genetic marker for analyzing the intraspecific diversity of T. vivax and T. congolense. Phylogenetic analysis of CatL sequences supported the phylogenetic relationship of trypanosomes based on the partial small subunit rRNA (V7-V8) gene sequence data (25). CatL genes have been successful in identifying nine clades of T. vivax (TviCatL1-9) throughout the West and East Africa and South America (25, 39), and five clades within the T. congolense savannah subgroup (SAV1-3, SAVna) (40). Trypanosoma vivax isolates from West Africa shared CatL clades with South America, including TviCatL1-4. Trypanosoma vivax CatL clades from East and Southern Africa showed divergent sequences, including TviCatL5–7 for isolates from Mozambique and TviCatL8–9 from Kenya (25). Trypanosoma vivax from Zambian cattle also formed one distinct clade and clustered with East African and Southern African sequences (39). Extending these studies to assess the genetic diversity of trypanosomes found in tsetse flies and cattle from the same ecosystem can enable a more in-depth understanding of the transmission of parasites and disease manifestation in cattle.
In this study, we aimed to elucidate the Trypanosoma spp. prevalence in tsetse flies and cattle from the Kafue ecosystem. Combined molecular methods were used for comprehensive detection and genotyping of African trypanosomes. ITS1 PCR was used to detect Trypanosoma spp. at the species level. CatL PCR coupled with next generation sequencing was used to illustrate the intraspecific diversity of Trypanosoma spp.
Materials and Methods
Study Area and Sample Collection
This study was performed in the Itezhi-Tezhi District, which is located in the south-western region of Central Province of Zambia, in the Kafue ecosystem. Kafue National Park is the oldest and largest national park in Zambia, covering an area of approximately 22,400 km2 (41). Kafue National Park is surrounded by several Game Management Areas (GMAs), which act as buffer zones for National Parks and mitigate the adverse effects of human activities. Licensed safari, subsistence hunting, and agricultural activities are permitted in the GMAs for local communities. The Nkala GMA covers an area of approximately 194 km2. There is no local population living in this area (42). Kafue National Park and the surrounding GMAs comprise the Kafue ecosystem, which is home to a wide variety of flora and fauna, including tsetse flies. Glossina morsitans centralis is the dominant tsetse fly species in this area, with a lower distribution of Glossina pallidipes (43). Therefore, in many communities adjacent to Nkala GMA, human settlement, cattle grazing areas, and tsetse infested areas overlap.
Cattle blood sampling was conducted in the five villages of Iyanda, New Ngoma, Ntubya, Kaminza, and Basanga in April and May of 2019 (Supplementary Figure 1). The major cattle breeds were crosses between local breeds (Tonga and Baila) and exotic breeds (mostly Boran and Brahman). The estimated cattle population within the community was 15,000 heads. Using Cochran's formula (44) with a 95% confidence level and a confidence interval of 5, the required sample size was computed to be 375 heads. Blood samples were collected from the jugular vein of the cattle using 18G needles and 5 mL syringes. Each sample was transferred to a heparin-lithium tube. A total of 498 blood samples were randomly collected from 65 farmers. The samples were immediately subjected to micro-hematocrit centrifugation to obtain packed cell volume (PCV) values (12,000 rpm, 5 min). Thin blood smears were also made, and Giemsa stained for microscopic observation of parasites. The remaining blood samples were preserved at 4°C until DNA extraction. Four epsilon traps were set for each sampling point to catch tsetse files. The traps were set once for 3 constructive days. The traps were visited each morning and afternoon to collect the captured flies. In areas with an insufficient number of catches, a mobile trap was used to supply the number of tsetse flies. Mobile trapping was conducted within a one-kilometer radius, which is within the average lifetime dispersal of morsitans group of tsetse flies (45). A total of 298 tsetse flies were captured from 10 sampling points (Supplementary Figure 2). The captured flies were inspected using a stereomicroscope for morphological identification of species and sex. The flies were stored in 2 mL sample tubes with silica beads to dry.
DNA Extraction
DNA extraction of cattle blood samples was conducted using QuickGene DNA whole blood kit S (Kurabo, Osaka, Japan), following the manufacturer's protocol. The dried tsetse flies were transferred to new tubes with beads and smashed using a Micro Smash MS−100 bead cell disrupter (Tomy, Tokyo, Japan) at 3,000 rpm for 45 s. DNA was extracted using a modified protocol with the DNA Isolation Kit for Mammalian Blood (Roche, Basel, Switzerland). Briefly, 330 μL of white cell lysis buffer was added directly into each tube, vortexed, and heated at 37°C for 30 min. Then, 170 μL of protein precipitation solution was added, vortexed thoroughly, and centrifuged at 15,000 rpm for 20 min. DNA was precipitated by the addition of ethanol. All extracted DNA was stored at −30°C until further use.
Tsetse Fly Species Confirmation Using Glossina Its PCR
For molecular confirmation of the tsetse fly species, GlossinaITS1_for (5′-GTG ATC CAC CGC TTA GAG TGA−3′) and GlossinaITS1_rev (5′-GCA AAA GTT GAC CGA ACT TGA−3′) primers were used to amplify the ITS1 region of ribosomal genes of tsetse flies (46). Reactions contained 1–10 ng of template DNA, 1 × Ampdirect Plus (Shimadzu Corp., Kyoto, Japan), 0.25 U BioTaq HS DNA Polymerase (Bioline, Memphis, TN, USA), 0.2 mM primers, and distilled water to a total volume of 10 μL. Amplification included an initial denaturation step at 95°C for 10 min, followed by 30 cycles each of 94°C for 30 s, 62°C for 1 min, 72°C for 2 min, and a final extension step at 72°C for 7 min. The band patterns were visually inspected after gel electrophoresis (G. pallidipes: 920 bp, G. m. centralis: 800 bp and 150 bp). There was a total of 212 G. pallidipes and 86 G. m. centralis samples.
Human Infective T. b. rhodesiense Detection Using Serum Resistance-Associated (SRA) PCR
SRA284F (5′-ATA GTG ACA AGA TGC GTA CTC AAC GC−3′) and SRA284R (5′-AAT GTG TTC GAG TAC TTC GGT CAC GCT−3′) primers was used to detect human infective T. b. rhodesiense (47). The PCR reagents were the same as those described above for Glossina ITS PCR. Amplification included an initial denaturation step at 95°C for 10 min, followed by 40 cycles each of 94°C for 30 s, 60°C for 1 min, 72°C for 1 min, and a final extension step at 72°C for 2 min.
ITS1 PCR for Trypanosoma spp. Detection and Species Identification
AITSF (5′-CGG AAG TTC ACC GAT ATT GC−3′) and AITSR (5′-AGG AAG CCA AGT CAT CCA TC−3′) primers were used to amplify the ITS1 region to analyze Trypanosoma spp. prevalence (12). The PCR reagents were the same as those of the Glossina ITS PCR described above. Amplification included an initial denaturation step at 95°C for 10 min, followed by 37 cycles of 94°C for 30 s, annealing at 58°C for 90 s, 72°C for 2 min, and a final extension step at 72°C for 7 min. Trypanosoma spp. were identified based on observation of gel electrophoresis.
CatL PCR and Sequencing on the Illumina MiSeq Platform
PCR amplification of the CatL region was carried out for the ITS1 positive samples using DTO154/DTO155 primers (33), customized by attaching adapter sequences (Illumina, San Diego, CA, USA) to the 5′ ends DTO154illumina (5′-ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT NNA CAG AAT TCC AGG GCC AAT GCG GCT CGT GCT GG−3′), DTO155illumina(5′-GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG ATC TNN TTA AAG CTT CCA CGA GTT CTT GAT GAT CCA GTA−3′). The reactions included 1–10 ng of genomic DNA, 0.2 mM primers, reagents from the KAPA Taq EXtra PCR Kit (Kapa Biosystems, Wilmington, MA, USA), using final concentrations of 1 × KAPA Taq EXtra buffer, 0.5 U KAPA Taq EXtra, 0.2 mM KAPA dNTP Mix, 1.5 mM MgCl2, and distilled water to a total volume of 10 μL. PCR grade water was used in place of genomic DNA as a negative control. Amplification included an initial denaturation step at 95°C for 10 min, followed by 15 cycles each of 94°C for 30 s, 60°C for 1 min, 72°C for 30 s, and a final extension step at 72°C for 10 min. The final PCR was done to attach a unique index to each sample to enable multiplexed sequencing using the Illumina platform. Reactions contained 1 mM Illumina dual-index primer mix, 1 × KAPA Taq EXtra buffer, 1 U KAPA Taq EXtra, 0.2 mM KAPA dNTP Mix, 1.5 mM MgCl2 and distilled water to a total volume of 20 μL. The PCR products were pooled in equal amounts into one library and analyzed using 2% agarose gel electrophoresis. The band of interest was cut and purified using the Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI, USA). Quantification of the library was done using a Qubit dsDNA HS assay kit and a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, USA), and adjusted to 4 nM using nuclease-free water as the final library. The library was then applied to the Illumina MiSeq platform (Illumina MiSeq System, RRID:SCR_016379). MiSeq Reagent Kit v3 (Illumina) was used for 300 base pairs, paired-end sequencing. PhiX DNA spike-in control (25%) was added to increase the diversity of the amplicon library.
Analysis and Assignment of CatL OTUs
The raw reads generated from CatL amplicon sequencing were polished, clustered at 95% identity, and the representative sequence was generated using the Amplicon Tool Kit (AMPtk) pipeline and default parameters of “amptk illumina,” “amptk dada2,” “amptk filter,” and “amptk lulu” (48). The representative sequence for each cluster was termed as an operational taxonomic unit (OTU). The generated CatL OTUs were manually filtered by excluding reads equal to or less than the negative control included in the analysis. The resulting CatL OTUs were used to construct a reference taxonomy database using the top hit result of BLASTn (BLASTN, RRID:SCR_001598) (49) above 90% identity and 100% query coverage. Finally, taxonomy was assigned to the CatL OTUs using “amptk taxonomy.” The generated CatL OTUs were assigned to each Trypanosoma spp., T. congolense subgroup (savannah, forest, kilifi) (13), and reported CatL clades based on the result of the top hit of the BLASTn homology search. Trypanosoma vivax CatL OTUs were assigned to their lineages (T. vivax and T. vivax-like) (21, 25–27) when they clustered with the reported sequences. All CatL OTU sequences generated in this study have been deposited to GenBank under Accession Numbers MT673751 to MT673783.
Phylogenetic Analyses
Nucleotide sequences of each CatL OTU and reference sequences were aligned using MAFFT online v7 (MAFFT, RRID:SCR_011811) (50). The aligned sequences with 257 nucleotides were then used to construct neighbor-joining trees (51) using MEGA X (MEGA Software, RRID:SCR_000667) (52). The evolutionary distances were computed using the Maximum Composite Likelihood method (53) and default parameters with 10,000 bootstraps. The tree was visualized and annotated using iTOL v5.5 (iTOL, RRID:SCR_018174) (54).
Statistical Analyses
Statistical analyses and visualization of other data were done in R v3.6.1 (R Project for Statistical Computing, RRID:SCR_001905) (55). For each pairwise comparisons of Trypanosoma spp. prevalence and PCV values, statistical significance (p < 0.05) was assessed using the Wilcoxon rank sum test with Bonferroni correction.
Ethics Statement
This study was conducted under ethics approval Ref. No. 2019-Feb-081 (ERES Converge IRB, Lusaka, Zambia).
Results
Trypanosoma spp. Prevalence Based on Microscopy, ITS1 PCR, and SRA PCR
Results for microscopy, ITS1 PCR, SRA PCR, and CatL sequencing for all individual samples can be found in Supplementary Table 1. In summary, seven of 105 (6.67%) cattle thin blood smears were positive for Trypanosoma spp. by microscopy in Ntubya village. All animals from other villages were microscopically negative for Trypanosoma spp., and Ntubya had significantly higher prevalence of Trypanosoma spp. by microscopy compared to New ngoma and Basanga (Table 1 and Supplementary Table 2). In ITS1 PCR, 63 of 498 cattle (12.65%) were positive for one or more Trypanosoma spp. (Table 1). When compared between villages, Ntubya had the highest prevalence (32 of 105 cattle, 30.48%), followed by New Ngoma (12/102, 11.77%), Kaminza (8/85, 9.41%), Basanga (7/116, 6.03%), and Iyanda (4/90, 4.44%). Ntubya had significantly higher prevalence of the total Trypanosoma spp. by ITS1 PCR compared to every other villages (Table 1 and Supplementary Table 2). Overall, the most abundant species in cattle samples was T. vivax (50/498, 10.04%), followed by T. congolense (8/498, 1.61%), and Trypanozoon (7/498, 1.41%). T. godfreyi and T. simiae were not detected from the cattle samples. Trypanosoma vivax was detected in all five villages, and was the most abundant species in Ntubya, Kaminza, and New Ngoma (Table 1 and Supplementary Figure 1). Ntubya had significantly higher prevalence of T. vivax by ITS1 PCR compared to every other villages (Table 1 and Supplementary Table 2). Trypanosoma congolense was detected in four villages, but not in Kaminza. Trypanozoon was detected in four villages, but not in New Ngoma, and was the most abundant species in Iyanda and Basanga (Table 1 and Supplementary Figure 1). Two multiple infections of different Trypanosoma spp. were found among the positive samples in Ntubya (2/32, 6.25%). They involved T. vivax/Trypanozoon and T. vivax/T. congolense (Table 1).
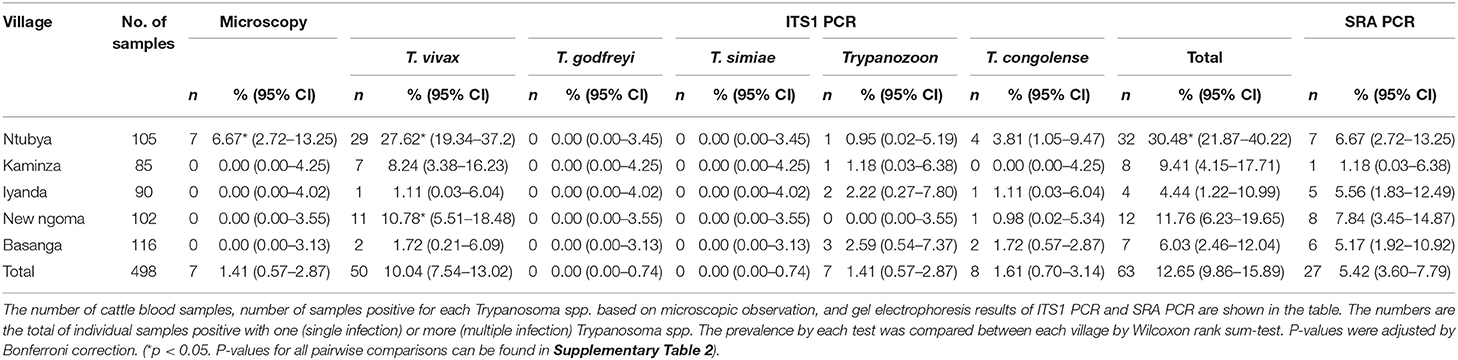
Table 1. Microscopy, ITS1 PCR, and SRA PCR results for cattle blood samples in each village.
In tsetse flies, 37 of 86 (43.02%) G. m. centralis were positive for one or more Trypanosoma spp. (Table 2). Of the 37 positive samples, seven were multiple infections of either T. vivax/T. simiae, T. vivax/T. congolense, T. simiae/T. congolense, or T. vivax/T. simiae/T. congolense (Table 2). Forty-three of 212 (20.28%) G. pallidipes were positive for one or more Trypanosoma spp. Of the 43 positive samples, five were multiple infections of either T. vivax/T. congolense, T. godfreyi/T. simiae and Trypanozoon/T. congolense (Table 2). Trypanosoma vivax was most abundant (52/298, 17.45%), followed by T. congolense (26/298, 8.73%), T. simiae (9/298, 3.02%), Trypanozoon (5/298, 1.68%), and T. godfreyi (1/298, 0.34%). Glossina morsitans centralis had significantly higher prevalence of the total Trypanosoma spp., T. vivax, and T. simiae by ITS1 PCR compared to G. pallidipes (Table 2 and Supplementary Table 3). The proportion of multiple infections in the positive samples was 18.92 and 11.63% for G. m. centralis and G. pallidipes, respectively (Table 2).
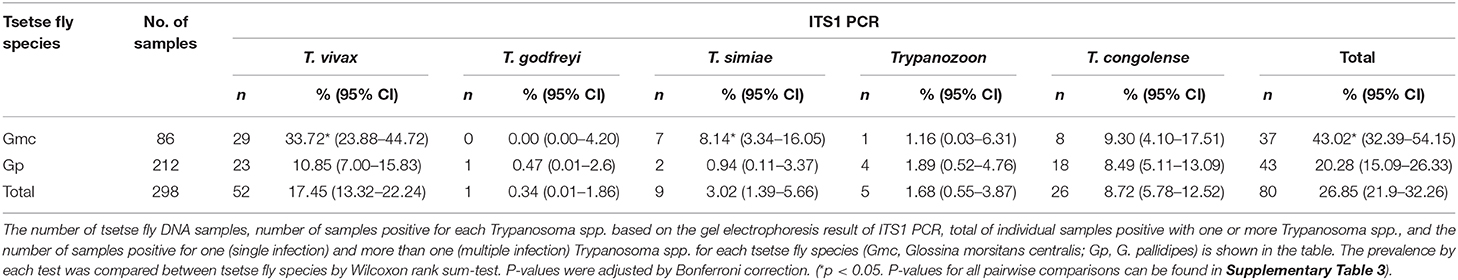
Table 2. ITS1 PCR for each tsetse fly species.
Twenty-seven of 498 cattle (5.42%) were positive for SRA PCR (Table 1). In descending order, the prevalence in the villages was New Ngoma (8/102, 7.84%), Ntubya (7/105, 6.67%), Iyanda (5/90, 5.56%), Basanga (6/116, 5.17%), and Kaminza (1/85, 1.18%). Multiple false-positive bands were identified from the tsetse fly samples after SRA PCR. Since no negative controls with PCR grade water turned positive in any batch, contamination during PCR and PCR preparation had not occurred. Therefore, contamination of the SRA gene during DNA extraction was suggested, and therefore all tsetse fly samples were excluded from the SRA PCR experiments.
The CatL OTU Diversity and Abundance Within Cattle and Tsetse Fly Samples
A total of 143 cattle and tsetse fly samples that were positive for ITS1 PCR were subjected to CatL PCR and MiSeq amplicon sequencing. Among the ITS1 PCR-positive samples, 91 were positive for CatL PCR, and resulted in using 63 samples for downstream analysis after sequencing. The reduction was caused presumably by the difference in sensitivity between the ITS1 PCR and CatL PCR, and by the final reads filtering using the negative control. As a result, 33 CatL OTUs were generated and assigned to each Trypanosoma spp. (Supplementary Figure 3). Three T. vivax CatL OTUs clustered with reported CatL clades (Figure 1). On the other hand, nine CatL OTUs, OTU_Tv4–12, did not cluster with any of the reference sequences and were considered to be included in novel CatL clades (Figure 1). The phylogenetic relationship of the CatL OTUs and reference sequences of all Trypanosoma spp. are shown in Supplementary Figure 3. The number of CatL OTUs assigned to T. vivax, T. congolense, T. simiae, T. godfreyi, and Trypanozoon were 12, 15, four, one, and one, respectively (Figure 2). Among them, OTU_Tv1, 2, 3, 4, 6, 7, and 10 (T. vivax), OTU_Tc1, 2, 3, and 11 (T. congolense), and OTU_Ts1 (T. simiae) were found in cattle and tsetse fly samples. The results represent simultaneous infection of samples by Trypanosoma spp. with different CatL OTUs. The most abundant CatL OTU in both cattle and tsetse flies was OTU_Tv2. OTU_Tb1 (Trypanozoon) was only detected in cattle. CatL OTUs found only in tsetse flies included OTU_Tv5, 8, 9, 11, and 12 (T. vivax), OTU_Tc4, 5, 6, 7, 8, 9, 10, 12, 13, 14, and 15 (T. congolense), OTU_Ts2, 3, and 4 (T. simiae), and OTU_Tg1 (T. godfreyi) (Figure 2 and Supplementary Figures 4, 5). Glossina morsitans centralis had the highest diversity of CatL OTUs, in which all CatL OTUs, except OTU_Tv10 and OTU_Tb1, were detected (Figure 2 and Supplementary Figure 5).
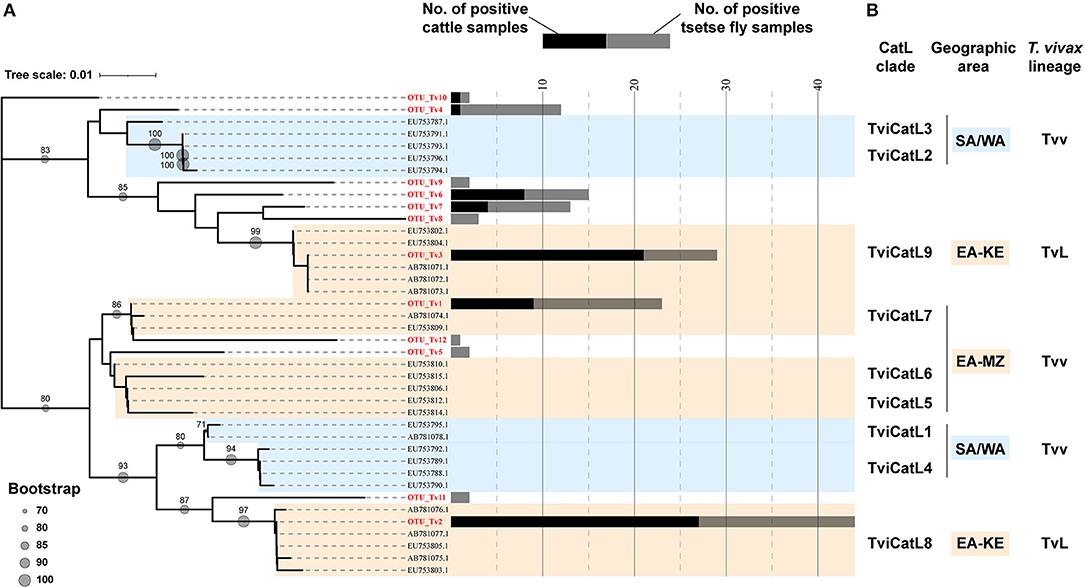
Figure 1. Neighbor-joining tree of 12 CatL OTUs and reference sequences for Trypanosoma vivax. (A) Phylogenetic relationship of the 12 CatL OTUs belonging to T. vivax. Bootstrap values above 70 are shown. The number of samples positive for each CatL OTUs are shown in bar graphs (black: number of cattle samples, gray: number of tsetse fly samples). (B) Nodes are defined with CatL clades (25) and T. vivax lineages (T. vivax and T. vivax-like) (27) reported for the corresponding reference sequences. SA/WA, isolates from South America and West Africa; EA-KE, isolates from Kenya and Zambia; EA-MZ, isolates from Mozambique and Zambia; Tvv, T. vivax lineage; TvL, T. vivax-like lineage.
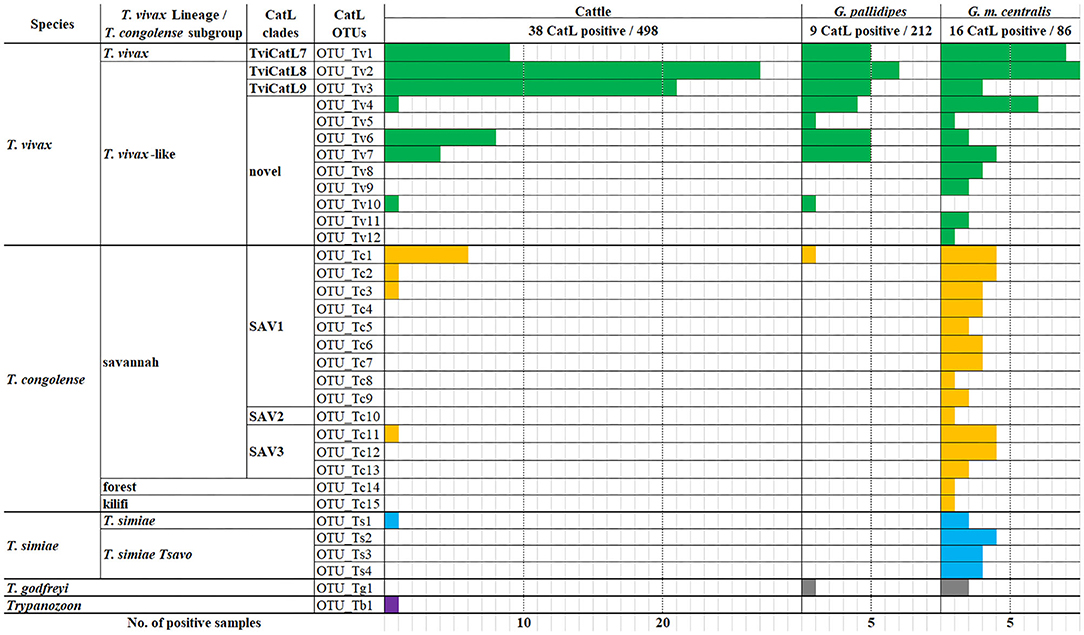
Figure 2. Number of samples positive for each CatL OTU. The number of samples positive for each CatL OTU is shown in the bar graph for cattle, G. pallidipes, and G. m. centralis.
Mean PCV Comparisons Between Villages, ITS1 PCR Result, and CatL OTU Result
The mean PCV value of cattle from each village, regardless of the Trypanosoma spp. infection status, was Ntubya 29.57 [standard deviation (SD): 6.80, 95% confidence interval (CI): 28.27–30.87], Kaminza 32.54 (SD: 5.40, 95% CI: 31.39–33.69), Iyanda 31.76 (SD: 4.31, 95% CI: 30.83–32.67), New Ngoma 33.18 (SD: 5.63, 95% CI: 32.08–34.27), and Basanga 32.75 (SD: 5.76, 95% CI: 31.70–33.80) (Supplementary Table 4). Ntubya had the lowest mean PCV, which was significantly lower than Kaminza, New ngoma, and Basanga (Supplementary Table 4). ITS1 PCR results grouped cattle samples into “infected” and “non-infected” groups. Infected cattle had significantly lower mean PCV in Ntubya and New Ngoma (Figure 3). When the cattle samples were grouped by the infection status according to each Trypanosoma spp., animals infected with T. vivax had significantly lower mean PCV compared with non-infected cattle (Figure 4A). Cattle infected with T. congolense and Trypanozoon also had a tendency of lower PCV compared to non-infected cattle, but no statistical significance was observed (Figures 4B,C). To check confounding effects, multiple regression analysis was conducted by including the village, sex, age, and the outcome of ITS1 PCR for each Trypanosoma spp. as coefficients. As a result, T. vivax infection significantly decreased PCV values even after excluding all other coefficients (p = 0.002). In addition, cattle with single infection of OTU_Tv2 (T. vivax-like, OTU pattern 2) tended to have lower mean PCV compared to negative cattle (OTU pattern 0) (Supplementary Figure 6).
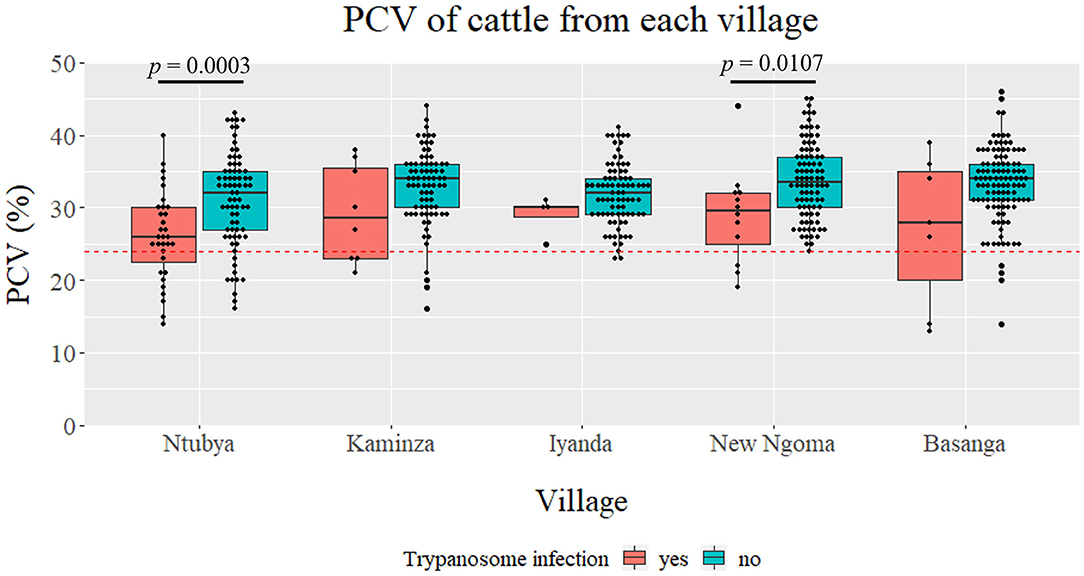
Figure 3. Comparison of packed cell volume (PCV) between villages. The PCV for ITS1 PCR positive (regardless of the species) and negative cattle are shown as boxplots for each village. Statistical significance was assessed by Wilcoxon rank sum-test (p < 0.05).
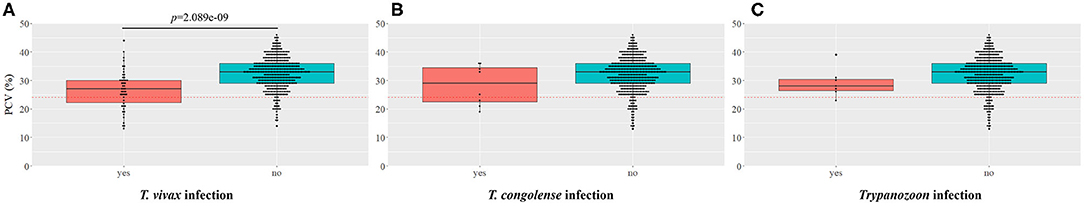
Figure 4. Comparison of packed cell volume (PCV) between Trypanosoma spp.-infected and non-infected cattle. The boxplot shows the comparison of the PCV between (A) T. vivax, (B) T. congolense, (C) Trypanozoon-positive and negative cattle samples (confirmed by ITS1 PCR). Statistical significance was assessed by Wilcoxon rank sum-test (p < 0.05).
Discussion
In our study, cattle infected with T. vivax had significantly lower mean PCV than non-infected cattle, indicating that T. vivax is the major Trypanosoma spp. causing anemia in this area. Moreover, since only seven cattle had a detectable level of parasitemia by microscopy, it is assumed that T. vivax causes chronic bovine trypanosomosis with low parasitemia. This assumption agrees with the report of prolonged T. vivax infections with long aparasitemic intervals (56). Moreover, there was a tendency toward a lower mean PCV for cattle infected with T. congolense compared with non-infected cattle, but no significant difference was observed. In East and Southern Africa, T. congolense has been reported to be more pathogenic to cattle than T. vivax (56), and there has been a focus on T. congolense as the agent of AAT. Our results re-emphasize the importance of T. vivax, along with T. congolense, as a source of anemia. Monitoring T. vivax would be especially crucial since it can be mechanically transmitted and become widespread in regions not infested with tsetse flies. Furthermore, bovine trypanosomosis research and control programs in tsetse fly infested areas should also include biting flies other than tsetse flies.
Notably, we have detected a variety of CatL OTUs, with pronounced diversity in T. vivax (T. vivax and T. vivax-like) and T. congolense savannah. Some of the T. vivax CatL OTUs detected in this study were consistent with the CatL clades (TviCatL7, TviCatL8, TviCatL9) reported in other East and southern African countries (21, 25, 39). The findings were expected considering the geographical location of the study area. TviCatL7 contains isolates from Mozambique nyala and Zambian cattle, which were confirmed as T. vivax lineages that are closely related to those from West Africa and South America (21, 39). We detected one CatL OTU in this clade (OTU_Tv1), which was found in cattle and tsetse flies. TviCatL8 (OTU_Tv2) and TviCatL9 (OTU_Tv3) is included in T. vivax-like lineages, which to date have only been detected from tsetse fly infested areas in East Africa. Cattle with single infection of OTU_Tv2 (T. vivax-like, clade TviCatL8) tended to have lower PCV values than those that were negative for all OTUs, and cattle with single infection of OTU_Tv1 (T. vivax, clade TviCatL7) had a mean PCV value that was comparable with cattle that were negative for all OTUs. Therefore, parasites with different CatL OTUs may differ in their pathogenesis against cattle.
The rich CatL OTU diversity noted presently within G. m. centralis was remarkable. Except for OTU_Tv10 and OTU_Tb1, all OTUs were detected. Similar results were shown in Mozambique, where the highest diversity based on gGAPDH and ITS rDNA were identified in tsetse fly (G. m. morsitans and G. pallidipes) samples, and less diversity within cattle and wild animal samples (27). Together with our results, these findings support the possibility of a range of different variants emerging through recombination in tsetse flies, of which some adapted to cattle and spread across East Africa and West Africa (27). Different host susceptibility between different Trypanosoma spp. and T. congolense subgroups have been reported (57), which may be occurring within the T. congolense subgroups or T. vivax lineages. The difference in susceptibility may be caused by the host preference of the parasite or the immune system of the animal host. However, more stringent classification of T. vivax taxonomy and population genomic studies on isolates will be needed to further assess these possibilities of recombination within tsetse flies and adaptation to cattle. On the contrary, the CatL OTU diversity in G. pallidipes was low and comparable with what was observed in cattle. The difference in bloodmeal host preference, host abundance, distribution, and dispersal rate between the two tsetse fly species may have affected the difference in the CatL OTU diversity. Different bloodmeal preferences have been reported within the morsitans group of tsetse flies, where G. pallidipes was more likely to take a bloodmeal from bovids, such as bushbuck, buffalo, and eland, and G. m. morsitans preferred suids (warthog) over bovids (bushbuck) (58). The distribution of G. pallidipes in the Kafue area may be strictly restricted (43), and the dispersal rates may be small compared to G. m. centralis (59). Therefore, it is hypothesized that the G. pallidipes sampled in this study may have had limited access to the variety of wildlife, and preferentially took their bloodmeal from cattle, which resulted in the similar CatL OTU diversity observed between G. pallidipes and cattle. However, careful consideration of this hypothesis is needed, since the decreased OTU diversity observed in G. pallidipes may have been affected by bias associated with trapping methods. G. pallidipes samples were all trapped by epsilon traps, whereas a majority of G. m. centralis samples were trapped by the mobile traps. Furthermore, we did not include other hematophagous flies, such as Tabanids or Stomoxys spp., which are likely to be responsible for mechanical transmission of T. vivax between cattle. Clarifying the CatL OTU diversity in these biting flies would increase the understanding of the dynamics of T. vivax within the ecosystem.
The overall prevalence of T. b. rhodesiense confirmed by SRA PCR was 5.42%, with the highest value in New Ngoma (7.84%) and the lowest in Kaminza (1.18%) (Table 1). This prevalence is surprising since there has been no recent official report of HAT in any of the sampled villages. The prevalence in this study was relatively high compared to other T. b. rhodesiense prevalence studies conducted in Zambia (60). In an extreme case of the HAT outbreak in the Soroti district of Uganda caused by the importation of cattle from HAT endemic area, the prevalence of T. b. rhodesiense in cattle assessed by SRA PCR was estimated to be 18% (61). Although the prevalence level in our case was not as high, the Kafue ecosystem is considered as an old HAT focus with a re-emerging risk since the last case in 2016 (4). A study conducted in the same Kafue ecosystem detected T. b. rhodesiense in buffalos, a sable antelope, and a vervet monkey using SRA PCR, with respective prevalence values of 9.40% (5/53), 12.50% (1/8), and 100% (1/1) (62). The collective findings indicate that T. b. rhodesiense is circulating within free-ranging wildlife and human-owned cattle. This could be a concern to the community and requires careful monitoring.
In summary, combining CatL PCR with next generation sequencing is useful in illustrating Trypanosoma spp. diversity, especially for T. vivax and T. vivax-like trypanosomes. Further studies using isolates belonging to different CatL clades, such as population genomic studies to test recombination or in vivo pathogenesis tests, could clarify the epidemiology and relationship of these parasites with disease manifestation in cattle.
Data Availability Statement
The datasets generated in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MT673751 to MT673783.
Ethics Statement
The animal study was reviewed and approved by ERES Converge IRB, Lusaka, Zambia. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author Contributions
YN, KH, and JY: conceptualization, study design, and drafting of manuscript. YN, VD, YQ, MS, and BN: field sampling. YN: laboratory experiment. YN and RO: data analysis. YN, KH, VD, YQ, RO, MS, CS, BN, and JY: revising and final approval of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Japan Program for Infectious Diseases Research and Infrastructure from Japan Agency for Medical Research and Development (AMED) under grant number JP20w m0125008; and the Global Leadership Training Programme in Africa (GLTP) of the University of United Nation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the members in Shandavu Camp for providing accommodation and support throughout the field sampling in Musungwa chiefdom. We also thank Mr. Jerry Banda and others from the Itezhi-Tezhi District Veterinary Office, and Dr. Cornelius Mweempwa from the Department of Veterinary Tropical Diseases, Faculty of Veterinary Science, University of Pretoria, for their reassuring assistance in fieldwork. We thank Dr. Hayato Harima and Mrs. Ami Soda from Hokudai Center for Zoonosis Control in Zambia (HUCZCZ) for their kind support in procurement and providing us laboratory capacity. Finally, we especially thank Olaf, Mirriam, Boyd, Kennedy, Felix, and Sylvester for organizing visits and interpreting between the farmers, and to all the farmers in Musungwa chiefdom who kindly cooperated with us in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2021.599815/full#supplementary-material
References
1. Desquesnes M, Dia ML. Mechanical transmission of Trypanosoma vivax in cattle by the African tabanid Atylotus fuscipes. Vet Parasitol. (2004) 119:9–19. doi: 10.1016/j.vetpar.2003.10.015
2. Connor RJ. Final Report of the Regional Trypanosomiasis Expert. Regional Tsetse and Trypanosomiasis Control Programme, Malawi, Mozambique, Zambia and Zimbabwe. Konigstein: FGU-Kronberg Consulting and Engineering GmbH (1989).
3. WHO. Investing to Overcome the Global Impact of Neglected Tropical Diseases. Third WHO Report on Neglected Tropical Diseases World Health Organization. WHO (2015).
4. Squarre D, Kabongo I, Munyeme M, Mumba C, Mwasinga W, Hachaambwa L. Human African trypanosomiasis in the Kafue National Park, Zambia. PLoS Negl Trop Dis. (2016) 10:e0004567. doi: 10.1371/journal.pntd.0004567
5. Simukoko H, Marcotty T, Phiri I, Geysen D, Vercruysse J, Van den Bossche P. The comparative role of cattle, goats and pigs in the epidemiology of livestock trypanosomiasis on the plateau of eastern Zambia. Vet Parasitol. (2007) 147:231–8. doi: 10.1016/j.vetpar.2007.04.005
6. Mbewe NJ, Namangala B, Sitali L, Vorster I, Michelo C. Prevalence of pathogenic trypanosomes in anaemic cattle from trypanosomosis challenged areas of Itezhi-tezhi district in central Zambia. Parasites Vectors. (2015) 8:638. doi: 10.1186/s13071-015-1260-0
7. Gardiner PR, Assoku RK, Whitelaw DD, Murray M. Haemorrhagic lesions resulting from Trypanosoma vivax infection in Ayrshire cattle. Vet Parasitol. (1989) 31:187–97. doi: 10.1016/0304-4017(89)90069-1
8. Woo PT. The haematocrit centrifuge for the detection of trypanosomes in blood. Can J Zoo. (1969) 47:921–3. doi: 10.1139/z69-150
9. Uilenberg G, Boyt WP. A Field Guide for the Diagnosis, Treatment and Prevention of African Animal Trypanosomosis. Food and Agriculture Org (1998). Available online at: http://www.fao.org/3/X0413E/X0413E00.htm
10. Desquesnes M, McLaughlin G, Zoungrana A, Dávila AM. Detection and identification of Trypanosoma of African livestock through a single PCR based on internal transcribed spacer 1 of rDNA. Internat J Parasitol. (2001) 31:610–4. doi: 10.1016/S0020-7519(01)00161-8
11. Njiru ZK, Constantine CC, Guya S, Crowther J, Kiragu JM, Thompson RC, et al. The use of ITS1 rDNA PCR in detecting pathogenic African trypanosomes. Parasitol Res. (2005) 95:186–92. doi: 10.1007/s00436-004-1267-5
12. Gaithuma AK, Yamagishi J, Martinelli A, Hayashida K, Kawai N, Marsela M, et al. A single test approach for accurate and sensitive detection and taxonomic characterization of Trypanosomes by comprehensive analysis of internal transcribed spacer 1 amplicons. PLoS Negl Trop Dis. (2019) 13:e0006842. doi: 10.1371/journal.pntd.0006842
13. Bengaly Z, Sidibé I, Ganaba R, Desquesnes M, Boly H, Sawadogo L. Comparative pathogenicity of three genetically distinct types of Trypanosoma congolense in cattle: clinical observations and haematological changes. Vet Parasitol. (2002) 108:1–9. doi: 10.1016/S0304-4017(02)00164-4
14. Mekata H, Konnai S, Simuunza M, Chembensofu M, Kano R, Witola WH, et al. Prevalence and source of trypanosome infections in field-captured vector flies (Glossina pallidipes) in southeastern Zambia. J Vet Med Sci. (2008) 70:923–8. doi: 10.1292/jvms.70.923
15. Masumu J, Marcotty T, Geysen D, Geerts S, Vercruysse J, Dorny P, et al. Comparison of the virulence of Trypanosoma congolense strains isolated from cattle in a trypanosomiasis endemic area of eastern Zambia. Internat J Parasitol. (2006) 36:497–501. doi: 10.1016/j.ijpara.2006.01.003
16. Van den Bossche P, Chitanga S, Masumu J, Marcotty T, Delespaux V. Virulence in Trypanosoma congolense Savannah subgroup. A comparison between strains and transmission cycles. Parasite Immunol. (2011) 33:456–60. doi: 10.1111/j.1365-3024.2010.01277.x
17. Morrison LJ, Tweedie A, Black A, Pinchbeck GL, Christley RM, Schoenefeld A, et al. Discovery of mating in the major African livestock pathogen Trypanosoma congolense. PLoS ONE. (2009) 4:e5564. doi: 10.1371/journal.pone.0005564
18. Holzmuller P, Herder S, Cuny G, De Meeûs T. From clonal to sexual: a step in T. congolense evolution? Trends Parasitol. (2010) 26:56–60. doi: 10.1016/j.pt.2009.11.006
19. Tihon E, Imamura H, Dujardin JC, Van Den Abbeele J, Van den Broeck F. Discovery and genomic analyses of hybridization between divergent lineages of Trypanosoma congolense, causative agent of Animal African Trypanosomiasis. Molec Ecol. (2017) 26:6524–38. doi: 10.1111/mec.14271
21. Rodrigues AC, Neves L, Garcia HA, Viola LB, Marcili A, Da Silva FM, et al. Phylogenetic analysis of Trypanosoma vivax supports the separation of South American/West African from East African isolates and a new T. vivax-like genotype infecting a nyala antelope from Mozambique. Parasitol. (2008) 135:1317–28. doi: 10.1017/S0031182008004848
22. Auty H, Anderson NE, Picozzi K, Lembo T, Mubanga J, Hoare R, et al. Trypanosome diversity in wildlife species from the Serengeti and Luangwa Valley ecosystems. PLoS Negl Trop Dis. (2012) 6:e1828. doi: 10.1371/journal.pntd.0001828
23. Malele I, Craske L, Knight C, Ferris V, Njiru Z, Hamilton P, et al. The use of specific and generic primers to identify trypanosome infections of wild tsetse flies in Tanzania by PCR. Infect Genet Evol. (2003) 3:271–9. doi: 10.1016/S1567-1348(03)00090-X
24. Cortez AP, Ventura RM, Rodrigues AC, Batista JS, Paiva F, Anez N, et al. The taxonomic and phylogenetic relationships of Trypanosoma vivax from South America and Africa. Parasitology. (2006) 133:159–69. doi: 10.1017/S0031182006000254
25. Cortez AP, Rodrigues AC, Garcia HA, Neves L, Batista JS, Bengaly Z, et al. Cathepsin L-like genes of Trypanosoma vivax from Africa and South America-characterization, relationships and diagnostic implications. Molec Cell Probes. (2009) 23:44–51. doi: 10.1016/j.mcp.2008.11.003
26. Adams ER, Hamilton PB, Rodrigues AC, Malele II, Delespaux V, Teixeira MM, et al. New Trypanosoma (Duttonella) vivax genotypes from tsetse flies in East Africa. Parasitology. (2010) 137:641–50. doi: 10.1017/S0031182009991508
27. Rodrigues CM, Garcia HA, Rodrigues AC, Costa-Martins AG, Pereira CL, Pereira DL, et al. New insights from Gorongosa National Park and Niassa National Reserve of Mozambique increasing the genetic diversity of Trypanosoma vivax and Trypanosoma vivax-like in tsetse flies, wild ungulates and livestock from East Africa. Parasites Vectors. (2017) 10:337. doi: 10.1186/s13071-017-2241-2
28. Adams ER, Hamilton PB, Gibson WC. African trypanosomes: celebrating diversity. Trends Parasitol. (2010) 26:324–8. doi: 10.1016/j.pt.2010.03.003
29. Fikru R, Matetovici I, Rogé S, Merga B, Goddeeris BM, Büscher P, et al. Ribosomal DNA analysis of tsetse and non-tsetse transmitted Ethiopian Trypanosoma vivax strains in view of improved molecular diagnosis. Vet Parasitol. (2016) 220:15–22. doi: 10.1016/j.vetpar.2016.02.013
30. Fikru R, Hagos A, Roge S, Reyna-Bello A, Gonzatti MI, Merga B, et al. A proline racemase based PCR for identification of Trypanosoma vivax in cattle blood. PLoS ONE. (2014) 9:e84819. doi: 10.1371/journal.pone.0084819
31. Fermino BR, Paiva F, Soares P, Tavares LE, Viola LB, Ferreira RC, et al. Field and experimental evidence of a new caiman trypanosome species closely phylogenetically related to fish trypanosomes and transmitted by leeches. Internat J Parasitol Parasites Wildlife. (2015) 4:368–78. doi: 10.1016/j.ijppaw.2015.10.005
32. Hamilton PB, Adams ER, Malele II, Gibson WC. A novel, high-throughput technique for species identification reveals a new species of tsetse-transmitted trypanosome related to the Trypanosoma brucei subgenus, Trypanozoon. Infect Genet Evol. (2008) 8:26–33. doi: 10.1016/j.meegid.2007.09.003
33. Lima AP, Tessier DC, Thomas DY, Scharfstein J, Storer AC, Vernet T. Identification of new cysteine protease gene isoforms in Trypanosoma cruzi. Molec Biochem Parasitol. (1994) 67:333–8. doi: 10.1016/0166-6851(94)00144-8
34. Ortiz PA, Da Silva FM, Cortez AP, Lima L, Campaner M, Pral EM, et al. Genes of cathepsin L-like proteases in Trypanosoma rangeli isolates: markers for diagnosis, genotyping and phylogenetic relationships. Acta Tropica. (2009) 112:249–59. doi: 10.1016/j.actatropica.2009.07.036
35. Rodrigues AC, Garcia HA, Ortiz PA, Cortez AP, Martinkovic F, Paiva F, et al. Cysteine proteases of Trypanosoma (Megatrypanum) theileri: cathepsin L-like gene sequences as targets for phylogenetic analysis, genotyping diagnosis. Parasitol Internat. (2010) 59:318–25. doi: 10.1016/j.parint.2010.03.002
36. Garcia HA, Rodrigues AC, Martinkovic F, Minervino AH, Campaner M, Nunes VL, et al. Multilocus phylogeographical analysis of Trypanosoma (Megatrypanum) genotypes from sympatric cattle and water buffalo populations supports evolutionary host constraint and close phylogenetic relationships with genotypes found in other ruminants. Internat J Parasitol. (2011) 41:1385–96. doi: 10.1016/j.ijpara.2011.09.001
37. Caffrey CR, Steverding D. Kinetoplastid papain-like cysteine peptidases. Molec Biochem Parasitol. (2009) 167:12–9. doi: 10.1016/j.molbiopara.2009.04.009
38. Sajid M, McKerrow JH. Cysteine proteases of parasitic organisms. Molec Biochem Parasitol. (2002) 120:1–21. doi: 10.1016/S0166-6851(01)00438-8
39. Nakayima J, Nakao R, Alhassan A, Hayashida K, Namangala B, Mahama C, et al. Genetic diversity among Trypanosoma (Duttonella) vivax strains from Zambia and Ghana, based on cathepsin L-like gene. Parasite. (2013) 20:24. doi: 10.1051/parasite/2013024
40. Rodrigues AC, Ortiz PA, Costa-Martins AG, Neves L, Garcia HA, Alves JM, et al. Congopain genes diverged to become specific to Savannah, Forest and Kilifi subgroups of Trypanosoma congolense, and are valuable for diagnosis, genotyping and phylogenetic inferences. Infect Genet Evolut. (2014) 23:20–31. doi: 10.1016/j.meegid.2014.01.012
41. Mwima HK. A brief history of Kafue National Park, Zambia. Koedoe. (2001) 44:57–72. doi: 10.4102/koedoe.v44i1.186
42. Mkanda FX, Mwakifwamba A, Simpamba T. Traditional stewardship and conservation in the Game Management Areas of Nkala and Namwala, Zambia. Oryx. (2014) 48:514–21. doi: 10.1017/S0030605313000574
43. Rogers DJ, Robinson TP. Tsetse distribution. In: Maudlin I, Holmes PH, Miles MA, editors. The Trypanosomiasis. Wallington: CABI Publishing (2004). p. 139–79.
44. Cochran WG. Estimation of sample size. In: Cochran WG, editor. Sampling Techniques. 3rd ed. New York, NY: John Wiley and Sons Inc. (1977). p. 72–88.
45. Jackson CHN. The analysis of a tsetse-fly population III. Ann Eugen Cambridge. (1948) 14:91–108. doi: 10.1111/j.1469-1809.1947.tb02381.x
46. Dyer NA, Lawton SP, Ravel S, Choi KS, Lehane MJ, Robinson AS, et al. Molecular phylogenetics of tsetse flies (Diptera: Glossinidae) based on mitochondrial (COI, 16S, ND2) and nuclear ribosomal DNA sequences, with an emphasis on the palpalis group. Molec Phylogenet Evol. (2008) 49:227–39. doi: 10.1016/j.ympev.2008.07.011
47. Radwanska M, Chamekh M, Vanhamme L, Claes F, Magez S, Magnus E, et al. The serum resistance-associated gene as a diagnostic tool for the detection of Trypanosoma brucei rhodesiense. Am J Trop Med Hyg. (2002) 6:684–90. doi: 10.4269/ajtmh.2002.67.684
48. Palmer JM, Jusino MA, Banik MT, Lindner DL. Non-biological synthetic spike-in controls and the AMPtk software pipeline improve mycobiome data. Peer J. (2018) 6:e4925. doi: 10.7717/peerj,.4925
49. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. (1990) 215:403–10. doi: 10.1016/S0022-2836(05)80360-2
50. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molec Biol Evol. (2013) 30:772–80. doi: 10.1093/molbev/mst010
51. Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molec Biol Evol. (1987) 4:406–25.
52. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Molec Biol Evol. (2018) 35:1547–9. doi: 10.1093/molbev/msy096
53. Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA. (2018) 101:11030–5. doi: 10.1073/pnas.0404206101
54. Letunic I, Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucl Acids Res. (2019) 47:256–9. doi: 10.1093/nar/gkz239
55. R Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing (2013). Available online at: http://www.R-project.org/ (accessed January 11, 2021).
56. Wilson AJ, Paris J, Luckins AG, Dar FK, Gray AR. Observations on a heard of beef cattle maintained in a tsetse area. Trop Anim Health Prod. (1976) 8:1. doi: 10.1007/BF02383357
57. Connor RJ, Van Den Bossche P. African animal trypanosomoses. In: Coetzer JAW, Tustin RC, editor. Infectious Diseases of Livestock Volume One. 2nd ed. Cape Town: Oxford University Press Southern Africa (2004). p. 251–96.
58. Leak SG. Behavioural ecology. In: Leak SG, editor. Tsetse Biology and Ecology: Their Role in the Epidemiology and Control of Trypanosomosis. Wallingford: CABI Publishing (1999). p.104–46.
59. Leak SG. Population dynamics. In: Leak SG, editor. Tsetse Biology and Ecology: Their Role in the Epidemiology and Control of Trypanosomosis. ILRI (aka ILCA and ILRAD) (1999). p.147–67.
60. Laohasinnarong D, Goto Y, Asada M, Nakao R, Hayashida K, Kajino K, et al. Studies of trypanosomiasis in the Luangwa valley, north-eastern Zambia. Parasites Vectors. (2015) 8:497. doi: 10.1186/s13071-015-1173-y
61. Welburn SC, Picozzi K, Fèvre EM, Coleman PG, Odiit M, Carrington M, et al. Identification of human-infective trypanosomes in animal reservoir of sleeping sickness in Uganda by means of serum-resistance-associated (SRA) gene. Lancet. (2001) 358:2017–9. doi: 10.1016/S0140-6736(01)07096-9
Keywords: Trypanosoma vivax, Trypanosoma vivax bovine trypanosomosis, Trypanosoma vivax-like, African animal trypanosomosis, cathepsin L-like cysteine protease, anemia
Citation: Nakamura Y, Hayashida K, Delesalle V, Qiu Y, Omori R, Simuunza M, Sugimoto C, Namangala B and Yamagishi J (2021) Genetic Diversity of African Trypanosomes in Tsetse Flies and Cattle From the Kafue Ecosystem. Front. Vet. Sci. 8:599815. doi: 10.3389/fvets.2021.599815
Received: 28 August 2020; Accepted: 04 January 2021;
Published: 27 January 2021.
Edited by:
David Modrý, University of Veterinary and Pharmaceutical Sciences Brno, CzechiaReviewed by:
Jan Votypka, Charles University, CzechiaSerena Cavallero, Sapienza University of Rome, Italy
Copyright © 2021 Nakamura, Hayashida, Delesalle, Qiu, Omori, Simuunza, Sugimoto, Namangala and Yamagishi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yukiko Nakamura, eW5ha2FtdXJhQGN6Yy5ob2t1ZGFpLmFjLmpw; Junya Yamagishi, anVueWFAY3pjLmhva3VkYWkuYWMuanA=