 Érica Bravo Sales1
Érica Bravo Sales1 Antônio Augusto Fonseca Jr1Cristina Magalhães Gonçalves1Andrey Pereira Lage2Giovanna Ivo Andrade2
Antônio Augusto Fonseca Jr1Cristina Magalhães Gonçalves1Andrey Pereira Lage2Giovanna Ivo Andrade2 Philip Noel Suffys3
Philip Noel Suffys3 Harrison Magdinier Gomes3Natanael Lamas Dias1
Harrison Magdinier Gomes3Natanael Lamas Dias1 José Soares Ferreira Neto4
José Soares Ferreira Neto4 Ana Marcia de Sá Guimarães4
Ana Marcia de Sá Guimarães4 Marcos Bryan Heinemann4*
Marcos Bryan Heinemann4*- 1Laboratório Federal de Defesa Agropecuária de Minas Gerais, Pedro Leopoldo, Minas Gerais, Brazil
- 2Escola de Veterinária, Universidade Federal de Minas Gerais, Belo Horizonte, Minas Gerais, Brazil
- 3FioCruz, Rio de Janeiro, Rio de Janeiro, Brazil
- 4Universidade de São Paulo, São Paulo, São Paulo, Brazil
The molecular typing of Mycobacterium bovis, which causes bovine tuberculosis, can be accomplished by combining different polymorphic markers, contributing to its epidemiological investigation. Multispacer sequence typing (MST) is a sequencing-based method that employs intergenic regions susceptible to higher mutation rates given the low selection pressure. It has been applied to M. tuberculosis, but not to M. bovis. The aim of this study was to evaluate a MST for M. bovis. A total of 58 strains isolated from tissues with lesions suggestive of bovine tuberculosis, coming from cattle herds in six Brazilian states and four standard samples of M. bovis were typified employing the MST technique. Fourteen intergenic regions were used, and four types of genetic events were reported: single nucleotide mutation (SNP), insertion, deletion, and tandem repeat (TR). Seven loci were chosen for typing. Twenty-eight type sequences (ST) were identified, indicating type sequences (ST) were identified, indicating a 92.9% HGDI (Hunter Gaston Discriminatory Index). The data were used to analyze the evolutionary patterns of these isolates and correlate them to phylogeographic lineages based on the formation of clonal complexes generated from eBURST software. Later, we associated the MST with spoligotyping technique, currently considered the gold standard for classification of M. bovis. The results support the MST as an alternative method for genotyping of M. bovis. The method has the advantage of sequencing and the availability of sequences analyzed in public databases, which can be used by professionals around the world as a tool for further analysis. This was the first study to identify the variability of isolates of M. bovis by the MST method.
Introduction
Mycobacterium bovis is the main pathogen of bovine tuberculosis (TB), an important disease of domestic cattle, which can also be associated with an underestimated number of human TB cases (zoonotic TB) and serious threats to wildlife conservation (1–4). Bovine TB is a disease of high health and economic relevance for livestock and is part of the group of notifiable diseases listed by the World Organization for Animal Health (OIE) (5). Infection routes in the cattle are influenced by factors such as age, environment, and hygiene practices, as the bacterium can be eliminated via various routes, such as respiratory droplets and aerosol, milk, feces, urine, vaginal secretions and/or semen (6). The presence of wildlife reservoirs in certain regions also hampers the control of the disease in livestock and translate into serious consequences for the preservation of wildlife species in multi-host systems (7). The disease directly affects livestock productivity, influencing international trade of animal products and, due to its zoonotic potential, is also of public health concern (8, 9).
Mycobacterium bovis belongs to the Mycobacterium tuberculosis complex (MTBC), a clonal group of mycobacterium species, lineages and/or ecotypes that cause similar diseases in a variety of mammalian hosts (10–12). Genomes of MTBC members are highly similar, with >99.9% identity over homologous regions, including the 16S rRNA gene, and absent remodeling through horizontal gene transfer or DNA recombination (10, 13, 14). This bacterial complex evolves only through SNPs (single nucleotide polymorphisms), short indels, large deletions, transposition of insertion sequence (IS) elements, and duplication of few paralogous gene families (10, 15, 16). Despite this high genetic similarity, species and lineages of the MTBC present variable phenotypes of host tropism and virulence (10–14).
Over the years, molecular genotyping techniques have significantly contributed to epidemiological studies of bovine TB, facilitating the knowledge of the spatial and temporal distribution of M. bovis strains, from populational structure to outbreak investigations (17, 18). Given the clonal nature of the MTBC, these techniques are based on few highly polymorphic regions of their genomes, including tandem repeats, a CRISPR locus, PGRS genes, and IS elements (17–21). The IS6110-based RFLP is a technique commonly used in M. tuberculosis genotyping, but of little application for M. bovis strains, as they carry only few IS6110 copies in their genomes (22). Among the remaining techniques, spacer oligonucleotide typing (spoligotyping; based on the CRISPR locus) and variable number tandem repeat- mycobacterial interspersed repetitive units (MIRU-VNTR) have been the preferred combination for typing isolates of M. bovis in several countries (17–21). However, both techniques have limitations. Spoligotyping does not provide good resolution at the region or farm level, which limits its use for pathogen's transmission detection (17, 18). Spoligotyping is also sensitive to homoplasy, i.e., unrelated lineages presenting identical spoligotypes (23). This happens because the loss of a spacer region at the spoligotyping locus is a common event that can occur in strains that are not phylogenetically related (23, 24). While MIRU-VNTR has been reported to better discriminate among isolates at the farm level, there are 24 loci to choose from, and laboratories must frequently test which loci would better fit their reality for better discriminatory power (25). MIRU-VNTR is also influenced by homoplasy (24) and depending on the laboratory infrastructure, a 24 loci evaluation of M. bovis isolates to avoid homoplasy bias can be very laborious and time consuming (17).
Recently, M. bovis whole-genome sequencing (WGS) using next-generation sequencing platforms has been suggested to replace these genotyping assays (17, 18). However, WGS still faces many challenges related to its financial burden and requirement of specialized personnel, particularly in low and middle-income countries in which bovine TB is highly endemic. Most reported M. bovis WGS studies have been conducted in developed countries or in developing countries by researchers from high-income nations (26–32). Thus, albeit likely ideal, WGS use is still unreachable to many programs of bovine TB control and eradication around the world. Thus, there is a need for the continuous development of genotyping techniques with high resolution power. Accordingly, a multispacer sequence typing (MST) scheme for M. tuberculosis based on the Sanger sequencing of eight PCR-amplified intergenic regions, including four regions previously reported as VNTR regions, has been proposed (33). The aim of this study was to investigate the usefulness of this MST scheme to genotype 58 M. bovis isolates obtained from different Brazilian states.
Materials and Methods
Bacterial Isolates
A total of 58 cultured isolates of M. bovis, deposited in the sample bank of the National Agricultural Laboratory of Minas Gerais (Lanagro/MG), Brazil, were selected. These isolates were obtained from cattle herds located in six Brazilian states [Minas Gerais (n = 48), Goiás (n = 3), Mato Grosso-MT (n = 1), Rio Grande do Sul-RS (n = 1), Paraná-PR (n = 1) and São Paulo (n = 4)] between the years 2006 and 2010. First isolation of these M. bovis from clinical samples were performed in Lanagro/MG according to previous publications (34). Isolated strains were identified as M. bovis by using a PCR assay described by Sales et al. (35). Four additional laboratory strains [M. bovis CR01; M. bovis AN5 (36); M. bovis BCG CR13; M. bovis México CR36] were also selected. All procedures were performed in a Biosafety Level 3+ Laboratory (BSL-3+) located at the Farming National Laboratory (LANAGRO) Minas Gerais, Ministry of Agriculture, Livestock and Supply, Brazil. Tubes containing DNA were properly disinfected, removed from BSL3+ and stored at −20°C until further analysis.
DNA Extraction and PCR Assays
A loop of each bacterial strain cultured in Stonebrink medium was subjected to a phenol-chloroform DNA extraction as previously described (37). Extracted DNA was subjected to conventional PCR assays of 14 intergenic spacer regions using previously described primer sequences (33). Each PCR assay was performed in a PX2 Thermal Cycler thermocycler (Milford, MA, USA). Briefly, 20 μL were prepared with the following concentrations of reagents: 10 pmol of each primer, 1.5 mM MgCl2, 10 mM dNTPs (Invitrogen, USA), 1 U Jump Start®Taq DNA Polymerase (Sigma, USA) and 1 X GoTaq Green® buffer (Promega, USA). DNA sample concentration added in each reaction was 100 ng/μL. As positive controls, we used DNA extracted from M. bovis strain AN5 (38), while negative controls consisted of ultrapure water. The conditions of the PCR assays determined for each pair of primers are shown in Supplementary Table 1. Amplification products were detected by gel visualization following electrophoresis.
Sequencing and Data Analysis
The PCR products were purified using the PureLink® PCR Purification Kit (Invitrogen, USA) and sequenced using Big Dye® Terminator V.3.1 Cycle Sequencing Kit (Applied Biosystems, USA) according to the manufacturer's recommendations. Sequencing was performed in both directions in the 3130 Genetic Analyzer (Applied Biosystems, USA) equipment.
Sequence assemblies were performed with CAP3 program (39) using nucleotide data with quality higher than PHRED 20. Sequences were edited in Bioedit v.7.2.5 program (40). Subsequently, all sequences obtained for each primer pair were subjected to a multiple sequence alignment using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) to verify the degree of identity between two or more sequences and the allelic diversity present in each spacer region. Each allele detected was assigned an identification number. For each individual DNA sample, the set of loci and their respective allele defined an allelic profile or sequence type (ST), which identified a clone. Bacterial isolates analyzed in this study were identified by numbers 1–58. The laboratory strains M. bovis CR01, AN5, CR13, CR36, and AF2122/97 (Genbank: NC002945.3) included in the analyses were represented by the numbers 59–63, respectively.
Spoligotyping
The spoligotyping was performed as described by Kamerbeek (41) using spolygotyping kit by Ocimum Biosolutions. The identification of individual spoligotypes and the appointment of unpublished patterns were made using Mbovis.org website (www.mbovis.org) (42).
Analysis of Genetic Relationship Among the Isolates
An evolutionary descent analysis of M. bovis isolates was performed using eBURST algorithm (43), available in Phyloviz software (44). Clonal complexes (CC) as defined by eBURST were identified and organized in dendrograms based on the comparison of the obtained allelic profiles of the tested M. bovis isolates. Briefly, CCs were determined by a simple model of clonal expansion and diversification based on the differences between these allelic profiles. Each CC was assigned a founder genotype that displays a more likely hypothetical pattern of evolutionary descent among the STs that make up this complex. The STs were organized into complexes by comparing the variation that exists among the loci of each founder genotype loci compared to other isolates (43). Accordingly, genotypic variations found in only one locus are called single locus variants (SLV), in two loci are called double locus variants (DLV), and in three loci are called triple locus variants (TLV) (43, 44). Confidence values for each SLV, DLV and TLV detected in each ST founder in a CC were assessed by bootstrapping (43). The geographic location and year of isolation of the strains were also included in the analysis.
Phylogenetic Analysis
The phylogenetic reconstruction of M. bovis isolates was performed by PubMLST program (http://pubmlst.org/) using the Neighbor-Joining model. The program uses PHILIP package to create Neighbor-Joining and UPGMA trees based on allelic profiles determined in the study.
Calculation of Allelic Diversity
The discrimination index (D) previously described by Hunter-Gaston (45) was used to calculate the allelic diversity at each locus and to assess genotypic diversity for each method employed (MST and spoligotyping).
Results
Multispacer Sequence Typing (MST)
Fourteen intergenic spacer regions were analyzed in the selected M. bovis isolates. Regions MST1, MST2, MST3, MST6, and MST8 showed no sequence variation in an initial evaluation of 15 randomly chosen M. bovis isolates of the collection and were then excluded from the study. In addition, after multiple attempts, the PCR assay of the MST10 region failed to amplify appropriate PCR products, and this region was also excluded from the study. For the MST14 region, Sanger sequencing was not able to cover the whole extent of the region, hampering the identification of possible polymorphic positions for some of the samples and was also excluded from further evaluation. Finally, the remaining regions MST4, MST5, MST7, MST9, MST11, MST12, and MST13 proved quite variable, with a total of 28 mutational events, including single nucleotide polymorphisms (SNP), insertions, deletions, and tandem repeats. Figure 1 shows the alleles identified for each of the seven evaluated spacer regions.

Figure 1. Alleles identified in seven intergenic spacers in Mycobacterium bovis. MST4 (ETR-B): eight alleles containing 1, 2, 3, 4, 5, and 7 copies of the 57 bp tandem repeat sequence (TR) and the presence of a SNP G47A (alleles 5 and 7). MST5: deletion of 67 bp. MST7: Five alleles containing from 2, 3, 4 and 5 copies of the 57 bp tandem repeat, a deletion of the first 11 bp repeat for all samples and a SNP G80A in allele 3. MST9: two alleles containing one and three repeat tandem copies of 54 bp, and a sequence represented by some polymorphisms in the genome of M. bovis, characterized by a different allele of a repeated region also present in the genome of M. avium. MST11: three alleles containing 3, 5, and 6 tandem repeats of 57 bp, the first and last repetition characterized by a deletion of 11 bp in all isolates. MST12: five alleles containing 3–5 repeat tandem copies of 77 bp with the first repetition characterized by a deletion of 37 bp in all isolates, the presence of a G76 base (allele 5), A424G SNP (allele 2–5), T485A SNP (allele 3) and a deletion of 18 bp after the last repeat (allele 1). MST13: three alleles containing from 1 to 3 copies of the 57 bp and a 23 bp deletion in the first repetition in all isolates.
Definition of Genetic Profiles
Multiple sequence alignment of the DNA sequences of all M. bovis isolates for each locus allowed the identification of up to 8 different alleles which received individual identifier numbers (Figure 1). The grouping of all loci for each individual defined an allelic profile that was represented by a sequence type (ST). For the 63 isolates of M. bovis studied, 28 STs were found, where eight were represented by more than one M. bovis isolate, and 20 were described as orphan STs, as shown in Table 1.
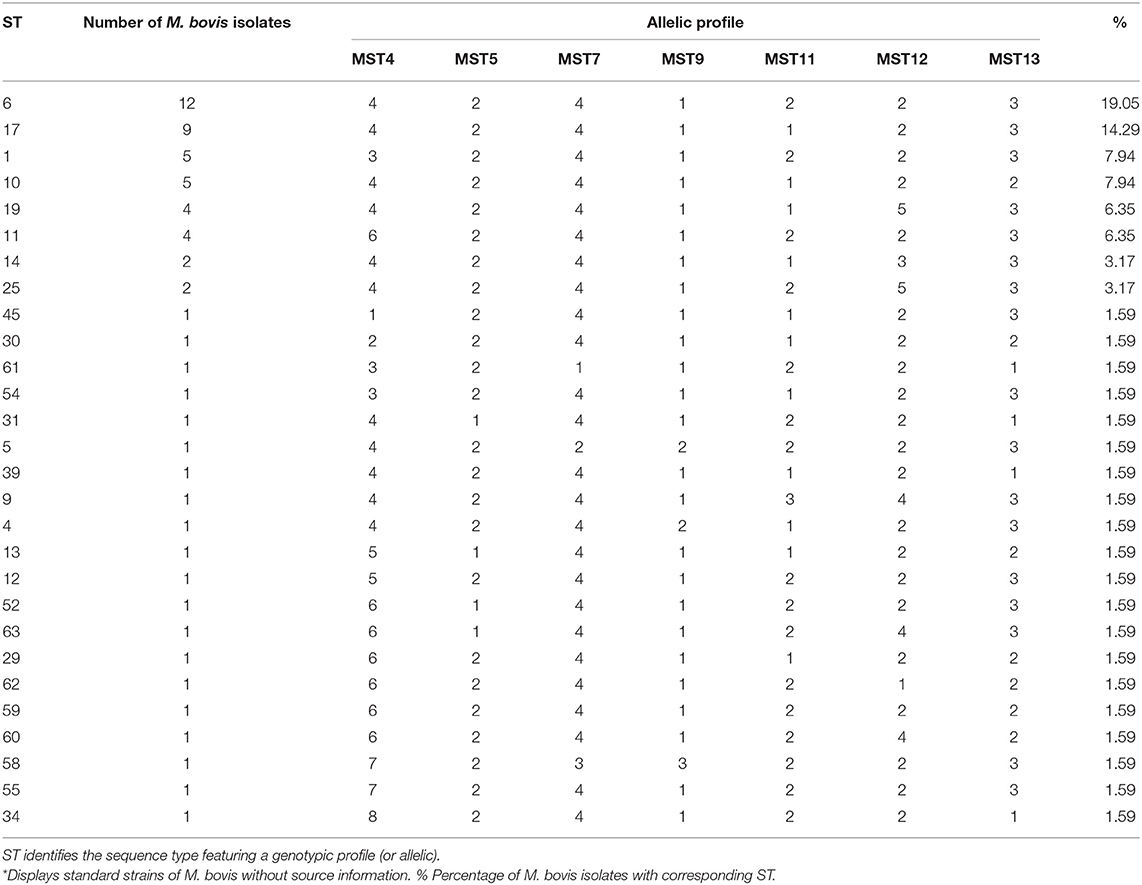
Table 1. Twenty-eight (28) ST identified by the comparison of genotypic profiles of 63 isolates of M. bovis.
Evolutionary Descent Analysis
The STs detected in the study were used to determine the genetic relationship of M. bovis isolates. A total of five CCs (ST6, ST10, ST11, ST17, and ST59) were detected (Figure 2). ST17, consisting of nine M. bovis isolates, being eight from Minas Gerais state and one from São Paulo state, was appointed as the founder CC of the whole group, as it had the highest number of SLVs (ST4, ST6, ST10, ST14, ST19, ST39, ST45, and ST54, including 24 M. bovis isolates from Minas Gerais, two from São Paulo and one from Mato Grosso) with diverging genotypes in only one of seven loci. ST6 was appointed the largest CC and presented the highest frequency of isolates with the same genotypic profile from different geographical locations: 10 M. bovis isolates from Minas Gerais, two from São Paulo, and one from Mato Grosso. This complex presented six SLVs, which expressed close relationship to ST1, ST11, ST12, ST17, ST25, and ST55, with 22 M. bovis isolates from the states of Minas Gerais (n = 19), São Paulo (n = 1), Goiás (n = 1), and Paraná (n = 1). ST11, consisting of four strains, coming from Minas Gerais and Goiás, is one of the SLVs of ST6 that has diversified producing their own SLV, characterized by members with unique genotypic profiles. Interestingly, the single M. bovis isolate from Rio Grande do Sul, the only state not bordering any of the states evaluated herein, having unique genotype (ST13) without associated SLVs, DLVs or TLVs.

Figure 2. Minimum spanning tree exhibiting the formation of clonal complexes according to genotypic profiles obtained by the multi sequence typing of Mycobacterium bovis. Distribution of 58 samples of M. bovis isolated from the years 2006 to 2010, four standard samples of M. bovis (CR01, CR02, CR13, and CR36) and the strain M. bovis (AF2122/97). The dendrogram reveals the formation of five clonal complexes ST6, ST10, ST11, ST17, and ST59 that formed from the comparison of 28 ST. In the subtitle “N” figure indicates the number of isolates with the same genotypic profile, as well as the size of the circles (the larger the circle, the larger the population represented). Circles with more than one color profile indicate the same genotype isolated from different regions and year of isolation of the pathogen.
Spoligotyping and MST
Spoligotyping was performed for 55 of the 63 M. bovis isolates that composed the study. A total of 22 spoligotypes were detected, 19 of which were already mentioned in the literature. A total of eight clusters (G1–G8) were formed, representing 74.54% (41/55) of the M. bovis isolates. The remaining 14 (24.45%) M. bovis isolates had the spoligotype described as orphans (Table 2). SB2211, SB2212, and SB2213, found in samples from Minas Gerais, São Paulo, and Mato Grosso, respectively, have never been described prior to this study. SB1135 was the most frequent spoligotype (n = 12), followed by SB0121 (n = 9), SB0295 (n = 6), and SB0881 (n = 5). SB1135 had samples only from Minas Gerais, while SB0121 and SB0295 were represented by Minas Gerais and São Paulo; and SB0881 by Minas Gerais and Goiás.

Table 2. Spoligotypes of 55 Mycobacterium bovis isolates.
A separate analysis contrasting MST and Spoligotyping was also performed using samples from Minas Gerais only, which corresponded to 43 of the 58 M. bovis. These samples came from 22 different towns and were isolated between 2006 and 2010. A total of 19 STs and 17 spoligotypes were identified. The eBURST displayed ST6 as the main clonal complex, showing for this CC three SLVs (ST17, ST11, and ST1). ST6 reported eight samples from seven different cities, and four spoligotypes (SB0295, SB0121, SB1135, and SB0333); ST17 reported seven samples from three different cities, and four spoligotypes (SB1135, SB0295, SB0333, and SB0881); ST1 composed five M. bovis isolates, from three cities, and three spoligotypes (SB1033, SB1050, and SB2212); and ST11, which depicted three samples, each one from a different city and showed orphans spoligotypes (SB0267, SB0849, and SB1802). The three most frequent spoligotypes were SB1135, represented by 12 samples isolated in 2007, all of them from a same town; SB0121 which was reported in eight samples, from seven cities and isolated in 2006, 2007, 2009, and 2010; and SB0295 reported in five samples, from four cities and isolated in 2008 and 2009 (Figure 3).
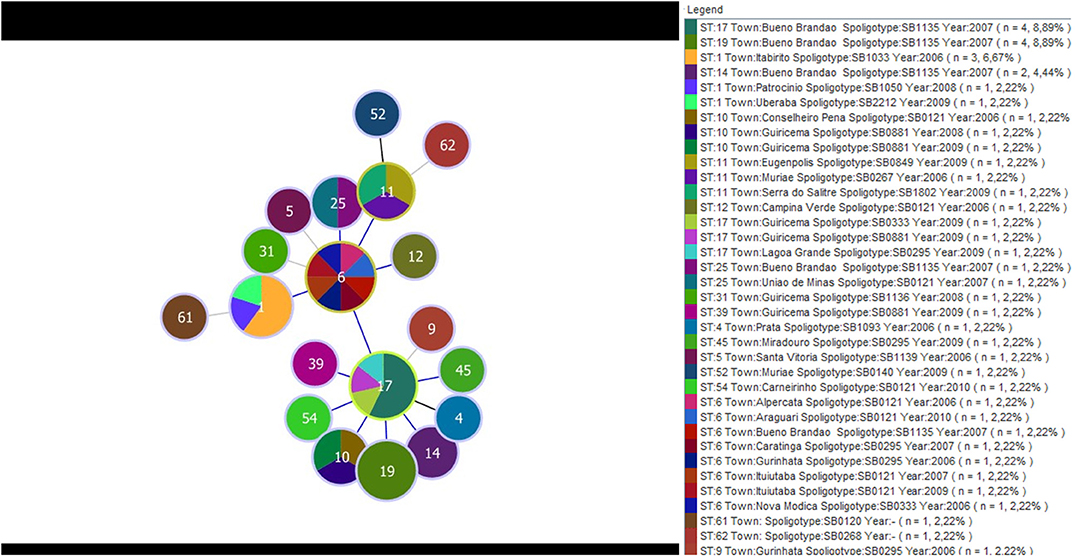
Figure 3. Minimum spanning tree displaying the distribution of 19 type sequences and 17 spoligotypes determined by the MST and Spoligotyping, respectively. The data were obtained from 43 isolates of M. bovis from 22 municipalities in MG and isolates between the years 2006 and 2010. The colors represent the different spolitypes and ‘n' indicates the number of isolates with the same genotype profile.
Phylogenetic Analysis
The phylogenetic tree was generated from 28 STs constituting a cladogram where each ST was represented by its number (Figure 4). Overall, M. bovis isolates were similarly clustered in the same groups formed by eBURST. It was observed that ST1, ST6, and ST12 were closely located in the center of the tree. When comparing this phylogenetic tree with the genotypic profiles of each ST, the genetic variation was limited by the difference in a single locus, admitting the close relationship between these isolates, which coincide with data observed previously by eBURST. The formation of five clades distributed in the tree edges was observed and the relationship between these clades was generated by variation of three or more loci identified by MST. Clade A was represented by the sequence type coming from Minas Gerais, São Paulo, and Paraná. Clade B was formed only by isolates from Minas Gerais. Clade C was represented by members of Minas Gerais and Goiás. And finally, clade D was represented by a single sample from Goiás and M. bovis BCG CR13, while clade E was formed by laboratory strains M. bovis CR01, CR02, AN5, CR36, and AF2122/97.
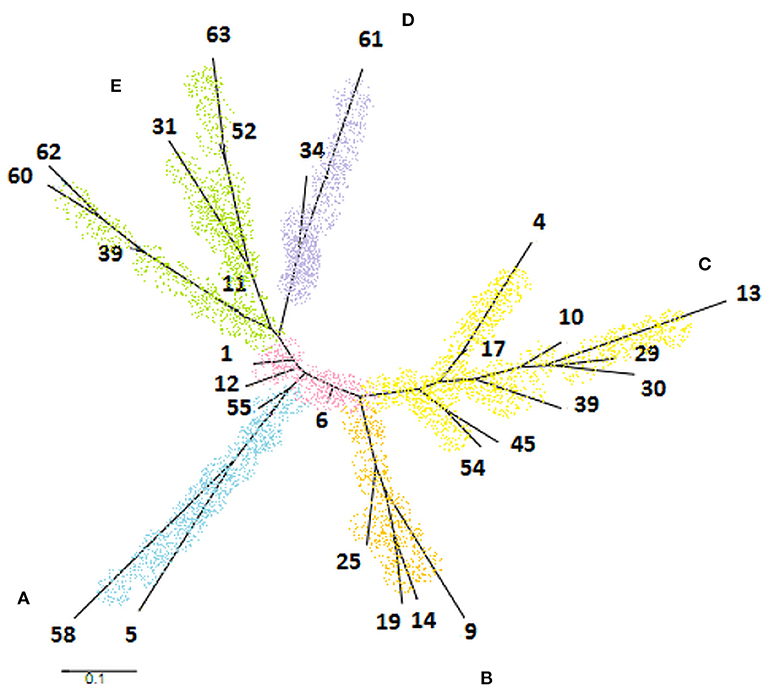
Figure 4. Phylogenetic tree generated by PubMLST program based on 28 sequence types of Mycobacterium bovis obtained in the study. It was observed the formation of five clades identified by letters and colors as follows: clade A (blue), clade B (orange), clade C (yellow), clade D (purple), and clade E (green).
The clade A, marked by the ST5 and ST58, showed the same pattern of spoligotype (SB1139), so as clade D, composed of ST34 and ST61 of spoligotype SB0120. The clade B, formed by ST9, ST14, ST19, and ST25, is composed of spoligotypes SB0295 and SB1135. These two spoligotypes differ only by the absence of the spacer eight in SB1135 compared to SB0295. Clade C includes eight different ST and four spoligotypes. The three ST located in the base of the tree were well-diversified in terms of spoligotypes patterns, corresponding to the high diversity of the ST6. Interestingly, although highly diverse in terms of ST types, these isolates differ only by SLVs. Clade E presented high diversity both of spoligotypes and of genotypic profiles described for ST (Figure 4).
Calculation Hunter and Gaston
The discriminatory power of MST and spoligotyping techniques were calculated according to the Hunter-Gaston discrimination index (45). The determination was based on the number of genotypic profiles defined during the test and the relative frequencies of each profile. For MST, the calculations were made for the 63 strains of M. bovis and 28 ST resulting in a 92.9% HGDI. Yet for spoligotype, reducing to 55 the number of isolates and to 22 the number of patterns, the index lowered to 91%.
Discussion
The 14 spacer regions analyzed in this study for isolates of M. bovis were first described by Djelouadji et al. (33) to characterize M. tuberculosis isolates. The decision of analyzing them was taken based on the genetic similarity corresponding to 99.95% between these two species. The markers used are from intergenic regions susceptible to higher mutation rates given the low selection pressure. As the bovine bacillus has a broad host range compared to human germ and evolution occur under various conditions for both, we studied the 14 regions to find the most variable in M. bovis.
Genotyping by the MST included the evaluation of 14 intergenic spacer regions, 58 representative samples of M. bovis, four standard samples and AF2122/97 strain obtained from GenBank. A total of seven spacers showed genetic variability for the analyzed isolates. Four of them were previously described as Exact Tandem Repeat (ETR) defining ETR-B (MST4), ETR-C (MST11), ETR-D also called MIRU4 (MST12) (46, 47) and Mtub21 (MST13) (48). The other three spacers used were MST5, MST7, and MST9. The results showed the occurrence of 28 genetic events, distributed among deletions, insertions, variation in the number of tandem repeats and point mutations. Sequencing shows the exact number of tandem repeats present in each sequence, and also determines stable markers within them such as SNPs (49) considered very important for studies of genetic events such as phylogeny.
Drancourt et al. (50) used ETR D-region (MST12) to differentiate eight M. tuberculosis complex members by sequencing. The region MST12 (ERT-D) in this study revealed the presence of five alleles characterized by a 40 bp sequence in all isolates, followed by 2–4 copies of the tandem repeat of 77 bp. It was also revealed G76 insertion of a base, an SNP A424G, an SNP T485A and a deletion of 18 bp after the last repetition of the first allele. These events were detected by comparison with the genome of the strain AF2122/97 analyzed in silico (Figure 1). These data, when compared to sequences characterized by (50) available in Genbank (EU180228- EU180234), showed that profiles found for M. bovis in this study were different from the profiles described by the author for the number of repetitions and for SNP.
MST performed for isolates of M. tuberculosis defined eight of 14 loci analyzed as variables (MST1, MST2, MST3, MST4, MST8, MST11, MST12, and MST13). In our study, four of these (MST1, MST2, MST3, and MST8) were excluded from our study due to low variability in the isolates of M. bovis. Djelouadji et al. (33) identified thirteen alleles in these regions that constituted genetic events of deletions and nucleotide substitutions. The other four spacers (MST4, MST11, MST12, and MST13) were characterized mainly by tandem repeats and showed a different profile found for M. bovis. A frequently observed event was the presence of a point mutation at the first base of many of the repetitions, which did not occur in any of the isolates of M. bovis studied in this work. MST12 and MST13 had a larger number of alleles for M. tuberculosis, while MST4 and MST12 were more variable for M. bovis.
The low genetic variability observed in the excluded MST may be related to the sampling bias in samples from Minas Gerais in relation to other states. The existing allelic diversity among M. bovis and M. tuberculosis may be due to the wide range of hosts of the bacillus of bovine, evolving in different environments and under different conditions (51). The population size and genetic diversity found in M. tuberculosis strains in relation to M. bovis can also be representative of these differences.
The most common spoligotypes in our study were SB1135 (21.8%), SB0121 (16.36%), SB0295 (10.9%) and SB0881 (9%) (Table 2). Of these, SB1135 has been described so far only in Brazil (Mbovis.org). Except for SB1135, the other three SB were also reported by Parreiras et al. (52), as well as SB0120, SB1802, SB1145, SB1033, and SB0267 described as an orphan or less frequent. Corroborating our findings, Rodriguez et al. (53) described SB0295 and SB0121 as the majority SB in a study conducted on a national level, as well as (38) which also recorded the occurrence of SB0295 spoligotype in the country. There have been reports of both in many Brazilian isolates and other Latin American countries (54). SB0881 was described previously in Brazil (55), France (56) and Spain (57). Haddad et al. (56) described the SB0295 and SB0121 in Holland in Belgium. SB0120 and SB0295 were also reported among animal wildlife and domestic species, in southern Spain (58), while SB0121 was identified in cattle and wild fauna in the Iberian Peninsula (59) and had also occurred in Mexico and Venezuela (54).
On the less frequent spoligotypes or orphans, SB0140 was described by Smith et al. (60, 61) as one of the most frequent patterns found throughout the world, particularly in Europe and the British Isles; however, the pattern was observed only in a single isolate in our study. SB0140 was also the most frequent pattern according to a study by (54) using isolates from Latin America. SB1145 and SB1802 were only isolated in Brazil, previously reported in Minas Gerais, also with low frequency (52).
CCs were formed and organized from the association of seven genetic regions. The use of clonal complexes as a tool for population analysis is important because it allows us to recognize specific subpopulations or clones that have unique genetic characteristics. Epidemiologically related isolates are derived from the clonal expansion of a single cell, resulting in common characteristics (62). By analyzing MST in association with spoligotyping we observed that some isolates were closely related with exclusive MST profiles and spoligotypes while others, coming from the same region and year, showed different profiles and patterns. The results identified 15 orphans' profiles for MST, 14 orphan spoligotypes and eight groups represented by more than one isolate with equal genotypes in both techniques (Figure 2). Similar results were obtained by Parreiras et al. (52) by associating spoligotyping and MIRU-VNTR. The procedure used for this kind of occurrence typed by VNTR in association with spoligotyping is to rearrange the isolates identified by the same VNTR profiles groups geographically located at spoligotyping (60), thereby obtaining more data on the phylogeny of the bovine bacillus.
The importance of finding an alternative method for genotyping M. bovis is a result of the occurrence of remarkably diverse populations in different countries, making it difficult to determine a set of useful markers that meets all regions (59). Regional differences in the discriminatory power of genetic markers make it necessary to define an optimal combination of markers that can be used in a particular region or country (63). The different patterns obtained indicate the variety of strains that infect cattle from different geographical areas.
In this study, seven polymorphic loci MST were characterized as variables for M. bovis. The observed molecular variation was explored to determine the evolutionary relationships of this pathogen. The results showed that the tested regions can be a promising tool in epidemiological studies. IS6110 RFLP techniques, spoligotyping, and VNTR have been used as a reference for studying the diversity of species of the M. tuberculosis complex (55). However, these methods do not recognize the genetic diversity of strains analyzed as sequencing-based methods capable of identifying all the genetic events present in the molecular marker. The MST method proposed offers the advantage of sequencing, which allows for the detection of different genetic events observed in one sequence. The typing methods performed based on sequencing provide important data on evolutionary forces that shape bacterial populations (64). Bing et al. (65) determined that a methodology based on sequencing of non-constotive genes (MLSA) can be used for molecular typing, in addition to being an excellent method for use in phylogenetic analyzes, for epidemiological and surveillance studies.
Our results demonstrate that the number of samples needs to be increased to provide additional data to monitor the formation and expansion of clonal complexes according to geographic location and year of isolation. This was the first study to identify the variability of isolates of M. bovis by the MST method. The results indicated that the method works to typify differentiate isolates of M. bovis using fewer than those used in VNTR or special reagents like the membranes used in spoligotyping.
Conclusions
Genotyping by MST can contribute to epidemiological studies of M. bovis. The method was efficient to detect the genetic variability present in the sequences analyzed and to infer evolutionary relationships in the short term. The method was also suitable to correlate geographical location and phylogenetic data of M. bovis isolates used in this study.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
ES, CG, GA, HG, ND, and PS performed the all-laboratory tests. ES, AF, and AL performed the interpretation of DNA sequencing results. AF, MH, and AL wrote the manuscript and AG did the translation. MH, AG, PS, and JF accurately reviewed the manuscript. All authors have read and approved the final version of the manuscript.
Funding
This study was financed in part by CAPES, Brasil–Finance Code 001.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
MH, AL, JF are grateful to CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) the fellowship. ES would like to thank CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) for the scholarship.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2021.666283/full#supplementary-material
References
1. Olea-Popelka F, Muwonge A, Perera A, Dean A, Mumford E, Erlacher-Vindel E, et al. Zoonotic tuberculosis in human beings caused by Mycobacterium bovis - a call for action. Lancet Infect Dis. (2017) 17:e21–5. doi: 10.1016/S1473-3099(16)30139-6
2. Miller RS, Sweeney SJ. Mycobacterium bovis (bovine tuberculosis) infection in North American wildlife: current status and opportunities for mitigation of risks of further infection in wildlife populations. Epidemiol Infect. (2013) 141:1357–70. doi: 10.1017/S0950268813000976
3. De Kantor IN, Ritacco V. An update on bovine tuberculosis programs in Latin American and Caribbean countries. Vet. Microbiol. (2006) 112:111–8. doi: 10.1016/j.vetmic.2005.11.033
4. Godfray HCJ, Donnelly CA, Kao RR, Macdonald DW, McDonald RA, Petrokofsky G, et al. A restatement of the natural science evidence base relevant to the control of bovine tuberculosis in Great Britain. Proc R Soc B Biol Sci. (2013) 280:20131634. doi: 10.1098/rspb.2013.1634
5. World Organizatio for Animal Health. Bovine Tuberculosis Chapter 3.4.6. In: Terrestrial Manual p1058-1074. 8th ed. (2018).
6. Domingo M, Vidal E, Marco A. Pathology of bovine tuberculosis. Res Vet Sci. (2014) 97:S20–9. doi: 10.1016/j.rvsc.2014.03.017
7. Hardstaff JL, Marion G, Hutchings MR, White PCL. Evaluating the tuberculosis hazard posed to cattle from wildlife across Europe. Res Vet Sci. (2014) 97:S86–93. doi: 10.1016/j.rvsc.2013.12.002
8. Acha PN, Szyfres B. Zoonosis y enfermidades transmisibles comunes al hombre y a los animales. Bacteriosis y micosis. Washington: OPAS. (2001). p. 416. doi: 10.1590/S1135-57272001000300009
9. Cosivi O, Grange JM, Daborn CJ, Raviglione MC, Fujikura T, Cousins D, et al. Zoonotic tuberculosis due to Mycobacterium bovis in developing countries. Emerg Infect Dis. (1998) 4:59–70. doi: 10.3201/eid0401.980108
10. Galagan JE. Genomic insights into tuberculosis. Nat Rev Genet. (2014) 15:307–20. doi: 10.1038/nrg3664
11. Gagneux S. Strain variation in the Mycobacterium tuberculosis complex: its role in biology, epidemiology and control. Adv Exp Med Biol. (2017) 1019:307. doi: 10.1007/978-3-319-64371-7
12. Brites D, Loiseau C, Menardo F, Borrell S, Boniotti MB, Warren R, et al. A new phylogenetic framework for the animal-adapted Mycobacterium tuberculosis complex. Front Microbiol. (2018) 9:2820. doi: 10.3389/fmicb.2018.02820
13. Hirsh AE, Tsolaki AG, DeRiemer K, Feldman MW, Small PM. Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc Natl Acad Sci. (2004) 101:4871–6. doi: 10.1073/pnas.0305627101
14. Gagneux S, Small PM. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis. (2007) 7:328–37. doi: 10.1016/S1473-3099(07)70108-1
15. Mulholland CV, Shockey AC, Aung HL, Cursons RT, O'Toole RF, Gautam SS, et al. Dispersal of Mycobacterium tuberculosis driven by historical European trade in the South Pacific. Front Microbiol. (2019) 4:2778. doi: 10.3389/fmicb.2019.02778
16. Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci. (2002): 99:3684–9. doi: 10.1073/pnas.052548299
17. Guimaraes AMS, Zimpel CK. Mycobacterium bovis: From genotyping to genome sequencing. Microorganisms. (2020) 8:667. doi: 10.3390/microorganisms8050667
18. Kao RR, Price-Carter M, Robbe-Austerman S. Use of genomics to track bovine tuberculosis transmission. Rev Sci Tech. (2016) 35:241–58. doi: 10.20506/rst.35.1.2430
19. Ramos DF, Tavares L, da Silva PEA, Dellagostin OA. Molecular typing of Mycobacterium bovis isolates: a review. Braz J Microbiol. (2014) 45:365–72. doi: 10.1590/S1517-83822014005000045
20. Haddad N, Masselot M, Durand B. Molecular differentiation of Mycobacterium bovis isolates. Review of main techniques and applications. Res Vet Sci. (2004) 76:1–18. doi: 10.1016/S0034-5288(03)00078-X
21. Merker M, Kohl TA, Niemann S, Supply P. The evolution of strain typing in the Mycobacterium tuberculosis complex. Adv Exp Med Biol. (2017) 1019:79–93. doi: 10.1007/978-3-319-64371-7_3
22. Gonzalo-Asensio J, Pérez I, Aguiló N, Uranga S, Picó A, Lampreave C, et al. New insights into the transposition mechanisms of IS6110 and its dynamic distribution between Mycobacterium tuberculosis Complex lineages. PLoS Genet. (2018) 14:e1007282. doi: 10.1371/journal.pgen.1007282
23. Warren RM, Streicher EM, Sampson SL, van der Spuy GD, Richardson M, Nguyen D, et al. Microevolution of the direct repeat region of Mycobacterium tuberculosis: Implications for interpretation of spoligotyping data. J Clin Microbiol. (2002) 40:4457–65. doi: 10.1128/JCM.40.12.4457-4465.2002
24. Reyes JF, Chan CHS, Tanaka MM. Impact of homoplasy on variable numbers of tandem repeats and spoligotypes in Mycobacterium tuberculosis. Infect Genet Evol. (2012) 12:811–8. doi: 10.1016/j.meegid.2011.05.018
25. Hauer A, Michelet L, De Cruz K, Cochard T, Branger M, Karoui C, et al. MIRU-VNTR allelic variability depends on Mycobacterium bovis clonal group identity. Infect Genet Evol. (2016) 45:165–9. doi: 10.1016/j.meegid.2016.08.038
26. Biek R, O'Hare A, Wright D, Mallon T, McCormick C, Orton RJ, et al. Whole genome sequencing reveals local transmission patterns of Mycobacterium bovis in sympatric cattle and badger populations. PLoS Pathog. (2012) 8: e1003008. doi: 10.1371/journal.ppat.1003008
27. Trewby H, Wright D, Breadon EL, Lycett SJ, Mallon TR, McCormick C, et al. Use of bacterial whole-genome sequencing to investigate local persistence and spread in bovine tuberculosis. Epidemics. (2016) 14:26–35. doi: 10.1016/j.epidem.2015.08.003
28. Ghebremariam MK, Hlokwe T, Rutten VPMG, Allepuz A, Cadmus S, Muwonge A, et al. Genetic profiling of Mycobacterium bovis strains from slaughtered cattle in Eritrea. PLoS Negl Trop Dis. (2018) 12:e0006406. doi: 10.1371/journal.pntd.0006406
29. Zimpel CK, Patané JSL, Guedes ACP, Souza RF, Pereira-Silva TT, Soler Camargo NC, et al. Global distribution and evolution of Mycobacterium bovis. Front Microbiol. (2020) 11:843. doi: 10.3389/fmicb.2020.00843
30. Otchere ID, van Tonder AJ, Asante-Poku A, Sánchez-Busó L, Coscollá M, Osei-Wusu S, et al. Molecular epidemiology and whole genome sequencing analysis of clinical Mycobacterium bovis from Ghana. PLoS ONE. (2019) 14:e0209395. doi: 10.1371/journal.pone.0209395
31. Salvador LCM, O'Brien DJ, Cosgrove MK, Stuber TP, Schooley AM, Crispell J, et al. Disease management at the wildlife-livestock interface: Using whole-genome sequencing to study the role of elk in Mycobacterium bovis transmission in Michigan, USA. Mol Ecol. (2019) 28:2192–205. doi: 10.1111/mec.15061
32. Zimpel CK, Brandão PE, Souza Filho AF, De Souza RF, Ykuta CY, Ferreira Neto JS, et al. Complete genome sequencing of Mycobacterium bovis SP38 and comparative genomics of Mycobacterium bovis and M. tuberculosis strains. Front Microbiol. (2017) 8:2389. doi: 10.3389/fmicb.2017.02389
33. Djelouadji Z, Arnold C, Gharbia S, Raoult D, Drancourt M. Multispacer sequence typing for Mycobacterium tuberculosis genotyping. PLoS ONE. (2008) 3:e2433. doi: 10.1371/journal.pone.0002433
34. De Azevedo Issa M, Martins Filho OS, Fonseca Júnior AA, Arrais MH, Santos LS, Reis JKP, et al. Comparative study of Mycobacterium bovis primary isolation methods. Braz J Microbiol. (2017) 48:139–44. doi: 10.1016/j.bjm.2016.07.026
35. Sales ML, Fonseca AA Jr, Orzil L, Alencar AP, Hodon MA, Issa MA, et al. Validation of two real-time PCRs targeting the PE-PGRS 20 gene and the region of difference 4 for the characterization of Mycobacterium bovis isolates. Genet Mol Res. (2014) 13:4607–16. doi: 10.4238/2014.June.18.3
36. Canevari Castelão AB, Nishibe C, Moura A, de Alencar AP, Issa MA, Hodon MA, et al. Draft genome sequence of Mycobacterium bovis strain AN5, used for production of purified protein derivative. Genome Announc. (2014) 2:e00277. doi: 10.1128/genomeA.00277-14
37. Sambrook J, Fritcsh EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd ed. New York, NY: Cold Spring Harbor Laboratory (1989). p. 3104.
38. Zanini MS, Moreira EC, Lopes MTP, Oliveira RS, Leão SC, Fioravanti RL, et al. Mycobacterium bovis: polymerase chain reaction identification in bovine lymph node biopsies and genotyping in isolates from Southeast Brazil by spoligotyping and restriction fragment length polymorphism. Mem Inst Oswaldo Cruz. (2001) 96:809–13. doi: 10.1590/S0074-02762001000600012
39. Huang X, Madan A. CAP3: A DNA sequence assembly program resource genome research. Genome Res. (1999) 9:868–77. doi: 10.1101/gr.9.9.868
40. Hall T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. (1999) 41:95–98.
41. Kamerbeek J, Schouls LEO, Kolk A, Kuijper S, Bunschoten A, Molhuizen H, et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. (1997) 35:907–14. doi: 10.1128/JCM.35.4.907-914.1997
42. Smith NH, Upton P. Naming spoligotype patterns for the RD9-deleted lineage of the Mycobacterium tuberculosis complex; www.Mbovis.org. Infect Genet Evol. (2012) 12:873–6. doi: 10.1016/j.meegid.2011.08.002
43. Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. (2004) 186:1518–30. doi: 10.1128/JB.186.5.1518-1530.2004
44. Francisco AP, Bugalho M, Ramirez M, Carriço JÁ. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinform. (2009) 10:152. doi: 10.1186/1471-2105-10-152
45. Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J Clin Microbiol. (1988) 26:2465–6. doi: 10.1128/JCM.26.11.2465-2466.1988
46. Supply P, Magdalena J, Himpens S, Locht C. Identification of novel intergenic repetitive units in a mycobacterial two-component system operon. Mol Microbiol. (1997) 26:991–1003. doi: 10.1046/j.1365-2958.1997.6361999.x
47. Frothingham R, Meeker-O'Connell WA. Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem DNA repeats. Microbiol. (1998) 144:1189–96. doi: 10.1099/00221287-144-5-1189
48. Le Flèche P, Fabre M, Denoeud F, Vergnaud G. High resolution, on-line identification of strains from Mycobacterium tuberculosis complex based on tandem repeat typing. BMC Microbiol. (2002) 2:37–48. doi: 10.1186/1471-2180-2-37
49. Gutacker MM, Mathema B, Soini HM, Shashkina E, Kreiswirth BN, Graviss EA, et al. Single-nucleotide polymorphism-based population genetic analysis of Mycobacterium tuberculosis strains from 4 geographic sites. J Infect Dis. (2006) 193:121–8. doi: 10.1086/498574
50. Djelouadji Z, Raoult D, Drancourt MA. Single-step sequencing method for the identification of Mycobacterium tuberculosis complex species. Plos Negl Trop Dis. (2008) 2:e253. doi: 10.1371/journal.pntd.0000253
51. Riojas MA, McGough KJ, Rider-Riojas CJ, Rastogi N, Hazbón MH. Phylogenomic analysis of the species of the Mycobacterium tuberculosis complex demonstrates that Mycobacterium africanum, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium microti and Mycobacterium pinnipedii are later heterotypic synonyms of Mycobacterium tuberculosis. Int J Syst Evol Microbiol. (2018) 68:324–32. doi: 10.1099/ijsem.0.002507
52. Parreiras PM, Andrade GI, Nascimento TF, Oelemann MC, Gomes HM, Alencar AP, et al. Spoligotyping and variable number tandem repeat analysis of Mycobacterium bovis isolates from cattle in Brazil. Mem Inst Oswaldo Cruz. (2012) 107:64–73. doi: 10.1590/S0074-02762012000100009
53. Rodriguez CAR, Zumárraga MJ, Oliveira EM, Cataldi AA, Romano MI, Otto HH, et al. Caracterização molecular de isolados de Mycobacterium bovis do Estado de São Paulo Brasil, utilizando a técnica de spoligotyping. Arq Inst Biolog. (2004) 71:277–82. Available online at: http://www.biologico.sp.gov.br/uploads/docs/arq/V71_3/rodriguez.PDF.
54. Zumárraga MJ, Arriaga C, Barandiaran S, Cobos-Marín L, de Waard J, Estrada-Garcia I, et al. Understanding the relationship between Mycobacterium bovis spoligotypes from cattle in Latin American countries. Res Vet Sci. (2013) 94:9–21. doi: 10.1016/j.rvsc.2012.07.012
55. Viana-Niero C, Rodriguez CAR, Bigi F, Zanini MS, Ferreira-Neto JS, Cataldi A, Leão SC. Identification of an IS6110 insertion site in plcD, the unique phospholipase C gene of Mycobacterium bovis. J Med Microbiol. (2006) 55:451–7. doi: 10.1099/jmm.0.46364-0
56. Haddad N, Ostyn A, Karoui C, Masselot M, Thorel MF, Hughes SL, et al. Spoligotype diversity of Mycobacterium bovis strains isolated in France from 1979 to 2000. J Clin Microbiol. (2001) 39:3623–32. doi: 10.1128/JCM.39.10.3623-3632.2001
57. Zanini MS, Moreira EC, Salas CE, Lopes MTP, Barouni AS, Roxo E, et al. Molecular typing of Mycobacterium bovis isolates from south-east Brazil by spoligotyping and RFLP. J Vet Med B Infect Dis Vet Public Health. (2005) 52:129–33. doi: 10.1111/j.1439-0450.2005.00835.x
58. García-Bocanegra I, Barranco I, Rodríguez-Gómez IM, Pérez B, Gómez-Laguna J, Rodríguez S, et al. Tuberculosis in alpacas (Lama pacos) caused by Mycobacterium bovis. J Clin Microbiol. (2010) 48:1960–4. doi: 10.1128/JCM.02518-09
59. Duarte EL, Domingos M, Amado A, Botelho A. Spoligotype diversity of Mycobacterium bovis and Mycobacterium caprae animal isolates. Vet Microbiol. (2008) 130:415–21. doi: 10.1016/j.vetmic.2008.02.012
60. Smith NH, Kremer K, Inwald J, Dale J, Driscoll JR, Gordon SV, et al. Ecotypes of the Mycobacterium tuberculosis complex. J Theor Biol. (2006) 239:220–5. doi: 10.1016/j.jtbi.2005.08.036
61. Smith NH. The global distribution and phylogeography of Mycobacterium bovis clonal complexes. Infect Genet Evol. (2012) 12:857–65. doi: 10.1016/j.meegid.2011.09.007
62. Milián-Suazo F, Banda-Ruíz V, Ramírez-Casillas C, Arriaga-Diaz C. Genotyping of Mycobacterium bovis by geographic location within Mexico. Prev Vet Med. (2002) 55:255–64. doi: 10.1016/S0167-5877(02)00015-6
63. Hilty M, Diguimbaye C, Schelling E, Baggi F, Tanner M, Zinsstag J. Evaluation of the discriminatory power of variable number tandem repeat (VNTR) typing of Mycobacterium bovis strains. Vet Microbiol. (2005) 109:217–22. doi: 10.1016/j.vetmic.2005.05.017
64. Comas I, Gagneux S. The past and future of tuberculosis research. PLoS Pathogens. (2009) 5:e1000600. doi: 10.1371/journal.ppat.1000600
Keywords: Mycobactereium bovis, bovine tuberculosis, genotype, epidemiology, MST
Citation: Sales ÉB, Fonseca AA Jr, Gonçalves CM, Lage AP, Andrade GI, Suffys PN, Gomes HM, Dias NL, Ferreira Neto JS, Guimarães AMdS and Heinemann MB (2021) Multispacer Sequence Typing for Mycobacterium bovis Genotyping. Front. Vet. Sci. 8:666283. doi: 10.3389/fvets.2021.666283
Received: 10 February 2021; Accepted: 30 March 2021;
Published: 26 April 2021.
Edited by:
Flabio R. Araujo, Brazilian Agricultural Research Corporation (EMBRAPA), BrazilReviewed by:
Martín José Zumárraga, National Agricultural Technology Institute (Argentina), ArgentinaEduardo Figueiredo, Federal University of Mato Grosso, Brazil
Copyright © 2021 Sales, Fonseca, Gonçalves, Lage, Andrade, Suffys, Gomes, Dias, Ferreira Neto, Guimarães and Heinemann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcos Bryan Heinemann, bWFyY29zYnJ5YW5AdXNwLmJy